

Summary
Engineering the genetic code of an organism has been proposed to provide a firewall from natural ecosystems by preventing viral infections and gene transfer1–6. However, numerous viruses and mobile genetic elements encode parts of the translational apparatus7–9, potentially rendering a genetic-code-based firewall ineffective. In this paper, we show that such mobile transfer RNAs (tRNAs) enable gene transfer and allow viral replication in Escherichia coli despite the genome-wide removal of three of the 64 codons and the previously essential cognate tRNA and release factor genes. We then establish a genetic firewall by discovering viral tRNAs that provide exceptionally efficient codon reassignment allowing us to develop cells bearing an amino-acid-swapped genetic code that reassigns two of the six serine codons to leucine during translation. This amino-acid-swapped genetic code renders cells resistant to viral infections by mistranslating viral proteomes and prevents the escape of synthetic genetic information by engineered reliance on serine codons to produce leucine-requiring proteins. As these cells may have selective advantage over wild organisms due to virus-resistance, we also repurpose a third codon to biocontain this virus-resistant host via dependence on an amino acid not found in nature10. Our results suggest a general strategy to make any organism safely resistant to all natural viruses and prevent genetic information flow into and out of genetically modified organisms.
The genetic code allows organisms to exchange functions through horizontal gene transfer (HGT) and enables recombinant gene expression in heterologous hosts. However, the shared language of the same code permits the undesired spread of antibiotic, herbicide, and pesticide-resistance genes and allows viruses to cause diseases. By exploiting the shared nature of the genetic code, recombinant DNA technologies revolutionized our ability to produce small molecules, peptides, biologics, and enzymes in vast quantities; however, the production cell cultures remained susceptible to viral contamination. To date, viral contamination in cell cultures exists as a real risk with severe consequences: Over the past four decades, dozens of viral contamination cases were documented in industry11–13. Horizontal gene transfer also threatens the safe use of Genetically Modified Organisms (GMOs) by enabling the spread of their engineered genetic information into natural ecosystems. Despite the impact of viral infections, HGT and the growing economic and societal role of GMOs and recombinant DNA, to date, no technology exists that could prevent viral infections and the escape of engineered genetic information from genetically modified biological systems. It is widely hypothesized that Genomically Recoded Organisms (GROs), whose genomes have been systematically redesigned to confer an alternate genetic code, would offer genetic isolation from natural ecosystems by obstructing the translation of horizontally transferred genetic material1–5,14, including both resistance to viral infections and horizontal gene transfer. Indeed, the genome-wide removal of TAG stop codons and release factor 1 (RF1) from E. coli, which abolishes cells’ ability to terminate translation at TAG stop codons, provides substantial but not complete resistance to bacteriophages2,3. Most recently, a strain of E. coli, Syn61Δ3, was created with a synthetic recoded genome in which all annotated instances of two serine codons, TCG and TCA (together TCR), and the TGA stop codon were replaced with synonymous alternatives, and the corresponding serine tRNA genes (serU and serT) and RF1 (prfA) have been deleted4,15. Although the compressed genetic code of Syn61Δ3 provided resistance to five viruses4, it could not prevent the escape of its engineered genetic information.
However, despite the potentially broad virus and gene transfer resistance of compressed genetic codes and these organisms’ widespread industrial applicability, how natural genetic material could breach genetic-code-based resistance remained unanswered. Numerous viruses and mobile genetic elements encode parts of the translational apparatus, ranging from single tRNA genes and release factors up to lacking only ribosomal genes7–9. These genes allow mobile genetic elements to reduce their dependency on host translational processes16–18 19–21. Therefore, the selection pressure posed by GROs’ compressed genetic code might facilitate the rapid evolution of viruses and mobile genetic elements capable of crossing a genetic-code-based barrier. Here we show that horizontally transferred tRNA genes can readily substitute cellular tRNAs and thus abolish genetic-code-based resistance to viral infections and HGT. We provide the first example of a virus-resistant, biocontained bacterial host that prevents both incoming and outgoing horizontal gene transfer, which paves the way toward engineering multi-virus-resistant cell lines from any organisms and the safe use of GMOs in natural environments by eliminating horizontal gene transfer.
Mobile tRNAs abolish virus resistance
We first investigated whether tRNAs of mobile genetic elements can substitute cellular tRNAs and support viral infection in cells with a compressed genetic code. We sampled the mobile tRNAome, tRNA genes encoded by horizontally transferred genetic elements, by computationally screening thousands of viral genomes for the presence of tRNA genes, and then synthesizing 1192 tRNA genes from phylogenetically diverse plasmids, transposable elements, and bacteriophages infecting members of the Enterobacteriaceae family (Supplementary Data 1). Next, we assayed these tRNAs for their ability to produce functional tRNAs in an E. coli host and substitute genomic tRNA genes to translate TCR codons. As depicted in Figure 1a, this high-throughput assay is based on an E. coli strain with a synthetic recoded genome in which all annotated instances of two sense serine codons (TCG, TCA) and a stop codon (TAG) were replaced with synonymous alternatives, and the corresponding serU, serT tRNA genes and release factor 1 (prfA) have been deleted. This strain, E. coli Syn61Δ3, thereby relies on a 61-codon genetic code and prevents the expression of protein-coding genes containing TCR codons. Candidate tRNAs have been synthesized and cloned into a plasmid carrying each tRNA under a strong constitutive promoter together with an nptII40TCA,68TCG,104TCA,251TCG aminoglycoside-O-phosphotransferase antibiotic resistance gene containing TCA codons at positions 40, 104 and TCG codons at positions 68 and 251. In wild-type E. coli cells bearing the canonical genetic code, nptII40TCA,68TCG,104TCA,251TCG confers resistance to kanamycin through serine incorporation at positions 40, 68, 104, and 251, and the production of full-length aph(3’)-II aminoglycoside-O-phosphotransferase. In Syn61Δ3, however, the production of this resistance-conferring gene product is inhibited due to the lack of serU and serT-encoded tRNA-SerUGA and -SerCGA needed for TCR codon decoding. Therefore, in our screen, only plasmid variants that are expressing tRNAs capable of decoding TCR codons will survive kanamycin selection. The transformation of this plasmid library into Syn61Δ3 and subsequent selection in the presence of kanamycin yielded thousands of colonies, indicating the presence of TCR translating tRNAs in our library. Pooled extraction of plasmid variants from kanamycin-resistant colonies followed by amplicon sequencing of their tRNA-insert identified 62 tRNA sequences capable of promoting nptII40TCA,68TCG,104TCA,251TCG expression (Figure 1b, Supplementary Data 1). These tRNAs represent 89% of all predicted TCR codon-recognizing tRNAs in our library and share 33.7–61.1% (median = 46.2%) similarity to the endogenous serU tRNA of E. coli. In agreement with the anticodon composition of mobile Ser tRNAs, most tRNA hits contained a UGA anticodon and carried the identity elements necessary for recognition by the host’s SerS serine-tRNA-ligase (Figure 1b).
