

Abstract
Cocaine exposure induces persistent changes in synaptic transmission and intrinsic properties of ventral tegmental area (VTA) dopamine neurons. Despite significant progress in understanding cocaine-induced plasticity, an effective treatment of cocaine addiction is lacking. Chronic cocaine potentiates excitatory and alters inhibitory transmission to dopamine neurons, induces dopamine neuron hyperexcitability, and reduces dopamine release in projection areas. Understanding how intrinsic and synaptic plasticity interact to control dopamine neuron firing and dopamine release could prove useful in the development of new therapeutics. In this review, we examine recent literature discussing cocaine-induced plasticity in the VTA and highlight potential therapeutic interventions.
Introduction
Despite an ever-increasing body of literature on brain mechanisms contributing to cocaine addiction, therapeutic treatments remain limited. Treating cocaine addiction is complex due to distinct and persistent changes — plasticity — in multiple brain regions, including the ventral tegmental area (VTA), prefrontal cortex (PFC), and ventral striatum [1]. Improving the odds of treating cocaine addiction lies in a greater understanding of cocaine-induced plasticity.
Exposure to cocaine results in two forms of changes: synaptic plasticity defined as changes in synaptic transmission and intrinsic plasticity defined as changes in the intrinsic electrical properties of neurons. Interplay between synaptic inputs and intrinsic properties control the ultimate outputs of the neuron, action potential (AP) firing and neurotransmitter release. In this review, we discuss recent animal model studies of cocaine-induced synaptic and intrinsic plasticity in the VTA, highlighting the findings thought to contribute to the vulnerability to relapse (Figure 1). We then illustrate the translational potential of some of these findings into the clinic.
Figure 1. Cocaine-induced plasticity in VTA dopamine neurons.
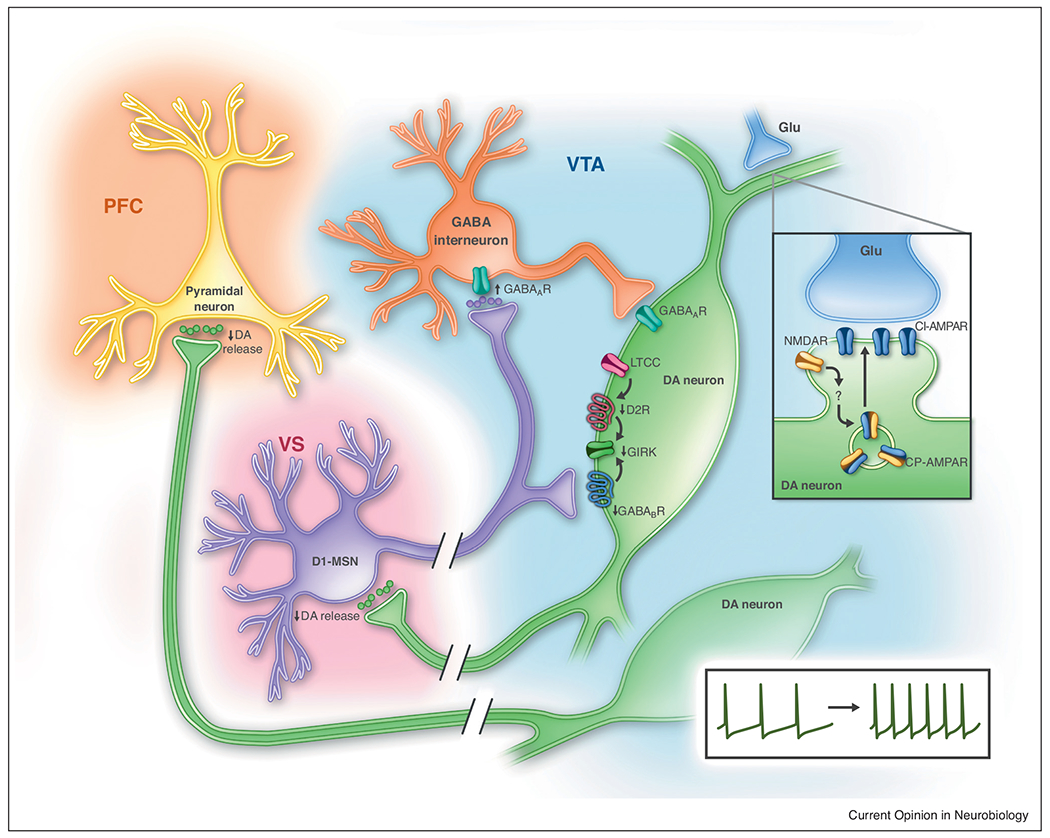
Cocaine enhances GABAA-R currents at the synapses between ventral striatal (VS) D1-MSNs and VTA GABA interneurons, while reducing inhibitory GABAB-R- and D2-R-activated GIRK conductance on VTA dopamine (DA) neurons. Calcium entry through L-type calcium channels (LTCC) regulates D2-R GIRK conductance and is an important player in cocaine-mediated behavior. Chronic cocaine induces LTP at the glutamatergic (Glu) synapses on VTA dopamine neurons (insert). This LTP is facilitated by activation of NMDA-Rs which drives CP-AMPA-R insertion into the synaptic membrane. These changes in GABAergic and glutamatergic synaptic transmission contribute to hyperexcitability of dopamine neurons. Cocaine decreases dopamine release on VS and prefrontal cortex (PFC) neurons.
