

Abstract

Polymerization of soluble amyloid beta (Aβ) peptide into protease-stable insoluble fibrillary aggregates is a critical step in the pathogenesis of Alzheimer’s disease (AD). The N-terminal (NT) hydrophobic central domain fragment 16KLVFF20 plays an important role in the formation and stabilization of β-sheets by self-recognition of the parent Aβ peptide, followed by aggregation of Aβ in the AD brain. Here, we analyze the effect of the NT region inducing β-sheet formation in the Aβ peptide by a single amino acid mutation in the native Aβ peptide fragment. We designed 14 hydrophobic peptides (NT-01 to NT-14) by a single mutation at 18Val by using hydrophobic residues leucine and proline in the natural Aβ peptide fragment (KLVFFAE) and analyzed its effect on the formation of Aβ aggregates. Among all these peptides, NT-02, NT-03, and NT-13 significantly affected the Aβ aggregate formation. When the NT peptides were coincubated with the Aβ peptide, a significant reduction in β-sheet formation and increment in random coil content of Aβ was seen, confirmed by circular dichroism spectroscopy and Fourier transform infrared spectroscopy, followed by the reduction of fibril formation measured by the thioflavin-T (ThT) binding assay. The aggregation inhibition was monitored by Congo red and ThT staining and electron microscopic examination. Moreover, the NT peptides protect the PC-12 differentiated neurons from Aβ-induced toxicity and apoptosis in vitro. Thus, manipulation of the Aβ secondary structure with protease-stable ligands that promote the random coil conformation may provide a tool to control the Aβ aggregates observed in AD patients.
Keywords: amyloid beta, Alzheimer’s disease, β-sheet breakers, neuroprotection, protease-stable ligands
Introduction
The amyloid beta (Aβ) peptide is the most important protein constituent in neuritic senile plaques1 and cerebrovascular amyloid deposition.2,3 Overproduction and polymerization of the Aβ peptide into protease-stable insoluble aggregates is a major step in the pathogenesis of Alzheimer’s disease (AD).4 In physiological conditions, the Aβ peptide easily aggregates into fibrillary β-sheets and then it forms insoluble aggregates.5 In the provision of this argument, particular mutations in the gene encoding the amyloid beta-precursor protein (APP),6 which leads to excess production of Aβ peptide with heterogenic structural variants (Aβ40, Aβ42, etc.), and these Aβ peptides are easily formed aggregates in cerebral amyloid angiopathy and AD brain.3,7 The Aβ aggregation process will proceed through the formation of a polymorphic oligomeric nucleus, and then it initiates the formation of protofibrils and followed by fibril elongation, and they individually show a range of cellular toxicities.8 Amyloid fibrils are larger, hydrophobic, and insoluble forms with toxic and cause neurotoxicity and dementia with common cytopathic effects that contribute to the pathogenesis of AD and another amyloidosis.9,10 Compounds, which can interfere with the aggregation process of the Aβ peptide, which may lead to effective and potent therapeutics for AD. Here, we have focused on (i) developing the metabolic protease-stable peptide molecules having D-configuration, (ii) identifying the peptides which interact with the hydrophobic core residues which responsible for initial nucleus formation, and (iii) finally studying the effects of these metabolically stable peptides on β-sheet formation in vitro. The Aβ peptide is amphiphilic, with a hydrophobic C-terminal residue 29GAIIGLMVGGVVIA42 and the N-terminal (NT) residue 10YEVHHQKLVFFAEDV24. The C-terminal residue inherently adopts β-sheet conformation and the NT residue 10YEVHHQKLVFFAEDV24, which allows the existence of a dynamic equilibrium between α-helix and β-sheet conformation. The Aβ peptide will exist in two conformations depending upon the secondary structure adopted by the N-terminus domain and its effects on its rate of β-sheet formation.11 The N-terminus amino acid residue 16KLVFF20 is a key fragment, which plays an important role in the self-recognition of the parent Aβ peptide, and then leads to the formation and stabilization of β-sheet fibrils, but for the Aβ peptide self-recognition, 18Val is not completely necessary.12 The alteration of the hydrophobic residue in the natural Aβ peptide structure shows the pipeline to the development of hydrophobic core-based β-sheet breakers for the therapy of AD.13 The substitution of hydrophobic amino acids in the N-terminus hydrophobic fragment (KLVFF) of the natural Aβ peptide reduces the formation of the β-sheet followed by the polymerization of the Aβ peptide into aggregates.14 In the present study, our approach to generating β-sheet breakers, for this Aβ self-recognition hydrophobic core residue 16KLVFFAE22, was used to achieve specificity and good binding affinity15 but the replacement of a key amino acid residue (18Val which is responsible for β-sheet formation) by an amino acid which is unable to fit with the hydrophobic core residue and may not involve in the β-sheet formation. Here, we have developed a small library of NT peptide molecules with the replacement of this 18Val by using a hydrophobic amino acid, leucine, which can increase α-helix stabilization,16 and proline, which can stabilize the α-helix and disrupt the β-sheet structure effectively.17 The peptides with Leu and Pro in their backbone make the particular structure, and they may interfere with the full length of the Aβ peptide and prevent self-recognition and inhibit the β-sheet-based Aβ aggregate formation. From this library, we found that peptides NT-02, NT-03, and NT-13 are potent β-sheet breakers. These newly designed NT peptides not only alter Aβ self-assembly but also reduce the Aβ toxicity in vitro. These NT peptides show significant activity in various regions which (i) selectively interact with the N-terminus hydrophobic region of Aβ and interfere with the formation of Aβ aggregates, (ii) are nontoxic to PC-12 derived neurons, showing remarkable neuroprotection against Aβ-induced toxicity in PC-12 derived neurons, (iii) have good serum stability, and (iv) peptides easily cross the blood–brain barrier (BBB) in mice.
Results and Discussion
Synthesis, Purification, and Characterization of Peptides
All the peptides (NT-01 to NT-14) were synthesized by using the solid-phase peptide synthesis (SPPS) method using Fmoc-protected rink amide resin. The synthetic scheme is described in Scheme 1. The conjugation of the peptide with fluorescein dye (5(6)-carboxyfluorescein) was achieved at the NT of the peptides to carry out the cellular uptake study. All the peptides were purified through RP-HPLC and characterized by high-resolution mass spectrometry (HRMS) and MALDI mass spectrometry (Tables 1 and S1, and Figures S7–S43).
Scheme 1. Synthetic Scheme for the Peptides by Using Rink Amide Resin Exemplified by Peptide NT-02.
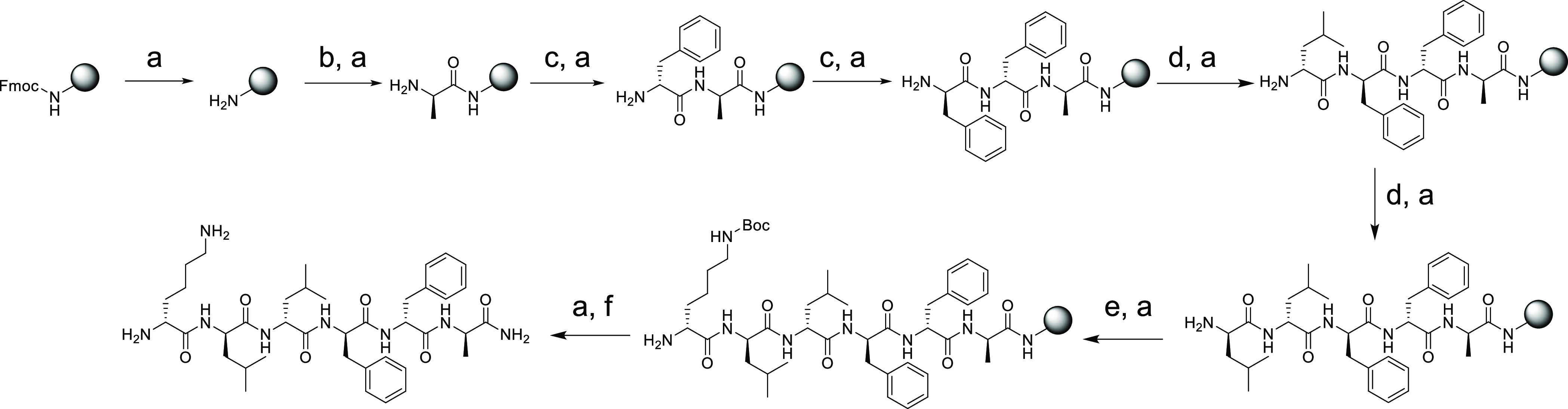
Reagents and conditions: (a) 20% piperidine in DMF, 3 min (two times); (b) Fmoc-d-Ala-OH (10 equiv), HBTU (10 equiv), DIPEA (10 equiv), DMF, 7 min; (c) Fmoc-d-Phe-OH (5 equiv), HBTU (5 equiv), DIPEA (10 equiv), DMF, 7 min; (d) Fmoc-d-Leu-OH (5 equiv), HBTU (5 equiv), DIPEA (10 equiv), DMF, 7 min; (e) Fmoc-d-Lys (Boc)-OH (5 equiv), HBTU (5 equiv), DIPEA (10 equiv), DMF, 7 min; (f) 92.5% TFA, 2.5% water, 2.5% phenol, 2.5% 1.2-ethylenedithiol, 12 h.
