

Abstract
Activity-based protein profiling (ABPP) is a versatile strategy for identifying and characterizing functional protein sites and compounds for therapeutic development. Yet, the vast majority of ABPP methods for covalent drug discovery target highly nucleophilic amino acids such as cysteine or lysine. Here, we report a methionine-directed ABPP platform using Redox-Activated Chemical Tagging (ReACT), which leverages a biomimetic oxidative ligation strategy for selective methionine modification. Application of ReACT to oncoprotein cyclin-dependent kinase 4 (CDK4) as a representative high-value drug target identified three new ligandable methionine sites. We then synthesized a methionine-targeting covalent ligand library bearing a diverse array of heterocyclic, heteroatom, and stereochemically-rich substituents. ABPP screening of this focused library identified 1oxF11 as a covalent modifier of CDK4 at an allosteric M169 site. This compound inhibited kinase activity in a dose-dependent manner on purified protein and in breast cancer cells. Further investigation of 1oxF11 found prominent cation-π and H-bonding interactions stabilizing the binding of this fragment at the M169 site. Quantitative mass-spectrometry studies validated 1oxF11 ligation of CDK4 in breast cancer cell lysates. Further biochemical analyses revealed crosstalk between M169 oxidation and T172 phosphorylation, where M169 oxidation prevented phosphorylation of the activating T172 site on CDK4 and blocked cell cycle progression. By identifying a new mechanism for allosteric methionine redox regulation on CDK4 and developing a unique modality for its therapeutic intervention, this work showcases a generalizable platform that provides a starting point for engaging in broader chemoproteomics and protein ligand discovery efforts to find and target previously undruggable methionine sites.
Keywords: activity-based protein profiling, bioconjugation, redox signaling, chemoproteomics, covalent fragment library
Graphical Abstract
INTRODUCTION
Covalent small molecules that target specific amino acid residues are powerful chemical tools that can reveal fundamental new protein function and identify lead candidates for accelerating drug discovery.1–8 Indeed, led by advances across broad fields encompassing organic chemistry, chemical biology, cell biology, and bioinformatics, covalent therapeutics now constitute approximately 30% of enzyme-targeting FDA-approved drugs.9 In this context, activity-based protein profiling (ABPP), where chemical probes measure protein function rather than protein abundance,10,11 has enabled new modalities for fragment-based drug discovery by applying small-molecule screening efforts in conjunction with chemoproteomics for target and site identification and characterization.12–35 These technologies rely on residue-specific covalent warheads that can be used from proteins to proteomes,5,36–42 yet the majority of reactive probe development to tackle this vast undruggable space has targeted cysteine43,44 or lysine,21,24,26,45 with relatively limited expansion of this chemical toolbox to other nucleophilic residues like tyrosine46 and glutamate/aspartate.47,48
Motivated to contribute to this area as part of a larger program in our laboratories to create and use activity-based chemical probes for biological applications,49–52 we and others have developed bioconjugation methods for methionine,53–59 one of two privileged sulfur-containing amino acids along with cysteine. Methionine is distinguished by its characteristic thioether moiety, which endows this hydrophobic amino acid with high redox activity and low nucleophilicity relative to its highly redox-active and nucleophilic cysteine congener. The methionine sulfur atom enables not only greater rotational freedom through lower strain gauche interactions but also provides the opportunity for unique single-atom post-translational modifications (PTM) through a reversible two-electron oxidation to generate both (R)- and (S)-methionine sulfoxide products.60 Akin to kinase writer and phosphatase eraser pairs for installing and removing phosphate PTMs, respectively, stereospecific reduction of methionine sulfoxide sites catalyzed by methionine sulfoxide reductase (Msr) eraser proteins can remove these single-oxygen PTMs. This reversible methionine thioether/sulfoxide cycle plays an integral part in the redox regulation of cell signaling events, antioxidant function, and other forms of protein regulation.61–63
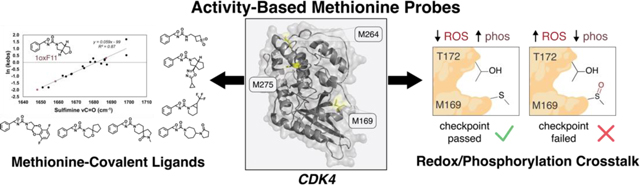
In contrast to cysteine and other protein nucleophiles, the reactivity of protein-bound methionine is dictated by its redox potential rather than by its pKa. As such, we developed Redox-Activated Chemical Tagging (ReACT), a versatile bioconjugation method that targets methionine through the use of oxaziridine reagents that promote selective nitrene fragment transfer reactivity that is isoelectronic to native methionine oxidation by oxygen atom transfer.53 Methionine functionalization with ReACT proceeds selectively and rapidly at physiological pH and generates stable, mass-spectrometry compatible sulfimine adducts, enabling further chemoproteomic characterization of putative protein targets and sites of modification. Indeed, ReACT has found utility in the context of synthesis of stapled cyclic peptides,56 production of antibody-drug conjugates (MetMAb),57 proximity-activated imaging reporters for protein function (PAIR),58 18F radioimaging tracers and probes for protein and nucleic acid biotinylation (BioReACT).64 Here, we present the development of ReACT as a versatile methionine-targeting ABPP platform for covalent ligand discovery. We highlight its application to the study of the cancer-driver protein cyclin-dependent kinase 4 (CDK4), a serine/threonine kinase which serves as a master regulator of mitogenic signaling responsible for G1-S phase progression of the cell cycle.65–67 CDK4 is a high-value therapeutic target that is commonly misregulated in a variety of cancers and is one of many CDKs targeted in cancer drug therapy efforts.68–70 ReACT ABPP identified three new hyperreactive, ligandable methionine residues on CDK4, including an allosteric M169 site that is proximal to an activating T172 phosphorylation site. We then designed and synthesized a focused 179-member oxaziridine fragment library featuring a diverse array of heterocyclic, heteroatom, and stereochemically-rich substituents. Gel-based ABPP screening and chemoproteomic characterization identified 1oxF11 as a covalent modifier of the CDK4/Cyclin-D1 (CDK4/CCND1) heterodimer at the M169 site. This compound inhibited the kinase activity of CDK4 in a dose-dependent manner on purified protein and in live cells. Structure-activity relationship (SAR) studies revealed that 1oxF11 is a privileged scaffold for CDK4 binding at the M169 site through prominent cation-π and H-bonding interactions. Quantitative chemoproteomic studies conducted with an alkynyl derivative of 1oxF11 revealed ligation of CDK4 in complex proteomes. Interestingly, further biochemical studies using 1D- and 2D-immunoblot analyses with a phospho-specific pT172 CDK4 antibody revealed reciprocal crosstalk between M169 oxidation and T172 phosphorylation, where M169 oxidation by 1oxF11 prevented phosphorylation of the activating T172 site, thus blocking cell cycle progression at the S-phase checkpoint. The collective data identify a new mechanism for allosteric methionine regulation on CDK4 and a unique redox vulnerability for therapeutic intervention. Owing to the generality of the ReACT ABPP workflow, this platform provides a starting point for expanding the repertoire of chemical tools for identifying and characterizing new functional methionine sites and developing methionine-targeting covalent ligands for accelerating drug discovery.
RESULTS AND DISCUSSION
ReACT Identifies New Ligandable Methionine Sites on the Cancer-Driver Protein CDK4.
As a starting point to develop a methionine-targeting ABPP platform for covalent ligand discovery, we chose to study CDK4 as a representative kinase and high-value drug target. Kinases present a unique challenge in the design of selective therapeutics as they share a highly conserved ATP-binding active site amongst other common domains.71,72 Indeed, off-target binding of promiscuous kinase inhibitors, many of which are substrate analogs of ATP, has led to the failure of numerous drug candidates in clinical trials due to undesirable side effects.73 As such, identification of allosteric sites distal from the ATP-binding active site pocket provides a promising alternative approach for tuning kinase selectivity.