Synaptic plasticity
It has been nearly two decades from our first report of cocaine-induced synaptic plasticity in the VTA. A single injection of cocaine causes an N-methyl-d-aspartate (NMDA) receptor (R)-dependent potentiation of glutamatergic synaptic transmission (i.e. α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-R-dependent) onto VTA dopamine neurons for at least five days [2]. This long-term potentiation (LTP) of glutamatergic (AMPA)-R-dependent transmission arises 3–5 hours after cocaine exposure, following a transient dopamine D1/D5 receptor-dependent strengthening of NMDA-R-dependent transmission [3,4]. Following chronic cocaine self-administration (>14 days), LTP of glutamatergic transmission persists for at least 3 months [5]. Expression of LTP coincides with greater insertion of Ca2+-permeable (CP) AMPA-Rs without a change in the total number of AMPA-Rs [6]. The higher conductance of these receptors contributes to the potentiation of excitatory transmission, and their Ca2+ permeability alters synaptic signaling to induce and maintain drug-related plasticity [7]. Positive modulation of metabotropic glutamate receptors (mGluR1s) can target and prevent this plasticity in the VTA and other brain regions [6,8•,9], providing a potential therapeutic target for cocaine addiction. Additionally, VTA NMDA-R signaling, which is required for excitatory LTP in the VTA [4], is also required for cocaine-induced ventral striatal plasticity and cocaine seeking [8•], suggesting VTA dopamine neuron LTP, CP-AMPA-Rs, and downstream adaptations are necessary for reinstatement. However, a direct link between cocaine-induced CP-AMPA-R insertion and NMDA-R signaling has not been shown and methods to examine this phenomenon have proven difficult [10]. Moreover, VTA CP-AMPAR plasticity requires insertion of the Ca2+-impermeable GluN3A NMDA-R subunit [11], suggesting an NMDA-R-independent source of Ca2+ is required for potentiation. Additional studies are necessary to fully describe the complex interaction between CP-AMPA-Rs and NMDA-Rs. Overall, cocaine potentiates glutamatergic transmission onto dopamine neurons first by causing a transient potentiation of NMDA-R signaling which in turn strengthens AMPA-R-dependent synaptic transmission.
