

Abstract

Poly(ethylene terephthalate) polyester represents the most common class of thermoplastic polymers widely used in the textile, bottling, and packaging industries. Terephthalic acid and ethylene glycol, both of petrochemical origin, are polymerized to yield the polyester. However, an earlier report suggests that polymerization of methoxyterephthalic acid with ethylene glycol provides a methoxy-polyester with similar properties. Currently, there are no established biobased synthetic routes toward the methoxyterephthalic acid monomer. Here, we show a viable route to the dicarboxylic acid from various tree species involving three catalytic steps. We demonstrate that sawdust can be converted to valuable aryl nitrile intermediates through hydrogenolysis, followed by an efficient fluorosulfation–catalytic cyanation sequence (>90%) and then converted to methoxyterephthalic acid by hydrolysis and oxidation. A preliminary polymerization result indicates a methoxy-polyester with acceptable thermal properties.
Keywords: lignocellulose, lignin valorization, reductive catalytic fractionation, fluorosulfation, palladium-catalyzed cyanation, terephthalic acid, methoxyterephthalic acid
Introduction
The global dependency on crude oil for the production of poly(ethylene terephthalate) (PET) plastics and fibers was estimated to reach 87.2 million metric tons of PET in 2022.1 PET plastics play a major role in the production of beverage bottles, food packaging, and textiles2,3 because of their highly desirable properties.4,5 As crude oil is a finite and nonrenewable resource, the continued manufacturing of PET products will not be compatible with the needs of a rising global population.6 Furthermore, the end-of-life PET plastic waste represents a global problem as recycling of this polymer only occurs to a low degree,7 with the majority ending up in landfills or incinerated. For example, in the U.S. alone, 57% of the PET plastic bottles and containers produced in 2018 were landfilled, while 14% were incinerated, and only 29% were recycled (see Scheme 1a).8 Gasification and pyrolysis represent alternative solutions, transforming the waste into smaller hydrocarbon compounds suitable for heating or as fuels.9 However, common for these solutions and the direct incineration of plastics, are their ultimate generation of CO2 from crude oil, thus exacerbating global warming. A renewable and carbon-neutral alternative is to replace crude oil with biomass as the starting material in PET production.10 Some solutions already exist as ethylene glycol can be produced from bio-ethylene via an epoxidation-hydrolysis approach or from sugars by a two-step pyrolysis-hydrogenation.11 For the other monomer, terephthalic acid (TA), only less elaborated biobased routes exist. These are multistep procedures that commonly rely on sugar fermentation, leading to a range of small-molecule compounds such as ethylene,12 isoprene,13iso-butanol,14 and 2,5-dimethylfurfural.15 Subsequently, these compounds can be transformed to p-xylene,16 representing the industrial precursor for TA in the AMOCO process.17 Another fermentation product, trans,trans-muconic acid, is also a substrate for TA synthesis, namely, via a Diels–Alder reaction followed by dehydrogenation.18
Scheme 1. Synthesis and Numbers on PET and a Lignin-Based Route to MTA.
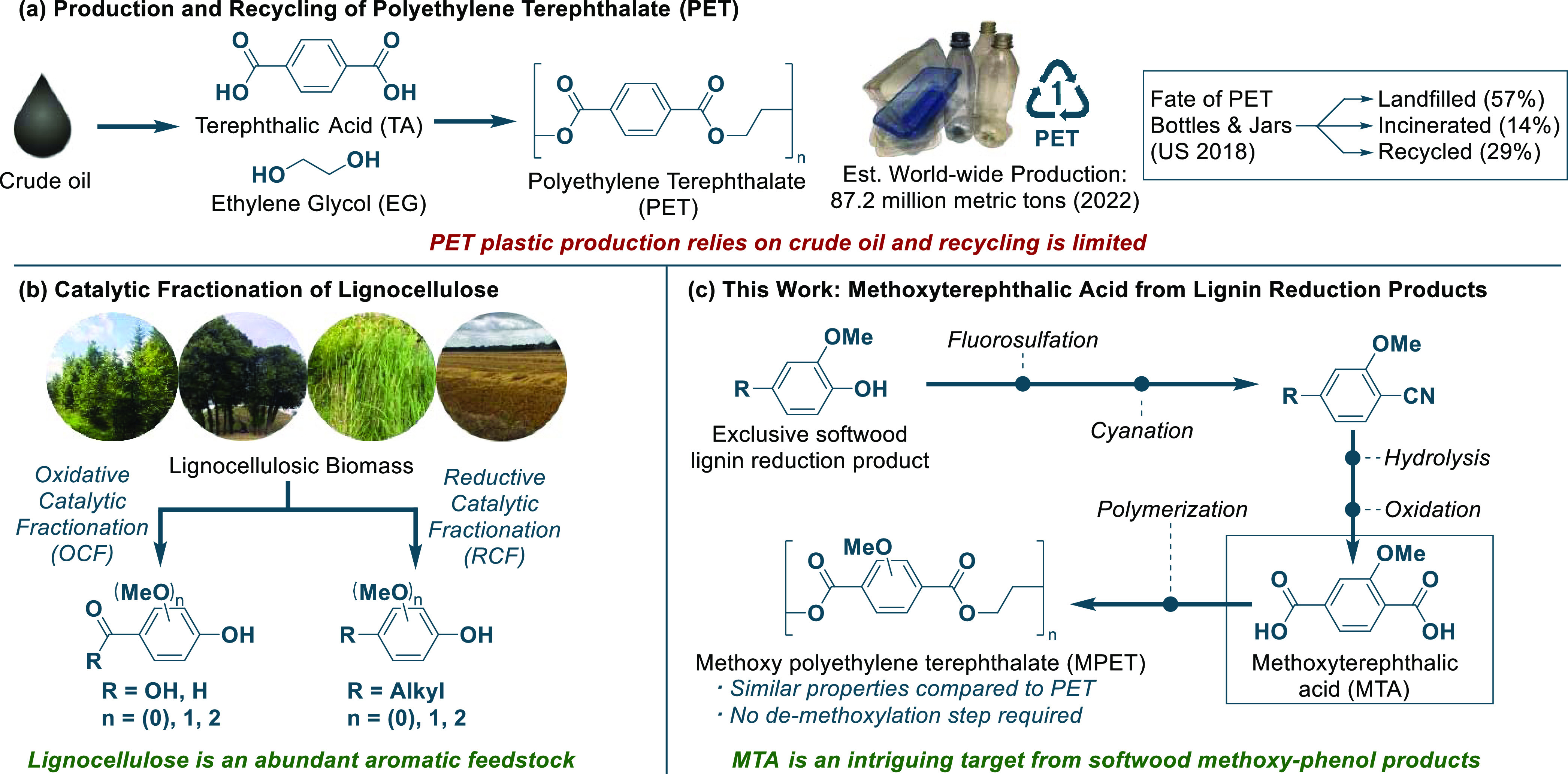
There is currently a high demand for biobased PET production, exemplified by the company goals of Coca-Cola and Virent, diverting toward biobased or recycled PET in the future.19 Synthetic procedures toward TA also exist, not relying on sugars as the main feedstock, including via limonene, which can be extracted from orange peels.20,21 However, a more desirable resource for TA synthesis is to rely on the omnipresent lignocellulose. This natural polymer is the most abundant renewable carbon resource, being composed of cellulose, hemicellulose, and lignin.22 While many applications exist for cellulose, there are only few uses of lignin, even though it has the potential to be a feedstock of the future for aromatic compounds.22 The main problem stems from the traditional pulping techniques that effectively separate the cellulose from lignocellulose, but in the process delivers a less reactive lignin polymer byproduct.23 More modern techniques such as oxidative and reductive catalytic fractionation (OCF and RCF) are on the contrary able to generate a useful lignin product stream, which is composed of methoxy-substituted phenols (see Scheme 1b).23−26 The highest yields are achieved with RCF, which depolymerizes lignocellulose under catalytic hydrogenative conditions applying a heterogeneous catalyst and a polar protic solvent, such as MeOH or H2O.23,25,26 Furthermore, applying hydrogen as a reagent aligns amiably with modern power-to-hydrogen strategies.27 The RCF method is believed to become a major industrial process in the future;25 thus, a synthetic route toward TA from lignin RCF products could be highly desirable.
