

Background:
Alcohol-associated liver disease (ALD) is caused by chronic use of alcohol and ranges from hepatic steatosis to fibrosis and cirrhosis. Bile acids are physiological detergents that also regulate hepatic glucose and lipid homeostasis by binding to several receptors. One such receptor, Takeda G protein–coupled receptor 5 (TGR5), may represent a therapeutic target for ALD. Here, we used a chronic 10-day + binge ethanol-feeding model in mice to study the role of TGR5 in alcohol-induced liver injury.
Methods:
Female C57BL/6J wild-type mice and Tgr5 −/− mice were pair-fed Lieber-DeCarli liquid diet with ethanol (5% v/v) or isocaloric control diet for 10 days followed by a gavage of 5% ethanol or isocaloric maltose control, respectively, to represent a binge-drinking episode. Tissues were harvested 9 hours following the binge, and metabolic phenotypes were characterized through examination of liver, adipose, and brain mechanistic pathways.
Results:
Tgr5 −/− mice were protected from alcohol-induced accumulation of hepatic triglycerides. Interestingly, liver and serum levels of Fgf21 were significantly increased during ethanol feeding in Tgr5 −/− mice, as was phosphorylation of Stat3. Parallel to Fgf21 levels, increased leptin gene expression in white adipose tissue and increased leptin receptor in liver were detected in Tgr5 −/− mice fed ethanol diet. Adipocyte lipase gene expression was significantly increased in Tgr5 −/− mice regardless of diet, whereas adipose browning markers were also increased in ethanol-fed Tgr5 −/− mice, indicating potential for enhanced white adipose metabolism. Lastly, hypothalamic mRNA targets of leptin, involved in the regulation of food intake, were significantly increased in Tgr5 −/− mice fed ethanol diet.
Conclusions:
Tgr5 −/− mice are protected from ethanol-induced liver damage and lipid accumulation. Alterations in lipid uptake and Fgf21 signaling, and enhanced metabolic activity of white adipose tissue, may mediate these effects.

INTRODUCTION
Alcohol use disorder (AUD) is estimated to affect 10%–15% of people living in the US, and although globally AUD is more common in men compared with women, evidence indicates this gap may be narrowing.1 Alcohol-associated liver disease (ALD) is a progressive condition that includes hepatic fat accumulation, metabolic imbalance, and inflammation, leading to steatosis, hepatitis, cirrhosis, and fibrosis.2 Although the liver is the primary site of alcohol metabolism and pathogenesis, adipose tissue is increasingly recognized as a contributor to hepatic steatosis. Chronic exposure to alcohol affects hepatic bile acid and hormone production, alters nutrient absorption, and leads to dysregulated interactions between the liver, gut, and other organs.2,3
Takeda G protein–coupled receptor 5 (Tgr5) is a bile acid receptor expressed in the gut, skeletal muscle, adipose tissue, brain, and elsewhere. Tgr5 is not expressed in hepatocytes; however, nonparenchymal cells of the liver, including Kupffer cells, sinusoidal endothelial cells, and cholangiocytes, express Tgr5.4 Tgr5 was the first discovered G protein–coupled receptor to bind bile acids,5 and physiologically, it exerts wide-ranging actions through intracellular cAMP-activated pathways. In the intestine, stimulation of Tgr5 induces glucagon-like peptide 1 secretion to improve glycemic control. In addition, Tgr5 stimulates gallbladder filling and increases thermogenesis in brown adipose tissue to maintain energy homeostasis.6 Tgr5 also mediates anti-inflammatory signals through suppression of Nf-κB signaling.7
Fibroblast growth factor 21 (Fgf21) is a hormone released by the liver under metabolic stress.8 Fgf21 induces browning of white adipose tissue (WAT), thus maintaining energy homeostasis by increasing metabolic activity.9 Several studies in mice have highlighted the positive outcomes of Fgf21 for fatty liver and obesity through enhancement of energy expenditure and futile cycling in adipocytes.10,11 Fgf21 knockout mice exposed to a chronic + binge model of alcohol consumption had reduced WAT lipolysis and were protected against alcohol-induced steatosis.12 Another important recent study demonstrated that Fgf21 and a human Fgf21 analog reduced alcohol preference in nonhuman primates,13 indicating a potentially therapeutic role of Fgf21 in AUD and ALD.
Our laboratory previously reported alterations in Fgf21 in male Tgr5 −/− mice.14 Here, we exposed female and male Tgr5 −/− mice to a chronic + binge model of ethanol feeding to discern the effects of Fgf21 as a mediator of liver-white adipose signaling in the development of alcohol-associated steatosis.
METHODS
Mice
Four- to 5-month-old female and male C57BL/6J wild-type and Tgr5 −/− mice (originally from Merck)15 (n = 5–7 per group) were bred and maintained in a temperature-controlled facility with a 12-hour light and 12-hour dark cycle at Northeast Ohio Medical University. The effects of alcohol on the liver-WAT axis were studied using the NIAAA chronic + binge model.16 Mice were acclimated to liquid Lieber-DeCarli control diet (BioServ #F1259; Flemington, NJ) ad libitum for 5 days. Mice were then divided into control-fed or ethanol-fed groups; ethanol-fed mice were allowed free access to ethanol-containing Leiber-Decarli diet (5% ethanol; BioServ #F1258) for 10 days, whereas control mice were pair-fed with an identical volume of the isocaloric control diet (0% ethanol). On the 11th day, ethanol-fed and control-fed mice were gavaged in the morning with a single isocaloric dose of ethanol (5 g/kg) or maltose dextrin (9 g/kg), respectively, to simulate a binge-drinking episode. Mice were euthanized 9 hour later (from ~3–5 pm), and tissues were frozen at −80 °C until analysis. All experiments were approved by the Institutional Animal Care and Use Committee of Northeast Ohio Medical University.
Real-time qPCR
RNA was isolated from tissues using Trizol. cDNA was synthesized using 2 µg of RNA, and TaqMan primers (Thermo Fisher, Waltham, MA) were used to determine relative mRNA expression. ΔΔCt values were calculated against the reference genes Gapdh and Hprt.
Western blotting
Total protein was isolated from liver using T-Per (Thermo Fisher). Microsomal protein was isolated from ~50 mg of liver tissue lysed in a Dounce homogenizer with 2-mL microsome isolation buffer. Samples were centrifuged to remove cellular debris, and the supernatant was centrifuged again at 35,000 rpm for 1 hour. Pellets were reconstituted in 200 µL of microsome isolation buffer to obtain microsomal proteins. Nuclear protein was isolated using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher #78833). Briefly, ~50 mg of liver was homogenized in CER-I buffer supplemented with HALT protease and phosphatase inhibitors. CER-II buffer was added, and the solution was stored as cytoplasmic protein. The remaining pellet was resuspended in NER buffer supplemented with HALT, and this solution was used for the analysis of nuclear proteins. Western blotting was performed by loading 20 µg of total, microsomal, or nuclear protein in 10% TGX gels (BioRad, Hercules, CA). Blots were incubated with antibodies against Adh (ab108203), Aldh2 (ab227021), Cyp2e1 (ab28146), p-Stat3(Tyr 705) (9145S), Stat3 (12640S), Pparγ (2443S), Lipe (ab63492), and Cd36 (ab64014). Loading controls were calnexin (microsomes, 4499s), histone (nuclear, 4499s), and gapdh (total protein, 5174s). Blots were rinsed, incubated in HRP-conjugated secondary antibody (BioRad 1721019), and were then imaged on a GBox Mini 6 Gel Documentation System (Syngene, Frederick, MD). Blots were quantified using ImageJ.