Persistent cocaine-induced synaptic plasticity is not limited to glutamatergic synaptic transmission but also involves gamma-aminobutyric acid (GABA) signaling including both GABAA-Rs and GABAB-Rs. Overall, GABAA-R signaling on dopamine neurons is reduced following cocaine [12] and opposes excitatory input to control dopamine neuron excitability. Methods to decrease excitation/inhibition ratios enhanced by cocaine reduces in vivo firing frequency of dopamine neurons [13,14] and enhances dopamine release in the ventral striatum [14] (see Dopamine release after chronic cocaine). In contrast, after chronic cocaine, GABA release is increased in the ventral striatum–VTA pathway, a pathway which originates predominantly from D1 receptor-expressing medium spiny neurons (MSNs) [15]. The D1-MSN axon terminals in the VTA synapse onto GABAergic VTA interneurons via GABAA-R [15]. Hence, by increasing GABA release in the ventral striatum–VTA pathway, chronic cocaine potentiates GABAA-R-mediated synaptic transmission between D1-MSNs and VTA GABA interneurons, thereby enhancing the excitability of VTA dopamine neurons through ‘disinhibition’ as shown in vivo by Bocklisch et al. [15]. However, GABAA-R input on dopamine neurons arrives from a variety of sources including non-VTA GABAergic input [16]. D1-MSNs also synapse onto dopamine neurons and signal via GABAB-Rs. This was initially suggested by data showing that D1 receptor agonists potentiate GABAB-R synaptic currents in VTA dopamine neurons [17,18], and later confirmed by projection-specific targeting studies [19•]. Therefore, D1-MSN GABAB-R signaling may play a more prominent role in reduced inhibition of VTA dopamine neurons. The effect of chronic cocaine on the GABAB-R-mediated synaptic transmission between MSNs and dopamine neurons has not been specifically examined; however, chronic cocaine reduces augmentation of GABAB-R synaptic currents by D1-R agonists [18] and reduced GABAB-R signaling in VTA dopamine neurons also enhances cocaine-induced locomotor sensitization [19•]. In addition to presynaptic changes in GABA release, chronic exposure to psychostimulants decreases the function of GABAB-Rs in the VTA through a postsynaptic mechanism [20], discussed further in the Intrinsic Plasticity section. Taken together, chronic cocaine enhances the excitability of dopamine neurons by enhancing excitatory synaptic transmission, suppressing GABAB-R inhibitory synaptic transmission, and reducing GABAA-R inhibition from VTA GABA interneurons and other non-VTA sources.
Intrinsic plasticity
Neuronal excitability of dopamine neurons is controlled by synaptic inputs and intrinsic electrical properties [21]. In addition to its effects on synaptic plasticity, recent studies reveal that chronic cocaine alters the intrinsic properties of dopamine neurons. In vivo, dopamine neurons fire APs tonically or in bursts [22,23]. Tonic firing consists of single APs at 1–6 Hz; these are intermittently interrupted by transient bursts of APs (i.e. phasic firing), which facilitate learning of reward-salient cues [24]. In vitro, tonic firing is preserved since it does not rely on synaptic inputs but rather the autonomous coordination of multiple intrinsic conductances [25]. Phasic firing on the other hand, involves glutamatergic synaptic input [22,23,26]. After chronic cocaine, dopamine neurons display both more frequent phasic bursts and faster tonic firing [27], indicating cocaine-induced intrinsic plasticity.
Early work established that cocaine-induced heightened excitability was in part attributable to decreased autoregulation by inhibitory dopamine D2 autoreceptors (D2-R) [28]. Along with GABAB-Rs, D2-Rs regulate AP firing by activating the hyperpolarizing G protein-coupled inwardly rectifying potassium (GIRK) conductance. Under physiological conditions, phasic firing in VTA dopamine neurons enhances GIRK conductance by inserting GIRK channels into the membrane [29•]. GIRK insertion depends on NMDA-Rs, intracellular Ca2+, and CaMKII activation [29•]. Increased GIRK conductance is also observed after potentiation of excitatory glutamatergic transmission (shown in CA1 pyramidal neurons in the hippocampus) [30]. These changes in GIRK conductance may be a form of homeostatic plasticity to maintain excitation-inhibition balance. However, following single or chronic cocaine or methamphetamine exposure, GIRK conductance in dopamine neurons is reduced for several days to a week [20,31,32], despite enhanced glutamatergic synaptic transmission and phasic firing. The loss of GIRK function involves a reduction in surface expression of the channel [31,32]. While the mechanism is not clear, buffering intracellular Ca2+ with chelators alleviates the reduction in GIRK conductance induced by methamphetamine [20], suggesting a potential therapy to restore this form of plasticity in patients. Although, the ability of in vitro intracellular calcium buffering to restore cocaine-induced reductions in GIRK conductance has not been tested.
