

Abstract
Intrinsically disordered regions of proteins often mediate important protein–protein interactions. However, the folding-upon-binding nature of many polypeptide–protein interactions limits the ability of modeling tools to predict the three-dimensional structures of such complexes. To address this problem, we have taken a tandem approach combining NMR chemical shift data and molecular simulations to determine the structures of peptide–protein complexes. Here, we use the MELD (Modeling Employing Limited Data) technique applied to polypeptide complexes formed with the extraterminal domain (ET) of bromo and extraterminal domain (BET) proteins, which exhibit a high degree of binding plasticity. This system is particularly challenging as the binding process includes allosteric changes across the ET receptor upon binding, and the polypeptide binding partners can adopt different conformations (e.g., helices and hairpins) in the complex. In a blind study, the new approach successfully modeled bound-state conformations and binding poses, using only protein receptor backbone chemical shift data, in excellent agreement with experimentally determined structures for moderately tight (Kd ~100 nM) binders. The hybrid MELD + NMR approach required additional peptide ligand chemical shift data for weaker (Kd ~250 μM peptide binding partners. AlphaFold also successfully predicts the structures of some of these peptide–protein complexes. However, whereas AlphaFold can provide qualitative peptide rankings, MELD can directly estimate relative binding affinities. The hybrid MELD + NMR approach offers a powerful new tool for structural analysis of protein–polypeptide complexes involving disorder-to-order transitions upon complex formation, which are not successfully modeled with most other complex prediction methods, providing both the 3D structures of peptide–protein complexes and their relative binding affinities.
Graphical Abstract
INTRODUCTION
Molecular modeling has become an essential toolset for predicting bound conformations in structural biology. These successes are attributable to advances in protein structure prediction, robust docking pipelines for small molecules, and accurate free energy methods for quantifying relative (and absolute) binding affinities.1 Despite these advances, the accuracy of these methods decreases rapidly for systems involving significant conformational changes upon complex formation, where receptors can accommodate multiple binding modes, and for highly charged systems.2,3 In particular, systems involving disorder-to-order transitions upon complex formation, including peptides that fold as they bind, challenge current protein–peptide docking methods. Machine learning tools have recently brought fast and accurate predictions for protein structures4,5 and are now being extended to predictions of complexes.5,6 More generally, three-dimensional structures of peptide–protein complexes provide essential information for understanding the mechanisms of multiprotein complex assembly and have the potential to inform drug discovery.
Here, we describe an integrative approach to structure determination for peptide–protein complexes combining NMR chemical shift data and molecular simulations. High-information-content NMR studies rely on extracting many distance and orientation restraints to solve the structure of the peptide–protein complex.7 At the other extreme, lower-information-content NMR studies, such as backbone chemical shift data, which are prerequisites to more extensive studies, provide valuable information about the binding epitope and (in some cases) the bound-state conformation of the peptide. But such methods do not usually provide enough data to characterize the binding mode reliably and structures of the complex involving disorder-to-order transitions upon binding.
Molecular simulations approach the problem of peptide binding by sampling the binding/unbinding landscape, including multiple binding modes and peptide conformations, relying on statistical mechanics to identify preferred conformations in the ensemble. Sampling multiple binding/unbinding events requires timescales much longer than the bound-state lifetime,8 entailing a significant computational effort even with special-purpose computers9 or advanced sampling technique strategies.10–14 Integrating experimental data reduces the conformational space, focusing sampling on structures that satisfy the physics and experimental data.15,16 This work identifies synergies between incomplete or “sparse” NMR data and simulations for structure prediction of peptide–protein complexes, focusing on applicability, transferability, and limitations. This pipeline allows a more rapid structure determination of complexes than conventional NMR approaches and can provide structures of complexes even for systems for which extensive NMR data cannot be obtained. We focus on the binding of polypeptides to the extraterminal domain (ET) of bromo and extraterminal domain (BET) proteins, which exhibit both disorder-to-order transitions of polypeptide binding partners and allosteric changes in the receptor and which also accommodate peptides binding in different conformations.7 This biologically important system exhibits a significant degree of plasticity in binding modes and peptide conformations,7,17–19 as well as a wide range of binding affinities, and poses challenges to current computational and experimental approaches.
The BET family of proteins plays important roles in eukaryotic gene regulation by recognizing and binding epigenetic signatures and recruiting other regulatory proteins. Structurally, BET proteins contain two bromodomains that recognize and bind acetylated signatures in histones and an extraterminal domain (ET) that serves as an anchor point to recruit other host regulatory and chromatin remodeling proteins.19 Some viruses, such as the murine leukemia virus (MLV) and Kaposi’s sarcoma-associated herpesvirus (KSHV), encode proteins that also bind to the ET domain. For retroviral integration, this effectively facilitates their location near active transcription start sites and CpG islands.20 The MLV integrase contains an intrinsically disordered C-terminal polypeptide “tail” segment that becomes structured upon binding the ET domain. KSVH virus has a latency-associated nuclear antigen (LANA) protein that binds ET. Understanding and predicting how different polypeptide sequences bind the ET domain can potentially lead to new cancer treatment and gene replacement therapy approaches.
The structured region of the ET domain adopts a 68-residue three-helix bundle (see Figure 1) with a binding site defined by a hydrophobic pocket flanked by a negatively charged loop region connecting helices α2 and α3. Proteins interact with the ET domain through short peptide epitopes that anchor hydrophobic residues in ET’s hydrophobic pocket and interlace positively charged residues of the ET-binding polypeptide segment with negatively charged residues of ET through a zipping mechanism.17 Interestingly, the binding mode, orientation of the bound polypeptide segment, and even secondary structure of the bound polypeptide can vary considerably for different polypeptide sequences, requiring the binding region of the ET receptor to adopt different conformations in different ET–polypeptide complexes (see Figure 1).
Figure 1.
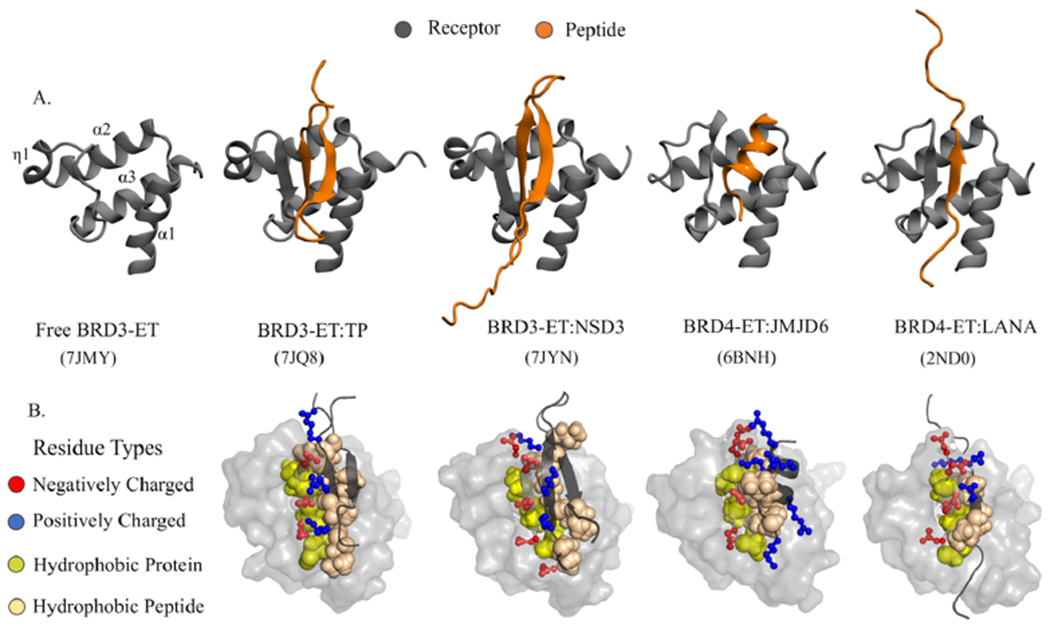
Plasticity of ET–peptide complex formation. (A) Experimental structures of the ET domain of BET proteins interacting with different peptides highlight differences across the binding modes observed in different ET–peptide complexes (PDB ID shown in parentheses). (B) A network of alternating positively (blue) and negatively (red) charged interactions between the peptide and protein residues in a zipper-like interaction mechanism that is further stabilized by hydrophobic packing between a cleft in the protein and hydrophobic peptide sidechains.
Several aspects have limited progress in characterizing ET complexes. First, many of these complexes involve disorder-to-order transitions of ET-binding regions of intrinsically disordered regions of protein partners, stabilized by transient interactions with medium to weak binding affinities, producing spectra with contributions from unbound and bound states. Second, despite the high homology between the ET domains of different BET proteins, there are marked differences in binding affinities (e.g., the host JMJD6 protein is a weaker binder to BRD3-ET than to BRD4-ET19). Third, the choice of sequence/length of the selected peptide epitope during experimental binding affinity assays also dramatically affects the measurements. For example, the ET-binding segment of NSD3 has been characterized in complexes with BRD3-ET and BRD4-ET using either short or long sequences that bind as a single strand or as a hairpin, respectively, where the short peptide is a weaker binder than the longer construct; the short peptide binds to BRD3-ET with Kd = 950 μM and to BRD4-ET with Kd = 140 μM, whereas the longer polypeptide binds to BRD3-ET with Kd = 250 ± 150 μM.21 This weak binding constant has made it difficult to determine the 3D structure of this complex.7 In this regime, both computational and experimental approaches are challenged.
The present work presents a solution to such challenging systems by combining computational tools and low-information-content NMR data (i.e., chemical shift data alone) to determine the structures of ET–peptide complexes (see Table 1 for peptides used in this work). We focus on peptides binding to BRD3-ET7 and BRD4-ET based on our available experimental data and those found in the literature.21,22 Future work will establish differences in binding affinities and mechanisms among ET domains from various BET proteins and how to exploit these differences for selectively targeting these different BET proteins.
Table 1.