Multiplex assay
A mouse cytokine/chemokine magnetic bead panel (EMD Millipore, Burlington, MA; cat# MCYTOMAG) was used to analyze the cytokines present in serum samples of wild-type (WT) and Tgr5 −/− control or ethanol-fed mice. Premixed beads, serum samples, and standards were loaded in a 96-well plate and were incubated for 2 hour. The plate was washed, and samples were incubated with biotinylated detection antibody. Streptavidin-PE conjugate was added, and each individual microsphere was identified using an xMAP bead-based technology in a Luminex MAGPIX analyzer (Luminex, Austin, TX).
Biochemical assays
Approximately 100 mg of liver tissue or feces was homogenized with 1 mL of 5% NP-40 in ddH2O using a Dounce homogenizer. Samples were heated to 90 °C in a water bath for 2–5 minutes. The samples were cooled to room temperature, and this process was repeated 2–3 times to ensure the dissolution of lipids. The samples were centrifuged at 13,000 rpm for 2 minutes, and the supernatant was collected and used for lipid quantification. Lipids were quantified in liver, feces, and serum using commercially available kits (cholesterol and triglycerides: Thermo Fisher #TR13421, #TR22421; free fatty acids: Abcam #65341).
Bile acid analysis
Bile acids were extracted from liver, whole intestine, gallbladder, and feces in ice-cold acetonitrile: methanol (1:1 v/v ratio). The samples were sonicated for an hour and centrifuged, and the supernatant was collected; the serum was quantified directly. Bile acid concentration was measured using a kit (Diazyme, Poway, CA). Total bile acid pool size was determined by adding bile acid concentrations in liver, intestine, and gallbladder.
Histology
Paraffin-embedded liver sections (8 µm) were probed for the macrophage marker F4/80. Sections were blocked in normal goat serum for 1 hour and were then incubated in primary antibody overnight (#70076) at 4 °C. Sections were incubated in SignalStain Boost IHC reagent (#8114s) for 30 minutes at room temperature. DAB chromagen concentrate (#11725s) was applied for 90 seconds, and the reaction was halted with water. Sections were visualized using an Olympus IX73 inverted microscope, and images were captured using Olympus cellSens Dimension software. Positive staining was quantified using ImageJ.
Statistical analysis
Statistical significance between multiple groups was determined using a 2-way ANOVA, followed by a Tukey multiple comparison post hoc test where appropriate. Data were analyzed using Prism (GraphPad Software, San Diego, CA) and are presented as mean ±SEM, with p < 0.05 considered statistically significant.
RESULTS
Tgr5 −/− mice are protected from liver injury induced by chronic + binge ethanol consumption
The Tgr5 knockout phenotype was confirmed in liver and WAT in female and male mice (Figure 1A, Supplemental Figure 1A, http://links.lww.com/HC9/A274). In male mice, body weight, WAT weight, and liver weight were unchanged by chronic + binge ethanol feeding or between genotypes (Supplemental Figure 1B, C, http://links.lww.com/HC9/A274). However, female Tgr5 −/− mice subjected to ethanol feeding weighed more than WT mice at the end of the experiment. Likewise, female Tgr5 −/− mice also had significantly increased white adipose tissue compared with ethanol-fed WT mice (Figure 1C). Liver weight was not altered by genotype or diet in female mice (Figure 1D). Hepatic mRNA analysis of aldehyde dehydrogenase (Adh) was significantly reduced in female ethanol-fed WT and Tgr5 −/− mice (Figure 1E), whereas the enzyme aldehyde dehydrogenase (Aldh2) was significantly reduced only in ethanol-fed WT mice (Figure 1E). These genes were not altered in either male WT or Tgr5 −/− mice (Supplemental Figure 1D, http://links.lww.com/HC9/A274). Western blotting confirmed Adh was significantly reduced by ethanol in female mice, whereas Aldh2 was significantly reduced in Tgr5 −/− mice (Figure 1F). Adh was unchanged in male mice, and Aldh2 was reduced in Tgr5 −/− mice and induced by ethanol diet (Supplemental Figure 1E, http://links.lww.com/HC9/A274). Liver Cyp2e1 was induced in female ethanol-fed WT and Tgr5 −/− mice, whereas in male mice, Cyp2e1 was only significantly induced in WT mice and tended to increase in Tgr5 −/− mice (Figure 1F, Supplemental Figure 1F, http://links.lww.com/HC9/A274). Lastly, serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST), hallmarks of liver injury, were significantly induced only in female WT mice, whereas Tgr5 −/− females were protected (Figure 1G). In male mice, serum ALT was induced in WT mice only and serum AST was unchanged (Supplemental Figure 1G, http://links.lww.com/HC9/A274). Overall, these data confirm increased susceptibility to ethanol-induced liver injury in female mice and suggest that female mice lacking Tgr5 may be protected against toxic ethanol intermediates and alcohol-induced liver damage.
FIGURE 1.
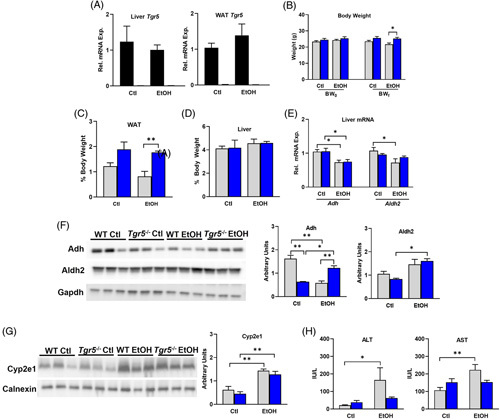
Tgr5 −/− mice are protected from liver injury induced by chronic + binge ethanol consumption. (A) Liver and WAT mRNA expression of Tgr5. (B) Body weight before (BW0) and after (BWf) administering the NIAAA chronic + binge ethanol model. (C) WAT weight. (D) Liver weight. (E) Hepatic mRNA expression of alcohol dehydrogenase (Adh) and aldehyde dehydrogenase (Aldh2). (F) Hepatic expression of Adh and Aldh2 protein. (G) Hepatic expression of Cyp2e1 protein. (H) Serum ALT and AST. *p < 0.05, **p < 0.01, ***p < 0.001. n = 5–6. Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; Ctl, control diet + maltose binge; EtOH, ethanol diet + ethanol binge; WAT, white adipose tissue.
