

Abstract

S-Adenosyl-l-methionine (SAM) analogs are adaptable tools for studying and therapeutically inhibiting SAM-dependent methyltransferases (MTases). Some MTases play significant roles in host–pathogen interactions, one of which is Clostridioides difficile-specific DNA adenine MTase (CamA). CamA is needed for efficient sporulation and alters persistence in the colon. To discover potent and selective CamA inhibitors, we explored modifications of the solvent-exposed edge of the SAM adenosine moiety. Starting from the two parental compounds (6e and 7), we designed an adenosine analog (11a) carrying a 3-phenylpropyl moiety at the adenine N6-amino group, and a 3-(cyclohexylmethyl guanidine)-ethyl moiety at the sulfur atom off the ribose ring. Compound 11a (IC50 = 0.15 μM) is 10× and 5× more potent against CamA than 6e and 7, respectively. The structure of the CamA–DNA–inhibitor complex revealed that 11a adopts a U-shaped conformation, with the two branches folded toward each other, and the aliphatic and aromatic rings at the two ends interacting with one another. 11a occupies the entire hydrophobic surface (apparently unique to CamA) next to the adenosine binding site. Our work presents a hybrid knowledge-based and fragment-based approach to generating CamA inhibitors that would be chemical agents to examine the mechanism(s) of action and therapeutic potentials of CamA in C. difficile infection.
Introduction
S-Adenosyl-l-methionine (SAM, SAMe, or AdoMet)1 is a metabolite that is biosynthesized naturally in almost all species (excepting only some obligate intracellular parasites that import it) and is the second most-used cofactor after ATP.2 SAM is involved in a wide range of biochemical reactions including (but not limited to) transmethylation reactions,3,4 radical SAM reactions,5−8 polyamine biosynthesis,9 and SAM-sensing riboswitches.10,11 SAM-dependent methyltransferases (MTases) catalyze methyl transfers onto a wide variety of substrate molecules, including both macromolecules (DNA, RNA, polysaccharides, lipids, and proteins) and micromolecules (such as catechol, arsenic, histamine, nicotinamide, and thiopurine). Given the significance of epigenetic methylation of macromolecules in cancer, immunity, cardiovascular and many other diseases, as well as methylation of anticancer drugs such as thiopurine,12,13 SAM-dependent MTases are increasingly becoming therapeutic targets.14−19
SAM analogs are versatile tools for studying and therapeutically inhibiting SAM-dependent MTases.20−22 Some of these compounds as inhibitors of human epigenetic enzymes,23,24 as well as inhibitors of viral RNA MTases from Zika virus and SARS-CoV-2,25−27 are in clinical trials. Interestingly, some SAM analogs designed as selective inhibitors of human protein arginine MTases (PRMTs)24,28,29 and human DOT1L (a histone H3 lysine 79 MTase)30 also inhibited the Clostridioides difficile-specific DNA adenine MTase (CamA) in vitro at low micromolar concentrations31 (Figure 1A). CamA exists in the genomes of all C. difficile sequenced to date, but closely related orthologs have not yet been found in any other bacteria.31,32 CamA is needed for normal sporulation and for persistent C. difficile infection (CDI) in an animal model.32 CDI is a leading cause of community-associated infection in children and of hospital-acquired diarrhea in older patients,33,34 can have lethal consequences, and has been identified as an urgent threat to human health (https://www.cdc.gov/drugresistance/biggest-threats.html).
Figure 1.
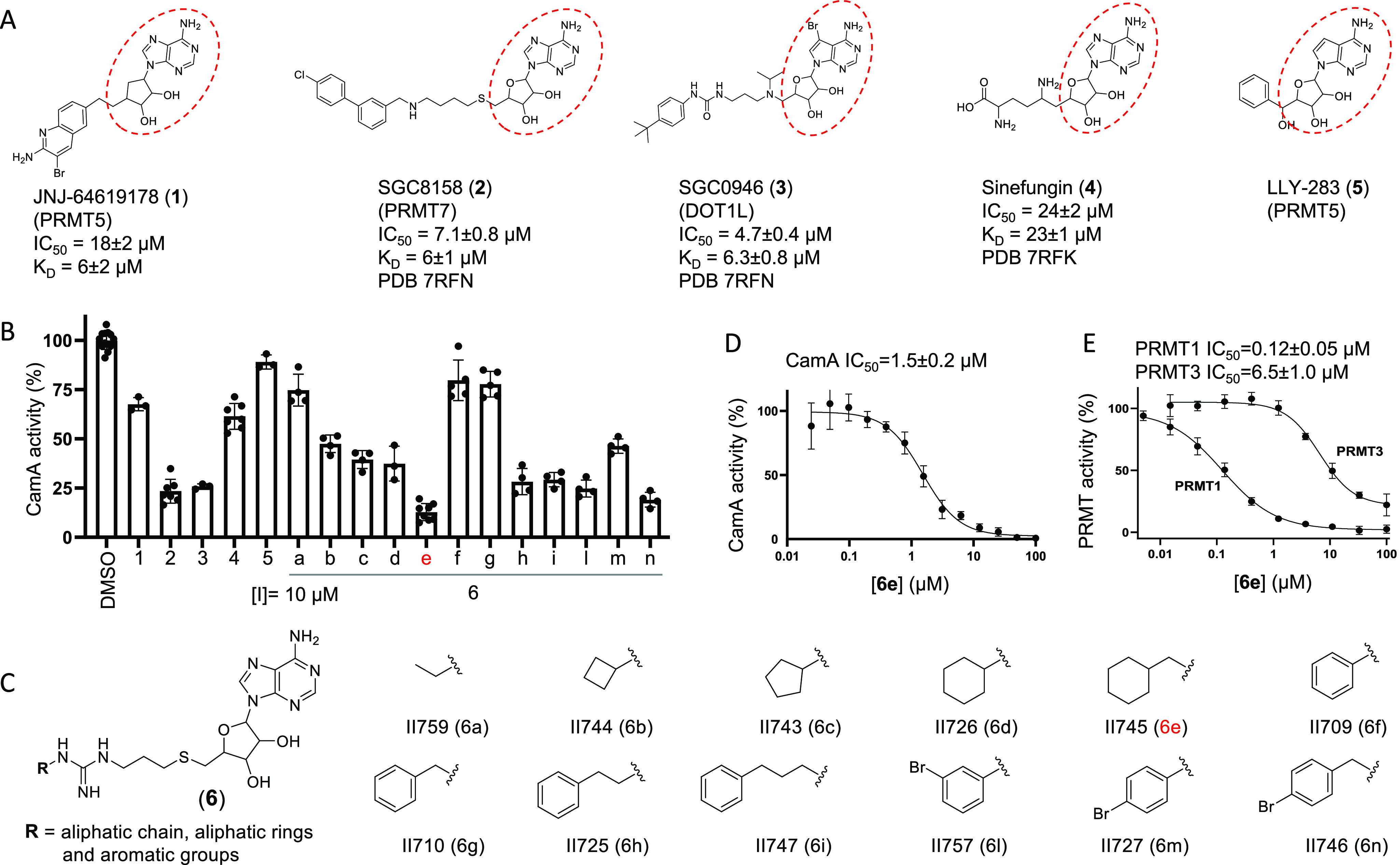
CamA inhibition by a pan-PRMT inhibitor (6e). (A) Chemical structures of five SAM-analogs. The IC50 and KD values against CamA, as well as PDB codes for the structures of CamA–DNA–inhibitor complexes, are extracted from Zhou et al.31 (B) Comparative inhibition of activity of CamA at a single concentration of 10 μM. These assays included 50 nM CamA and 40 μM SAM. (C) Chemical structures of compounds 6a–6n carrying substituents at the R group, taken from Iyamu et al.48 [6j and 6k in ref (48) were no longer available at the time of testing.] (D, E) IC50 values of 6e against CamA (panel D) or PRMT1 and PRMT3 (panel E).
We showed that CamA35 shares a conserved SAM binding site with PRMTs36−39 and DOT1L40 and many other Rossman-fold Class I MTases.41,42 Given that similarity, CamA has an unexpectedly low binding affinity for the methyl donor SAM (Km > 17 μM).35 For comparison, most other MTases have much higher SAM affinity; for example, the E. coli DNA adenine MTase Dam has a SAM Km of 3–6 μM,43−45 for human PCIF1 it is 0.7 μM, and for MettL5 it is 1.0 μM.46 The unusually low SAM affinity of CamA might be related to a coupled conformational rearrangement of the two N-terminal helices.35 Whatever the explanation may prove to be, this unusual property triggered us to investigate if SAM analogs are specifically potent inhibitors of CamA activity in competition with the weakly bound SAM cosubstrate.
In the present study, we took advantage of recent advances in development of molecular tools and in the growing number of clinically used compounds capable of potently and selectively inhibiting PRMTs.47 These PRMT inhibitors can work as competitors of SAM or substrate polypeptide or allosteric inhibitors.17 We started from a small set of pan-PRMT inhibitors48 together with our recently developed CamA-specific adenosine analogs.49 The combination of unique moieties in these two chemotypes led us to design compound 11a (YD905), which represents a more potent lead compound against CamA, having a half-maximal inhibitory concentration (IC50) of ∼0.15 μM.
Results
CamA Inhibition by a Pan-PRMT Inhibitor
We previously determined that a few known compounds showed in vitro inhibition of CamA at low micromolar concentrations.31 These included the PRMT5 inhibitor JNJ-64619178 (1),24 the PRMT7 inhibitor SGC8158 (2),29 and the DOT1L inhibitor SGC0946 (3).30 These compounds are all SAM-competitive inhibitors, all contain an adenosyl moiety (Figure 1A), and all of them occupy the individual enzymes’ SAM binding site. The three compounds demonstrated improved potency of CamA inhibition relative to sinefungin (4), a pan inhibitor of SAM-dependent MTases, starting from IC50 values of 24 μM for sinefungin and progressively decreasing to 18 μM (JNJ-64619178), 7.1 μM (SGC8158), and 4.7 μM (SGC0946). We also made use of the observation that LLY-283 (5), a SAM analog developed as a PRMT5 inhibitor,50 has the shortest branch off its ribose moiety compared to compounds 1–3 and displayed much weaker inhibition of CamA31 (Figure 1B), suggesting a path to explore in further improving these inhibitors.