Six Systems Investigated with MELD + NMR and Their Corresponding Peptide Sequences and Public Database Identifiers, Where Peptide Residue Numbers Correspond to the Sequence Numbering Used for These Studies
| peptide acronyms used in this article | PDB/BMRB ID | Uniprot ID (residue range) | residue range (MELD) | complete name of the peptides / Sequence |
|---|---|---|---|---|
| TP7 | 7JQ8/30,786 |
P03355 (1716–1738) |
69–91 | C-terminal tail peptide of region of murine leukemia virus integrase |
| SRLTWRVQRSQNPLKIRLTREAP | ||||
| NSD37 | 7JYN/30,790 | Q9BZ95-1 (151–184) |
69–102 | histone-lysine N-methyltransferase nuclear receptor binding SET domain protein 341 |
| PEIKLKITKTIQNGRELFESSLCGDLLNEVQASE | ||||
| LANA17 | 2ND0/26,242 |
Q9QR71 (1098–1116) |
69–87 | latency-associated nuclear antigen region of Kaposi’s sarcoma-associated herpesvirus |
| NLQSSIVKFKKPLPLTQPG | ||||
| JMJD618 | 6BNH/30,373 | Q6NYC1-1 (84–96) |
69–81 | short fragment of Jumonji domain-containing protein 6, a member of Jumonji C family of oxygenases |
| KWTLERLKRKYRN | ||||
| CHD421 | 6BGG/30,367 | Q14839-1 (290–301) |
69–80 | chromodomain-helicase DNA-binding protein 4, a core ATPase subunit of Nucleosome Remodeling and Deacetylation (NuRD) complex42 |
| KVAPLKIKLGGF | ||||
| BRG121 | 6BGH/30,368 | P51532-1 (1591–1602) |
69–80 | Brahma-related gene 1, a catalytic ATPase subunit of human Brg1 associated factor (BAF)43 |
| RSVKVKIKLGRK | ||||
We formulated this study in two stages. First, we conducted blind computational modeling with the MELD approach15 for two peptide–protein complex structures followed by an assessment of model accuracy based on high-quality NMR structures of these complexes.7 Here, the experimental team provided NMR data sets for peptide–protein complexes for which structures were not yet deposited in the Protein Data Bank and not available to the prediction team, with increasing information content and collected predictions from the computational group at each stage (see Figure 2). The lowest-tier data used only backbone 15N-1H chemical shift perturbation (CSP) data measured on the receptor protein, effectively identifying possible binding hotspots. For ET, these CSP data reflect changes in nuclear environments at the peptide binding site and changes throughout the structure due to allosteric conformational shifts resulting from peptide binding.7 At the highest tier of experimental information, in addition to the backbone 15N-1H CSPs measured on the receptor protein, we also used dihedral angle restraints based on backbone chemical shift data for the isotope-enriched receptor-bound peptide.23,24 Although the principle regime of study is for tight (Kd < 1 μM) and weak (1 μM < Kd < 500 μM) complexes, in order to explore the impact of a few key contacts and the potential for docking very weak complexes (Kd > 500 μM), in addition to these chemical shift data, a few peptide–protein contacts based on the strongest NOEs observed between the protein and bound peptide were also provided in a third sparse-NMR data set. In the second stage of the study, we extended the method to other peptide–ET domain complexes for which three-dimensional structures were already published to assess the generality of our methods.
Figure 2.
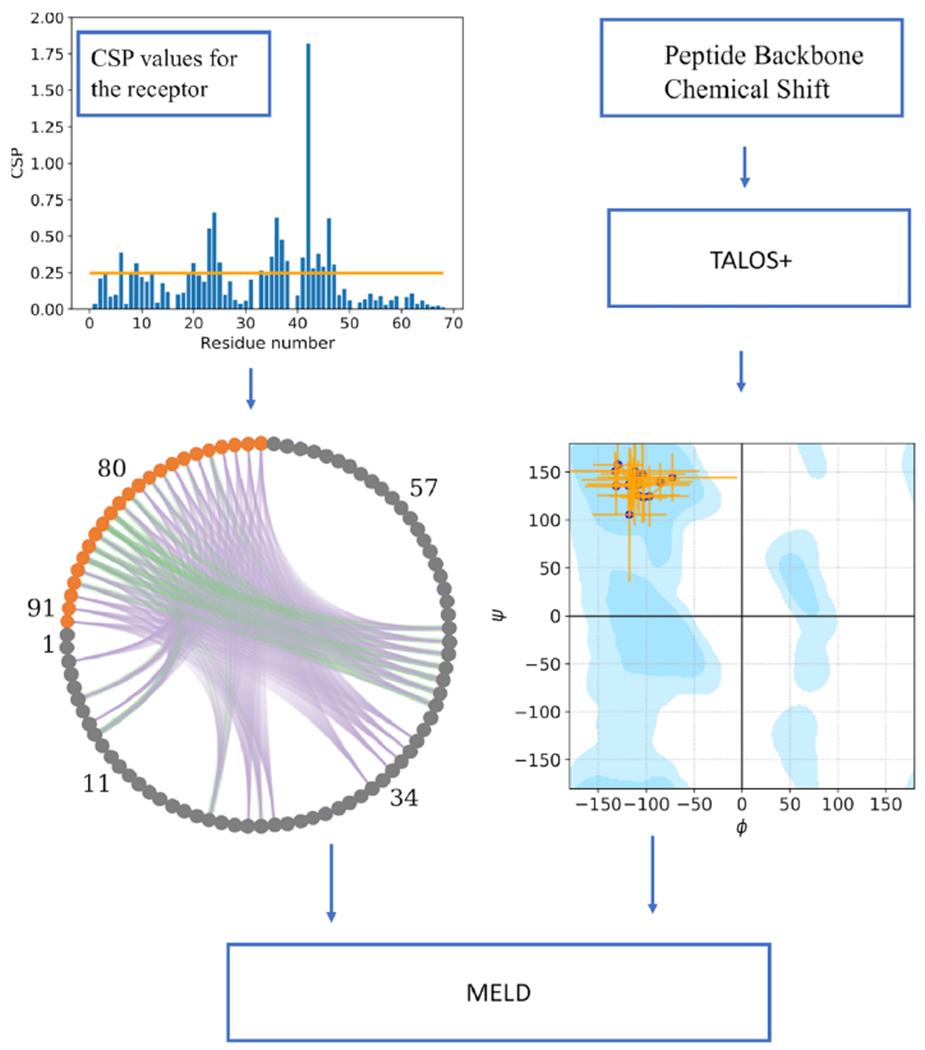
General flowchart of the current work. The upper left plot shows NMR CSP values for each residue in the receptor, with the threshold cutoff used to define active residues shown as an orange horizontal line. Given the active residues, the lower left circular plot represents the combinatorics of possible restraints between the peptide (orange circles) and the receptor (gray circles); each line between pairs of residues is a potential contact. Purple lines represent contacts that are not present in the native structure, and green lines show the ones present (true positives). For some active residues, there are no green lines, indicating that their high CSP values were due to an allosteric or propagated structural changes. The lower right plot shows the dihedral restraints on the binding peptide conformation, with their uncertainties (orange lines) used in MELD with TALOS data. The Ramachandran plot is made using https://github.com/gerdos/PyRAMA.
METHODS
Experimental Methods.
NMR Data.
Experimental NMR data were generated for the murine BRD3-ET domain (residues 554 to 640) and its complexes with the 23-residue C-terminal tail peptide (TP) of MLV IN (residues 1716 to 1738 of the Gag-Pol polyprotein), and the ET-binding polypeptide segments of murine NSD3 (residues 151 to 184) as summarized in Table 1. 13C,15N-enriched samples of ET were produced using standard methods.7,25 Isotope-enriched peptides were produced as fusion proteins followed by proteolytic cleavage, as described elsewhere.7 Sequence-specific resonance assignments for peptide complexes (for both the ET protein and for the bound polypeptides) were determined using standard triple-resonance NMR methods, also described elsewhere.7 NMR data for ET-binding domains of CHD421 (BMRB ID 30367), BRG121 (BMRB ID 30368), LANA17 (BMRB ID 26042), and JMJD618 (BMRB ID 30373) were obtained from the BioMagResDataBank (BMRB26). Backbone amide 15N-1H chemical shift perturbations (CSPs) of the apo BRD3 ET domain relative to values in the complex were calculated using ΔδN,H = ((ΔδN/6)2 + (ΔδH)2)0.5 and plotted as a function of BRD3 ET sequence. The threshold for defining a significant CSP was determined by iterative analysis.27 The standard deviation (σ) of the shift changes ΔδN,H was first calculated. To prevent biasing the distribution by including the small number of residues with very large shift changes, any residues for which the shift change is greater than 3σ were excluded. The standard deviation (σ) of the remaining ΔδN,H values was then recalculated. Iteration of these calculations was performed until no further residues were excluded. The threshold value for a minimal CSP was then set to ΔδN,H = 3σ = 0.02 ppm. 13C- and 15N-edited 3D NOESY spectra for uniformly 13C,15N-enriched ET-TP and ET-NSD3 complexes were recorded with NOE mixing times of 120 ms.
Isothermal Titration Calorimetry.
Isothermal titration calorimetry was carried out using a MicroCal VP-ITC Isothermal Titration Calorimeter in the Analytical Biochemistry Core Facility of the Center for Biotechnology and Interdisciplinary Sciences (CBIS) at Rensselaer Polytechnic Institute. TP (SRLTWRVQRSQNPLKIRLTREAP) was prepared by commercial solid-phase peptide synthesis and purified by reverse-phase HPLC (Peptide 2.0). Recombinant NSD3 (EFTGSPEIKLKITKTIQNGRELFESSLCGDLLNEVQASE) was prepared as described previously.7 Samples of ET (~2.4 mL) and peptide binding partners (~300 μL) were prepared for ITC studies by dialyzing together in separate dialysis bags placed in the same beaker of the ITC buffer containing 20 mM Tris, 150 mM NaCl, and 1 mM TCEP at pH 7.5. The ET and peptide binding partners were first dialyzed in 1 L ITC buffer at 4 °C for 8 h and then dialyzed into a new 1 L ITC buffer at 4 °C overnight. Protein and peptide concentrations were determined after dialysis by absorbance spectroscopy at 280 or 205 nm28 using extinction coefficients for ET (ε280 = 4470 M−1 cm−1), TP (ε280 = 5500 M−1 cm−1), and NSD3 (ε205 = 126,480 M−1 cm−1, contains no Tyr or Trp) calculated from their respective amino acid sequences.
Data Sets Used for the Blind Study.
We use three different experimental NMR data sets to mimic different levels of experimental NMR information: (1) CSP, (2) CSP + TALOS, and (3) CSP + TALOS + NOE. For the lowest-information-content data set, experimental docking data include only backbone 15N-1H CSP data for the protein receptor in the presence/absence of a bound peptide. We call these CSP data sets. This approach uses the comparison of the [15N-1H]-HSQC spectra of apo and peptide-bound receptor and has the advantage that the peptide binding partner does not require isotope enrichment. Residues of the receptor (in this case, ET) for which there is a chemical shift perturbation upon complex formation may be directly involved in binding or indirectly affected by allosteric conformational changes. Hence, these CSP data do not provide information about which specific protein atoms (e.g., backbone vs sidechain) are involved in the interaction with the peptide or which peptide residues might be involved in the binding. The CSP threshold for significant perturbations (calculated as described above) indicates that the 15N-1H chemical shifts of the majority of residues in the ET receptor are perturbed upon complex formation, with a broad range of CSP values (Figure S1). On the basis of the distribution of CSPs across the ET structure, for both the BRD3-ET:TP and BRD3-ET:NSD3 complexes, we assigned only residues with relatively large CSP values ΔδN,H > 0.25 ppm as “active residues”, likely to be in or near the peptide binding site. This conservative threshold is significantly larger than the estimated value of a minimal CSP, ΔδN,H = 0.02 ppm (cited above). These CSPs include 20 and 22 H-N sites, respectively, for the two complexes (Figure S1).
