

Abstract
Purpose of Review
Obesity has increased worldwide recently and represents a major global health challenge. This review focuses on the obesity-associated cellular senescence in various organs and the role of these senescent cells (SnCs) in driving complications associated with obesity. Also, the ability to target SnCs pharmacologically with drugs termed senotherapeutics as a therapy for these complications is discussed.
Recent Findings
Several studies have shown a positive correlation between obesity and SnC burden in organs such as adipose tissue, liver, and pancreatic-β-cells. These SnCs produce several secretory factors which affect other cells and tissues in a paracrine manner resulting in organ dysfunction. The accumulation of SnCs in adipocytes affects their lipid storage and impairs adipogenesis. The inflammatory senescence-associated secretory phenotype (SASP) of SnCs downregulates the antioxidant capacity and mitochondrial function in tissues. Senescent hepatocytes cannot oxidize fatty acids, which leads to lipid deposition and senescence in β-cells decrease function. These and other adverse effects of SnCs contribute to insulin resistance and type-2 diabetes. The reduction in the SnC burden genetically or pharmacologically improves the complications associated with obesity.
Summary
The accumulation of SnCs with age and disease accelerates aging. Obesity is a key driver of SnC accumulation, and the complications associated with obesity can be controlled by reducing the SnC burden. Thus, senotherapeutic drugs have the potential to be an effective therapeutic option.
Introduction
Obesity is characterized by abnormal or excessive fat accumulation in the body. A combination of hormonal, genetic, and metabolic determinants drive the obesogenic phenotype affecting major components of nutrient intake and energy expenditure [1]. At present, one-third of the global population is overweight, of which ten percent are classified as obese [2]. Obesity shortens the life span by 20 years, and it is estimated that at least 2.8 million people die every year from obesity-associated complications [3]. Like aging, obesity increases organ deterioration and accelerates aging-associated pathologies such as type-2 diabetes (T2D), cardiovascular diseases (CVD), non-alcoholic fatty liver diseases (NAFLD), and multiple types of cancers [4–6]. Obesity promotes these complications, at least in part, by inducing premature cellular senescence in various tissues [7]. Telomeres shortening, a hallmark of cellular senescence, is observed in obese children [8–12] and obese adults [13–16].
The senescent cell (SnC) burden increases during obesity in tissues such as adipose tissue (AT), pancreas, liver, and brain [17]. These SnCs secrete inflammatory factors, proteases, growth factors, and more, known as senescence-associated secretory phenotype (SASP), that can spread senescence to other cells and multiple organs [17]. Recent studies show that targeting cellular senescence, either genetically or pharmacologically, lessens obesity-related dysfunction. This review focuses on the mechanisms of obesity-induced premature senescence in different organs, the role of these SnCs in driving disease, and the therapeutic options for targeting SnCs.
Hallmarks of Cellular Senescence
Cellular senescence is an irreversible cell cycle arrest driven in response to different types of cellular stress. Senescence can be triggered by DNA damage, telomere attrition, oxidative damage, mitotic stress, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, and by oncogene activation. Although transient cellular senescence can play a beneficial role (e.g., during embryonic development, wound healing, tissue remodeling, and tumor suppression following oncogene activation), aberrant accumulation of SnCs accelerates aging and plays a causative role in age-related diseases.
DNA damage is one of the major inducers of cellular senescence. DNA double-strand breaks (DSBs) activate the DNA-damage response (DDR) pathway, which increases the levels of phosphorylated histone H2AX, p53 binding protein 1 (53BP1) in the chromatin as well as activation of DDR signaling kinases ATM, ATR, CHK1, and CHK2 which eventually activates the p53/p21CIP1 axis [18]. p53 induces the expression of a cyclin-dependent kinase inhibitor, p21CIP1, which results in inhibition of cyclin-dependent kinase-2 (CDK2) activity and cell-cycle exit [19]. p21CIP1 also functions through cell cycle-independent pathways to regulate signal transduction pathways. Persistent activation of the DDR pathway also increases the expression of p16INK4a, an inhibitor of CDK4 and CDK6, contributing to long-lasting growth arrest [19]. The DDR pathway can be triggered by either intrinsic (e.g., oxidative stress, telomere shortening, oncogene activation) or external factors (e.g., UV-, γ- irradiation, chemotherapeutics) [20].
Most SnCs develop an inflammatory SASP that can confer adverse effects through paracrine and endocrine pathways. The SASP includes cytokines, chemokines, extracellular matrix proteases, growth factors, extracellular vesicles, and other signaling molecules [21]. The activity of the transcription factors NF-κB, CCAAT/enhancer-binding protein-β (C/EBPβ), and GATA4 is increased with senescence, binding to the promoters of SASP genes to upregulate their transcription [22–24]. The JAK-STAT3 pathway [25], mTOR pathway [26], bromodomain-containing protein 4 (BRD4) [27], NOTCH1 activity [28], and HMGB proteins [29, 30] are also reported to modulate the composition of SASP with HMGB1 being secreted by SnCs are part of the SASP. In addition, the pro-survival factors are upregulated in SnCs, termed senescent cell anti-apoptotic pathways (SCAPs), leading to SnCs becoming apoptotic resistant. However, SnCs also upregulate certain pro-apoptotic genes, making them prone to apoptosis following treatment with drugs targeting the SCAPs [31].
Since SnCs chronically express the SASP, they are metabolically active with a high AMP: ATP ratio [32]. Reduction in intracellular ATP levels leads to allosteric activation of AMPK, which regulates cellular energy metabolism by activating energy-producing pathways and inhibiting energy-consuming pathways [33]. Though the increased activity of AMPK has been reported in senescent endothelial cells [34], recent studies suggest that activation of AMPK can reduce senescence in AT stromal cells [35] and protects against diet-induced obesity [36]. This suggests that the role of AMPK and other signaling pathways in SnC may vary with the cell type.
Mitochondrial dysfunction-associated senescence (MiDAS) is driven by attenuated electron transport chain NADH oxidation leading to increased cytosolic NADH levels. The lowering NAD+ /NADH ratio inhibits a key glycolytic enzyme, GAPDH, thereby blunting substrate-level phosphorylation resulting in lower ATP levels. This subsequently causes AMPK activation and cell cycle arrest [37]. SnCs have increased ER stress and unfolded protein response (UPR) leading to the export of misfolded proteins, enlargement of ER, and reduction in protein synthesis [38]. A contributing factor to the increased demand for SASP production is UPR [39]. Another feature of SnCs is increased cell size and altered morphology, partially driven by increased mTOR activity. The activation of the mTOR pathway during cellular senescence induces hypertrophy in endothelial cells [40]. The rearrangement of vimentin filaments also contributes to the altered morphology in SnCs [41]. Elevated lysosomal proteins are another characteristic feature of SnCs, particularly the lysosomal enzyme senescence-associated β-galactosidase (SA-β-gal) [42, 43]. SnCs also display reduced mitochondrial fission and increased fusion [44], which contributes to the increased accumulation of old mitochondria and protects the cell from apoptosis [44]. Loss of lamin B, a structural protein of nuclear lamina, is another marker of cellular senescence [45]. These features of SnCs promote multiple organelle dysfunctions and in turn drive aging hallmarks such as chronic inflammation, proteostatic dysfunction, nutrient signaling disruption, and stem cell exhaustion.