A CamA inhibition screen was carried out using a set of 12 compounds designed as bisubstrate analogs of pan-PRMT inhibitors. These compounds join a thioadenosine to various substituted guanidinium groups (which mimic the target arginine side chain of protein substrates), through a hydrophobic propyl linker (Figure 1C).48 The 12 bisubstrate compounds vary at the guanidium end, having either an aliphatic or an aromatic group (Figure 1C). Among them, compound 6e (II745), which contains an aliphatic ring, most potently inhibited ∼85% activity of CamA at 10 μM concentration and in the presence of 40 μM SAM (Figure 1B). We made the following two observations: (i) eliminating the one-carbon linker between the aliphatic ring and guanidine group (from 6e to 6d) decreased CamA inhibition (from ∼85% to ∼60%); and (ii) substituting the aliphatic ring in 6e with the aromatic analog in 6g nearly eliminated inhibitory activity (∼10% inhibition). These two features, the one-carbon linker and aliphatic ring, might contribute to the flexibility of compound 6e.
We compared the inhibitory activity of compound 6e for CamA with PRMT1 and PRMT3. With an IC50 value of 1.5 μM, 6e has 12.5× weaker inhibition of CamA than of PRMT1 (0.12 μM), but >4× stronger inhibition than of PRMT3 (6.5 μM) (Figure 1D-1E). Nevertheless, 6e demonstrated >3× improved potency of CamA inhibition relative to SGC0946 (4.7 μM).
Design and Synthesis of Compounds 11a and 11b
To further develop more potent inhibitors of CamA, we introduced a substituent onto the amino group (N6) of adenosine (Figure 2A). The rationale of this design stemmed from our recent structural characterization of CamA–DNA complexes in the presence and absence of methyl donor cofactor (SAM or its end product, S-adenosyl-l-homocysteine, SAH), which revealed a unique solvent-exposed edge of the SAM/SAH adenosine moiety (Figure 2B).31,35 The N6-modified compounds bind in the pocket normally occupied by the SAM adenosine, with the N6-addition directed outward and resting against a mostly hydrophobic surface (see Discussion), making a unique mixture of interactions with CamA involving van der Waals, aromatic, and/or polar contacts.49 Among them, compound 7 (MC4741) contains a 3-phenylpropyl moiety at the N6-amino group of adenosine and demonstrated an IC50 value of 0.7 μM against CamA49 [Note compound MC4741 was numbered as 14 in ref (49)]. Importantly, for potential therapeutic use in humans, compound 7 does not inhibit either PRMT1 or PRMT3 (Figures 2C and S1). Significantly, in PRMT3 there is no equivalent solvent-exposed surface next to the SAM/SAH adenosine moiety (Figure 2D; see Discussion on PRMT1). We reasoned that combining the unique features of 6e and 7 could lead to synergistic CamA inhibition.
Figure 2.
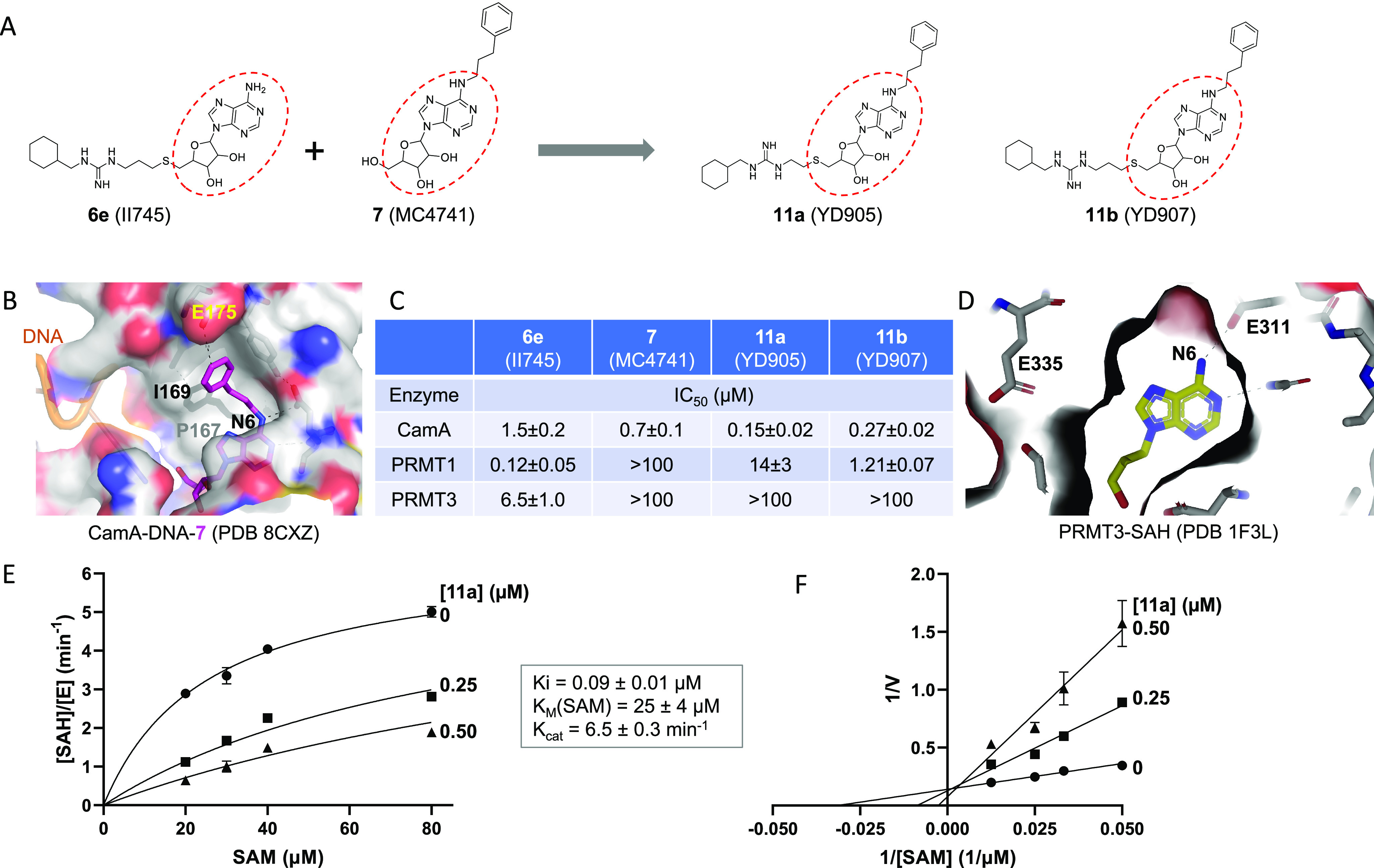
Compound 11a (YD905) is competitive with SAM. (A) Chemical structures of 6e (II745), 7 (MC4741), 11a (YD905), and 11b (YD907). 11a and 11b differ in the length of the linker (2C vs 3C) off of the sulfur atom. (B) Structure of CamA–DNA in complex with compound 7 (magenta) (PDB 8CXZ). The 3-phenylpropyl moiety at the N6-amino of adenosine is bound to a largely hydrophobic surface (colored in gray). (C) Summary of inhibition (IC50 values) of CamA, PRMT1, and PRMT3 by the four compounds shown in panel A (see Figure S1 in Supporting Information). (D) Structure of PRMT3 in complex with SAH (yellow), which is completely enclosed by PRMT3 residues (PDB 1F3L). (E) Michaelis–Menten saturation curves of CamA kinetics varying SAM concentrations (20, 30, 40, and 80 μM) and inhibitor 11a concentrations (0, 0.25, and 0.5 μM). (F) Lineweaver–Burk double reciprocal plot suggests competitive inhibition by 11a with respect to SAM.
We thus designed and synthesized two new compounds, 11a (YD905) and 11b (YD907), respectively, having a 2-carbon ethyl or 3-carbon propyl linker between the guanidinium group and the sulfur atom. Compound 11a exhibited enhanced inhibition against CamA relative to the two parental compounds, with either 10× improvement from 6e (IC50 of 0.15 μM vs. 1.5 μM) or 5× improvement from 7 (IC50 of 0.15 μM vs. 0.7 μM) (Figure 2C). However, increasing the linker from 2-carbon in 11a to 3-carbon in 11b resulted in 1.8× reduced inhibition of CamA relative to 11a (i.e., the IC50 value increased from 0.15 μM to 0.27 μM).
As expected, both 11a and 11b exhibit reduced inhibition of PRMT1 and PRMT3, as one of the parental compounds (7) does not significantly inhibit either of the two PRMTs. 11a has a remarkable reduction (∼116×) in inhibition of PRMT1 from that of 6e (IC50 of 14 μM vs. 0.12 μM) (Figure 2C). The same magnitude of negative effect on PRMT3 activity made the inhibition unmeasurable (IC50 > 100 μM). However, compound 11b has a much milder effect on PRMT1 inhibition than 6e, resulting in a 10× reduction of PRMT1 inhibition (IC50 increased to 1.2 μM from 0.12 μM), instead of the >100× reduction by 11a. The lesser reduction of PRMT1 inhibition by 11b is probably owing to the 3-carbon linker, given the reported optimal length between the thioadenosine and guanidinium group for PRMT inhibition by bisubstrate analogs.51
To further understand the mode of CamA inhibition by compound 11a, we measured the inhibition constant (Ki = 90 nM under our experimental conditions) by varying SAM concentration in the absence and presence of inhibitor at two concentrations (Figure 2E). A Lineweaver–Burk plot suggests that compound 11a is a competitive inhibitor of SAM, as expected (Figure 2F). In agreement with our previous measurements (Km > 17 μM),35 CamA has a weak binding affinity (as reflected by the Km value) of 25 μM for SAM (Figure 2E).