Ample evidence points to a critical role of VTA L-type Ca2+ channels (LTCC) in cocaine-induced synaptic plasticity and cocaine-related behaviors. The vast majority of work on LTCCs in dopamine neurons has been conducted in the substantia nigra pars compacta (SNc) dopamine neurons where low-threshold activating LTCCs contribute to rhythmic dendritic Ca2+ oscillations, which are not present in VTA dopamine neurons [33]. In VTA dopamine neurons, pharmacological activation of LTCCs induces burst firing [34,35] and genetic deletion of Cav1.3 LTCC slows tonic firing [35], suggesting under some conditions, Ca2+ entry into VTA dopamine neurons via LTCCs may promote faster tonic firing and more frequent phasic-firing events. Direct measurement of LTCC conductance in dopamine neurons after cocaine is still lacking. Nevertheless, VTA LTCCs are necessary and sufficient for cocaine-related behaviors in animal models. For example, pharmacological stimulation of LTCCs in the VTA enhances cocaine-related behaviors [36] and mimics behavioral sensitization [37], while intra-VTA administration of an LTCC blocker prevents cocaine-induced conditioned place preference (CPP) and prevents reinstatement of cocaine-CPP [38,39]. We speculate that VTA LTCC blockers may alleviate the reduction in GIRK conductance after cocaine, or desensitization of D2 autoreceptors induced by psychostimulants akin to Ca2+ chelators, as enhancement of D2 receptor-activated GIRK conductance by LTCC blockers is observed in SNc dopamine neurons in naïve animals [40,41] and GIRK is directly inhibited by intracellular Ca2+ [42•].
The role of the VTA LTCCs in modulating neuronal excitability and mediating cocaine-related behaviors poise them as a unique target in the efforts to reverse changes in the reward circuitry after exposure to drugs. Beyond NMDA-R and LTCCs, we predict that studies on the interplay between synaptic and intrinsic mechanisms will become a greater point of focus and greatly advance the understanding of cocaine-induced plasticity.
Dopamine release after chronic cocaine
Withdrawal from chronic cocaine results in imbalanced excitatory and inhibitory synaptic transmission and alters intrinsic conductances, resulting in hyperexcitability of VTA dopamine neurons. Yet, after chronic cocaine exposure, tonic extracellular dopamine level is reduced in the ventral striatum and PFC in both rodent [43–46] and human studies [47]. These studies support the dopamine depletion hypothesis of cocaine addiction [48], proposing that reduced dopamine levels contribute to cocaine addiction. The disconnect between dopamine neurons hyperexcitability measured at the cell body region and dopamine release in projection regions after chronic cocaine is difficult to reconcile. Data from animal and human studies offer multiple explanations. These include decreased dopamine synthesis, decreased vesicular loading, decreased dopamine release through functional increase in D2 autoreceptors, or increased dopamine clearance (i.e. reuptake or intracellular degradation). A possible yet unsubstantiated explanation for the disconnect between dopamine neuron excitability and release could be due to failure of AP propagation or reduced terminal excitability after chronic cocaine; increased expression of the potassium channel Kv1.2 in dopamine axon terminals could reduce dopamine release by reducing the terminal excitability. Interestingly, chronic cocaine has been shown to upregulate Kv1.2 potassium channel surface expression in ventral striatal MSNs resulting in reduced excitability of MSNs [49]. Further, genetic deletion or pharmacological blockade of Kv1.2 channels in dopamine neurons enhances dopamine release and reduces axonal D2 autoreceptor-mediated inhibition of dopamine release in the striatum [50•]. Whether Kv1.2 expression is increased in dopamine axon terminals after chronic cocaine remains to be explored.
By increasing the signal-to-noise ratio (phasic dopamine in response to drug-related cues vis-à-vis tonic dopamine level), low basal dopamine in the ventral striatum after chronic cocaine may play a critical role in the reinstatement of drug seeking [51]. While phasic dopamine release in the ventral striatum is necessary and sufficient for cocaine seeking [39,52], replenishing dopamine release with a systemic injection of the dopamine precursor l-DOPA prevents escalation of cocaine intake [45] and blocks cue-induced reinstatement [53]. Similarly, increasing dopamine levels in the ventral striatum by infusion of LTCC blockers into the VTA inhibits reinstatement of cocaine seeking [39]. Thus, therapies aimed to restore basal dopamine levels, by reducing the phasic dopamine signal-to-noise ratio, may blunt the salience of drug-related cues and therefore inhibit relapse to drug seeking. Nevertheless, a better fundamental understanding of the regulation of tonic and phasic dopamine release under physiological conditions and after cocaine exposure will aid in the development of effective therapies.
Clinical perspectives
More than a hundred randomized clinical trials (RCTs) investigating the efficacy of more than sixty medications have been conducted so far, but an efficacious treatment for cocaine addiction remains elusive [54]. With the exception of metabotropic glutamate receptors, several drug classes have been tested including Ca2+ channel blockers, GABAB-R agonists, monoamine modulators (e. g. dopamine and serotonin reuptake blockers) and several others [54].