Previously, two multistep routes from lignin to TA have been disclosed by the groups of Zhu and Yan.28,29 In the first, TA was prepared from vanillic and syringic acid from the oxidative degradation of lignin. However, these two products only account for 5 wt % of all of the oxidation products.16 Furthermore, harsh reaction conditions were necessary to overcome the challenging demethoxylation and carboxylation steps, which are both conducted at a minimum of 400 °C and a pressure of 40 bar CO2/H2 using noncommercial catalysts.28 In the subsequent work by Yan et al., a reductive catalytic fractionation was developed, providing significantly higher yields of lignin monomer products.29 The challenges of this multistep protocol also lie in the harsh conditions necessary for a productive demethoxylation step, operating at 320 °C with a pressure of 30 bar H2 that results in a 71% yield, while the remainder consists of undesired side-products.29 Furthermore, the subsequent triflation of the RCF phenol products suffers from the use of reactive and expensive triflic anhydride, which is not compatible with a crude lignin product mixture that may hold aliphatic alcohol functionalities and water.30
In a patent from 1959, the General Electric Company (GE) reported the synthesis of polyesters including methoxy poly(ethylene terephthalate) (MPET) from methoxyterephthalic acid (MTA) and ethylene glycol.31 However, these materials have been largely overshadowed by conventional PET. This is explained by the conventional and direct production of TA from crude-oil-derived p-xylene.4,17 With a future potentially relying on renewable lignocellulose as one resource for creating polyester materials, the methoxylated feedstock could represent an advantageous starting point for preparing a biobased MTA, which could be useful for an eventual MPET production. Here, we envisage a route from the methoxy-phenol products of RCF to MTA applying a two-step fluorosulfation, catalytic cyanation strategy with industrially viable reagents. MTA is then reached from the aryl nitrile cyanation products by two well-described industrial processes, after which a polymerization with ethylene glycol is able to yield MPET (see Scheme 1c).
Results and Discussion
To explore the efficacy of the proposed route for synthesizing MTA from lignin, we collected lignocellulose samples (sawdust) from eight different tree species and subjected them to the reported RCF conditions by Sels and co-workers,32 which was previously shown to effectively depolymerize various lignocelluloses via the Ru/C-catalyzed hydrogenolysis reaction under a hydrogen atmosphere (30 bar) in MeOH at a temperature of 250 °C. The resulting methoxy-phenol products were obtained in up to 75 mg/g of sawdust (for maple sawdust, see p. S14 of the SI), which corresponds to 27 wt % with respect to the lignin weight. This is acceptable with respect to the results obtained by Sels and co-workers32 and other main contributors in the RCF field, such as Beckham, Román-Leshkov, and co-workers (regarding results with Ru/C and various conditions and tree species, 21–32 wt % monomer yields).33−35 However, it should also be noted that the monomer yields depend to a great extent on the specific lignocellulose composition of the wood sample, which can vary according to the age, growth period, and home region.36,37 The obtained phenol products were subsequently converted to aryl nitriles via sequential fluorosulfation and catalytic cyanation (see Scheme 2a). Sulfuryl fluoride is the ideal reagent for the activation of the RCF phenol fraction because of its industrial-scale production of 3000 tons/year and its use for phenol activation while stable toward hydrolysis (see Scheme 2b).38,39 Recently, related C–O activation strategies have been reported for lignin phenol monomers in the context of electrochemical reductive coupling and phenol deoxygenation.40,41 The specific RCF phenol products, 1 and 2, have not before been subjected to fluorosulfation; however, gratifyingly, we observed quantitative yields of the corresponding aryl fluorosulfates when the reaction was conducted with ex situ generated sulfuryl fluoride in a two-chamber reactor system (see Scheme 2b).42 The selection of K2CO3 instead of the widely used NEt3 as base for fluorosulfation39,42 does not only give quantitative yields of the reaction but is also more viable from an industrial viewpoint, representing a less expensive and greener option.43−45 Furthermore, the use of a heterogeneous base facilitates purification through simple filtration.
Scheme 2. Three-Step Route to Aryl Nitriles via Reductive Catalytic Fractionation, Fluorosulfation, and Cyanation.
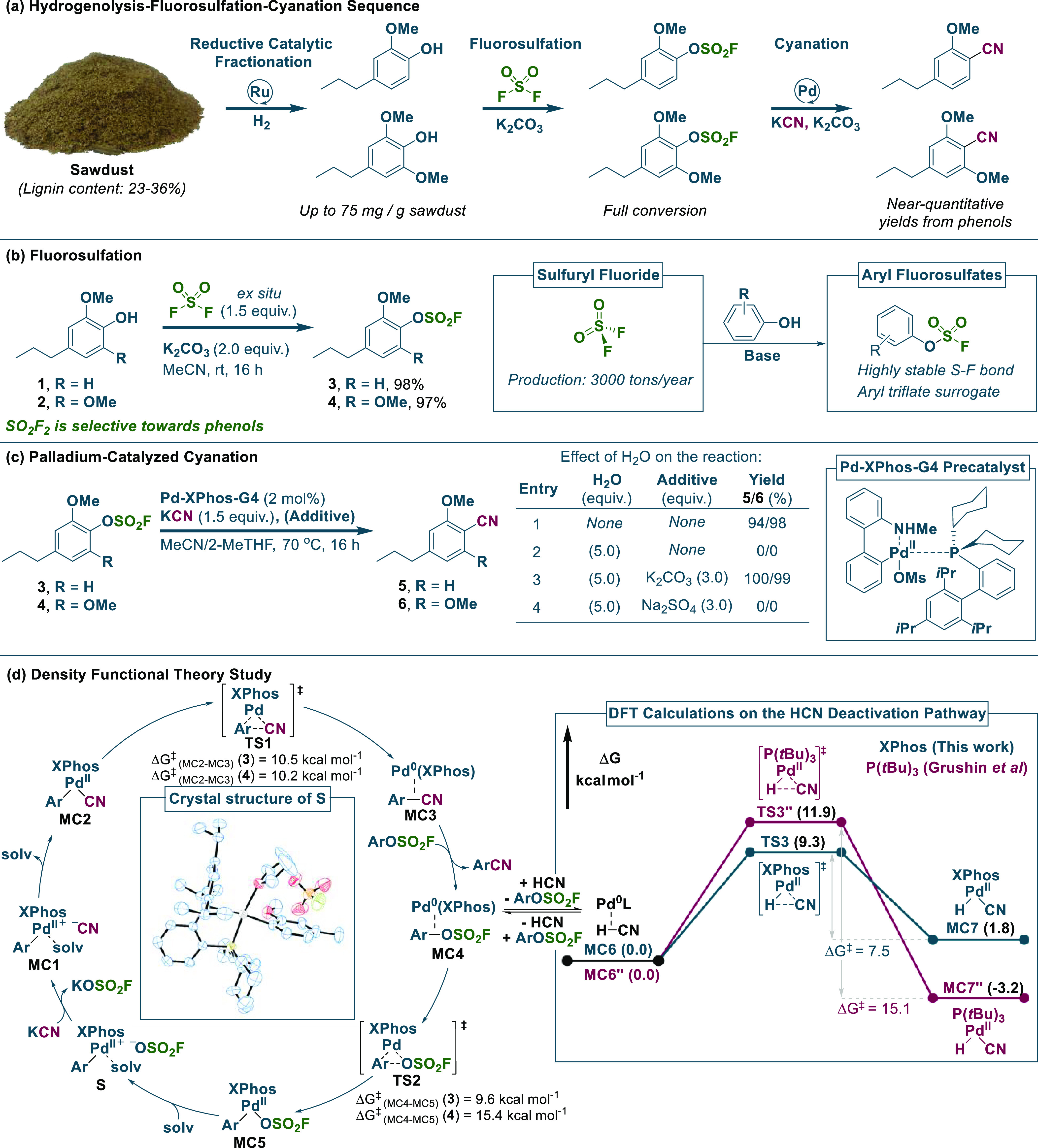
Fluorosulfation reactions were conducted on a scale of 5–10 mmol in a two-chamber reactor system using ex situ generated sulfuryl fluoride (see pp. S4–S5 of the SI). The palladium-catalyzed cyanation of aryl fluorosulfates was optimized on a 0.2–0.4 mmol scale (see the full optimization in Tables S1–S3 in the SI). All of the presented DFT results were calculated at the PBE-D3/Def2-TZVPP//PBE-D3/Def2-SVP level of theory with SMD (acetonitrile).
