

Abstract
Terpenoids constitute a structurally diverse class of secondary metabolites with wide applications in the pharmaceutical, fragrance and flavor industries. Desarmillaria tabescens CPCC 401429 is a basidiomycetous mushroom that could produce anti-tumor melleolides. To date, no studies have been conducted to thoroughly investigate the sesquiterpenes biosynthetic potential in Desarmillaria or related genus. This study aims to unravel the phylogeny, terpenome, and functional characterization of unique sesquiterpene biosynthetic genes of the strain CPCC 401429. Herein, we report the genome of the fungus containing 15,145 protein-encoding genes. MLST-based phylogeny and comparative genomic analyses shed light on the precise reclassification of D. tabescens suggesting that it belongs to the genus Desarmillaria. Gene ontology enrichment and pathway analyses uncover the hidden capacity for producing polyketides and terpenoids. Genome mining directed predictive framework reveals a diverse network of sesquiterpene synthases (STSs). Among twelve putative STSs encoded in the genome, six ones are belonging to the novel minor group: diverse Clade IV. In addition, RNA-sequencing based transcriptomic profiling revealed differentially expressed genes (DEGs) of the fungus CPCC 401429 in three different fermentation conditions, that of which enable us to identify noteworthy genes exemplified as STSs coding genes. Among the ten sesquiterpene biosynthetic DEGs, two genes including DtSTS9 and DtSTS10 were selected for functional characterization. Yeast cells expressing DtSTS9 and DtSTS10 could produce diverse sesquiterpene compounds, reinforced that STSs in the group Clade IV might be highly promiscuous producers. This highlights the potential of Desarmillaria in generating novel terpenoids. To summarize, our analyses will facilitate our understanding of phylogeny, STSs diversity and functional significance of Desarmillaria species. These results will encourage the scientific community for further research on uncharacterized STSs of Basidiomycota phylum, biological functions, and potential application of this vast source of secondary metabolites.
Keywords: Desarmillaria, genome mining, phylogeny, sesquiterpene synthases, RNA sequencing, functional identification
1. Introduction
Sesquiterpenes are synthesized from the prenyl precursors farnesyl diphophate (FPP) by a family of enzymes known as sesquiterpene synthases (STSs) [1]. Basidiomycete mushrooms are particularly adept at producing a wide range of structurally complexified sesquiterpenes [2]. Exploration of the medicinal and pharmacological potential of these underexplored mushroom species could be scientifically and pharmacologically attractive. Continuing secondary metabolites discovery targeting rare basidiomycetes, previous investigations have reported a number of active terpenoids or meroterpenoids, such as antroalbocin A [3], melleolides [4], illudin S [5], conosilane A [6], sterhirsutins [7], many of which exhibit some antibiotic and cytotoxic properties. The ever-increasing genomic information has revealed great potential for mining of putative STS homologues, indicating that the phylum Basidiomycota terpenoids and their synthases represent abundant but largely untapped natural resources [8,9]. Specially, each fungus of the phylum Basidiomycota has an average of 10–20 putative STSs encoding genes. However, only few sesquiterpenes are reportedly characterized [4,7]. Intriguingly, the basidiomycetous STSs have been shown to be readily expressed in well-established laboratory hosts, i.e., Escherichia coli [8,10] and Saccharomyces cerevisiae [11]. Moreover, as many basidiomycetes are relatively slow growing in laboratory conditions. Thus, all this makes heterologous expression an attractive approach for accessing the basidiomycete terpenoid chemistry.
Basidiomycetous Armillaria, belonging to the family Physalacriaceae family, are ubiquitous as destructive forest pathogens of forests, orchards, and plantations [12]. Species from this genus cause the Armillaria butt and root disease that weakens and often kills woody perennials [12,13]. Armillaria species were considered to be polymorphic, which impact on forest populations has both ecological and economic significance [13]. There are approximately more than 70 officially described species [14], and accurate species delimitation and pathogenicity levels assessment of Armillaria genus is pivotal for forestry conservation [15]. As saprotrophs, Armillaria species produce fleshy fruiting bodies that appear in large clumps around contaminated plants [16]. The fungus is beneficial in some ways, as it serves as a natural recycler of nutrients in wooded ecosystems, breaking down woody materials to increase soil health. Interestingly, Armillaria species are also considered as source of nutrition in Europe and Asia and some species are associated with traditional Chinese medicine for their pharmaceutical properties [17]. Armillaria spp. were demonstrated to yield a wealth of biologically active metabolites, exemplified as sterols, sesquiterpene aryl esters, and many other components exhibiting therapeutic or dietary values [17,18,19]. Thus, it is obligated to fully elucidate the molecular basis or mechanism of Armillaria at the genomic level.
The technological revolution in genome sequencing and bioinformatics have markedly increased the number of sequenced Basidiomycetous genomes. There are already nine Armillaria genomes that have been published to date, and those for A. mellea DSM3731, A. ostoyae C18/9, A. solidipes 28-4, A. borealis FPL87.14 v1.0, A. fuscipes CMW2740, A. cepistipes B5, A. altimontana 837-10, two A. gallica strains Ar21-2 and 012m were included. Analyses of these genomic and transcriptomic information have provided insights into evolution, genetics, developmental biology, plant-cell-wall-degrading enzymes (PCWDE) repertoire, and related pathogenicity factors [20,21,22,23,24]. Sipos and coworkers performed omics sequencing to provide genetic and regulatory insights into toolkits for wood-decaying, morphogenesis and complex multicellularity including rhizomorph and fruiting bodies development of the strain A. ostoyae C18 [23]. The taxonomy of Armillaria has raised controversial debate, recently, Armillaria was narrowed down to include only annulate species, and Desarmillaria was introduced to accommodate two exannulate mushroom-forming armillarioid species (D. ectypa and D. tabescens) [25,26]. The lineage to diverge is that composed of annulate mushroom-forming armillarioid species, in what A. mellea (Vahl: Fr.) P. Kumm is the type species [27]. Koch et al. (2017) conducted six-locus phylogenetic analysis among Armillaria and related affinities, demonstrated that Guyanagaster and Desarmillaria are close lineages [25]. On the delimitation and characterization of Armillaria species, the genome-based phylogenomics and comparative genomics were invaluable to understand the phylogeny of Armillaria and sister groups.
In this study, we report a 50.36 Mb draft genome sequence of D. tabescens CPCC 401429. MLST-based phylogeny and synteny analysis at the genomic level were investigated, which reinforced that the D. tabescens was supposed to belong to the genus of Desarmillaria instead of Armillaria as it used to. Genome profiling and transcript on three different fermentation conditions shed light on the secondary metabolism. Notably, guided by STSs phylogeny based predictive framework, a diverse network of twelve putative sesquiterpene biosynthetic genes were obtained and half of them were categorized into the new subfamily, i.e., diverse Clade IV. Intriguingly, two genes including DtSTS9 and DtSTS10 were codon optimized and cloned for heterologous expression in yeast host. Through gas chromatography-mass spectrometry (GC-MS) analysis, yeast cells expressing DtSTS9 and DtSTS10 produce diverse sesquiterpenes. This work provides the genomic basis to understand and investigate the new genus Desarmillaria. We exemplify the exploitation on uncharacterized STSs of basidiomycetes and help to broaden our knowledge of sesquiterpenes biosynthesis.