Table 1. Sequence, HRMS, and HPLC Data of the Synthesized Peptides.
| mass |
HPLC |
|||||
|---|---|---|---|---|---|---|
| s. no. | peptide sequence | molecular formula | calculated | obtained (M + H) | tR (min) | purity (%) |
| 1 | H2N-KLLFFAE-NH2 | C44H67N9O9 | 865.5062 | 865.6390 | 5.196 | 100 |
| 2 | H2N-KLLFFA-NH2 | C39H60N8O6 | 736.4636 | 737.7675 | 5.275 | 100 |
| 3 | H2N-KLLFF-NH2 | C36H55N7O5 | 665.4265 | 666.7092 | 5.298 | 100 |
| 4 | H2N-KLLF-NH2 | C27H46N6 | 518.3581 | 519.3088 | 5.303 | 100 |
| 5 | H2N-LLFFAE-NH2 | C38H55N6 | 737.4112 | 738.5652 | 5.301 | 100 |
| 6 | H2N-LLFFA-NH2 | C33H48N6 | 608.3686 | 609.4024 | 5.280 | 100 |
| 7 | H2N-LLFF-NH2 | C30H43N5 | 537.3315 | 538.5833 | 5.232 | 100 |
| 8 | H2N-LFFAE-NH2 | C32H44N6 | 624.3271 | 625.3097 | 5.247 | 99.12 |
| 9 | H2N-LFFA-NH2 | C27H37N5 | 495.2846 | 496.2403 | 5.156 | 100 |
| 10 | H2N-KLPFFAE-NH2 | C43H63N9 | 849.4749 | 850.7135 | 5.201 | 100 |
| 11 | H2N-KLPFFA-NH2 | C38H56N8 | 720.4323 | 720.6613 | 5.190 | 98.93 |
| 12 | H2N-KLPFF-NH2 | C35H51N7 | 649.3952 | 650.2834 | 5.251 | 100 |
| 13 | H2N-LPFFAE-NH2 | C37H51N7 | 721.3799 | 721.1677 | 5.203 | 100 |
| 14 | H2N-LPFFA-NH2 | C32H44N6 | 592.3373 | 593.5271 | 5.178 | 99.58 |
| 15 | H2N-KLVFF-NH2 | C35H53N7 | 651.4108 | 652.3820 | 5.222 | 98.98 |
| 16 | Aβ42 | NH2-1DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA42-COOH | ||||
MTT Cell Viability Assay
For the initial screening of newly synthesized NT peptides against Aβ peptide aggregation, we performed the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) cell viability assay to evaluate the effect of NT peptides against Aβ aggregation and Aβ-induced toxicity in PC-12-derived neuronal cells. PC-12 (rat pheochromocytoma cells) cells have been previously well-developed models for neuronal toxicity studies and to show their sensitivity to the toxic effects of Aβ aggregates.18 Reduction of MTT to formazan takes place in live cells by the NAD(P)H-dependent oxidoreductase enzyme, but in dead cells, this reduction process will not take place because of the absence of the NAD(P)H-dependent oxidoreductase enzyme; by this experiment, we can measure the cell proliferation and cellular viability after the treatment of the compound on PC-12 cells in drug screening.19,20 First, we checked the cellular toxicity of peptides. For this, we treated different concentrations (5, 10, 20, 40, 80, and 160 μM) of NT peptides with differentiated PC-12-derived neurons. The results indicated that a maximum number of NT peptides did not show any toxic effect on the PC-12-derived neurons (Figures 1 and S1). However, only peptides NT-06, NT-07, and NT-08 showed 71.68, 42.54, and 50.07% of cell viability at a 160 μM concentration, respectively, as shown in Figure S1 and Table S2. Then, we check the neuroprotection capability of newly synthesized hydrophobic NT peptides against Aβ42 peptide-induced toxicity in PC-12-derived neurons by performing the MTT cell viability assay. It has been described before that the amino acid residues Aβ (16–20) (KLVFF) show strong activity against Aβ toxicity and prevent fibril formation.14 In the MTT assay, KLVFF (NT-15) is taken as a positive control. The Aβ42-untreated PC-12-derived neurons were taken as 100% viable. First, PC-12-derived neurons were treated with the Aβ42 monomer peptide alone and NT peptides with the Aβ42 peptide and incubated for 24 h. Since formazan formation takes place by viable cells only. When the Aβ42 peptide was treated alone, a reduction in the number of viable cells along with a decrease in the formation of formazan was observed, indicating cytotoxicity induced by the Aβ42 peptide. However, NT-01, NT-02, NT-03, NT-04, NT-05, NT-06, NT-07, NT-08, NT-09, NT-10, NT-11, NT-12, NT-13, and NT-14 peptides showed 43.20, 75.13, 76.64, 73.46, 74.38, 62.61, 35.54, 44.35, 40.33, 50.19, 60.08, 36.10, 76.08, and 60.58% of cell viability against Aβ42-induced toxicity with 5 μM Aβ42 and 5 μM NT peptides, respectively (1:1 ratio) (Figures 1E–G, S2, and Table S3). When PC-12-derived neurons were treated with the Aβ42 peptide alone, the cells died and the cell viability was 38.05% only, followed by positive control KLVFF (NT-15) that showed a 72.08% cell viability (Figure 1H). Moreover, we checked for the inhibitory effects on the Aβ42 peptide aggregation at different concentrations, 5, 10, 20, 40, 80, and 160 μM, of the NT peptide (Figures 1E–H and S2). The results show that the maximum number of NT peptides rescue the PC-12 neurons from Aβ42-induced toxicity. From the experimental results, peptides NT-02, NT-03, and NT-13 showed high neuroprotection against Aβ42-induced toxicity and showed a cell viability of nearly 85–90% by inhibiting the aggregation and reducing the neurotoxicity (Figure 1E–G). When compared with the positive control KLVFF peptide, the cell viability and neuroprotection of NT-02, NT-03, and NT-13 were higher, but those of the remaining peptides NT-01, NT-04, NT-05, NT-06, NT-07, NT-08, NT-09, NT-10, NT-11, NT-12, and NT-014 were less or nearly the same as those of the positive control. Furthermore, we incubated the NT peptides with Aβ42 for 7 days with constant stirring at 37 °C to form fibrillary aggregates. Then, the fibrillary Aβ42 aggregates were subjected to PC-12 derived neurons to check the fibrillary Aβ42 aggregate-induced toxicity in PC-12 neuronal cells. Then, we performed the MTT assay, with results indicating that peptides NT-02, NT-03, and NT-13 were more potent than all other peptides (Figures 1I–K), showing a 94.30, 92.41, and 92.16% viability, respectively, at a 160 μM concentration. This MTT assay clearly shows that the peptides NT-02, NT-03, and NT-13 are highly potent and active against Aβ peptide aggregation and protect the neurons from Aβ-induced toxicity. Then, we checked the cellular uptake of our peptides using PC-12-derived neurons. For this, PC-12-derived neurons were treated with 5(6)-carboxyfluorescein-conjugated NT peptides. The experimental results show that (Figure 2) neurons were healthy in morphology, and cell bodies and dendrites were found to stain with green color 5(6)-carboxyfluorescein-conjugated NT-02, NT-03, and NT-13 peptides, and these outcomes suggest that our peptides show sufficient cellular uptake in the PC-12-derived neurons, and this opens the path to performing in vitro cellular studies.
Figure 1.

Cellular viability of peptides (A) NT-02, (B) NT-03, (C) NT-13, and (D) positive control NT-15 (KLVFF) in PC-12-derived neurons by using the MTT assay. Effects of NT peptides on the Aβ42 peptide-induced cytotoxicity in PC-12-derived neurons. (E) PC-12-derived neurons were treated with the Aβ42 peptide (5 μM) alone and Aβ42 peptide with (E) NT-02, (F) NT-03, (G) NT-13 peptide, and (H) positive control NT-15 for 24 h after which their ability to reduce MTT was measured. Cellular viability of PC12-derived neurons after being treated with Aβ42 aggregates (5 μM) alone and Aβ42 aggregates with (I) NT-02, (J) NT-03, (K) NT-13 peptides, and (L) positive control NT-15 for 24 h after which their ability to reduce MTT was measured. Error bars represent mean ± standard deviation (SD), n = 3. Statistical data were analyzed by a one-way ANOVA test by the multiple comparison tests (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, vs Aβ42) using software (GraphPad Prism, ISI, San Diego, CA).
Figure 2.
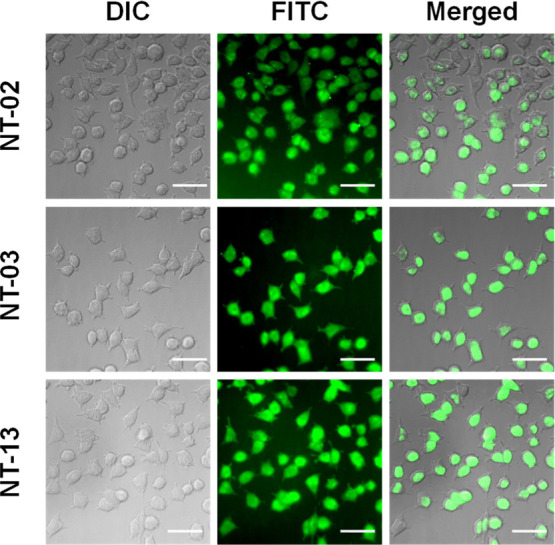
Cellular uptake images of 5(6)-carboxyfluorescein-attached NT-02, NT-03, and NT-13 peptides in differentiated PC-12-derived neurons reveal a significant cellular uptake. The scale bar corresponds to 50 μm.
Modulation of Neuronal Apoptosis
NT peptides protect the neurons from Aβ42-induced toxicity by preventing aggregation. Here, we assessed the neuronal toxicity by using the cell apoptosis assay. For this, PC-12-derived neurons were treated with Aβ42 (10 μM) alone and Aβ42 (10 μM) with NT-02, NT-03, and NT-13 peptides (10 μM) for 24 h. For the analysis of live cells and dead cells, we stained the neurons with Calcein AM and propidium iodide (PI). The reduction of apoptosis in PC-12-derived neurons by NT peptides was confirmed by using PI staining, which can stain late apoptotic cells and show red fluorescence. Figure 3 shows the effective rescue of PC-12-derived neurons from Aβ42 induced apoptosis by NT peptides. The fluorescence intensity of PI (apoptosis cells) was quantified and is shown in Figure 4A. The Aβ42-only-treated neurons showed 31.04-fold apoptosis after 24 h of incubation, and NT-02, NT-03, and NT-13 peptides showed significant protection and rescued the neurons from apoptosis with a reduction to 2.84-, 6.21-, and 4.13-fold apoptotic cells, respectively. These results suggest that NT peptides are nontoxic in nature and insignificant apoptosis inducers.
Figure 3.