Aside from pharmacotherapy, neuromodulation tools such as transcranial magnetic stimulation (TMS) and transcranial direct current stimulation (tDCS) are currently being explored to treat cocaine addiction [55••,56]. These efforts, while preliminary, appear very promising. TMS, currently FDA-approved in the treatment of depression, involves rapid magnetic pulses delivered non-invasively, which generate electric currents in neuronal circuits. High and low frequency TMS are thought to increase and decrease cortical excitability, respectively [57]. tDCS involves applying a direct current between an anodal and a cathodal electrodes placed on the scalp, and alters neuronal excitability [58]. Both TMS and tDCS of the PFC can be used to activate the VTA and increase dopamine release in the PFC (i.e. anterior cingulate and orbitofrontal cortex) [58,59], an area that supports and drives burst firing of VTA dopamine neurons [60]. tDCS also enhances VTA-PFC connectivity [58]. Interestingly, several pilot studies reported that repetitive high frequency TMS (rTMS) of the left dorsolateral PFC reduces craving to drugs of abuse including cocaine [57,61] and a pilot clinical trial of high frequency rTMS of the left dorsolateral PFC showed reduced relapse rates in cocaine addicts [55••]. While the therapeutic effect of TMS could be due to reversing the reduced PFC excitability observed in animal models after chronic cocaine [62], it is also possible that the TMS effect is related to increasing dopamine release in the PFC [59]. Further research is warranted to explore the TMS-induced neurobiological changes in VTA-PFC and VTA-ventral striatum connectivity and the TMS effects on dopamine release in cocaine-dependent subjects.
Conclusions
In recent years, the understanding of cocaine-induced intrinsic and synaptic plasticity has progressed significantly and promising therapies are emerging. Yet, we still face obstacles and fundamental knowledge gaps to effectively translate animal model research to the clinic. First, we hope that the next few years will bring a greater, much needed mechanistic understanding of how chronic cocaine exposure affects the interplay between intrinsic and synaptic mechanisms at the level of VTA dopamine neurons and within local circuits within the VTA. Next, standardized cocaine administration models to best mimic human addiction (e.g. self-administration) may provide greater clinical translation of electrophysiological findings. Lastly, advancing the mechanistic understanding of noninvasive neuromodulation therapy, ideally delivered in combination with pharmacological treatments, will provide additional insight into how these therapies can be optimized.
Acknowledgement
This work was funded by the National Institute on Drug Abuse Intramural Research Program.
Footnotes
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Wolf ME: Synaptic mechanisms underlying persistent cocaine craving. Nat Rev 2016, 17:351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ungless MA, Whistler JL, Malenka RC, Bonci A: Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 2001, 411:583–587. [DOI] [PubMed] [Google Scholar]
- 3.Schilstrom B, Yaka R, Argilli E, Suvarna N, Schumann J, Chen BT, Carman M, Singh V, Mailliard WS, Ron D et al. Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J Neurosci 2006, 26:8549–8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Argilli E, Sibley DR, Malenka RC, England PM, Bonci A: Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci 2008, 28:9092–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A: Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron 2008, 59:288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellone C, Luscher C: Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent longterm depression. Nat Neurosci 2006, 9:636–641. [DOI] [PubMed] [Google Scholar]
- 7.Wolf ME, Tseng KY: Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Front Mol Neurosci 2012, 5:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.•.Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR,Spanagel R, Luscher C: Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci 2009, 12:1036–1041. [DOI] [PubMed] [Google Scholar]; This study demonstrates the necessity of VTA DA neuron plasticity in driving downstream cocaine plasticity and cue-induced cocaine seeking.