2. Materials and Methods
2.1. Strains and Cultivation Conditions
The fungus D. tabescens CPCC 401429 was cultured on potato dextrose medium (PDA; BD company) and incubated at 28 °C for two weeks [4]. To produce mycelium for genomic DNA (gDNA) extraction, the strain was grown in liquid MEP medium (malt extract broth 2.0%, BD, soy bean powder 0.2%). For RNA sequencing, the strain CPCC 401429 was cultivated in PDB broth (BD), YMEG broth (malt extract 1%, yeast extract 0.4%, glucose 0.4%, CaCO3 0.2%), and F1 broth (corn steep liquor 1.0%, soy bean powder 0.2%, glucose 2.0%, peptone 0.5%, ammonium sulfate 0.2%), respectively. The cultures were cultivated on a rotatory shaker (130 rpm) for 15-day.
Standard DNA cloning experiments were conducted using E. coli Trans1-T1. E. coli cultures carrying respective plasmid were grown in Luria-Bertani (LB) medium supplemented with ampicillin (100 μg/mL). S. cerevisiae BJ5464 was used as the host for heterologous expression [28]. Individual transformant of the yeast strain was grown in shaking cultures in 10 mL synthetic dextrose minimal medium containing 6.7 g/L of yeast nitrogen base without amino acids, 20 g/L of dextrose, 1.3 g/L of amino acid dropout powder. For production of the sesquiterpenes, a total of 200 mL YPD broth (BD) was inoculated with 5 mL of the starter culture and incubated for 96 h at 30 °C under shaking conditions (200 rpm). The strains, plasmids, primers, and genetically engineered cells are listed in Table 1.
Table 1.
Strains, primers, genetically engineered yeast in the study.
| Strains | Description | Source |
|---|---|---|
| D. tabescens CPCC 401429 | Melleolides producing strain | [4] |
| E. coli Trans T1 | F-φ80 lac ZΔM15 Δ(lacZYA-arg F) U169 endA1 recA1 hsdR17(rk-,mk+) supE44λ- thi -1 gyrA96 relA1 phoA | Transgen |
| S. cerevisiae BJ5464 | (MATα ura3-52 his3-Δ200 leu2- Δ1 trp1 pep4::HIS3 prb1 Δ1.6R can1 GAL | [29] |
| BJ-CK | S. cerevisiae BJ 5464, carrying plasmid YET | This study |
| BJ-DtSTS9 | S. cerevisiae BJ 5464, carrying plasmid YET-DtSTS9 | This study |
| BJ-DtSTS10 | S. cerevisiae BJ 5464, carrying plasmid YET-DtSTS10 | This study |
| Plasmids | Description | Source |
| YET | tryptophan auxotrophic expressing plasmid of yeast | [29] |
| YET-DtSTS9 | YET expressing plasmid with gene DtSTS9 | This study |
| YET-DtSTS10 | YET expressing plasmid with gene DtSTS10 | This study |
| Primers | Sequence | Size (bp) |
| DtSTS9-F | atacaatcaactatcaactattaactatatcgtaataccatatgacactatctacagca | 963 bp |
| DtSTS9-R | cttgataatggaaactataaatcgtgaaggcatgtttaaacttataatcccagctcgct | |
| DtSTS10-F | atacaatcaactatcaactattaactatatcgtaataccatatgactttatctacttcg | 1005 bp |
| DtSTS10-R | cttgataatggaaactataaatcgtgaaggcatgtttaaacctatatgccaagctcgct |
2.2. Sequencing, De Novo Assembly, and Bioinformatic Tools
Genomic DNA from Desarmillaria was isolated using HP Fungal DNA Kit (Omega, Guangzhou, China) according to the manufacturer’s instructions. Further purification was conducted using RNase digestion, chloroform extraction, and isopropanol precipitation [29]. The high molecular weight gDNA was washed twice with 75% ethanol, and dissolved in 200 μL RNase-free water. Integrity and amount of the extracted gDNA were detected by 1% agarose gel electrophoresis and using the NanoDrop ND-2000 Spectrophotometer (Thermo Fisher, Waltham, MA, USA). Shotgun sequencing was performed in the laboratory of at Shanghai Majorbio Bio-pharm Technology Co. Ltd. (Shanghai, China) using Illumina Hiseq 2000 platform. The PE150 (pair-end) libraries with a mean insert size 400 bp were subjected to PacBio sequencing. Adapter sequences were trimmed and short reads were formatted using Trimmomatic v. 0.36 [30] with minimum quality of 19 and minimum length of 30 bp. Quality was assessed using SMRT Analysis software [31].
The obtained sequence reads were assembled into contigs and scaffolds using SOAPdenovo v. 2.04 [32] and Canu v. 1.7 [33]. K-mer values were automatically selected based on read length and data type. The final scaffold set was verified for miss-assemblies using the BUSCO v3.0 and corrected if necessary [34]. Gene annotations of the coding sequences were formatted to GenBank database for BLASTP alignment or obtained via AUGUSTUS program [35] and verified on the basis of homologous proteins in the NCBI database. The gene ontology (GO) functional annotation was performed using Blast2go [36]. In parallel, protein annotations were also performed using KEGG (Kyoto Encyclopedia of Genes and Genomes), Pfam [37], and Swiss-prot databases [38].
2.3. RNA Purification, Library Construction, and RNA-Sequencing
The total RNA was extracted from PDB broth, YMEG broth, and F1 broth using PureLinkTM RNA Mini Kit (Invitrogen, USA) and gDNA was removed using DNaseI (Takara). The RNA quality and concentration measurements were performed using 2100 Bioanalyzer (Agilent) and quantified using the NanoDrop ND-2000, and only high-quality purified RNA was processed for construction sequencing library (OD260/280 = 1.8–2.2, OD260/230 ≥ 2.0, RIN ≥ 8.0, 28S:18S ≥ 1.0, >1 μg). RNA purification, reverse transcription, library construction and sequencing were performed at Shanghai Majorbio Bio-pharm Biotechnology Co., Ltd. (Shanghai, China) based on the manufacturer’s instructions (Illumina). Double-stranded cDNA libraries construction were carried out using the TruSeqTM RNA sample preparation Kit using 1.0 μg of total RNA (Illumina). Libraries were size selected for cDNA target fragments of 300 bp on 2% Low Range Ultra Agarose followed by PCR amplified using Phusion DNA polymerase (NEB) for 15 PCR cycles. Sequencing was conducted on Illumina NovaSeq 6000 sequencer instrument (2 × 150 bp read length).
2.4. Quality Control, Read Mapping, and Transcriptome Analysis
The raw paired end reads were trimmed and quality controlled by FASTQ preprocessor with default parameters [39]. Then clean reads were separately aligned to reference genome with orientation mode using HISAT2 software [40]. The mapped reads of each sample were assembled by StringTie in a reference-based strategy [41]. To identify differential expression genes (DEGs) between two different samples, the expression level of each gene was calculated according to the transcripts per million reads (TPM) approach. RSEM was used to quantify gene abundances [42]. Functional-enrichment analysis including GO, KEGG were performed to identify which DEGs were significantly enriched in GO terms and metabolic pathways at P-adjust ≤ 0.05 compared with the whole-transcriptome background. GO functional enrichment and KEGG pathway analysis were performed by Goatools and KOBAS [43,44].