Compound 11a Binds to a Solvent-Exposed, Largely Hydrophobic Surface
To define the basis for the enhanced binding and inhibition of CamA by compounds 11a (YD905) and 11b (YD907) relative to the parent compounds, we cocrystallized the CamA–DNA complex with each of the two compounds. The two complexes both crystallized in space group P212121, and very similar structures (with root-mean-square deviation of 0.7 Å) were determined to resolutions of 2.74 Å (11a) and 2.59 Å (11b) (Table S1). While 11a was fully observed in the electron density, the portion of 11b where the 3-carbon linker located was partially disordered (Figure 3A). In the following section, we describe CamA interactions with 11a (Figure 3B), with a focus on the interactions involving the two branches off the guanidino group and the amino group of adenosine.
Figure 3.

Structure of CamA–DNA–11a complex. (A) Omit Fo – Fc electron densities of 11a and 11b contoured at 3σ above the mean, from PDB 8FS1 and PDB 8FS2, respectively. (B) Structure of CamA–DNA with bound compound 11a (in stick) within a dashed green box (PDB 8FS1). Surface representation is shown with carbon atoms (light gray), oxygen atoms (red), and nitrogen atoms (blue). The bound DNA substrate is shown in a cartoon model. (C) Enlarged solvent-exposed surface where 11a (in green) binds. The surface is formed by aromatic residues Y30 (left), Y178 (right), and I115 (front), hydrophobic residues P167, I169, L174, L196, and F200 in the back (see panel H), and acidic residues (D149 and E174). In this view, the 11a adenosine is partially obscured by I115. (D) Superimposition of CamA-bound compound 11a (green; PDB 8FS1), SAH (PDB 7LT5), or sinefungin (yellow; PDB 7RFK), highlighting the U-shape of 11a. (E) The aromatic side chain of Y30 conformationally changes between the SAH/sinefungin-bound state (yellow) and the 11a-bound state (green). (F) The N-terminal helices (αA and αB) are disordered in the 11a-bound state. (G, H) Compound 11a contacts Y30 (left), E175, Y178, and D149 (right), and hydrophobic residues in the back (panel H). (I) The hydrophobic side chain of I169 packs against the aliphatic ring and the aromatic ring located, respectively, at the two ends of compound 11a. (J) The sulfur atom of 11a is 3.3 Å away from the N6 atom of the target DNA adenine. (K) Superimposition of CamA-bound compound 11a (green; PDB 8FS1) and compound 7 (magenta; PDB 8CXZ). (L, M) Two views of superimposition of three CamA-bound inhibitors, 11a (green; PDB 8FS1), GSK0946 (cyan; PDB 7RFL), and sinefungin (yellow; PDB 7RFK).
The adenosyl moiety (adenine and ribose rings) of compound 11a occupies the exact same binding site as the corresponding moieties of SAH and sinefungin (Figure 3C,D). However, unlike the homocysteine moiety of SAH and sinefungin, which exhibits an extended conformation, 11a adopts a U-shaped conformation, with the two branches folded toward each other, yielding a 3.5 Å separation between the aliphatic ring off the guanidinium group and the aromatic ring off the adenosine (Figure 3D). This separation of 3.5 Å reflects the optimum distance between the two ring carbon atoms with a van der Waals radius of 1.7 Å. Compound 11a is bordered by two tyrosine residues, Y30 adjoining the guanidinium branch and Y178 next to the adenosine branch (Figure 3D). Y30, one of the two residues that sandwich the target DNA adenine, rotates its aromatic ring ∼90° toward the guanidinium group (Figure 3E). The binding of compound 11a also resulted in disorder of the two N-terminal helices (residues 1–28), owing to the collision between the guanidinium branch and the helix αB (Figure 3F).
The two branches of 11a form extensive interactions with CamA and the bound substrate DNA target adenine (Ade), via a mixture of interactions involving van der Waals, aromatic, and polar contacts (Figure 3G,H). The branch off the guanidinium group contacts Y30, the target Ade, P167, as well as I169 (Figure 3H). The hydrophobic side chains of I169 and L174 pack against the aliphatic ring and the aromatic ring located at the ends of the two branches of 11a (Figure 3H,I). The sulfur atom interacts with the DNA target Ade N6 amino group (Figure 3J). The 3-phenylpropyl moiety attached to the Ade N6-amino of 11a has the same conformation as that of compound 7 (Figure 3K). In sum, compound 11a has a ∼520 A2 interface with CamA (>100 A2 larger than that of compound 7), occupying nearly the entire hydrophobic surface as shown in Figure 3C. As oriented in Figure 3C, the hydrophobic surface is formed by Y30 (left), Y178 (right), I115 (front), and P167, I169, L174, L196, and F200 in the back (Figure 3H). Finally, superimposition of three CamA-bound inhibitors, sinefungin (4), SGC0946 (3), and 11a, indicates that the extension off the ribose ring points in three different directions and engages in three different sets of interactions (Figure 3L,M). The 11a-mediated interactions involve the predominantly hydrophobic surface and help to explain its most potent inhibition by 11a.
Selectivity
Next, we examined four compounds, two parental compounds (6e and 7) and two derived compounds (11a and 11b), against a collection of related SAM-dependent MTases acting on nucleic acids (Figure 4), containing Escherichia coli Dam, Caulobactor vibrioides CcrM, and four human RNA (or dual RNA/DNA) adenine MTases (MettL3–MettL14, MettL5–Trm112, MettL16, and PCIF1) that have been enzymatically characterized in our laboratory.46,52−58 While these four compounds showed robust (≥80%) inhibition of CamA activity (Figure 4A) at 10 μM concentration in the presence of 40 μM SAM, none of them showed >50% inhibition of the other tested MTases (Figure 4B–G). Compound 6e came close to the 50% threshold with complexes of MettL3–MettL14 and MettL5–Trm112 (Figure 4D,E), and 11b was close for MettL5–Trm112 (Figure 4E), but the difference from CamA inhibition was dramatic. As noted above, compound 6e was developed as a pan-inhibitor of PRMT enzymes,48 and it does inhibit CamA activity with an IC50 of 1.5 μM (this study). Thus, it is not too surprising that 6e also modestly inhibits the activity of MettL3–MettL14 (with an estimated IC50 > 10 μM), as analogs of adenosine with N-substituted amide of ribofuranuronic acid off the ribose ring have been developed as inhibitors of MettL3–MettL14.59
Figure 4.
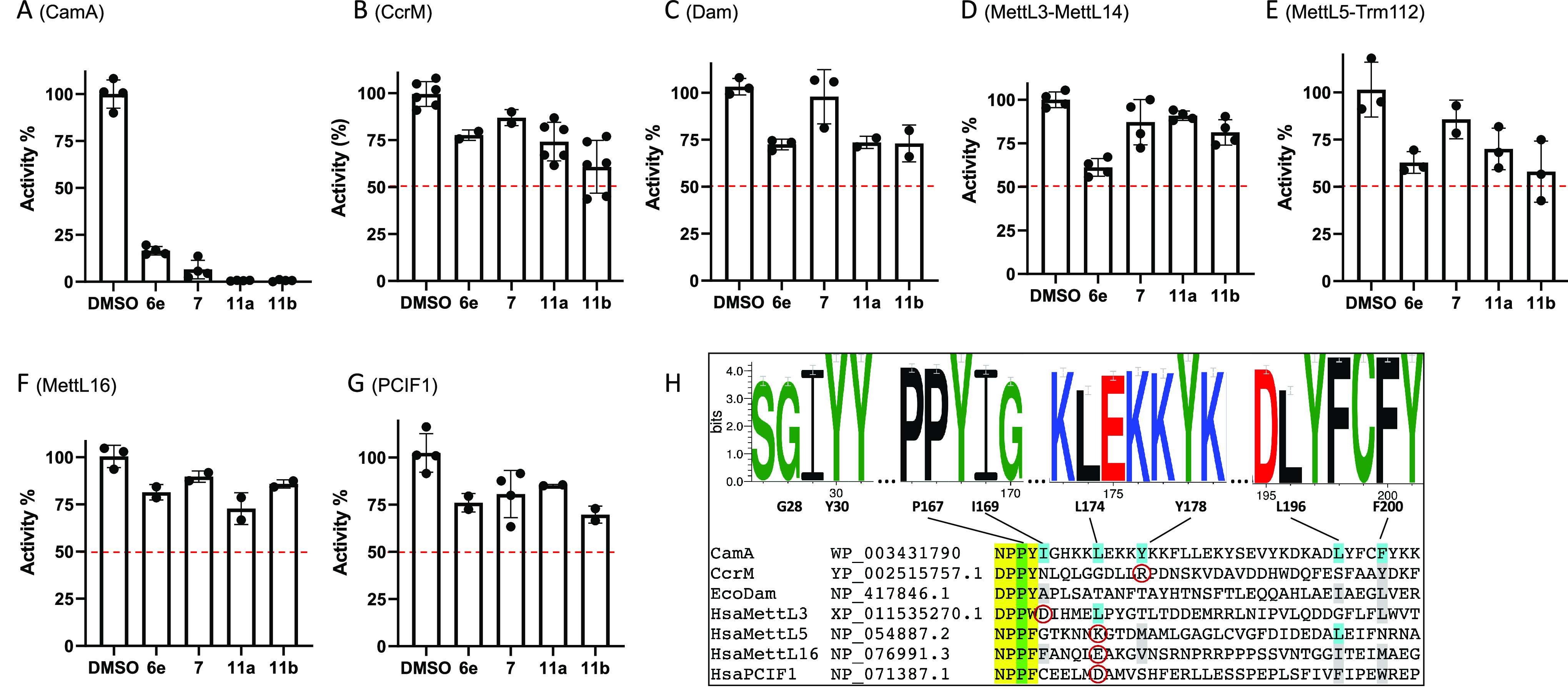
Selectivity of compounds 6e, 7, 11a, and 11b against activities of (A) CamA, (B) CcrM, (C) Dam, (D) MettL3–MettL14, (E) MettL5–Trm112, (F) MettL16, and (G) PCIF1. The oligonucleotide substrates and reaction conditions are listed in Table S2. Compound 7 used in the selectivity experiments was from ref (49). (H) Hydrophobic patch in CamA. The sequence logo shows that the eight residues forming this surface patch are very highly conserved among 58 nonidentical CamA orthologs, but they are not conserved among other DNA/RNA adenine MTases. The catalytic motif IV (D/N)PP(Y/F/W) is highlighted in yellow, the relevant P within the motif in green, and the other patch residues in cyan. In the non-CamA MTases, aligned different (but still hydrophobic) residues are shown in gray, and charged residues are shown in red circles.