- 9.Loweth JA, Tseng KY, Wolf ME: Using metabotropic glutamate receptors to modulate cocaine’s synaptic and behavioral effects: MGluR1 finds a niche. Curr Opin Neurobiol 2013, 23:500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guzman D, Carreira MB, Friedman AK, Adachi M, Neve RL, Lisa M, Han M, Cowan CW, Self DW, Neve RL et al. Inactivation of NMDA receptors in the ventral tegmental area during cocaine self-administration prevents GluA1 up-regulation but with paradoxical increases in cocaine-seeking behavior. J Neurosci 2018, 38:575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan T, Mameli M, Connor ECO, Dey PN, Verpelli C, Sala C, Perez-otano I, Lu C: Expression of cocaine-evoked synaptic plasticity by GluN3A-containing NMDA receptors. Neuron 2013, 80:1025–1038. [DOI] [PubMed] [Google Scholar]
- 12.Liu Q, Pu L, Poo M: Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature 2005, 437:1027–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, Zhong P, Vickstrom C, Li Y, Liu QS: PDE4 inhibition restores the balance between excitation and inhibition in VTA dopamine neurons disrupted by repeated in vivo cocaine exposure. Neuropsychopharmacology 2017, 42:1991–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Li Y, Yu L, Vickstrom CR, Liu Q: VTA mTOR signaling regulates dopamine dynamics, cocaine-induced synaptic alterations, and reward. Neuropsychopharmacology 2017, 43:1066–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bocklisch C, Pascoli V, Wong JC, House DR, Yvon C, de Roo M, Tan KR, Luscher C: Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science 2013, 341:1521–1525. [DOI] [PubMed] [Google Scholar]
- 16.Oliva I, Wanat MJ: Ventral tegmental area afferents and drug-dependent behaviors. Front Psychiatry 2016, 7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cameron DL, Williams JT: Dopamine D1 receptors facilitate transmitter release. Nature 1993, 366:344–347. [DOI] [PubMed] [Google Scholar]
- 18.Bonci A, Williams JT: A common mechanism mediates longterm changes in synaptic transmission after chronic cocaine and morphine. Neuron 1996, 16:631–639. [DOI] [PubMed] [Google Scholar]
- 19.•.Edwards NJ, Tejeda HA, Pignatelli M, Zhang S, McDevitt RA, Wu J, Bass CE, Bettler B, Morales M, Bonci A: Circuit specificity in the inhibitory architecture of the VTA regulates cocaine-induced behavior. Nat Neurosci 2017, 20:438–448. [DOI] [PubMed] [Google Scholar]; This is the first paper to establish direct evidence of circuit-specific GABAA-R and GABAB-R receptor currents from ventral striatum projections to VTA GABA interneurons and dopamine neurons, respectively. These subcircuits are critical in understanding GABA regulation of dopamine neuron excitability.
- 20.Sharpe AL, Varela E, Bettinger L, Beckstead MJ: Methamphetamine self-administration in mice decreases GIRK channel-mediated currents in midbrain dopamine neurons. Int J Neuropsychopharmacol, 2014, 18 10.1093/ijnp/pyu073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paladini CA, Roeper J: Generating bursts (and pauses) in the dopamine midbrain neurons. Neuroscience 2014, 282:109–121. [DOI] [PubMed] [Google Scholar]
- 22.Grace AA, Bunney BS: The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci 1984, 4:2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grace AA, Bunney BS: The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci 1984, 4:2866–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper DC: The significance of action potential bursting in the brain reward circuit. Neurochem Int 2002, 41:333–340. [DOI] [PubMed] [Google Scholar]
- 25.Gantz SC, Ford CP, Morikawa H, Williams JT: The evolving understanding of dopamine neurons in the substantia nigra and ventral tegmental area. Annu Rev Physiol 2018, 80:219–241. [DOI] [PubMed] [Google Scholar]
- 26.Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA et al. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci U S A 2009, 106:7281–7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marinelli M, Rudick CN, Hu XT, White FJ: Excitability of dopamine neurons: modulation and physiological consequences. CNS Neurol Disord Drug Targets 2006, 5:79–97. [DOI] [PubMed] [Google Scholar]
- 28.White FJ, Kalivas PW: Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend 1998, 51:141–153. [DOI] [PubMed] [Google Scholar]
- 29.•.Lalive AL, Munoz MB, Bellone C, Slesinger PA, Luscher C, Tan KR: Firing modes of dopamine neurons drive bidirectional GIRK channel plasticity. J Neurosci 2014, 34:5107–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using brain slice electrophysiology to induce tonic or burst firing of dopamine neurons by current injection, Lalive and colleagues demonstrate that burst firing enhances while tonic firing depresses GABAB-R-GIRK conductance. This mechanism is unexplored in the context of cocaine exposure.