2.5. Multi-Locus Sequence Typing (MLST)-Based Phylogenetic Analysis
To determine the phylogeny and identity of the strain D. tabescens CPCC 401429, we compiled a dataset composed of sequence data from twenty-five sequenced basidiomycetes including eight Armillaria spp. and sixteen Agaricales encompassing white and brown rot wood decomposers and ectomycorrhizal fungi, species of Heterobasidion irregulare TC 32-1 from Russulales served as outgoup [23]. Eleven loci including 18S-ITS-28S region, elongation factor 1-α (EF1α), RNA polymerase II (RPB2), actin-1 (ACT1), glyceraldehyde-3-phosphate dehydrogenase (GPD), beta-tubulin (TUB), plasma membrane H+-ATPase (ATP), glucose-6-phosphate isomerase (PGI), 60S ribosomal protein L2 (RPL), 60S ribosomal protein L18 (RPK), and polyubiquitin (UBQ) coding genes were performed, to delimit Armillaria systematic relationship through phylogenetic analysis [25,45]. The sequences used are compiled in Supplementary file II.
Sequences alignment were conducted using MAFFT v7.471 with FFT-NS-2 algorithm [46], followed by MEGA 7 to refine the alignment done manually [47]. The introns of the housekeeping genes were excluded. ModelFinder was used to predict the best-fitted substitution model according to Akaike Information Criterion (AIC) [48]. The model VT was selected for Bayesian inference (BI) analyses. Bayesian trees were constructed with MrBayes v3.2.6 under the GTR+G+I model [49]. Two independent runs were conducted for 10,000,000 generations with sampling every 100 generations using Markov Chain Monte Carlo (MCMC) algorithm. The first 25% of the samples were discarded as burn-in fraction. The Bayesian tree generated was visualized in FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/, accessed on 25 November 2018) and edited using Adobe Illustrator software.
2.6. Bioinformatic Annotation for STSs and Phylogenetic Tree Construction
The genome of D. tabescens CPCC 401429 was further bioinformatically analyzed using antiSMASH database. All the predicted amino acid sequences of protein-coding genes present in the genome of the strain CPCC 401429 have been searched for homologues by BLASTP tool in NCBI database. The predicted STSs genes used for the functional analysis were annotated and verified for a coherent exon/intron structure. In addition, several characterized fungal STSs from Clitopilus pseudopinsitus, Omphalotus olearius, Agrocybe aegerita, Coniophora puteana, Coprinopsis cinerea, Postia placenta, Lignosus rhinocerotis, Stereum hirsutum, Termitoymces sp., Armillaria gallica, Fomitopsis pinicola, and Heterobasidion annosum were incorporated for phylogenetic analyses. Bayesian trees of STSs were constructed with MrBayes v3.2.6 using the same parameters during MLST-based phylogenetic analysis [49].
2.7. Gene Synthesis, Cloning and Expression of STSs in Yeast
Candidate STSs coding genes from the strain CPCC 401429 were synthesized by Shanghai Generay Biotech Co., Ltd. and codon-optimized for heterologous expression in host S. cerevisiae. The oligonucleotide primers used for constructing the genes are listed in Table 1. PCR reactions were performed using Q5 High-Fidelity DNA Polymerase (New England Biolabs) according to the manufacturer’s protocol at an annealing temperature of 56 °C. In vivo yeast recombination cloning strategy was performed using a Frozen-EZ Yeast Transformation II Kit™ (Zymo Research) where competent cell S. cerevisiae BJ5464 was transformed with the individual DNA fragment (PCR product) of the respective STS gene and linearized vector backbone derived from YET plasmid [28,29]. Yeast transformants grown on synthetic dropout agar plate lacking tryptophan were screened by diagnostic PCR. Plasmid DNA was isolated from positive transformants using a E.Z.N.A® Yeast Plasmid Miniprep Kit (Omega, Guangzhou, China) and used to transform chemically competent E. coli Trans1-T1 (TransGen Biotech, Beijing, China). Plasmid DNA was isolated from the positive clones grown on LB broth with ampicillin using E.Z.N.A® Plasmid DNA Mini Kit (Omega, Guangzhou, China) and further verified by DNA sequencing.
2.8. GC-MS Analysis of Terpenoids
After fermentation, the S. cerevisiae liquid cultures (5.0 g each) were harvested in the headspace (20 mL) and the samples were placed at 55 °C for 15 min and volatile compounds were sampled at 55 °C for 40 min by SPME with a DVB/CAR/PDMS fiber. Compounds were desorbed in the split/splitless inlet of an Agilent 7890B/5977A gas chromatography equipped with an Agilent 7200 accurate-mass quadrupole time-of-flight (GC-MS-TOF; Agilent Technologies, Santa Clara, CA, USA) and analyzed at 250 °C for 5 min. The GC/MS-TOF was equipped with a DB-WAX column (Agilent Technologies; 60 m × 0.25 mm ID, 0.25 μm film thickness) [8,11,12]. The system was operated under the following condition: compounds were detected in split mode at split ratio of 0:1; the GC oven temperature was programmed to ramp from 50 °C (held for 5 min) to 90 °C at 3 °C min−1, to 150 °C at 2 °C min−1, then to 230 °C at 8 °C min−1, and finally to 230 °C (held for 5 min). Full scan mass spectra were acquired in the mass range of 50–350 m/z, and ionization was performed by electron impact at 70 eV with an electrospray ionization source temperature set at 230 °C.
3. Results
3.1. General Genome Features of D. tabescens CPCC 401429
With the Illumina PE150-based HiSeq 2500 sequencer and PacBio Sequel, raw data containing 871,317 reads and 6147.6 million bases were obtained. The high-quality reads were assembled by SOAPdenovo 2.04 and Canu 1.7 to generate 713 polished scaffolds. According to the coverage and the GC content, the assembly were performed and optimized to afford the draft genome sequence of D. tabescens CPCC 401429. The genome was 50.36 Mb in size and included 705 scaffolds with an N50 of 126 kb and a GC content of 47.14% (Table 2). There were approximately 0.06% (29,142 bp) repetitive sequences in the genome. A total of 15,415 protein-coding genes were predicted in the genome by AUGUSTUS program [35]. The genome completeness was 92.4% based on fungal lineage employed by the BUSCO 3.0 assessment [50]. In addition, 255 tRNAs and 4 rRNAs were defined with tRNA-scan-SE 2.0 and Barrnap V 0.8, respectively. The genome size and number of predicted genes are markedly different with eight other Armillaria mushrooms (Table S1, supporting information I). All the assembly data indicated that whole genome supports a detailed investigation of the gene content and sesquiterpene synthases-encoding pathway of sequenced Armillaria strains. Among the sequenced strains, the D. tabescens is the smallest, while others are >55 Mb, and even the strain of A. gallica 012m with 87.31 Mb. Significantly, a number of STSs encoding genes were detected among the genus, in which the strain CPCC 401429 represented the median level with twelve genes (Table S1, supporting information I).
Table 2.