Ethanol-induced alterations in hepatic lipid and cholesterol metabolism in Tgr5 −/− mice
Ethanol consumption induces hepatic lipogenesis and fatty liver disease.17 Here, hepatic cholesterol was unchanged in female mice, though ethanol consumption significantly increased hepatic triglycerides and free fatty acids only in WT mice (Figure 2A). Serum cholesterol and triglycerides were not altered, though serum free fatty acids were induced by ethanol consumption in WT mice only (Figure 2B). Fecal excretion of cholesterol tended to increase in Tgr5 −/− mice, fecal triglycerides were not affected, and excretion of free fatty acid in feces was significantly reduced in WT fed ethanol diet (Figure 2C). In male mice fed chronic + binge ethanol, hepatic cholesterol and triglycerides were unchanged by ethanol or genotype, as were serum cholesterol and triglycerides (Supplemental Figure 2, http://links.lww.com/HC9/A274). Hepatic expression of genes involved in lipogenesis was investigated. Sterol regulatory element binding protein 1 (Srebp1c) was significantly reduced only in WT mice fed ethanol, whereas fatty acid synthase (Fasn) and acetyl CoA carboxylase (Acc) were unchanged (Figure 2D). The expressions of fatty acid translocase (Cd36) and hepatic lipase (Lipc) were not altered in Tgr5 −/− mice; however, ethanol feeding significantly upregulated Cd36 and significantly reduced Lipc in WT mice (Figure 2D). Lastly, hormone-sensitive lipase (Lipe) was downregulated in ethanol-fed mice in both genotypes (Figure 2D). Overall, these data suggest that Tgr5 −/− mice may be protected from alcohol-induced hepatic lipid accumulation.
FIGURE 2.
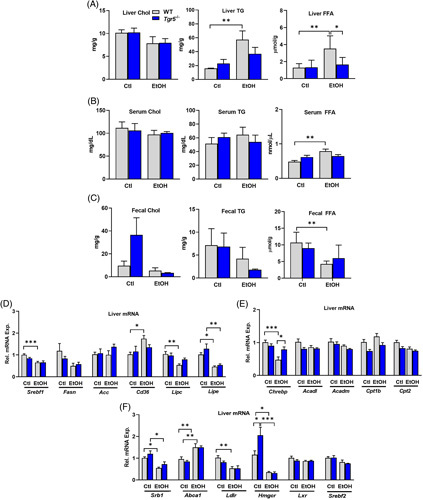
Ethanol-induced alterations in hepatic lipid and cholesterol metabolism in Tgr5 −/− mice. (A) Hepatic Chol, TG, and FFA content. (B) Serum Chol, TG, and FFA. (C) Fecal Chol, TG, and FFA. (D). Hepatic expression of genes involved in lipogenesis. (E) Hepatic expression of genes involved in fatty acid oxidation. (F) Hepatic expression of genes involved in cholesterol synthesis and transport. *p < 0.05, **p < 0.01, ***p < 0.001. n = 5–6. Abbreviations: Chol, cholesterol; Ctl, control diet + maltose binge; EtOH, ethanol diet + ethanol binge; FFA, free fatty acid; TG, triglyceride; WT, wild type.
Carbohydrate response element binding protein (Chrebp), a transcription factor typically upregulated during de novo lipogenesis,18 was significantly reduced by ethanol in WT mice but remained elevated in Tgr5 −/− mice (Figure 2E). Acyl-CoA dehydrogenase medium chain (Acadm) and long chain (Acadl) and carnitine palmitoyltransferase 1β (Cpt1β) and Cpt2 were not affected by ethanol (Figure 2E). In addition, hepatic expression of scavenger receptor B1 (Srb1), involved in HDL cholesterol uptake, was attenuated by ethanol diet, whereas ATP-binding cassette subfamily A member 1 (Abca1), involved in HDL efflux, was upregulated in both WT and Tgr5 −/− mice (Figure 2F). Low-density lipoprotein receptor (Ldlr) was reduced only in WT mice fed ethanol. Interestingly, the rate-limiting enzyme that directs cholesterol biosynthesis, HMG-CoA reductase (Hmgcr), was upregulated in Tgr5 −/− mice and was then suppressed after ethanol feeding. However, liver X receptor (Lxr) and Srebp2, a regulator of cholesterol and fatty acid homeostasis, were not altered (Figure 2F).
Ethanol suppresses bile acid synthesis and reduces the bile acid pool in Tgr5 −/− mice
Serum bile acids were significantly increased in Tgr5 −/− mice, and chronic + binge ethanol led to a significant suppression of serum bile acid content (Figure 3A). Liver bile acids remained unchanged regardless of diet or genotype (Figure 3B), whereas intestinal bile acids were significantly increased by ethanol in WT mice and were reduced by ethanol in Tgr5 −/− mice (Figure 3C). Gallbladder bile acid content and volume were reduced in the absence of Tgr5 (Figure 3D, E), consistent with the role of Tgr5 in gallbladder filling.19 Reduced gallbladder bile acids may increase serum bile acid content in Tgr5 −/− mice. Fecal bile acid excretion was not affected by chronic + binge ethanol, whereas the bile acid pool, defined as the sum of bile acids in the liver, gallbladder, and intestine, was significantly reduced in Tgr5 −/− mice fed ethanol (Figure 3G). These data indicate that chronic + binge ethanol consumption differentially affects bile acid homeostasis in Tgr5 −/− mice.
FIGURE 3.
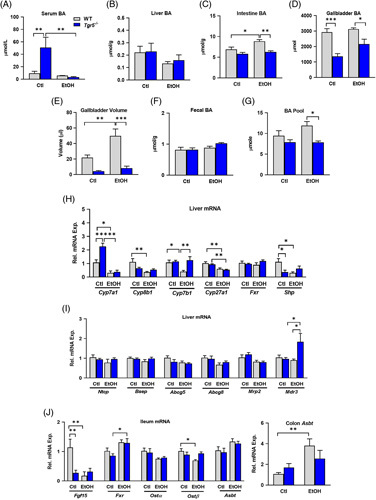
Ethanol suppresses bile acid synthesis and reduces the bile acid pool in Tgr5 −/− mice. Bile acids (BA) were quantified in (A) Serum, (B) Liver, (C) Intestine, and (D) Gallbladder. (E) Gallbladder volume. (F) Fecal bile acids. (G) Bile acid pool, summed from total liver, intestine, and gallbladder bile acids. (H and I) Hepatic expression of genes involved in bile acid synthesis and transport. (J) Ileum and colon expression of genes involved in bile acid regulation and transport. *p < 0.05, **p < 0.01, ***p < 0.001. n = 5–6. Abbreviations: Ctl, control diet + maltose binge; EtOH, ethanol diet + ethanol binge; WT, wild type.