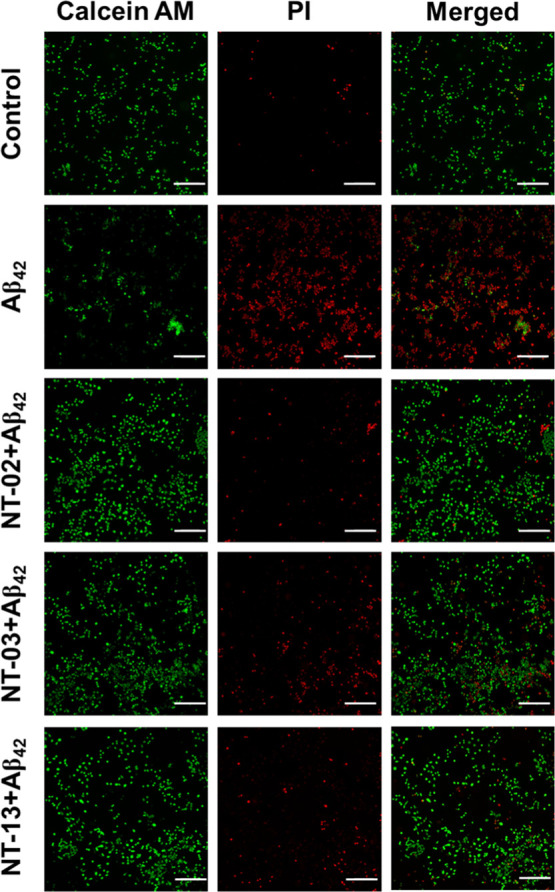
Cell apoptosis. Fluorescence images of differentiated PC-12-derived neuronal cells stained with Calcein AM and PI for normal and late apoptotic cell visualization. Initiation of apoptosis by Aβ42 peptide treatment compared to the control and rescue by NT-02, NT-03, and NT-13 peptides. The scale bar corresponds to 200 μm.
Figure 4.

(A) Quantification of percentage apoptotic cells of different treatments shows the significant rescue of cells from Aβ42-induced apoptosis by NT-02, NT-03, and NT-13 peptides. Effects of NT peptides on Aβ42 aggregation measured by the ThT fluorescence assay. (B) Peptides NT-02, (C) NT-03, and (D) NT-13 in Aβ42 aggregation. The control sample represents the Aβ42 peptide (5 μM) alone. (E–G) Effects of peptides NT-02, NT-03, and NT-13 on Aβ42 aggregation for 7 days. Aβ42 (5 μM) and peptide at a 10 μM concentration. Control sample represents the Aβ42 peptide (5 μM) alone. (H) Serum stability of NT peptides in human serum up to 24 h. Error bars represent mean ± standard deviation (SD), n = 3. Statistical data were analyzed by a one-way ANOVA test by the multiple comparison tests (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, vs Aβ42) using software GraphPad Prism (ISI, San Diego, CA).
ThT Assay
The Aβ peptide has different morphological forms in the AD brain. The Aβ peptide monomer can form higher-order assemblies ranging from the low molecular weight of dimers, trimers, and tetramers to higher-range molecular weight oligomers, protofibrils, and fibrils. Aβ fibrils are insoluble and they induce membrane disruption, synaptic dysfunction, and finally neuronal death. Thioflavin-T (ThT) is a benzothiazole dye; it shows enhancement of fluorescence intensity upon binding to Aβ fibril aggregates, and it is commonly used to monitor the Aβ fibrillary aggregation, both in vitro and in vivo.21 Additionally, the fluorescence intensity of ThT indicates the quantity of Aβ aggregates present in the solution. The ThT assay was executed to evaluate the inhibition of Aβ aggregation using all the peptides. In the ThT study, the control experiment Aβ42 alone showed the increment of fluorescence intensity, but in the case of Aβ42 incubated with NT peptides, we did not find the increment of fluorescence intensity. From the experimental results, all the designed peptides have the potential ability to interact with the Aβ peptide and inhibit self-aggregation. First, we recorded the fluorescence intensity of the alone Aβ42. Taking the Aβ42 aggregation Aβ42 alone as 100%, the % aggregates were calculated for the samples Aβ42 treated with the NT peptides. For the positive control peptide, KLVFF (NT-15) was used. Figures 4B–D and S4 show the % of Aβ42 aggregates present in the Aβ42 peptide incubated alone, Aβ42 with NT peptides, and Aβ42 with KLVFF. From the ThT assay, the maximum of the NT peptides shows 45–60% inhibition at a concentration of 5 μM. However, peptides NT-02 (57.61%), NT-03 (54.26%), and NT-13 (48.41%) showed higher inhibition than the other peptides as well as a positive control NT-15(41.87). Then, we executed the dose-dependent ThT assay with Aβ42 peptide by using various concentrations of NT peptides. The positive control experimental results show that the % aggregation is 58.13, 55.02, 48.04, 39.18, 35.98, and 30.29%, but in the presence of peptide NT-02, the % aggregation is 42.39, 36.33, 29.77, 25.57, 22.14, and 21.04%, peptide NT-03 shows 45.74, 35.71, 25.34, 21.56, 20.63, and 19.37%, and peptide NT-13 shows 51.59, 35.61, 24.49, 23.15, 22.25, and 20.12% when Aβ42 was incubated with various concentrations (5, 10, 20, 40, 80, and 160 μM) of NT peptides, respectively (Figures 4B–D and Table S5). This means that the peptides inhibited about 78.96, 80.63, and 79.88% of the Aβ42 peptide aggregation at a 160 μM concentration of peptides NT-02, NT-03, and NT-13, respectively. This ThT assay was well supported by the MTT assay and clearly showed that NT-02, NT-03, and NT-13 peptides inhibited Aβ42 peptide aggregation. Moreover, using the ThT assay, we also checked if the peptides NT-02, NT-03, and NT-13 were in a time-dependent manner against the inhibition of Aβ42 peptide aggregation. Figures 4E–G shows that the Aβ42 peptide with NT peptides and Aβ42 alone. We can see that in the Aβ42 peptide alone, there was a dramatic increment in the ThT fluorescence intensity that could be ascribed to the aggregation state of the Aβ42 peptide. When the 12 h time represents the lag phase shows the absence of larger Aβ42 aggregates. After 12 h, the dynamic increase in the fluorescence intensity corresponds to the growing phase. The fluorescence intensity reached maximum saturation after about 96 h. The saturated fluorescence intensity was taken as 100%. Then, % aggregation was calculated for peptides NT-02, NT-03, and NT-13, which showed 35, 46, and 31% at 96 h of incubation, respectively. The NT peptides were incubated with the Aβ42 peptide, showing that the fluorescence intensity reached saturation at 72 h. Since the fluorescence intensity corresponded to the aggregation state of the Aβ42 peptides, a complete reduction of fluorescence intensity was observed in the presence of peptides NT-02, NT-03, and NT-13. This indicates that NT peptides can interfere with the Aβ42 peptide and in the self-assembly. In the meantime, the peptides showed their inhibitory effects on Aβ42 aggregation even after a long-time incubation. Next, we performed an isothermal titration calorimetry (ITC) study to know the binding affinity of NT peptides with the Aβ42 peptide. This study reveals that the binding affinities (Ka) of NT-02, NT-03, and NT-13 are about 2.25 × 106 ± 1.023 × 105, (3.47 ± 1.23) × 106, and 1.42 × 105 ± 3.42 × 104 M–1, respectively (Figure 5A–C). This result shows that peptides have a good binding affinity with the Aβ42 peptide. From the ThT fluorescence results, it was found that the peptides NT-02, NT-03, and NT-13 are more potent than all other peptides. The ThT experimental results further support the results we saw in the MTT assay.
Figure 5.

ITC experiment of NT peptides showing interaction with the Aβ42 peptide. (A) NT-02 peptide shows an affinity of 2.25 × 106 ± 1.023 × 105 M–1, (B) NT-03 peptide shows an affinity of (3.47 ± 1.23) × 106 M–1, and (C) NT-13 peptide shows an affinity of 1.42 × 105 ± 3.42 × 104 M–1. Molecular docking images of NT peptides with the Aβ42 peptide (PDB ID: 1Z0Q). (D) Docking studies of the NT-02 peptide with the Aβ42 peptide show H-bonding interaction with a −5.2 kcal/mol binding energy. (E) Docking studies of NT-03 with Aβ42 and showing H-bonding interactions with −5.3 kcal/mol binding energy. (F) Docking studies of NT-13 with Aβ42 and showing H-bonding interaction with a −4.5 kcal/mol binding energy.
Molecular Docking Studies
According to the amino acid analysis of the Aβ peptide by the Chou–Fasman22 and Garnier–Osguthorpe–Robson methods,23 the probability of β-sheet conformation is higher in the Aβ peptide within the regions that consist of the hydrophobic C-terminal region after residue 28 (residues 29–42) and the NT region (residues 10–24). The structure of the NT domain is important for Aβ fibril formation, and this region will show a dynamic equilibrium between the α-helix and β-sheet conformations of the Aβ peptide.24 In the hydrophobic segment in the C-terminus of the Aβ peptide, invariably, it adopts β-sheet conformation in aqueous solutions at independent pH and various temperatures. However, the N-terminus hydrophobic domain will show different conformations depending on the conditions. While at pH = 1–4 and greater than pH = 7, the Aβ peptide will show the α-helix conformation, at pH = 4–7, it rapidly forms and shows the β-sheet conformation.25−27 Moreover, the Aβ peptide will show the α-helix when the membrane mimics solvents that promotes an intramolecular hydrogen bonding with the Aβ peptide. Because of this, the Aβ peptide will exist in two conformations depending upon the secondary structure adopted by the N-terminus domain. Here, we perform molecular docking studies so that we can see how our peptide will interact with Aβ peptides and how it can reduce the β-sheet formation followed by reducing the Aβ aggregate formation. Molecular docking studies have been widely used for studying protein–ligand interactions and their binding modes. Here, we carried out molecular docking studies with the Aβ42 monomer structure (PDB ID: 1Z0Q)28 to find out the binding modes of our peptides with Aβ42. The molecular docking studies results show that there are multiple binding sites in Aβ42 for the binding of peptides. However, the most favorable binding site is shown in Figure 5D–F. From the docking study, all the peptides have selectively binding interactions with N-terminus hydrophobic region residues of Aβ42. Peptides NT-02 and NT-03 were bound with the Aβ42 peptide (PDB ID: 1Z0Q) at Glu11, His14, Gln15, and Asp23 and Glu3, Asp7, and His14 through hydrogen bonding interaction. Peptide NT-13 shows hydrogen bonding interaction with His14 and Tyr10 residues. The molecular docking studies reveal that the peptides NT-02, NT-03, and NT-13 can interact with the N-terminus region of the Aβ42 peptide and then prevent the self-recognition and β-sheet formation, followed by aggregation of the Aβ42 peptide. These results indicate that the N-terminus hydrophobic core residues 10 to 24 of Aβ are crucial for the formation of β-sheets. This result further supports the results we saw in the MTT assay and ThT assay.