As noted earlier, and discussed below, CamA differs from the other MTases in having a hydrophobic surface patch adjacent to the adenosine-binding pocket, and the N6-linked substituent in compounds 7, 11a, and 11b lies against this patch. The patch is conserved among CamA orthologs but is not seen in the other MTases (Figure 4H). For comparison, MettL16 has hydrophobic residues in five of the six aligned positions forming the CamA patch (Figure 4H), yet 11a shows just ∼25% inhibition (Figure 4F), possibly because a negative charged glutamate is in the place of L174. 11a has about the same effect on CcrM (Figure 4B), which has hydrophobic residues in just two of the six positions and also has a positively charged arginine in the place of Y178. E. coli Dam has no charged residues at the CamA hydrophobic patch positions, but has polar threonine residues at two of those positions, and shows about the same 25% inhibition as MettL16 and CcrM (Figure 4C).
Discussion
The majority (if not all) of SAM-dependent methyltransfer reactions involve a direct transfer of the methyl group from the charged methylsulfonium center to substrate, with an SN2-like mechanism.60 The close juxtaposition between the sulfur atom of SAM (the methyl donor) and substrate (the methyl acceptor) allowed design and synthesis of bivalent (or bisubstrate) inhibitors61 that have an effective linker between the two fragments, thus mimicking the transition state of the methyl transfer reaction. Compound 6e is one such example, and it inhibits PRMT1 (IC50 = 0.12 μM), CamA (IC50 = 1.5 μM), PRMT3 (IC50 = 6.5 μM) and MettL3–MettL14 (IC50 > 10 μM). The common feature of the substrates of these enzymes is the amino group (NH2) of protein arginine (Nε) or DNA/RNA adenine (N6). Variation in these bivalent inhibitors usually involves the extensions off the ribose ring, because of where the methylsulfonium atom is located. We modeled compound 6e onto the existing structures of MettL3–MettL14 complexed with known adenosine analogs having varied IC50 values59 (∼9 μM to >250 μM) (Figure 5A). It is evident that 6e could be easily accommodated in the surface groove extended from the adenosine binding site (Figure 5B).
Figure 5.

Models of compounds 6e or 11b in binding MettL3, PRMT1, and MettL5. (A) Structures of MettL3–MettL14 bound with two inhibitors (PDB 6TTW and PDB 6TTX). (B) A model of 6e binding MettL3 by superimposing the adenosine moiety. (C) Structure of PRMT1–SAH (PDB 1ORI) where the SAH binding site is exposed while the N-terminal residues are disordered in the absence of bound arginine substrate (circled). (D) Structure of PRMT1–SAH–GSK3368715 (PDB 6NT2) where SAH is largely buried in the presence of substrate-competitor inhibitor. The arrow indicates a narrow opening near the adenine N6-amino group. (E) The distance is 3.9 Å between the sulfur atom of SAH and the GSK inhibitor which occupied the substrate arginine binding site. (F) A model of compound 11b binding PRMT1 in the open conformation. (G, H) A model of compound 11b binding PRMT1 in the closed conformation. Compound 6e could be modeled the same way without the N6-addition. (I) Structure of MettL5–Trm112 in complex with SAM (PDB 6H2U). A model of 11e binding MettL5 by superimposing the adenosine moiety, and the aliphatic and aromatic rings (circled) are visible from the surface.
On the other hand, Ade N6-substituted SAH/SAM analogs have also been explored, initially targeted toward a mutant of the S. cerevisiae PRMT called RMT1.62 Recently we characterized 42 adenosine analogs as potential inhibitors of CamA, which incorporated substituents at the adenosine C6-amino group (N6), including arylalkyl groups with a growing carbon-number alkyl chain, or variously substituted aminoalkyl groups.49 Among them, compound 7 contains a 3-phenylpropyl moiety with the three-carbon aliphatic chain connecting the Ade N6 atom with the phenyl ring, and which inhibits CamA with an IC50 of 0.7 μM. However, compound 7 inhibits neither PRMT1 nor PRMT3 (Figure S1), nor does it significantly reduce the activities of a few other tested DNA/RNA adenine MTases (Figure 4).
Combining features of 6e and 7 gave rise to compounds 11a and 11b, with extensions projecting in two directions from ribose and adenine, respectively. We show here that 11a and 11b have synergistic effects on specificity, increasing inhibition of CamA and at the same time decreasing that of PRMT1 (Figure 2C). In addition to the direction of the effects differing between 11a and 11b on CamA and PRMT1, the strength of these respective effects also differs, which might be attributed to the length of hydrophobic linker joining the thioadenosine and guanidinium group. For CamA, compound 11a has 1.8× increased inhibition of CamA relative to 11b; the 3-carbon length in 11b might disturb the optimal distance between the aliphatic ring and the aromatic ring seen in 11a, and resulted in a partially disordered aliphatic ring (Figure 3A). For PRMT1, both compounds showed reduced inhibition from that of the parental compound 6e; but compound 11b, with an IC50 value of 1.2 μM under the conditions tested (Figure 2C), has >11× stronger inhibition than 11a, in agreement with a previous study showing that the 3-carbon linker is preferred for the PRMT bisubstrate inhibitors.51
PRMT1–SAH structures have been determined to be in an open conformation in the absence of bound substrate37 (Figure 5C) and in a closed conformation in the presence of a substrate-competitive inhibitor63 (Figure 5D,E). While 6e and 11b could be easily modeled onto the open conformation (Figure 5F), the 3-carbon optimum length in 11b for PRMT151 might force PRMT1 to undergo conformational change to accommodate the substituent at the N6-amino group (i.e., to open up a narrow opening near the N6-amino group indicated by an arrow in Figure 5D,G). Compound 11b links a portion of SAH to a part of the substrate-competitive inhibitor (Figure 5H). In addition, compound 11b, which at a concentration of 10 μM showed close to 50% inhibition of MettL5–Trm112 (Figure 4E), could be modeled with two branches occupying the SAM and target adenine binding sites, respectively (Figure 5I).
Here, we present compound 11a [1-(cyclohexylmethyl)-3-(2-((((2S,3S,4R,5R)-3,4-dihydroxy-5-(6-((3-phenylpropyl)amino)-9H-purin-9-yl)tetrahydrofuran-2-yl)methyl)thio)ethyl)guanidine] as a selective and potent CamA inhibitor. Like SAM/SAH, the inhibitor occupies the adenosine binding site with the N6-addition and the S-linked substitution directed outward and resting against a mainly hydrophobic surface, engaging in a mixture of interactions involving van der Waals, aromatic, and polar contacts (Figure 3C). The compound fully occupied the entire pocket-adjacent hydrophobic surface that appears to be unique to CamA among amino MTases examined in this study. We suggest that this hydrophobic surface patch contributes to CamA specific targetability.
To further improve our lead compound’s ability, we will turn our attention to the space occupied by the homocysteine moiety of SAH/sinefungin, which points in a different direction from that of the S-linked substitute in 11a (Figure 3D). A branched extension at or near the sulfur atom could be designed to merge 11a and sinefungin or 11a and SGC0946 (Figure 3L,M). For example, derivatives of AzaSAM, in which the nitrogen that replaces the SAM sulfur atom is coupled to various alkylamino groups, were developed as inhibitors of Set7/9 (a protein lysine MTase).64 In sum, our work on discovering CamA inhibitors would specifically add to continuing exploration for other antimicrobial and/or antivirulence agents targeting C. difficile and could yield synergistic therapeutic approaches in clinical settings.
Methods
Chemistry
Starting materials, reagents, and solvents were purchased commercially (Sigma Chemical Co. and Ambeed Inc.). Analytic and preparative high-pressure liquid chromatography (RP-HPLC) were performed on an Agilent 1260 Series system, which was run with a 5–95% acetonitrile/water gradient with 0.1% trifluoroacetic acid (TFA). Flash chromatography was performed on a Teledyne CombiFlash Rf 100 system, run with a 0–5% methanol/dichloromethane gradient. The reaction yields were estimated based on the weight of the desired products. All 1H and 13C NMR spectra were recorded on a Bruker 500 MHz spectrometer. Based on HPLC analysis, all compounds used for IC50 determination possessed a purity of >95%. High-resolution mass spectrometry (HRMS) spectra were logged on an Agilent high resolution 6550 quadrupole time-of-flight (Q-TOF) LC-MS instrument.
The desired compounds 11a and 11b were synthesized following the reported methods48 (Scheme 1). Briefly, the commercially available 6-chloropurine ribonucleoside reacted with 3-phenylpropylamine to yield N6-phenylpropyladenosine (7), which was then subjected to the Mitsunobu reaction to produce compound 8. Then the acetyl group of 8 was removed, followed by its substitution with Boc protected ethyl or propyl amine. Next, the Boc group was removed in the presence of 50% formic acid in water. Under vacuum, the volatiles were removed, and the residues were lyophilized to produce compounds 9a and 9b as a pale-yellow solid. Compound 10 was made as reported in quantitative yields,48 and was then subjected to the reaction with 9a–9b, followed by final Fmoc deprotection to generate 11a–11b.
Scheme 1.