Obesity-associated Senescence and Organ Disorders
Senescence in Adipose Tissue and Impaired Adipogenesis
Adipose tissue (AT) is a complex and metabolically active endocrine organ. There are three distinct types of adipose tissues: brown adipose tissue (BAT), white adipose tissue (WAT) [46], and beige adipose tissue [47]. Through a process called thermogenesis, BAT converts chemical energy into heat energy and serves an important role in maintaining body temperature. WAT stores excess fatty acids (FA) into triglycerides (TG) and acts as a major energy reservoir. Beige adipose tissue is inducible and can function to dissipate energy under conditions of excess demand such as thermal challenge. WAT is subdivided into visceral adipose tissue (VAT) and subcutaneous adipose tissue (SCAT). VAT is located around the internal organs and represents 20% of total fat. SCAT locates under the skin and is accountable for storing 80% of total fat [6]. AT is comprised of mature adipocytes, preadipocytes, endothelial cells, fibroblasts, and immune cells [48]. Adipocytes are the most prevalent cell type in the AT and are responsible for lipogenesis. During an abundant energy supply, the lipids are stored in the form of TG [48]. Adipocytes secrete various proteins and other molecules that are collectively known as adipokines or adipocytokines [49]. In AT, adipokines modulate adipogenesis, immune cell migration, and adipocyte metabolism and function. Adipokines are also involved in regulating systemic actions in the brain, liver, muscle, vasculature, heart, and pancreas [49]. Beige and brown fat also secrete a variety of signaling factors, many of which are small molecule metabolites and participate in inter-organ signaling [47].
Obesity alters AT size, function, cellular composition, and distribution. The hypertrophic expansion of adipocytes elevates proinflammatory adipokines [50]. Macrophage infiltration is a causative factor for insulin resistance and T2D in obese subjects. Macrophages infiltrating the AT surround necrotic adipocytes and form crown-like structures. In obese conditions, the proinflammatory M1 macrophage subpopulation increases, whereas the anti-inflammatory M2 population diminishes. This imbalance between M1 and M2 macrophages may promote a pro-inflammatory state. AT inflammation results in dysregulated adipocytokine production and reduced AT lipid storing ability, thereby inducing ectopic lipid accumulation and insulin resistance in peripheral tissues [51].
An increase in cellular senescence and inflammation of VAT [52–55] and SCAT [55, 56] is observed in the obese animal model and humans. Excessive caloric intake in mice leads to increased reactive oxygen species (ROS) [57] and ATP production [58], which triggers the senescence of AT. Elevated ROS activates p53/p21 pathway and promotes senescent phenotype and the SASP, including secretion of TNFα and IL6 [57]. Increased secretion of SASP attracts immune cells leading to low-grade inflammation and decreased formation of white and beige adipocytes. AT with a high SnC burden shows impaired lipid handling, impaired thermogenesis, insulin resistance, and aberrant adipokine production.
Senescent Preadipocytes
Preadipocytes expand via hyperplasia and differentiate to mature adipocytes by a process called adipogenesis. This is essential to sequester lipids efficiently to avoid lipotoxicity in other tissues, such as the liver, muscle, and heart [59]. Adipocyte size is inversely associated with the polyunsaturated FA composition in fat depots. In contrast, AT and dietary saturated FA positively correlated with adipocyte size and number [60]. A recent study suggests that treating human preadipocytes with saturated FA such as palmitic acid results in hypertrophy, increased DNA damage, and p16INK4a positive nuclei [61].
Impaired adipogenesis in SCAT is a major contributor to obesity-associated metabolic complications [62]. Though the number of adipogenic progenitor cells is not reduced, the increased senescent progenitor cell impairs adipogenesis in SCAT of humans with hypertrophic obesity [63]. Activation of two critical signaling pathways, the p53/p21CIP1 and p16INK4a/pRb tumor suppressor pathways, contributes to cellular senescence in adipogenic progenitor stem cells [64]. The activity of key adipogenic transcription factors, such as PPARγ2 and C/EBPα, was reduced in senescent preadipocytes derived from human abdominal subcutaneous fat pads [64]. Oxidative phosphorylation and glycolysis were increased in preadipocytes derived from both SCAT and VAT of HFD-fed mice, along with ATP overproduction [58]. Cellular senescence of progenitor cells also contributes to impaired adipogenesis, TG storage, and adipokine secretion [64]. Primary human senescent fat progenitors secrete high levels of activin-A and directly inhibit adipogenesis in non-senescent progenitors [65]. In addition, the JAK/STAT pathway plays an important role in the SASP [66], with inhibition of the JAK pathway suppressing SASP production and improving adipogenesis in old mice [65, 67].
Senescence of Matured Adipocytes
Adipocyte senescence plays a causative role developing obesity and insulin resistance in DNA repair enzyme-defective mouse models [68]. Obese patients with hyperinsulinemia show an increased p16INK4a and SA-β-gal activity in mature adipocytes. These senescent adipocytes are hypertrophic and have elevated SASP factors (e.g., IL-6, IL-8, and MCP1) secretion [69]. Upregulation of p53 in adipose tissue triggers cellular senescence and insulin resistance [57]. Treatment with a p53 inhibitor and adipocyte-specific ablation of p53 effectively reduces obesity-induced cellular senescence and its associated insulin resistance [68].
Senescence of Endothelial Cells in Adipose Tissue
FA stored in AT needs to be transported, and endothelium cells (EC) play an essential role in the lipid handling. In addition, interactions between EC and adipocytes are vital to maintaining WAT homeostasis [70]. A recent study stated that polyamines produced by EC stimulate adipocyte lipolysis and maintain WAT homeostasis [71]. Adipocytes secrete both pro-angiogenic and anti-angiogenic factors with a balance between pro-angiogenic and anti-angiogenic molecules required for normal angiogenesis. However, in obesity, the expansion of AT affects tissue vascularization. The hypoxic condition created by the AT expansion induces inadequate vessel maintenance/growth and inflammation [70]. Hypoxia-related gene expressions were higher in VAT than in SCAT adipocytes isolated from obese subjects. In addition, VAT displays higher vascular density and EC abundance, along with a higher number of SA-β-gal positive cells. Thus, the chronic pro-angiogenic and pro-inflammatory microenvironment may accelerate the VAT-EC premature senescence resulting in endothelial dysfunction [72]. PPARγ (peroxisome proliferator-activated receptor-gamma) regulates FA uptake by regulating the transcription of FA transporters and certain FA binding proteins (e.g., FATP1, CD36, and FABP4). Cellular senescence modulates the activity of PPARγ. The nuclear translocation of PPARγ was impaired, and the positive regulation of FA transport by PPARγ agonists was abolished in senescent cells, resulting in AT dysfunction and redistribution of lipids [73].
Senescence of Immune Cells in Adipose Tissue
Excessive stimulation of AT by diet induces inflammation that can take its toll on endogenous immune cells. As discussed previously, high caloric diet consumption can lead to obesity and insulin resistance. The excessive consumption of nutrients induces a process that initiates an inflammatory response that left unresolved is deleterious to normal tissue function. This process involves the production of PPARγ through the stimulation of CD36 both on adipocytes and endogenous immune cells [74–76]. The in-situ production of inflammatory cytokines like TNF-α and chemokines like MCP-1 leads to increased macrophages (MF) either through the conversion of circulating monocytes into MF or the proliferation of endogenous MF [77, 78].