Genome feature of the Desarmillaria tabescens CPCC 401429.
| Genome Feature | No./Value |
|---|---|
| Genome size (Mb) | 50.36 |
| Number of scaffolds | 713 |
| Scaffold N50 (bp) | 125,925 |
| Scaffold N90 (bp) | 33,591 |
| Longest scaffold (bp) | 839,203 |
| Shortest scaffold (bp) | 526 |
| GC content (%) | 47.14% |
| Protein-coding genes | 15,145 |
| Average gene length (bp) | 1959 |
| Complete BUSCOs (%) | 92.4% |
| Genes of KEGG | 3441 |
| Genes of COG | 9507 |
| tRNA No. | 255 |
| rRNA No. | 4 |
| TE No. | 438 |
3.2. Phylogenetic Analysis of the Strain CPCC 401429
The tef-1α DNA sequence retrieved from the genome of CPCC 401429 strain was used to preliminarily determine the identity in a BLAST nucleotide search of NCBI and exactly matched the tef-1α region of D. tabescens HKAS86603 (GenBank accession KT822441) with 99.6% similarity. A phylogenetic dendrogram of tef-1α recovered from CPCC 401429 and other Armillaria species or lineages (Figure 1) was generated, as shown in this phylogenetic tree, the D. tabescens is more closely clustered to D. ectypa species and formed a distinct group (clade V). Interestingly, Koch et al. (2017) performed six loci (28S, EF1α, RPB2, TUB, actin-1 and gpd) phylogenetic analysis to investigate and resolve the evolutionary status of diverging lineages Guyanagaster and Armillaria subgenus Desarmillaria [25]. To further investigate the armillarioid phylogeny, 18S-ITS-28S regions and ten housekeeping genes (tef-1α, rpb2, act1, gpd, tub, atp, pgi, rpl, rpk and ubq) were selected for MLST-based phylogenetic tree construction. As shown in Figure 2, markedly, the species D. tabescens CPCC 401429 and the species Guyanagaster necrorhizus MCA 3950 clustered as a well-supported branch. However, Armillaria sensu stricto includes annulate mushroom-forming armillarioid species, exemplified as A. mellea, A. ostoyae, A. borealis, A. cepistipes, A. solidipes, A. fuscipes, A. altimontana, and A. gallica species. Our result reinforced that D. tabescens was supposed to belong to the genus of Desarmillaria [25,51].
Figure 1.

Dendrogram generated from elongation factor 1-α (Tef-1α) DNA sequence data showing the phylogenetic relationships of Armillaria species and lineages. The phylogenetic tree was constructed by the Neighbor-Joining method with MEGA 7.0 programme [47]. The bootstrap scores are based on 1000 reiterations. The Tef-1α DNA sequence of A. fuscipes CMW2740 acts as the outgroup.
Figure 2.

MLST-based phylogeny of the strain D. tabescens CPCC 401429 and twenty-four sequenced basidiomycetes encompassing white and brown rot fungi. The species of Heterobasidion irregulare TC 32-1 served as outgoup [23].
3.3. Genome Annotation and Bioinformatic Analysis
The data of genomic annotations were summarized in Table S1. During the analysis of GO classification, 9044 genes were annotated with the three main categories: cellular component, molecular function, and biological process, respectively (Figure 3A). The identified coding proteins associated with molecular function are more than other two groups. In the KEGG pathway annotation, the 8300 predicated genes could be divided into 46 categories based on their functions (Figure 3B). Among these categories, 94 genes are associated with the metabolism of polyketides and terpenoids, which indicates that D. tabescens was a potential natural resource for polyketides and terpenoids biosynthesis. In the carbohydrate-active enzymes (CAZymes) annotation, 509 candidate CAZymes coding genes were identified in the genome of the strain CPCC 401429. Significantly, 199 glycoside hydrolases (GH) dominated in the classes of CAZymes, followed by 145 auxiliary activities proteins (AA), carbohydrate esterases (CE), glycosyl transferases (GT), polysaccharide lyases (PL), and carbohydrate-binding modules (CBM). Compared with the genome of other close Armillaria species, the strain CPCC 401429 had less CAZymes in which the GHs, GTs, and CBMs represented the smallest sets among six Armillaria strains (Table S2, supporting information I). In addition, as saprotrophic wood-rotting fungi, Armillaria species exhibit higher quantities of CEs and AAs than other Agaricales exemplified as Agaricus bisporus, Pleurotus ostreatus, Coprinopsis cinerea, Schizophyllum commune, Lentinula edodes, and Laccaria bicolor. Interestingly, most CEs and AAs could be functioning as lignin-, cellulose-, pectin- and hemicellulose-degrading enzymes, indicating the strong potential to degrade plant cell wall (PCW) components. Cytochrome P450s, as multifunctional oxygenases, play a key role in adaptation to nutritional niche or secondary metabolites biosynthesis. Based on domain and pfam prediction, a total of 404 cytochrome P450s coding genes were screened in the genome. The number exceeded those of A. gallica 012m (n = 271), A. cepistipes B5 (n = 324) and A. gallica Ar21-2 (n = 330). These P450s might be closely related to the formation of basic metabolism and secondary metabolites biosynthesis in D. tabescens [4]. There are three major bioactive compounds in D. tabescens, exemplified as melleolides, armillamide, and ergosterol [4]. The synthesis of orsellinate-derived melleolides and armillamide are catalyzed by a cascade of redox reactions through the combined action of cytochrome P450s and oxidoreductases, respectively [4].
Figure 3.

Gene function annotation of D. tabescens CPCC 401429 genome. (A) GO enrichment analysis of annotated genes from the strain CPCC 401429. (B) KEGG pathway annotation of the genome.
3.4. The Composition of STSs Homologues and Phylogeny of STSs
Bioinformatic analysis using antiSMASH database and a BLASTP search for putative sesquiterpene synthases (STSs) present in the genome of the strain D. tabescens CPCC 401429 revealed twelve candidate sesquiterpene biosynthetic genes (DtSTS1–DtSTS12, accession no. OQ442799-OQ442810) coding for the STSs (Figure 4). Investigation of the flanking regions of the STSs gene allowed the discovery of other genes coding cytochrome P450 monoxygenase or SDR reductase. As shown in Figure 5, In the phylogenetic analysis, the STSs are clustered in three categories: (i) Δ6 -protoilludene type (Clade III), this group includes DtSTS3, DtSTS6, and DtSTS7, which cluster with Pro1 from A. gallica [52], PRO1_ARMOS from A. ostoyae, Arg6 and Arg7 from Agrocybe aegerita [10]. This specificity also counts for protoilludene synthases from Omphalotus olearius and S. hirsutum. It has been demonstrated that Δ6-protoilludene is the precursor for melleolides that have shown antitumor and antimicrobial activities [4]. (ii) DtSTS5, DtSTS11, and DtSTS12 are closely related to Agr1 and Agr3 from A. aegerita [10], Cop1-Cop3 from C. cinereus [8], and Omp3 from Omphalotus olearius [9]. These STSs have been characterized in previous genome-wide studies, in which S. cerevisiae and/or E. coli served as heterologous systems [11]. The STSs of this group could utilize a 1,10-cyclization of (2E, 6E)-farnesyl pyrophosphate (FPP) to generate sesquiterpenes derived from an (E, E)-germacradienyl cation. Cop1 and Cop2 synthesize germacrene A as the major product, while Cop3 and Omp1 are α-muurolene synthases [8,9]. Omp3 accumulated several sesquiterpenes including α-muurolene, β-elemene, δ-cadinene, and selina-4, 7-diene [8,9]. (iii) The residual six STSs were grouped into Clade IV, this group includes few members, i.e., Cop6 from C. cinereus, ShSTS1 from S. hisutum, Omp9 and Omp10 from O. olearius. Cop6 is α-cuprenene synthase [8], Omp9 could synthesize α-barbatene (57%) and β-barbatene (21%), while Omp10 synthesizes daucene (21%), trans-dauca-4 (11),8-diene (71%) [9]. This implied that STSs in Clade IV might produce diverse sesquiterpene compounds.
Figure 4.

The genes (gene clusters) encoding sesquiterpene synthases (STSs) obtained from D. tabescens CPCC 401429. Note: sesquiterpene synthases DtSTS1-DtSTS1. DH, dehydrogenase; α-KGD, α-KG dependent dioxygenase; P450, P450 monooxygenase. The arrows represent the genes.
Figure 5.