The ensuing catalytic cyanation step takes advantage of the excellent reactivity of aryl fluorosulfates in transition-metal-catalyzed cross-coupling reactions, being generally viewed as aryl triflate surrogates.39,42 Initially, we explored the industrial source of cyanide, HCN, with a two-chamber setup,46 in a palladium-catalyzed cyanation of aryl fluorosulfates 3 and 4 relying on XPhos as the ligand. Quantitative yields of both aryl nitriles 5 and 6 were achieved (see Supporting Tables S1 and S2). As deactivation of palladium catalysts has been reported from the presence of minute quantities of HCN for the cyanation of aryl electrophiles,47,48 we were surprised by the efficiency of this protocol, which suggests an unusual stability for the Pd–XPhos complexes. Subsequently, it was discovered that both KCN and NaCN in lieu of HCN rendered equally quantitative transformations (see Scheme 2c and Supporting Table S3), being more desirable from a safety perspective. Through optimization, we found that the successful transformation of aryl fluorosulfates 3 and 4 necessitates 0.5–2 mol % of a Pd–XPhos catalyst generated from the Buchwald G4-precatalyst49 using industrially viable solvents such as MeCN and biomass-derived 2-MeTHF.50 Furthermore, for the previously reported cyanation reactions with aryl fluorosulfates, lower yields are observed with sterically hindered ortho-methoxy-substituted substrates.51,52 However, the Pd–XPhos catalyst employed in this transformation reacts amiably with even the dimethoxy-substituted substrate, 4 (see Scheme 2c). Another strong point of this reaction is clearly illustrated by its ability to tolerate small amounts of water (see Scheme 2c), which can be detrimental to palladium-catalyzed cyanation reactions as illustrated by the seminal work from the groups of Beller, Grushin, and Macgregor.47,48
We postulate that the addition of K2CO3 is crucial to the reaction, as it serves to deprotonate and remove HCN formed in situ from the reaction of KCN and water. Trace HCN has been reported to deactivate palladium catalysts by oxidative addition, yielding off-cycle H–PdII–CN and eventually inactive palladium(II) cyanide complexes.47,48 In the presence of K2CO3, reductive elimination of HCN from the H–PdII–CN complex becomes favored by an ensuing deprotonation of the HCN. Furthermore, we were able to support our claim on the effect of K2CO3 being more than just a dehydration agent of the solvent mixture, as the same reaction with an equal amount of the drying agent, Na2SO4 instead of carbonate resulted in no conversion when conducted in the presence of 5 equiv of H2O (see Scheme 2c). Furthermore, we hypothesize that the success of 2-MeTHF and MeCN as a solvent mixture could be due to an ideal solubility of cyanide, which has been suggested before for the Pd-catalyzed cyanation of aryl bromides using a similar THF/MeCN solvent mixture.53
A mechanism for the catalytic cyanation reaction is depicted in Scheme 2d, which follows the generally accepted catalytic cycle for oxidative addition (MC4–MC5), cyanation (MC5–MC2), and reductive elimination (MC2–MC3).47,48 Additionally, it is likely that a solvent-coordinated complex S is formed after the dissociation of fluorosulfate counterion from MC5, which is supported by the obtained crystal structure for S (see Supporting Figure S21). Energies of intermediates and barriers in the catalytic cycle were determined with DFT calculations (PBE-D3,SMD, see Supporting Figures S11 and S13). The calculations identify MC2–MC3 (reductive elimination) as the rate-limiting step for the reaction with 3 (ΔG‡ = 10.5 kcal mol–1), and MC4–MC5 (oxidative addition) for 4 (ΔG‡ = 15.4 kcal mol–1). The overall reaction free energy for the catalytic cyanation with 3 was found to be −58.7 kcal mol–1 (343.15 K), while for 4 it was −60.1 kcal mol–1 (343.15 K). To better elucidate the underlining reasons for the stability of the catalytic system against HCN deactivation, further DFT studies were undertaken. From the Pd0-intermediate MC4, oxidative addition of HCN leads to MC6 of the deactivation pathway. From the calculations, it is evident that the conversion of MC6–MC7 with HCN is plausible due to an energy barrier of ΔG‡ = 9.3 kcal mol–1. However, the reverse reaction (MC7–MC6) has a lower barrier (ΔG‡ = 7.5 kcal mol–1) and is thus likely to occur as well. For comparison, we calculated the energies for the same deactivation mechanism with the Pd–P(tBu)3 system reported by Grushin and co-workers (see Scheme 2d).53 This system is known to be effective for the cyanation of aryl bromides with KCN but requires dry conditions to circumvent HCN formation from trace water. Our calculations for the Pd–P(tBu)3 system revealed a higher vulnerability toward the HCN deactivation pathway as the back reaction from MC7″–MC6″ has an energy barrier, being twice the height of our system (ΔG‡ = 15.1 vs ΔG‡ = 7.5 kcal mol–1). Furthermore, MC7″ is −3.2 kcal mol–1 lower in energy compared to MC6″, thereby providing a thermodynamic driving force for the formation of MC7″. Another postulation as to why our reported system is less susceptible to HCN deactivation is that the catalyst resting state is likely to resemble the PdII–complex S or MC5, as deduced by monitoring the progression of the reaction with time-resolved 31P–NMR spectroscopy (see Supporting Figure S8).
With the optimized fluorosulfation and cyanation conditions at hand, we explored the sequential transformation of sawdust originating from different tree species to the corresponding aryl nitriles. By qualitative observations made with GCMS, we assessed that the fluorosulfation step required 3 equiv of sulfuryl fluoride and K2CO3 each for a full conversion in all cases. In the case of the cyanation reaction, a slightly higher catalyst loading of 5 mol % and 3 equiv of K2CO3 were required for high conversion with all tree species (see Scheme 3), although eventual industrialization of the process would require a significantly lower catalyst loading. From exploring the scope of sawdust samples, best yields were obtained with hardwoods, such as birch, willow, oak, hazel, and maple, compared to softwood species including spruce, grandis, and noble fir, which is in line with the general observations made in earlier studies on RCF.25,32 However, interestingly, with softwood in contrast to the hardwood samples, the 2-methoxy-substituted phenol product was exclusively isolated, being precisely the precursor required for MTA synthesis.31 This product distribution is accounted for by the different composition of H-, G-, and S-lignin subunits present in softwoods versus hardwoods.22,23 As such, these observations obviate eventual separation of nitriles 5 and 6 when relying on softwoods, and as this tree sort is generally faster growing than the corresponding hardwoods,54 this could be advantageous from an industrial viewpoint. A scale-up experiment (10 g) was also conducted with noble fir sawdust, and we observed an acceptable 6.1 wt % yield of the aryl nitrile 5, which is, however, a lower yield in comparison with the small-scale yield (9.1 wt %) due to not achieving full conversion in the cyanation step. Instead, if 5 was purified before the cyanation reaction, we could observe quantitative isolated yields on 2–10 mmol scales with a low catalyst loading of 0.5–2 mol % (see Table S3 in the SI). With the isolated 2-methoxy-substituted aryl nitrile from the various sawdust samples studied, the corresponding carboxylic acid product was finally produced by an initial hydrolysis with NaOH (Scheme 4a). Subsequently, the conditions of the AMOCO process17,29 promoted the synthesis of MTA by oxidation, and the product as a colorless solid was isolated in high purity as deduced from NMR spectroscopic analysis. Besides the AMOCO process, there are many other excellent strategies toward C(sp3)–H oxidation that have been recently reviewed.55−57 Furthermore, the research group of Maes also studied the benzylic oxidation of lignin-derived propylguaiacol and syringol by using Na2S2O8 in order to obtain aryl ketones.58 An alternatively interesting pathway for reaching MTA involves a carboxylation strategy, which we showcased from spruce sawdust using a nickel-catalyzed reductive carboxylation system inspired by the work of Mei and co-workers.59 Nonetheless, a major limitation to this strategy is the necessity of a stoichiometric reductant as exemplified by the need for manganese metal.