Reagents and conditions of analog synthesis: (a) n-butanol, 3-phenylpropylamine, reflux, 12 h, 83%; (b) thioacetic acid, triphenylphosphine, diisopropyl azodicarboxylate, tetrahydrofuran, 0 °C to room temperature, overnight, 71%; (c) sodium methoxide, MeOH, room temperature, overnight; (d) HCOOH, H2O, 0 °C to room temperature, 2 d, 75–77% in two steps; (e) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, N,N-diisopropylethylamine, dichloromethane, room temperature, overnight; (f) piperidine, dichloromethane, room temperature, 30 min, 53–57% in two steps.
Synthesis of Compound 7
6-Chloropurine ribonucleoside (2.86 g, 10.0 mmol) and 3-phenylpropylamine (6.76 g, 50.0 mmol), in 200 mL of anhydrous n-butanol, were stirred under reflux for 12 h. After cooling down to RT and under reduced pressure, the volatiles were removed, and the residue was purified by flash chromatography (DCM/MeOH, 95/5) to give 7 (3.20 g, yield 83%).
Synthesis of Compound 8
In a chilly solution of triphenylphosphine (2.62 g,10.0 mmol) in dry THF (100 mL), diethyl azodicarboxylate (1.6 mL, 10 mmol) was added drop by drop over 5 min and stirred for 30 min. Then 7 (1.92 g, 5.0 mmol) was added and continued to stir for 2 h. To the resulting yellow suspension was slowly added a solution of thioacetic acid (0.7 mL, 10.0 mmol) in dry THF (5 mL); the mixture was stirred overnight while the temperature increased slowly to RT. The crude oil was purified by flash chromatography (DCM/MeOH, 97/3) to yield 8 (1.57 g, yield 71%), after the removal of volatiles under reduced pressure.
Synthesis of Compounds 9a and 9b
Under an argon atmosphere, to a stirring solution of compound 8 (445 mg, 1.0 mmol) and tert-butyl (2-bromoethyl)carbamate or tert-butyl (3-bromopropyl)carbamate (1.5 mmol) in dry methanol (20 mL) was added sodium methoxide (30% in methanol, 0.6 mL, 3.0 mmol). The volatiles were removed under reduced pressure, after stirring overnight at RT, and the residue was purified by flash chromatography (DCM/MeOH, 97/3). The fractions were collected and dried under vacuum. The generated oil was dissolved in water (5.0 mL) and formic acid (5.0 mL) at 0 °C and stirred at RT for 2 days. Under reduced pressure, the volatiles were removed and the residues were lyophilized to afford the desired product 9a (333 mg, yield 75%) and 9b (352 mg, yield 77%), each as a pale-yellow solid.
Synthesis of Thiourea (10)
To a stirring solution of Fmoc-isothiocyanate (281 mg, 1.0 mmol) in 25 mL at 0 °C, DCM was slowly added cyclohexylmethanamine (113 mg, 1.0 mmol). The reaction mixture was stirred for 1 h at RT. The volatiles were removed under reduced pressure to yield crude 10, which was directly used for the reaction.
Synthesis of Compounds 11a and 11b
To a stirring solution of amine 9a or 9b (0.1 mmol) in 10 mL DCM and thiourea 10 (59 mg, 0.15 mmol) were added 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (23 mg, 0.15 mmol) and N,N-diisopropylethylamine (19 mg, 0.15 mmol). The reaction mixture was stirred at RT overnight and was then diluted with 50 mL water and extracted with dichloromethane (3 × 20 mL). The combined organic layers were washed with water and brine and dried with anhydrous sodium sulfate. The volatiles were removed under reduced pressure to afford a crude oil, which was subsequently treated with 20% piperidine in 10 mL dichloromethane for 30 min. Then, the reaction was concentrated and separated with preparative HPLC (MeCN/H2O, 10–40%) to yield 11a (31 mg, yield 53%) and 11b (34 mg, yield 57%).
11a
HRMS m/z calcd for C29H42N8O3S [M + H]+: 583.3173. Found: 583.3185. 1H NMR (500 MHz, CD3OD) δ 8.53–8.32 (m, 1H), 8.30 (s, 1H), 7.26–7.15 (m, 4H), 7.15–7.09 (m, 1H), 6.02 (d, J = 4.9 Hz, 1H), 4.75 (s, 1H), 4.33 (t, J = 5.1 Hz, 1H), 4.25–4.17 (m, 1H), 3.58 (s, 2H), 3.38 (t, J = 6.6 Hz, 2H), 3.11–2.91 (m, 4H), 2.75 (ddd, J = 15.4, 10.5, 6.5 Hz, 4H), 2.03 (s, 2H), 1.81–1.59 (m, 5H), 1.52 (ddd, J = 11.4, 7.9, 4.0 Hz, 1H), 1.31–1.11 (m, 3H), 0.93 (tt, J = 12.1, 6.3 Hz, 2H). 13C NMR (126 MHz, CD3OD) δ 156.20, 152.55, 149.45, 147.88, 141.23, 140.52, 128.06, 128.03, 125.61, 119.57, 88.96, 84.46, 73.64, 72.62, 40.76, 37.21, 33.87, 32.61, 31.43, 30.20, 25.98, 25.41.
11b
HRMS m/z calcd for C30H44N8O3S [M + H]+: 597.3330. Found: 597.3342. 1H NMR (500 MHz, CD3OD) δ 8.32 (s, 1H), 8.28 (s, 1H), 7.30–7.17 (m, 4H), 7.16–7.10 (m, 1H), 6.01 (d, J = 4.9 Hz, 1H), 4.75 (s, 1H), 4.32 (t, J = 5.0 Hz, 1H), 4.25–4.16 (m, 1H), 3.58 (s, 1H), 3.25 (t, J = 6.9 Hz, 2H), 3.09–2.87 (m, 4H), 2.74 (dd, J = 8.6, 6.8 Hz, 2H), 2.63 (t, J = 7.1 Hz, 2H), 2.03 (t, J = 7.6 Hz, 2H), 1.84 (q, J = 6.9 Hz, 2H), 1.80–1.59 (m, 6H), 1.52 (dt, J = 7.8, 4.0 Hz, 1H), 1.35–1.10 (m, 4H), 0.94 (tt, J = 12.0, 6.3 Hz, 2H). 13C NMR (126 MHz, CD3OD) δ 156.20, 153.23, 150.38, 141.33, 140.12, 128.04, 125.58, 119.50, 88.84, 84.30, 73.63, 72.65, 39.88, 37.28, 33.90, 32.65, 30.21, 29.21, 28.34, 25.99, 25.42.
MTases Used in the Study
The MTases used in the current study were characterized previously and prepared in our laboratories: Clostridioides difficile CamA (pXC2184),35Caulobacter crescentus [now called C. vibrioides] CcrM (pXC2121),65Escherichia coli Dam (pXC1612),52,53 human PCIF1 (pXC2055),46,58 human MettL16 (pXC2210),46 human MettL5–Trm112 (pXC2062–pXC2076),46 and human MettL3–MettL14.55,56 Human PRMT1 (residues 10–352)66 and human PRMT3 (residues 211–531)67 were purified according to published procedures.
Inhibition Assay on PRMT1 and PRMT3
To measure the inhibitory effect of the compounds on MTase activities of PRMT1 and PRMT3, a fluorescence-based SAHH-coupled assay68,69 was utilized. A BMG CLARIOstar microplate reader measured the fluorescence signal for 18 min with excitation at 400 nm and emission at 465 nm. All experiments were performed in duplicate, and GraphPad Prism software 8.0 was used to process data.
For PRMT1, the assay was performed in 100 μL including 0.1 μM PRMT1, 10 μM SAM, 5 μM SAHH, and 10 μM ThioGlo4 in 2.5 mM HEPES (pH 7.0), 25 mM NaCl, 25 μM EDTA, 50 μM tris(2-carboxyethyl)phosphine (TCEP), 0.01% Triton X-100. For PRMT3, the assay was performed in 100 μL including 0.25 μM PRMT3, 30 μM SAM, 5 μM SAHH, and 15 μM ThioGlo4 in 20 mM Tris (pH, 7.5), 0.01% Triton X-100. After incubation with the inhibitors for 10 min at 37 °C (PRMT1) or 30 °C (PRMT3), the addition of 5 μM histone H4 peptide (residues 1–21) initiated reactions.
Inhibition Assay of CamA-Mediated Methylation
The CamA methylation assay was performed in 100 mM NaCl, 50 mM Tris-HCl, pH 7.5, 1 mM dithiothreitol (DTT), 0.25% DMSO, and 0.1 mg mL–1 BSA. CamA (50 nM final concentration) was preincubated with varied concentration of compound at RT (∼22 °C) for 5 min followed by addition of 2.5 μM double-stranded DNA (final concentration) (Table S2) and 40 μM SAM (final concentration) (Figures 1B,D and S1). To determine the Ki of compound 11a against CamA, the methylation reaction was carried out with varying concentrations of SAM (20, 30, 40, and 80 μM) and inhibitor 11a (0, 0.25, and 0.5 μM) (Figure 2E). Reactions were incubated for 3 min at ∼22 °C (RT), and quenched by adding TFA to 0.1%. The final reaction mixture (5 μL) was transferred to a low-volume 384-well plate, and the SAH concentration was measured by Promega luminescence assay (MTase-Glo).70 A Synergy 4 Multi-Mode Microplate Reader (BioTek) was used to detect the luminescence signal.
Compound Selectivity Assays
Table S2 lists the MTases used in the selectivity assays (Figure 4), their respective substrates, and the reaction conditions under 10 μM inhibitor concentration. Integrated DNA Technologies (IDT) synthesized oligonucleotides, and the mRNA cap analog (catalog number N-7113) was purchased from TriLink BioTechnologies. Inhibition activities at RT (∼22 °C) were measured by Promega luminescence assay (MTase-Glo).
X-ray Crystallography
An Art Robbins Gryphon Crystallization Robot set up the sitting drop crystals screening by vapor diffusion method. Crystals of CamA–DNA–11a (or 11b) grew under similar conditions after 3–4 days at ∼19 °C (RT): 0.28 M potassium citrate, 0.1 M Tris-HCl, pH 7.0–7.5, and 21–24% (w/v) polyethylene glycol 3350. Crystals were quickly soaked in reservoir solution supplemented with 20% (v/v) ethylene glycol and flash frozen by liquid nitrogen.