The second data set adds conformational restraints for phi/psi dihedral angle ranges for the bound peptide based on the backbone chemical shifts measured for the bound peptide in the complex, determined with the program TALOS 23,24 (CSP + TALOS data sets). These data require isotope-enrichment of the peptide ligand and NMR assignments for the bound peptide carried out as described previously.7 TALOS-generated peptide dihedral torsion restraints were used in MELD simulations with maximum and minimum values shown in Tables S1 and S2.
The third data set includes the same CSP and dihedral restraint information together with three additional restraints derived from three strong interchain NOEs among backbone and relatively easy to assign sidechain methyl or aromatic resonances (CSP + TALOS + NOE data sets). Generally speaking, significant additional effort is needed to obtain such NOE data, and this third approach was explored largely for the sake of exploring the limits of the MELD + NMR method. Specifically, in this third data set, distance restraints were imposed between Val 24 CG1/Trp 73 HH2, Ile 44 CD1/Ile 84 CD1, and Glu 47 CG/Leu 82 CD2 for the BRD3-ET:TP system and between Ile 44 H/Leu 73 H, Ile 42 H/Ile 75 H, and Val 24 CG1/Phe 86 CD1 for the BRD3-ET:NSD3 system. Simulations were performed by trimming off 19 unstructured residues in the N-terminal region of ET (which are distant from the peptide binding site) and renumbering the resulting trimmed domain sequence to start at residue 1. Thus, the residue numbering convention used in the MELD + NMR calculations uses residues 1 to 68 for the ET receptor (corresponding to residues 573 to 640 of the BRD3 protein), with residues of the peptide binding partner numbered from 69 onward (corresponding residue numbers shown in Table 1).
Computational Methods.
MELD Approach.
MELD is a plugin to the OpenMM29 molecular simulations engine. It integrates data and simulations through Bayesian inference.15 The main advantage is that it accommodates data sources that are ambiguous and/or noisy, making it well-suited for the analysis of protein NMR data. For example, CSP information on the ET receptor protein identifies possible sites of interaction between the protein and peptide but does not distinguish which residues are at the interface and in contact with residues in the bound peptide and which are never in direct contact (e.g., CSPs distant from the binding site can arise from allosteric or propagated structural changes). We thus include all possibilities, knowing that only a small subset will be present in the structure of the complex (see Figure 2). MELD samples through conformations and different implementations (subsets) of the data to produce an ensemble that is compatible with a subset of data and the physics model (given a force field).
MELD Simulations.
As in previous studies, we used an H, T-REMD protocol30 to explore the energy landscape efficiently. We used 30 replicas running for 1.5 μs starting from an unbound folded receptor and extended peptide conformation placed 30 Å away from the receptor. The temperature increases geometrically from 300 K at the lowest replica to 500 K at the 12th replica and is kept constant at 500 K afterward. The Hamiltonian changes according to how strongly we enforce the data [e.g., strongly, k = 350 kJ/(mol nm2), below replica 12th, and with no restraints at the 30th replica—changing nonlinearly in between the 12th and 30th replicas]. The physics model uses the GBNeck2 implicit solvent model31 and a combination of the ff14SB32 (side chains) and ff99SB33 (backbone) force fields.
The CSP data were modeled by all possible combinations between the set of all peptide residues and the set of ET active residues with CSP above the ΔδN,H = 0.25 ppm threshold using a flat-bottom harmonic restraint between Cβ’s of each pair. The restraints added no energy penalty up to 8 Å, and then the energy penalty increased quadratically until 10 Å and linearly beyond, with a force constant of 350 kJ/(mol nm2). The combinatorics leads to many possible restraints, but only a few are present in the bound structure; hence, we assigned a 4% confidence level to this data set. Which restraints are enforced is deterministic: at each timestep, all restraint energies are evaluated, and the 4% lowest energy restraints are enforced until the next timestep. Different replicas can satisfy different sets of restraints. An advantage of MELD is that each different data source can have different confidence values. For example, a second protocol created combinatoric restraints between only the hydrophobic residues in the peptide and the active protein residues. This resulted in a lower number of overall restraints and a higher confidence level (10%).
For the CSP + TALOS data sets, we modeled bound-peptide chemical shift data in a similar manner by enforcing phi and psi dihedral angle restraints based on the minimum and maximum values of backbone dihedral angles consistent with NMR data, as provided by TALOS (see Figure 2 and Tables S1–S6). Modeling of dihedral restraints is based on the TALOS analysis of the chemical shift values for each residue and is therefore of higher accuracy than the ambiguous modeling of CSP data. We set the confidence on this data to 80%. Finally, for CSP + TALOS + NOE data sets, we modeled NOE data with a 5 Å flat-bottom potential between backbone–backbone hydrogens. For NOEs between side-chain–sidechain or backbone–sidechain hydrogens, we mapped the sidechain hydrogen to the corresponding heavy atom and added a 6 Å fat-bottom potential. We used 100% confidence on this data set.
We also applied internal distance restraints to the ET structure in all simulations to avoid protein unfolding at high temperatures in the replica exchange. For this purpose, we calculated all Cα–Cα distances in the apo-protein, selected those closer than 8 Å, and created a restraining potential for each using an 8 Å flat-bottom harmonic potential. To allow for possible conformational changes during binding, both locally to the binding site due to binding plasticity and distant from the binding site due to allosteric changes, we set this data set with a 90% confidence inside MELD. The accuracy parameter in the different data sources plays a critical role in guiding the search in MELD, but it is unknown a priori. Enforcing higher accuracy values results in more restrained systems and thus faster convergence (shorter simulation times). However, if the accuracy value is set too high, the restrained protein structure might not be compatible with all the data, leading to incorrect simulations.
Competitive Binding Study.
Relative binding affinities can be calculated from MELD simulations in which two (or more) peptides compete for the binding site. For this step, we presuppose that the individual structures of both peptide–protein complexes are known or have been determined with the previous approach. The relative binding affinities for BRD3-ET were assessed for the TP and NSD3 peptide sequences shown in Table 1. We selected three possible restraints common to both systems based on the experimentally determined structures of these complexes. In the starting conformation, both peptides were 30 Å away from each other and from the receptor. In this case, the contacts we enforced had to be satisfied by either peptide at lower replicas, whereas both were unbound at higher replicas. We also added restraints to keep the two peptides from interacting with each other. The ratio of the populations of peptide molecules bound in the binding site is related to the relative binding free energy.34
Clustering.
We cluster the last 1 μs of the five lowest temperature replicas using RMSD as a similarity metric with a hierarchical agglomerative clustering algorithm implemented in cpptraj.35 All protocols for complexes use average linkage to calculate the distance between clusters with a 1.5 Å cutoff. For all cases, we have two clustering protocols: one using LRMSD (ligand root mean square deviation)36 calculated considering the whole peptide and another using LRMSD calculated on only the core region of the peptide (excluding floppy terminal regions). We report the centroid of the highest populated cluster as our prediction.
AlphaFold Predictions.
We used the AlphaFold4 advanced colab version (https://github.com/sokrypton/ColabFold)37 in its early implementation with a 30-residue glycine linker as well as a newer protocol with no linker and with the AlphaFold multimer6 to predict the structures of these six complexes.38 From each of these AlphaFold models, five structural models were predicted for each complex and ranked according to their pLDDT scores.4 Related ET-peptide structures already published in the PDB were excluded in the input AlphaFold structure prediction. Each complex model was then refined with the ff99SB forcefield.33 Predictions were in agreement except for JMJD6, which remained unbound in the absence of a linker. Hence, we reported results only from the glycine linker protocol.
Analysis Methods.
Predicted structures were assessed using different state-of-the-art metrics used in the protein–protein or protein–peptide docking communities, including IRMSD, fnat, and ILRMSD.2,36,39 Experimental structures determined by NMR methods were reported as ensembles with 20 models. The medoid conformer of each NMR ensemble was used to calculate each of these metrics. The medoid, or representative, structure for each complex was calculated using the PDBstat software package.7,40
RESULTS
Isothermal Titration Calorimetry Measurements.
Binding affinities of TP and NSD3 peptides to BRD3 ET, Kd of 13 ± 10 nM and 250 ± 150 μM at 10 °C, respectively, were estimated at pH 7.5 and 10 °C by isothermal titration calorimetry measurements (Figure S2). The binding affinity of TP to BRD3 ET at pH 7.5 and 25 °C, Kd of 90 ± 10 nM (Figure S2), is similar to that reported previously, Kd ~150 nM, for BRD4-ET:TP binding at pH 7.0 and 25 °C.22 There is a ~19,000-fold difference in affinities for BRD3 ET between TP and NSD3 peptides at 10 °C. For both cases, the stoichiometries of complex formation were 1:1. The NSD3 peptide showed signs of aggregation at 25 °C, requiring that we restrict our estimate of Kd by ITC to 10 °C.
Blind Studies Distinguish Different Binding Modes.
Figure 3 highlights our results using different data sets for the two blind studies (TP and NSD3 peptides binding to the BRD3 ET domain). In all cases, we report the centroid of the top population cluster from the ensembles. Interestingly, each peptide requires a different amount of data to determine the complex structure accurately. MELD + NMR using CSP for the ET receptor and TALOS backbone dihedral-angle restraints for the bound peptide (i.e., only chemical shift data) successfully identifies both peptides binding through antiparallel strands, with TP forming intermolecular antiparallel beta-sheet interactions with ET along its C-terminal region and NSD3 forming intermolecular beta-sheet interactions through its N-terminal region, resulting in flipped orientations for the peptide hairpin with respect to each other (see Figure 3). These binding modes are in excellent agreement with the experimental NMR structures of the two complexes (IRMSD = 2.28 and 1.97 Å, respectively). For the stronger-binding peptide (TP), CSP on ET data alone were sufficient to provide an accurate binding mode (see Figure 3A), and no data for the bound peptide were needed. Using the CSP on ET data from TP for predicting the structure of the BRD3-ET:NSD3 complex also did not change the predictions (see the Supporting Information (SI)), demonstrating that even with a significantly different binding mode, a single study of CSPs on the receptor can be used to guide binding of other binding peptides using MELD successfully. For both complexes, adding the three distance restraints based on specific, strong intermolecular NOEs does not increase the accuracy of the prediction.
Figure 3.