Against this backdrop, combining the synergistic approaches of fragment-based drug discovery74–76 and covalent ligand development2,5 we sought to expand the scope of proteins that can be targeted by small molecules. This bottom-up approach involves screening of covalent ligand fragment libraries against broadly reactive, but amino acid-specific, bioconjugation warheads to discover novel binding pockets for covalent modification.77 SARs can then be interrogated to further optimize selectivity and potency after identification of promising fragment leads. To identify potential new ligandable methionine sites in CDK4, we applied a set of three unique ReACT oxaziridine probes to this target: Ox1-alkyne, Ox1-azide, and Ox32-alkyne (Figure 1a). Interestingly, each ABPP probe showed a different pattern of covalent methionine labeling on our protein of interest, presaging that these sites can be preferentially targeted. Ox1-azide modifies three reactive sites: M169, M264, and M275 (Figure 1b, Figure S1). In contrast, Ox1-alkyne labeled only M169, whereas Ox32-alkyne engaged only M264. Sites of modification were determined via shotgun proteomics (Figure 1c). The frequency of modifications observed on each methionine site correlated with the solvent accessibility of that residue. M169 displayed the highest reactivity, followed by M264, with M275 being the least solvent accessible and least reactive. Notably, the newly identified hyperreactive M169 site is proximal to a known T172 phosphorylation site on CDK4 that activates protein function.78,79
Figure 1.

Activity-based protein profiling (ABPP) using oxaziridine probes for Redox-Activated Chemical Tagging (ReACT) identifies new hyperreactive, ligandable methionine sites on CDK4. (a) Structures of Ox1-alkyne, Ox32-alkyne, and Ox1-azide ABPP probes. (b) Ribbon diagram of CDK4 (PDB: 2W9Z). Methionine residues modified by oxaziridines are highlighted, with colored squares representing the corresponding ReACT reagents found to modify each site. Each residue is additionally labeled with its calculated solvent accessibility. (c) Shotgun proteomics general workflow. Isolated protein is first incubated with compound. After excess compound is neutralized, protein is tryptic-digested and analyzed via MS/MS to reveal compound site(s) of modification.
Design and Synthesis of a Methionine-Targeting Covalent Ligand Library.
After identifying hyperreactive methionine residues as potentially new ligandable sites for CDK4, we next sought to develop a methionine-directed covalent ligand platform to engage this protein target through the design and synthesis of a focused oxaziridine fragment library. To this end, we first optimized a suite of synthetic routes to such products, which included three methods for imine synthesis followed by oxidation via mCPBA (Scheme 1). As oxaziridine synthesis limited available functional groups to those stable to the final oxidation of the imine intermediate, and the oxaziridine itself can show sensitivity to acid, base, reductants, metals, and thiols, we sought to prepare a focused library to achieve sufficient structural diversity under these constraints.
Scheme 1.

General synthetic routes to oxaziridine compounds for creating a focused covalent ligand fragment library. All syntheses began with primary or secondary amine synthons. Three routes were utilized to generate imine intermediates depending on starting amine. All imines were converted to oxaziridines using the same convergent method outlined.
The design of the oxaziridine library was initiated through a triaging selection of the key diversity element, namely the corresponding amine component. 43,950 amines were available for selection (Enamine); utilizing ICM Chemist Pro (Molsoft LLC) sub-structure selections were made driven by learnings from our initial study.56 Clustering analysis for each selected amine set was conducted using ICM Chemist Pro (Molsoft LLC) generating the detailed clusters for each sub-type (SI methods). Thereafter, rounds of selections based on diversity and removal of compounds containing foreseen chemoselectivity issues, resulted in a preferred set of 234 amines. These amines were derivatized to the corresponding sulfimine or urea in silico and a 3D conformational search allowed C=O stretching frequencies to be determined. In order to ensure formation of N-transfer sulfimine over O-transfer sulfoxide products upon reaction with methionine, we employed principal component analysis (PCA) methods to optimize this parameter and found that rates of sulfimine hydrolysis correlated with the calculated sulfimine adduct νC=O values (Figure 2a). The finalized library of 179 unique oxaziridine fragments featured a diverse array of functional groups, including spirocycles, halogens, azoles, ethers, and amides (Figure 2c, Figure S2).
Figure 2.

Design, synthesis, and evaluation of an oxaziridine-based covalent ligand library for targeting methionine sites and identification of 1oxF11 as a covalent modifier of CDK4 via gel-based ABPP screening. (a) Design of oxaziridine fragments that favor formation of N-transfer sulfimine over O-transfer sulfoxide products upon reaction with methionine using principal component analysis (PCA). Rates of sulfimine hydrolysis correlate with calculated νC=O values. (b) Schematic of gel-based ABPP screening workflow. The protein target is preincubated with covalent ligand followed by treatment with Ox1-azide probe. Excess oxaziridine is then quenched with N-acetyl methionine (NAM) and the sample treated with DBCO-Cy3 overnight for fluorescence detection. Samples are finally separated by SDS-PAGE. Loss of fluorescent signal suggests competitive ligand binding to the protein. Silver stain is used to identify and exclude samples where signal loss corresponds to overall protein loss, likely due to aggregation induced by the ligand. (c) Representative structure types within the oxaziridine fragment library, organized by common functional groups. (d) Representative data from gel-based ABPP screen. CDK4 and ligand incubated at equimolar doses. (e) Structure of 1oxF11 fragment identified in gel-based ABPP screens as a competitive ligand for CDK4. Dose-dependent treatment of 1oxF11 against CDK4, CDK1, and CDK6 shows selective loss of fluorescent signal only with CDK4, suggesting isoform-specific engagement of this target.
Methionine-Directed ABPP Screen Against CDK4 Identifies Lead Compound Fragment 1oxF11.
With this oxaziridine fragment library in hand, we proceeded to screen for methionine-directed modifiers of CDK4 via a gel-based ABPP platform (Figure 2b). We chose the Ox1-azide ReACT probe as it was the most promiscuous and could engage three hyperreactive methionine sites in competitive binding assays with potential covalent oxaziridine ligands. Briefly, isolated CDK4 was treated either with DMSO (vehicle) or a covalent ligand from the oxaziridine library (ligand-treated). Samples were then treated with Ox1-azide probe followed by a quench step with N-acetyl methionine (NAM) to remove any excess oxaziridine. DBCO-Cy3 was then introduced by strain-promoted click chemistry to provide a fluorescence readout. The samples were subsequently separated via SDS-PAGE and fluorescence signals were normalized using a silver stain to triage any covalent ligands that induced general protein aggregation, which would generate a false positive Cy3 signal (Figure 2b).
Using ReACT, ABPP screening of the focused oxaziridine fragment library on CDK4 revealed 1oxF11 as a candidate for further study (Figure 2d, Figure S3). This fragment showed competition with the Ox1-azide probe for CDK4 binding in a dose-dependent manner. We then tested the isoform specificity with two closely related congeners, CDK1 and CDK6. CDK1 is ubiquitously expressed and essential for healthy cell function, and CDK6 shows the highest structural similarity to CDK4.80 Indeed, all CDK4 inhibitors currently in therapeutic use also display activity against CDK6. Along these lines, we were pleased to observe that 1oxF11 displayed no dose-dependent labeling against either CDK1 or CDK6 by gel analysis (Figure 2e). Additionally, we confirmed that this compound did not induce general aggregation in cell lysates (Figure S4).
1oxF11 is a Covalent Modifier of CDK4 at the M169 Site and Inhibits Activity on Purified Protein.