At Argonne National Laboratory, X-ray diffraction data were collected at the SER-CAT beamline 22ID of the Advanced Photon Source. Crystallographic data sets were indexed, integrated, and scaled with HKL2000.71 The difference Fourier method was used to solve the structures of the ternary CamA–DNA–11a (or 11b) complex, using the binary CamA–DNA structure as a template (PDB 7LNJ). Crystals were isomorphous essentially. In the first refinement cycle, rigid body refinement was used for positioning the new structures in the unit cell, and difference electron density maps (2Fo – Fc and Fo – Fc) were used for locating bound inhibitor molecules. The grade web server (http://grade.globalphasing.org) was used to generate CIF file restraints for 11a and 11b and for model building and refinement. COOT72 and PHENIX REFINE73 were used respectively and iteratively for model building and refinement of the structure. Table S1 summarizes the statistics of diffraction data collection and refinement. The PDB validation server74 was used for validation of the final structural models. PyMol version 2.5.2 (Schrödinger, LLC) was used to prepare structure images.
Computer Modeling of Compounds 6e and 11b Binding in MettL3, MettL5, and PRMT1
COOT75 was utilized to superimpose the structure of 11b-bound CamA (PDB 8FS2) with structures of other MTases (MettL3,59 MettL5,76 PRMT137,63) having bound SAM/SAH or compounds also containing an adenosine moiety. The secondary-structure matching (SSM)77 and least-squares (LSQ) options were primarily used for the superimposition. SSM aligns proteins based on their topology which works fairly well with the seven-stranded Class I MTases,41 and LSQ allowed additional fine-tuning of the superimposition. Small rotations and translations of 11b were made such that the adenosine moiety of 11b exactly overlaid with that moiety in the other MTase structures. Finally, the edit chi angle function in COOT allowed rotation of permissible torsional angles to place the extensions of 11b so as to eliminate clashes with the MTase and permit potential interactions. The 6e-bound MettL3 was generated by deleting the 3-phenylpropyl moiety from 11b.
Sequence Alignment of Residues Forming a Hydrophobic Patch in CamA
CamA is the only known DNA adenine MTase with a hydrophobic surface patch adjacent to the SAM-binding pocket. A WebLogo analysis78 of 58 nonidentical CamA orthologs32 shows that the eight hydrophobic residues forming this surface patch are very highly conserved among CamA orthologs (the upper panel of Figure 4H). However, these residues are not conserved among other DNA/RNA adenine MTases, CcrM from Caulobacter vibrioides (formerly crescentus) (YP_002515757.1), Dam from E. coli (NP_417846.1), and four human RNA MTases, MettL3 (XP_011535270.1) (the catalytic subunit of MettL3–MettL14), MettL5 (NP_054887.2) (the catalytic subunit of MettL5–Trm112), MettL16 (NP_076991.3), and PCIF1 (NP_071387.1). Only the more carboxyl-proximal six of the eight patch-forming residues are shown in the lower panel of Figure 4H, as the sequences in that region can be unambiguously aligned using the conserved (N/D)PP(Y/F/W) motif IV79 as an anchor point.
Acknowledgments
We thank Y. Cao of MD Anderson Cancer Center for technical assistance. We thank the beamline scientists of Southeast Regional Collaborative Access Team (SER-CAT) at the Advanced Photon Source (APS), Argonne National Laboratory, USA. The work was supported by U.S. National Institutes of Health grant R35GM134744 (to X.C.), Cancer Prevention and Research Institute of Texas grant RR160029 (to X.C., who is a CPRIT Scholar in Cancer Research), and Purdue University Faculty Scholar program from the Ralph W. and Grace M. Showalter Trust (to R.H.), Italian Ministry of University FISR2019_00374 MeDyCa (to A.M.) and Progetto di Ateneo Sapienza 2021 no. RM12117A61C811CE (to D.R.). The use of SER-CAT is supported by its member institutions and equipment grants (S10_RR25528, S10_RR028976, and S10_OD027000) from the US National Institutes of Health. Use of the APS was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract W-31-109-Eng-38.
Glossary
Abbreviations
- CamA
Clostridioides difficile-specific DNA adenine methyltransferase
- CDI
Clostridioides difficile infections
- IC50
half maximal inhibitory concentration
- KD
dissociation constant
- MTase
methyltransferase
- PRMT
protein arginine methyltransferase
- SAH
S-adenosyl-l-homocysteine
- SAM
S-adenosyl-l-methionine
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.3c00035.
Summary of X-ray data collection and refinement statistics (Table S1); Inhibition reaction conditions (Table S2); Raw data of IC50 plots (Figure S1); NMR spectra of compounds 11a and 11b; HR-MS spectra of compounds 11a and 11b; Purity of compounds 11a and 11b (PDF)
Molecular formula strings (SMILES) and associated data (XLSX)
Accession Codes
The experimental data that support the findings of this study are contained within the article. The X-ray structure (coordinates) and the source data (structure factor file) of CamA–DNA with bound inhibitors have been deposited to the PDB, and these will be released upon article publication under accession numbers PDB 8FS1 (11a) and PDB 8FS2 (11b).
Author Contributions
▽ J.Z. and Y.D. contributed equally to this work. J.Z. performed CamA and PCIF1 purification, crystallization, and inhibition assays; Y.D. performed compound synthesis (7–10) and PRMT inhibition; I.D.I. provided compounds of 6 series; J.R.H. helped with X-ray data collection, provided Dam enzyme, and performed compound modeling; D.Y. purified MettL16 and MettL5; T.H. and M.V. purified MettL3–MettL14 complex; D.R. and A.M. provided compound 7 used in the selectivity assays; R.M.B. performed bioinformatics, assisted in preparing the manuscript, and participated in discussion; X.Z. provided supervision, conceptualization, and project administration; R.H. and X.C. designed and organized the study, performed writing, reviewing, and editing of the manuscript, and provided conceptualization and funding acquisition.
The authors declare no competing financial interest.
Supplementary Material
References
- Cantoni G. L. S-Adenosylmethionine; a new intermediate formed enzymatically from L-methionine and adenosinetriphosphate. J. Biol. Chem. 1953, 204 (1), 403–16. 10.1016/S0021-9258(18)66148-4. [DOI] [PubMed] [Google Scholar]
- Cheng X.; Blumenthal R. M.. S-Adenosylmethionine-Dependent Methyltransferases: Structures and Functions; World Scientific Publishing Co: River Edge, NJ, 1999. [Google Scholar]
- Cantoni G. L. Biological methylation: selected aspects. Annu. Rev. Biochem. 1975, 44, 435–51. 10.1146/annurev.bi.44.070175.002251. [DOI] [PubMed] [Google Scholar]
- Abdelraheem E.; Thair B.; Varela R. F.; Jockmann E.; Popadic D.; Hailes H. C.; Ward J. M.; Iribarren A. M.; Lewkowicz E. S.; Andexer J. N.; Hagedoorn P. L.; Hanefeld U. Methyltransferases: Functions and Applications. Chembiochem 2022, 23, e202200212. 10.1002/cbic.202200212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozarich J. W. S-adenosylmethionine-dependent enzyme activation. Biofactors 1988, 1, 123–8. [PubMed] [Google Scholar]
- Sofia H. J.; Chen G.; Hetzler B. G.; Reyes-Spindola J. F.; Miller N. E. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001, 29 (5), 1097–106. 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick J. B.; Broderick W. E.; Hoffman B. M. Radical SAM enzymes: Nature’s choice for radical reactions. FEBS Lett. 2023, 597 (1), 92–101. 10.1002/1873-3468.14519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyachandran V. R.; Boal A. K. Structural insights into auxiliary cofactor usage by radical S-adenosylmethionine enzymes. Curr. Opin Chem. Biol. 2022, 68, 102153. 10.1016/j.cbpa.2022.102153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae D. H.; Lane D. J. R.; Jansson P. J.; Richardson D. R. The old and new biochemistry of polyamines. Biochim Biophys Acta Gen Subj 2018, 1862 (9), 2053–2068. 10.1016/j.bbagen.2018.06.004. [DOI] [PubMed] [Google Scholar]
- Winkler W. C.; Nahvi A.; Sudarsan N.; Barrick J. E.; Breaker R. R. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat. Struct. Biol. 2003, 10 (9), 701–7. 10.1038/nsb967. [DOI] [PubMed] [Google Scholar]
- Zheng L.; Song Q.; Xu X.; Shen X.; Li C.; Li H.; Chen H.; Ren A. Structure-based insights into recognition and regulation of SAM-sensing riboswitches. Sci. China Life Sci. 2023, 66 (1), 31–50. 10.1007/s11427-022-2188-7. [DOI] [PubMed] [Google Scholar]
- Ramos K. N.; Gregornik D.; Ramos K. S. Pharmacogenomics insights into precision pediatric oncology. Curr. Opin Pediatr 2021, 33 (6), 564–569. 10.1097/MOP.0000000000001065. [DOI] [PubMed] [Google Scholar]
- Franca R.; Braidotti S.; Stocco G.; Decorti G. Understanding thiopurine methyltransferase polymorphisms for the targeted treatment of hematologic malignancies. Expert Opin Drug Metab Toxicol 2021, 17 (10), 1187–1198. 10.1080/17425255.2021.1974398. [DOI] [PubMed] [Google Scholar]