Scheme 3. Scope of the Three-Step Synthesis of Aryl Nitriles Directly from Sawdusts.
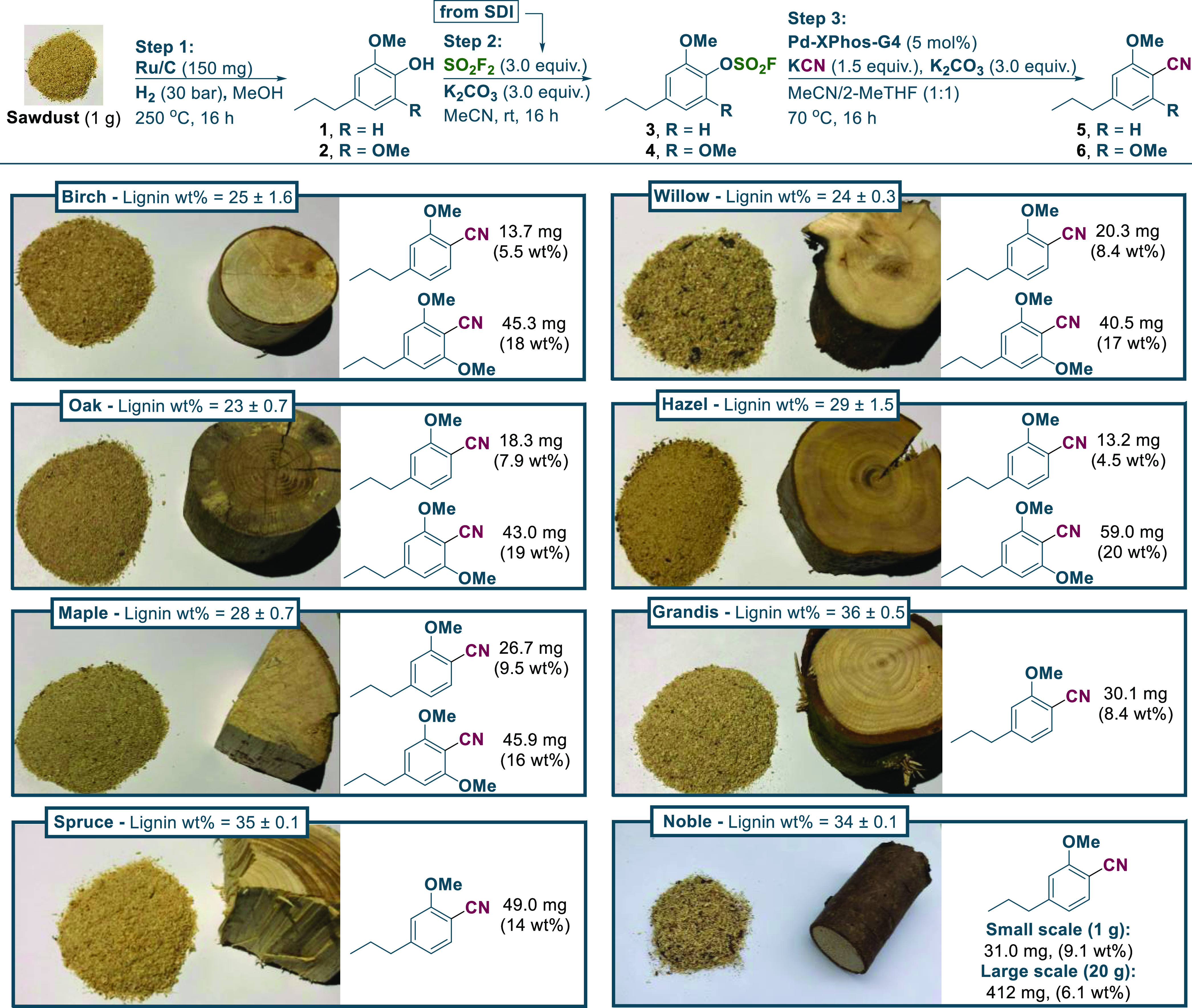
Lignin weight percentages were determined by the Klason lignin method and are based on triplicate results (see Supporting Figure S3 and Table S4). Mass and wt % yields for aryl nitrile products 5 and 6 are given as averages of duplicate reactions and are reported relative to the mass of lignin (see the Supporting Information pp. S12–S15). SO2F2 was generated ex situ from SDI (1,1′-sulfonylbis(1H-imidazole)) and potassium fluoride in TFA. The large-scale reaction with noble fir required Pd–XPhos–G4 (6.5 mol %), K2CO3 (3.9 equiv), and KCN (2.0 equiv). See the General Procedures section pp. S11–S12 of the SI. Qualitative assessment by 1H- and 13C-NMR indicates a high purity of the respective compounds 5 and 6 after purification by flash column chromatography, however with the co-elution of minute amounts of ethyl-substituted aryl nitriles (see the SI page S12). Purification methods used: Step 1 (filtration through celite, then filtration through silica), Step 2 (filtration through celite), Step 3 (flash column chromatography); see the General Procedures A and B, pp. S11–S12.
Scheme 4. Synthesis and Polymerization of MTA.