We next characterized the sites of modification and subsequent effect on protein activity on CDK4 by 1oxF11 treatment in vitro. To start, we performed shotgun proteomics with 1oxF11 on the purified and active CDK4/CCND1 heterodimer. We identified that the primary site of modification on CDK4 by 1oxF11 was M169, with minor labeling at M264 (Figure 3a, Figure S5). We then mutated the M169 site to leucine in order to further investigate the effects of M169 ligation. When this site is mutated, the primary site of modification was identified as M264 (Figure 3b). Next, we demonstrated that 1oxF11 inhibits activity of the CDK4/CCND1 complex in a dose-dependent manner using a luciferase-based activity assay as a proxy for kinase activity. Here, we observed an IC50 value of 1.89 μM for the WT form and 3.00 μM for the M169L variant (Figure 3c). We posit that the primary negative modulator for kinase activity is M169 ligation, while M264 ligation may also affect protein activity through another allosteric mechanism.
Figure 3.

1oxF11 is a covalent modifier of CDK4 at M169 and inhibits its activity on purified protein. (a) MS2 spectrum of 1oxF11-modified CDK4 showing covalent ligation at M169. (b) MS2 spectrum of 1oxF11-modified M169L mutant CDK4 showing covalent ligation at M264. (c) Kinase activity assay on purified WT and M169L CDK4/CCND1 protein variants showing dose-dependent inhibition in response to 1oxF11 treatment.
1oxF11 is a Privileged Scaffold for Binding the M169 Site of CDK4.
To better understand the propensity for 1oxF11 to bind the M169 site, we performed molecular dynamics studies on 1oxF11 (Figure 4a, Movie S2). These studies revealed that 1oxF11 binds the M169 site through prolonged cation-π interactions with R139/R181 of CDK4 at the phenyl moiety. Critically, the R181 residue has a strong, pronounced hydrogen-bonding interaction with the carbonyl present on the urea moiety of 1oxF11 which is further stabilized by transient H-bonding interactions with V174 (Figure 4b). We determined the strength of these interactions by calculating the percent interaction time with each residue of CDK4 across the simulation (Figure 4c). Furthermore, we observed that the tetrahydrofuranyl moiety of 1oxF11 is conformationally locked in a position that enables a hydrogen bonding interaction with the amide backbone of V174. Together, these data inform the binding mode of 1oxF11 in the M169 pocket, positioning the oxaziridine for proximal M169 ligation (Figure 4d). We also performed molecular dynamics simulations on two other CDK4 binders, 1oxA6 and 1oxH2, and found that their noncovalent interactions are much less pronounced, leading to reduced occupancy time (Figure S6, Movie S1, Movie S3).
Figure 4.

Molecular dynamics of 1oxF11 bound to CDK4. (a) full structure of CDK4 with 1oxF11 noncovalently bound. (b) Expansion of M169 binding pocket. (c) Ligand-residue interaction diagram over time. (d) Schematic of 1oxF11 binding.
1oxF11 Decreases Cell Viability and Inhibits Cellular CDK4 Activity.
Turning our attention to cellular studies, we next sought to identify a model cell line that displayed heightened sensitivity to 1oxF11. We screened 1oxF11 across a small panel of cancer cell lines with sensitivity to ribociclib, a clinically-approved CDK4/6 inhibitor (Figure 5a). We selected the human breast adenocarcinoma line MCF-7 as a model for further study due to its pronounced sensitivity to 1oxF11 treatment (Figure 5a). Additionally, the contributions of CDK4 to uncontrolled cell division in the MCF-7 line have been well-documented.81,82 Indeed, we observed a dose-dependent decrease in cell viability of MCF-7 cells in response to 1oxF11, with an EC50 of 329 μM (Figure 5b). This lower EC50 observed in cells compared to in vitro is likely the result of several factors including cell permeability, non-productive consumption by thiols, and/or off-target effects. We also investigated the effects of 1oxF11 treatment specific to the M169 site in cellulo by generating constructs for transient mammalian transfection of both FLAG-tagged WT and M169L variants of CDK4. We began by constitutively knocking down endogenous CDK4 expression, followed by transient expression of both variants to control for background inhibition of endogenously expressed CDK4. We observed EC50 values of 189 and 296 μM for transfected WT and M169L variants, respectively (Figure 5b). This complements our in vitro results suggesting modification of M169 leads to decreased MCF-7 cell viability. We attribute the decreased EC50 values for transfected cells compared to the untransfected cells to the increased sensitivity of mammalian cells following treatment with harsh transfection reagents.
Figure 5.

Decrease in cell viability and inhibition of cellular CDK4 activity by 1oxF11 and its 1oxF11yne analog in various cancer cell models. (a) Screening of ribociclib-sensitive cell lines by treatment with 500 μM 1oxF11 showing differential cell viability profiles across cancer types. The MCF-7 cell line displays the highest sensitivity to 1oxF11. Error bars represent standard deviation of n = 3 biological replicates. (b) Left: Immunoblot timecourse of constitutive knockdown and overexpression of transiently expressed CDK4. Right: Dose-response curves of untransfected and transfected MCF-7 cells transiently expressing CDK4 treated with 1oxF11. Error bars represent standard deviation of n = 3 biological replicates. (c) Synthetic route of 1oxF11yne bearing an alkyne handle for bio-orthogonal detection and enrichment purposes. (d) MS2 data showing that 1oxF11yne is a covalent modifier of CDK4 at the same M169 site as the parent 1oxF11 fragment. (e) Simplified scheme of cellular CDK4 activity, where Rb represents the native substrate of CDK4. Immunoblot analyses measuring the extent of Rb phosphorylation provides a method to detect intracellular CDK4 activity. (f) Immunoblot analysis of MCF-7 cells treated with a titration of 1oxF11 and 1oxF11yne to assess changes in cellular CDK4 activity. (g) Competition binding assay between 1oxF11 and 1oxF11yne, providing evidence for target engagement in MCF-7 cells.
To further probe how 1oxF11 contributes to the observed cellular phenotype, we synthesized a 1oxF11 analog, 1oxF11yne, containing an alkyne handle for detection and enrichment using click chemistry (Figure 5c). Shotgun proteomics experiments revealed that the 1oxF11yne probe modifies CDK4 selectively at the same M169 site as the parent 1oxF11 fragment (Figure 5d). We then sought to test the effects of both probes on CDK4 activity within a cellular context. To achieve this goal, we turned to immunoblot analysis, monitoring the phosphorylation status of retinoblastoma protein (Rb), the main cellular substrate of CDK4. When active, CDK4 in complex with its cognate cyclin partner phosphorylates Rb at one of 14 sites (Figure 5e).83 We used phospho-responsive antibodies specific for three of these sites, S780, S807, and S811, as a method to assess CDK4 activity in cellulo. To control for increased background levels of pRb due to cells being in different stages of the cell cycle, we serum-synchronized MCF-7 cells to the G0/G1 phase. Indeed, incubation of serum-synchronized MCF-7 cells with either 1oxF11 or 1oxF11yne at a dose of 500 μM resulted in a marked decrease in signal from the pRb antibodies, suggesting a decrease in cellular CDK4 activity under these conditions (Figure 5f). Finally, we confirmed CDK4 target engagement in cells using a competition binding assay between 1oxF11 and 1oxF11yne (Figure 5g). To overcome detection challenges with the low endogenous expression levels of CDK4, even in cells known to upregulate the protein such as MCF-7, we transfected cells with WT CDK4 to achieve transient expression of in these models. Treatment of cells with a titration of 1oxF11 prior to incubation with 500 μM 1oxF11yne probe showed a dose-dependent decrease in signal in eluted proteins and a corresponding increase in signal in the respective supernatant, indicating competition between the two compounds and engagement of CDK4 in a cellular context (Figure S7). Additionally, 1oxF11yne displayed lower promiscuity in lysate compared to Ox32-alkyne, further suggesting its heightened selectivity in a complex milieu of proteins (Figure S8).