Fourier Transform Infrared Spectroscopy Studies
The results from MTT assay, ThT assays, and docking studies attracted us to further study the peptides. The molecular docking experiment results show that peptides have a binding interaction at the N-terminus region of the Aβ42 peptide. When the peptide binds to the N-terminus region of Aβ42, it may reduce the stabilization of β-sheet conformation, followed by its aggregation. Here, we check the interaction of peptides with Aβ42 and their antiaggregation effect by a reduction in β-sheet formation by Fourier transform infrared (FTIR) spectroscopy.29,30 Moreover, the Aβ42 peptide will show a secondary structure like the α-helix and β-sheet. FTIR spectroscopy provides information about the secondary structure content of the Aβ42 peptide in a solution. The Aβ42 contains amide bonds and shows a characteristic absorption in the infrared (IR) spectrum. The C=O and N–H bonds both are involved in the hydrogen bonding interaction between the different elements of the secondary structure of Aβ42. The α-helix and β-sheet structure were resolved by systematically the shape of the C=O amide bond present in the secondary structure of Aβ42. The amide bond C=O has its characteristic absorption frequency in the IR region, α-helix (1640–1660 cm–1), β-sheet (1610–1640 cm–1), and random coil (1640–1650 and 1660–1685 cm–1). The Aβ peptide is self-aggregated and forms β-sheet fibrils, which are toxic. Here, we checked the effect of our newly designed NT peptides against Aβ peptide aggregation to form β-sheet fibrils. First, we checked the Aβ peptide secondary structure content by subjecting the Aβ peptide alone to FTIR spectroscopy without any incubation (Figure 6A). The IR spectrum shows the C=O amide stretching frequency at 1642.84 cm–1, which indicates that the Aβ peptide was in the form of α-helix. Then, we incubated the Aβ42 peptide alone for 7 days and then subjected it to FTIR spectroscopy. The IR spectrum (Figure 6B) shows the C=O amide bond stretching frequency at 1638.89 cm–1, and it clearly shows the formation of β-sheets. By these two IR spectra, we have confirmed the formation of β-sheets after 7 days of incubation of the Aβ peptide. After these, we checked the effect of our peptides if they can or cannot prevent the formation of β-sheets. Then, we incubated the Aβ peptide with our newly designed peptides NT-02, NT-03, and NT-13; the IR spectrum (Figure 6C–H) did not show the β-sheet characteristic peak in the range from 1610 to 1640 cm–1, and IR spectra indicated that the solutions did not contain the β-sheet content, which means that the peptides inhibited β-sheet formation in the solution. When Aβ42 was treated with NT-02 and NT-13, Aβ42 was in the form of α-helix, observing peaks at 1642.76 and 1645.94 cm–1, respectively, but when Aβ42 was treated with NT-03, it was in the form of a random coil by observing a peak at 1699.71 cm–1. The results of FTIR spectroscopy show that peptides can prevent the self-recognition of parent peptides by binding at the N-terminus region and stabilizing the α-helix conformation and disrupting the β-sheet structure formation followed by reducing the Aβ induced toxicity. This means that our designing concept of the NT strategy is efficient and they bind with the NT region of the Aβ42 peptide and increase α-helix stabilization and inhibit β-sheet-based Aβ aggregate formation. These FTIR spectroscopy results further well support the results we saw in the molecular docking studies by inhibition of fibril aggregation by binding at the N-terminus of the Aβ42 peptide and neuroprotection effects in PC-12-derived neurons and the results seen in the ThT assay.
Figure 6.

Analysis of β-sheet content by FTIR spectroscopy. FTIR spectrum of the (A) Aβ42 peptide alone for 0 day incubation. The spectrum shows the Aβ42 peptide in α-helix conformation. (B) Aβ42 peptide alone upon 7 days of incubation. The spectrum shows that the Aβ42 peptide converted into β-sheet conformation. (C,E,G) FTIR spectrum of peptide NT-02, 03, and 13 alone for 7 days of incubation. The spectrum shows that NT peptides in α-helix conformation. (D,F,H) FTIR spectrum of Aβ42 with NT-02, 03, and 13 for 7 days of incubation. The spectrum shows the Aβ42 peptide in α-helix conformation.
Circular Dichroism Spectroscopy Studies
In the aqueous solution (pH = 7), the Aβ42 peptide adopts a structure as a mixture of α-helix, β-sheet, and random coil, and when it forms Aβ aggregates, the confirmations will be in the form of the β-sheet structure. Here, we analyzed the effect of our peptides on the formation of fibrillary β-sheets by using circular dichroism (CD) spectroscopy. CD spectroscopy is a technique that is widely used for the quantitative identification of structural aspects of proteins and peptides.31 The Aβ42 peptide bonds are optically active, and the ellipticity they exhibit may change based on the confirmation of the peptide bond. The secondary structure of proteins can be analyzed by CD spectroscopy by using the far ultraviolet region (far UV 180–260 nm). The protein secondary structural conformations (β-sheet, α-helix, and random coil) have characteristic spectra; on the basis of the unique spectra, protein structure analyses may be done. We performed CD spectroscopy to analyze the effect of our NT peptides on the secondary structural conformation changes of the Aβ42 peptide by observing the residual molar ellipticity at 217 nm. The negative ellipticity at 217 nm indicates the β-sheet content in proteins and peptides, and the deep negative ellipticity will increase at 217 nm as the content of the β-sheet increase. For this, we incubated the Aβ peptide with three different concentrations (5, 10, and 20 μM) of NT peptides for 7 days and the secondary structure was analyzed by CD spectroscopy. First, we analyzed the secondary structure of the Aβ42 peptide alone at 0 days of incubation and we found negative ellipticity at 197 nm which means that Aβ42 is in a random coil. Figure 7A–C shows the peak (green color) negative ellipticity at 197 nm. Then, we incubated the Aβ42 peptide alone for 7 days and analyzed the secondary structure, and the CD spectrum showed negative ellipticity at 217 nm. Figure 7A–C shows the peak (block color) negative ellipticity at 217 nm. This means that Aβ42 forms the β-sheet structure after 7 days of incubation. Then, we incubated NT peptides with the Aβ42 peptide for 7 days and analyzed the effect of the NT peptide on β-sheet formation. The results indicate that in the presence of our NT peptides, a reduction in β-sheet content was observed by a reduction in negative ellipticity at 217 nm and an increase in the random coil content was observed upon increasing the negative ellipticity at 197 nm. Figure 7A–C shows negative ellipticity at 217 nm by order of Aβ42 alone and Aβ42 with NT peptides after incubation for 7 days. CD spectroscopy reveals that the peptides can interact with Aβ42 and prevent the formation of β-sheet formation, followed by aggregation of the Aβ42 peptide.
Figure 7.

CD spectroscopic studies showing β-sheet content and the inhibitory effects of peptides (A) NT-02, (B) NT-03, and (C) NT-13. CD spectra show the Aβ42 sample at t = 0 h (green), and the Aβ42 sample at t = 7 days (black). Dot blot assay for monitoring Aβ42 aggregation. (D) Dot blot image for Aβ42 content detected by 6E10. (E) Dot blot image for Aβ42 fibrillary aggregation inhibition in the presence of NT peptides detected by OC. (F) Bar diagram of Aβ42 fibrillary aggregation inhibition by NT peptides. (G) Dot blot image for Aβ42 content detected by 6E10. (H) Dot blot image for Aβ42 oligomeric aggregation inhibition in the presence of NT peptides detected by A11. (I) Bar diagram of Aβ42 oligomer inhibition by NT peptides. Error bars represent mean ± standard deviation (SD), n = 3. Statistical data were analyzed by a one-way ANOVA test by the multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, vs control) using software (GraphPad Prism, ISI, San Diego, CA).
Dot Blot Experiment to Monitor the Inhibition of Aβ Aggregation
Furthermore, we move toward a blotting experiment to confirm the inhibition activity of peptides NT-02, NT-03, and NT-13. In this dot blot assay, we have used three different antibodies 6E10, OC, and A11 for the detection of a different form of the Aβ peptide. Antibody 6E10 can bind to all kinds of Aβ peptides like monomer, oligomer, and fibril aggregates and gives a positive trend; because of this, we used 6E10 as a control experiment to detect the Aβ peptide content in the sample. For the specific detection of an oligomeric form of amyloid species content antibody A11 and for the detection of amyloid fibril aggregates, we used antibody OC.32 First, the Aβ42 peptide was incubated alone and Aβ42 with different concentrations (5, 10, and 20 μM) of NT peptide for the oligomer and fibril aggregate formation, separately. After incubation of the Aβ peptide samples, the samples were spotted on three different nitrocellulose membranes. Then, the membranes were treated with three respective primary antibodies and secondary antibodies. Then, the chemiluminescence intensity of the spots on the membrane was measured. Here, we checked the antibody 6E10 staining of all the incubated samples. Since the 6E10 antibody detects all forms of Aβ. The experimental results clearly showed that all the samples have Aβ peptide content indicated by the spot observed on the antibody 6E10-treated membrane. Figure 7D shows the Aβ content in fibril aggregate samples, and Figure 7G shows Aβ in oligomeric aggregate samples. Then, we checked the effect of the NT peptide on the formation of Aβ42 aggregates, and the experimental results show that the Aβ42 peptide alone formed the oligomers and fibrils, which are detected by antibodies A11 and OC with high intensity (Figure 7H,E). However, in the case of the Aβ42 peptide with the NT peptide, a drastic reduction of spot intensity was observed (Figure 7H,E). When Aβ42 was incubated with a 20 μM concentration of NT peptides, the formation of oligomers and fibrils was completely inhibited and the spot completely disappeared. The spot intensity of the control sample with Aβ42 alone was taken as 100%, and % inhibition was calculated for the samples treated with the NT peptides. Subsequently, the percentage of Aβ aggregates in each test peptide was calculated by using the formula: % aggregates = {[intensity of the test spot]/[intensity of the control spot]} × 100. The bar diagram showed (Figure 7F,I) the % of aggregates present in the samples when compared with the control. This dot blot experiment clearly shows the antiamyloid aggregation effect of NT peptides against the Aβ42 peptide.