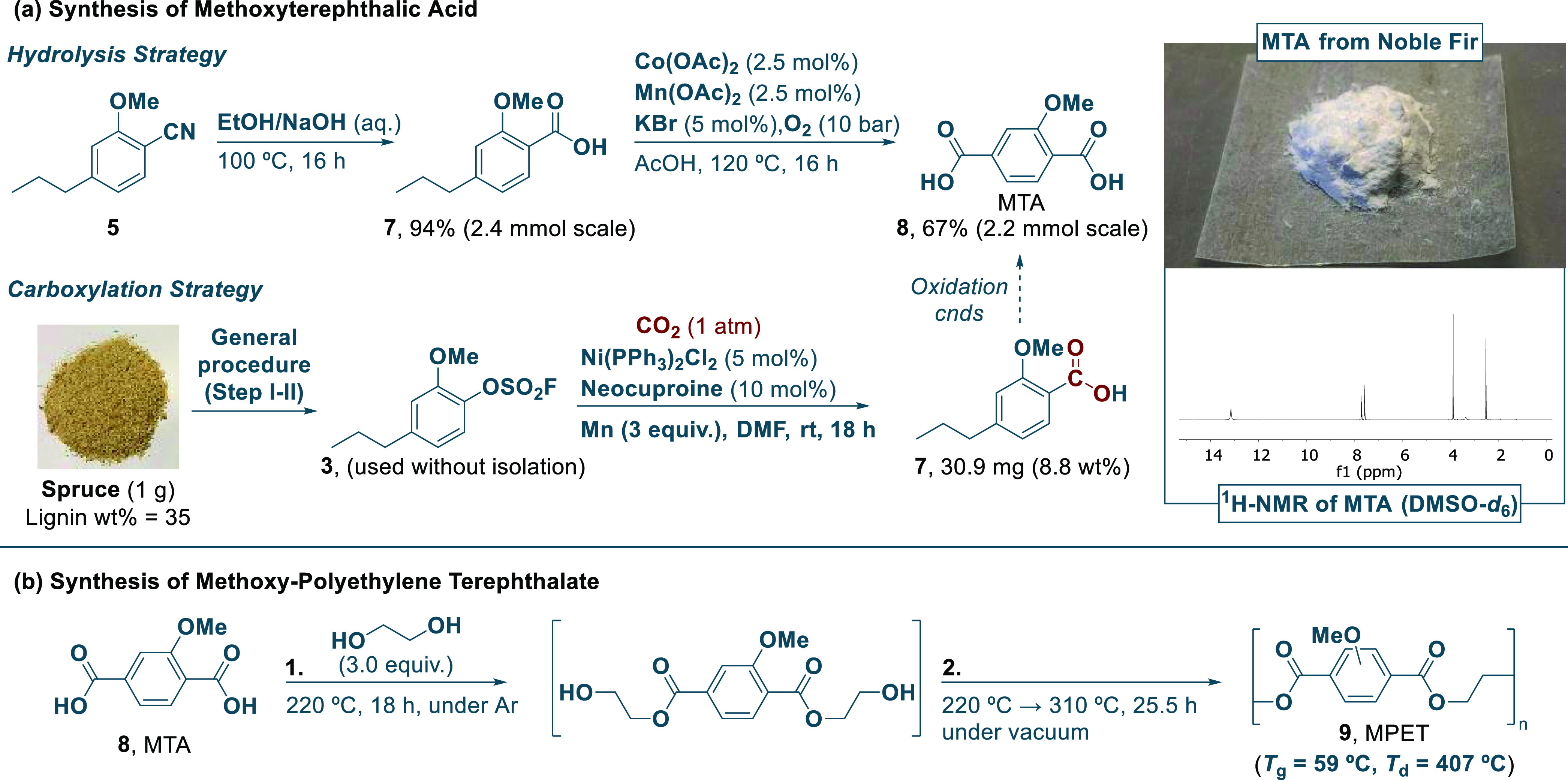
Experimental procedure for nitrile hydrolysis (see the SI, pp. S16–S17), oxidation (see the SI, pp. S17–S18), carboxylation (see the SI, p. S17), and polymerization (see the SI, p. S18). The 2.4 mmol scale hydrolysis of 5 to 7 was conducted with 5 obtained from noble fir, and the following oxidation step was conducted with the noble fir-derived 7. The MTA used for the polymerization was obtained from a reported procedure.61 Qualitative assessment by 1H- and 13C-NMR analysis of the hydrolysis, oxidation, and carboxylation steps indicate a high purity of the respective compounds 7 and 8. Purification methods used: hydrolysis step (acid–base wash), oxidation step (decantation with AcOH, H2O, and pentane), carboxylation step (acid–base wash), polymerization (no purification). The glass-transition temperature was evaluated with differential scanning calorimetry (DSC), while the decomposition temperature was analyzed with thermogravimetric analysis (TGA) (see the SI, Figures S6 and S7). The error was measured to be ±1.0 °C for DSC analysis and ±1.7 °C for TGA.
Lastly, we attempted a preliminary un-optimized synthesis of MPET via the depolymerization of MTA with ethylene glycol. The reaction conditions were inspired from that described in the patent,31 and they deviate slightly from standard PET synthesis, in which catalysts such as antimony oxides or titanium alkoxides are frequently applied.60 MTA was first reacted with 3 equiv of ethylene glycol at 220 °C for 18 h in a closed system under an argon atmosphere. The subsequent polymerization step was conducted under vacuum, heating at 220 °C for 4 h, then at 270 °C for 3.5 h, and finally at 310 °C for 18 h, to yield MPET as a dark polymer, which was characterized by infrared spectroscopy (IR), thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC) (see Supporting Figures S5–S7). The polymer product displayed a glass-transition temperature (Tg) of 59 °C and a decomposition temperature (Td) of 407 °C, indicating acceptable thermal properties. Future work should include further optimization of MPET polymerization, while also expanding the characterization to include mechanical properties, crystallinity, gas permeability, chemical resistance, and rheological parameters.
Conclusions
In summary, we have shown the first biobased synthesis of methoxyterephthalic acid directly from lignocellulose. Our developed multistep procedure displays high-yielding reaction steps, such as for fluorosulfation, cyanation, and hydrolysis, while also a good yield for the final oxidation. The developed conditions for the palladium-catalyzed cyanation, consisting of a Pd–XPhos system, display an unusual stability toward cyanide-mediated deactivation, and investigations on the mechanism indicate a favorable PdII catalyst resting state and a thermodynamically unfavored oxidative addition of HCN to Pd0–XPhos, which are potential reasons for the observed stability. A preliminary polymerization reaction showed the possible synthesis of MPET from MTA and ethylene glycol, and future work should further optimize the polymerization and also focus on a more in-depth characterization of the polymer, to evaluate whether it has industrial potential as a new polyester material.
Methods
General Procedure for Aryl Nitrile Synthesis from Lignocellulose Samples
Representative Example with Birch Sawdust
To each of the two duplicate Parr autoclave reactors fitted with Teflon inlays (30 mL total volume) were added Ru/C (5 wt % Ru, 75 mg), Birch sawdust (500 mg), and MeOH (10 mL). The reactors were closed, evacuated five times with H2, and subjected to a H2 pressure of 30 bar at room temperature. The autoclaves were then stirred at 250 °C for 16 h and thereafter filtered through celite into a round-bottom flask, rinsed with EtOAc, and concentrated in vacuo. EtOAc (25 mL) was added to the concentrate followed by sonification for 5 min. Pentane (25 mL) was then introduced, whereafter the mixture was passed through a short silica dry column (5 cm silica), rinsing with EtOAc/pentane (1:1, 50 mL) into a cone-shaped flask (100 mL). The concentrate was used in the subsequent step without further purification. Quantification via GC-MS using dodecane as an internal standard was carried out for the monomer products, 2-methoxy-4-propylphenol 1 (13.9 mg) and 2,6-dimethoxy-4-propylphenol 2 (51.3 mg). This provides a reaction scale of 0.345 mmol for the subsequent fluorosulfation and cyanation reaction steps.
To chamber A of a two-chamber reactor (20 mL total volume) were added K2CO3 (143 mg, 1.03 mmol, 3.0 equiv) and the concentrate from the prior hydrogenolysis reaction (Step I), which was transferred with MeCN (4 mL × 1 mL). The chamber was sealed with a screwcap fitted with a Teflon-coated silicone seal. 1,1′-Sulfonylbis(1H-imidazole) (205 mg, 1.03 mmol, 3.0 equiv) and KF (160 mg, 2.76 mmol, 8.0 equiv) were added to chamber B of the two-chamber reactor. The chamber was sealed with a screwcap fitted with a pierceable Teflon-coated silicone seal. Trifluoroacetic acid (0.5 mL) was then added to the closed chamber B via the Teflon-coated silicone seal and sulfuryl fluoride release was observed within a minute. The reactor was stirred at room temperature for 16 h, after which the reaction mixture was passed through celite into a cone-shaped flask and concentrated in vacuo. Qualitative GC-MS analysis showed a full conversion of 1 and 2 to aryl fluorosulfates 3 and 4 (see the SI, Figure S4). The concentrate was used in the subsequent step without further purification.