ReDiMe-ABPP Identifies CDK4 as a Target of 1oxF11yne in MCF-7 Lysates.
Following our promising immunoblotting and substrate promiscuity results, we next sought to apply quantitative chemoproteomics to identify targets of 1oxF11yne in a proteome-wide context. With the 1oxF11yne probe in hand, we enriched for protein targets of 1oxF11yne by first treating MCF-7 lysates expressing CDK4 with 500 μM of the probe followed by CuAAC to a desthiobiotin azide, enabling streptavidin pulldown of labeled targets. In order to exclude proteins that non-specifically bind to the streptavidin interface, we also treated MCF-7 lysates with a vehicle control followed by CuAAC and enrichment. We incorporated reductive dimethylation (ReDiMe) labeling to our ABPP platform, enabling sample multiplexing of 1oxF11yne targets against non-specific binders and providing an isotopic signature to distinguish between the two protein groups (Figure 6a). Through these studies, we identified 59 protein targets that are engaged by 1oxF11yne including CDK4 (Figure 6b) with no overlap to the DMSO control. A full list of these protein targets was also generated (Sheet S2). We further analyzed the subcellular distribution of these targets and found that the majority of these proteins are cytosolically or nuclearly localized, providing further evidence of probe quenching in the cytosolic environment through off-target effects (Figure 6c). We also assessed the function of these protein targets through PANTHER GO analyses, demonstrating a clear enrichment in proteins involved in protein-protein/protein-small molecule binding as well as catalysis (Figure 6d). Together, these data provide a promising avenue for future structure optimization to increase the selectivity of 1oxF11 for CDK4 and thereby increase its potency in a proteome-wide context.
Figure 6.

ReDiMe-ABPP platform to identify targets of 1oxF11yne in MCF-7 lysates. (a) Schematic of the chemoproteomic platform to identify protein targets of 1oxF11yne. MCF-7 lysates transiently expressing CDK4 were treated with either 500 μM 1oxF11yne or vehicle followed by CuAAC to a DTB-N3. Samples were then separately enriched with streptavidin-agarose beads and tryptically digested. Following peptide elution, the samples were differentially labeled with light or heavy isotopes at their N-termini through reductive dimethylation to enable identification of 1oxF11yne targets compared to proteins that non-specifically bind to the streptavidin-agarose interface. Samples were multiplexed and analyzed across n = 3 biological replicates. (b) Unique protein targets identified by ReDiMe-ABPP. A total of 72 unique protein targets with n = 2 statistical representation were identified as 1oxF11yne targets including CDK4, with some overlap to non-specific binders. (c) Subcellular distribution of identified 1oxF11yne targets. (d) PANTHER GO protein function of identified 1oxF11yne targets.
Biochemical Studies of CDK4 Inhibition by 1oxF11 Reveal Reciprocal Crosstalk Between M169 Oxidation and T172 Phosphorylation.
With data establishing 1oxF11 as a covalent modifier of CDK4 at a newly identified allosteric M169 site, along with its ability to inhibit CDK4 activity on purified protein and in cells and decrease cell viability in a dose-dependent manner, combined with chemoproteomic verification of target engagement in cellulo, we next sought to further interrogate its potential mechanism of action at the biochemical level. In this context, CDK4 plays a key role in the cell cycle in clearing the cell for division, only allowing for passage through the S-phase checkpoint when properly activated. In particular, this signaling pathway relies on proper binding of CDK4 to its respective cyclin, as well as phosphorylation at T172 by cyclin-dependent activating kinase (CAK) to activate the protein.84,85 Owing to the proximity of M169 to this activating T172 phosphorylation site, we hypothesized that M169 may act as an allosteric redox regulatory switch at this S-phase checkpoint.
Oxidation can transform the normally hydrophobic methionine residue into a hyper-polarized and sterically-demanding methionine sulfoxide congener, which vastly modulates the electrostatic and steric environment of this residue. We hypothesized that oxidation could block access of CAK to T172 and prevent its phosphorylation, thus causing the cell to fail the S-phase checkpoint (Figure 7a). This type of crosstalk between methionine oxidation and adjacent phosphorylation sites has been reported for other systems.86,87 Indeed, M169 and T172 lie in a flexible region of CDK4 that can come within 7 angstroms of each other, a distance observed to undergo this phenomenon previously (Figure 7b).88 We performed 2D-immunoblot analyses utilizing a pCDK4-Thr172 specific-antibody to investigate the effects of 1oxF11 treatment on CDK4 activation. Through 2D-analysis we were able to separate distinct proteoforms of pCDK4, with spot 3 corresponding to CDK4 in its T172 phosphorylated state, and spot 1 to unphosphorylated CDK4. We observed that treatment with 1oxF11 was indeed able to diminish T172 phosphorylation status on CDK4 in MCF-7 cells in a dose-dependent manner (Figure 7c). To control for differences in protein expression, we normalized pCDK4-T172 levels to unphosphorylated CDK4 (spot 3 normalized to spot 1). These data, along with supporting evidence showing engagement of CDK4 through chemoproteomics and immunoblotting, as well as inhibition of its activity both in vitro and in these same MCF-7 cell models, support a model in which M169 oxidation/T172 phosphorylation crosstalk offers a potential new redox vulnerability where oxidative modification of CDK4 at an allosteric M169 by 1oxF11 inhibits CDK4 activity by hindering phosphorylation at its activating T172 site.
Figure 7.

The ReACT covalent ligand probe platform enables discovery of a reciprocal oxidation/phosphorylation crosstalk pathway in CDK4 through proximal allosteric M169 and T172 sites, where M169-targeted oxidation can inhibit CDK4 activity by preventing phosphorylation at T172. (a) Schematic outlining oxidation/phosphorylation crosstalk between CDK4 M169 and T172. Under low oxidative conditions, T172 of CDK4 is phosphorylated by cyclin-dependent activating kinase (CAK) as part of a critical activating step leading to cell division to pass the S-phase checkpoint. High oxidative conditions can lead to oxidation at M169, which blocks the T172 phosphorylation site, thus preventing cell division via S-phase checkpoint failure. This crosstalk identifies a methionine redox-dependent vulnerability for potential therapeutic intervention. (b) Ribbon structure (PDB: 2W9Z) highlighting proximity of M169 and T172. (c) Monitoring CDK4 phosphorylation status using 2D-immunoblot analyses with phospho-specific CDK4 antibodies. Phosphorylation at T172 decreases with increasing concentrations of added 1oxF11 in MCF-7 cells. Spots 1, 2, 3, and 4 represent different phosphorylation states of CDK4, with spot 1 being unphosphorylated and spot 3 being monophosphorylated at T172. Peaks represent changes in intensity of spots normalized to peak 1 intensity. A clear decrease in spot 3 is observed upon treatment with increasing doses of 1oxF11, consistent with a model where this covalent ligand inhibits CDK4 activity by promoting M169 oxidation to block T172 phosphorylation.