The effects of chronic + binge ethanol on the hepatobiliary system in Tgr5 −/− mice were further investigated. mRNA expression of the rate-limiting enzyme for bile acid synthesis, cholesterol 7α-hydroxylase (Cyp7a1), was significantly upregulated in Tgr5 −/− mice and was suppressed by ethanol in both WT and Tgr5 −/− mice (Figure 3H). Ethanol diet suppressed the expression of the cholic acid synthesis gene sterol 12α-hydroxylase (Cyp8b1) in WT mice only, whereas the alternative pathway enzyme oxysterol 7α-hydroxylase (Cyp7b1) was suppressed in WT mice fed ethanol and induced in Tgr5 −/− mice fed ethanol. Lastly, sterol 27-hydroxylase (Cyp27a1) was suppressed by ethanol in both WT and Tgr5 −/− mice (Figure 3H). Hepatic expression of farnesoid X receptor (Fxr) was not altered, but small heterodimer partner (Shp), a negative regulator of Cyp7a1, was reduced in Tgr5 −/− mice, corresponding to enhanced Cyp7a1 gene expression (Figure 3H). Furthermore, hepatic mRNA expressions of the bile acid transporters sodium taurocholate cotransporting polypeptide (Ntcp) and bile salt export pump (Bsep) were not altered nor was expression of the cholesterol efflux transporters Abcg5 or Abcg8. Expression of multidrug resistance protein 3 (Mdr3), involved in phospholipid export to the biliary system, was significantly upregulated in Tgr5 −/− mice fed ethanol (Figure 3I). Thus, these data imply Tgr5 −/− mice may exhibit adaptation of the hepatobiliary system to protect against alcohol-induced liver damage.
Intestinal expression of Fgf15, another negative regulator of Cyp7a1, was suppressed by ethanol in both WT and Tgr5 −/− mice, also consistent with elevated Cyp7a1 expression. Conversely, intestinal Fxr, which controls ileal release of Fgf15, was increased with ethanol diet in Tgr5 −/− mice but remained unaffected in WT mice (Figure 3J). The expression of organic solute and steroid transporter (Ostα) was not altered, but Ostβ was significantly reduced in ethanol-fed WT mice (Figure 3J). Lastly, intestinal expression of apical sodium-dependent bile-acid transporter (Asbt) was significantly upregulated in colon of WT mice fed ethanol, consistent with increased intestinal bile acids and indicative of potentially upregulated extrahepatic circulation of bile acids.
Ethanol-induced inflammation in Tgr5 −/− mice
Hepatic gene expression of macrophage activation markers that relay inflammatory signals, such as inducible nitric oxide synthase (Nos2) and Il-6, was upregulated in the absence of Tgr5 but decreased with ethanol diet (Figure 4A), suggesting the possibility that ethanol could be immunosuppressive in the absence of Tgr5. Further, mRNA expression of markers of inflammation, such as cluster of differentiation 14 (Cd14), Il-1β, Tnfα, interferon γ (Ifnγ), and Il-12α, did not change with genotype or diet (Figure 4B). Similarly, gene expressions of anti-inflammatory markers such as arginase 1 (Arg1) and IL-10 were unchanged (Figure 4C). Serum samples collected 9 hour after ethanol binge were analyzed by multiplex ELISA. Serum levels of proinflammatory cytokines such as IL-1α, IL-13, IL-17, monocyte chemoattractant protein 1 (MCP-1), macrophage inflammatory protein 1B (MIP-1β), and TNFα remained unchanged with genotype or diet (Figure 4D). Liver sections were probed for F4/80, a marker of macrophage recruitment. F4/80 expression was suppressed by ethanol in both WT and Tgr5 −/− mice (Supplemental Figure 3, http://links.lww.com/HC9/A274). With only 20%–30% of total hepatic cells expressing Tgr5, the lack of Tgr5 in the liver may not result in a significant inflammatory phenotype after 10 days of ethanol feeding.
FIGURE 4.
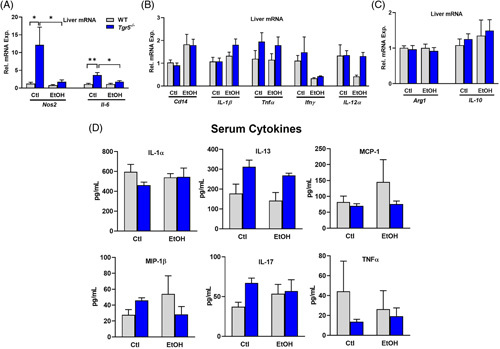
Ethanol-induced inflammation in Tgr5 −/− mice. (A and B) Hepatic expression of inflammatory genes. (C) Hepatic expression of inflammation resolution genes. (D) Serum cytokines quantified by multiplex ELISA. *p < 0.05, **p < 0.01, ***p < 0.001. n = 5–6. Abbreviations: Ctl, control diet + maltose binge; EtOH, ethanol diet + ethanol binge; MCP-1, monocyte chemoattractant protein 1.
Tgr5 −/− mice have increased ethanol-induced Fgf21
Fgf21 is a fasting-induced master regulator of carbohydrate and lipid metabolism10 and is known to protect the liver from ethanol-induced injury.8 Both liver Fgf21 mRNA and serum Fgf21 were significantly increased in ethanol-fed Tgr5 −/− mice compared with control diet and compared with WT mice fed ethanol diet (Figure 5A). Fgf21 expression in the liver is under the control of transcription factors such as peroxisome proliferator-activated receptor (Pparα) and signal transducer and activator of transcription 3 (Stat3),20 whereas adipocytes serve as a secondary source of local Fgf21 production likely under the control of Pparγ. Interestingly, hepatic mRNA expression of Pparγ was significantly enhanced in Tgr5 −/− mice fed ethanol diet; however, hepatic expressions of Pparα and its coactivator (Pgc1α), which mediates β-oxidation of fatty acids, were significantly reduced in both WT and Tgr5 −/− mice fed ethanol (Figure 5B). The increase in Pparγ was confirmed by western blot (Figure 5C), as was increased phosphorylation of Stat3 (Figure 5D). Altogether, these data suggest that Tgr5 −/− mice have enhanced ethanol-induced expression of Fgf21 that may be independent of Pparα.
FIGURE 5.
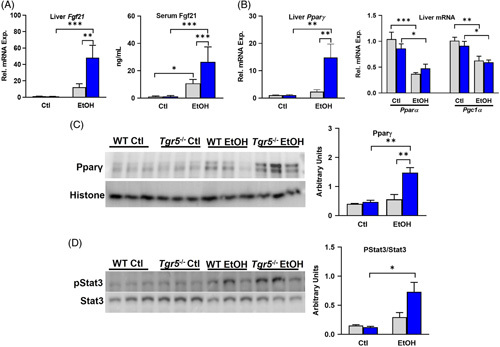
Tgr5 −/− mice have increased ethanol-induced Fgf21. (A) Hepatic expression of Fgf21 and serum Fgf21. (B). Hepatic mRNA expression of Ppars. (C) Hepatic Pparγ protein expression (n = 3). (D) Hepatic phosphorylation of Stat3 normalized to total Stat3 (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. n = 5–6. Abbreviations: Ctl, control diet + maltose binge; EtOH, ethanol diet + ethanol binge; WT, wild type.