Phylogeny and distribution of sesquiterpene synthases obtained from D. tabescens CPCC 401429 with other basidiomycetous STSs. Rooted phylogenetic tree was constructed using MrBayes v3.2.6 [49]. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (10,000,000 replicates) are shown next to the branches. The selected STSs were obtained from C. pseudopinsitus (CpSTS), O. olearius (Omp), C. aegerita (Agr), C. puteana (Copu), C. cinerea (Cop), L. rhinocerotus (GME), S. hirsutum (ShSTS), protoilludene synthases described from A. ostoyae (PRO1_ARMOS) and A. gallica (Pro1).
3.5. RNA-Sequencing Based Transcriptome Analysis of the Strain CPCC 401429
To further investigate and evaluate the differently expressed genes (DEGs), we constructed three DEGs libraries to compare YMEG to F1, PDB to YMEG, and PDB to F1 (Figures S1–S3, supporting information I). Overall, there are 10,003, 10,623, and 9895 genes which were transcribed in F1, PDB, and YMEG fermentation broth, respectively. Among the transcribed genes, 9367 genes were commonly shared (Figure 6A). Additionally, we detected 1721, 1531, and 1616 up-regulated genes, and 1948, 2271, and 2026 down-regulated genes between PDB and YMEG libraries, PDB and YMEG libraries, PDB and F1 libraries, respectively (Figure 6B, Figures S3 and S4, supporting information I). In organisms, different genes accomplish in a synchronizing mode to perform the biological functions. Pathway-directed analysis supports to further characterize the molecular functions of genes. Functional enrichment analysis was conducted using all DEGs against the GO database to investigate DEGs involved in metabolism and development. The enrichment data supports the hypothesis that the DEGs are approximately related to glycoside metabolism, fruiting body formation, membrane transporters, and secondary metabolites (SMs) production (Tables S3–S6, supporting information I). The latter includes SMs biosynthesis, exemplified as meroterpenoid melleolides, polyketides, and sesquiterpenes. It is worth noting that the DEGs functioning in the basal metabolic process, e.g., glycoside metabolism and transmembrane transport, which could promote the biological process of secondary metabolism (Tables S3 and S4, Supplementary Information I). Moreover, KEGG pathway analysis indicated that mevalonate and acetyl-CoA and metabolic pathways were also up-regulated, which facilitate the biosynthesis of sesquiterpenes and melleolides. Ten genes associated with GO terms related to sesquiterpene biosynthesis were identified. Among them, one gene DtSTS3 exhibited up-regulation in F1 fermentation broth, which is responsible for melleolides biosynthesis. While five genes (DtSTS4, DtSTS7, DtSTS9, DtSTS11, DtSTS12) and four genes (DtSTS2, DtSTS5, DtSTS8, and DtSTS10) showed up-regulated expression in PDB broth and YMEG broth, respectively (Figure 6C). We envisage that these transcriptome analysis results would be conductive to the further study of STSs characterization in Desarmillaria or close affinities.
Figure 6.

Mining transcriptome data for sesquiterpene synthases encoding genes in D. tabescens CPCC 401429. (A) Venn analysis of DEGs in three fermentation media. (B) Differentiated expression statistics of the transcriptome data of the strain CPCC 401429 in three fermentation media. (C) Heatmap of three samples including F1, YMEG, and PDB fermentation broth obtained by RNA−seq. F1−1, F1−2, and F1−3 are samples of F1 fermentation medium; YMEG−1, YMEG−2, and YMEG−3 are samples of YMEG fermentation medium; PDB−1, PDB−2, and PDB−3 are samples of PDB fermentation medium.
3.6. Heterologous Expression of STSs Encoding Genes in Yeast Produced Diverse Sesquiterpenes
To elucidate the sesquiterpenes (STs) potentially produced by the strain CPCC 401429, we first attempted to reconstitute STs production using the S. cerevisiae BJ5464 strain as a heterologous host. Two predicted STSs (DtSTS9, DtSTS10) were codon optimized, cloned into the YET vector and verified by sequencing. The expression plasmids were transformed into the BJ5464 strain [28]. For this, 100 mL of cultures were grown to mid log phase and then solid phase micro-extraction (SPME) coupled with Gas Chromatography Mass Spectrometry (GC-MS) was used to extract and analyze the volatiles in the headspace. Subsequent analyses of terpenoids produced by the resulting recombinant transformants carrying corresponding STSs expression cassette were performed. The two DtSTSs gave rise to diverse sesquiterpenes in liquid cultures of the corresponding yeast transformant (Table 3), while no sesquiterpenes was detected from the control strain BJ-CK. Terpene compounds were identified by comparing retention indices and mass spectra to National Institute of Standards and Technology (NIST) Standard Reference Database (v20, 2023-01, https://webbook.nist.gov/, accessed on 25 November 2018). Comparison of the GC–MS profiles of the yeast cultures expressing DtSTS9 and DtSTS10 against the empty vector yeast control revealed the putative sesquiterpene (m/z = 204, C15H24), or sesquiterpene alcohols (m/z = 240, C15H26O) as products (Table 3). Wide varieties of sesquierpenes were detected in the yeast culture expressing DtSTS9 and DtSTS10. Besides the noticeable product cis-thujopsene (11.25% of the peak area), β-himachalene (4.82%) as well as small amounts of α-cubebene (4.4%), acoradiene, α-bulnesene, 4,10-dimethyl-7-isopropyl [4,4,0]-bicyclo-1,4-decadiene, β-chamigrene, thujopsene-(I2), and widdrol were also detected in the headspace of DtSTS9 cultures. In addition, S. cerevisiae strain expressing DtSTS10 gave rise to α-himachalene as the main product (83.59% of the peak area), together with small amounts of α-bisabolol (4.62%), β-sesquiphellandrene (2.9%), (+)-cuparene (2.0%), β-himachalene, cis-thujopsene, α-cedrene, acoradiene, γ-muurolene and δ-elemene. Interestingly, minor widdrol (2.14%) and bisabolol (4.62%) were detected in DtSTS10 culture based on GC-MS analysis, respectively. Yap et al., characterized the structures of (+)-torreyol, α-cardinol, and 1-napthalenol in genetically engineered S. cerevisiae cultures derived from L. rhinocerotis STSs [11]. The results suggested that the sesquiterpene alcohols might be generated from sesquiterpene precursors with the aid of endogenous cytochrome P450 monooxygenase of yeast [11].
Table 3.