Figure 1. Discovery of mobile TCR codon translating tRNAs in E. coli Syn61Δ3.
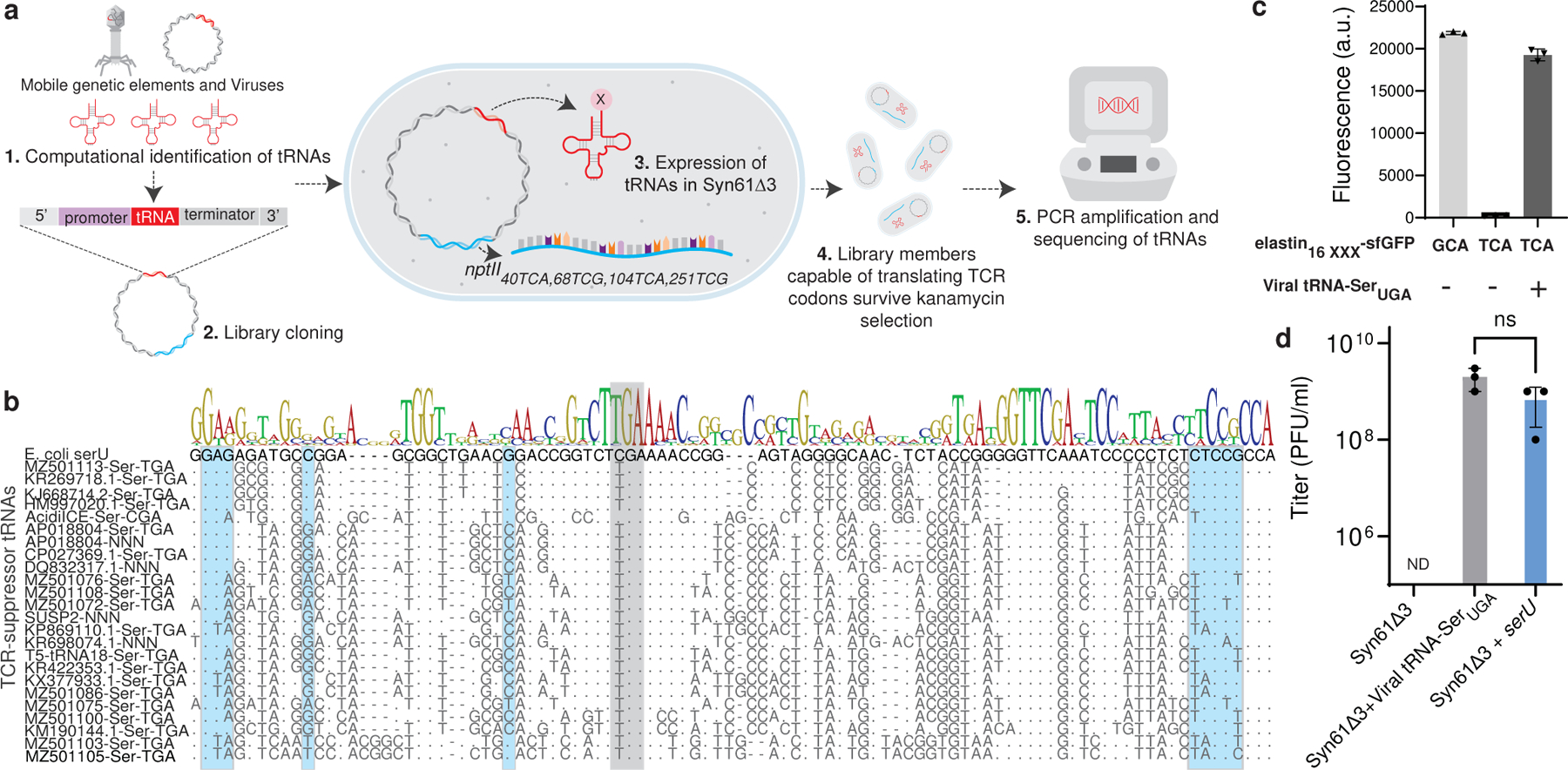
(a). We screened the mobile tRNAome for tRNAs that can simultaneously translate TCA and TCG (together TCR) codons by computationally identifying tRNA genes in mobile genetic elements (1.) and then synthesizing select candidates as an oligonucleotide library and cloning these variants into a plasmid vector carrying a nptII40TCA,68TCG,104TCA,251TCG marker (conferring kanamycin resistance) (2.). Following the transformation of this library into Syn61Δ3 (3.), in which the deletion of serU (encoding tRNA-SerCGA) and serT (encoding tRNA-SerUGA) makes TCG and TCA codons unreadable, only variants carrying functional TCR suppressor tRNAs survive kanamycin selection (4.). Finally, high-throughput sequencing of the tRNA inserts from kanamycin-resistant clones identified suppressor tRNAs (5.).
(b). Multiple sequence alignment of mobile TCR codon translating tRNAs. Grey shading indicates the anticodon region, while the host’s serine-tRNA-ligase identity elements are shown in blue.
(c). Viral serine tRNAUGA translates the TCA codon. Syn61Δ3 expressing elastin16 GCA(alanine)-sfGFP-His6 served as wild-type expression control, and the elastin16 TCR-sfGFP-His6 expression was compared with and without the coexpression of the tRNA-SerUGA of Escherichia phage IrisVonRoten24. xxx marks the analyzed codon, TCA or GCA. Bar graph represents the mean; error bars represent SD based on n=3 independent experiments; a.u. denotes arbitrary fluorescence units.