Insulin was shown to induce the activation of mTORC2 through which RICTOR induces the production of MCP-1. It has been demonstrated that insulin resistance precedes the onset of inflammation. Mouse models of high-fat–diet-induced obesity, adipose targeted mTORC2, and RICTOR depleted mice were used to demonstrate that insulin resistance is established before the accumulation of MF and onset of inflammation with the accumulation of MF is driven through MCP-1 [78]. A reduction of insulin/mTORC2 signaling triggers MCP-1 expression, which results in inflammation characterized by the accumulation of pro-inflammatory MF. Obesity-induced inflammation can switch the functional phenotype from an anti-inflammatory (M2) into a pro-inflammatory (M1) phenotype [79]. These polarization states represent opposite ends of an activation spectrum, and it is now recognized that macrophages in vivo are exposed to multiple factors within the local microenvironment and thus have a more diverse phenotype [80, 81]. Obesity-associated inflammation has also been shown to affect the function of other immune cells. In a clinical study, patients between 40 and 55 years of age with a body mass index (BMI) greater than 35 kg/m2 showed the presence of B cells characterized by a SASP in adipose tissue biopsies, which were compared to bloodborne B cells [82]. These B cells expressed a higher transcriptional level of senescent markers p16INK4a, p21CIP1, and p53 as compared to B cells from the blood. Similar to T cells, an increase of B cells exhibiting a memory phenotype appears in the AT with a simultaneous loss of naïve B cells. Naturally, aged mice show an increase in B cells. In addition, impaired maturation of B cells linked to Oct coactivator expression is dependent on the NLRP3-induced inflammasome activation while impacting the metabolic health of surrounding cells [83, 84]. Despite the phenotype of B cells, the expression of B-cell-activating factor (BAFF) was linked with aging-associated insulin resistance in 10-month-old mice [85]. BAFF KO mice gained less weight and exhibited tolerances to insulin and glucose. Thus, there appears that antigen presentation and cytokine and antibody production by B cells plays a key role in diabetes [86].
The inflammatory SASP factors TNFα, IFNγ, and IL-1β each have the capacity to downregulate numerous genes in adipocytes, most notably pathways linked to mitochondrial function and oxidative stress control [87]. Indeed, inflammatory cytokines downregulate several key antioxidants such as Gpx4, Gsta4, Prdx3, and Aldh2 that in sum result in defective electron transport and mitochondrial dysfunction [88]. Concomitant elevation in ROS in turn leads to broad impacts, including attenuated insulin signaling, increased lipid oxidation, and ER stress.
Significant changes occur to T cells in response to obesity and aging. A study in which mice were placed on a HFD for 14 weeks drove the accumulation of CD44hiCD62Llo T cells that co-expressed PD-1 and CD153, a member of the TNF superfamily [89]. These T cells accumulate in crown-like structures in VAT. Further analysis demonstrated that a subset of these cells develops a senescent signature characterized by staining for SA-β-gal staining and the DNA damage marker a significant amount of osteopontin (OPN). In a cohort of T2D patients, there was an expansion of effector T cells expressing the memory marker CD45RA (EMRA T cells), similar to that was seen in mice [90]. Similarly, there is an abundance of regulatory CD4+ T cells in AT of lean mice but reduced in AT of obese mice [91].
Furthermore, PPARγ expression in Tregs maintains not only the number of these cells but also insulin sensitivity in AT [92]. Collectively, AT Tregs appear to play a positive role in both maintaining tolerance and insulin sensitivity [93]. Other immune cells have been shown to contribute to AT homeostasis by proper surveillance and deletion of SnCs. Mice fed a normal caloric diet showed that natural killer (NK) T cells help to maintain a normal level of SnCs in AT compared to mice fed an HFD, which accumulated more SnCs at the same age and duration of analysis [94]. Targeted activation of NKT cells helps limit the number of SnCs. Thus, a properly functioning immune system not only helps limit the accumulation of SnCs in situ but also maintains the normal function of peripheral tissues.
Senescence in Liver and Non-Alcoholic Fatty Liver Disease
The liver is a key metabolic organ and an accessory digestive organ that synthesizes and secretes many molecules that have endocrine, exocrine (digestive), and clotting functions [95]. It comprises hepatocytes, hepatic stellate cells (HSC), Kupffer cells, and liver sinusoidal endothelial cells [96]. Obesity increases the SnC accumulation in the liver [52, 97, 98]. The increased p16INK4a and p21CIP1 levels in the liver are associated with elevated triacylglycerol accumulation in obese rats. The severity of NAFLD is increased with SnC accumulation [98]. Induction of hepatocyte senescence in mice with liver-specific deletion of the DNA repair protein XPG results in increased fat deposition [97]. Patients with NAFLD displayed elevated p21Cip1, shorter hepatocyte telomere length, and a higher hepatocyte nuclear area which are indicators of senescence [99]. Mitochondria of senescent hepatocytes show the inability to oxidize fatty acid, resulting in increased fat deposition [97]. Post-translational modifications, especially acetylation, regulate many enzymes in metabolic pathways including, gluconeogenesis, TCA cycle, and β-oxidation. This acetylation is partly regulated by sirtuins (SIRT) class III NAD+ -dependent deacetylases (HDACs). SIRT3 is a mitochondrial sirtuin that regulates lipid and glucose metabolism in the liver [100]. Reduced SIRT3 activity and NAD+ levels contribute to impaired FA and lipid accumulation in the liver of obese mice [101].
Senescence in Pancreatic-β-cells and β-cell Dysfunction
The pancreatic-β-cells are endocrine cells that synthesize and secrete insulin depending on blood glucose concentration. The function of pancreatic-β-cell plays a critical role in determining whether people with obesity develop T2D. Obese individuals show increased β-cell mass [102, 103] as well as increased pancreatic fat content [104]. In addition, both plasma insulin concentrations and β-cell insulin secretion are increased in basal and postprandial conditions in obese populations who do not have T2D [105]. In contrast, T2D subjects show a loss of functional β-cell mass [106]. The increase in plasma insulin level and insulin secretion rate is often able to overcome the insulin resistance and thus maintains the fasting blood glucose and glucose tolerance. A gradual decrease in β-cell function causes a progressive decline in glycemic control, resulting in prediabetes and eventually T2D [107]. HFD mice exhibit increased cellular senescence and decreased β-cell proliferation [108]. The decrease in Ki-67-positive β-cells and increased SA-β-gal-positive area were detected in these mice on HFD. Interestingly, the SA-β-gal positive area negatively correlates with insulin levels. Moreover, p38, a mediator of cellular senescence, was found in β-cells of HFD animals [108]. Aging and SASP indices were elevated along with SA-β-gal positive cell accumulation[109]. Senescent β-cells exhibit downregulation of hallmark identity genes and upregulation of non-identify genes including proinflammatory cytokines [110]. These inflammatory pathway proteins diminish β-cell-specific transcriptional factors and insulin secretion. The long-term accumulation of senescent β-cells induces metabolic dysfunction through loss of β-cell mass and function [110], which impairs glucose tolerance and leads to insulin resistance.