Heterologous production of sesquiterpenes in yeast expressing STSs.
| Rt (min) | Peak Area (%) | Name/Structure | Rt (min) | Peak Area (%) | Name/Structure |
|---|---|---|---|---|---|
| BJ-At_STS9 | BJ-At_STS10 | ||||
| 33.563 | 2.61 |
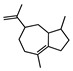 α-bulnesene |
35.938 | 1.86 |
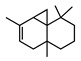 cis-thujopsene |
| 34.167 | 1.29 |
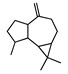 (+)-aromadendrene |
38.618 | 0.54 |
 acoradiene |
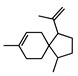
| 34.866 | 3.6 |
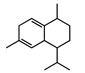 4,10-dimethyl-7-isopropyl [4,4,0]-bicyclo-1,4-decadiene |
38.87 | 1.55 |
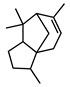 α-cedrene |
| 35.971 | 11.25 |
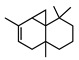 cis-thujopsene |
39.39 | 0.88 |
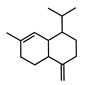 γ-muurolene |
| 38.627 | 3.68 |
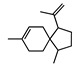 acoradiene |
39.959 | 0.37 |
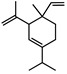 δ-elemene |
| 39.699 | 4.4 |
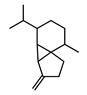 α-cubebene |
41.567 | 83.59 |
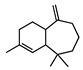 α-himachalene |
| 40.243 | 1.66 |
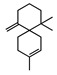 β-chamigrene |
41.924 | 2.0 |
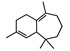 β-himachalene |
| 41.511 | 0.68 |
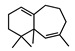 thujopsene-(I2) |
43.573 | 2.9 |
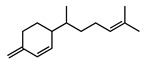 β-sesquiphellandrene |
| 41.917 | 4.82 |
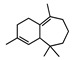 β-himachalene |
45.864 | 2.0 |
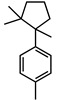 (+)-cuparene |
| 45.865 | 1.41 |
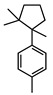 (+)-cuparene |
54.483 | 4.62 |
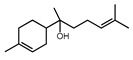 α-bisabolol |
| 53.606 | 2.14 |
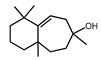 widdrol |
|||
4. Discussion
Armillaria spp. are devastating forest pathogens and they forage for hosts and achieve growing dispersal via root-like multicellular rhizomorphs [23]. Compared with other Armillaria species, natural melanized rhizomorphs of D. tabescens are rarely observed in laboratory or nature [25,26]. Herein, we sequenced and investigated the genome, performed RNA sequencing and directed discovery of sesquiterpene synthases in the strain CPCC 401429. The genome information would be helpful to investigate this unusual fungus. Armillaria species encode a large proportion of carbohydrate metabolism genes, and this could supply a strategy to bypass competition with other microbes of Armillaria spp. [23]. Compared to the genomes of other Armillaria fungi, the strain CPCC 401429 had a smaller genomic size (Table 2). The genomic features of D. tabescens had its own unique characteristics. There were 507 genes associated with carbohydrate metabolism, resulting in less genes associated with GHs, GTs, and CBMs among sequenced Armillaria strains. Slipos et al. (2017) reported that Armillaria species usually include large genomes for white rot saprotrophs evolved mostly by gene family diversification driven by transposable elements (TEs) proliferation [23,53,54]. Likewise, the D. tabescens exhibited relatively small proportion of TEs coding genes (2.89%), which is much lower than Armillaria species including A. solidipes (20.9%), A. gallica (32.6%), and A. cepistipes (34.8%) [23]. Meanwhile, the fungus CPCC 401429 encodes a wide array of genes involved in secondary metabolism of terpenoids and polyketides, which indicates that D. tabescens was a potential resource strain for polyketides and terpenoids production. Genome mining-based analysis of the sequenced Armillaria spp. and closely related species including Guyanagaster necrorhizus MCA 3950 and Floccularia luteovirens C10, demonstrated that melleolides biosynthetic gene cluster is commonly shared [4,55]. This indicated that these melleolide-coding BGCs is an interesting and evolutionary conserved feature of Armillaria genus or closely related species. This might provide crucial defense against predators or other microbes in adapting to a soil-borne lifestyle [23]. Conspicuously, the mel locus in D. tabescens is a shortest, delicate, and tightly clustered BGC responsible for melleolides biosynthesis [4]. Additionally, as to phylogenetic analysis of D. tabescens CPCC 401429, with the results of NR annotation, MLST-based phylogeny, and synteny analysis, we considered that D. tabescens was supposed to belong to the genus of Desarmillaria instead of Armillaria as it used to. It is noteworthy that the D. tabescens species formed a distinct group with G. necrorhizus MCA 3950, suggesting it would support the proposal of reattribution of A. tabescens and A. ectypa species in Armillaria subgenus Desarmillaria [25,26].
Basidiomycetous mushrooms are particularly adept at synthesizing a broad range of structurally diversified sesquiterpenes. Up to now, many Basidiomycetous sesquiterpene synthases have been characterized, exemplified as putative STSs from O. olearius, C. puteana, L. rhinocerotus, and A. aegerita [8,9,10,11]. With the recent revolution in genome-sequencing technology, a large number of Basidiomycetous genomes have been completed. Bioinformatic mining indicates that most genes/clusters of the phylum Basidiomycota are silent or cryptic under laboratory conditions [4,9,10]. The terpenoids and their synthases represent largely untapped “dark matter” that awaits to be characterized [1,2,4]. What are the ecological functions of STSs that play important roles in secondary metabolism during the lifecycle of mushrooms? What is the underlying mechanism that controls the functional evolution of STSs? Are these STSs unique to specific evolutionary lineage (taxa) or widely distributed? To answer these questions, functional characterization of increasing STSs encoded in the fungal genome is essential and indispensable.
As a cutting-edge discipline that drives original breakthroughs in the field of biopharmaceuticals, the rise of synthetic biology can promote drug leads discovery by designing new biosynthetic pathways. Therefore, mining and discovering of biosynthetic elements are urgently needed in synthetic biology technology [10]. Also, bioinformatic analysis and functional prediction of specific STSs would facilitate the screening of ideal candidate synthetic elements or bioprospecting for new sesquiterpenes. Since most Basidiomycetous mushrooms are difficult to manipulate in genetic transformation, S. cerevisiae yeast has emerged as a powerful host cell for production of fungal secondary metabolites and pathways reconstitution [11,56]. To our knowledge, this is the first report to document the endeavor of the yeast expression for mining the sesquiterpene repertoire in Desarmillaria species and Armillaria affinities. Phylogenetic analysis of characterized STSs reinforced that certain group of STSs shares highly conserved sequences, suggesting that they might catalyze the likely cyclization mechanism [9,10]. Thus, phylogenetic analysis might be useful to mine novel terpenoid synthases. In this study, noteworthy, half members STSs from the strain CPCC 401429 were distributed in minor group Clade IV. This group of STSs belongs to a new subfamily and only few STSs have been characterized. Our report on STSs derived from D. tabescens enriches the number of STSs of this subfamily and expands our knowledge of STSs diversity for Desarmillaria or close genus. Yeast cells expressing DtSTS9 and DtSTS10 yielded diverse sesquiterpene molecules, suggesting that this subfamily might be highly promiscuous producers. This highlights the potential of Armillaria in generating novel sesquiterpenoids. Accessing the basidiomycetous pool of STSs can be an immediate source of novel biocatalysts, or yield STSs that could be further specialized by directed evolution or combinatorial engineering. This also lays foundation for downstream in-depth deciphering catalytic mechanism of novel STSs.
5. Conclusions
In summary, we sequenced the genome of the fungus D. tabescens CPCC 401429 and identified twelve STSs encoding genes. Bioinformatic analysis and MLST-based phylogeny allowed us to support the reclassifying the fungus D. tabescens in Desarmillaria subgenus. Also, the large number of STS genes in the genome highlights the potential of the strain CPCC 401429 in generating varying sesquiterpenoids. In combination with transcriptomic analysis, two new STSs encoding genes were selected for yeast heterologous expression. Based on phylogenetic analysis and GC-MS identification, DtSTS9 and DtSTS10 belonging to the subfamily IV, are novel and very interesting sesquiterpene synthases with high potential for downstream research. This study also demonstrated that yeast based heterologous expression is an attractive approach for accessing the basidiomycete terpenoid chemistry.