- 30.Huang YZ, Edwards MJ, Rounis E, Bhatia KP, Rothwell JC: Theta burst stimulation of the human motor cortex. Neuron 2005, 45:201–206. [DOI] [PubMed] [Google Scholar]
- 31.Padgett CL, Lalive AL, Tan KR, Terunuma M, Munoz MB, Pangalos MN, Martinez-Hernandez J, Watanabe M, Moss SJ, Lujan R et al. Methamphetamine-evoked depression of GABA (b) receptor signaling in GABA neurons of the VTA. Neuron 2012, 73:978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arora D, Hearing M, Haluk DM, Mirkovic K, Fajardo-Serrano A, Wessendorf MW, Watanabe M, Lujan R, Wickman K: Acute cocaine exposure weakens GABA(B) receptor-dependent G-protein-gated inwardly rectifying K+ signaling in dopamine neurons of the ventral tegmental area. J Neurosci 2011, 31:12251–12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hage TA, Khaliq ZM: Tonic firing rate controls dendritic Ca2+ signaling and synaptic gain in substantia nigra dopamine neurons. J Neurosci 2015, 35:5823–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Dore J, Chen X: Calcium influx through L-type channels generates protein kinase M to induce burst firing of dopamine cells in the rat ventral tegmental area. J Biol Chem 2007, 282:8594–8603. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y, Harding M, Pittman A, Dore J, Striessnig J, Rajadhyaksha A, Chen X: Ca(v)1.2 and Ca(v)1.3 L-type calcium channels regulate dopaminergic firing activity in the mouse ventral tegmental area. J Neurophysiol 2014, 112:1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez-Rivera A, Hao J, Tropea TF, Giordano TP, Kosovsky M, Rice RC, Lee A, Huganir RL, Striessnig J, Addy nA et al. Enhancing VTA Cav1.3 L-type Ca(2+) channel activity promotes cocaine and mood-related behaviors via overlapping AMPA receptor mechanisms in the nucleus accumbens. Mol Psychiatry 2017, 22:1735–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Licata SC, Freeman AY, Pierce-Bancroft AF, Pierce RC: Repeated stimulation of L-type calcium channels in the rat ventral tegmental area mimics the initiation of behavioral sensitization to cocaine. Psychopharmacology (Berl) 2000, 152:110–118. [DOI] [PubMed] [Google Scholar]
- 38.Degoulet M, Stelly CE, Ahn KC, Morikawa H: L-type Ca(2)(+) channel blockade with antihypertensive medication disrupts VTA synaptic plasticity and drug-associated contextual memory. Mol Psychiatry 2016, 21:394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Addy NA, Nunes EJ, Hughley SM, Small KM, Baracz SJ, Haight JL, Rajadhyaksha AM: The L-type calcium channel blocker, isradipine, attenuates cue-induced cocaine-seeking by enhancing dopaminergic activity in the ventral tegmental area to nucleus accumbens pathway. Neuropsychopharmacology 2018:1–12 10.1038/s41386-018-0080-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dragicevic E, Poetschke C, Duda J, Schlaudraff F, Lammel S, Schiemann J, Fauler M, Hetzel A, Watanabe M, Lujan R et al. Cav1.3 channels control D2-autoreceptor responses via NCS-1 in substantia nigra dopamine neurons. Brain 2014, 137:2287–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gantz SC, Robinson BG, Buck DC, Bunzow JR, Neve RL, Williams JT, Neve KA: Distinct regulation of dopamine D2S and D2L autoreceptor signaling by calcium. eLife 2015, 4 10.7554/eLife.09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.•.Kramer PF, Williams JT: Calcium release from stores inhibits GIRK. Cell Rep 2016, 17:3246–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors establish that intracellular calcium directly inhibits GIRK conductance, whether activated by GABAB-R or independent of receptor activation. This mechanism is unexplored in the context of cocaine exposure.