Insulin Resistance—a Metabolic Consequence of Obesity-induced Senescence
The excessive fat accumulation in the body induces many metabolic abnormalities, including insulin resistance, NAFLD, β-cell dysfunction, and T2D. Obesity increases the prevalence of T2D by influencing both insulin action and β-cell function. Insulin regulates blood glucose level by promoting protein and glycogen synthesis in muscle and liver, facilitating lipid synthesis and storage in AT and liver, and by inhibiting glycogenolysis, gluconeogenesis, and FA oxidation in insulin-responsive tissues. Excessive lipid accumulation in tissues other than AT is known as ectopic lipid accumulation, which is the major contributor of insulin resistance.
Obesity triggers senescence in pre-adipocytes as well as matured adipocytes resulting in dysfunctional adipocytes and impaired adipogenesis [111]. Dysfunctional adipocytes generate more inflammatory cytokines and adipokines such as TNF-α, IL-6, leptin, visfatin, resistin, angiotensin II, and PAI-1 than healthy adipocytes [112]. Plasma PAI-1 levels were higher in the obese insulin-resistant group and were inversely correlated with hepatic and skeletal muscle insulin sensitivity [113]. TNF-α and IL-6, abate adipocyte differentiation, reduces lipogenesis, and increases lipolysis [114]. This redirects lipids toward peripheral tissues, including the pancreas, liver, skeletal muscle, and heart.
The oversupply of free fatty acids (FFAs) and their metabolites like long-chain acyl-CoA (LC-CoA), diacylglycerol (DAG), and ceramides are deleterious to the cell [114]. These FFA metabolites activate serine/threonine kinases such as protein kinase C (PKC) isoforms, c-Jun N-terminal kinase (JNK) pathway, and I-kappa B kinase β (IKKβ)/NF-κB pathway [115, 116]. In skeletal muscle, serine-phosphorylated forms of insulin receptor substrate-1 (IRS1) cannot activate its downstream molecules, subsequently leading to decreased translocation of glucose transporter-4 (GLUT4) vesicles to the cell membrane [117]. People with obesity and T2D have impaired insulin-stimulated glucose oxidation and glycogen synthesis due to defects in insulin receptor and post-receptor insulin signaling [118]. In the liver, DAG accumulation activates PKCε, which reduces IRS2 tyrosine phosphorylation. This impairs insulin-stimulated glycogen synthase activity and reduces the ability of insulin to suppress gluconeogenesis [119, 120]. The increase in FFAs also can have harmful effects on β-cells, for example, prolonged infusion of FFAs decreases glucose-stimulated insulin secretion [121]. The combined deleterious effects of increased FFAs and glucose on β-cell survival and function is termed as glucolipotoxicity, which induces ER stress, mitochondrial dysfunction, and oxidative damage, resulting in β-cell dysfunction and death [122].
Senotherapeutics—a Novel Therapeutic Strategy for Obesity
Senotherapeutics selectively kill SnCs (senolytics) or suppress the SASP (senomorphics), to extend healthspan and lifespan. Accumulation of p16high cells is higher in the pancreas and p21high cells are higher in VAT [109, 123]. Clearance of p16high cells improves glucose metabolism, insulin secretion, and decreased expression of aging, senescence, and SASP genes in islets of obese mice [109]. Targeting p21CIP1 highly expressing cells using the senolytic cocktail of dasatinib plus quercetin (D + Q) improves glucose tolerance and insulin sensitivity in obese mice [123]. This D + Q cocktail also removes p21high cells ex vivo in VAT explants from humans with obesity and alleviates insulin resistance following xenotransplantation into mice [123].
Removal of SnCs using the senolytic Bcl-2 inhibitor ABT263 reduced hyperglycemia and improved β-cell gene expression in aged mice [109]. Freshly isolated human omental adipose tissue obtained from obese individuals treated with D + Q had significantly less TAF+, p16INK4A, and SA-βgal+ cells as well as reduced proinflammatory cytokines IL6, IL8, MCP1, and PAI1. The treatment also improved PPARγ and CEBPα, which are essential transcription factors for adipogenesis [124]. Treatment of the D + Q cocktail in AT explants prepared from SCAT biopsies, restored the elevated SA-β-gal activity, and reduced the release of leptin, PAI1, IGFBP3, CCL2, and IL-6 [56]. Thus, targeting SnCs improves adipogenesis, and β-cell function and thereby reduces hyperglycemia.
Conclusion
Cellular senescence is a crucial driver for obesity-associated complications. They create an inflammatory microenvironment that could damage tissues and causes dysfunction (Fig. 1). SnCs accumulate in both subcutaneous and visceral AT. AT inflammation results in abnormal adipokine production and directly inhibits adipogenesis in non-senescent progenitors. Consequently, lipid deposition occurs in peripheral tissues and results in insulin resistance. Recent studies exhibit the beneficial effects of senolytics. Targeting SnCs genetically or pharmacologically effectively improves adipogenesis, reduces hyperglycemia, and improves glucose tolerance and insulin sensitivity. Thus, treatment with a senotherapeutic could be a beneficial approach to restoring obesity-associated complications.
Fig. 1.