CONCLUSIONS
We have presented a methionine-directed ABPP platform for identifying and developing covalent ligands for new functional methionine sites. We highlighted the potential value of this approach using CDK4 as a representative high-value target to showcase the application of ReACT probes to reveal fundamental new chemical function on proteins and accelerate drug discovery efforts by expanding covalent ligand development beyond the more common cysteine and lysine protein nucleophiles. Indeed, we applied methionine-directed ReACT probes with broad reactivity to identify novel hyperreactive, ligandable methionine sites on CDK4. We then optimized the synthesis of oxaziridine probes and used these methods to design and synthesize a focused covalent ligand library of ca. 180 oxaziridine fragments bearing chemically diverse functional groups, including spirocycles, halogens, azoles, ethers, and amides. Synthesis of the fragment library was guided by computational design to ensure efficient N-transfer rates and sulfimine stability of the subsequent products with methionine. We moved on to establish that this ReACT ABPP platform was useful for fragment-based screening efforts against the representative oncoprotein CDK4. Chemoproteomic experiments revealed that fragment 1oxF11 was a covalent modifier of CDK4 that selectively labeled its allosteric M169 site with isoform specificity over CDK1 and CDK6. We derived structure activity relationships from molecular dynamics studies that suggest that the tetrahydrofuranyl moiety on 1oxF11 uniquely positions it toward a hydrogen bonding interaction with the amide backbone of V174 in addition to cation- π interactions with the phenyl moiety alpha to the oxaziridine. Biochemical and cell-based assays showed that 1oxF11 can inhibit CDK4 activity on purified protein and in cells and decrease cell viability in a dose-dependent manner, with detection of target engagement in cells enabled by the synthesis of a 1oxF11yne probe bearing an alkyne handle for detection and enrichment. Quantitative chemoproteomics studies demonstrated that 1oxF11yne engages CDK4 in a proteome context with 59 other unique protein targets identified. Additionally, biochemical studies uncovered a novel redox regulatory mechanism for kinase inactivation through reciprocal oxidation/phosphorylation crosstalk between proximal M169 and T172 residues in CDK4, where M169 oxidation hinders phosphorylation at the protein-activating T172 site. Indeed, use of a phospho-specific pT172-CDK4 antibody in 1D- and 2D-immunoblot analyses established that treatment with the M169-modifying 1oxF11 covalent ligand diminished T172 phosphorylation and CDK4 activity. The resulting loss of CDK4 function prevented downstream Rb phosphorylation at S780 and S807/811, leading to cell cycle arrest by failure at the S-phase checkpoint. Our findings thus support a role for M169 as a redox sensor site at the S-phase checkpoint, preventing cell division under highly oxidative conditions by sterically preventing phosphorylation at T172. This newly discovered redox vulnerability in CDK4 provides an alternative modality for therapeutic intervention. Current and future efforts are geared to increase the size and diversity of methionine-directed activity-based probes and covalent ligand platforms to improve potency and selectivity, pursue other high-value targets and therapeutic modalities, as well as expand these approaches to other native amino acids in the proteome beyond cysteine, lysine, and methionine.
Supplementary Material
ACKNOWLEDGMENT
We thank Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies (NB-CPACT) for supporting this work. We also thank the NIH (R01 GM139245 and ES28096 to C.J.C. and R35 GM118190 to F.D.T.) for funding. A.G.R., A.G-V., and A.H.C. thank the NSF Graduate Fellowship Program for financial support. A.G.R. and A.G-V. were partially supported by a Chemical Biology Training Grant from the NIH (T32 GM066698). P.J.M. thanks the Natural Sciences and Engineering Research Council of Canada (NSERC) for a postdoctoral fellowship. S.W.M.C. is supported by the AGBT-Elaine R. Mardis Fellowship in Cancer Genomics from the Damon Runyon Cancer Research Foundation and The Genome Partnership, Inc. (DRG-2395-20). X.X. and D.H. are Tang Distinguished Scholars of the University of California, Berkeley. P.Z.M. thanks the American Cancer Society for a postdoctoral fellowship (PF-18-132-01-CDD). C.J.C. is a CIFAR Fellow. P.P.R. and K.C. thank WALInnov (CICLIBTEST 1710166) and the FRS-FNRS (J.0141.19) for funding. P.P.R. is a Senior Research Associate of the FRS-FNRS. We thank Alison Killilea (UC Berkeley Tissue Culture Facility) and Prof Daniel Nomura for technical assistance, Dr. Hasan Celik for assistance with NMR spectroscopy (NIH S10OD024998), and Drs. Kathleen Durkin and David Small for computational support (NIH S10OD023532).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at http://pubs.acs.org.
Experimental details, including chemoproteomic, biochemical, molecular dynamics, and cell-based assay methods, general methods for synthesis of oxaziridine probes, and supplemental data figures (PDF). Raw LC-MS/MS data (PDF).
J.M.M., R.A.L., E.F., D.D., Y.L., L.M.M., M.S., and J.A.T. are employees of Novartis Institutes for BioMedical Research.
REFERENCES
- (1).Schreiber SL; Kotz JD; Li M; Aubé J; Austin CP; Reed JC; Rosen H; White EL; Sklar LA; Lindsley CW; Alexander BR; Bittker JA; Clemons PA; de Souza A; Foley MA; Palmer M; Shamji AF; Wawer MJ; McManus O; Wu M; Zou B; Yu H; Golden JE; Schoenen FJ; Simeonov A; Jadhav A; Jackson MR; Pinkerton AB; Chung TDY; Griffin PR; Cravatt BF; Hodder PS; Roush WR; Roberts E; Chung D-H; Jonsson CB; Noah JW; Severson WE; Ananthan S; Edwards B; Oprea TI; Conn PJ; Hopkins CR; Wood MR; Stauffer SR; Emmitte KA; Brady LS; Driscoll J; Li IY; Loomis CR; Margolis RN; Michelotti E; Perry ME; Pillai A; Yao Y.Advancing Biological Understanding and Therapeutics Discovery with Small-Molecule Probes. Cell 2015, 161 (6), 1252–1265. DOI: 10.1016/j.cell.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Johnson DS; Weerapana E; Cravatt BF Strategies for Discovering and Derisking Covalent, Irreversible Enzyme Inhibitors. Future Med. Chem 2010, 2 (6), 949–964. DOI: 10.4155/fmc.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Spradlin JN; Zhang E; Nomura DK Reimagining Druggability Using Chemoproteomic Platforms. Acc. Chem. Res 2021, 54 (7), 1801–1813. DOI: 10.1021/acs.accounts.1c00065. [DOI] [PubMed] [Google Scholar]
- (4).McGregor LM; Jenkins ML; Kerwin C; Burke JE; Shokat KM Expanding the Scope of Electrophiles Capable of Targeting K-Ras Oncogenes. Biochemistry 2017, 56 (25), 3178–3183. DOI: 10.1021/acs.biochem.7b00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lu W; Kostic M; Zhang T; Che J; Patricelli MP; Jones LH; Chouchani ET; Gray NS Fragment-Based Covalent Ligand Discovery. RSC Chem. Biol 2021, 2, 254–367. DOI: 10.1039/D0CB00222D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cuesta A; Taunton J.Lysine-Targeted Inhibitors and Chemoproteomic Probes. Annu. Rev. Biochem 2019, 88 (1), 365–381. DOI: 10.1146/annurev-biochem-061516-044805. [DOI] [PubMed] [Google Scholar]