Microscopic Examination of the Aβ42 Aggregation Process
It is well known that the formation of Aβ fibril aggregates occurs by a nucleation-dependent elongation mechanism. However, because of the insufficient analytical methods, the detailed structural features of various intermediates in the fibrillation pathway, including the initial structure and mechanism of nucleus formation, remain elusive. The fluorescent dyes ThT and Congo red (CR) are supposed to be used for specific imaging of the fibrillary structure. Here, we check the effect of the NT peptide on the fibrillation rate of the Aβ peptide by using the microscopic examination. For this, we incubated the Aβ peptide (10 μM) solution alone and the Aβ peptide (10 μM) with NT peptides (10 μM) for 7 days. For the visualization of Aβ peptide aggregation and the effect of NT peptides on its rate formation, we took aliquots from incubated solutions every 24 h and analyzed them by fluorescence microscopy. The incubated solutions were taken, and to this fluorescent dye, ThT or CR was added and kept aside for 1 min in the dark condition. Then, the fibrillary structures were imaged by fluorescence microscopy in the presence of the fluorescent dyes ThT and CR (Figure 8). The images clearly show the progression of Aβ fibrillary aggregates when the Aβ peptide is incubated alone. In the case of the Aβ42 peptide alone, fluorescence spots were observed after 24 h of incubation. Then, a drastic increase after 72 h was observed and spots became highly fluorescent with an increase in the aggregation of the Aβ42 peptide. After 92 h, saturation was observed and the Aβ42 peptide showed mature fibrils with high fluorescence intensity. However, when the Aβ peptide was coincubated with NT peptides, the progression of Aβ aggregates was reduced and images clearly showed that NT peptides inhibited Aβ fibrillation. In the case of NT peptides, fluorescent spots were observed at 24 h of incubation, but they were very less compared to Aβ42 alone, and after 96 h, the fluorescent spots were saturated and they were very less and small in size when compared with the Aβ42 peptide alone. The fluorescence microscopy images well support the above in vitro assays.
Figure 8.

CR and ThT binding to Aβ42 aggregates. Fluorescence microscopical images show the presence of Aβ42 aggregates in all samples. Fluorescence microscopic images of 10 μM Aβ42 peptide in the presence of 1 μM CR (column 1) and ThT (column 2). Images clearly indicate that NT peptides inhibit the formation of Aβ42 aggregates. The scale bar corresponds to 200 μm.
High-Resolution Transmission Electron Microscopy Studies
Transmission electron microscopy (TEM) provides detailed images of the macromolecular structures by passing an electron beam through the sample such that light will be absorbed and scattered and it produces contrast and images. TEM is a pivotal tool widely used to identify the presence of Aβ42 fibrils and study their morphology and size. Here, we used high-resolution-TEM (HR-TEM) microscopy to visually investigate the effects of NT peptides on the morphology, size, and distribution of Aβ42 aggregates. For the control experiment, Aβ42 was incubated alone for 7 days at 37 °C. After incubation of the Aβ42 peptide, we observed numerous long thick rod-like cylindrical fibrils. Figure 9A,E shows the fibril aggregation state of the Aβ42 peptide. However, after incubation of Aβ42 with NT peptides, outstanding changes were observed in the morphology and distribution of the Aβ42 peptides. The aggregates were completely disappeared when the Aβ42 peptide incubated with NT peptides (Figure 9B–D), and NT peptides did not form any aggregates and fibrils when incubated alone under the same conditions (Figure 9F–H). All these results again prove that our NT peptides play a key role in the Aβ42 aggregation process and prevent the formation of higher-order assemblies.
Figure 9.

HR-TEM images. (A,E) Aβ42 peptide fibrils after 7 days of incubation of Aβ42 alone at 37 °C. Absence of the fibril structure after coincubation of the Aβ42 peptide with (B) NT-02, (C) NT-03, and (D) NT-13 peptides at 37 °C for 7 days. Peptides (F) NT-02, (G) NT-03, and (H) NT-13 incubated alone for 7 days, and no fibril formation was observed.
Serum Stability Studies of NT Peptides
All the above studies indicate that peptides NT-02, NT-03, and NT-13 show a significant potential effect against Aβ aggregation and protect neuronal cells from Aβ-induced toxicity. However, any therapeutics designed for targeting the brain need to be stable in the physiological conditions and it should cross the BBB. For this, we check the stability of peptides in the human serum and find out that the peptides are potential candidates for performing in vivo experiments. Here, we studied the stability of peptides in human serum solution up to 72 h at 37 °C, and at different time intervals, aliquots were taken out from the incubated solution and we checked the percentage of safe peptides present in serum by using reverse-phase high-performance liquid chromatography (RP-HPLC). The HPLC data clearly shows that our peptides were stable in human serum solution and show 95.47, 91.57, and 90.44% of the intact peptide remaining in the serum after 72 h of incubation (Figure 4H) of NT-02, NT-03, and NT-13 respectively. Finally, our peptides show good stability in serum and this indicates that peptides have a greater bioavailability in physiological conditions. This means that the cleavage site of the peptide by serum protease was changed by using d-amino acid and increased the stability of the peptide in the serum.33 These NT peptides are potential candidates for evaluation in an animal model and for the development of anti-AD therapeutics.
BBB Crossing Studies
The brain has a natural barrier called the BBB; it helps to preserve the homeostasis of the brain’s internal environment. It is made of neuronal cells, astrocytes, pericytes, and brain capillary endothelial cells. The tight connections between brain capillary endothelial cells prevent the transport of paracellular substances from blood to the brain. Therefore, more than 98% of organic small molecules do not cross the BBB. Therefore, a BBB crossing experiment was performed to check whether NT peptides can penetrate the BBB or not. We performed the BBB penetration test by following a previously well-established procedure.34 Here, the peptides were injected intraperitoneally, and after 6 h, the mice were sacrificed through transcardial perfusion. The brains were collected and homogenized in an acetonitrile solution. Then, homogenized solutions were inspected by HRMS. The mass spectrum (Figure S5) shows the molecular mass peak of our peptides; as a negative control, sucrose was used, and we did not find the molecular peak in the mass spectrum. This BBB penetration experiment clearly shows that our peptides cross the BBB and show good biostability, which means that they have a greater potential in anti-AD drug development.
Conclusions
In summary, our newly designed proteolytically stable NT peptides were synthesized by replacing the hydrophobic amino acid 18Val with proline and leucine. By the evaluation of these peptides as an anti-Aβ aggregating agent by in vitro and in vivo experiments, we identified the peptides NT-02, NT-03, and NT-13, which are more potent than the other peptides are more potent than the other peptides. These NT peptides reduce Aβ fibrillation in the range of 60–80% observed in the ThT assay. In the cellular system, these peptides are noncytotoxic and show a significant neuroprotection effect (85–90%) from Aβ-induced toxicity and protects the neurons from Aβ-induced apoptosis. Moreover, the effect of peptides on the formation of the β-sheet structure was analyzed by FT-IR and CD spectroscopies, and inhibition of aggregate formation was observed by fluorescence microscopy and HR-TEM electron microscopic images. Finally, these peptides NT-02, NT-03, and NT-13 can cross the BBB, have significant stability in serum, and maintain healthy morphology of PC-12-derived neurons. From above, all the results suggest that proteolytically stable peptides could be a potential lead for further drug development against anti-AD and opens a new opportunity for treatment against AD.
Experimental Section
General Information
All the Fmoc-protected amino acids, Fmoc-d-Lys (Boc)-OH (≥98%), Fmoc-d-Leu-OH (≥99.5%), Fmoc-d-Val-OH (≥98%), Fmoc-d-Phe-OH (≥99.8%), Fmoc-d-Ala-OH (≥99.5%), Fmoc-d-Glu (OtBu)-OH (≥99.5%), and Fmoc-d-Pro-OH (≥99.5%), and Rink Amide AM resin were purchased from Novabiochem (Merck, Germany). All chemicals including N,N-diisopropylethylamine (DIPEA), piperidine, 1,1,1,3,3,3-hexafluoroisopropanol (HFIP), 2,4-ethanedithiol, phenol, trifluoroacetic acid (TFA), 5(6)-carboxyfluorescein, and 3-[dis(dimethylamino)methyliumyl]-3H-benzotriazole-1-oxide hexafluorophosphate (HBTU) were purchased from Sigma-Aldrich and used without further purification unless specified. All the solvents including N,N′-dimethylformamide (DMF), methanol, acetonitrile (ACN), dichloromethane (DCM), and diethyl ether were purchased from Merck (Germany) and used for synthesis. All the solvents were of analytical grade and used without further purification unless otherwise stated. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), cell culture-grade DMSO, and Dulbecco’s modified Eagle’s medium (DMEM) were purchased from Sigma-Aldrich. Aβ (1–42) was purchased from Auotec (Sweden). Human recombinant nerve growth factor (NGF) was purchased from Sigma (St. Louis, MO, USA), and anti-NGF was obtained from Abcam. Primary antibodies 6E10, A11, and OC were obtained from Bio Legend, Thermo Scientific, and Millipore, respectively. Nitrocellulose membranes were obtained from Merck Millipore. The fluorescence spectra were recorded using a Photon Technology International (PTI) Quanta Master fluorometer QM-40. RP-HPLC (Waters 1290 Infinity II Prep LC model using a Phenomenex TM column, LC-18, 250 × 21.2 mm, 10 Am) which was used for the purification of peptides and characterized for identity by HRMS and MALDI mass spectroscopy. Mass spectra were obtained using Agilent, G6530 Q-TOF HRMS, Applied Biosystems MDS SCIEX, and MALDI TOF/TOF analyzer (4800) mass spectrometer.