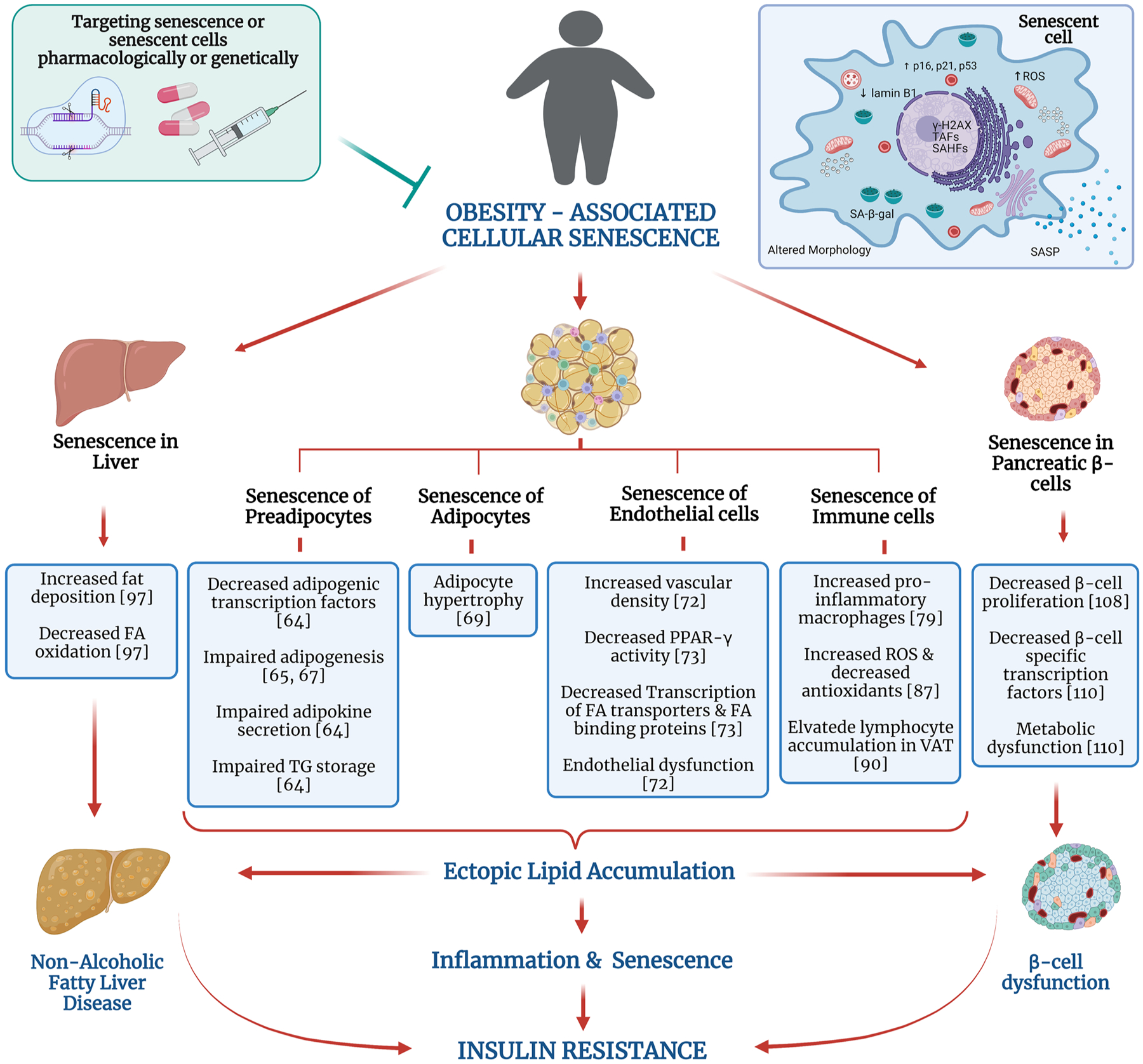
Obesity-associated cellular senescence. Cellular senescence is an irreversible cell cycle arrest with senescent cells (SnCs) displaying characteristics such as elevated expression of p16INK4a, p21CIP1, p53 proteins, increased lysosomal senescence associated β-galactosidase activity (SA-β-gal), altered morphology, decreased lamin B1, elevated ROS levels, and senescence-associated secretory phenotype (SASP) production. Obesity is associated with accumulation of SnCs in various organs such as liver, adipose tissue (AT), and pancreatic β-cells and plays a major role in driving obesity-associated complications. SnC accumulation in AT impairs their ability to store lipids, elevates pro-inflammatory macrophages, and impairs adipogenesis process. Improper lipid storage in AT leads to fat deposition in other organs. Senescent liver cells loss the ability to oxidize fatty acids, resulting in excessive lipid accumulation. Elevated senescent burden in pancreatic β-cells decreases its proliferation capacity and causes metabolic dysfunction. Altogether, these affect the insulin-signaling pathway and glucose homeostasis. Thus, obesity-associated cellular senescence is a major contributor to the development of insulin resistance and impaired glucose tolerance. Targeting the SnCs either genetically or pharmacologically reduces the complications associated with obesity. Thus, senotherapeutic drugs may be an effective therapeutic option. Abbreviations: FA, fatty acid; ROS, reactive oxygen species; SASP, senescence-associated secretory phenotype; SAHF, senescence-associated heterochromatin foci; TAF, telomere-associated foci. Figure created with Biorender.com
Funding
This work was supported by NIH grants RO1 AG063543-02S1, P01 AG043376, U19 AG056278, RO1 AG063543, P01 AG062413, U54 AG076041, R01 AG069819, R01 AG063543-S1, and R00 AG058800; an Aligning Science Across Parkinson’s grant ASAP-000592 from the Michael J. Fox Foundation; and the Glenn Foundation for Medical Research.
Footnotes
Competing Interests L.J.N. and P.D.R. have a financial interest related to this research. The other authors declare they have no financial interest.
References:
- 1.Cheng L, Wang J, Dai H, Duan Y, An Y, Shi L, et al. Brown and beige adipose tissue: a novel therapeutic strategy for obesity and type 2 diabetes mellitus. Adipocyte. 2021;10(1):48–65. 10.1080/21623945.2020.1870060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheryl D MDC Fryar, and Ogden Cynthia L.. Prevalence of overweight, obesity, and severe obesity among adults aged 20 and over: United States, 1960–1962 through 2017–2018. NCHS Health E-Stats. 2020. [Google Scholar]
- 3.Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond). 2008;32(9):1431–7. 10.1038/ijo.2008.102. [DOI] [PubMed] [Google Scholar]
- 4.Kawai T, Autieri MV, Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol. 2021;320(3):C375–91. 10.1152/ajpcell.00379.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberto CA, Swinburn B, Hawkes C, Huang TT, Costa SA, Ashe M, et al. Patchy progress on obesity prevention: emerging examples, entrenched barriers, and new thinking. Lancet. 2015;385(9985):2400–9. 10.1016/s0140-6736(14)61744-x. [DOI] [PubMed] [Google Scholar]
- 6.Santos AL, Sinha S. Obesity and aging: molecular mechanisms and therapeutic approaches. Ageing Res Rev. 2021;67:101268. 10.1016/j.arr.2021.101268. [DOI] [PubMed] [Google Scholar]
- 7.Burton DGA, Faragher RGA. Obesity and type-2 diabetes as inducers of premature cellular senescence and ageing. Biogerontology. 2018;19(6):447–59. 10.1007/s10522-018-9763-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin L, Qin K, Chen D, Lu C, Chen W, Guo VY. Systematic review and meta-analysis of the association between paediatric obesity and telomere length. Acta Paediatr. 2021;110(10):2695–703. 10.1111/apa.15971. [DOI] [PubMed] [Google Scholar]
- 9.Al-Attas OS, Al-Daghri N, Bamakhramah A, Shaun Sabico S, McTernan P, Huang TT. Telomere length in relation to insulin resistance, inflammation and obesity among Arab youth. Acta Paediatr. 2010;99(6):896–9. 10.1111/j.1651-2227.2010.01720.x. [DOI] [PubMed] [Google Scholar]
- 10.Clemente DBP, Maitre L, Bustamante M, Chatzi L, Roumeliotaki T, Fossati S, et al. Obesity is associated with shorter telomeres in 8 year-old children. Sci Rep. 2019;9(1):18739. 10.1038/s41598-019-55283-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamprokostopoulou A, Moschonis G, Manios Y, Critselis E, Nicolaides NC, Stefa A, et al. Childhood obesity and leucocyte telomere length. Eur J Clin Invest. 2019;49(12):e13178. 10.1111/eci.13178. [DOI] [PubMed] [Google Scholar]
- 12.Licea-Cejudo RC, Arenas-Sandoval LK, Salazar-León J, Martínez-Martínez MV, Carreón-Rodríguez A, Pedraza-Alva G, et al. A dysfunctional family environment and a high body fat percentage negatively affect telomere length in Mexican boys aged 8–10 years. Acta Paediatr. 2020;109(10):2091–8. 10.1111/apa.15234. [DOI] [PubMed] [Google Scholar]
- 13.Welendorf C, Nicoletti CF, Pinhel MAS, Noronha NY, de Paula BMF, Nonino CB. Obesity, weight loss, and influence on telomere length: new insights for personalized nutrition. Nutrition. 2019;66:115–21. 10.1016/j.nut.2019.05.002. [DOI] [PubMed] [Google Scholar]
- 14.García-Calzón S, Moleres A, Marcos A, Campoy C, Moreno LA, Azcona-Sanjulián MC, et al. Telomere length as a biomarker for adiposity changes after a multidisciplinary intervention in over-weight/obese adolescents: the EVASYON study. PLoS One. 2014;9(2):e89828. 10.1371/journal.pone.0089828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee M, Martin H, Firpo MA, Demerath EW. Inverse association between adiposity and telomere length: the fels longitudinal study. Am J Hum Biol. 2011;23(1):100–6. 10.1002/ajhb.21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rode L, Nordestgaard BG, Weischer M, Bojesen SE. Increased body mass index, elevated C-reactive protein, and short telomere length. J Clin Endocrinol Metab. 2014;99(9):E1671–5. 10.1210/jc.2014-1161. [DOI] [PubMed] [Google Scholar]
- 17.Palmer AK, Jensen MD, Tchkonia T, Kirkland JL. Chapter 11 - Senescence in obesity: causes and consequences. In: Serrano M, Muñoz-Espín D, editors. Cellular senescence in disease. Academic Press; 2022. p. 289–308. [Google Scholar]
- 18.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426(6963):194–8. 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 19.Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. Embo j. 2003;22(16):4212–22. 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.d’Adda di Fagagna F Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8(7):512–22. 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 21.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. 10.1146/annurevpathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–31. 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 23.Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006–18. 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 24.Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349(6255):aaa5612. 10.1126/science.aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9(1):75–89. 10.1016/j.celrep.2014.08.044. [DOI] [PubMed] [Google Scholar]