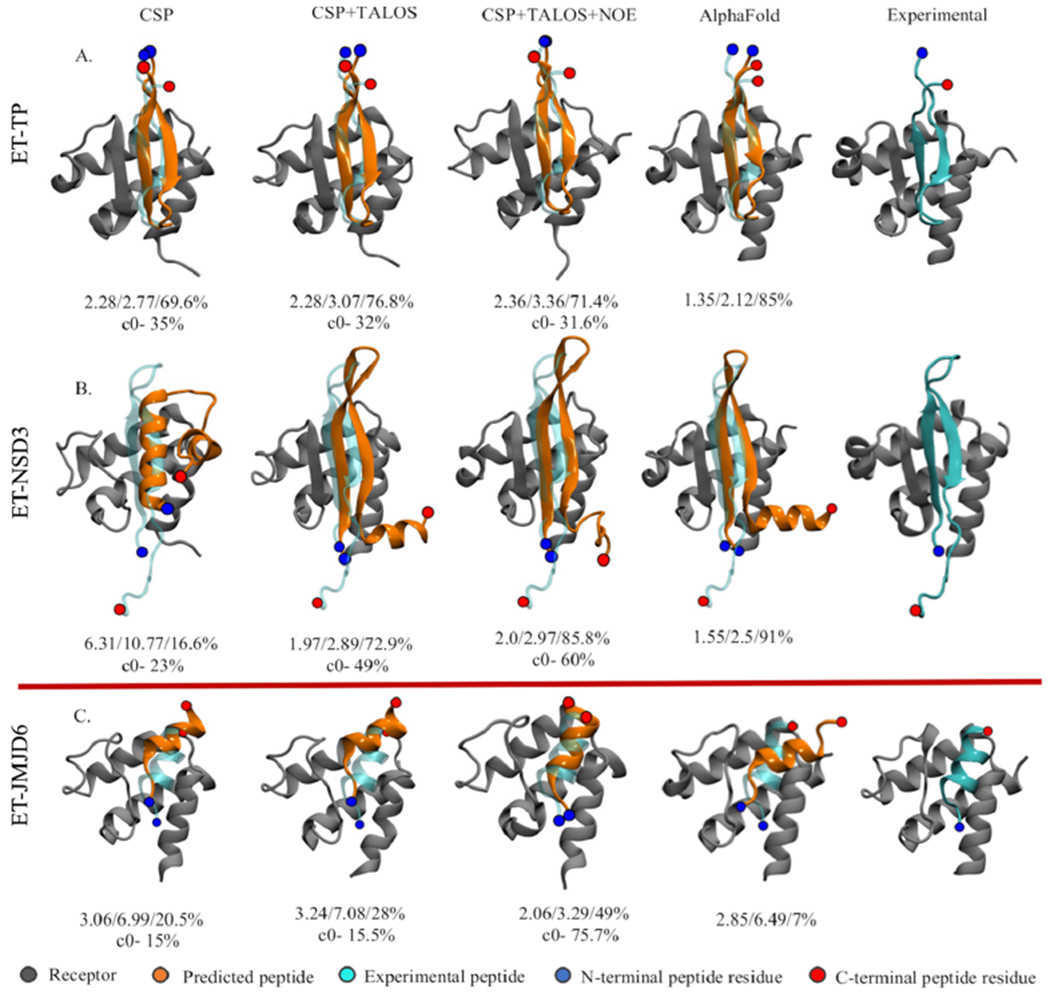
Predicted binding modes. The top panel shows the top MELD prediction using different experimental data sets (first three columns) for the blind study along with AlphaFold (fourth column) predictions overlaid over the experimental structure (cyan): (A) BRD3-TP and (B) BRD3-NSD3. (C) Predictions for the known helical binder BRD4-JMJD6. The numbers below each structure represent IRMSD/ILRMSD/fnat in the first row and the population of the top clusters in the second row.
Interestingly, using no experimental data for these two complexes, AlphaFold performs similarly to chemical-shift guided MELD (IRMSD = 1.35 and 1.55 Å, respectively). For the BRD3-ET:NSD3 complex, AlphaFold also predicts an additional alpha helix in the tail of NSD3, which is unstructured in the experimental NMR structure (see Figure 3 and Figure S3). These results demonstrate the potential to use AlphaFold either to screen for potential binding poses prior to beginning experimental studies, such as NMR-guided MELD modeling, or for providing a validation of the results of MELD + NMR studies.
Funneling of the Binding Landscape in MELD + NMR Ensembles Is Informative of the Expected Accuracy of the Predictions.
Funneling plots capture the ability of MELD ensembles to sample and direct toward the native complex. Each funneling plot shows all the cluster centers in the ensemble as a function of the RMSD to the complex structure (see Figure 4). MELD simulates multiple binding/unbinding events driven by the data. Unbound states are represented as low population clusters sampled at a high replica index (red). At lower replica indexes, the simulation samples bound (green clusters) and misbound (blue clusters) states. We seek approaches that have funneling toward the green regions as seen for the TP peptide. Although the final top representative structure is identified based on clustering on the lower replicas, these funneling plots clustering on all replicas are useful to identify confidence in the results of MELD + NMR and can be used to determine if a docking study can provide a reliable result. When the experimental structure is unknown (e.g., during these blind studies), we use the top scoring cluster as a reference for RMSD calculations. Typically, we run independent simulations satisfying different amounts of contacts between the peptide and protein and use funneling plots to determine the best protocol. We select protocols that lead to more funneled landscapes and higher populations of the top cluster.44 For the TP peptide, different protocols agreed in modeling the same bound conformation, increasing the confidence in our predictions (see Figure S4). For the NSD3 peptide, using CSP data alone, different protocols were not in agreement (see the higher number of misfolded states in Figure 4), with top predictions from different protocols differing by more than 5 Å backbone RMSD between them. Adding backbone dihedral restraints for the bound peptide ligand, determined from chemical shift data on the bound peptide using TALOS,24 narrowed the number of clusters, allowing the identification of a core region of the NSD3 peptide that was bound with a flexible terminal region. This flexible region was responsible for a higher diversity of binding modes and lower populations of the top cluster; clustering on the core region rapidly identified a top cluster with the highest confidence (49% population of the top cluster). A posteriori analysis looking back at the CSP-only data set shows that the correct binding mode was identified as the fifth cluster, what in the docking field is considered a scoring failure. Adding dihedral restraints for the bound peptide reduces the number of clusters by 50% concerning the initial protocol (see Figure S4). Similarly, a posteriori analysis for TP reveals that excluding the disordered terminal region residues from the clustering calculation improved the confidence score for the correct prediction (populations above 55%, see Figure S4). In our experience, when the population of the first cluster is above 30%, MELD predictions correctly represent experimental structures.45
Figure 4.
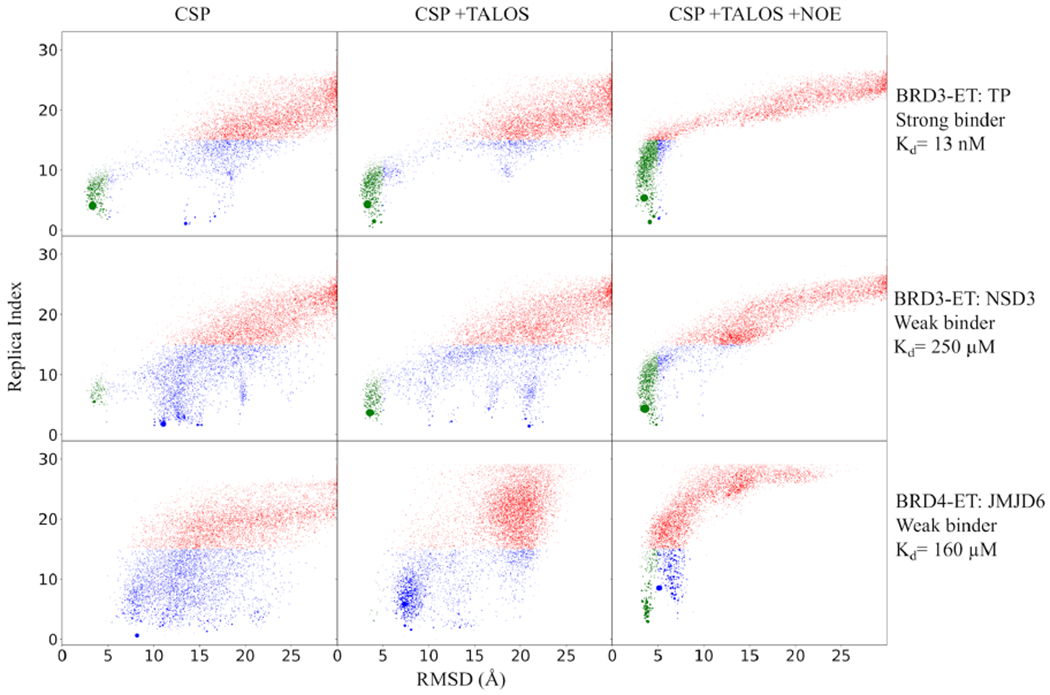
Binding funnel plot. Each point represents the centroid of a cluster center arising from the clustering of MELD ensembles. The size of the cluster is proportional to the population. Each cluster centroid is represented at the average replica index of the cluster to which they belong and the LRMSD of the centroid structure with respect to the experimental one. Clusters sampled at a high replica index are shown in red; clusters sampled at lower replicas are shown in green (LRMSD <5 Å) or blue. Blue represents misbound conformations. Successful simulations have strong funneling behavior and high populations of green clusters. Reported values for Kd for TP and NSD3 at 10 °C based on the current study and for JMJD6 based on literature18 at 20 °C.
Studies on Known Peptide–ET Complexes.
Four additional peptide–ET complexes have been previously experimentally characterized (BRD4-ET:JMJD6, BRD4-ET:-LANA, BRD3-ET:CHD4, and BRD3-ET:BRG1). These four peptides are all much weaker binders than TP (Kd ~16018 ~635,17 ~95,21 and ~7 μM,21 respectively). Like NSD3, binding affinity measurements with ITC for these weaker-binding peptides generally have large uncertainty (see, for example, Figure S2C). Of these, JMJD6 binds as an alpha-helix,18 with the rest binding as single antiparallel β-strands. MELD + NMR calculations were carried out for these published complexes to further explore the interplay between binding affinity, the information content of the data, and MELD’s ability to predict structures of weak binding to very weak binding complexes such as JMJD6 and LANA. JMJD6 is the only solved structure binding as a helix,18 illustrating the broad binding mode plasticity of the ET domain. However, JMJD6 is also a special case, as access to the ET-binding epitope (amino-acid residues 84–96)18 requires a conformational change in the JMJD6 protein structure to expose these residues for binding into the ET cleft.