Leptin signaling is altered in Tgr5 −/− mice fed ethanol
The adipokine leptin stimulates β-oxidation of fat and suppresses appetite through central pathways in the hypothalamus.21 Leptin levels increase proportionally with body weight, and patients with obesity may be hyperleptinemic and exhibit leptin resistance.22 In addition, leptin regulates Fgf21 expression, and leptin along with Fgf21 may act synergistically to restore glucose tolerance.23,24 Serum concentration and mRNA expression of leptin were increased in Tgr5 −/− mice, which was then reduced by ethanol (Figure 6A, B). Conversely, hepatic expression of the leptin receptor (Lepr) was significantly increased in Tgr5 −/− mice fed ethanol diet compared with control Tgr5 −/− mice and ethanol-fed WT mice (Figure 6C). Further analysis indicated both lipogenic genes like lipoprotein lipase (Lpl) and Cd36 and lipolytic genes such as adipose triglyceride lipase (Atgl), Lipe, and perilipin 1 (Plin1) were enhanced in Tgr5 −/− adipose tissue and remained unchanged by ethanol (Figure 6D). WAT Lipe and Cd36 protein levels were upregulated by ethanol feeding only in Tgr5 −/− mice (Supplemental Figure 4, http://links.lww.com/HC9/A274). In addition, Tgr5 −/− mice fed ethanol diet had increased expression of browning genes in white adipose, including uncoupling protein 1 (Ucp1) and cell death-inducing DFFA-like effector A (Cidea), whereas iodothyronine deiodinase 2 (Dio2) and PR domain zinc finger protein 16 (Prdm16) were not altered (Figure 6E).
FIGURE 6.

Leptin metabolism is altered in Tgr5 −/− mice fed ethanol. (A) Serum leptin. (B) mRNA expression of leptin in WAT. (C) mRNA expression of leptin receptor in liver. (D) Expression of lipogenic and lipolytic genes in WAT. (E) Expression of browning genes in WAT. (F) Expression of genes involved in glucose regulation in WAT. (G) Expression of Fgf21 and regulatory genes in WAT. *p < 0.05, **p < 0.01, ***p < 0.001. n = 5–6. Abbreviations: Ctl, control diet + maltose binge; EtOH, ethanol diet + ethanol binge; WAT, white adipose tissue; WT, wild type.
Furthermore, expression of insulin-dependent glucose transporter (Glut4) and insulin receptor (Insr) was enhanced in Tgr5 −/− mice regardless of diet. Similarly, CCAAT enhancer-binding protein α (Cebpα), which regulates adipocyte differentiation and Fgf21 production, was significantly enhanced in the absence of Tgr5 (Figure 6F). Lastly, Fgf21 expression in white adipose was not altered, though Pparα expression was significantly induced in Tgr5 −/− mice fed ethanol and Pparγ was significantly reduced. Adiponectin, an adipokine involved in maintaining and improving energy homeostasis, was unchanged (Figure 6G). These data indicate alcohol-fed Tgr5 −/− mice may be under the influence of metabolic futile cycles in white adipose that ultimately protect against the development of alcohol-associated fatty liver.
Hypothalamic leptin signaling is altered in Tgr5 −/− mice
Leptin exerts many of its effects within the arcuate nucleus of the hypothalamus. Hypothalamic expression of leptin receptor was enhanced during ethanol feeding in both WT and Tgr5 −/− mice, whereas expression of proopiomelanocortin (Pomc), an anorexigenic gene downstream of leptin signaling, tended to increase in the absence of Tgr5 and was significantly increased in ethanol-fed Tgr5 −/− mice compared with ethanol-fed WT mice. Neuropeptide Y (Npy), a leptin target gene that stimulates food intake, was significantly increased in ethanol-fed WT mice and significantly reduced in ethanol-fed Tgr5 −/− mice (Figure 7A).
FIGURE 7.
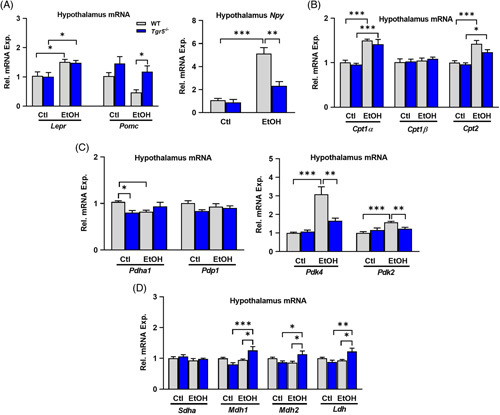
Hypothalamic leptin signaling is altered in Tgr5 −/− mice fed ethanol. (A) Hypothalamic expression of genes downstream of leptin signaling. (B) Hypothalamic expression of genes involved in fatty acid metabolism. (C and D) Hypothalamic expression of genes involved in glycolysis and the tricarboxylic acid cycle. *p < 0.05, **p < 0.01, ***p < 0.001. n = 5–6. Abbreviations: Ctl, control diet + maltose binge; EtOH, ethanol diet + ethanol binge; WT, wild type.
In addition, genes involved in lipid and glucose metabolism were analyzed. Ethanol diet increased gene expression of Cpt1α, Cpt1β, and Cpt2 in both WT and Tgr5 −/− mouse hypothalamus (Figure 7B). Pyruvate dehydrogenase (Pdh) links glycolysis to mitochondrial oxidative metabolism. Expression of Pdh was significantly reduced in ethanol-fed WT mice but remained unchanged in Tgr5 −/− mice. Pyruvate dehydrogenase phosphatase (Pdp1), which activates Pdh, was not changed, but the negative regulators of Pdh, pyruvate dehydrogenase kinases (Pdk4 and Pdk2), were significantly increased in ethanol-fed WT mice and were significantly reduced in Tgr5 −/− mice fed ethanol (Figure 7C). The increased Pdk4 expression observed in ethanol-fed WT mice did not correlate with altered expression of TCA intermediate enzymes such as succinate dehydrogenase (Sdha) and malate dehydrogenases (Mdh1 and Mdh2). Interestingly, Tgr5 −/− mice on ethanol diet had significantly elevated expression of Mdh1 and Mdh2 (Figure 7D). These data indicate that Tgr5 −/− mice may have underlying neuroendocrine alterations in appetite and propensity for food intake that are differentially affected by ethanol. Further investigation is required to analyze whether leptin signaling in the hypothalamus interferes with voluntary food intake in these mice.