Synthesis of NT Peptides
All the peptides were synthesized by the SPPS method using Fmoc-protected rink amide resin in our laboratory. For the synthesis of peptides, we used 200 mg of Fmoc-protected rink amide resin in a reaction vessel and swelled the resin overnight in DMF/DCM (5 mL/5 mL) solvents at 4 °C. After 12 h of incubation, the reaction vessel was taken out and kept outside to bring it to room temperature. For the synthesis of peptides, we used Fmoc-protected d-amino acids. For the coupling of amino acid Fmoc-protected rink amide resin was deprotected with 20% piperidine in DMF and first amino acid was coupled to the resin was performed. Then, five equivalent Fmoc-protected amino acids were taken, five equivalents of DIPEA, and five equivalent HBTU were used as an activator base and DMF was used as a solvent in the CEM microwave peptide synthesizer (Liberty 1). All the amino acids were coupled successively and followed by Fmoc-deprotection was done by using 20% piperidine in DMF. After coupling of amino acids to resin and deprotection of amino acids after attaching them to resin washed by DMF and DCM solvents. All the fragment sequences were synthesized by the same protocol. Cleavage of prepared peptides from the resin was done by 92.5% TFA, 2.5% Milli Q water, 2.5% phenol, and 2.5% EDT (1,2-ethanedithiol). The reaction was subjected to 6 h of constant shaking (Labnet international). After completion of the reaction, the resin suspension was filtered, and the filtrate was collected, followed by the removal of TFA from the filtrate by nitrogen gas flow. The remaining filtrate was added to cold diethyl ether dropwise to ensure complete precipitation. Finally, the precipitate was separated by centrifugation at 4 °C at 7000 rpm. All the peptides were purified by using a C-18 reverse-phase HPLC column and characterized using MALDI and HRMS mass spectrometry and stored in a −20 °C freezer for further use.
Purification of Peptides by Using HPLC and Characterization
The crude peptides (1 mg/mL) were dissolved in a solution of water and methanol (1:1; v/v) and loaded into a preparative RP-HPLC (Waters 1290 Infinity II Prep LC model using a Phenomenex TM column, LC-18, 250 × 21.2 mm, 10 Am), operating at 254 nm with a flow rate of 1 mL/min. The peptides were analyzed by using a mobile-phase solvent system (A) of 0.1% TFA in acetonitrile (CH3CN) and (B) 0.1% TFA in water. Then, the system was run for 10 min as a linear gradient by using 10–70% of the mobile-phase B system. The desired fractions of the pure peptide were collected and concentrated. The peptides were dissolved in a minimum amount of ACN/water (1:1; v/v) solution and lyophilized. A fluffy white powder was collected, and the purity of the peptides was checked by analytical RP-HPLC (Waters 1290 Infinity II Prep LC model using a Phenomenex TM column, LC-18, 250 × 21.2 mm, 10 Am) operating at 254 nm with a flow rate of 1 mL/min. The mobile-phase solvent system was the same as the one used before, and the system was run for 20 min using a gradient of 10–60% of mobile-phase B. The purity of the peptides was in the range of 98–100%. Then, peptides were characterized by using HRMS.
Preparation of the Aβ42 Solution
We purchased synthetic Amyloid Beta Met—1–42 (1.0 mg) human from ALEXOTECH, with a purity of 95% characterized by HPLC and sodium dodecyl-sulfate polyacrylamide gel electrophoresis. The Aβ42 peptide (1 mg) was dissolved in 400 μL of HFIP and stored at −20 °C to maintain the monomer state of the Aβ42 peptide. At the time of the experiment. the Aβ42 peptide solution in HFIP was taken out, HFIP was removed through gentle nitrogen gas flow, and a white solid residue was collected. The white solid Aβ42 peptide was dissolved in a minimum amount of a 1% NH4OH solution. Then, a phosphate-buffered saline (PBS) (pH = 7.4) solution was used to make the required concentration of the Aβ42 peptide.
Preparation of Aβ42 Fibril Aggregates
The solution of the Aβ42 peptide (4 μL) in HFIP was taken out and dried using a gentle flow of nitrogen gas. Then, the remaining solid Aβ42 peptide was dissolved in a 1% NH4OH solution which was then diluted in 30 μL of PBS solution (pH = 7.4) to 80 μM. For the fibril aggregates, the Aβ42 solution was incubated at 37 °C for 72 h with gentle and constant shaking. The formation of Aβ42 fibril aggregates was confirmed by the ThT assay.
ThT Assay
For the confirmation of fibrils by the ThT assay, we used the incubated Aβ42 peptide solution in PBS (pH = 7.4). For the control experiment, the Aβ42 peptide (10 μM in final conc.) monomer solution in PBS was taken and ThT solution (2 μM in final conc.) was added to it; then, the fluorescence spectrum was recorded (λex = 435 nm and λem = 445–800 nm). For the Aβ42 fibril aggregates, the incubated Aβ42 solution (10 μM in final conc.) in PBS was taken, the ThT solution (2 μM in final conc.) was added to it, and the fluorescence spectrum was recorded (λex = 435 nm and λem = 445–800 nm).
Aβ42 Peptide Aggregation Inhibition
For Aβ42 peptide aggregation inhibition, we used a 5 μM concentration of the Aβ42 peptide in the final volume and three different concentrations of the peptide (5, 10, 20, 40, 80, and 160 μM) were used in the final volume. First, the Aβ42 peptide (5 μM in final conc.) monomer solution in PBS was taken, and the peptide solution in PBS was added to it and incubated at 37 °C for 72 h with constant shaking. Then, the incubated solutions were taken out and the ThT solution (1 μM in final volume) was added to it, followed by recording the fluorescence spectrum at λex = 435 nm and λem = 445–800 nm. For time-dependent aggregation inhibition, the Aβ42 peptide (5 μM in final conc.) monomer solution in PBS was taken and the peptide (10 μM in final volume) solution in PBS was added to it and incubated at 37 °C for 7 days continuously with constant shaking. At the time intervals of 24, 48, 72, 96, 120, 144, and 168 h, the incubated solution aliquots were taken out and ThT solution (1 μM in final volume) was added to it, followed by recording the fluorescence spectrum at λex = 435 nm and λem = 445–800 nm.
ITC Analysis
The ITC experiment was performed at a fixed temperature (298 K) in the PBS solution (0.1 mM) for the Aβ42 (20 μM) and NT peptides. At every 5 min interval, 10 μL of the peptide was added by a computer control program with continuous stirring. For this ITC experiment, we performed 28 injections for each experiment. Every peak in the binding isotherm represents the interaction of the peptide with the Aβ42 peptide. Finally, with the successive addition of the peptide, heat was released and analyzed. Then, the amount of heat released was plotted against the molar ratio of the peptide to the Aβ42 peptide.
Dot Blot Assay
For the dot blot assay, the Aβ42 (10 μM in final volume) solution was taken, and to this, various concentrations of the peptide (5, 10, and 20 μM in final volume) solution were added. Then, all the solutions were incubated at 37 °C for 72 h with gentle and constant shaking. After completion, 2 μL of the incubated solution was spotted on the nitrocellulose membrane and kept under air for 5 min or more until the spots had completely dried on the nitrocellulose membrane. For blocking of nonspecific sites, the nitrocellulose membrane was incubated in a solution of 5% bovine serum albumin in a TBS-T (Tris-buffered saline with 0.1% Tween 20 Detergent) buffer solution for 1 h at room temperature. Then, the nitrocellulose membranes were treated with primary antibody 6E10 (1:3000) to detect all forms of the Aβ peptide, A11 (1:1000) to detect the Aβ oligomers, and antibody OC (1:1000) to detect Aβ fibrillary aggregates, followed by incubating the membranes overnight at 4 °C. After overnight incubation, the membranes were washed with TBS-T buffer three times for 3–5 min each. Then, horseradish peroxidase-conjugated antimouse secondary antibody (1:10,000) and antirabbit secondary antibody (1:10,000) were added to them and they were incubated for 2 h at room temperature followed by three washes with TBS-T buffer for 3–5 min each. Finally, an enhanced chemiluminescence reagent was added to the membrane and it was incubated, and readings of chemiluminescence were taken at several different lengths of exposure using ChemiDoc Azure Biosystem C400.
Molecular Docking
For the docking experiment, we took the Aβ42 peptide (PDB ID: 1Z0Q) from the Protein Data Bank and the protein file was made by using Auto Dock Tools 1.5.6 software. Then, we drew the NT peptide structure using Avogadro software and optimized the NT peptide structure by using the CHARMM36 force field method. Then, we made the ligand files by using Auto Dock Tools 1.5.6 software. The optimized NT peptide assists as the input ligand, and the rigid protein Aβ42 (PDB ID: 1Z0Q) was used as the macromolecule receptor. The grid box centered on the protein was defined with a dimension of 52 × 26 × 26 using a 1 Å grid step, which is great enough to cover the whole Aβ42 peptide and leave enough space for docking the ligand on the surface. Finally, we performed the molecular docking procedure using the AutoDock Vina (version 1.1.2) according to the Lamarckian genetic algorithm method. The binding modes were studied by using PyMol 2.5.