(d). The expression of viral TCR suppressor tRNAs and serU (tRNA-SerCGA) restores the replication of T6 bacteriophage in Syn61Δ3. Dots represent data from n=3 independent experiments, error bars represent SD, and the bar graph represents the mean. ND represents below the detection limit (i.e., <103 PFU/ml); ns indicates lack of significance (p = 0.116) based on unpaired two-sided Student’s t-test.
Notable examples include the UAG anticodon-containing serine tRNA of the laboratory model coliphage T5, tRNAs from plasmids of multidrug-resistant E. coli isolates (GenBank IDs AP018804 and CP023851), and the Ser-tRNACGA of the integrative conjugative element of Acidithiobacillus ferrooxidans. The presence of mobile tRNAs in integrative conjugative elements is especially concerning as these mobile genetic elements can carry up to 38 tRNAs corresponding to all 20 amino acids in a single operon and are capable of excision and transfer into neighboring bacterial cells20,22. In agreement with prior studies7,8,23, our computational screen also showed that mobile tRNA genes are not limited to mobile genetic elements of bacteria. Computational analysis of viruses infecting Vertebrates and Archaea revealed the presence of sense and stop codon suppressor tRNAs in both groups, suggesting that mobile tRNAs are prevalent across viruses infecting prokaryotic, archaeal, and eukaryotic hosts (Supplementary Data 2).
We confirmed the TCR codon-recognizing tRNAs’ predicted serine amino acid identity by coexpressing a selected tRNA hit with an elastin16TCA-sfGFP-His6 construct harboring a single TCA codon at position 16. The coexpression of the tRNA-SerUGA of Escherichia phage IrisVonRoten24 together with the elastin16TCA-sfGFP-His6 construct conferred near wild-type level expression (Figure 1c) and tryptic digest followed by reverse-phase liquid chromatography and tandem mass spectrometry (LC/MS-MS) confirmed serine incorporation at the TCA position (Extended Data Figure 1a).
Next, we investigated whether mobile tRNAome-derived tRNAs could promote viral replication. A previous study demonstrated that Syn61Δ3 resists infection by multiple bacteriophages, including Enterobacteria phage T64. Infecting Syn61Δ3 with T6 phage recapitulated these results. In contrast, the infection of Syn61Δ3 harboring a bacteriophage-derived Ser-tRNAUGA gene with T6 resulted in rapid lysis, indicating that tRNA genes that reside in viral genomes can substitute cellular tRNAs and promote phage infection (Figure 1d).
The discovery of diverse TCR codon translating tRNAs on horizontally transferred genetic elements indicates that mobile tRNA genes are widespread and can readily complement the lack of cellular tRNAs to promote viral replication and horizontal gene transfer.
Viruses infecting a recoded organism
We next investigated whether lytic viruses of Syn61Δ3 exist. We infected Syn61Δ3 cells with eleven coliphages whose genome harbor TCR translating tRNA genes based on our plasmid-based screen (Figure 1b). Surprisingly, none of these eleven phages could overcome the recoded host’s genetic isolation, indicating that the presence of tRNA genes on viral genomes does not directly rescue viral replication in recoded organisms (Extended Data Figure 1b).
We next attempted to isolate lytic viruses from diverse environmental samples by performing a standard two-step enrichment-based phage isolation protocol and using Syn61Δ3 as host. First, bacteria-free filtrates of environmental and wastewater samples (n=13, from Massachusetts (USA), Extended Data Table 1a) were mixed with Syn61Δ3 and grown until stationary phase. Next, bacterial cells were removed, and we analyzed the presence of lytic phages by mixing sample supernatants with Syn61Δ3 in soft-agar overlays. Five samples produced visible lysis. Viral plaque isolation from these samples followed by DNA sequencing and de novo genome assembly identified 12 novel phage strains. All identified phages belong to the Caudovirales order and the Myoviridae family, taxa rich in tRNA-encoding bacteriophages8 (Extended Data Table 1b). Computational identification of tRNA genes revealed the presence of tRNA operons in all phage isolates, with 10 to 27 tRNA genes in each genome (Supplementary Data 1). Surprisingly, all isolates harbored TCR codon translating serine tRNAs with a UGA anticodon that we identified in our earlier nptII40TCA,68TCG,104TCA,251TCG suppressor screen (Figure 1b). One isolate, REP1, also harbored a predicted homing endonuclease (Figure 2c). Homing endonucleases encoded in tRNA operons have been shown to be responsible for the horizontal transfer of tRNA gene clusters25. Phage isolates showed more than two orders of magnitude difference in viral titers after replication on recoded cells (Figure 2a). One of the most virulent isolates, REP12, required 60 minutes to complete a replication cycle at 37 °C in Syn61Δ3 (Figure 2b).
Figure 2. Lytic phages of Syn61Δ3.