Acknowledgments
We are grateful to Yi Tang (Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, California, United States) for sharing the YET plasmid and strain S. cerevisiae BJ5464. We would like to thank Jian Li (Beijing Engineering and Technology Research Center of Food Additives, Beijing Technology and Business University) for GC-MS analysis of the genetically engineered yeast.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jof9040481/s1, File S1: Supporting Information I; Text S1: Supporting Information II, Sequences for MLST-based phylogeny of twenty-five higher fungus.
Author Contributions
T.Z. conceived the study, obtained initial funding, and supervised experiments. T.Z., J.F. and X.R. carried out experiments and bioinformatic analysis. W.H., H.L., J.L., H.W., X.L. and L.W. advised on the design and interpretation of data in Figure 2 and Figure 6. T.Z. wrote the draft manuscript. T.Z., L.Z. and L.Y. edited the draft manuscript. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the raw sequence files are available in the National Center for Biotechnology Information under the OQ442799-OQ442810.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This work was financially supported by the National Natural Science Foundation of China (No. 31872617), the CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-055, 2019-I2M-1-005), and the central level, scientific research institutes for basic R & D fund business (3332018097).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Christianson D.W. Unearthing the roots of the terpenome. Curr. Opin. Chem. Biol. 2008;12:141–150. doi: 10.1016/j.cbpa.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu J., Yang X., Duan Y., Wang P., Qi J., Gao J., Liu C. Biosynthesis of sesquiterpenes in basidiomycetes: A review. J. Fungi. 2022;8:913. doi: 10.3390/jof8090913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li W., He J., Feng T., Yang H., Ai H., Li Z., Liu J. Antroalbocin A, an antibacterial sesquiterpenoid from higher fungus Antrodiella albocinnamomea. Org. Lett. 2018;20:8019–8021. doi: 10.1021/acs.orglett.8b03595. [DOI] [PubMed] [Google Scholar]
- 4.Zhang T., Cai G., Rong X., Wang Y., Gong K., Liu W., Wang L., Pang X., Yu L. A combination of genome mining with an OSMAC approach facilitates the discovery of and ccontributions to the biosynthesis of melleolides from the basidiomycete Armillaria tabescens. J. Agric. Food Chem. 2022;70:12430–12441. doi: 10.1021/acs.jafc.2c04079. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann V.K., Huang A., Ibanez-Calero S., Wilson G.R., Rinehart K.L. Illudin S, the sole antiviral compound in mature fruiting bodies of Omphalotus illudens. J. Nat. Prod. 2003;66:1257–1258. doi: 10.1021/np030205w. [DOI] [PubMed] [Google Scholar]
- 6.Yang X.Y., Feng T., Li Z., Sheng Y., Yin X., Leng Y., Liu J. Conosilane A, an unprecedented sesquiterpene from the cultures of basidiomycete Conocybe siliginea. Org. Lett. 2012;14:5382–5384. doi: 10.1021/ol302468a. [DOI] [PubMed] [Google Scholar]
- 7.Qi Q.Y., Bao L., Ren J., Han J., Zhang Z., Li Y., Yao Y., Cao R., Liu H. Sterhirsutins A and B, two new heterodimeric sesquiterpenes with a new skeleton from the culture of Stereum hirsutum collected in Tibet Plateau. Org. Lett. 2014;16:5092–5095. doi: 10.1021/ol502441n. [DOI] [PubMed] [Google Scholar]
- 8.Agger S., Lopez-Gallego F., Schmidt-Dannert C. Diversity of sesquiterpene synthases in the basidiomycete Coprinus cinereus. Mol. Microbiol. 2009;72:1181–1195. doi: 10.1111/j.1365-2958.2009.06717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wawrzyn G.T., Quin M.B., Choudhary S., Lopez-Gallego F., Schmidt-Dannert C. Draft genome of Omphalotus olearius provides a predictive framework for sesquiterpenoid natural product biosynthesis in Basidiomycota. Chem. Biol. 2012;19:772–783. doi: 10.1016/j.chembiol.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang C., Chen X., Orban A., Shukal S., Birk F., Too H.P., Ruhl M. Agrocybe aegerita serves as a gateway for identifying sesquiterpene biosynthetic enzymes in higher fungi. ACS Chem. Biol. 2020;15:1268–1277. doi: 10.1021/acschembio.0c00155. [DOI] [PubMed] [Google Scholar]
- 11.Yap H.Y., Muria-Gonzalez M.J., Kong B.H., Stubbs K.A., Tan C.S., Ng S.T., Tan N.H., Solomon P.S., Fung S.Y., Chooi Y.H. Heterologous expression of cytotoxic sesquiterpenoids from the medicinal mushroom Lignosus rhinocerotis in yeast. Microb. Cell Factories. 2017;16:1–13. doi: 10.1186/s12934-017-0713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Onsando J.M., Wargo P.M., Waudo S.W. Distribution, severity, and spread of Armillaria root disease in Kenya tea plantations. Plant Dis. 1997;81:133–137. doi: 10.1094/PDIS.1997.81.2.133. [DOI] [PubMed] [Google Scholar]
- 13.Baumgartner K., Coetzee M.P., Hoffmeister D. Secrets of the subterranean pathosystem of Armillaria. Mol. Plant Pathol. 2011;12:515–534. doi: 10.1111/j.1364-3703.2010.00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fradj N., de Montigny N., Merindolm N., Awwad F., Boumghar Y., Germain H., Desgagne-Penix I. A first insight into North American plant pathogenic fungi Armillaria sinapina transcriptome. Biology. 2020;9:153. doi: 10.3390/biology9070153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo T., Wang H.C., Xue W.Q., Zhao J., Yang Z.L. Phylogenetic analyses of Armillaria reveal at least 15 phylogenetic lineages in China, seven of which are associated with cultivated Gastrodia elata. PLoS ONE. 2016;11:e0154794. doi: 10.1371/journal.pone.0154794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baumgartner K., Travadon R., Bruhn J., Bergemann S.E. Contrasting patterns of genetic diversity and population structure of Armillaria mellea sensu stricto in the eastern and western United States. Phytopathology. 2010;100:708–718. doi: 10.1094/PHYTO-100-7-0708. [DOI] [PubMed] [Google Scholar]
- 17.Muszynska B., Sulkowska-Ziaja K., Wolkowska M., Ekiert H. Chemical, pharmacological, and biological characterization of the culinary-medicinal honey mushroom, Armillaria mellea (Vahl) P. Kumm. (Agaricomycetideae): A review. Int. J. Med. Mushrooms. 2011;13:167–175. doi: 10.1615/IntJMedMushr.v13.i2.90. [DOI] [PubMed] [Google Scholar]
- 18.Hobbs C. Medicinal Mushrooms: An Exploration of Tradition, Healing, and Culture. Botanica Press; Summertown, TN, USA: 1986. [Google Scholar]
- 19.Zhang T., Du Y., Liu X., Sun X., Cai E., Zhu H., Zhao Y. Study on antidepressant-like effect of protoilludane sesquiterpenoid aromatic esters from Armillaria mellea. Nat. Prod. Res. 2021;35:1042–1045. doi: 10.1080/14786419.2019.1614577. [DOI] [PubMed] [Google Scholar]
- 20.Heinzelmann R., Rigling D., Sipos G., Munsterkotter M., Croll D. Chromosomal assembly and analyses of genome-wide recombination rates in the forest pathogenic fungus Armillaria ostoyae. Heredity. 2020;124:699–713. doi: 10.1038/s41437-020-0306-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akulova V.S., Sharov V.V., Aksyonova A.I., Putintseva Y.A., Oreshkova N.V., Feranchuk S.I., Kuzmin D.A., Pavlov I.N., Litovka Y.A., Krutovsky K.V. De novo sequencing, assembly and functional annotation of Armillaria borealis genome. BMC Genom. 2020;21:534. doi: 10.1186/s12864-020-06964-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhan M., Tian M., Wang W., Li G., Lu X., Cai G., Yang H., Du G., Huang L. Draft genomic sequence of Armillaria gallica 012m: Insights into its symbiotic relationship with Gastrodia elata. Braz. J. Microbiol. 2020;51:1539–1552. doi: 10.1007/s42770-020-00317-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sipos G., Prasanna A.N., Walter M.C., O’Connor E., Balint B., Krizsan K., Kiss B., Hess J., Varga T., Slot J., et al. Genome expansion and lineage-specific genetic innovations in the forest pathogenic fungi Armillaria. Nat. Ecol. Evol. 2017;1:1931–1941. doi: 10.1038/s41559-017-0347-8. [DOI] [PubMed] [Google Scholar]