Cell Uptake and Cell Viability
For the cellular uptake of the NT peptides, PC-12 cells were plated in 35 mm glass-bottom confocal dishes. The cell was cultured for 24 h in a 5% CO2 incubator at 37 °C. After reaching 60–70% of the population, the cells were differentiated into neurons by treating the cells with 100 ng/mL NGF prepared in DMEM with reduced horse serum. A differentiated neuron morphology was observed (Figure S6) by immunocytochemistry experiments through microtubule staining in PC12-derived neurons using corresponding primary and secondary antibodies following our previously standardized protocol.35 Then, the differentiated PC-12 derived neurons were treated with 5(6)-carboxyfluorescein-attached NT peptides and incubated for another 6 h, then washed gently once with 1× PBS, and fixed for 45 min using 4% formaldehyde in an incubator. Again, after washing once with 1× PBS, finally, imaging was done using a fluorescence microscope (OLYMPUS IX83) using a 40× objective. Images were processed using Image J software. The cellular toxicity of our peptides was checked in differentiated PC-12-derived neurons using the MTT reduction method. For this experiment, PC-12 cells were seeded at a low density of 1 × 104 cells per well in a 96-well plate and incubated for 24 h at 37 °C under 5% CO2 conditions. After 50–60% confluence, the cells were differentiated into neurons by treating the cells with 100 ng/mL NGF prepared in DMEM with reduced horse serum. Then, the differentiated PC-12-derived neurons were treated with three different concentrations of the peptide (5, 10, 20, 40, 80, and 160 μM) and incubated for 24 h at 37 °C under 5% CO2 conditions. Then, to check the toxicity effect of the peptides in PC-12 neurons, 50 μL of the MTT solution (5 mg/mL) in PBS was added into each well and further incubated for 4 h at 37 °C. Then, the medium was discarded and 50 μL of DMSO/methanol (1:1 v/v) was added to each well to dissolve the formazan. The absorbance was measured using a microplate reader (Thermo Scientific; Varioskan LUX) at 570 nm. Using these absorbance data, the percent viability of the cells was calculated following the method given below.
Neuroprotection Effect of Peptides
For the neuroprotection effect of the peptides or restoring the cell viability in the presence of Aβ42 mediated toxicity was performed in PC-12 derived neurons. For this, these PC-12 cells were seeded at a low density of 1 × 104 cells per well in a 96-well plate and incubated for 24 h at 37 °C under 5% CO2 conditions. After 50–60% confluence, the cells were differentiated into neurons by treating the cells with 100 ng/mL NGF prepared in DMEM with reduced horse serum. Then, the cells were treated with Aβ42 (5 μM) and individual peptides (5, 10, 20, 40, 80, and 160 μM) and incubated for 24 h at 37 °C under 5% CO2 conditions. Then, to check the neuroprotection effect of the peptides in PC-12 neurons, 50 μL of MTT solution (5 mg/mL) in PBS was added into each well and further incubated for 4 h at 37 °C. After 4 h, the media were discarded, and then formazan was dissolved with DMSO/MeOH (1:1 v/v). Finally, the absorption intensity of formazan was determined as a function of viable cells at 570 nm using a microplate reader (Thermo Scientific; Varioskan LUX). The absorbance data were used to calculate the percent viability of the cells by previously following the equation used in the MTT assay.
Apoptosis Assay
In a 96-well plate, PC-12 cells were seeded (1 × 104 cells per well) and incubated for 24 h at 37 °C; after 50–60% confluence, the cells were treated with 100 ng/mL NGF for differentiation into neurons. Then, the differentiated neuronal cells were treated with Aβ42 (10 μM) alone and Aβ42 (10 μM) with NT peptides (10 μM) and then incubated for 24 h. After 24 h, neurons were washed with PBS buffer. Then, the neurons were stained with Calcein AM and PI. The fluorescence images were obtained using an Olympus fluorescence microscope. Apoptosis was analyzed and quantified using ImageJ software.
FTIR Spectroscopy Experiment
For the FTIR spectroscopy experiment, we used the Aβ42 (20 μM) peptide monomer solution in PBS (pH = 7.4) and individual peptides (20 μM) were added and incubated at 37 °C for 1 week. For the control experiment, the Aβ42 peptide (20 μM) was incubated alone. After incubation, solutions were centrifuged at 12,000 rpm for 10 min, the supernatant solution was taken out, and the remaining small quantity of the precipitate was collected and freeze-dried. Then, the precipitate was dissolved in acetonitrile and subjected to FTIR spectroscopy (PerkinElmer Spectrum Version 10.5.3).
CD Spectroscopy
To check the β-sheet content by the CD experiment, we took the Aβ42 (5 μM) peptide monomer solution in PBS (pH = 7.4), and to these, individual peptides (5, 10, and 20 μM) were added and incubated at 37 °C for 1 week with gentle and constant shaking. For the control experiment, the Aβ42 (5 μM) peptide was incubated alone. After completion of incubation, the solutions were inspected by CD spectroscopy using a JASCO (J-810) spectrometer.
Fluorescence Microscopy
For fluorescence microscopy imaging, we took 5 μL of the solution from the Aβ42 (10 μM) alone and Aβ42 (10 μM) with NT peptides (10 μM) incubated for 7 days. For the analysis, incubated solutions were taken at interval times of 24, 48, 72, 96, 120, 144, and 168 h; the incubated solution was taken out and spotted on a glass slide separately. Then, we added a 4 μM solution of ThT or CR on the slide, and a cover slip was immediately placed over the sample. Images were captured using a fluorescence microscope (OLYMPUS IX83) using a 10× objective.
Serum Stability
For the stability of peptides in the physiological environment or serum condition, the peptides (150 μM in final volume) were incubated with 50% human serum at 37 °C. For the analysis of peptides, at different time intervals (0, 6, 12, 24, 48, and 72 h), 100 μL aliquots were taken out from the incubated solution and diluted with 100 μL of acetonitrile for precipitating serum proteins; then, the solution was incubated at 4 °C for 30 min to stop the protease activity. After 30 min of incubation, samples were taken out and centrifuged at 10,000 rpm for 30 min at 4 °C. Then, the supernatant solution was collected. For the blank experiment, we used only the free serum solution without any compound. Finally, these supernatant solutions were analyzed by reverse-phase HPLC. The same procedure was used for all the time intervals.
BBB Crossing
In our laboratory, we used female pathogen-free C57BL/6J mice (8–10 weeks) for the BBB crossing experiment. All animal experiments were conducted following the laws and regulations of the regulatory authorities of our Institutional Animal Ethics Committee. For the BBB permeability test, we used 12 mice and divided them into 4 groups (3 mice/group). Peptide NT-02, NT-03, and NT-13 (100 μL) (dosage of 5 mg/kg body weight of mice) solutions were injected intraperitoneally into the three different groups of mice separately (Table S7). For the control experiment, sucrose was dissolved in a normal saline solution and 100 μL was injected into the last group of mice. After 6 h, mice were anesthetized with Avertin (IP) and sacrificed. Transcardial perfusion was performed to remove the blood from the body. Then, we removed the blood vessel and meninges, after which the brains were collected in PBS solutions. The brain cortical region was dissected and used for further analysis. Then, the cortex was homogenized in liquid nitrogen and extracted in acetonitrile. The acetonitrile suspension was centrifuged, the supernatant was collected, and then it was inspected using HRMS.
Data Analysis
Dot blots were analyzed by ImageJ software. For the measurement of spectroscopic data and histograms, we used Origin 8.5 pro software. Error bars represent mean ± standard deviation (SD), n = 3. Statistical data were analyzed by the one-way ANOVA test by the multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, vs control or Aβ42) using software GraphPad Prism (ISI, San Diego, CA).
Acknowledgments
R.M. thanks the NIPER for providing his fellowship; J.K. and V.G. thank DST-INSPIRE for their fellowship; P.K.G. thanks the CSIR for his fellowship; R.R. thanks IIT Jodhpur for providing his fellowship. S.G. kindly acknowledges the DST-SERB, India (CRG/2019/000670), for financial support. We thank CSIR-IICB Kolkata and IIT Jodhpur for providing infrastructure.
Glossary
Abbreviations
- AD
Alzheimer’s disease
- APP
amyloid beta precursor protein
- Aβ
amyloid beta
- PS1
presenilin 1
- BBB
blood–brain barrier
- ThT
thioflavin-T
- PDB
Protein Data Bank
- SD
standard deviation
- PBS
phosphate-buffered saline
- DMEM
Dulbecco’s modified Eagle medium
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- DCM
dichloromethane
- ACN
acetonitrile
- THF
tetrahydrofuran
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- NH4OH
ammonium hydroxide
- HFIP
1,1,1,3,3,3-hexafluoro-2-propanol
- HRMS
high-resolution mass spectroscopy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.2c00067.
Experimental details, figures, HRMS, and MALDI mass spectra (PDF)
Author Contributions
R.M. performed the synthesis, purification, and characterization of all peptides and all the in vitro assays, dot blot experiments, and molecular docking studies. J.K. performed the BBB experiment and cell-based assays. V.G. performed the cell uptake experiment. P.K.G and R.R. helped with the conduction of various experiments and helped to analyze the data and various experiments. S.G. conceived the idea, supervised the project, and wrote the manuscript. CRediT: Rathnam Mallesh conceptualization (supporting), data curation (equal), formal analysis (equal), investigation (equal), methodology (equal), visualization (equal), writing-original draft (equal), writing-review & editing (equal); Juhee Khan data curation (supporting), formal analysis (supporting), investigation (supporting), methodology (supporting), writing-original draft (supporting), writing-review & editing (supporting); Prabir Kumar Gharai data curation (supporting), methodology (supporting), writing-original draft (supporting); Varsha Gupta data curation (supporting), investigation (supporting); Rajsekhar Roy data curation (supporting), formal analysis (supporting), investigation (supporting), methodology (supporting), writing-review & editing (supporting); Surajit Ghosh conceptualization (lead), formal analysis (lead), funding acquisition (lead), investigation (lead), methodology (lead), project administration (lead), resources (lead), supervision (lead), validation (lead), writing-original draft (lead), writing-review & editing (lead).
This work was supported by the DST-SERB, India (CRG/2019/000670).
The authors declare no competing financial interest.