DISCUSSION
ALD comprises a spectrum of liver diseases that begins with accumulation of fat depots. Steatosis occurs in more than 90% of patients who consume 60 g of alcohol per day25 and may be reversible with abstinence but can progress to hepatitis and cirrhosis if left untreated. Current treatment regimens, including corticosteroids, IL-1 receptor antagonism, and nutritional support, provide limited benefits. Obeticholic acid, a bile acid analog and FXR agonist, was in a phase 2 clinical trial to treat alcohol-associated hepatitis but was terminated because of drug-induced hepatotoxicity.26 Fgf21 may represent a viable treatment option, as ALD was more severe in Fgf21 knockout mice and exogenous Fgf21 reduced alcohol-induced steatosis.27 In addition, elevated Fgf21 is associated with the reduction of preference for both alcohol13 and sugar,28 and polymorphisms of the Fgf21 coreceptor β-Klotho are associated with alcohol consumption in humans.29 Previously, our laboratory demonstrated that Tgr5 −/− mice were protected against starvation-induced hepatic steatosis.14 Here, we investigated the role of Tgr5 in alcohol-induced steatosis and revealed that hepatokines and adipokines were significantly enhanced in Tgr5 −/− mice, posing a protective response to ethanol-induced pathology.
In addition to maintaining glucose and lipid homeostasis, the liver is a major site of alcohol metabolism. Alcohol consumption causes metabolic alterations that can occur as soon as 2 weeks in rodents.17 Leiber-DeCarli ethanol + binge diet-induced complications involve increased ALT and AST levels, reduced fatty acid oxidation, and enhanced lipogenesis. Similar to these observations, we demonstrated increased ALT and AST levels in ethanol-fed WT mice; however, these enzymes were not altered in Tgr5 −/− mice, nor did Tgr5 −/− mice exhibit increased lipid accumulation in the liver. De novo lipogenesis was not affected, but lipid uptake and metabolism may have been altered as indicated by increased hepatic TG and free fatty acid, increased Cd36, and decreased Lipc gene expression in WT mice.
Alcohol also impairs bile acid homeostasis, resulting in increased serum bile acids and reduced bile acid synthesis.30 Chronic + binge ethanol consumption reduced Cyp7a1 expression in mice,31 and mice lacking Cyp7a1 were more susceptible to liver injury.32 Here, serum bile acids and Cyp7a1 expression were significantly reduced in Tgr5 −/− mice fed ethanol. Interestingly, ethanol increased intestinal bile acid content and gallbladder volume in WT mice, whereas these remained reduced in Tgr5 −/− mice fed ethanol. Bile acid pool size was also significantly reduced in Tgr5 −/− mice after chronic + binge ethanol. Only a few studies have examined the role of Tgr5 in ALD. Tgr5 −/− mice fed Lieber-DeCarli ethanol diet without a binge had more steatosis and liver injury. This model resulted in increased levels of unconjugated bile acids in both WT and Tgr5 −/− mice and a significant reduction in secondary bile acids in Tgr5 −/− mice, though total levels of serum bile acids were unchanged by ethanol.33 This contrasts with another study in Tgr5 −/− mice, in which chronic + binge alcohol feeding resulted in increased serum bile acids in both WT and Tgr5 −/− mice, and lack of Tgr5 did not contribute to alcohol-induced changes in bile acid composition.34 Although both studies found that Tgr5 −/− mice had increased inflammation and liver damage after ethanol feeding, they highlight the need for investigation into other factors such as sex, route of ethanol administration, time of tissue collection, influence of the gut microbiome, and differential feeding methods, which may partially explain differences in results. Bile acids have both proinflammatory and anti-inflammatory effects, and secondary bile acids are associated with increased inflammation.35 Tgr5 mediates anti-inflammatory pathways through suppression of NF-κB7 but induces proinflammatory cytokines in Kupffer cells.36 Ethanol has proinflammatory or immunosuppressive effects depending on dose, route of administration, and sex,37 and the gut microbiome affects bile acid composition and hydrophobicity of the bile acid pool,38 both of which contribute to inflammatory responses. Therefore, further investigation into the role that bile acid-activated Tgr5 plays in the liver-gut axis within the context of ALD is warranted.
Several reports have shown that hepatic Fgf21 upregulation is associated with protection from alcohol-induced hepatic steatosis and could be a promising therapeutic target for ALD.8,27 Fgf21 is produced during starvation,14 ethanol consumption,8 and hepatic stress.39 Our study revealed that liver Fgf21 was highly increased in response to ethanol in Tgr5 −/− mice and this may involve Stat3. Research also indicates that leptin mediates its effects by phosphorylating Stat3, which increases Fgf21.23,40 These results correspond to a study demonstrating that Fgf21 increases futile cycling and energy expenditure, which could protect against hepatic steatosis.10 We also detected upregulation of white adipose browning gene expression in Tgr5 −/− mice. Both Tgr5 and ethanol play roles in adipocyte browning; however, the synergistic browning response to ethanol in Tgr5 −/− mice requires further study.
Leptin is a fat-derived hormone that exerts its effects on metabolic pathways in the liver and brain. First known as a satiety hormone, leptin is now recognized as a regulator of lipid, glucose, and immune function.41 Leptin levels are proportional to fat mass and exert action on hypothalamic neurons, which express NPY/agouti-related protein (AgRP) and POMC. The effects of alcohol on circulating leptin are controversial. External leptin administration was shown to reduce alcohol-induced fatty acid accumulation in mice, demonstrating a protective role of leptin in alcohol-associated liver injury.42 Another study found that chronic plus binge ethanol feeding to mice deficient in lipolysis resulted in increased circulating leptin and leptin receptor expression,43 though ethanol did not alter circulating leptin levels in control wild-type mice.43,44 In humans, binge drinking in males was associated with increased circulating leptin, whereas female binge drinkers exhibited increased leptin receptor expression.45 Circulating leptin was positively associated with steatosis and cirrhosis in patients who consumed alcohol,46 though another small study demonstrated reduced circulating leptin after acute ethanol consumption in healthy individuals,47 highlighting the importance of studying sex-specific responses to alcohol use and the use of multiple models of alcohol administration in animal studies. Although serum leptin was reduced by ethanol in Tgr5 -/- mice, leptin gene expression was significantly upregulated, as was expression of the leptin receptor gene in liver, indicating a possible compensatory response to amplify leptin signaling. This is supported by a previous study demonstrating that Fgf21 administration acutely reduced circulating leptin to enhance leptin sensitivity in mice.48 Leptin-induced stimulation of the hypothalamus inhibits the drive to consume food and increases energy expenditure to maintain body weight.49 Here, we used a pair-fed model in which mice received identical amounts of isocaloric diets, which may mask alterations in the behavioral drive for food or alcohol. The role of Tgr5 in the brain is largely unknown, and the arcuate nucleus represents only a small portion of the entire hypothalamus. Therefore, more site-specific examinations, such as immunohistochemistry or single-cell RNA-seq, would provide detailed insight into the interactions of leptin, Fgf21, and Tgr5.