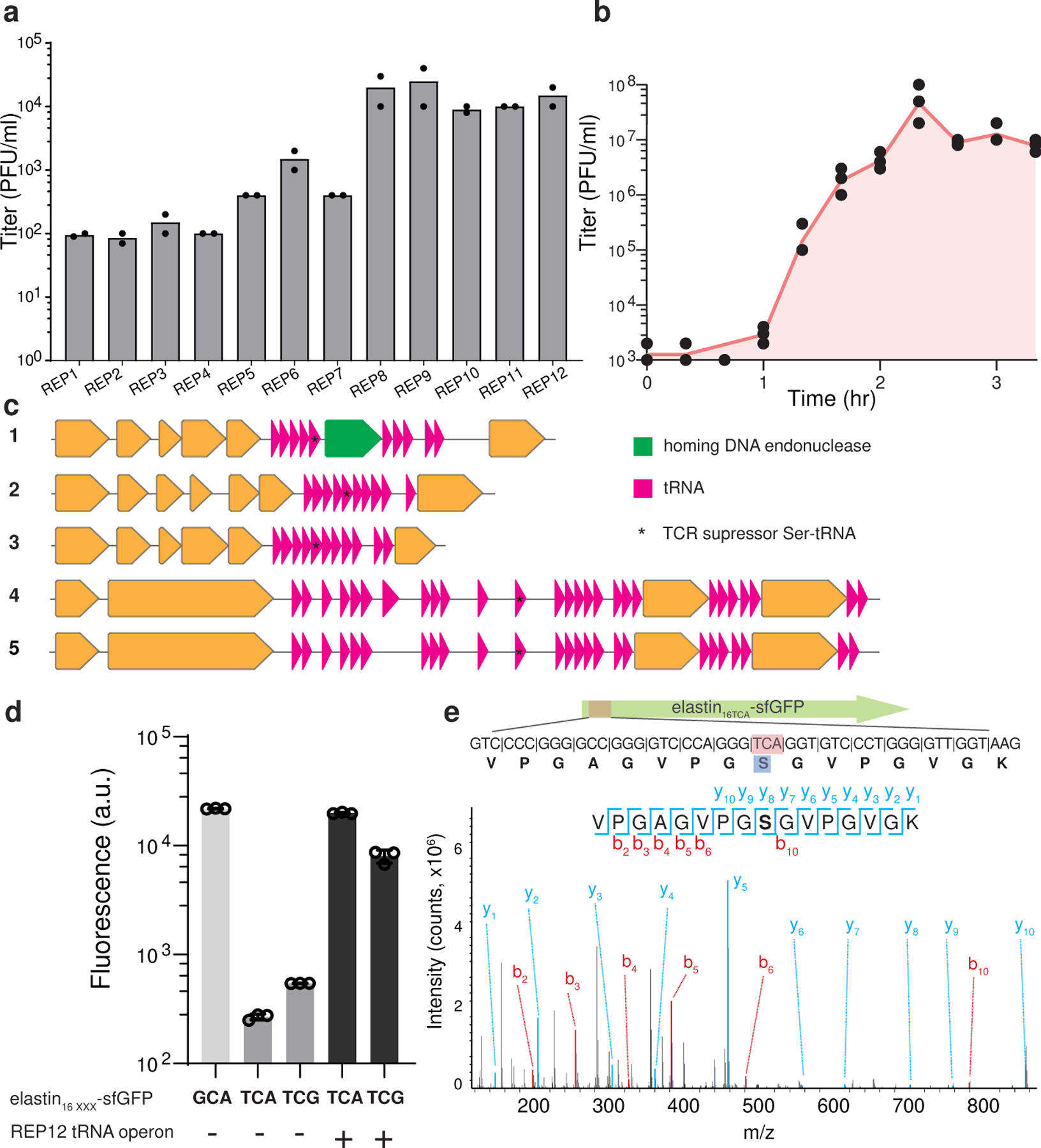
(a). Titer of Syn61Δ3 phage isolates after replication on Syn61Δ3. Dots represent data from n=2 independent experiments; bar graphs represent the mean.
(b). Single-step growth curve of REP12 lytic Syn61Δ3 phage. Single-step growth was performed in n=3 independent experiments, red line represents the mean, and dots represent the total viral titer.
(c). Genomic maps of tRNA operons in lytic Syn61Δ3 phages. Magenta arrows represent predicted tRNA genes; * denotes tRNA genes identified in our earlier TCR codon suppressor screen (Figure 1b); green arrow represents homing endonuclease gene, while orange arrows represent protein-coding genes. Phage operon numbers correspond to the following REP phages: 1.=REP1; 2.=REP2; 3.=REP4; 4.=REP6; 5.=REP12.
(d). Viral tRNA operon-expressed tRNAs translate TCR codons. Syn61Δ3 expressing elastin16GCA(alanine)-sfGFP-His6 served as wild-type expression control, and the elastin16 TCR-sfGFP-His6 expression was compared with and without the coexpression of the REP12 viral tRNA operon. xxx marks the analyzed codon, TCA, TCG, or GCA. A.u. denotes arbitrary fluorescence units; bar graphs represent the mean. Error bars represent SD based on n=3 independent experiments.
(e). Viral tRNA operon-expressed tRNAs decode TCR codons as serine. The amino acid identity of the translated TCA codon within elastin16 TCA-sfGFP-His6 was confirmed by tandem mass spectrometry from Syn61Δ3 cells containing the REP12 tRNA operon and its cognate promoter. The figure shows the amino acid sequence and MS/MS spectrum of the analyzed elastin16 TCR peptide. MS/MS data was collected once.
The isolated viral strains infecting Syn61Δ3 show that bacteriophages that can overcome sense codon recoding-based viral resistance exist and are widespread in environmental samples. However, the presence of TCR codon translating serine tRNAs on viral genomes does not directly rescue viral replication in the recoded strain.
Viral tRNAs substitute cellular tRNAs
We next investigated how tRNA-encoding viruses evade genetic-code-based resistance. Time-course transcriptome analysis of REP12 phage-infected Syn61Δ3 cells during the viral replication cycle revealed early and high-level expression of the viral tRNA operon (Supplementary Figure 1). In agreement with this observation, the computational prediction of bacterial promoters driving the tRNA array indicated the presence of multiple strong constitutive promoters upstream of the tRNA operon region. We then investigated the time-course kinetics of tRNA expression in Syn61Δ3 cells that were infected with our REP12 phage by performing tRNA sequencing (tRNAseq). Time-course tRNAseq experiments revealed remarkably high-level expression of the viral tRNA-SerUGA immediately after phage attachment (i.e., a relative viral tRNA-SerUGA abundance of 56.1% (±5%) compared to the host serV tRNA) (Extended Data Figure 2). Throughout the entire phage replication cycle, the phage tRNA-SerUGA remained one of the most abundant viral tRNA species inside infected Syn61Δ3 cells (Extended Data Figure 2). We next investigated whether phage tRNA-SerUGA participates in translation by analyzing the presence of their mature form. The gene encoding the tRNA-SerUGA in REP12’s genome does not encode the universal 5’-CCA tRNA tail, which allows for amino acid attachment as well as for interaction with the ribosome. Therefore, CCA tail addition must happen before these tRNAs can participate in translational processes. The sequencing-based analysis of phage tRNA-SerUGA ends detected CCA tail addition in 62.9% (±1.9%) of all tRNA sequencing reads immediately after phage attachment, indicating that mature tRNA-SerUGAs are instantly produced after host infection (Extended Data Figure 3).
We also investigated transcriptomic changes in Syn61Δ3 during phage replication. Analysis of the host transcriptome after phage infection revealed upregulation in genes responsible for tRNA maturation and modification. Upregulated genes include queG, encoding epoxyqueuosine reductase that catalyzes the final step in the de novo synthesis of queuosine in tRNAs26, and trmJ, tRNA Cm32/Um32 methyltransferase27, which introduces methyl groups at the 2’-O position of U32 of several tRNAs, including tRNA-SerUGA, suggesting the potential posttranscriptional modification of phage-derived tRNAs (Extended Data Figure 4).