- Siklos M.; Kubicek S. Therapeutic targeting of chromatin: Status and opportunities. FEBS J. 2022, 289 (5), 1276–1301. 10.1111/febs.15966. [DOI] [PubMed] [Google Scholar]
- Tatekawa S.; Ofusa K.; Chijimatsu R.; Vecchione A.; Tamari K.; Ogawa K.; Ishii H. Methylosystem for Cancer Sieging Strategy. Cancers (Basel) 2021, 13 (20), 5088. 10.3390/cancers13205088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Sun C.; Cai W.; Li J.; Rosen B. P.; Chen J. Insights into S-adenosyl-l-methionine (SAM)-dependent methyltransferase related diseases and genetic polymorphisms. Mutat Res. Rev. Mutat Res. 2021, 788, 108396. 10.1016/j.mrrev.2021.108396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A. S. M.; Li F.; Eram M. S.; Bolotokova A.; Dela Sena C. C.; Vedadi M. Chemical probes for protein arginine methyltransferases. Methods 2020, 175, 30–43. 10.1016/j.ymeth.2019.11.017. [DOI] [PubMed] [Google Scholar]
- Jarrold J.; Davies C. C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret?. Trends Mol. Med. 2019, 25 (11), 993–1009. 10.1016/j.molmed.2019.05.007. [DOI] [PubMed] [Google Scholar]
- Yoo C. B.; Jones P. A. Epigenetic therapy of cancer: past, present and future. Nat. Rev. Drug Discov 2006, 5 (1), 37–50. 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Zheng Y. G. SAM/SAH Analogs as Versatile Tools for SAM-Dependent Methyltransferases. ACS Chem. Biol. 2016, 11 (3), 583–97. 10.1021/acschembio.5b00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X. C.; Kapilashrami K.; Luo M. Synthesis and Assays of Inhibitors of Methyltransferases. Methods Enzymol 2016, 574, 245–308. 10.1016/bs.mie.2016.01.009. [DOI] [PubMed] [Google Scholar]
- Rudenko A. Y.; Mariasina S. S.; Sergiev P. V.; Polshakov V. I. Analogs of S-Adenosyl-L-Methionine in Studies of Methyltransferases. Mol. Biol. 2022, 56 (2), 229–250. 10.1134/S002689332202011X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q.; Schapira M.; Arrowsmith C. H.; Barsyte-Lovejoy D. Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov 2021, 20 (7), 509–530. 10.1038/s41573-021-00159-8. [DOI] [PubMed] [Google Scholar]
- Villar M. V.; Spreafico A.; Moreno V.; Braña I.; Hernandez T.; Razak A. A.; Wang J.; Haddish-Berhane N.; Mehta J.; Johnson A.; Maes A.; Haslam J.; Mistry P.; Kalota A.; Lenox L.; Infante J.; Lorenzi M.; Xie H.; Lauring J.; Patel M. R. First-in-human study of JNJ-64619178, a protein arginine methyltransferase 5 (PRMT5) inhibitor, in patients with advanced cancers. Annals of Oncology 2020, 31, S470. 10.1016/j.annonc.2020.08.651. [DOI] [Google Scholar]
- Jain R.; Butler K. V.; Coloma J.; Jin J.; Aggarwal A. K. Development of a S-adenosylmethionine analog that intrudes the RNA-cap binding site of Zika methyltransferase. Sci. Rep. 2017, 7, 1632. 10.1038/s41598-017-01756-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed-Belkacem R.; Sutto-Ortiz P.; Guiraud M.; Canard B.; Vasseur J. J.; Decroly E.; Debart F. Synthesis of adenine dinucleosides SAM analogs as specific inhibitors of SARS-CoV nsp14 RNA cap guanine-N7-methyltransferase. Eur. J. Med. Chem. 2020, 201, 112557. 10.1016/j.ejmech.2020.112557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota K.; Schapira M.; Perveen S.; Khalili Yazdi A.; Li F.; Chau I.; Ghiabi P.; Hajian T.; Loppnau P.; Bolotokova A.; Satchell K. J. F.; Wang K.; Li D.; Liu J.; Smil D.; Luo M.; Jin J.; Fish P. V.; Brown P. J.; Vedadi M. Probing the SAM Binding Site of SARS-CoV-2 Nsp14 In Vitro Using SAM Competitive Inhibitors Guides Developing Selective Bisubstrate Inhibitors. SLAS Discov 2021, 26 (9), 1200–1211. 10.1177/24725552211026261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H.; Yan X.; Zhu K.; Zhang H. Discovery of Novel PRMT5 Inhibitors by Virtual Screening and Biological Evaluations. Chem. Pharm. Bull. (Tokyo) 2019, 67 (4), 382–388. 10.1248/cpb.c18-00980. [DOI] [PubMed] [Google Scholar]
- Szewczyk M. M.; Ishikawa Y.; Organ S.; Sakai N.; Li F.; Halabelian L.; Ackloo S.; Couzens A. L.; Eram M.; Dilworth D.; Fukushi H.; Harding R.; Dela Sena C. C.; Sugo T.; Hayashi K.; McLeod D.; Zepeda C.; Aman A.; Sanchez-Osuna M.; Bonneil E.; Takagi S.; Al-Awar R.; Tyers M.; Richard S.; Takizawa M.; Gingras A. C.; Arrowsmith C. H.; Vedadi M.; Brown P. J.; Nara H.; Barsyte-Lovejoy D. Pharmacological inhibition of PRMT7 links arginine monomethylation to the cellular stress response. Nat. Commun. 2020, 11, 2396. 10.1038/s41467-020-16271-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.; Chory E. J.; Wernimont A. K.; Tempel W.; Scopton A.; Federation A.; Marineau J. J.; Qi J.; Barsyte-Lovejoy D.; Yi J.; Marcellus R.; Iacob R. E.; Engen J. R.; Griffin C.; Aman A.; Wienholds E.; Li F.; Pineda J.; Estiu G.; Shatseva T.; Hajian T.; Al-Awar R.; Dick J. E.; Vedadi M.; Brown P. J.; Arrowsmith C. H.; Bradner J. E.; Schapira M. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Horton J. R.; Yu D.; Ren R.; Blumenthal R. M.; Zhang X.; Cheng X. Repurposing epigenetic inhibitors to target the Clostridioides difficile-specific DNA adenine methyltransferase and sporulation regulator CamA. Epigenetics 2022, 17 (9), 970–981. 10.1080/15592294.2021.1976910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira P. H.; Ribis J. W.; Garrett E. M.; Trzilova D.; Kim A.; Sekulovic O.; Mead E. A.; Pak T.; Zhu S.; Deikus G.; Touchon M.; Lewis-Sandari M.; Beckford C.; Zeitouni N. E.; Altman D. R.; Webster E.; Oussenko I.; Bunyavanich S.; Aggarwal A. K.; Bashir A.; Patel G.; Wallach F.; Hamula C.; Huprikar S.; Schadt E. E.; Sebra R.; van Bakel H.; Kasarskis A.; Tamayo R.; Shen A.; Fang G. Epigenomic characterization of Clostridioides difficile finds a conserved DNA methyltransferase that mediates sporulation and pathogenesis. Nat. Microbiol 2020, 5 (1), 166–180. 10.1038/s41564-019-0613-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guh A. Y.; Mu Y.; Winston L. G.; Johnston H.; Olson D.; Farley M. M.; Wilson L. E.; Holzbauer S. M.; Phipps E. C.; Dumyati G. K.; Beldavs Z. G.; Kainer M. A.; Karlsson M.; Gerding D. N.; McDonald L. C. Emerging Infections Program Clostridioides difficile Infection Working, G., Trends in U.S. Burden of Clostridioides difficile Infection and Outcomes. New England journal of medicine 2020, 382 (14), 1320–1330. 10.1056/NEJMoa1910215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams D. J.; Barone J. B.; Nylund C. M. Community-Associated Clostridioides difficile Infection in Children: A Review of Recent Literature. J. Pediatric Infect Dis Soc. 2021, 10, S22–S26. 10.1093/jpids/piab064. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Horton J. R.; Blumenthal R. M.; Zhang X.; Cheng X. Clostridioides difficile specific DNA adenine methyltransferase CamA squeezes and flips adenine out of DNA helix. Nat. Commun. 2021, 12, 3436. 10.1038/s41467-021-23693-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Zhou L.; Cheng X. Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J. 2000, 19 (14), 3509–19. 10.1093/emboj/19.14.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Cheng X. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 2003, 11 (5), 509–20. 10.1016/S0969-2126(03)00071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue W. W.; Hassler M.; Roe S. M.; Thompson-Vale V.; Pearl L. H. Insights into histone code syntax from structural and biochemical studies of CARM1 methyltransferase. EMBO J. 2007, 26 (20), 4402–12. 10.1038/sj.emboj.7601856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.; Wang M.; Lv Z.; Yang N.; Liu Y.; Bao S.; Gong W.; Xu R. M. Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (51), 20538–43. 10.1073/pnas.1106946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q.; Wang H.; Ng H. H.; Erdjument-Bromage H.; Tempst P.; Struhl K.; Zhang Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 2002, 12 (12), 1052–8. 10.1016/S0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- Schubert H. L.; Blumenthal R. M.; Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 2003, 28 (6), 329–35. 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Collins R. E.; Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 267–94. 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urig S.; Gowher H.; Hermann A.; Beck C.; Fatemi M.; Humeny A.; Jeltsch A. The Escherichia coli dam DNA methyltransferase modifies DNA in a highly processive reaction. J. Mol. Biol. 2002, 319 (5), 1085–96. 10.1016/S0022-2836(02)00371-6. [DOI] [PubMed] [Google Scholar]