- 26.Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17(9):1205–17. 10.1038/ncb3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 2016;6(6):612–29. 10.1158/2159-8290.Cd-16-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoare M, Ito Y, Kang TW, Weekes MP, Matheson NJ, Patten DA, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol. 2016;18(9):979–92. 10.1038/ncb3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aird KM, Iwasaki O, Kossenkov AV, Tanizawa H, Fatkhutdinov N, Bitler BG, et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol. 2016;215(3):325–34. 10.1083/jcb.201608026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, et al. p53-dependent release of alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201(4):613–29. 10.1083/jcb.201206006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Pitcher LE, Prahalad V, Niedernhofer LJ, Robbins PD. Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. Febs j. 2022. 10.1111/febs.16350. [DOI] [PubMed] [Google Scholar]
- 32.James EL, Michalek RD, Pitiyage GN, de Castro AM, Vignola KS, Jones J, et al. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J Proteome Res. 2015;14(4):1854–71. 10.1021/pr501221g. [DOI] [PubMed] [Google Scholar]
- 33.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472(7342):230–3. 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zu Y, Liu L, Lee MY, Xu C, Liang Y, Man RY, et al. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ Res. 2010;106(8):1384–93. 10.1161/circresaha.109.215483. [DOI] [PubMed] [Google Scholar]
- 35.Le Pelletier L, Mantecon M, Gorwood J, Auclair M, Foresti R, Motterlini R, et al. Metformin alleviates stress-induced cellular senescence of aging human adipose stromal cells and the ensuing adipocyte dysfunction. Elife. 2021;10. 10.7554/eLife.62635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pollard AE, Martins L, Muckett PJ, Khadayate S, Bornot A, Clausen M, et al. AMPK activation protects against diet induced obesity through Ucp1-independent thermogenesis in subcutaneous white adipose tissue. Nat Metab. 2019;1(3):340–9. 10.1038/s42255-019-0036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23(2):303–14. 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, Zhu H, Yan X, Gu H, Gu Z, Liu F. Endoplasmic reticulum stress participates in the progress of senescence and apoptosis of osteoarthritis chondrocytes. Biochem Biophys Res Commun. 2017;491(2):368–73. 10.1016/j.bbrc.2017.07.094. [DOI] [PubMed] [Google Scholar]
- 39.Cormenier J, Martin N, Deslé J, Salazar-Cardozo C, Pourtier A, Abbadie C, et al. The ATF6α arm of the unfolded protein response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E(2) intracrine pathway. Mech Ageing Dev. 2018;170:82–91. 10.1016/j.mad.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Bent EH, Gilbert LA, Hemann MT. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016;30(16):1811–21. 10.1101/gad.284851.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hwang ES, Yoon G, Kang HT. A comparative analysis of the cell biology of senescence and aging. Cell Mol Life Sci. 2009;66(15):2503–24. 10.1007/s00018-009-0034-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5(2):187–95. 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 43.Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113(Pt 20):3613–22. [DOI] [PubMed] [Google Scholar]
- 44.Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine. 2017;21:7–13. 10.1016/j.ebiom.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sadaie M, Salama R, Carroll T, Tomimatsu K, Chandra T, Young AR, et al. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2013;27(16):1800–8. 10.1101/gad.217281.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen P, Spiegelman BM. Cell biology of fat storage. Mol Biol Cell. 2016;27(16):2523–7. 10.1091/mbc.E15-10-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitehead A, Krause FN, Moran A, MacCannell ADV, Scragg JL, McNally BD, et al. Brown and beige adipose tissue regulate systemic metabolism through a metabolite interorgan signaling axis. Nat Commun. 2021;12(1):1905. 10.1038/s41467-021-22272-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsiloulis T, Watt MJ. Exercise and the regulation of adipose tissue metabolism. Prog Mol Biol Transl Sci. 2015;135:175–201. 10.1016/bs.pmbts.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 49.Fasshauer M, Blüher M. Adipokines in health and disease. Trends Pharmacol Sci. 2015;36(7):461–70. 10.1016/j.tips.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 50.Skurk T, Alberti-Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab. 2007;92(3):1023–33. 10.1210/jc.2006-1055. [DOI] [PubMed] [Google Scholar]
- 51.Suganami T, Tanaka M, Ogawa Y. Adipose tissue inflammation and ectopic lipid accumulation. Endocr J. 2012;59(10):849–57. 10.1507/endocrj.ej12-0271. [DOI] [PubMed] [Google Scholar]
- 52.Shirakawa K, Yan X, Shinmura K, Endo J, Kataoka M, Katsumata Y, et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J Clin Invest. 2016;126(12):4626–39. 10.1172/jci88606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khan S, Chan YT, Revelo XS, Winer DA. The immune landscape of visceral adipose tissue during obesity and aging. Front endocrinol Lausanne. 2020;11:267. 10.3389/fendo.2020.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamada T, Kamiya M, Higuchi M, Nakanishi N. Fat depot-specific differences of macrophage infiltration and cellular senescence in obese bovine adipose tissues. J Vet Med Sci. 2018;80(10):1495–503. 10.1292/jvms.18-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alessio N, Acar MB, Demirsoy IH, Squillaro T, Siniscalco D, Bernardo GD, et al. Obesity is associated with senescence of mesenchymal stromal cells derived from bone marrow subutaneous and visceral fat of young mice. Aging Albany NY. 2020;12(13):12609–21. 10.18632/aging.103606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rouault C, Marcelin G, Adriouch S, Rose C, Genser L, Ambrosini M, et al. Senescence-associated β-galactosidase in subcutaneous adipose tissue associates with altered glycaemic status and truncal fat in severe obesity. Diabetologia. 2021;64(1):240–54. 10.1007/s00125-020-05307-0. [DOI] [PubMed] [Google Scholar]
- 57.Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15(9):1082–7. 10.1038/nm.2014. [DOI] [PubMed] [Google Scholar]