Finally, we also validated the role of phage tRNA-SerUGA tRNAs in decoding TCR codons. We first cloned the REP12 viral tRNA operon containing the hypothetical tRNA-SerUGA and its predicted promoter into a plasmid vector. Coexpression of this tRNA operon with an elastin16 TCA-sfGFP-His6 and elastin16 TCG-sfGFP-His6 construct, harboring either a single TCA or TCG codon at position 16, respectively, resulted in high-level elastin-sfGFP-His6 expression (Figure 2d). Next, tryptic digestion followed by LC/MS-MS analysis confirmed serine incorporation in response to both the TCA and TCG codon in these elastin16 TCR-sfGFP-His6 samples (Figure 2e,Supplementary Figure 2). As expected, the coexpression of the same elastin16 TCA-sfGFP-His6 construct with the only tRNA-SerUGA of the viral tRNA operon conferred a similar effect, and LC/MS-MS analysis confirmed the role of this tRNA in decoding viral TCR codons as serine (Supplementary Figure 2).
Together these results show that lytic phages of Syn61Δ3 overcome genetic-code-based viral resistance by rapidly complementing the cellular tRNA pool with virus-encoded tRNAs.
Swapped genetic code resists viruses
We predicted that establishing an artificial genetic code, in which TCR codons encode an amino acid different from their natural serine identity, would create a genetic firewall that safeguards cells from horizontal gene transfer and infection by tRNA-encoding viruses. In an amino-acid-swapped genetic code, viral tRNAs would compete with host-expressed tRNAs that decode TCR codons as a non-serine amino acid resulting in the mistranslation of viral proteins. As the genetic isolation provided by amino-acid-swapped genetic codes is expected to correlate with the hydrophobicity and polarity distance between exchanged amino acids28, we sought to reassign TCR serine codons to leucine, the amino acid most distant from TCR codons’ natural serine meaning. The more distant Trp, Phe, Tyr, and Ile exchanges were not investigated as their cognate aminoacyl-tRNA synthetases are nonpermissive to anticodon mutations and expected to require extensive enzyme and/or tRNA engineering to function29–31. Although alternative forms of swapped codes emerged in the past, these codes either targeted the exchange of chemically more similar amino acids (i.e., Ser to Ala, His, and Pro) or remained tested in vitro only6,28,32,33.
To establish a serineTCR-to-leucine swapped genetic code (Figure 3a), we utilized Syn61Δ3, which genome-wide lacks annotated instances of TCR codons and their corresponding tRNA genes, and sought to identify tRNAs capable of efficiently translating TCR codons as leucine. We modified our high throughput tRNA library screen (Figure 1a) to evolve TCR suppressors from the endogenous E. coli leuU tRNA carrying a TCA and TCG decoding anticodon. We coexpressed a 65,536-member mutagenized library of the anticodon-swapped leuU tRNA gene in which the anticodon loop of both tRNAs has been fully randomized, together with an aph3Ia29×Leu→TCR, a kanamycin resistance-conferring gene in which all 29 instances of leucine codons were replaced with TCR serine codons. In this system, only anticodon-swapped leuU variants capable of simultaneously translating all 29 TCR codons as leucine would confer resistance to kanamycin. High kanamycin selection pressure in combination with 29 instances of TCR codons in aph3Ia was expected to select tRNA variants that provide wild-type level translation efficiency for TCR codons. We identified two distinct leuU variants by applying “high” kanamycin concentration (i.e., 200 μg/ml) as selection pressure to Syn61Δ3 cells carrying the anticodon-swapped tRNA library. These variants, carrying tRNAs containing distinct anticodon loop mutations (Extended Data Figure 5a), were then infected with a cocktail of all twelve phage isolates (Extended Data Table 1) that are capable of lysing Syn61Δ3 at a 10:1 cell-to-phage ratio (i.e., a Multiplicity of Infection (MOI) of 0.1). Instead of plaque and efficiency-of-plating assays34,35 with selected phage isolates and assaying viral titer on the resistant strain, we quantified virus resistance using a mixture of all twelve Syn61Δ3-infecting viral isolates, and measured virion release from infected cells after 24 hours by plating culture-supernatants on a virus-susceptible wild-type E. coli host. This can reveal even slow or non-plaque-forming viral replication in the target cells35,36 (Supplementary Note).
Figure 3. An amino-acid-swapped genetic code provides multi-virus resistance.