- Bergerat A.; Guschlbauer W. The double role of methyl donor and allosteric effector of S-adenosyl-methionine for Dam methylase of E. coli. Nucleic Acids Res. 1990, 18 (15), 4369–75. 10.1093/nar/18.15.4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzabal S.; DuBois S.; Thielking V.; Cano A.; Eritja R.; Guschlbauer W. Dam methylase from Escherichia coli: kinetic studies using modified DNA oligomers: hemimethylated substrates. Nucleic Acids Res. 1995, 23 (18), 3648–55. 10.1093/nar/23.18.3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.; Kaur G.; Blumenthal R. M.; Zhang X.; Cheng X. Enzymatic characterization of three human RNA adenosine methyltransferases reveals diverse substrate affinities and reaction optima. J. Biol. Chem. 2021, 296, 100270. 10.1016/j.jbc.2021.100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guccione E.; Schwarz M.; Di Tullio F.; Mzoughi S. Cancer synthetic vulnerabilities to protein arginine methyltransferase inhibitors. Curr. Opin Pharmacol 2021, 59, 33–42. 10.1016/j.coph.2021.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyamu I. D.; Al-Hamashi A. A.; Huang R. A Pan-Inhibitor for Protein Arginine Methyltransferase Family Enzymes. Biomolecules 2021, 11 (6), 854. 10.3390/biom11060854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Horton J. R.; Menna M.; Fiorentino F.; Ren R.; Yu D.; Hajian T.; Vedadi M.; Mazzoccanti G.; Ciogli A.; Weinhold E.; Huben M.; Blumenthal R. M.; Zhang X.; Mai A.; Rotili D.; Cheng X. Systematic Design of Adenosine Analogs as Inhibitors of a Clostridioides difficile-Specific DNA Adenine Methyltransferase Required for Normal Sporulation and Persistence. J. Med. Chem. 2023, 66 (1), 934–950. 10.1021/acs.jmedchem.2c01789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonday Z. Q.; Cortez G. S.; Grogan M. J.; Antonysamy S.; Weichert K.; Bocchinfuso W. P.; Li F.; Kennedy S.; Li B.; Mader M. M.; Arrowsmith C. H.; Brown P. J.; Eram M. S.; Szewczyk M. M.; Barsyte-Lovejoy D.; Vedadi M.; Guccione E.; Campbell R. M. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS medicinal chemistry letters 2018, 9 (7), 612–617. 10.1021/acsmedchemlett.8b00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hamashi A. A.; Chen D.; Deng Y.; Dong G.; Huang R. Discovery of a potent and dual-selective bisubstrate inhibitor for protein arginine methyltransferase 4/5. Acta Pharm. Sin B 2021, 11 (9), 2709–2718. 10.1016/j.apsb.2020.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton J. R.; Liebert K.; Hattman S.; Jeltsch A.; Cheng X. Transition from nonspecific to specific DNA interactions along the substrate-recognition pathway of dam methyltransferase. Cell 2005, 121 (3), 349–61. 10.1016/j.cell.2005.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton J. R.; Liebert K.; Bekes M.; Jeltsch A.; Cheng X. Structure and substrate recognition of the Escherichia coli DNA adenine methyltransferase. J. Mol. Biol. 2006, 358 (2), 559–70. 10.1016/j.jmb.2006.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton J. R.; Woodcock C. B.; Opot S. B.; Reich N. O.; Zhang X.; Cheng X. The cell cycle-regulated DNA adenine methyltransferase CcrM opens a bubble at its DNA recognition site. Nat. Commun. 2019, 10, 4600. 10.1038/s41467-019-12498-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock C. B.; Yu D.; Hajian T.; Li J.; Huang Y.; Dai N.; Correa I. R. Jr.; Wu T.; Vedadi M.; Zhang X.; Cheng X. Human MettL3–MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discov 2019, 5, 63. 10.1038/s41421-019-0136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.; Horton J. R.; Yang J.; Hajian T.; Vedadi M.; Sagum C. A.; Bedford M. T.; Blumenthal R. M.; Zhang X.; Cheng X. Human MettL3-MettL14 RNA adenine methyltransferase complex is active on double-stranded DNA containing lesions. Nucleic Acids Res. 2021, 49 (20), 11629–11642. 10.1093/nar/gkab460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.; Dai N.; Wolf E. J.; Correa I. R. Jr.; Zhou J.; Wu T.; Blumenthal R. M.; Zhang X.; Cheng X. Enzymatic characterization of mRNA cap adenosine-N6 methyltransferase PCIF1 activity on uncapped RNAs. J. Biol. Chem. 2022, 298 (4), 101751. 10.1016/j.jbc.2022.101751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.; Zhou J.; Chen Q.; Wu T.; Blumenthal R. M.; Zhang X.; Cheng X. Enzymatic Characterization of In Vitro Activity of RNA Methyltransferase PCIF1 on DNA. Biochemistry 2022, 61 (11), 1005–1013. 10.1021/acs.biochem.2c00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi R. K.; Huang D.; Eberle S. A.; Wiedmer L.; Sledz P.; Caflisch A. Small-Molecule Inhibitors of METTL3, the Major Human Epitranscriptomic Writer. ChemMedChem. 2020, 15 (9), 744–748. 10.1002/cmdc.202000011. [DOI] [PubMed] [Google Scholar]
- Coward J. K.Chemical mechanisms of methyl transfer reactions: comparison of methylases with nonenzymic ‘model reactions’. In The Biochemistry of Adenosylmethionine; Salvatore F., Ed.; Columbia University Press, 1977; pp 127–144. [Google Scholar]
- Huang R.; Martinez-Ferrando I.; Cole P. A. Enhanced interrogation: emerging strategies for cell signaling inhibition. Nat. Struct Mol. Biol. 2010, 17 (6), 646–9. 10.1038/nsmb0610-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q.; Jiang F.; Schultz P. G.; Gray N. S. Design of allele-specific protein methyltransferase inhibitors. J. Am. Chem. Soc. 2001, 123 (47), 11608–13. 10.1021/ja011423j. [DOI] [PubMed] [Google Scholar]
- Fedoriw A.; Rajapurkar S. R.; O’Brien S.; Gerhart S. V.; Mitchell L. H.; Adams N. D.; Rioux N.; Lingaraj T.; Ribich S. A.; Pappalardi M. B.; Shah N.; Laraio J.; Liu Y.; Butticello M.; Carpenter C. L.; Creasy C.; Korenchuk S.; McCabe M. T.; McHugh C. F.; Nagarajan R.; Wagner C.; Zappacosta F.; Annan R.; Concha N. O.; Thomas R. A.; Hart T. K.; Smith J. J.; Copeland R. A.; Moyer M. P.; Campbell J.; Stickland K.; Mills J.; Jacques-O’Hagan S.; Allain C.; Johnston D.; Raimondi A.; Porter Scott M.; Waters N.; Swinger K.; Boriack-Sjodin A.; Riera T.; Shapiro G.; Chesworth R.; Prinjha R. K.; Kruger R. G.; Barbash O.; Mohammad H. P. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019, 36 (1), 100–114. 10.1016/j.ccell.2019.05.014. [DOI] [PubMed] [Google Scholar]
- Mori S.; Iwase K.; Iwanami N.; Tanaka Y.; Kagechika H.; Hirano T. Development of novel bisubstrate-type inhibitors of histone methyltransferase SET7/9. Bioorg. Med. Chem. 2010, 18 (23), 8158–66. 10.1016/j.bmc.2010.10.022. [DOI] [PubMed] [Google Scholar]
- Woodcock C. B.; Yakubov A. B.; Reich N. O. Caulobacter crescentus Cell Cycle-Regulated DNA Methyltransferase Uses a Novel Mechanism for Substrate Recognition. Biochemistry 2017, 56 (30), 3913–3922. 10.1021/acs.biochem.7b00378. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Xie N.; Jin M.; Stahley M. R.; Stivers J. T.; Zheng Y. G. A transient kinetic analysis of PRMT1 catalysis. Biochemistry 2011, 50 (32), 7033–44. 10.1021/bi200456u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siarheyeva A.; Senisterra G.; Allali-Hassani A.; Dong A.; Dobrovetsky E.; Wasney G. A.; Chau I.; Marcellus R.; Hajian T.; Liu F.; Korboukh I.; Smil D.; Bolshan Y.; Min J.; Wu H.; Zeng H.; Loppnau P.; Poda G.; Griffin C.; Aman A.; Brown P. J.; Jin J.; Al-Awar R.; Arrowsmith C. H.; Schapira M.; Vedadi M. An allosteric inhibitor of protein arginine methyltransferase 3. Structure 2012, 20 (8), 1425–35. 10.1016/j.str.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Richardson S. L.; Mao Y.; Zhang G.; Hanjra P.; Peterson D. L.; Huang R. Kinetic mechanism of protein N-terminal methyltransferase 1. J. Biol. Chem. 2015, 290 (18), 11601–10. 10.1074/jbc.M114.626846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G.; Yasgar A.; Peterson D. L.; Zakharov A.; Talley D.; Cheng K. C.; Jadhav A.; Simeonov A.; Huang R. Optimization of High-Throughput Methyltransferase Assays for the Discovery of Small Molecule Inhibitors. ACS Comb. Sci. 2020, 22 (8), 422–432. 10.1021/acscombsci.0c00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K.; Zegzouti H.; Goueli S. A. Methyltransferase-Glo: a universal, bioluminescent and homogenous assay for monitoring all classes of methyltransferases. Epigenomics 2016, 8 (3), 321–39. 10.2217/epi.15.113. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Borek D.; Majewski W.; Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr. A 2003, 59, 228–34. 10.1107/S0108767303005488. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–32. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. Towards automated crystallographic structure refinement with phenix.. refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–67. 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read R. J.; Adams P. D.; Arendall W. B. 3rd; Brunger A. T.; Emsley P.; Joosten R. P.; Kleywegt G. J.; Krissinel E. B.; Lutteke T.; Otwinowski Z.; Perrakis A.; Richardson J. S.; Sheffler W. H.; Smith J. L.; Tickle I. J.; Vriend G.; Zwart P. H. A new generation of crystallographic validation tools for the protein data bank. Structure 2011, 19 (10), 1395–412. 10.1016/j.str.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tran N.; Ernst F. G. M.; Hawley B. R.; Zorbas C.; Ulryck N.; Hackert P.; Bohnsack K. E.; Bohnsack M. T.; Jaffrey S. R.; Graille M.; Lafontaine D. L. J. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019, 47 (15), 7719–7733. 10.1093/nar/gkz619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2256–68. 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Crooks G. E.; Hon G.; Chandonia J. M.; Brenner S. E. WebLogo: a sequence logo generator. Genome Res. 2004, 14 (6), 1188–90. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone T.; Blumenthal R. M.; Cheng X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 1995, 253 (4), 618–32. 10.1006/jmbi.1995.0577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.