Some low-affinity complexes exhibit weaker CSP effects on the ET receptor. This challenge was addressed, assuming competitive binding of these peptides with TP, using the CSP data for the BRD3-ET:TP complex in MELD + NMR modeling of these other complexes. Similarly, fast or intermediate exchange on the NMR chemical shift timescale also results in ensemble-averaging of the bound-state peptide chemical shift data, precluding the straightforward determination of the bound-state peptide backbone dihedral angles from these chemical shift data for weakly binding complexes using TALOS (see Tables S7 and S8). In principle, in the intermediate or fast exchange regime of very weak complexes, bound-state peptide backbone chemical shifts can be determined using more sophisticated NMR experiments such as relaxation dispersion (e.g., Carr–Purcell-Meiboom–Gill46 (CPMG)), chemical exchange by saturation transfer (CEST),47 and/or peptide titration experiments. To mimic such NMR data for testing MELD + NMR’s applicability even for weakly binding systems, for these very weak binders, we obtained phi/psi restraint ranges directly from the experimental structures using MDTraj48 (see Tables S3–S6). For consistency with the blind study, we also simulated three strong NOEs based on the experimental structures (see Table S9) for generating intermolecular distance restraints for the CSP + TALOS+NOE data set.
Of the four peptides, MELD recovers the experimental binding modes for tighter-binding CHD4 and BRG1 complexes (Kd < 100 μM) using only CSP on ET data and no data for the bound peptide (see SI and Figure S5), as was observed for the BRD3-ET:TP complex. LANA is the weakest binding peptide in the set (Kd ~635 μM), and using only the CSP on ET data produces incorrect binding modes and peptide conformations. Adding peptide backbone dihedral angle restraints for the LANA peptide in the BRD4-ET:LANA complex suffices to allow MELD + NMR to model a binding mode quite similar to the experimental structure, as was also observed (above) in modeling the BRD3-ET:NSD3 complex (Kd ~250 μM). The accuracy of the BRD4-ET:LANA binding pose is further improved by adding a few simulated NOE restraints (backbone–backbone contacts, see SI and Figure S5), demonstrating the value of such data if they can be obtained.
However, the weak and unique BRD4-ET:JMJD6 complex was more challenging. Here, we selected the BRD4-ET variant over BRD3-ET because previous studies detect weak or no binding to the latter.19 JMJD6 was found to bind as a helix using each of the three different data sets, with a bound-state structure similar to that observed in the experimental structure of this complex.18 However, the helix is displaced from the experimental binding mode when using CSP and CSP + TALOS data. Interestingly, AlphaFold predicts a helical structure for this system for the bound peptide. However, this binding pose is also significantly different from the reported experimental structure (Figure S3). The top five AlphaFold predictions are diverse in binding mode and more closely agree with the MELD predictions than the experimental BRD4-ET:JMJD6 complex structure. As expected, adding three strong-NOE-based distance restraints simulated from the experimental structure to MELD + NMR simulations recovers the experimentally observed binding pose (see Figure 4).
Competitive Binding Simulations Identify TP as a Stronger Binder than NSD3, Consistent with Experimental ITC Measurements.
We recently introduced a competitive binding assay with AlphaFold49 that is able to correctly rank order the strongest binders in the set of peptides studied here. However, this modeling approach does not provide any information about their binding affinities. Although more computationally demanding, MELD calculations sample multiple states, relying on a statistical–mechanical analysis to assess their relative importance; in competitive binding simulations, the populations of each peptide in the active site allow us to calculate the relative binding affinity. Previous results on 20 peptides binding MDM2 and MDMX show that this MELD-based method predicts relative binding affinities for medium and strong binders.34,50 We used this strategy for comparing the binding affinities of the TP and NSD3 peptides for the ET in our blind study. For these simulations, we chose a common set of information to guide each peptide to the binding site (see Methods and Figure 5A). Figure 5B summarizes the population of each peptide in the binding site along each replica. Whereas, at high replica indexes, both peptides are unbound early in the binding process, there is a marked preference for TP binding over NSD3. The latter peptide can sample the binding site multiple times, especially in intermediate replicas, but it is rarely sampled at the lowest replica. On the basis of these results, we predict a relative binding affinity of TP and NSD3 to BRD3-ET, ΔΔGTP/NSD3, of −2.45 ± 0.20 kcal/mol (see SI). The ITC experiments at pH 7.5 demonstrate that the TP peptide is a better binder, with experimental relative binding free energy (at 10 °C) of −5.5 ± 1 kcal/mol (Figure S2) obtained using the Kd values in Table S10. Both experiments and computations agree that the TP peptide is a significantly stronger binder. Although there is good qualitative agreement between calculated and observed binding affinities, the difference reflects in part the high experimental and computational uncertainties associated with estimating the Kd’s of such weak binders (e.g., NSD3).
Figure 5.
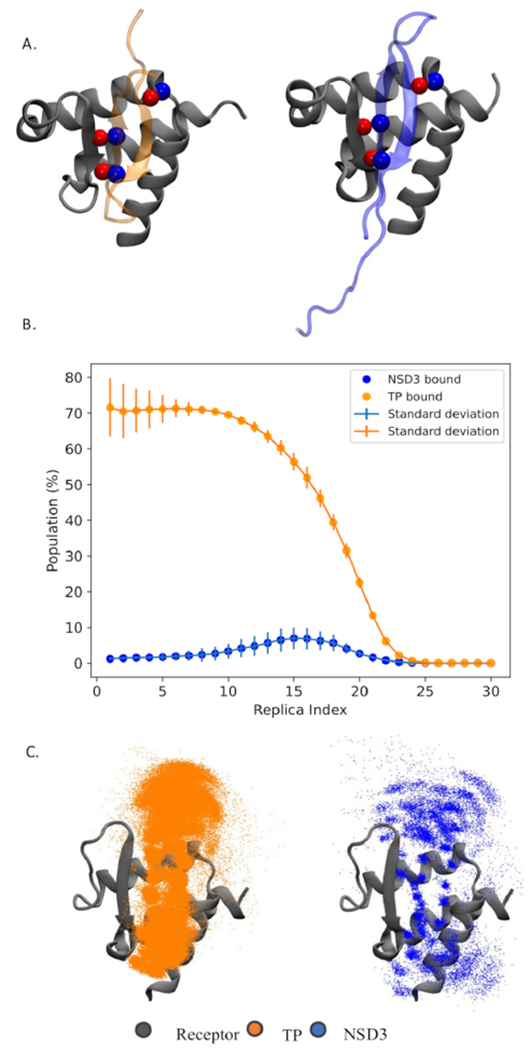
Competitive binding simulations. (A) Three equivalent handpicked contacts used to guide sampling for competitive binding. Red and blue spheres represent oxygen and nitrogen, respectively. (B) Population of native bound conformation for each peptide at each of the 30 replicas (replica index 0 represents the lowest temperature and flat-bottom harmonic restraints fully enforced). The error bars represent the variation in population across three independent simulations. (C) Superposition of snapshots in which the TP peptide (orange) or NSD3 (blue) is found binding the protein in the lowest replica ensemble. Only the Cα’s of the peptide are represented (as dots) for clarity.
DISCUSSION
The protein–protein and peptide–protein docking field has significantly advanced thanks to community efforts such as CAPRI (Critical Assessment of Prediction of Interaction).51,52 However, it is an unsolved challenge to reliably predict binding poses without any experimental data when a large degree of conformational flexibility is involved, which is generally the case for peptides binding to receptors. Such docking studies are particularly challenging for binding receptors like ET that exhibit binding plasticity, where peptides can bind with significantly different conformations. Moreover, although docking can be more successful when the bound-state peptide conformation is known, such docking studies do not provide insights about the entropic cost for peptide folding and, as such, challenge the understanding of how likely it is that a particular peptide sequence would fold into the required conformation for binding.
Our strategy is to develop a reliable physics-based approach for predicting structures and relative energetics of protein receptor–peptide complexes that is guided by low-information-content NMR data, such as backbone chemical shift data. Chemical shift data are particularly attractive for this application because they are (i) a prerequisite for further NMR studies to provide higher-information-content data and (ii) obtained by either solution-state or solid-state NMR studies. Backbone chemical shifts can be determined by solution-state NMR for systems as large as 60–80 kDa53 and potentially for even large complexes using solid-state NMR. For very weak binding systems, these data may need to be supplemented with the strongest backbone/backbone, backbone/methyl, backbone/aromatic, or methyl/methyl NOEs, which can generally be obtained even for modestly large protein–peptide complexes. However, only backbone CSP data for the receptor were required for most of the complexes studied here. These backbone CSP data provide a general guide to MELD for locating the binding hotspot on the protein surface, and they need not be very complete or accurate. This information could even be transferred from one complex to another.
Methods like HADDOCK already use this general CSP approach,54,55 addressing the challenge in interpreting the ambiguity in the CSP data by creating ambiguous contact restraints, and are successful in predicting docking between folded proteins guided by such data. The key advantage of HADDOCK is speed because it is based on docking and heuristic scoring functions. However, the folding-upon-binding nature of protein–peptide complexes makes them particularly challenging systems for docking predictions with all available methods, including HADDOCK, and especially difficult when the receptor can accommodate peptides in different binding modes and different peptide conformations (e.g., helices and strands as observed for ET–peptide complexes). We find that providing peptide ensembles and CSP or CSP + TALOS data is not enough to predict the conformation of the complex with HADDOCK for either TP (see Figure S6) or NSD3 (see Figure S7). When we provide the conformation of the bound peptide (single structure docking), HADDOCK predicts the conformation of the BRD3-ET:TP complex but not that of BRD3-ET:NSD3. In contrast, the MELD + NMR approach uses simulations and statistical mechanics to identify low free energy states and is thus suitable to account for the entropic component of folding upon binding. The main advantage of such an approach is the production of models that agree with both a physical model and experimental data, providing valuable biophysical parameters such as relative free energies of binding.
A unique strength of the MELD + NMR method is that the energy funneling plots (Figure 4) provide an assessment of the reliability of any particular docking study. Given a particular set of experimental chemical shift perturbation data, docking trajectory landscapes that are highly funneled lead to successful predictions. For examples, in Figure 4, the BRD3-ET:TP CSP, BRD3-ET:TP CSP + TALOS, and BRD3-ET:NSD3 CSP + TALOS trajectories are highly funneled and result in accurate docking models, whereas the BRD3-ET:NSD3 CSP trajectory is not highly funneled and does not provide a correct docking model. All of the trajectories in Figure 4 that include some intermolecular contacts are highly funneled and provide reliable docking models. However, it may not generally be possible to obtain such interchain NOE data for challenging protein–peptide complexes for which the CSP + TALOS method, using chemical shift data for both the protein receptor and the bound peptide, is not successful.
In this study, we also observe that AlphaFold can successfully predict structures of these ET–peptide complexes. Similar applications of AlphaFold for peptide docking have also been recently reported by other groups.56 However, the deep-learning-based AlphaFold models do not provide quantitative information about relative binding affinities,49 which is a natural product of MELD binding simulations. Novel research on the applicability and limitations of AlphaFold is starting to provide qualitative insights into binding affinities.49,57 In the case of the BRD4-ET:JMJD6 complex, AlphaFold returns multiple models with different binding poses. In this sense, NMR-guided MELD + NMR and data-independent AlphaFold calculations provide both validating and complementary information for accurately modeling receptor–peptide complexes.