- 24.Collins C., Keane T.M., Turner D.J., O’Keeffe G., Fitzpatrick D.A., Doyle S. Genomic and proteomic dissection of the ubiquitous plant pathogen, Armillaria mellea: Toward a new infection model system. J. Proteome Res. 2013;12:2552–2570. doi: 10.1021/pr301131t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koch R.A., Wilson A.W., Sene O., Henkel T.W., Aime M.C. Resolved phylogeny and biogeography of the root pathogen Armillaria and its gasteroid relative, Guyanagaster. BMC Evol. Biol. 2017;17:33. doi: 10.1186/s12862-017-0877-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang J., Pecoraro L., Cai L., Yuan Z., Zhao P., Tsui C.K.M., Zhang Z. Phylogenetic relationships, speciation, and origin of Armillaria in the Northern hemisphere: A lesson based on rRNA and elongation factor 1-alpha. J. Fungi. 2021;7:1088. doi: 10.3390/jof7121088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watling R., Kile G.A., Gregory N.M. The genus Armillaria-nomenclature, typification, the identity of Armillaria mellea and species differentiation. Trans. Br. Mycol. Soc. 1982;78:271–285. doi: 10.1016/S0007-1536(82)80011-9. [DOI] [Google Scholar]
- 28.Zhang T., Gu G., Liu G., Su J., Zhan Z., Zhao J., Qian J., Cai G., Cen C., Zhang D., et al. Late-stage cascade of oxidation reactions during the biosynthesis of oxalicine B in Penicillium oxalicum. Acta Pharm. Sin. B. 2023;13:256–270. doi: 10.1016/j.apsb.2022.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang T., Pang X., Zhao J., Guo Z., He W., Cai G., Su J., Cen S., Yu L. Discovery and activation of the cryptic cluster from Aspergillus sp. CPCC 400735 for asperphenalenone biosynthesis. ACS Chem. Biol. 2022;17:1524–1533. doi: 10.1021/acschembio.2c00204. [DOI] [PubMed] [Google Scholar]
- 30.Bolger A.M., Lohse M. Usadel B: Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen T., Tagett R., Diaz D., Draghici S. A novel approach for data integration and disease subtyping. Genome Res. 2017;27:2025–2039. doi: 10.1101/gr.215129.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li R., Zhu H., Ruan J., Qian W., Fang X., Shi Z., Li Y., Li S., Shan G., Kristiansen K., et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simao F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 35.Stanke M., Morgenstern B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33:W465–W467. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gotz S., Garcia-Gomez J.M., Terol J., Williams T.D., Nagaraj S.H., Nueda M.J., Robles M., Talon M., Dopazo J., Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G.A., Sonnhammer E.L.L., Tosatto S.C.E., Paladin L., Raj S., Richardson L.J., et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021;49:D412–D419. doi: 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peitsch M.C., Wilkins M.R., Tonella L., Sanchez J.C., Appel R.D., Hochstrasser D.F. Large-scale protein modelling and integration with the SWISS-PROT and SWISS-2DPAGE databases: The example of Escherichia coli. Electrophoresis. 1997;18:498–501. doi: 10.1002/elps.1150180326. [DOI] [PubMed] [Google Scholar]
- 39.Chen S., Zhou Y., Chen Y., Gu J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim D., Langmead B., Salzberg S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li B., Dewey C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klopfenstein D.V., Zhang L., Pedersen B.S., Ramirez F., Vesztrocy A.W., Naldi A., Mungall C.J., Yunes J.M., Botvinnik O., Weigel M., et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018;8:10872. doi: 10.1038/s41598-018-28948-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bu D., Luo H., Huo P., Wang Z., Zhang S., He Z., Wu Y., Zhao L., Liu J., Guo J., et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021;49:W317–W325. doi: 10.1093/nar/gkab447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnston P.R., Quijada L., Smith C.A., Baral H.O., Hosoya T., Baschien C., Partel K., Zhuang W.Y., Haelewaters D., Park D., et al. A multigene phylogeny toward a new phylogenetic classification of Leotiomycetes. IMA Fungus. 2019;10:1–22. doi: 10.1186/s43008-019-0002-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katoh K., Rozewicki J., Yamada K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20:1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar S., Stecher G., Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., von Haeseler A., Jermiin L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ronquist F., Teslenko M., van der Mark P., Ayres D.L., Darling A., Hohna S., Larget B., Liu L., Suchard M.A., Huelsenbeck J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manni M., Berkeley M.R., Seppey M., Zdobnov E.M. BUSCO: Assessing genomic data quality and beyond. Curr. Protoc. 2021;1:e323. doi: 10.1002/cpz1.323. [DOI] [PubMed] [Google Scholar]
- 51.Koch R.A., Herr J.R. Global distribution and richness of Armillaria and related species inferred from public databases and amplicon sequencing datasets. Front. Microbiol. 2021;12:733159. doi: 10.3389/fmicb.2021.733159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Engels B., Heinig U., Grothe T., Stadler M., Jennewein S. Cloning and characterization of an Armillaria gallica cDNA encoding protoilludene synthase, which catalyzes the first committed step in the synthesis of antimicrobial melleolides. J. Biol. Chem. 2011;286:6871–6878. doi: 10.1074/jbc.M110.165845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raffaele S., Kamoun S. Genome evolution in filamentous plant pathogens: Why bigger can be better. Nat. Rev. Microbiol. 2012;10:417–430. doi: 10.1038/nrmicro2790. [DOI] [PubMed] [Google Scholar]
- 54.Haas B.J., Kamoun S., Zody M.C., Jiang R.H., Handsaker R.E., Cano L.M., Grabherr M., Kodira C.D., Raffaele S., Torto-Alalibo T., et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009;461:393–398. doi: 10.1038/nature08358. [DOI] [PubMed] [Google Scholar]
- 55.Liu Z., Lu H., Zhang X., Chen Q. The genomic and transcriptomic analyses of Floccularia luteovirens, a rare edible fungus in the Qinghai-Tibet Plateau, provide insights into the taxonomy placement and fruiting body formation. J. Fungi. 2021;7:887. doi: 10.3390/jof7110887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen R., Jia Q., Mu X., Hu B., Sun X., Deng Z., Chen F., Bian G., Liu T. Systematic mining of fungal chimeric terpene synthases using an efficient precursor-providing yeast chassis. Proc. Natl. Acad. Sci. USA. 2021;118:e2023247118. doi: 10.1073/pnas.2023247118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the raw sequence files are available in the National Center for Biotechnology Information under the OQ442799-OQ442810.