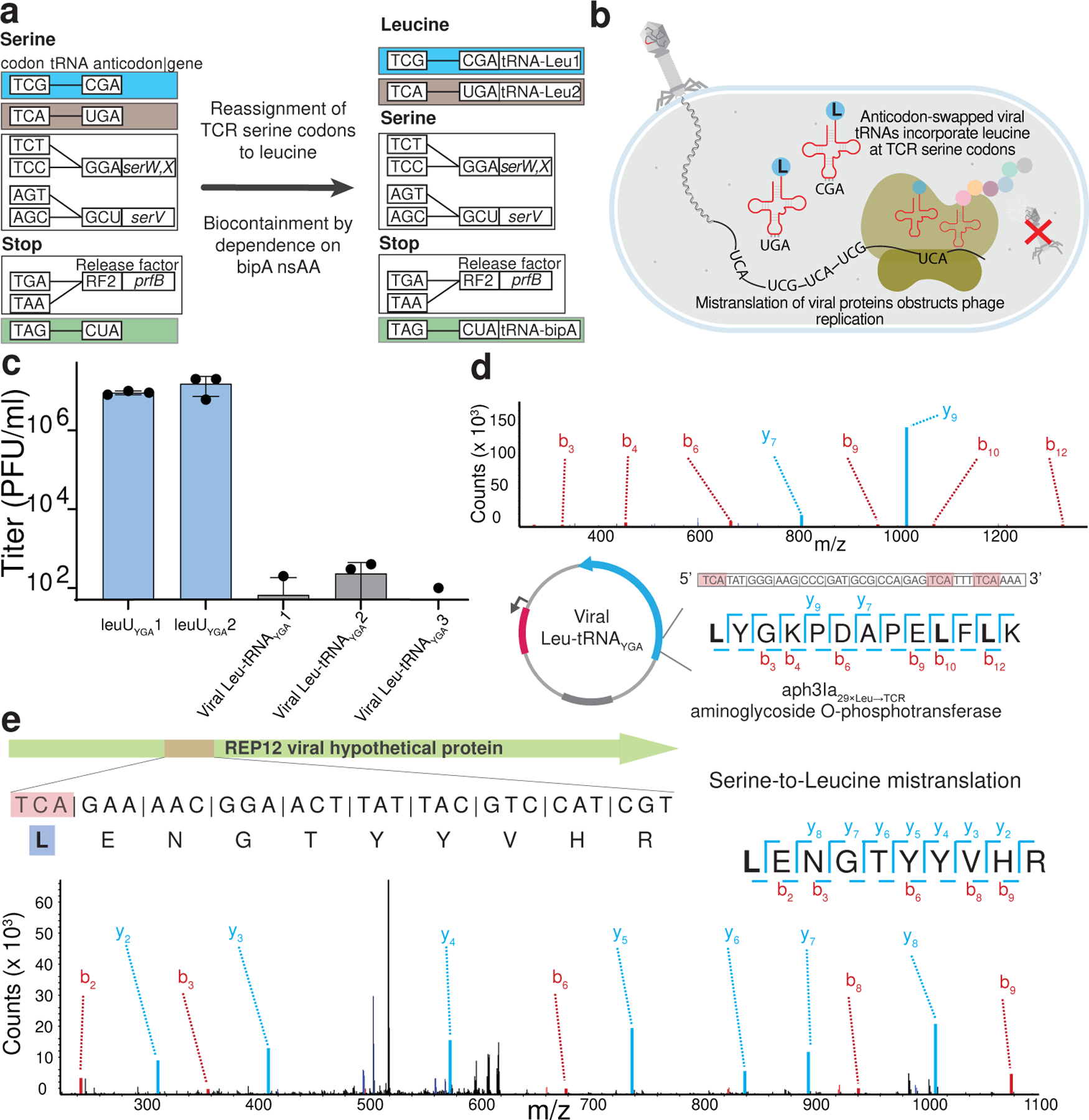
(a). The creation of an E. coli GRO, Ec_Syn61Δ3-SL, in which both TCA and TCG—naturally serine-meaning—codons are translated as leucine. The introduction of bacteriophage-derived Leu-tRNAUGA and Leu-tRNACGA to Syn61Δ3 reassigns TCA and TCG codons to leucine, while the reassignment of the TAG stop codon to encode L-4,4’-biphenylalanine (bipA) in an essential gene of the host ensures the biocontainment of Ec_Syn61Δ3-SL.
(b). Schematic of viral infection in Ec_Syn61Δ3-SL. The reassignment of sense codons TCA and TCG to leucine in Ec_Syn61Δ3-SL provides multivirus resistance by mistranslating the viral proteome.
(c). Bacteriophage-derived Leu-tRNAUGA and Leu-tRNACGA expression in Syn61Δ3 provides multivirus resistance. The figure shows the titer of lytic Syn61Δ3 phages following the infection of the corresponding Leu-tRNAYGA-expressing Syn61Δ3 strain with a mixture of twelve distinct REP Syn61Δ3 phages (Extended Data Table 1, Supplementary Data 3). All experiments were performed in three independent replicates; dots represent data from n=3 independent experiments; bar graphs represent the mean; the error bars represent SD.
(d). The reassignment of TCR codons to leucine within the coding sequence of aph3Ia29×Leu→TCR in Syn61Δ3-LS was confirmed by tandem mass spectrometry. The figure shows the amino acid sequence and MS/MS spectrum of the detected aph3Ia29×Leu→TCR peptide and its coding sequence. MS/MS data was collected once.
(e). Mistranslated viral protein synthesis in Ec_Syn61Δ3-SL. The figure shows the amino acid sequence and MS/MS spectrum of a bacteriophage-expressed protein, together with its viral genomic sequence, in which the naturally serine-coding TCA codon is mistranslated as leucine. The experiment was performed by infecting Ec_Syn61Δ3-SL cells, expressing Leu9-tRNAYGA from Escherichia phage OSYSP, with the REP12 phage and the proteome of infected cells was analyzed by tandem mass spectrometry. MS/MS data was collected once.
Surprisingly, all selected leuU library members allowed robust phage replication, with phage titers reaching ~107 PFU/ml after 24 hours (Figure 3c). We hypothesized that viral replication in the presence of TCR suppressing leuU variants is due to these tRNAs lower suppression efficiency compared to competing phage-carried serine tRNAs, which leads to rapid viral takeover. Viral tRNA-SerYGA, that are tRNA-SerUGA and tRNA-SerCGA, might have i.) higher aminoacylation efficiency by their corresponding E. coli aminoacyl-tRNA-ligase than our selected leuU variants, ii.) higher affinity towards the bacterial ribosome, and/or iii.) better evade phage- and host-carried tRNA-degrading effector proteins16,37,38.
Based on this observation, we hypothesized that bacteriophage-encoded tRNAs might provide much higher suppression efficiencies for their cognate codons than their native E. coli counterpart. Therefore, and to exploit this superior translation efficiency of bacteriophage-encoded tRNAs and their resistance to phage-carried tRNA degrading enzymes, we next constructed a small, focused library that coexpressed YGA anticodon-swapped mutants of 13 phage-encoded leucine tRNAs, together with the aph3Ia29×Leu→TCR aminoglycoside O-phosphotransferase gene. The transformation of this library into Syn61Δ3 cells and subsequent “high” concentration (i.e., 200 μg/ml) kanamycin selection identified three distinct tRNAs enabling robust growth. Identified tRNAs showed only 48.3–37.9% similarity to E. coli leuU but carried most of the canonical E. coli leucine-tRNA ligase identity elements (Extended Data Figure 5b). Furthermore, the analysis of the total tRNA content of these cells by tRNAseq confirmed the presence of synthetic phage Leu-tRNAYGA tRNAs with similar abundances as the cellular endogenous serine tRNAs (i.e., a relative expression level of 172% and 140% for Leu-tRNAUGA and Leu-tRNACGA respectively, compared to serV (Extended Data Figure 6)).