Here, we demonstrate a successful approach for accurately modeling protein–peptide complexes by combining NMR backbone chemical shift data and MELD simulations. The goals of the blind study are multiple: (i) determine if the method is successful, (ii) assess its sensitivity to sequence and conformation, (iii) determine the amount of data needed for confident determination of the structures, and (iv) assess if MELD + NMR simulations can provide reliable relative free energies of binding. We observe that for tight (Kd < 1 μM) and even for some of the weak ET–peptide complexes, MELD + NMR can reliably predict bound-state conformations of the peptide using only backbone chemical shift data for the protein receptor (CSP). Specifically, in the best cases of the BRD3-ET:TP, BRD3-ET:BRG1, and BRD3-ET:CHD4 complexes, only CSP data on the receptor (i.e., ET) side of the complex were sufficient for an accurate NMR-guided MELD docking. For weaker complexes, some additional data are also required for the bound-state conformation of the peptide; e.g., for the BRD3-ET:NSD3 and BRD3-ET:LANA complexes, accurate modeling also requires backbone chemical shifts for the bound peptide, which are used to define backbone dihedral restraints with TALOS. Significantly, the method is successful in blind binding studies involving very different binding modes; e.g., the TP peptide and NSD3 peptide both bind ET as beta hairpins but bind in “flipped” orientations (see Figure 1). However, for some of the weaker complexes studied here, a few additional intermolecular distance restraints improve the modeling of the complex. The JMJD6 peptide is an outlier in this set, as it is reported to bind BRD4-ET more strongly than NSD3 and LANA bind BRD3-ET, yet our methods do not accurately predict some details of this complex (see below). Finally, MELD provides a framework to calculate relative free energies of binding for TP versus NSD3. Although we observe good agreement across replicas from our calculations (ΔΔGTP/NSD3 of −2.45 ± 0.20), the accuracy with respect to experiments is not great (3.05 kcal/mol difference). The discrepancy in accuracy is not surprising, as even the accuracy in state-of-the-art calculations for small molecule binders is around 1–2 kcal/mol,58 and methods for flexible binders, such as those studied here, have not yet achieved this level of accuracy. Despite this, it is encouraging to find qualitative agreement for this system, as we have observed for other systems using MELD competitive binding strategies.34,50
In binding to peptides, the ET domain exhibits CSPs throughout the domain structure, reflecting an allosteric conformational change propagated across most domains.7 The biological significance of these structural changes resulting from partner binding is not yet understood. As expected, the unstructured N-terminal segment of ET, which remains unstructured in the complex, does not exhibit CSPs due to peptide binding and/or allosteric changes.7 The CSP data in Figure S1 show insignificant chemical shift changes in this N-terminal unstructured region of ET, not involved in binding, where ΔδN,H chemical shift changes between apo and peptide-bound forms are all <0.02 ppm. For peptide docking, we set the threshold CSP as >0.25 ppm, a threshold value that is enough to capture the highest perturbations, while bearing in mind that smaller ΔδN,H CSPs (0.02–0.25 ppm) are also significant changes arising from allosteric changes, protein–peptide interaction, or both effects. As seen in Figure 2 and Figure S1, with this CSP threshold, we identify ~20 residues as potentially involved in peptide binding, but MELD can correctly identify that only a subset of these contacts is present in the peptide binding epitope of ET, with the rest of these CSPs corresponding to conformational changes of ET (both in and distant from the binding site) that accompany peptide binding. Hence, MELD + NMR does not require complete or highly accurate backbone CSP data to guide the modeling of the complex.
The ET-JMJD6 complex was especially challenging to model. Using CSP data transferred from the BRD3-ET:TP system to the BDR4-ET:JMJD6 complex (Kd ~160 μM18), MELD + NMR docking simulations result in a binding pose similar to the experimental structure, except that the binding mode is shifted by ~6 Å RMSD relative to the native pose. Similar results were obtained using the BRD3-ET domain in the MELD + NMR docking calculations and for models of this complex returned by AlphaFold (Figure S8). We also performed explicit solvent molecular dynamics simulations starting from the experimental NMR structure (without restraints). These simulations also exhibited shifting of the peptide out of the hydrophobic cleft of ET after 100 ns of simulation (Figure S8). Whereas the experimental structure shows a tryptophan residue of the JMJD6 buried in the hydrophobic cleft of the highly homologous BRD4-ET domain, this is not observed in the MELD + NMR, AlphaFold, or explicit solvent simulations starting from the experimental structure. These results suggest that the displaced pose observed in both MELD + NMR and AlphaFold might represent a true conformational state of the complex that is not modeled by the published experimental NMR structure analysis. This discrepancy could arise from well-known challenges in interpreting ensemble-averaged NMR data. Simulations provide the structure with the most weight in the Boltzmann ensemble. NOESY experiments yield peaks in the spectra when two atoms are close in space. The intensity of the peaks is an ensemble average over the experimental observable, which rapidly decays with increasing distance between the two atoms (as <1/r6>).59,60 In practical terms, short distances between two atoms have a stronger weight in conventional NMR structure determination protocols than longer distances. A structure exhibiting a long distance between two atoms 70% of the time and a close distance 30% of the time may be interpreted by conventional modeling methods as a short distance in one single (or dominant) conformation present in the sample. We believe that this could be of special importance in the case of weak peptide binders, such as JMJD6. If multiple binding modes are present, some of the NOEs observed may arise from a minor population with short interproton distances. Satisfying these NOE-based distance restraints could then overweight the representation of these structures in the final ensemble reported by traditional NMR-based modeling methods. This is a general challenge for docking studies using NOE data. It provides a good illustration of why NMR-guided MELD + NMR protocols using exclusively chemical shift data may be even better suited for modeling the dominant poses of peptide–protein complexes than conventional methods using such ensemble-averaged NOE data.
In developing MELD + NMR, we carefully considered the accessibility of experimental NMR data needed to model complexes successfully. The method assumes extensive (though not necessarily complete) backbone 15N-1H CSPs for the receptor protein, which can generally be obtained for systems of up to 60–80 kDa. For studies of a series of peptides binding to a common receptor, it may often be sufficient to use the CSP data from one complex in modeling a second complex with a peptide known to compete for binding with the peptide of the first complex, as shown here for the BRD3-ET:TP and BRD3-ET:NSD3 complexes. In some cases, it is also helpful to have backbone chemical shift data for the bound peptide for generating backbone dihedral angle restraints, requiring the production of isotope-enriched peptides. Several recently described peptide–protein fusion systems facilitate the high-level production of isotope-enriched peptides.7 Although not the focus of the present study, other types of NMR data, such as residue dipolar couplings (RDCs) or paramagnetic relaxation enhancements (PREs), could be valuable to orient the peptide concerning the protein secondary structure elements,61 albeit with the same caveats for the effects of dynamic averaging in weaker binding complexes discussed above for chemical shift and NOE data. The nature of such data requires new restraint types and replica exchange optimizations in MELD, which are planned developments for future work.
A few key caveats should be emphasized. For some flexible, dynamic protein–peptide complexes, it may be difficult to determine backbone resonance assignments needed for CSP measurements even when using a perdeuterated 15N,13C,2H-enriched protein sample. It is one step more challenging to determine backbone 15N,13C,1H resonance assignments of a bound isotope-enriched peptide needed to determine dihedral angle restraints in the CSP + TALOS method required for weaker binding peptides. Hence, the approach outlined here is limited to cases where extensive backbone (though generally not sidechain) resonance assignments can be obtained for the protein receptor and (for weaker complexes) the isotope-enriched bound peptide. It remains to be seen how many systems meet these requirements. For complexes that do not provide strongly funneled trajectories using chemical shift data alone, like the ET–JMJD6 complex (Figure 4), more sharply funneled docking trajectories may sometimes be obtained given a few strong intermolecular NOE-based contacts. However, this is not a generally usable approach for the MELD + NMR method, as it may in fact be difficult to obtain required sidechain resonance assignments, or any experimental distance restraints at all, for very weak and/or large protein–peptide complexes.
The eruption of machine learning into the field of structural biology has many exciting prospects. We took advantage of the AlphaFold pipeline as an orthogonal computational approach. Although AlphaFold was not originally designed for the purpose of peptide binding, a recent report shows that a tethered protein–peptide method using AlphaFold, similar to the approach used in this work, is about ~40% accurate in peptide–protein docking benchmarks.56 The AlphaFold predictions for three of the six systems analyzed in detail here were remarkably good matches to the MELD + NMR models. In particular, both methods provide accurate models of both the BRD3-ET:TP and BRD3-ET:NSD3 complexes (see Figure 3). For the JMJD6 peptide, both methods modeled a helix as the bound-state peptide conformation, consistent with the experimental structure, and both predicted a binding mode displaced with respect to the experimentally reported binding site (Figure S3). AlphaFold can capture the overall native structure for the three other systems studied. However, caution is needed as some of the complexes predicted with AlphaFold exhibit a one- or two-residue register shift in the strand pairing between the protein and the peptide with respect to the experimental structure (Figure S9). MELD + NMR, however, provided models for these complexes in excellent agreement with the experimental structures. Overall, we take these predictions with optimism that deep-learning approaches such as AlphaFold can be used to filter and identify where and how peptides are likely to interact with a protein receptor. However, as they are predictions, they need to be compared with at least some experimental data before building studies based on these models. The MELD + NMR approach used here is more computationally expensive at run time but produces models that are already compatible with experimental NMR data.
MELD + NMR accounts for peptide conformational entropic preferences through sampling and can be exploited for virtual competitive binding studies to assess binding affinities. These types of calculations are, however, subject to convergence issues. We have previously found that when competing peptides with large differences in binding affinities or competing with weak binders, the calculations are more qualitative than quantitative.34,50 In our blind study, MELD predicted the TP to be a stronger binder than NSD3, in good agreement with experiments. However, for the large difference in experimental binding affinities (−5.5 ± 1 kcal/mol), we should not expect quantitative predictions from MELD.34 Recently, we have shown the ability to carry competitive binding simulations with AlphaFold as a higher-throughput methodology.49 In this mode, AlphaFold provides a qualitative answer when comparing two peptides: either similar binding affinities or one peptide favored for binding. Powerful strategies will combine both methods in assessing peptide ligand candidates, where AlphaFold can first filter peptides predicted to bind and then perform competitive binding to evaluate the expected top binders. Finally, MELD can provide quantitative ranking among the top binders. This strategy will thus be useful to inform which peptides are worth exploring with more expensive experimental NMR studies.