In an argon-filled glovebox, Pd–XPhos–G4 (14.8 mg, 0.017 mmol, 5 mol %), K2CO3 (143 mg, 1.03 mmol, 3.0 equiv.), and KCN (33.7 mg, 0.517 mmol, 1.5 equiv) were added to a pressure tube (9 mL total volume). The concentrate from the prior fluorosulfation reaction (Step II) was also transferred to the tube with MeCN (2 mL × 1 mL) and 2-MeTHF (2 mL × 1 mL). The chamber was sealed with a screwcap fitted with a Teflon-coated silicone seal and stirred at 70 °C for 16 h. The reaction mixture was then evaporated onto celite and purified by flash column chromatography to yield aryl nitrile products 5 (13.6 mg, 5.4 wt %) as a colorless liquid (eluted with pentane to pentane/EtOAc 95:5) and 6 (44.4 mg, 18 wt %) as a colorless crystalline solid (eluted with Pentane/EtOAc 80:20).
General Procedure for Methoxyterephthalic Acid Synthesis
2-Methoxy-4-propylbenzonitrile 5 (412 mg, 2.35 mmol (from noble fir)), EtOH (20 mL), and aq. NaOH (34%, 20 mL) were added to a pressure tube (120 mL total volume). The tube was sealed with a screwcap fitted with a Teflon-coated silicone seal and then heated to 100 °C for 16 h. After the reaction, the EtOH was removed in vacuo, after which EtOAc (100 mL) was added to the concentrate and then extracted with aq. NaOH (1 M, 2 mL × 50 mL). The combined aqueous phases were acidified carefully with conc. aq. HCl until pH < 3 and then extracted with EtOAc (3 mL × 100 mL). The organic phases were combined, dried over Na2SO4, filtered, and concentrated in vacuo to yield 2-methoxy-4-propylbenzoic acid 7 as an off-white solid (430 mg, 94%).
A suspension of the obtained 2-methoxy-4-propylbenzoic acid 7 from noble fir (430 mg, 2.21 mmol), Co(OAc)2 (9.8 mg, 0.055 mmol, 2.5 mol %), Mn(OAc)2 (9.6 mg, 0.055 mmol, 2.5 mol %), and KBr (13 mg, 0.11 mmol, 5.0 mol %) in AcOH (2 mL) was stirred in a vial and heated gently with a heat gun until homogeneous. The solution was transferred to a Parr autoclave reactor fitted with a Teflon inlay (30 mL total volume), rinsed with AcOH (1 mL), which was then closed, evacuated 10 times with O2 at a pressure of 5 bar, and then pressurized to an O2 pressure of 10 bar at room temperature. The reaction was heated to 120 °C for 16 h under slow stirring (100 rpm), whereafter the reaction mixture was transferred to a round-bottom flask, in which a colorless precipitate would form over time. The colorless precipitate was decanted with AcOH (8 mL), H2O (2 mL × 8 mL), and pentane (8 mL). The precipitate was dried in vacuo to yield methoxyterephthalic acid 8 as a colorless solid (289 mg, 67%).
General Procedure for Methoxy Poly(ethylene terephthalate) Synthesis
Methoxyterephthalic acid (obtained from a literature protocol)61 (3.54 g, 18.1 mmol) was stirred in ethylene glycol (3.0 mL, 54 mmol, 3.0 equiv) under Ar in a pressure tube (20 mL total volume) at 220 °C for 18 h, after which the reaction mixture was subjected to vacuum heating at 220 °C for 4 h, then at 270 °C for 3.5 h, and finally at 310 °C for 18 h. The resulting polymer was isolated without further manipulation and characterized by IR, TGA, and DSC analyses (see Figures S5–S7 in the SI).
Acknowledgments
The authors appreciate financial support from the Danish National Research Foundation (grant no. DNRF118), NordForsk (grant no. 85378), the Independent Research Fund Denmark/Technology and Production Sciences, and Aarhus University. Support from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 862179 and Marie Sklodowska-Curie grant agreement no. 859910 is also gratefully acknowledged. This publication reflects the views only of the authors, and the Commission cannot be held responsible for any use, which may be made of the information contained therein. K.H. thanks the Research Council of Norway (no. 300769) and Sigma2 (no. nn9330k). The authors are also highly thankful to CSCAA for the computing hours for the DFT study, and to Anders Peder Steffen Pedersen for the supply of sawdust samples used in this work.
Glossary
Abbreviations
- TA
terephthalic acid
- MTA
methoxyterephthalic acid
- PET
poly(ethylene terephthalate)
- MPET
methoxy poly(ethylene terephthalate)
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00092.
Author Contributions
S.P. and T.S. designed the project. S.P. optimized fluorosulfation and cyanation steps, and G.B. aided in optimizing the cyanation reaction with sawdust. S.P. and G.B. evaluated the sawdust scope. G.B. determined lignin weight percentages and conducted the carboxylation of spruce-derived fluorosulfate. G.B. investigated the mechanism experimentally and with DFT. K.H. directed the DFT calculations. H.H. conducted X-ray crystallography. S.P. optimized and conducted nitrile hydrolysis and AMOCO oxidation steps. M.H. and S.P. conducted the polymerization of MTA to MPET. M.H. conducted TGA and DSC analyses of MPET. S.P. and T.S. wrote the manuscript, with comments from all co-authors. T.S. directed the project. CRediT: Simon Steffen Pedersen conceptualization, investigation, methodology, writing-original draft; Gabriel Batista investigation, methodology.
The authors declare the following competing financial interest(s): Troels Skrydstrup is co-owner of SyTracks a/s, which commercializes the two-chamber reactor, COware.
Supplementary Material
References
- Global Polyethylene Terephtalate Market Report 2017 – By End-Use Industries, Products & Regions – Research and Markets. https://www.businesswire.com/news/home/20170914005775/en/Global-Polyethylene-Terephtalate-Market-Report-2017---By-End-Use-Industries-Products-Regions---Research-and-Markets (accessed 13 February, 2023).
- de Cort S.; Godts F.; Moreau F.. Packaging Materials 1: Polyethylene Terephthalate (PET) for Food Packaging Applications. ILSI Europe Report Series. https://ilsi.eu/publication/packaging-materials-1-polyethylene-terephthalate-pet-for-food-packaging-applications-updated-version/ (accessed 13 February, 2023).
- Park S. H.; Kim S. H. Poly (ethylene terephthalate) recycling for high value added textiles. Fash. Text. 2014, 1, 1 10.1186/s40691-014-0001-x. [DOI] [Google Scholar]
- Wittcoff H. A.; Reuben B. G.; Plotkin J. S.. Industrial Organic Chemicals, 3rd ed.; Wiley: New York, 2012; pp 397–407. [Google Scholar]
- Srithep Y.; Javadi A.; Pilla S.; Turng L.-S.; Gong S.; Clemons C.; Peng J. Processing and Characterization of Recycled Poly(ethylene terephthalate) Blends With Chain Extenders, Thermoplastic Elastomer, and/or Poly(butylene adipate-co-terephthalate). Polym. Eng. Sci. 2011, 51, 1023–1032. 10.1002/pen.21916. [DOI] [Google Scholar]
- Adam D. How far will global population rise? Researchers can’t agree. Nature 2021, 597, 462–465. 10.1038/d41586-021-02522-6. [DOI] [PubMed] [Google Scholar]
- Tullo A.Will plastics recycling meet its deadline? C&EN. https://cen.acs.org/environment/recycling/plastics-recycling-meet-deadline/99/i37 (accessed 13 February, 2023).
- United States Environmental Protection Agency: Advancing Sustainable Materials Management: 2018 Fact Sheet. https://www.epa.gov/sites/default/files/2021-01/documents/2018_ff_fact_sheet_dec_2020_fnl_508.pdf (accessed 13 February, 2023).