Next, similarly to our previous infection assay, phage tRNAYGA expressing cells were infected with a mixture of twelve distinct, lytic phages of Syn61Δ3 at a MOI = 0.1. The analysis of phage titer in culture supernatants after 24 hours showed a marked drop compared to the input phage inoculum, suggesting that anticodon-swapped viral leucine tRNAs block phage replication (Figure 3c).
We then investigated the mechanism of phage resistance in E. coli cells carrying virus-derived tRNA-LeuYGA tRNAs (Ec_Syn61Δ3 Ser→Leu Swap, or Ec_Syn61Δ3-SL in short, Figure 3b) by performing total proteome analysis. Untargeted deep proteome analysis of uninfected cells by tandem mass spectrometry validated the translation of TCR codons as leucine in Ec_Syn61Δ3-SL (Figure 3d). Time-course untargeted proteome analysis after bacteriophage infection revealed extensive mistranslation at TCR codons in newly synthesized phage proteins (Figure 3e, Supplementary Figure 3), indicating that an amino-acid-swapped genetic code broadly obstructs viral protein synthesis. In agreement with earlier reports that showed the partial recognition of TCT codons by tRNAUGA39,40 and due to the extreme sensitivity of our untargeted proteomics assay, we also detected serine-to-leucine mistranslation at TCT codon positions in Ec_Syn61Δ3-SL cells (Extended Data Figure 7). The recognition of TCT codons by phage tRNA-LeuYGA tRNAs might also be responsible for the fitness decrease of Ec_Syn61Δ3-SL cells compared to its ancestor strain (i.e., a doubling time of 69.3 minutes, compared to 44.29 minutes for the parental Syn61Δ3 strain in rich 2×YT media (Extended Data Figure 8)). Alternatively, the fitness decrease of Ec_Syn61Δ3-SL might also be attributable to the presence of TCR codons in essential genes of Syn61Δ3. According to our genome analysis, at least four essential genes of Syn61Δ3, mukE, ykfM, yjbS, safA, contain TCR codons and become mistranslated in Ec_Syn61Δ3-SL (Supplementary Data 4).
Finally, we also sought to develop a tightly biocontained version of Ec_Syn61Δ3-SL because a virus-resistant strain might have a competitive advantage in natural ecosystems due to the lack of predating bacteriophages. Synthetic auxotrophy based on the engineered reliance of essential proteins on human-provided nonstandard amino acids (nsAAs), e.g., L-4,4’-biphenylalanine (bipA), offers tight biocontainment that remains stable under long-term evolution10,41,42. Therefore, we generated a recombination deficient (i.e., ΔrecA), biocontained version of Ec_Syn61Δ3-SL bearing a bipA-dependent essential adk gene and the bipA aminoacyl-tRNA synthetase/tRNA-bipACUA system, by first performing adaptive laboratory evolution on a recA knock-out Syn61Δ3, and then replacing the genomic adk copy with its bipA-dependent variant43 (Methods). This strain maintained the low escape frequency of previously reported singe-gene synthetic auxotrophs10 (i.e., 2.9×10−6 (±5.9×10−7) escape frequency) and provided robust growth. We also tested the viral resistance of Ec_Syn61Δ3-SL under mock environmental conditions by repeating our phage enrichment and isolation process with a mixture of 12 environmental samples, including fresh sewage (Extended Data Table 1, Supplementary Note), but could not detect plaque-forming phages in culture supernatants (Extended Data Figure 9).
Together, these results demonstrate that reassigning sense codons TCA and TCG to leucine in vivo provides broad protection against viruses, including novel mixtures of viruses directly from environmental samples, and the TAG stop codon can be simultaneously utilized to biocontain this virus-resistant strain via dependence on an amino acid not found in nature.
Genetic-code-based bidirectional firewall
Finally, we developed a set of plasmid vectors that we systematically addicted to an amino-acid-swapped genetic code in which leucine is encoded as TCR codons. GMOs are increasingly deployed for large-scale use in agriculture, therapeutics, bioenergy, and bioremediation. Consequently, it is critical to implement robust biocontainment strategies that prevent the unintended proliferation of GMOs and protect natural ecosystems from engineered genetic information. Although efficient biocontainment strategies exist (e.g., bipA nsAA-based synthetic auxotrophy, as in Ec_Syn61Δ3-SL), current methods fail to prevent the horizontal gene transfer (HGT)-based escape of engineered genetic information. Synthetic addiction to a swapped genetic code offers a solution to this problem. Using our virus-derived tRNA-LeuYGA expressing Ec_Syn61Δ3-SL cells, we, therefore, developed a set of plasmid vectors that depend on TCR codons to express leucine-containing proteins and thus can only function in cells that efficiently translate TCR codons as leucine (Figure 4a). These plasmids, called the pLS plasmids, offer four widely used, orthogonal antibiotic resistance markers in combination with four mutually orthogonal low- to high-copy-number origins-of-replication for stable maintenance in Ec_Syn61Δ3-SL cells (Figure 4b, Supplementary Data 3). Antibiotic resistance genes and proteins necessary for pLS plasmid replication encode leucine as TCR—naturally serine-meaning—codons and, therefore, fail to function in cells bearing the canonical genetic code. The addiction of resistance markers and replication proteins to an artificial genetic code ensures that pLS plasmids can stably and safely maintain synthetic genetic functions but restrict these genes’ functionality to Ec_Syn61Δ3-SL cells. Using our pLS plasmids, we achieved serine-to-leucine codon reassignment simultaneously at ~14,000 codon positions in a single cell, thus demonstrating our viral tRNA-based system’s exceptional codon reassignment efficiency.
Figure 4. Addiction of synthetic genetic information to a genetic code in which TCR codons encode leucine prevents horizontal gene transfer.