Altogether, our study reveals unique cross-talk between the liver, WAT, and the hypothalamus in the absence of Tgr5 (Figure 8). Ethanol-mediated Fgf21 production by the liver is overcompensated in mice lacking Tgr5, which led to improved liver health by modulating the liver-adipose-brain axis. Increased leptin may contribute to increased Fgf21 and increased energy expenditure through futile cycling in WAT, which feeds-forward to improve metabolism. The impact of increased Fgf21 and alterations in leptin on food intake and nutrient preference in Tgr5 −/− mice is worthy of further investigation.
FIGURE 8.
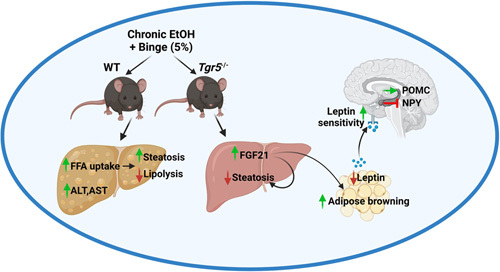
Ethanol-induced Fgf21 in Tgr5 −/− mice. Chronic + binge ethanol typically induces steatosis and liver damage. In Tgr5 −/− mice, ethanol increased Fgf21, which exerts metabolic effects in the liver, adipose tissue, and elsewhere and is implicated in mediating preference for ethanol and carbohydrate consumption. This may contribute to Fgf21-induced alterations in leptin sensitivity and downstream leptin signaling in Tgr5 −/− mice. Figure created with Biorender.com. Abbreviations: EtOH, ethanol diet + ethanol binge; FFA, free fatty acid; WT, wild type.
Supplementary Material
Acknowledgments
FUNDING INFORMATION
This study was supported by grants AA015951, DK058379, and CCF22706432 (subaward of 2P50AA024333-07).
CONFLICTS OF INTEREST
The authors have no conflicts to report.
Footnotes
Abbreviations: Abca1, ATP-binding cassette subfamily A member 1; AUD, alcohol use disorder; ALD, alcohol-associated liver disease; ADH, alcohol dehydrogenase; AgRP, agouti-related protein; ALDH, aldehyde dehydrogenase; Atgl, adipose triglyceride lipase; Arg1, arginase 1; BAT, brown adipose tissue; Bsep, bile salt export pump; Cd14, cluster of differentiation 14; Cebpα, CCAAT enhancer-binding protein α; Cidea, cell death-inducing DFFA-like effector A; Chrebp, carbohydrate response element binding protein; Ctl, control diet + maltose binge; CYP2E1, cytochrome P450 2E1; Dio2, deiodinase 2; EtOH, ethanol diet + ethanol binge; FFA, free fatty acid Fxr, farnesoid X receptor; Ifnγ, interferon γ; Insr, insulin receptor; Ldlr, low-density lipoprotein receptor; Lpl, lipoprotein lipase; Mdh, malate dehydrogenases; MCP-1, monocyte chemoattractant protein 1; MIP-1β, macrophage inflammatory protein 1B; Ntcp, sodium taurocholate cotransporting polypeptide; Npy, neuropeptide Y; Pdh, pyruvate dehydrogenase; Pdp1, pyruvate dehydrogenase phosphatase; PE, Phycoerythrin; Plin1, perilipin 1; Prdm16, PR domain zinc finger protein 16; Pomc, proopiomelanocortin; Shp, small heterodimer partner; Srb1, scavenger receptor B1; Stat3, signal transducer and activator of transcription 3; Sdha, succinate dehydrogenase; Tgr5, Takeda G protein–coupled receptor 5; Ucp1, uncoupling protein 1; WAT, white adipose tissue; WT, wild type.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal's website, www.hepcommjournal.com.
Contributor Information
Sabita Pokhrel, Email: pokhrel@neomed.edu.
Matthew Dilts, Email: mdilts@neomed.edu.
Zachary Stahl, Email: zstahl@neomed.edu.
Shannon Boehme, Email: sboehme@neomed.edu.
Gabrielle Frame, Email: gframe1@kent.edu.
John Y.L. Chiang, Email: jchiang@neomed.edu.
Jessica M. Ferrell, Email: jfrancl@neomed.edu.
REFERENCES
- 1. Carvalho AF, Heilig M, Perez A, Probst C, Rehm J. Alcohol use disorders. Lancet. 2019;394:781–92. [DOI] [PubMed] [Google Scholar]
- 2. Subramaniyan V, Chakravarthi S, Jegasothy R, Seng WY, Fuloria NK, Fuloria S, et al. Alcohol-associated liver disease: a review on its pathophysiology, diagnosis and drug therapy. Toxicol Reports. 2021;8:376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Poole LG, Dolin CE, Arteel GE. Organ–organ crosstalk and alcoholic liver disease. Biomolecules. 2017;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pathak P, Liu H, Boehme S, Xie C, Krausz KW, Gonzalez F, et al. Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. J Biol Chem. 2017;292:11055–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Duboc H, Taché Y, Hofmann AF. The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig Liver Dis. 2014;46:302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. [DOI] [PubMed] [Google Scholar]
- 7. Guo C, Qi H, Yu Y, Zhang Q, Su J, Yu D, et al. The G-protein-coupled bile acid receptor Gpbar1 (TGR5) inhibits gastric inflammation through antagonizing NF-κB signaling pathway. Front Pharmacol. 2015;6:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Desai BN, Singhal G, Watanabe M, Stevanovic D, Lundasen T, Fisher FM, et al. Fibroblast growth factor 21 (FGF21) is robustly induced by ethanol and has a protective role in ethanol associated liver injury. Mol Metab. 2017;6:1395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26:271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coskun T, Bina HA, Schneider MA, Dunbar JD, Hu CC, Chen Y, et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–27. [DOI] [PubMed] [Google Scholar]
- 11. Yang C, Lu W, Lin T, You P, Ye M, Huang Y, et al. Activation of liver FGF21 in hepatocarcinogenesis and during hepatic stress. BMC Gastroenterol. 2013;13:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao C, Liu Y, Xiao J, Liu L, Chen S, Mohammadi M, et al. FGF21 mediates alcohol-induced adipose tissue lipolysis by activation of systemic release of catecholamine in mice. J Lipid Res. 2015;56:1481–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flippo KH, Trammell SAJ, Gillum MP, Aklan I, Perez MB, Yavuz Y, et al. FGF21 suppresses alcohol consumption through an amygdalo-striatal circuit. Cell Metab. 2022;34:317–28.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Donepudi AC, Boehme S, Li F, Chiang JY. G-protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis in mice. Hepatology. 2017;65:813–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, Yang S, et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J. 2006;398:423–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc. 2013;8:627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeon S, Carr R. Alcohol effects on hepatic lipid metabolism. J Lipid Res. 2020;61:470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ortega-Prieto P, Postic C. Carbohydrate sensing through the transcription factor ChREBP. Front Genet. 2019;10:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li T, Holmstrom SR, Kir S, Umetani M, Schmidt DR, Kliewer SA, et al. The G protein-coupled bile acid receptor, TGR5, stimulates gallbladder filling. Mol Endocrinol. 2011;25:1066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–37. [DOI] [PubMed] [Google Scholar]