- (7).Peeler JC; Weerapana E.Chemical Biology Approaches to Interrogate the Selenoproteome. Acc. Chem. Res 2019, 52 (10), 2832–2840. DOI: 10.1021/acs.accounts.9b00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jones LH Reactive Chemical Probes: Beyond the Kinase Cysteinome. Angew. Chem. Int. Ed 2018, 57 (30), 9220–9223. DOI: 10.1002/anie.201802693. [DOI] [PubMed] [Google Scholar]
- (9).Roberston JG Mechanistic Basis of Enzyme-Targeted Drugs. Biochemistry 2005, 44 (15), 5562–5569. [DOI] [PubMed] [Google Scholar]
- (10).Deng H; Lei Q; Wu Y; He Y; Li W.Activity-Based Protein Profiling: Recent Advances in Medicinal Chemistry. Eur. J. Med. Chem 2020, 191, 112151. DOI: 10.1016/j.ejmech.2020.112151. [DOI] [PubMed] [Google Scholar]
- (11).Sadaghiani AM; Verhelst SH; Bogyo M.Tagging and Detection Strategies for Activity-Based Proteomics. Curr. Opin. Chem. Biol 2007, 11 (1), 20–28. DOI: 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]
- (12).Bachovchin DA; Cravatt BF The Pharmacological Landscape and Therapeutic Potential of Serine Hydrolases. Nat. Rev. Drug Discov 2012, 11 (1), 52–68. DOI: 10.1038/nrd3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Deu E; Verdoes M; Bogyo M.New Approaches for Dissecting Protease Functions to Improve Probe Development and Drug Discovery. Nat. Struct. Mol. Biol 2012, 19 (1), 9–16. DOI: 10.1038/nsmb.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Liu Y; Patricelli MP; Cravatt BF Activity-Based Protein Profiling: The Serine Hydrolases. Proc. Natl. Acad. Sci 1999, 96 (26), 14694–14699. DOI: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kumar S; Zhou B; Liang F; Wang W-Q; Huang Z; Zhang Z-Y Activity-Based Probes for Protein Tyrosine Phosphatases. Proc. Natl. Acad. Sci 2004, 101 (21), 7943–7948. DOI: 10.1073/pnas.0402323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Saghatelian A; Jessani N; Joseph A; Humphrey M; Cravatt BF Activity-Based Probes for the Proteomic Profiling of Metalloproteases. Proc. Natl. Acad. Sci 2004, 101 (27), 10000–10005. DOI: 10.1073/pnas.0402784101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vocadlo DJ; Bertozzi CR A Strategy for Functional Proteomic Analysis of Glycosidase Activity from Cell Lysates. Angew. Chem. Int. Ed 2004, 43 (40), 5338–5342. DOI: 10.1002/anie.200454235. [DOI] [PubMed] [Google Scholar]
- (18).Kato D; Boatright KM; Berger AB; Nazif T; Blum G; Ryan C; Chehade KAH; Salvesen GS; Bogyo M.Activity-Based Probes That Target Diverse Cysteine Protease Families. Nat. Chem. Biol 2005, 1 (1), 33–38. DOI: 10.1038/nchembio707. [DOI] [PubMed] [Google Scholar]
- (19).Patricelli MP; Szardenings AK; Liyanage M; Nomanbhoy TK; Wu M; Weissig H; Aban A; Chun D; Tanner S; Kozarich JW Functional Interrogation of the Kinome Using Nucleotide Acyl Phosphates. Biochemistry 2007, 46 (2), 350–358. DOI: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- (20).Ostrem JM; Peters U; Sos ML; Wells JA; Shokat KM K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503 (7477), 548–551. DOI: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shannon DA; Banerjee R; Webster ER; Bak DW; Wang C; Weerapana E.Investigating the Proteome Reactivity and Selectivity of Aryl Halides. J. Am. Chem. Soc 2014, 136 (9), 3330–3333. DOI: 10.1021/ja4116204. [DOI] [PubMed] [Google Scholar]
- (22).Backus KM; Correia BE; Lum KM; Forli S; Horning BD; González-Páez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; Wolan DW; Cravatt BF Proteome-Wide Covalent Ligand Discovery in Native Biological Systems. Nature 2016, 534 (7608), 570–574. DOI: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Matthews ML; He L; Horning BD; Olson EJ; Correia BE; Yates JR; Dawson PE; Cravatt BF Chemoproteomic Profiling and Discovery of Protein Electrophiles in Human Cells. Nat. Chem 2017, 9 (3), 234–243. DOI: 10.1038/nchem.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hacker SM; Backus KM; Lazear MR; Forli S; Correia BE; Cravatt BF Global Profiling of Lysine Reactivity and Ligandability in the Human Proteome. Nat. Chem 2017, 9 (12), 1181–1190. DOI: 10.1038/nchem.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Parker CG; Galmozzi A; Wang Y; Correia BE; Sasaki K; Joslyn CM; Kim AS; Cavallaro CL; Lawrence RM; Johnson SR; Narvaiza I; Saez E; Cravatt BF Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 2017, 168 (3), 527–541. DOI: 10.1016/j.cell.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ward CC; Kleinman JI; Nomura DK NHS-Esters As Versatile Reactivity-Based Probes for Mapping Proteome-Wide Ligandable Hotspots. ACS Chem. Biol 2017, 12 (6), 1478–1483. DOI: 10.1021/acschembio.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhao Q; Ouyang X; Wan X; Gajiwala KS; Kath JC; Jones LH; Burlingame AL; Taunton J.Broad-Spectrum Kinase Profiling in Live Cells with Lysine-Targeted Sulfonyl Fluoride Probes. J. Am. Chem. Soc 2017, 139 (2), 680–685. DOI: 10.1021/jacs.6b08536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mortenson DE; Brighty GJ; Plate L; Bare G; Chen W; Li S; Wang H; Cravatt BF; Forli S; Powers ET; Sharpless KB; Wilson IA; Kelly JW “Inverse Drug Discovery” Strategy To Identify Proteins That Are Targeted by Latent Electrophiles As Exemplified by Aryl Fluorosulfates. J. Am. Chem. Soc 2018, 140 (1), 200–210. DOI: 10.1021/jacs.7b08366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chaikuad A; Koch P; Laufer SA; Knapp S.The Cysteinome of Protein Kinases as a Target in Drug Development. Angew. Chem. Int. Ed 2018, 57 (16), 4372–4385. DOI: 10.1002/anie.201707875. [DOI] [PubMed] [Google Scholar]
- (30).Wang Y; Dix MM; Bianco G; Remsberg JR; Lee H-Y; Kalocsay M; Gygi SP; Forli S; Vite G; Lawrence RM; Parker CG; Cravatt BF Expedited Mapping of the Ligandable Proteome Using Fully Functionalized Enantiomeric Probe Pairs. Nat. Chem 2019, 11 (12), 1113–1123. DOI: 10.1038/s41557-019-0351-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Resnick E; Bradley A; Gan J; Douangamath A; Krojer T; Sethi R; Geurink PP; Aimon A; Amitai G; Bellini D; Bennett J; Fairhead M; Fedorov O; Gabizon R; Gan J; Guo J; Plotnikov A; Reznik N; Ruda GF; Díaz-Sáez L; Straub VM; Szommer T; Velupillai S; Zaidman D; Zhang Y; Coker AR; Dowson CG; Barr HM; Wang C; Huber KVM; Brennan PE; Ovaa H; von Delft F; London N.Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc 2019, 141 (22), 8951–8968. DOI: 10.1021/jacs.9b02822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Brulet JW; Borne AL; Yuan K; Libby AH; Hsu K-L Liganding Functional Tyrosine Sites on Proteins Using Sulfur–Triazole Exchange Chemistry. J. Am. Chem. Soc 2020, 142 (18), 8270–8280. DOI: 10.1021/jacs.0c00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Huang T; Hosseinibarkooie S; Borne AL; Granade ME; Brulet JW; Harris TE; Ferris HA; Hsu K-L Chemoproteomic Profiling of Kinases in Live Cells Using Electrophilic Sulfonyl Triazole Probes. Chem. Sci 2021, 12 (9), 3295–3307. DOI: 10.1039/D0SC06623K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lin Z; Wang X; Bustin KA; Shishikura K; McKnight NR; He L; Suciu RM; Hu K; Han X; Ahmadi M; Olson EJ; Parsons WH; Matthews ML Activity-Based Hydrazine Probes for Protein Profiling of Electrophilic Functionality in Therapeutic Targets. ACS Cent. Sci 2021, 7 (9), 1524–1534. DOI: 10.1021/acscentsci.1c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Grams RJ; Hsu K-L Reactive Chemistry for Covalent Probe and Therapeutic Development. Trends Pharmacol. Sci 2022, 43 (3), 249–262. DOI: 10.1016/j.tips.2021.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Stephanopoulos N; Francis MB Choosing an Effective Protein Bioconjugation Strategy. Nat. Chem. Biol 2011, 7 (12), 876–884. DOI: 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]
