

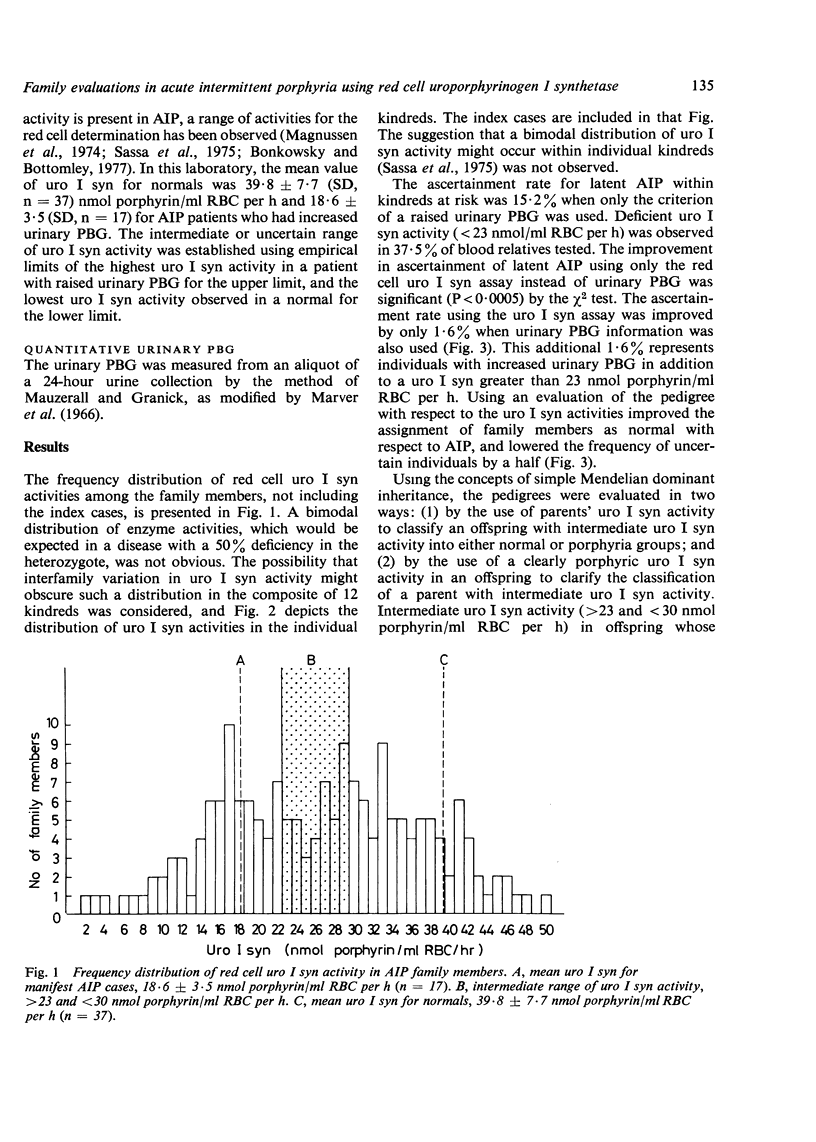
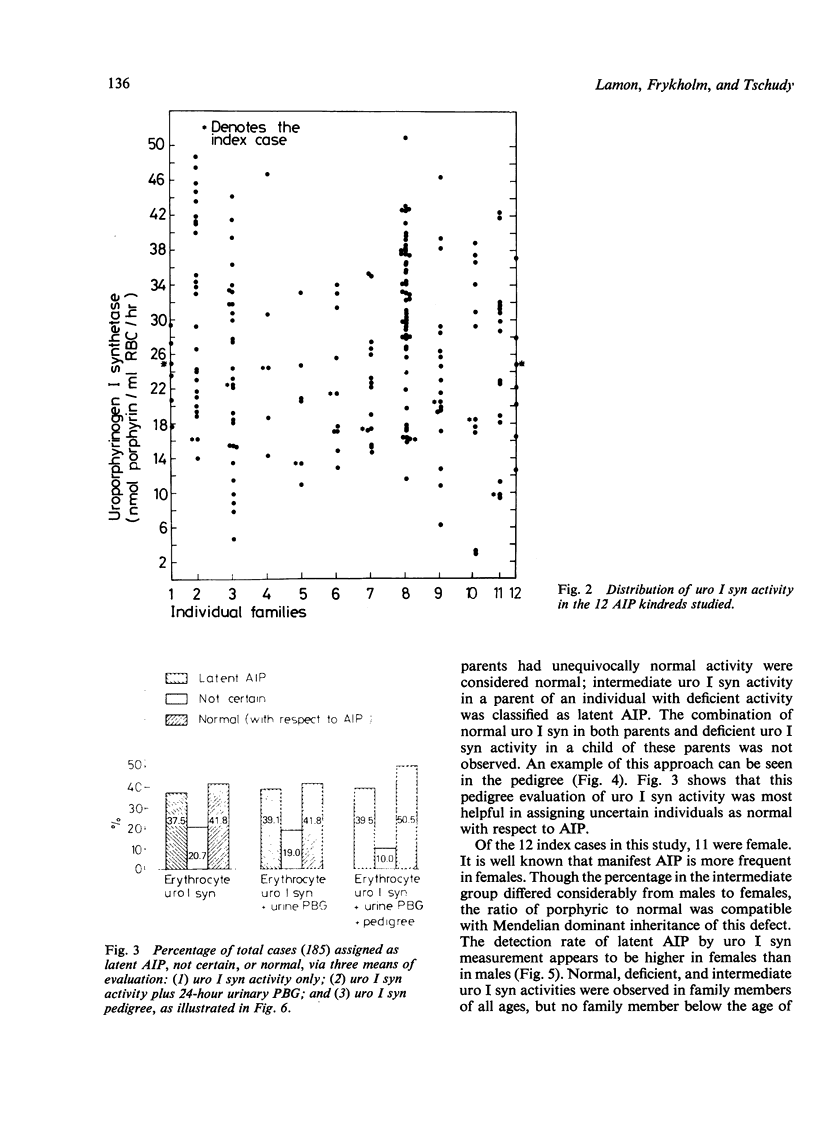
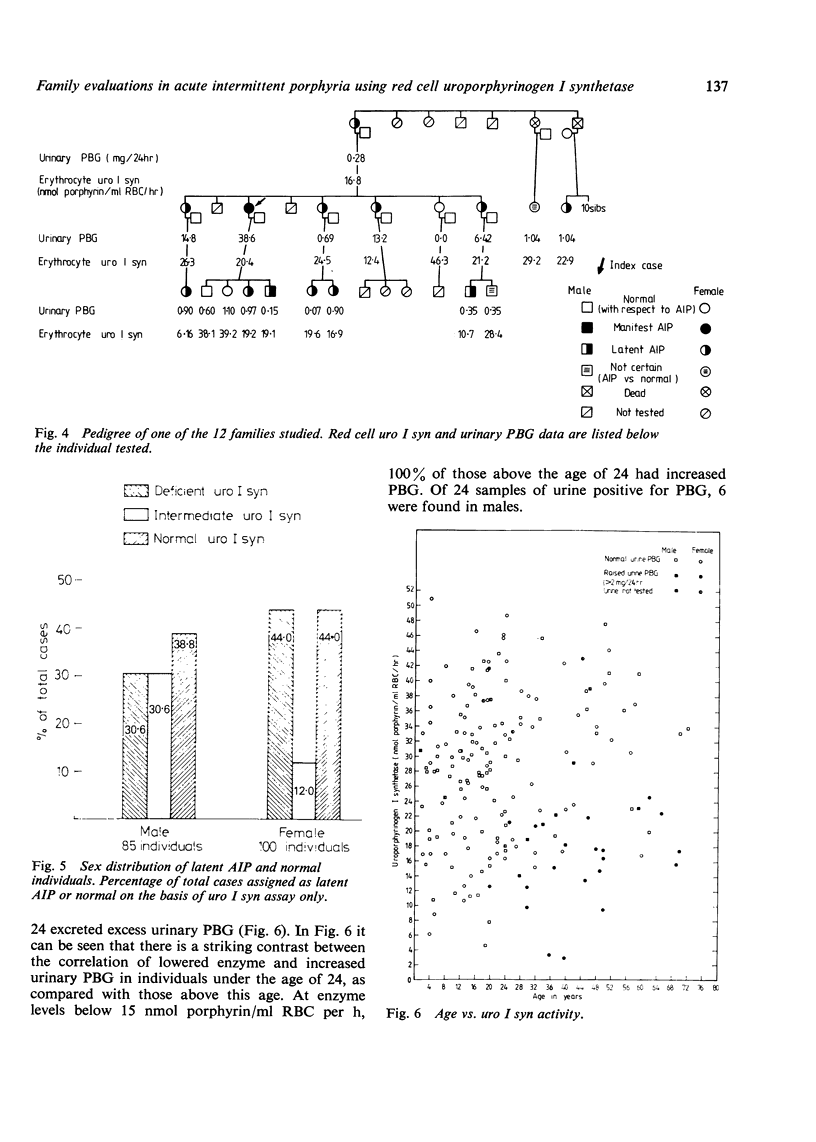
Abstract
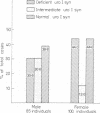
Acute intermittent porphyria (AIP) is a primary disorder of haem biosynthesis that is chemically characterised by raised urinary porphobilinogen (PBG). A defect in the biochemical pathway at the step of PBG conversion to uroporphyrinogen has been shown to be a result of a partial deficiency of the enzyme uroporphyrinogen I synthetase (uro I syn). The ascertainment rate of latent AIP (that is, chemically manifest but clinically asymptomatic) was examined in 185 individuals from 12 AIP kindreds using three parameters: red cell uro I syn, quantitative urinary PBG, and pedigree analysis with respect to uro I syn. Approximately 80% of individuals could be assigned as normal or latent AIP on the basis of the uro I syn assay alone. The remaining 20% could not be assigned because of an intermediate range of activity for the red cell assay