Supplementary Material
References
- Wong C. W.; Quaranta V.; Glenner G. G. Neuritic Plaques and Cerebrovascular Amyloid in Alzheimer Disease Are Antigenically Related. Proc. Natl. Acad. Sci. 1985, 82, 8729–8732. 10.1073/pnas.82.24.8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner G. G.; Wong C. W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Roher A. E.; Lowenson J. D.; Clarke S.; Woods A. S.; Cotter R. J.; Gowing E.; Ball M. J. Beta-Amyloid-(1-42) Is a Major Component of Cerebrovascular Amyloid Deposits: Implications for the Pathology of Alzheimer Disease. Proc. Natl. Acad. Sci. 1993, 90, 10836–10840. 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J. Amyloid β-Protein and the Genetics of Alzheimer’s Disease. J. Biol. Chem. 1996, 271, 18295–18298. 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- Terzi E.; Hölzemann G.; Seelig J. Self-Association of β-Amyloid Peptide (1–40) in Solution and Binding to Lipid Membranes. J. Mol. Biol. 1995, 252, 633–642. 10.1006/jmbi.1995.0525. [DOI] [PubMed] [Google Scholar]
- Kang J.; Lemaire H.-G.; Unterbeck A.; Salbaum J. M.; Masters C. L.; Grzeschik K.-H.; Multhaup G.; Beyreuther K.; Müller-Hill B. The Precursor of Alzheimer’s Disease Amyloid A4 Protein Resembles a Cell-Surface Receptor. Nature 1987, 325, 733–736. 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- Tamaoka A.; Odaka A.; Ishibashi Y.; Usami M.; Sahara N.; Suzuki N.; Nukina N.; Mizusawa H.; Shoji S.; Kanazawa I. APP717 Missense Mutation Affects the Ratio of Amyloid Beta Protein Species (A Beta 1-42/43 and a Beta 1-40) in Familial Alzheimer’s Disease Brain. J. Biol. Chem. 1994, 269, 32721–32724. 10.1016/s0021-9258(20)30050-8. [DOI] [PubMed] [Google Scholar]
- Lomakin A.; Chung D. S.; Benedek G. B.; Kirschner D. A.; Teplow D. B. On the Nucleation and Growth of Amyloid Beta-Protein Fibrils: Detection of Nuclei and Quantitation of Rate Constants. Proc. Natl. Acad. Sci. 1996, 93, 1125–1129. 10.1073/pnas.93.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychaudhuri R.; Yang M.; Hoshi M. M.; Teplow D. B. Amyloid β-Protein Assembly and Alzheimer Disease. J. Biol. Chem. 2009, 284, 4749–4753. 10.1074/jbc.r800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsbury C. S.; Wirtz S.; Müller S. A.; Sunderji S.; Wicki P.; Aebi U.; Frey P. Studies on the in Vitro Assembly of Aβ 1–40: Implications for the Search for Aβ Fibril Formation Inhibitors. J. Struct. Biol. 2000, 130, 217–231. 10.1006/jsbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- Pike C. J.; Walencewicz-Wasserman A. J.; Kosmoski J.; Cribbs D. H.; Glabe C. G.; Cotman C. W. Structure-Activity Analyses of β-Amyloid Peptides: Contributions of the Β25–35 Region to Aggregation and Neurotoxicity. J. Neurochem. 1995, 64, 253–265. 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- Soto C.; Castaño E. M.; Frangione B.; Inestrosa N. C. The α-Helical to β-Strand Transition in the Amino-Terminal Fragment of the Amyloid β-Peptide Modulates Amyloid Formation. J. Biol. Chem. 1995, 270, 3063–3067. 10.1074/jbc.270.7.3063. [DOI] [PubMed] [Google Scholar]
- Hilbich C.; Kisters-Woike B.; Reed J.; Masters C. L.; Beyreuther K. Substitutions of Hydrophobic Amino Acids Reduce the Amyloidogenicity of Alzheimer’s Disease ΒA4 Peptides. J. Mol. Biol. 1992, 228, 460–473. 10.1016/0022-2836(92)90835-8. [DOI] [PubMed] [Google Scholar]
- Tjernberg L. O.; Näslund J.; Lindqvist F.; Johansson J.; Karlström A. R.; Thyberg J.; Terenius L.; Nordstedt C. Arrest of -Amyloid Fibril Formation by a Pentapeptide Ligand (*). J. Biol. Chem. 1996, 271, 8545–8548. 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- Tjernberg L. O.; Lilliehöök C.; Callaway D. J.; Näslund J.; Hahne S.; Thyberg J.; Terenius L.; Nordstedt C. Controlling Amyloid Beta-Peptide Fibril Formation with Protease-Stable Ligands. J. Biol. Chem. 1997, 272, 12601–12605. 10.1074/jbc.272.19.12601. [DOI] [PubMed] [Google Scholar]
- Lyu P. C.; Sherman J. C.; Chen A.; Kallenbach N. R. Alpha-Helix Stabilization by Natural and Unnatural Amino Acids with Alkyl Side Chains. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 5317–5320. 10.1073/pnas.88.12.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. C.; Goto N. K.; Williams K. A.; Deber C. M. Alpha-Helical, but Not Beta-Sheet, Propensity of Proline Is Determined by Peptide Environment. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 6676–6681. 10.1073/pnas.93.13.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman M. S.; Ragan C. I.; Iversen L. L. Inhibition of PC12 Cell Redox Activity Is a Specific, Early Indicator of the Mechanism of Beta-Amyloid-Mediated Cell Death. Proc. Natl. Acad. Sci. 1994, 91, 1470–1474. 10.1073/pnas.91.4.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Peterson D. A.; Kimura H.; Schubert D. Mechanism of Cellular 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide (MTT) Reduction. J. Neurochem. 1997, 69, 581–593. 10.1046/j.1471-4159.1997.69020581.x. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Levine H. III Thioflavine T Interaction with Synthetic Alzheimer’s Disease β-Amyloid Peptides: Detection of Amyloid Aggregation in Solution. Protein Sci. 1993, 2, 404–410. 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou P. Y.; Fasman G. D. Empirical Predictions of Protein Conformation. Annu. Rev. Biochem. 1978, 47, 251–276. 10.1146/annurev.bi.47.070178.001343. [DOI] [PubMed] [Google Scholar]
- Garnier J.; Osguthorpe D. J.; Robson B. Analysis of the Accuracy and Implications of Simple Methods for Predicting the Secondary Structure of Globular Proteins. J. Mol. Biol. 1978, 120, 97–120. 10.1016/0022-2836(78)90297-8. [DOI] [PubMed] [Google Scholar]
- Soto C.; Brañes M. C.; Alvarez J.; Inestrosa N. C. Structural Determinants of the Alzheimer’s Amyloid β-Peptide. J. Neurochem. 1994, 63, 1191–1198. 10.1046/j.1471-4159.1994.63041191.x. [DOI] [PubMed] [Google Scholar]
- Zagorski M. G.; Barrow C. J. NMR Studies of Amyloid .Beta.-Peptides: Proton Assignments, Secondary Structure, and Mechanism of an .Alpha.-Helix .Fwdarw. .Beta.-Sheet Conversion for a Homologous, 28-Residue, N-Terminal Fragment. Biochemistry 1992, 31, 5621–5631. 10.1021/bi00139a028. [DOI] [PubMed] [Google Scholar]
- Barrow C. J.; Zagorski M. G. Solution Structures of β Peptide and Its Constituent Fragments: Relation to Amyloid Deposition. Science 1991, 253, 179–182. 10.1126/science.1853202. [DOI] [PubMed] [Google Scholar]
- Hilbich C.; Kisters-Woike B.; Reed J.; Masters C. L.; Beyreuther K. Aggregation and Secondary Structure of Synthetic Amyloid ΒA4 Peptides of Alzheimer’s Disease. J. Mol. Biol. 1991, 218, 149–163. 10.1016/0022-2836(91)90881-6. [DOI] [PubMed] [Google Scholar]
- Tomaselli S.; Esposito V.; Vangone P.; van Nuland N. A. J.; Bonvin A. M. J. J.; Guerrini R.; Tancredi T.; Temussi P. A.; Picone D. The α-to-β Conformational Transition of Alzheimer’s Aβ-(1–42) Peptide in Aqueous Media Is Reversible: A Step by Step Conformational Analysis Suggests the Location of β Conformation Seeding. ChemBioChem 2006, 7, 257–267. 10.1002/cbic.200500223. [DOI] [PubMed] [Google Scholar]
- Surewicz W. K.; Mantsch H. H. New Insight into Protein Secondary Structure from Resolution-Enhanced Infrared Spectra. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1988, 952, 115–130. 10.1016/0167-4838(88)90107-0. [DOI] [PubMed] [Google Scholar]
- Byler D. M.; Susi H. Examination of the Secondary Structure of Proteins by Deconvolved FTIR Spectra. Biopolymers 1986, 25, 469–487. 10.1002/bip.360250307. [DOI] [PubMed] [Google Scholar]
- Adler A. J.; Greenfield N. J.; Fasman G. D. [27] Circular Dichroism and Optical Rotatory Dispersion of Proteins and Polypeptides. Methods Enzymol. 1973, 27, 675–735. 10.1016/s0076-6879(73)27030-1. [DOI] [PubMed] [Google Scholar]
- Pradhan K.; Das G.; Mondal P.; Khan J.; Barman S.; Ghosh S. Genesis of Neuroprotective Peptoid from Aβ30–34 Inhibits Aβ Aggregation and AChE Activity. ACS Chem. Neurosci. 2018, 9, 2929–2940. 10.1021/acschemneuro.8b00071. [DOI] [PubMed] [Google Scholar]
- Hong S. Y.; Oh J. E.; Lee K. H. Effect of D-Amino Acid Substitution on the Stability, the Secondary Structure, and the Activity of Membrane-Active Peptide. Biochem. Pharmacol. 1999, 58, 1775–1780. 10.1016/s0006-2952(99)00259-2. [DOI] [PubMed] [Google Scholar]
- Mallesh R.; Khan J.; Pradhan K.; Roy R.; Jana N. R.; Jaisankar P.; Ghosh S. Design and Development of Benzothiazole-Based Fluorescent Probes for Selective Detection of Aβ Aggregates in Alzheimer’s Disease. ACS Chem. Neurosci. 2022, 13, 2503–2516. 10.1021/acschemneuro.2c00361. [DOI] [PubMed] [Google Scholar]
- Adak A.; Das G.; Barman S.; Mohapatra S.; Bhunia D.; Jana B.; Ghosh S. Biodegradable Neuro-Compatible Peptide Hydrogel Promotes Neurite Outgrowth, Shows Significant Neuroprotection, and Delivers Anti-Alzheimer Drug. ACS Appl. Mater. Interfaces 2017, 9, 5067–5076. 10.1021/acsami.6b12114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.