- 58.Pini M, Czibik G, Sawaki D, Mezdari Z, Braud L, Delmont T, et al. Adipose tissue senescence is mediated by increased ATP content after a short-term high-fat diet exposure. Aging Cell. 2021;20(8):e13421. 10.1111/acel.13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hafidi ME, Buelna-Chontal M, Sánchez-Muñoz F, Carbó R. Adipogenesis: a necessary but harmful strategy. Int J Mol Sci. 2019;20(15):3657. 10.3390/ijms20153657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garaulet M, Hernandez-Morante JJ, Lujan J, Tebar FJ, Zamora S. Relationship between fat cell size and number and fatty acid composition in adipose tissue from different fat depots in overweight/obese humans. Int J Obes (Lond). 2006;30(6):899–905. 10.1038/sj.ijo.0803219. [DOI] [PubMed] [Google Scholar]
- 61.Ishaq A, Tchkonia T, Kirkland JL, Siervo M, Saretzki G. Palmitate induces DNA damage and senescence in human adipocytes in vitro that can be alleviated by oleic acid but not inorganic nitrate. Exp Gerontol. 2022;163:111798. 10.1016/j.exger.2022.111798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hammarstedt A, Gogg S, Hedjazifar S, Nerstedt A, Smith U. Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol Rev. 2018;98(4):1911–41. 10.1152/physrev.00034.2017. [DOI] [PubMed] [Google Scholar]
- 63.Gustafson B, Nerstedt A, Smith U. Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nat Commun. 2019;10(1):2757. 10.1038/s41467-019-10688-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mitterberger MC, Lechner S, Mattesich M, Zwerschke W. Adipogenic differentiation is impaired in replicative senescent human subcutaneous adipose-derived stromal/progenitor cells. J Gerontol A Biol Sci Med Sci. 2014;69(1):13–24. 10.1093/gerona/glt043. [DOI] [PubMed] [Google Scholar]
- 65.Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife. 2015;4:e12997. 10.7554/eLife.12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meyer SC, Levine RL. Molecular pathways: molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin Cancer Res. 2014;20(8):2051–9. 10.1158/1078-0432.Ccr-13-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A. 2015;112(46):E6301–10. 10.1073/pnas.1515386112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen YW, Harris RA, Hatahet Z, Chou KM. Ablation of XP-V gene causes adipose tissue senescence and metabolic abnormalities. Proc Natl Acad Sci U S A. 2015;112(33):E4556–64. 10.1073/pnas.1506954112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Q, Hagberg CE, Silva Cascales H, Lang S, Hyvönen MT, Salehzadeh F, et al. Obesity and hyperinsulinemia drive adipocytes to activate a cell cycle program and senesce. Nat Med. 2021;27(11):1941–53. 10.1038/s41591-021-01501-8. [DOI] [PubMed] [Google Scholar]
- 70.Herold J, Kalucka J. Angiogenesis in adipose tissue: the inter-play between adipose and endothelial cells Front Physiol 2021;11:624903 10.3389/fphys.2020.624903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Monelli E, Villacampa P, Zabala-Letona A, Martinez-Romero A, Llena J, Beiroa D, et al. Angiocrine polyamine production regulates adiposity. Nat Metab. 2022;4(3):327–43. 10.1038/s42255-022-00544-6. [DOI] [PubMed] [Google Scholar]
- 72.Villaret A, Galitzky J, Decaunes P, Estève D, Marques MA, Sengenès C, et al. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes. 2010;59(11):2755–63. 10.2337/db10-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Briot A, Decaunes P, Volat F, Belles C, Coupaye M, Ledoux S, et al. Senescence alters PPARγ (peroxisome proliferator-activated receptor gamma)-dependent fatty acid handling in human adipose tissue microvascular endothelial cells and favors inflammation. Arterioscler Thromb Vasc Biol. 2018;38(5):1134–46. 10.1161/atvbaha.118.310797. [DOI] [PubMed] [Google Scholar]
- 74.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7(1):48–52. [DOI] [PubMed] [Google Scholar]
- 75.Kennedy DJ, Kuchibhotla S, Westfall KM, Silverstein RL, Morton RE, Febbraio M. A CD36-dependent pathway enhances macrophage and adipose tissue inflammation and impairs insulin signalling. Cardiovasc Res. 2011;89(3):604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Science Signaling. 2009;2(72):re3. 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, et al. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2014;19(1):162–71. 10.1016/j.cmet.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shimobayashi M, Albert V, Woelnerhanssen B, Frei IC, Weissenberger D, Meyer-Gerspach AC, et al. Insulin resistance causes inflammation in adipose tissue. J Clin Investig. 2018;128(4):1538–50. 10.1172/JCI96139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage sub-types. Diabetes. 2008;57(12):3239–46. 10.2337/db08-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci U S A. 2018;115(22):E5096–105. 10.1073/pnas.1802611115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Silva HM, Báfica A, Rodrigues-Luiz GF, Chi J, Santos PDA, Reis BS, et al. Vasculature-associated fat macrophages readily adapt to inflammatory and metabolic challenges. J Exp Med. 2019;216(4):786–806. 10.1084/jem.20181049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frasca D, Romero M, Diaz A, Garcia D, Thaller S, Blomberg BB. B Cells with a senescent-associated secretory phenotype accumulate in the adipose tissue of individuals with obesity. LID. Int J Mol Sci. 2021;22(4):1839. 10.3390/ijms22041839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carter S, Miard S, Caron A, Sallé-Lefort S, St-Pierre P, Anhê FF, et al. Loss of OcaB prevents age-induced fat accretion and insulin resistance by altering b-lymphocyte transition and promoting energy expenditure. Diabetes. 2018;67(7):1285–96. 10.2337/db17-0558. [DOI] [PubMed] [Google Scholar]
- 84.Camell CD, Günther P, Lee A, Goldberg EL, Spadaro O, Youm YH, et al. Aging induces an Nlrp3 inflammasome-dependent expansion of adipose B cells that impairs metabolic homeostasis. Cell Metab. 2019;30(6):1024–39.e6. 10.1016/j.cmet.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim B, Hyun CK. B-cell-activating factor depletion ameliorates aging-dependent insulin resistance via enhancement of thermogenesis in adipose tissues. LID Int J Mol Sci. 2020;21(14):5121. 10.3390/ijms21145121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Winer DA, Winer S, Chng MH, Shen L, Engleman EG. B Lymphocytes in obesity-related adipose tissue inflammation and insulin resistance. Cell Mol Life Sci. 2014;71(6):1033–43. 10.1007/s00018-013-1486-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Curtis JM, Grimsrud PA, Wright WS, Xu X, Foncea RE, Graham DW, et al. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes. 2010;59(5):1132–42. 10.2337/db09-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Long EK, Olson DM, Bernlohr DA. High-fat diet induces changes in adipose tissue trans-4-oxo-2-nonenal and trans-4-hydroxy-2-nonenal levels in a depot-specific manner. Free Radic Biol Med. 2013;63:390–8. 10.1016/j.freeradbiomed.2013.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shirakawa K, Yan X, Shinmura K, Endo J, Kataoka M, Katsumata Y, et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J Clin Investig. 2016;126(12):4626–39. 10.1172/JCI88606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lau EYM, Carroll EC, Callender LA, Hood GA, Berryman V, Pattrick M, et al. Type 2 diabetes is associated with the accumulation of senescent T cells. Clin Exp Immunol. 2019;197(2):205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486(7404):549–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Becker M, Levings MK, Daniel C. Adipose-tissue regulatory T cells: critical players in adipose-immune crosstalk. Eur J Immunol. 2017;47(11):1867–74. 10.1002/eji.201646739. [DOI] [PubMed] [Google Scholar]