- 43.Parsons LH, Smith AD, Justice JB Jr: Basal extracellular dopamine is decreased in the rat nucleus accumbens during abstinence from chronic cocaine. Synapse 1991, 9:60–65. [DOI] [PubMed] [Google Scholar]
- 44.Weiss F, Markou A, Lorang MT, Koob GF: Basal extracellular dopamine levels in the nucleus accumbens are decreased during cocaine withdrawal after unlimited-access self-administration. Brain Res 1992, 593:314–318. [DOI] [PubMed] [Google Scholar]
- 45.Willuhn I, Burgeno LM, Groblewski PA, Phillips PEM: Excessive cocaine use results from decreased phasic dopamine signaling in the striatum. Nat Neurosci 2014, 17:704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karcum F, Suddath RL, Wyatt RJ: Chronic cocaine and rat brain catecholamines: long-term reduction in hypothalamic and frontal cortex dopamine metabolism. Eur J Pharmacol 1990, 186:1–8. [DOI] [PubMed] [Google Scholar]
- 47.Ashok AH, Mizuno Y, Volkow ND, Howes OD: Association of stimulant use with dopaminergic alterations in users of cocaine, amphetamine, or methamphetamine: a systematic review and meta-analysis. JAMA Psychiatry 2017, 74:511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dackis CA, Gold MS: New concepts in cocaine addiction: the dopamine depletion hypothesis. Neurosci Biobehav Rev 1985, 9:469–477. [DOI] [PubMed] [Google Scholar]
- 49.Kourrich S, Hayashi T, Chuang JY, Tsai SY, Su TP, Bonci A: Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell 2013, 152:236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.•.Fulton S, Thibault D, Mendez JA, Lahaie N, Tirotta E, Borrelli E, Bouvier M, Tempel BL, Trudeau LE: Contribution of Kv1.2 voltage-gated potassium channel to D2 autoreceptor regulation of axonal dopamine overflow. J Biol Chem 2011, 286:9360–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that axonal D2-R activation increases Kv1.2 conductance and this mechanism contributes to D2-R autoinhibition of DA release. This mechanism is unexplored in the context of cocaine exposure.
- 51.Wanat MJ, Willuhn I, Clark JJ, Phillips PE: Phasic dopamine release in appetitive behaviors and drug addiction. Curr Drug Abuse Rev 2009, 2:195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phillips PEM, Stuber GD, Heien MLAV, Wightman RM, Carelli RM: Subsecond dopamine release promotes cocaine seeking. Nature 2003, 422:614. [DOI] [PubMed] [Google Scholar]
- 53.Antinori S, Fattore L, Saba P, Fratta W, Gessa GL, Devoto P: Levodopa prevents the reinstatement of cocaine self-administration in rats via potentiation of dopamine release in the medial prefrontal cortex. Addict Biol 2018, 23:556–568. [DOI] [PubMed] [Google Scholar]
- 54.Czoty PW, Stoops WW, Rush CR: Evaluation of the “pipeline” for development of medications for cocaine use disorder: a review of translational preclinical, human laboratory, and clinical trial research. Pharmacol Rev 2016, 68:533–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.••.Terraneo A, Leggio L, Saladini M, Ermani M, Bonci A, Gallimberti L: Transcranial magnetic stimulation of dorsolateral prefrontal cortex reduces cocaine use: a pilot study. Eur Neuropsychopharmacol 2016, 26:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is the first clinical trial to show that TMS of the dorsolateral PFC reduces cocaine use. The study was based on intrinsic excitability findings of Ref. [62], providing promise for targeting plasticity for the effective treatment of cocaine addiction.
- 56.Pettorruso M, Spagnolo PA, Leggio L, Janiri L, Di Giannantonio M, Gallimberti L, Bonci A, Martinotti G: Repetitive transcranial magnetic stimulation of the left dorsolateral prefrontal cortex may improve symptoms of anhedonia in individuals with cocaine use disorder: a pilot study. Brain Stimul 2018, 11 (5):1105–1197. [DOI] [PubMed] [Google Scholar]
- 57.Dunlop K, Hanlon CA, Downar J: Noninvasive brain stimulation treatments for addiction and major depression. Ann N Y Acad Sci 2017, 1394:31–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chib VS, Yun K, Takahashi H, Shimojo S: Noninvasive remote activation of the ventral midbrain by transcranial direct current stimulation of prefrontal cortex. Transl Psychiatry 2013, 3:e268–e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cho SS, Strafella AP: rTMS of the left dorsolateral prefrontal cortex modulates dopamine release in the ipsilateral anterior cingulate cortex and orbitofrontal cortex. PLoS One 2009, 4:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murase S, Grenhoff J, Chouvet G, Gonon FG, Svensson TH: Prefrontal cortex regulates burst firing and transmitter release in rat mesolimbic dopamine neurons studied in vivo. Neurosci Lett 1993, 157:53–56. [DOI] [PubMed] [Google Scholar]
- 61.Salling MC, Martinez D: Brain stimulation in addiction. Neuropsychopharmacology 2016, 41:2798–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen BT, Yau HJ, Hatch C, Kusumoto-Yoshida I, Cho SL, Hopf FW, Bonci A: Rescuing cocaine-induced prefrontal cortex hypoactivity prevents compulsive cocaine seeking. Nature 2013, 496:359–362. [DOI] [PubMed] [Google Scholar]