- Royte E.Is burning plastic waste a good idea? National Geographic. https://www.nationalgeographic.com/environment/article/should-we-burn-plastic-waste (accessed 13 February, 2023). [Google Scholar]
- Hatti-Kaul R.; Nilsson L. J.; Zhang B.; Rehnberg N.; Lundmark S. Designing Biobased Recyclable Polymers for Plastics. Trends Biotechnol. 2020, 38, 50–67. 10.1016/j.tibtech.2019.04.011. [DOI] [PubMed] [Google Scholar]
- Tullo A. New route planned to biobased ethylene glycol. Chem. Eng. News 2017, 95, 10. [Google Scholar]
- Lyons T. W.; Guironnet D.; Findlater M.; Brookhart M. Synthesis of p-Xylene from Ethylene. J. Am. Chem. Soc. 2012, 134, 15708–15711. 10.1021/ja307612b. [DOI] [PubMed] [Google Scholar]
- Miller K. K.; Zhang P.; Nishizawa-Brennen Y.; Frost J. W. Synthesis of Biobased Terephthalic Acid from Cycloaddition of Isoprene with Acrylic Acid. ACS Sustainable Chem. Eng. 2014, 2, 2053–2056. 10.1021/sc5003038. [DOI] [Google Scholar]
- Peters M. W.; Taylor H. D.; Jenni M.; Manzer L. E.; Henton D. E.. Integrated Process to Selectively Convert Renewable Isobutanol to P-Xylene.U.S. Patent US2011/0087000A1, 2010.
- Williams C. L.; Chang C. C.; Do P.; Nikbin N.; Caratzoulas S.; Vlachos D. G.; Lobo R. F.; Fan W.; Dauenhauer P. J. Cycloaddition of Biomass-Derived Furans for Catalytic Production of Renewable p-Xylene. ACS Catal. 2012, 2, 935–939. 10.1021/cs300011a. [DOI] [Google Scholar]
- Wan Y.; Lee J.-M. Toward Value-Added Dicarboxylic Acids from Biomass Derivatives via Thermocatalytic Conversion. ACS Catal. 2021, 11, 2524–2560. 10.1021/acscatal.0c05419. [DOI] [Google Scholar]
- Tomás R. A. F.; Bordado J. C. M.; Gomes J. F. P. p-Xylene Oxidation to Terephthalic Acid: A Literature Review Oriented toward Process Optimization and Development. Chem. Rev. 2013, 113, 7421–7469. 10.1021/cr300298j. [DOI] [PubMed] [Google Scholar]
- Lu R.; Lu F.; Chen J.; Yu W.; Huang Q.; Zhang J.; Xu J. Production of Diethyl Terephthalate from Biomass-Derived Muconic Acid. Angew. Chem., Int. Ed. 2016, 55, 249–253. 10.1002/anie.201509149. [DOI] [PubMed] [Google Scholar]
- Tullo A. H.Coca-Cola looks to make plastic bottles from wood. C&EN. https://cen.acs.org/business/biobased-chemicals/Coca-Cola-looks-make-plastic/99/i40 (accessed 13 February, 2023). [Google Scholar]
- Colonna M.; Berti C.; Fiorini M.; Binassi E.; Mazzacurati M.; Vannini M.; Karanam S. Synthesis and radiocarbon evidence of terephthalate polyesters completely prepared from renewable resources. Green Chem. 2011, 13, 2543–2548. 10.1039/c1gc15400a. [DOI] [Google Scholar]
- Neaţu F.; Culica G.; Florea M.; Parvulescu V. I.; Cavani F. Synthesis of Terephthalic Acid by p-Cymene Oxidation using Oxygen: Toward a More Sustainable Production of Bio-Polyethylene Terephthalate. ChemSusChem 2016, 9, 3102–3112. 10.1002/cssc.201600718. [DOI] [PubMed] [Google Scholar]
- Kärkäs M. D.; Matsuura B. S.; Monos T. M.; Magallanes G.; Stephenson C. R. J. Transition-metal catalyzed valorization of lignin: the key to a sustainable carbon-neutral future. Org. Biomol. Chem. 2016, 14, 1853–1914. 10.1039/C5OB02212F. [DOI] [PubMed] [Google Scholar]
- Sun Z.; Fridrich B.; de Santi A.; Elangovan S.; Barta K. Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev. 2018, 118, 614–678. 10.1021/acs.chemrev.7b00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H.; Weeda E. P.; Alherech M.; Anson C. W.; Karlen S. D.; Cui Y.; Foster C. E.; Stahl S. S. Oxidative Catalytic Fractionation of Lignocellulosic Biomass under Non-alkaline Conditions. J. Am. Chem. Soc. 2021, 143, 15462–15470. 10.1021/jacs.1c08635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renders T.; Van den Bossche G.; Vangeel T.; van Aelst K.; Sels B. Reductive catalytic fractionation: state of the art of the lignin-first biorefinery. Curr. Opin. Biotechnol. 2019, 56, 193–201. 10.1016/j.copbio.2018.12.005. [DOI] [PubMed] [Google Scholar]
- Abu-Omar M. M.; Barta K.; Beckham G. T.; Luterbacher J. S.; Ralph J.; Rinaldi R.; Román-Leshkov Y.; Samec J. S. M.; Sels B. F.; Wang F. Guidelines for performing lignin-first biorefining. Energy Environ. Sci. 2021, 14, 262–292. 10.1039/D0EE02870C. [DOI] [Google Scholar]
- Hu G.; Chen C.; Lu H. T.; Wu Y.; Liu C.; Tao L.; Men Y.; He G.; Li K. G. A Review of Technical Advances, Barriers, and Solutions in the Power to Hydrogen (P2H) Roadmap. Engineering 2020, 6, 1364–1380. 10.1016/j.eng.2020.04.016. [DOI] [Google Scholar]
- Bai Z.; Phuan W. C.; Ding J.; Heng T. H.; Luo J.; Zhu Y. Production of Terephthalic Acid from Lignin-Based Phenolic Acids by a Cascade Fixed-Bed Process. ACS Catal. 2016, 6, 6141–6145. 10.1021/acscatal.6b02052. [DOI] [Google Scholar]
- Song S.; Zhang J.; Gözaydin G.; Yan N. Production of Terephthalic Acid from Corn Stover Lignin. Angew. Chem., Int. Ed. 2019, 58, 4934–4937. 10.1002/anie.201814284. [DOI] [PubMed] [Google Scholar]
- European Chemicals Agency: Trifluoromethanesulphonic anhydride Hydrolysis. https://echa.europa.eu/registration-dossier/-/registered-dossier/22478/5/2/3 (accessed 13 February, 2023).
- Burkhard C. A.Linear Polyesters, 1959, US2902469.