- 94.Arora S, Thompson PJ, Wang Y, Bhattacharyya A, Apostolopoulou H, Hatano R, et al. Invariant natural killer T cells coordinate removal of senescent cells. Med (New York, NY). 2021;2(8):938–50. 10.1016/j.medj.2021.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Piñeiro-Carrero VM, Piñeiro EO. Liver Pediatrics. 2004;113(4 Suppl):1097–106. [PubMed] [Google Scholar]
- 96.Bogdanos DP, Gao B, Gershwin ME. Liver immunology. Compr Physiol. 2013;3(2):567–98. 10.1002/cphy.c120011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. 2017;8:15691. 10.1038/ncomms15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang X, Zhou D, Strakovsky R, Zhang Y, Pan YX. Hepatic cellular senescence pathway genes are induced through histone modifications in a diet-induced obese rat model. Am J Physiol Gastrointest Liver Physiol. 2012;302(5):G558–64. 10.1152/ajpgi.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aravinthan A, Mells G, Allison M, Leathart J, Kotronen A, Yki-Jarvinen H, et al. Gene polymorphisms of cellular senescence marker p21 and disease progression in non-alcohol-related fatty liver disease. Cell Cycle. 2014;13(9):1489–94. 10.4161/cc.28471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27(24):8807–14. 10.1128/mcb.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Choudhury M, Jonscher KR, Friedman JE. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging Albany NY. 2011;3(2):175–8. 10.18632/aging.100289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36(1):111–7. 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kou K, Saisho Y, Jinzaki M, Itoh H. Relationship between body mass index and pancreas volume in Japanese people. Jop. 2014;15(6):626–7. 10.6092/1590-8577/2858. [DOI] [PubMed] [Google Scholar]
- 104.van Raalte DH, van der Zijl NJ, Diamant M. Pancreatic steatosis in humans: cause or marker of lipotoxicity? Curr Opin Clin Nutr Metab Care. 2010;13(4):478–85. 10.1097/MCO.0b013e32833aa1ef. [DOI] [PubMed] [Google Scholar]
- 105.van Vliet S, Koh HE, Patterson BW, Yoshino M, LaForest R, Gropler RJ, et al. Obesity is associated with increased basal and postprandial β-cell insulin secretion even in the absence of insulin resistance. Diabetes. 2020;69(10):2112–9. 10.2337/db20-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–10. 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 107.Klein S, Gastaldelli A, Yki-Järvinen H, Scherer PE. Why does obesity cause diabetes? Cell Metab. 2022;34(1):11–20. 10.1016/j.cmet.2021.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia. 2005;48(1):58–67. 10.1007/s00125-004-1605-2. [DOI] [PubMed] [Google Scholar]
- 109.Aguayo-Mazzucato C, Andle J, Lee TB Jr, Midha A, Talemal L, Chipashvili V, et al. Acceleration of β cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab. 2019;30(1):129–42.e4. 10.1016/j.cmet.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aguayo-Mazzucato C, Midha A. β-cell senescence in type 2 diabetes. Aging Albany NY. 2019;11(22):9967–8. 10.18632/aging.102502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu Z, Wu KKL, Jiang X, Xu A, Cheng KKY. The role of adipose tissue senescence in obesity- and ageing-related metabolic disorders. Clin Sci (Lond). 2020;134(2):315–30. 10.1042/cs20190966. [DOI] [PubMed] [Google Scholar]
- 112.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85–97. 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fuchs A, Samovski D, Smith GI, Cifarelli V, Farabi SS, Yoshino J, et al. Associations among adipose tissue immunology, inflammation, exosomes and insulin sensitivity in people with obesity and nonalcoholic fatty liver disease. gastroenterology. 2021;161(3):968–81. e12. 10.1053/j.gastro.2021.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Snel M, Jonker JT, Schoones J, Lamb H, de Roos A, Pijl H, et al. Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. Int J Endocrinol. 2012;2012:983814. 10.1155/2012/983814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55(Supplement_2):S9–S15. 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pederson TM, Kramer DL, Rondinone CM. Serine/threonine phosphorylation of IRS-1 triggers its degradation: possible regulation by tyrosine phosphorylation. Diabetes. 2001;50(1):24–31. 10.2337/diabetes.50.1.24. [DOI] [PubMed] [Google Scholar]
- 117.Tanti JF, Jager J. Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharmacol. 2009;9(6):753–62. 10.1016/j.coph.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 118.Mitrakou A, Kelley D, Veneman T, Jenssen T, Pangburn T, Reilly J, et al. Contribution of abnormal muscle and liver glucose metabolism to postprandial hyperglycemia in NIDDM. Diabetes. 1990;39(11):1381–90. 10.2337/diab.39.11.1381. [DOI] [PubMed] [Google Scholar]
- 119.Schmitz-Peiffer C Deconstructing the role of PKC Epsilon in glucose homeostasis. Trends Endocrinol Metab. 2020;31(5):344–56. 10.1016/j.tem.2020.01.016. [DOI] [PubMed] [Google Scholar]
- 120.Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology. 2014;59(2):713–23. 10.1002/hep.26672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Paolisso G, Gambardella A, Amato L, Tortoriello R, D’Amore A, Varricchio M, et al. Opposite effects of short- and long-term fatty acid infusion on insulin secretion in healthy subjects. Diabetologia. 1995;38(11):1295–9. 10.1007/BF00401761. [DOI] [PubMed] [Google Scholar]
- 122.Prentki M, Peyot M-L, Masiello P, Madiraju SRM. Nutrient-induced metabolic stress, adaptation, detoxification, and toxicity in the pancreatic β-cell. Diabetes. 2020;69(3):279–90. 10.2337/dbi19-0014. [DOI] [PubMed] [Google Scholar]
- 123.Wang L, Wang B, Gasek NS, Zhou Y, Cohn RL, Martin DE, et al. Targeting p21(Cip1) highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab. 2022;34(1):75–89. e8. 10.1016/j.cmet.2021.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24(8):1246–56. 10.1038/s41591-018-0092-9. [DOI] [PMC free article] [PubMed] [Google Scholar]