- (37).Spicer CD; Davis BG Selective Chemical Protein Modification. Nat. Commun 2014, 5 (1), 4740. DOI: 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- (38).Shannon DA; Weerapana E.Covalent Protein Modification: The Current Landscape of Residue-Specific Electrophiles. Curr. Opin. Chem. Biol 2015, 24, 18–26. DOI: 10.1016/j.cbpa.2014.10.021. [DOI] [PubMed] [Google Scholar]
- (39).Roberts AM; Ward CC; Nomura DK Activity-Based Protein Profiling for Mapping and Pharmacologically Interrogating Proteome-Wide Ligandable Hotspots. Curr. Opin. Biotechnol 2017, 43, 25–33. DOI: 10.1016/j.copbio.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).deGruyter JN; Malins LR; Baran PS Residue-Specific Peptide Modification: A Chemist’s Guide. Biochemistry 2017, 56 (30), 3863–3873. DOI: 10.1021/acs.biochem.7b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hoyt EA; Cal PMSD; Oliveira BL; Bernardes GJL Contemporary Approaches to Site-Selective Protein Modification. Nat. Rev. Chem 2019, 3 (3), 147–171. DOI: 10.1038/s41570-019-0079-1. [DOI] [Google Scholar]
- (42).Zanon PRA; Yu F; Musacchio PZ; Lewald L; Zollo M; Krauskopf K; Chang CJ; Toste FD; Nesvizhskii AI; Hacker SM Profiling the Proteome-Wide Selectivity of Diverse Electrophiles. ChemRxiv. 2021, DOI: 10.26434/chemrxiv-2021-w7rss-v2. [DOI] [Google Scholar]
- (43).Weerapana E; Simon GM; Cravatt BF Disparate Proteome Reactivity Profiles of Carbon Electrophiles. Nat. Chem. Biol 2008, 4 (7), 405–407. DOI: 10.1038/nchembio.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Weerapana E; Wang C; Simon GM; Richter F; Khare S; Dillon MBD; Bachovchin DA; Mowen K; Baker D; Cravatt BF Quantitative Reactivity Profiling Predicts Functional Cysteines in Proteomes. Nature 2010, 468 (7325), 790–795. DOI: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Abbasov ME; Kavanagh ME; Ichu T-A; Lazear MR; Tao Y; Crowley VM; am Ende CW; Hacker SM; Ho J; Dix MM; Suciu R; Hayward MM; Kiessling LL; Cravatt BF A Proteome-Wide Atlas of Lysine-Reactive Chemistry. Nat. Chem 2021, 13 (11), 1081–1092. DOI: 10.1038/s41557-021-00765-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hahm HS; Toroitich EK; Borne AL; Brulet JW; Libby AH; Yuan K; Ware TB; McCloud RL; Ciancone AM; Hsu K-L Global Targeting of Functional Tyrosines Using Sulfur-Triazole Exchange Chemistry. Nat. Chem. Biol 2019, 16 (2), 150–159. DOI: 10.1038/s41589-019-0404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ma N; Hu J; Zhang Z-M; Liu W; Huang M; Fan Y; Yin X; Wang J; Ding K; Ye W; Li Z.2 H -Azirine-Based Reagents for Chemoselective Bioconjugation at Carboxyl Residues Inside Live Cells. J. Am. Chem. Soc 2020, 142 (13), 6051–6059. DOI: 10.1021/jacs.9b12116. [DOI] [PubMed] [Google Scholar]
- (48).Bach K; Beerkens BLH; Zanon PRA; Hacker SM Light-Activatable, 2,5-Disubstituted Tetrazoles for the Proteome-Wide Profiling of Aspartates and Glutamates in Living Bacteria. ACS Cent. Sci 2020, 6 (4), 546–554. DOI: 10.1021/acscentsci.9b01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Chan J; Dodani SC; Chang CJ Reaction-Based Small-Molecule Fluorescent Probes for Chemoselective Bioimaging. Nat. Chem 2012, 4 (12), 973–984. DOI: 10.1038/nchem.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bruemmer KJ; Crossley SWM; Chang CJ Activity‐Based Sensing: A Synthetic Methods Approach for Selective Molecular Imaging and Beyond. Angew. Chem. Int. Ed 2020, 59 (33), 13734–13762. DOI: 10.1002/anie.201909690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Iovan DA; Jia S; Chang CJ Inorganic Chemistry Approaches to Activity-Based Sensing: From Metal Sensors to Bioorthogonal Metal Chemistry. Inorg. Chem 2019, 58 (20), 13546–13560. DOI: 10.1021/acs.inorgchem.9b01221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Jia S; He D; Chang CJ Bioinspired Thiophosphorodichloridate Reagents for Chemoselective Histidine Bioconjugation. J. Am. Chem. Soc 2019, 141 (18), 7294–7301. DOI: 10.1021/jacs.8b11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Lin S; Yang X; Jia S; Weeks AM; Hornsby M; Lee PS; Nichiporuk RV; Iavarone AT; Wells JA; Toste FD; Chang CJ Redox-Based Reagents for Chemoselective Methionine Bioconjugation. Science 2017, 355 (6325), 597–602. DOI: 10.1126/science.aal3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Taylor MT; Nelson JE; Suero MG; Gaunt MJ A Protein Functionalization Platform Based on Selective Reactions at Methionine Residues. Nature 2018, 562 (7728), 563–568. DOI: 10.1038/s41586-018-0608-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Kim J; Li BX; Huang RY-C; Qiao JX; Ewing WR; MacMillan DWC Site-Selective Functionalization of Methionine Residues via Photoredox Catalysis. J. Am. Chem. Soc 2020, 142 (51), 21260–21266. DOI: 10.1021/jacs.0c09926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Christian AH; Jia S; Cao W; Zhang P; Meza AT; Sigman MS; Chang CJ; Toste FD A Physical Organic Approach to Tuning Reagents for Selective and Stable Methionine Bioconjugation. J. Am. Chem. Soc 2019, 141 (32), 12657–12662. DOI: 10.1021/jacs.9b04744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Elledge SK; Tran HL; Christian AH; Steri V; Hann B; Toste FD; Chang CJ; Wells JA Systematic Identification of Engineered Methionines and Oxaziridines for Efficient, Stable, and Site-Specific Antibody Bioconjugation. Proc. Natl. Acad. Sci 2020, 117 (11), 5733–5740. DOI: 10.1073/pnas.1920561117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ohata J; Krishnamoorthy L; Gonzalez MA; Xiao T; Iovan DA; Toste FD; Miller EW; Chang CJ An Activity-Based Methionine Bioconjugation Approach To Developing Proximity-Activated Imaging Reporters. ACS Cent. Sci 2020, 6 (1), 32–40. DOI: 10.1021/acscentsci.9b01038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Hall YD; Gross EJ; El-Shaffey HM; Ohata J.Methionine-Selective Protein Bioconjugation. Dojin News 2021, 177, 2. [Google Scholar]