CONCLUSIONS
The novel MELD + NMR protocol produces valuable insights and predictions for the problem of polypeptide folding upon binding, an important and challenging area of computational modeling. We have shown reliable predictions for tight and even for relatively weak peptide–protein complexes using only backbone chemical shift perturbation data for the receptor. For weaker complexes, the method is enabled by bound-state chemical shift data, which provide dihedral angle restraints. However, a few limitations are apparent. The current protocols struggle to find the correct bound-state conformation for some weaker binders without at least some NOE-based distance restraints. This is attributable, at least in part, to conformational averaging between the bound and free state in very weak complexes, which confounds the interpretation of NMR data. Generally speaking, weak peptide binders are very challenging for structural analysis using any experimental method. However, future work will improve performance in weak binders where several conformations might be contributing to the binding affinity. This tandem experimental/computational approach can also be useful for rational peptide design, where a large number of peptide designs directed to a common receptor can be rapidly screened for potential complex formation with AlphaFold and then tested more rigorously using MELD + NMR.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Theresa Ramelot for useful suggestions in the course of this study.
Funding
This work was supported by National Institutes of Health grants R35 GM122518 (to M.J.R.), R01 GM120574 (to G.T.M.), and R35 GM141818 (to G.T.M.). A.P. is grateful for funding provided by the American Cancer Society (AWD11297, GM01432).
ABBREVIATIONS
- BET
bromo and extraterminal domain protein
- BRD3 and BRD4
specific BET proteins
- CSP
chemical shift perturbation
- ET
extraterminal domain (residues 554–640) of human BRD3
- MELD
Modeling Employing Limited Data
- NSD3
peptide fragment (residues 148–184) of the human nuclear receptor binding SET domain protein 3
- TP
peptide corresponding to 22 C-terminal residues of murine ET
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.2c01595
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.2c01595.
Supplementary results; CSP values for each residue of the ET domain (in this case, BRD3 ET) (Figure S1); ITC results for TP and NSD3 binding to BRD3 ET receptor (Figure S2); similarity between MELD and AlphaFold result for NSD3 and JMJD6 binding (Figure S3); confidence and diversity in the prediction of TP and NSD3 binding (Figure S4); MELD and AlphaFold predictions for LANA, BRG1, and CHD4 binding to ET domain (Figure S5); MELD and HADDOCK prediction comparison for TP and NSD3 binding (Figure S6 and S7); comparison of the orientation of tryptophan in JMJD6 in MELD, AF2, experimental and explicit solvent simulation of experimental conformation (Figure S8); residue pairing comparison of MELD and AF2 results (Figure S9); dihedral angle information of TP peptide, predicted from CSP (Table S1); dihedral angle information of NSD3 peptide, predicted from CSP (Table S2); dihedral angle information of JMJD6 peptide, calculated from the native structure (Table S3); dihedral angle information of LANA peptide, calculated from the native structure (Table S4); dihedral angle information of BRG1 peptide, calculated from the native structure (Table S5); dihedral angle information of CHD4 peptide, calculated from the native structure (Table S6); dihedral angle information of JMJD6 peptide, predicted from CSP (Table S7); dihedral angle information of LANA peptide, predicted from CSP (Table S8); NOEs used in MELD simulation for JMJD6 (Table S9); and thermodynamic properties of TP and NSD3 obtained from ITC experiment (Table S10) (PDF)
The authors declare the following competing financial interest(s): GTM is a founder of Nexomics Biosciences, Inc. This affiliation is not a competing interest with respect to this study. The remaining authors declare no competing interests.
Contributor Information
Arup Mondal, The Quantum Theory Project, Department of Chemistry, University of Florida, Gainesville, Florida 32611, United States.
G.V.T. Swapna, Department of Pharmacology, Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, United States; Department of Chemistry and Chemical Biology, Center for Biotechnology and Interdisciplinary Sciences, Rensselaer Polytechnic Institute, Troy, New York 12180, United States
Maria M. Lopez, Department of Chemistry and Chemical Biology, Center for Biotechnology and Interdisciplinary Sciences, Rensselaer Polytechnic Institute, Troy, New York 12180, United States
Laura Klang, Department of Chemistry and Chemical Biology, Center for Biotechnology and Interdisciplinary Sciences, Rensselaer Polytechnic Institute, Troy, New York 12180, United States.
Jingzhou Hao, Department of Chemistry and Chemical Biology, Center for Biotechnology and Interdisciplinary Sciences, Rensselaer Polytechnic Institute, Troy, New York 12180, United States.
LiChung Ma, Department of Pharmacology, Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, United States.
Monica J. Roth, Department of Pharmacology, Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, United States
Gaetano T. Montelione, Department of Chemistry and Chemical Biology, Center for Biotechnology and Interdisciplinary Sciences, Rensselaer Polytechnic Institute, Troy, New York 12180, United States
Alberto Perez, The Quantum Theory Project, Department of Chemistry, University of Florida, Gainesville, Florida 32611, United States.
Data Availability Statement
The MELD software used in this work is freely available on github (https://github.com/maccallumlab/meld), and experimental NMR data are accessible at the BioMagResDataBank with the accession codes indicated in the manuscript. Trajectories are available from the authors on request.
REFERENCES
- (1).Pu C; Yan G; Shi J; Li R Assessing the Performance of Docking Scoring Function, FEP, MM-GBSA, and QM/MM-GBSA Approaches on a Series of PLK1 Inhibitors. Medchemcomm 2017, 8, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Weng G; Gao J; Wang Z; Wang E; Hu X; Yao X; Cao D; Hou T Comprehensive Evaluation of Fourteen Docking Programs on Protein–Peptide Complexes. J. Chem. Theory Comput 2020, 16, 3959–3969. [DOI] [PubMed] [Google Scholar]
- (3).Agrawal P; Singh H; Srivastava HK; Singh S; Kishore G; Raghava GPS. Benchmarking of Different Molecular Docking Methods for Protein-Peptide Docking. Bmc Bioinformatics 2019, 19, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Baek M; DiMaio F; Anishchenko I; Dauparas J; Ovchinnikov S; Lee GR; Wang J; Cong Q; Kinch LN; Schaeffer RD; Millán C; Park H; Adams C; Glassman CR; DeGiovanni A; Pereira JH; Rodrigues AV; van Dijk AA; Ebrecht AC; Opperman DJ; Sagmeister T; Buhlheller C; Pavkov-Keller T; Rathinaswamy MK; Dalwadi U; Yip CK; Burke JE; Garcia KC; Grishin NV; Adams PD; Read RJ; Baker D Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Evans R; O’Neill M; Pritzel A; Antropova N; Senior A; Green T; Žídek A; Bates R; Blackwell S; Yim J; Ronneberger O; Bodenstein S; Zielinski M; Bridgland A; Potapenko A; Cowie A; Tunyasuvunakool K; Jain R; Clancy E; Kohli P; Jumper J; Hassabis D Protein Complex Prediction with AlphaFold-Multimer. Biorxiv 2021, 2021, 04.463034. [Google Scholar]
- (7).Aiyer S; Swapna GVT; Ma L-C; Liu G; Hao J; Chalmers G; Jacobs BC; Montelione GT; Roth MJ A Common Binding Motif in the ET Domain of BRD3 Forms Polymorphic Structural Interfaces with Host and Viral Proteins. Structure 2021, 29, 1–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bowman GR Accurately Modeling Nanosecond Protein Dynamics Requires at Least Microseconds of Simulation. J. Comput. Chem 2016, 37, 558–566. [DOI] [PubMed] [Google Scholar]
- (9).Pan AC; Xu H; Palpant T; Shaw DE Quantitative Characterization of the Binding and Unbinding of Millimolar Drug Fragments with Molecular Dynamics Simulations. J. Chem. Theory Comput 2017, 13, 3372–3377. [DOI] [PubMed] [Google Scholar]
- (10).Paul F; Wehmeyer C; Abualrous ET; Wu H; Crabtree MD; Schöneberg J; Clarke J; Freund C; Weikl TR; Noé F Protein-Peptide Association Kinetics beyond the Seconds Timescale from Atomistic Simulations. Nat. Commun 2017, 8, 1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ribeiro; Tiwary P. Toward Achieving Efficient and Accurate Ligand-Protein Unbinding with Deep Learning and Molecular Dynamics through RAVE. J. Chem. Theory Comput. 2019, 15, 708–719. [DOI] [PubMed] [Google Scholar]
- (12).Zhou G; Pantelopulos GA; Mukherjee S; Voelz VA Bridging Microscopic and Macroscopic Mechanisms of P53-MDM2 Binding with Kinetic Network Models. Biophys. J 2017, 113, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dickson A. Mapping the Ligand Binding Landscape. Biophys. J 2018, 115, 1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zwier MC; Pratt AJ; Adelman JL; Kaus JW; Zuckerman DM; Chong LT Efficient Atomistic Simulation of Pathways and Calculation of Rate Constants for a Protein-Peptide Binding Process: Application to the MDM2 Protein and an Intrinsically Disordered P53 Peptide. J. Phys. Chem. Lett 2016, 7, 3440–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).MacCallum JL; Perez A; Dill K Determining Protein Structures by Combining Semireliable Data with Atomistic Physical Models by Bayesian Inference. Proc. Natl. Acad. Sci 2015, 112, 6985–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Seffernick JT; Lindert S Hybrid Methods for Combined Experimental and Computational Determination of Protein Structure. J Chem Phys 2020, 153, 240901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang Q; Zeng L; Shen C; Ju Y; Konuma T; Zhao C; Vakoc CR; Zhou M-M Structural Mechanism of Transcriptional Regulator NSD3 Recognition by the ET Domain of BRD4. Structure 2016, 24, 1201–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Konuma T; Yu D; Zhao C; Ju Y; Sharma R; Ren C; Zhang Q; Zhou M-M; Zeng L Structural Mechanism of the Oxygenase JMJD6 Recognition by the Extraterminal (ET) Domain of BRD4. Sci. Rep 2017, 7, 16272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rahman S; Sowa ME; Ottinger M; Smith JA; Shi Y; Harper JW; Howley PM The Brd4 Extraterminal Domain Confers Transcription Activation Independent of PTEFb by Recruiting Multiple