in which the diagnosis cannot be certain. When the pedigree was used in the evaluation of the uro I syn data, the number of uncertain individuals, with respect to AIP, decreased to 10%. The enzyme method detected latent AIP in 37.5% of blood relatives, whereas quantitative urinary PBG alone detected only 15.2%. The pattern of inheritance for the uro I syn deficiency is consistent with Mendelian dominant inheritance, and it is likely that it is the basic inherited defect in AIP.
Full text
PDF


Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bonkowsky H. L., Tschudy D. P., Weinbach E. C., Ebert P. S., Doherty J. M. Porphyrin synthesis and mitochondrial respiration in acute intermittent porphyria: studies using cultured human fibroblasts. J Lab Clin Med. 1975 Jan;85(1):93–102. [PubMed] [Google Scholar]
- HAEGER B. Urinary delta-aminolaevulic acid and porphobilinogen in different types of porphyria. Lancet. 1958 Sep 20;2(7047):606–608. doi: 10.1016/s0140-6736(58)90334-9. [DOI] [PubMed] [Google Scholar]
- Magnussen C. R., Levine J. B., Doherty J. M., Cheesman J. O., Tschudy D. P. A red cell enzyme method for the diagnosis of acute intermittent porphyria. Blood. 1974 Dec;44(6):857–868. [PubMed] [Google Scholar]
- Sassa S., Granick S., Kappas A. Effect of lead and genetic factors on heme biosynthesis in the human red cell. Ann N Y Acad Sci. 1975 Apr 15;244:419–440. doi: 10.1111/j.1749-6632.1975.tb41546.x. [DOI] [PubMed] [Google Scholar]
- Strand L. J., Felsher B. F., Redeker A. G., Marver H. S. Heme biosynthesis in intermittent acute prophyria: decreased hepatic conversion of porphobilinogen to porphyrins and increased delta aminolevulinic acid synthetase activity. Proc Natl Acad Sci U S A. 1970 Nov;67(3):1315–1320. doi: 10.1073/pnas.67.3.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschudy D. P., Valsamis M., Magnussen C. R. Acute intermittent porphyria: clinical and selected research aspects. Ann Intern Med. 1975 Dec;83(6):851–864. doi: 10.7326/0003-4819-83-6-851. [DOI] [PubMed] [Google Scholar]