- 21. Dodd GT, Decherf S, Loh K, Simonds SE, Wiede F, Balland E, et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell. 2015;160:88–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Polyzos SA, Kountouras J, Mantzoros CS. Leptin in nonalcoholic fatty liver disease: a narrative review. Metabolism. 2015;64:60–78. [DOI] [PubMed] [Google Scholar]
- 23. Asrih M, Veyrat-Durebex C, Poher AL, Lyautey J, Rohner-Jeanrenaud F, Jornayvaz FR. Leptin as a potential regulator of FGF21. Cell Physiol Biochem. 2016;38:1218–25. [DOI] [PubMed] [Google Scholar]
- 24. Véniant MM, Sivits G, Helmering J, Komorowski R, Lee J, Fan W, et al. Pharmacologic effects of FGF21 are independent of the “browning” of white adipose tissue. Cell Metab. 2015;21:731–8. [DOI] [PubMed] [Google Scholar]
- 25. Singal AK, Bataller R, Ahn J, Kamath PS, Shah VH. ACG clinical guideline: alcoholic liver disease. Am J Gastroenterol. 2018;113:113–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tornai D, Szabo G. Emerging medical therapies for severe alcoholic hepatitis. Clin Mol Hepatol. 2020;26:686–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y, Zhao C, Xiao J, Liu L, Zhang M, Wang C, et al. Fibroblast growth factor 21 deficiency exacerbates chronic alcohol-induced hepatic steatosis and injury. Sci Rep. 2016;6:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Søberg S, Sandholt CH, Jespersen NZ, Toft U, Madsen AL, von Holstein-Rathlou S, et al. FGF21 is a sugar-induced hormone associated with sweet intake and preference in humans. Cell Metab. 2017;25:1045–53.e6. [DOI] [PubMed] [Google Scholar]
- 29. Schumann G, Liu C, O’Reilly P, Gao H, Song P, Xu B, et al. KLB is associated with alcohol drinking, and its gene product β-Klotho is necessary for FGF21 regulation of alcohol preference. Proc Natl Acad Sci U S A. 2016;113:14372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brandl K, Hartmann P, Jih LJ, Pizzo DP, Argemi J, Ventura-Cots M, et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J Hepatol. 2018;69:396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kong B, Zhang M, Huang M, Rizzolo D, Armstrong LE, Schumacher JD, et al. FXR deficiency alters bile acid pool composition and exacerbates chronic alcohol induced liver injury. Dig Liver Dis. 2019;51:570–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Donepudi AC, Ferrell JM, Boehme S, Choi HS, Chiang JYL. Deficiency of cholesterol 7alpha-hydroxylase in bile acid synthesis exacerbates alcohol-induced liver injury in mice. Hepatol Commun. 2018;2:99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Spatz M, Ciocan D, Merlen G, Rainteau D, Humbert L, Gomes-rochette N, et al. Bile acid-receptor TGR5 deficiency worsens liver injury in alcohol-fed mice by inducing intestinal microbiota dysbiosis. JHEP Reports. 2021;3:100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fan M, Wang Y, Jin L, Fang Z, Peng J, Tu J, et al. Bile acid–mediated activation of brown fat protects from alcohol-induced steatosis and liver injury in mice. Cell Mol Gastroenterol Hepatol. 2022;13:809–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Song P, Zhang Y, Klaassen CD. Dose-response of five bile acids on serum and liver bile acid concentrations and hepatotoxicty in mice. Toxicol Sci. 2011;123:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lou G, Ma X, Fu X, Meng Z, Zhang W, Wang YD, et al. GPBAR1/TGR5 mediates bile acid-induced cytokine expression in murine Kupffer cells. PLoS One. 2014;9:e93567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barr T, Helms C, Grant K, Messaoudi I. Opposing effects of alcohol on the immune system. Prog Neuro-Psychopharmacology Biol Psychiatry. 2016;65:242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Winston JA, Theriot CM. Diversification of host bile acids by members of the gut microbiota. Gut Microbes. 2020;11:158–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Habegger KM, Stemmer K, Cheng C, Muller TD, Heppner KM, Ottaway N, et al. Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes. 2013;62:1453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ, et al. Fibroblast growth factor-21 regulates PPARγ activity and the antidiabetic actions of thiazolidinediones. Cell. 2012;148:556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harris RBS. Is leptin the parabiotic “satiety” factor ? Past and present interpretations. Appetite. 2013;61:111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tan X, Sun X, Li Q, Zhao Y, Zhong W, Sun X, et al. Leptin deficiency contributes to the pathogenesis of alcoholic fatty liver disease in mice. Am J Pathol. 2012;181:1279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mathur M, Yeh Y-T, Arya RK, Jiang L, Pornour M, Chen W, et al. Adipose lipolysis is important for ethanol to induce fatty liver in the National Institute on Alcohol Abuse and Alcoholism murine model of chronic and binge ethanol feeding. Hepatology. 2022. doi: 10.1002/hep.32675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Degroat AR, Fleming CK, Dunlay SM, Hagood KL, Moorman JP, Peterson JM. The sex specific effect of alcohol consumption on circulating levels of CTRP3. PLoS One. 2018;13:e0207011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bouna-Pyrrou P, Muehle C, Kornhuber J, Weinland C, Lenz B. Body mass index and serum levels of soluble leptin receptor are sex-specifically related to alcohol binge drinking behavior. Psychoneuroendocrinology. 2021;127:105179. [DOI] [PubMed] [Google Scholar]
- 46. Naveau S, Perlemuter G, Chaillet M, Raynard B, Balian A, Beuzen F, et al. Serum leptin in patients with alcoholic liver disease. Alcohol Clin Exp Res. 2006;30:1422–8. [DOI] [PubMed] [Google Scholar]
- 47. Röjdmark S, Calissendorff J, Brismar K. Alcohol ingestion decreases both diurnal and nocturnal secretion of leptin in healthy individuals. Clin Endocrinol (Oxf). 2001;55:639–647. [DOI] [PubMed] [Google Scholar]
- 48. Claflin KE, Sullivan AI, Naber MC, Flippo KH, Morgan DA, Neff TJ, et al. Pharmacological FGF21 signals to glutamatergic neurons to enhance leptin action and lower body weight during obesity. Mol Metab. 2022;64:101564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martínez-Uña M, López-Mancheño Y, Diéguez C, Fernández-Rojo MA, Novelle MG. Unraveling the role of leptin in liver function and its relationship with liver diseases. Int J Mol Sci. 2020;21:1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.