- Van den Bosch S.; Schutyser W.; Vanholme R.; Driessen T.; Koelewijn S.-F.; Renders T.; De Meester B.; Huijgen W. J. J.; Dehaen W.; Courtin C. M.; Lagrain B.; Boerjan W.; Sels B. F. Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps. Energy Environ. Sci. 2015, 8, 1748–1763. 10.1039/C5EE00204D. [DOI] [Google Scholar]
- Facas G. G.; Brandner D. G.; Bussard J. R.; Román-Leshkov Y.; Beckham G. T. Interdependence of Solvent and Catalyst Selection on Low Pressure Hydrogen-Free Reductive Catalytic Fractionation. ACS Sustainable Chem. Eng. 2023, 11, 4517–4522. 10.1021/acssuschemeng.2c07394. [DOI] [Google Scholar]
- Kenny J. K.; Brandner D. G.; Neefe S. R.; Michener W. E.; Román-Leshkov Y.; Beckham G. T.; Medlin J. W. Catalyst choice impacts aromatic monomer yields and selectivity in hydrogen-free reductive catalytic fractionation. React. Chem. Eng. 2022, 7, 2527–2533. 10.1039/D2RE00275B. [DOI] [Google Scholar]
- Anderson E. M.; Katahira R.; Reed M.; Resch M. G.; Karp E. M.; Beckham G. T.; Román-Leshkov Y. Reductive Catalytic Fractionation of Corn Stover Lignin. ACS Sustainable Chem. Eng. 2016, 4, 6940–6950. 10.1021/acssuschemeng.6b01858. [DOI] [Google Scholar]
- Klein I.; Saha B.; Abu-Omar M. M. Lignin depolymerization over Ni/C catalyst in methanol, a continuation: effect of substrate and catalyst loading. Catal. Sci. Technol. 2015, 5, 3242–3245. 10.1039/C5CY00490J. [DOI] [Google Scholar]
- Studer M. H.; DeMartini J. D.; Davis M. F.; Sykes R. W.; Davison B.; Keller M.; Tuskan G. A.; Wyman C. E. Lignin content in natural Populus variants affects sugar release. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6300–6305. 10.1073/pnas.1009252108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulbaek Andersen M. P.; Blake D. R.; Rowland F. S.; Hurley M. D.; Wallington T. J. Atmospheric Chemistry of Sulfuryl Fluoride: Reaction with OH Radicals, Cl Atoms and O3, Atmospheric Lifetime, IR Spectrum, and Global Warming Potential. Environ. Sci. Technol. 2009, 43, 1067–1070. 10.1021/es802439f. [DOI] [PubMed] [Google Scholar]
- Dong J.; Krasnova L.; Finn M. G.; Sharpless K. B. Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. 10.1002/anie.201309399. [DOI] [PubMed] [Google Scholar]
- Su Z.-M.; Twilton J.; Hoyt C. B.; Wang F.; Stanley L.; Mayes H. B.; Kang K.; Weix D. J.; Beckham G. T.; Stahl S. S. Ni- and Ni/Pd-Catalyzed Reductive Coupling of Lignin-Derived Aromatics to Access Biobased Plasticizers. ACS Cent. Sci. 2023, 9, 159–165. 10.1021/acscentsci.2c01324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smet G.; Bai X.; Mensch C.; Sergeyev S.; Evano G.; Maes B. U. W. Selective Nickel-Catalyzed Hydrodeacetoxylation of Aryl Acetates. Angew. Chem., Int. Ed. 2022, 61, e202201751 10.1002/anie.202201751. [DOI] [PubMed] [Google Scholar]
- Veryser C.; Demaerel J.; Bieliunas V.; Gilles P.; De Borggraeve W. M. Ex Situ Generation of Sulfuryl Fluoride for the Synthesis of Aryl Fluorosulfates. Org. Lett. 2017, 19, 5244–5247. 10.1021/acs.orglett.7b02522. [DOI] [PubMed] [Google Scholar]
- Triethylamine. https://www.sigmaaldrich.com/DK/en/substance/triethylamine10119121448 (accessed 2023-02-13).
- Potassium Carbonate. https://www.sigmaaldrich.com/DK/en/substance/potassiumcarbonate13821584087 (accessed 2023-02-13).
- Henderson R. K.; Hill A. P.; Redman A. M.; Sneddon H. F. Development of GSK’s acid and base selection guides. Green Chem. 2015, 17, 945–949. 10.1039/C4GC01481B. [DOI] [Google Scholar]
- Kristensen S. K.; Eikeland E. Z.; Taarning E.; Lindhardt A. T.; Skrydstrup T. Ex situ generation of stoichiometric HCN and its application in the Pd-catalysed cyanation of aryl bromides: evidence for a transmetallation step between two oxidative addition Pd-complexes. Chem. Sci. 2017, 8, 8094–8105. 10.1039/C7SC03912C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anbarasan P.; Schareina T.; Beller M. Recent developments and perspectives in palladium-catalyzed cyanation of aryl halides: synthesis of benzonitriles. Chem. Soc. Rev. 2011, 40, 5049–5067. 10.1039/c1cs15004a. [DOI] [PubMed] [Google Scholar]
- Erhardt S.; Grushin V. V.; Kilpatrick A. H.; Macgregor S. A.; Marshall W. J.; Roe D. C. Mechanisms of Catalyst Poisoning in Palladium-Catalyzed Cyanation of Haloarenes. Remarkably Facile C–N Bond Activation in the [(Ph3P)4Pd]/[Bu4N]+ CN– System. J. Am. Chem. Soc. 2008, 130, 4828–4845. 10.1021/ja078298h. [DOI] [PubMed] [Google Scholar]
- Bruno N. C.; Niljianskul N.; Buchwald S. L. N-Substituted 2-Aminobiphenylpalladium Methanesulfonate Precatalysts and Their Use in C–C and C–N Cross-Couplings. J. Am. Chem. Soc. 2014, 79, 4161–4166. 10.1021/jo500355k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace V.; Hoyos P.; Castoldi L.; de Maria P. D.; Alcántara A. R. 2-Methyltetrahydrofuran (2-MeTHF): A Biomass-Derived Solvent with Broad Application in Organic Chemistry. ChemSusChem 2012, 5, 1369–1379. 10.1002/cssc.201100780. [DOI] [PubMed] [Google Scholar]
- Zhao C.; Fang W.-Y.; Rakesh K. P.; Qin H.-L. Pd-Catalyzed one-pot dehydroxylative coupling of phenols with K4[Fe(CN)6] mediated by SO2F2: a practical method for the direct conversion of phenols to aryl nitriles. Org. Chem. Front. 2018, 5, 1835–1839. 10.1039/C8QO00295A. [DOI] [Google Scholar]
- Gan Y.; Wang G.; Xie X.; Liu Y. Nickel-Catalyzed Cyanation of Phenol Derivatives with Zn(CN)2 Involving C–O Bond Cleavage. J. Org. Chem. 2018, 83, 14036–14048. 10.1021/acs.joc.8b02498. [DOI] [PubMed] [Google Scholar]
- Ushkov A. V.; Grushin V. V. Rational Catalysis Design on the Basis of Mechanistic Understanding: Highly Efficient Pd-Catalyzed Cyanation of Aryl Bromides with NaCN in Recyclable Solvents. J. Am. Chem. Soc. 2011, 133, 10999–11005. 10.1021/ja2042035. [DOI] [PubMed] [Google Scholar]
- Kulachenko A.Is Hardwood harder than the softwood? http://fibrenet.eu/index.php?id=blog-post-nine (accessed 13 February, 2023).
- Sterckx H.; Morel B.; Maes B. U. W. Catalytic Aerobic Oxidation of C(sp3) −H Bonds. Angew. Chem., Int. Ed. 2019, 58, 7946–7970. 10.1002/anie.201804946. [DOI] [PubMed] [Google Scholar]
- White M. C.; Zhao J. Aliphatic C–H Oxidations for Late-Stage Functionalization. J. Am. Chem. Soc. 2018, 140, 13988–14009. 10.1021/jacs.8b05195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. Y.; Oh K. Recent advances in the copper-catalyzed aerobic Csp3–H oxidation strategy. Org. Biomol. Chem. 2021, 19, 3569–3583. 10.1039/D1OB00081K. [DOI] [PubMed] [Google Scholar]
- Blondiaux E.; Bomon J.; Smolén M.; Kaval N.; Lemière F.; Sergeyev S.; Diels L.; Sels B.; Maes B. U. W. Bio-based Aromatic Amines from Lignin-Derived Monomers. ACS Sustainable Chem. Eng. 2019, 7, 6906–6916. 10.1021/acssuschemeng.8b06467. [DOI] [Google Scholar]
- Ma C.; Zhao C.-Q.; Xu X.-T.; Li Z.-M.; Wang X.-Y.; Zhang K.; Mei T.-S. Nickel-Catalyzed Carboxylation of Aryl and Heteroaryl Fluorosulfates Using Carbon Dioxide. Org. Lett. 2019, 21, 2464–2467. 10.1021/acs.orglett.9b00836. [DOI] [PubMed] [Google Scholar]
- MacDonald W. A. New advances in poly(ethylene terephthalate) polymerization and degradation. Polym. Int. 2002, 51, 923–930. 10.1002/pi.917. [DOI] [Google Scholar]
- Vermoortele F.; Vandichel M.; Van de Voorde B.; Ameloot R.; Waroquier M.; et al. Electronic Effects of Linker Substitution on Lewis Acid Catalysis with Metal-Organic Frameworks. Angew. Chem., Int. Ed. 2012, 51, 4887–4890. 10.1002/anie.201108565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.