- (60).Aledo JC Methionine in Proteins: The Cinderella of the Proteinogenic Amino Acids. Protein Sci. 2019, 28 (10), 1785–1796. DOI: 10.1002/pro.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Lee BC; Péterfi Z; Hoffmann FW; Moore RE; Kaya A; Avanesov A; Tarrago L; Zhou Y; Weerapana E; Fomenko DE; Hoffmann PR; Gladyshev VN MsrB1 and MICALs Regulate Actin Assembly and Macrophage Function via Reversible Stereoselective Methionine Oxidation. Mol. Cell 2013, 51 (3), 397–404. DOI: 10.1016/j.molcel.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Ciorba MA; Heinemann SH; Weissbach H; Brot N; Hoshi T.Modulation of Potassium Channel Function by Methionine Oxidation and Reduction. Proc. Natl. Acad. Sci 1997, 94 (18), 9932–9937. DOI: 10.1073/pnas.94.18.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Levine RL; Mosoni L; Berlett BS; Stadtman ER Methionine Residues as Endogenous Antioxidants in Proteins. Proc. Natl. Acad. Sci 1996, 93 (26), 15036–15040. DOI: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Cotton AD; Wells JA; Seiple IB Biotin as a Reactive Handle to Selectively Label Proteins and DNA with Small Molecules. ACS Chem. Biol 2021, DOI: 10.1021/acschembio.1c00252. [DOI] [PubMed] [Google Scholar]
- (65).Matsushime H; Ewen ME; Strom DK; Kato J-Y; Hanks SK; Roussel MF; Sherr CJ Identification and Properties of an Atypical Catalytic Subunit (P34PSK-J3/Cdk4) for Mammalian D Type G1 Cyclins. Cell 1992, 71, 323–334. [DOI] [PubMed] [Google Scholar]
- (66).Malumbres M; Barbacid M.Cell Cycle, CDKs and Cancer: A Changing Paradigm. Nat. Rev. Cancer 2009, 9 (3), 153–166. DOI: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- (67).Asghar U; Witkiewicz AK; Turner NC; Knudsen ES The History and Future of Targeting Cyclin-Dependent Kinases in Cancer Therapy. Nat. Rev. Drug Discov 2015, 14 (2), 130–146. DOI: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Kim S; Tiedt R; Loo A; Horn T; Delach S; Kovats S; Haas K; Engstler BS; Cao A; Pinzon-Ortiz M; Mulford I; Acker MG; Chopra R; Brain C; di Tomaso E; Sellers WR; Caponigro G.The Potent and Selective Cyclin-Dependent Kinases 4 and 6 Inhibitor Ribociclib (LEE011) Is a Versatile Combination Partner in Preclinical Cancer Models. Oncotarget 2018, 9 (81), 35226–35240. DOI: 10.18632/oncotarget.26215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).O’Leary B; Finn RS; Turner NC Treating Cancer with Selective CDK4/6 Inhibitors. Nat. Rev. Clin. Oncol 2016, 13 (7), 417–430. DOI: 10.1038/nrclinonc.2016.26. [DOI] [PubMed] [Google Scholar]
- (70).Malumbres M; Carnero A.Cell Cycle Deregulation: A Common Motif in Cancer. Prog. Cell Cycle Res 2003, 5, 5–18. [PubMed] [Google Scholar]
- (71).Zhang J; Yang PL; Gray NS Targeting Cancer with Small Molecule Kinase Inhibitors. Nat. Rev. Cancer 2009, 9 (1), 28–39. DOI: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Goldsmith EJ; Akella R; Min X; Zhou T; Humphreys JM Substrate and Docking Interactions in Serine/Threonine Protein Kinases. Chem. Rev 2007, 107 (11), 5065–5081. DOI: 10.1021/cr068221w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Liao JJ-L Molecular Recognition of Protein Kinase Binding Pockets for Design of Potent and Selective Kinase Inhibitors. J. Med. Chem 2007, 50 (3), 409–424. DOI: 10.1021/jm0608107. [DOI] [PubMed] [Google Scholar]
- (74).Scott DE; Coyne AG; Hudson SA; Abell C.Fragment-Based Approaches in Drug Discovery and Chemical Biology. Biochemistry 2012, 51 (25), 4990–5003. DOI: 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- (75).Hopkins AL; Keserü GM; Leeson PD; Rees DC; Reynolds CH The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discov 2014, 13 (2), 105–121. DOI: 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- (76).Erlanson DA; Fesik SW; Hubbard RE; Jahnke W; Jhoti H.Twenty Years on: The Impact of Fragments on Drug Discovery. Nat. Rev. Drug Discov 2016, 15 (9), 605–619. DOI: 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- (77).Weerapana E; Speers AE; Cravatt BF Tandem Orthogonal Proteolysis-Activity-Based Protein Profiling (TOP-ABPP)—a General Method for Mapping Sites of Probe Modification in Proteomes. Nat. Protoc 2007, 2 (6), 1414–1425. DOI: 10.1038/nprot.2007.194. [DOI] [PubMed] [Google Scholar]
- (78).Kato JY; Matsuoka M; Strom DK; Sherr CJ Regulation of Cyclin D-Dependent Kinase 4 (Cdk4) by Cdk4-Activating Kinase. Mol. Cell Biol 1994, 14 (4), 2713–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Bisteau X; Paternot S; Colleoni B; Ecker K; Coulonval K; De Groote P; Declercq W; Hengst L; Roger PP CDK4 T172 Phosphorylation Is Central in a CDK7-Dependent Bidirectional CDK4/CDK2 Interplay Mediated by P21 Phosphorylation at the Restriction Point. PLoS Genet. 2013, 9 (5), 1–21. DOI: 10.1371/journal.pgen.1003546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Malumbres M.Cyclin-Dependent Kinases. Genome Biol. 2014, 15 (6), 1–10. DOI: 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Grillo M; Bott MJ; Khandke N; McGinnis JP; Miranda M; Meyyappan M; Rosfjord EC; Rabindran SK Validation of Cyclin D1/CDK4 as an Anticancer Drug Target in MCF-7 Breast Cancer Cells: Effect of Regulated Overexpression of Cyclin D1 and SiRNA-Mediated Inhibition of Endogenous Cyclin D1 and CDK4 Expression. Breast Cancer Res. Treat 2006, 95 (2), 185–194. DOI: 10.1007/s10549-005-9066-y. [DOI] [PubMed] [Google Scholar]
- (82).Choi EJ Hesperetin Induced G1-Phase Cell Cycle Arrest in Human Breast Cancer MCF-7 Cells: Involvement of CDK4 and P21. Nutr. Cancer 2007, 59 (1), 115–119. DOI: 10.1080/01635580701419030. [DOI] [PubMed] [Google Scholar]
- (83).Narasimha AM; Kaulich M; Shapiro GS; Choi YJ; Sicinski P; Dowdy SF Cyclin D Activates the Rb Tumor Suppressor by Mono-Phosphorylation. eLife 2014, 3, e02872. DOI: 10.7554/eLife.02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Schachter MM; Merrick KA; Larochelle S; Hirschi A; Zhang C; Shokat KM; Rubin SM; Fisher RP A Cdk7-Cdk4 T-Loop Phosphorylation Cascade Promotes G1 Progression. Mol. Cell 2013, 50 (2), 250–260. DOI: 10.1016/j.molcel.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Bockstaele L; Kooken H; Libert F; Paternot S; Dumont JE; de Launoit Y; Roger PP; Coulonval K.Regulated Activating Thr172 Phosphorylation of Cyclin-Dependent Kinase 4(CDK4): Its Relationship with Cyclins and CDK “Inhibitors.” Mol. Cell. Biol 2006, 26 (13), 5070–5085. DOI: 10.1128/MCB.02006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Rao RSP; Møller IM; Thelen JJ; Miernyk JA Convergent Signaling Pathways—Interaction between Methionine Oxidation and Serine/Threonine/Tyrosine O-Phosphorylation. Cell Stress Chaperones 2015, 20 (1), 15–21. DOI: 10.1007/s12192-014-0544-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Oien DB; Carrasco GA; Moskovitz J.Decreased Phosphorylation and Increased Methionine Oxidation of α-Synuclein in the Methionine Sulfoxide Reductase A Knockout Mouse. J. Amino Acids 2011, 2011, 1–6. DOI: 10.4061/2011/721094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Veredas FJ; Cantón FR; Aledo JC Methionine Residues around Phosphorylation Sites Are Preferentially Oxidized in Vivo under Stress Conditions. Sci. Rep 2017, 7 (1), 40403. DOI: 10.1038/srep40403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.