Proteins, Including NSD3. Mol. Cell. Biol 2011, 31, 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Aiyer S; Swapna GVT; Malani N; Aramini JM; Schneider WM; Plumb MR; Ghanem M; Larue RC; Sharma A; Studamire B; Kvaratskhelia M; Bushman FD; Montelione GT; Roth MJ Altering Murine Leukemia Virus Integration through Disruption of the Integrase and BET Protein Family Interaction. Nucleic Acids Res. 2014, 42, 5917–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wai DCC; Szyszka TN; Campbell AE; Kwong C; Wilkinson-White LE; Silva APG; Low JKK; Kwan AH; Gamsjaeger R; Chalmers JD; Patrick WM; Lu B; Vakoc CR; Blobel GA; Mackay JP The BRD3 ET Domain Recognizes a Short Peptide Motif through a Mechanism that is Conserved across Chromatin Remodelers and Transcriptional Regulators. J. Biol. Chem 2018, 293, 7160–7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Crowe BL; Larue RC; Yuan C; Hess S; Kvaratskhelia M; Foster MP Structure of the Brd4 ET Domain Bound to a C-Terminal Motif from γ-Retroviral Integrases Reveals a Conserved Mechanism of Interaction. Proc. Natl. Acad. Sci. (USA) 2016, 113, 2086–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Shen Y; Bax A Protein Structural Information Derived from NMR Chemical Shift with the Neural Network Program TALOS-N. Artif. Neural Networks 2015, 1260, 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Shen Y; Delaglio F; Cornilescu G; Bax A TALOS+: A Hybrid Method for Predicting Protein Backbone Torsion Angles from NMR Chemical Shifts. J. Biomol. NMR 2009, 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jansson M; Li Y-C; Jendeberg L; Anderson S; Montelione GT; Nilsson B High-Level Production of Uniformly 15N-and 13C-Enriched Fusion Proteins in Escherichia Coli. J. Biomol. NMR 1996, 7, 131–141. [DOI] [PubMed] [Google Scholar]
- (26).Ulrich EL; Akutsu H; Doreleijers JF; Harano Y; Ioannidis YE; Lin J; Livny M; Mading S; Maziuk D; Miller Z; Nakatani E; Schulte CF; Tolmie DE; Wenger RK; Yao H;Markley JL BioMagResBank. Nucleic Acids Res. 2007, 36, D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ma L-C; Guan R; Hamilton K; Aramini JM; Mao L; Wang S; Krug RM; Montelione GT A Second RNA-Binding Site in the NS1 Protein of Influenza B Virus. Structure 2016, 24, 1562–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Anthis NJ; Clore GM Sequence-specific Determination of Protein and Peptide Concentrations by Absorbance at 205 Nm. Protein Sci. 2013, 22, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y ; Beauchamp KA; Wang L-P; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput. Biol 2017, 13, No. e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sugita Y; Okamoto Y Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- (31).Nguyen H; Roe DR; Simmerling C Improved Generalized Born Solvent Model Parameters for Protein Simulations. J. Chem. Theory Comput 2013, 9, 2020–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins: Struct., Funct., Bioinf 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Morrone JA; Perez A; MacCallum J; Dill KA Computed Binding of Peptides to Proteins with MELD-Accelerated Molecular Dynamics. J. Chem. Theory Comput 2017, 13, 870–876. [DOI] [PubMed] [Google Scholar]
- (35).Roe DR; Cheatham TE III PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- (36).Lensink MF; Méndez R; Wodak SJ Docking and Scoring Protein Complexes: CAPRI 3rd Edition. Proteins Struct. Funct. Bioinform 2007, 69, 704–718. [DOI] [PubMed] [Google Scholar]
- (37).Mirdita M; Schütze K; Moriwaki Y; Heo L; Ovchinnikov S ; Steinegger M ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tsaban T; Varga JK; Avraham O; Ben-Aharon Z; Khramushin A; Schueler-Furman O Harnessing Protein Folding Neural Networks for Peptide–Protein Docking. Nat. Commun 2022, 13, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lensink MF; Brysbaert G; Nadzirin N; Velankar S; Chaleil RAG; Gerguri T; Bates PA; Laine E; Carbone A; Grudinin S; Kong R; Liu R; Xu X; Shi H; Chang S; Eisenstein M; Karczynska A; Czaplewski C; Lubecka E; Lipska A; Krupa P; Mozolewska M; Golon Ł; Samsonov S; Liwo A; Crivelli S; Pagès G; Karasikov M; Kadukova M; Yan Y; Huang S; Rosell M; Rodríguez-Lumbreras LA; Romero-Durana M; Díaz-Bueno L; Fernandez-Recio J; Christoffer C; Terashi G; Shin W; Aderinwale T; Subraman SRMV; Kihara D; Kozakov D; Vajda S; Porter K; Padhorny D; Desta I; Beglov D; Ignatov M; Kotelnikov S; Moal IH; Ritchie DW; de Beauchêne IC; Maigret B; Devignes M; Echartea MER; Barradas-Bautista D; Cao Z; Cavallo L; Oliva R; Cao Y; Shen Y; Baek M; Park T; Woo H; Seok C; Braitbard M; Bitton L; Scheidman-Duhovny D; Dapkūnas J; Olechnovič K; Venclovas Č; Kundrotas PJ; Belkin S; Chakravarty D; Badal VD; Vakser IA; Vreven T; Vangaveti S; Borrman T; Weng Z; Guest JD; Gowthaman R; Pierce BG; Xu X; Duan R; Qiu L; Hou J; Merideth BR; Ma Z; Cheng J; Zou X; Koukos PI; Roel-Touris J; Ambrosetti F; Geng C; Schaarschmidt J; Trellet ME; Melquiond ASJ; Xue L; Jiménez-García B; Noort CW; Honorato RV; Bonvin AMJJ; Wodak SJ Blind Prediction of Homo- and Hetero-protein Complexes: The CASP13-CAPRI Experiment. Proteins Struct Funct Bioinform 2019, 87, 1200–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Tejero R; Snyder D; Mao B; Aramini JM; Montelione GT PDBStat: A Universal Restraint Converter and Restraint Analysis Software Package for Protein NMR. J. Biomol. NMR 2013, 56, 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).He C; Li F; Zhang J; Wu J; Shi Y The Methyltransferase NSD3 Has Chromatin-Binding Motifs, PHD5-C5HCH, that are Distinct from Other NSD (Nuclear Receptor SET Domain) Family Members in Their Histone H3 Recognition*. J Biol Chem 2013, 288, 4692–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nitarska J; Smith JG; Sherlock WT; Hillege MMG; Nott A; Barshop WD; Vashisht AA; Wohlschlegel JA; Mitter R; Riccio A A Functional Switch of NuRD Chromatin Remodeling Complex Subunits Regulates Mouse Cortical Development. Cell Rep. 2016, 17, 1683–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Tang L; Nogales E; Ciferri C Structure and Function of SWI/SNF Chromatin Remodeling Complexes and Mechanistic Implications for Transcription. Prog. Biophysics. Mol. Biol 2010, 102, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Mondal A; Perez A Simultaneous Assignment and Structure Determination of Proteins From Sparsely Labeled NMR Datasets. Front. Mol. Biosci 2021, 8, 774394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Robertson JC; Perez A; Dill K MELD × MD Folds Nonthreadables, Giving Native Structures and Populations. J. Chem. Theory Comput 2018, 14, 6734–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Vallurupalli P; Hansen DF; Lundström P; Kay LE CPMG Relaxation Dispersion NMR Experiments Measuring Glycine 1Hα and 13Cα Chemical Shifts in the ‘Invisible’ Excited States of Proteins. J. Biomol. NMR 2009, 45, 45–55. [DOI] [PubMed] [Google Scholar]
- (47).Vallurupalli P; Bouvignies G; Kay LE Studying “Invisible” Excited Protein States in Slow Exchange with a Major State Conformation. J. Am. Chem. Soc 2012, 134, 8148–8161. [DOI] [PubMed] [Google Scholar]
- (48).McGibbon RT; Beauchamp KA; Harrigan MP; Klein C; Swails JM; Hernández CX; Schwantes CR; Wang L-P; Lane TJ; Pande VS MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J 2015, 109, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Chang L; Perez A Ranking Peptide Binders by Affinity with AlphaFold. Angew. Chem. Int. Ed 2023, 62, e202213362. [DOI] [PubMed] [Google Scholar]
- (50).Morrone JA; Perez A; Deng Q; Ha SN; Holloway MK; Sawyer TK; Sherborne BS; Brown FK; Dill KA Molecular Simulations Identify Binding Poses and Approximate Affinities of Stapled α-Helical Peptides to MDM2 and MDMX. J. Chem. Theory Comput 2017, 13, 863–869. [DOI] [PubMed] [Google Scholar]
- (51).Nadaradjane AA; Quignot C; Traoré S; Andreani J; Guerois R Docking Proteins and Peptides under Evolutionary Constraints in Critical Assessment of PRediction of Interactions Rounds 38 to 45. Proteins Struct. Funct. Bioinform 2020, 88, 986–998. [DOI] [PubMed] [Google Scholar]
- (52).Janin J; Henrick K; Moult J; Eyck LT; Sternberg MJE; Vajda S; Vakser I; Wodak SJ CAPRI: A Critical Assessment of PRedicted Interactions. Proteins Struct. Funct. Bioinform 2003, 52, 2–9. [DOI] [PubMed] [Google Scholar]
- (53).Foster MP; McElroy CA; Amero CD Solution NMR of Large Molecules and Assemblies. Biochemistry 2007, 46, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).de Vries SJ; van Dijk M; Bonvin AMJJ The HADDOCK Web Server for Data-Driven Biomolecular Docking. Nat. Protoc 2010, 5, 883–897. [DOI] [PubMed] [Google Scholar]
- (55).Dominguez C; Boelens R; Bonvin AMJJ HADDOCK: A Protein-Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc 2003, 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- (56).Ko J; Lee J Can AlphaFold2 Predict Protein-Peptide Complex Structures Accurately? Biorxiv 2021, DOI: 10.1101/20210727453972. [DOI] [Google Scholar]
- (57).Roney JP; Ovchinnikov S State-of-the-Art Estimation of Protein Model Accuracy Using AlphaFold. Phys. Rev. Lett 2022, 129, 238101. [DOI] [PubMed] [Google Scholar]
- (58).Schindler CEM; Baumann H; Blum A; Böse D; Buchstaller H-P; Burgdorf L; Cappel D; Chekler E; Czodrowski P; Dorsch D; Eguida MKI; Follows B; Fuchß T; Grädler U; Gunera J; Johnson T; Lebrun CJ; Karra S; Klein M; Knehans T; Koetzner L; Krier M; Leiendecker M; Leuthner B; Li L; Mochalkin I; Musil D; Neagu C; Rippmann F; Schiemann K;Schulz R; Steinbrecher T; Tanzer E-M; Lopez AU; Follis AV; Wegener A; Kuhn D Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects. J. Chem. Inf. Model 2020, 60, 5457–5474. [DOI] [PubMed] [Google Scholar]
- (59).Gáspári Z; Perczel A Chapter 2 Protein Dynamics as Reported by NMR. Annu. Rep. NMR Spectrosc 2010, 71, 35–75. [Google Scholar]
- (60).Makriyannis A; Pavlopoulos S Encyclopedia of Spectroscopy and Spectrometry. Magnetic Reson Article Titles S 2017, 306–315. [Google Scholar]
- (61).Lipsitz RS; Tjandra N. Residual Dipolar Couplings in NMR Structure Analysis. Annu. Rev. Biophys 2004, 33, 387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MELD software used in this work is freely available on github (https://github.com/maccallumlab/meld), and experimental NMR data are accessible at the BioMagResDataBank with the accession codes indicated in the manuscript. Trajectories are available from the authors on request.