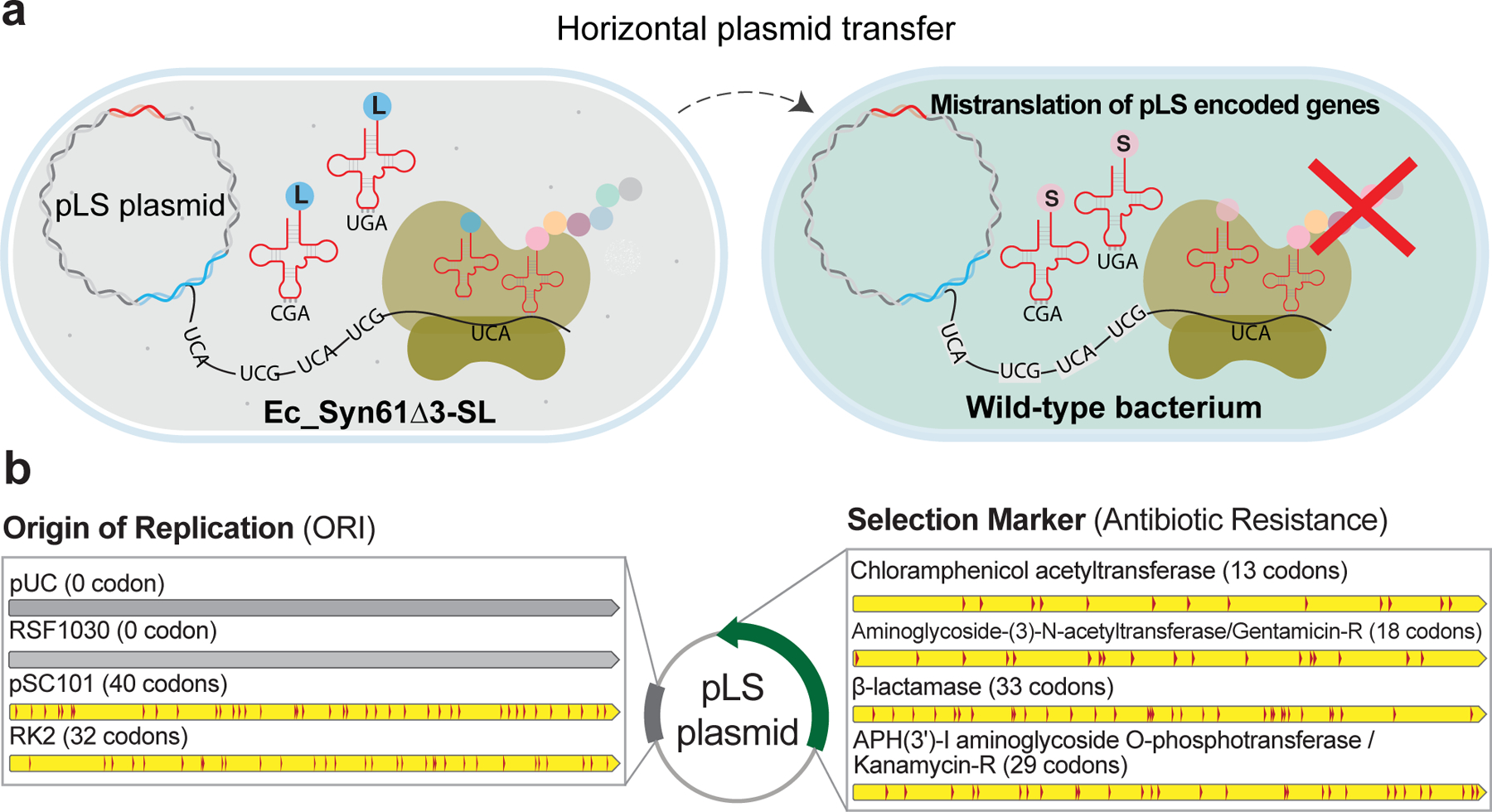
(a). We developed a set of plasmid vectors, termed the pLS plasmids, that rely on TCR codons to express leucine-containing proteins. pLS plasmids only function in Ec_Syn61Δ3-SL expressing bacteriophage-derived synthetic tRNA-LeuYGA tRNAs, and the encoded proteins of pLS plasmids become mistranslated in cells bearing the canonical genetic code.
(b). The pLS plasmids offer multiple mutually orthogonal antibiotic resistance markers together with low to high copy-number origins-of-replication (ORI) that are addicted to an artificial genetic code in which leucine is encoded as TCR codons. Number in parenthesis marks the number of LeuTCR codons in each gene. Detailed sequence information and the description of pLS plasmids are available in Supplementary Data 3.
We then tested the ability of our pLS vectors to function in cells bearing the standard genetic code cells by electroporating plasmid DNA from six variants (i.e., pLS1–6) into wild-type E. coli K-12 MG1655 cells but could not detect escapees carrying pLS plasmids (i.e., an electroporation efficiency of <1 CFU/μg, while the electroporation efficiency the recipients cells was 3×109 ±1.1×109 CFU/μg (±s.d., n = 3 independent experiments) based on a pUC-KanR control plasmid). The escape of pLS plasmids was similarly prevented when the phage tRNA-LeuYGA expression cassette was encoded within the plasmid backbone (i.e., pLS1 and pLS2), indicating that anticodon-swapped viral tRNAs are severely toxic to wild-type cells. Based on these results, we also expect that, similarly to pLS’ genes, any leucine-requiring protein, up to entire chromosomes, can be addicted to Ec_Syn61Δ3-SL by recoding target genes to encode one or more leucine positions as TCR codons.
In sum, the addiction of pLS plasmids to an artificial genetic code in which leucine is encoded as TCR codons, in combination with nsAA-based synthetic auxotrophy, promises escape-free biocontainment for engineered genetic information.
Discussion
Previous works provided support for using rational genetic code engineering to isolate GMOs from natural ecosystems by preventing viral infections and horizontal gene transfer2–4; however, how natural viruses and transferred genes could breach genetic-code-based resistance remained unanswered. By systematically investigating horizontal gene transfer into E. coli Syn61Δ3, a synthetic organism with a compressed genetic code4, we demonstrated that tRNAs expressed by bacteriophages, plasmids, and integrative conjugative elements, readily substitute cellular tRNAs and abolish genetic-code-based isolation. We discovered twelve viruses in environmental samples that can infect E. coli Syn61Δ3, despite its compressed genetic code (Figure 2a). These bacteriophages express up to 27 tRNAs, including a functional tRNA-SerUGA needed to replace the host’s deleted tRNAs. These findings suggest that the selection pressure posed by compressed genetic codes can facilitate the rapid evolution of viruses and mobile genetic elements capable of crossing a genetic-code-based barrier. This hypothesis is further supported by the co-localization of tRNAs and homing endonucleases (Figure 2c) that catalyze copy-and-paste tRNA operon transfer25. We have also shown that mobile tRNAs are not limited to bacteria, as multiple archaeal and eukaryotic viruses also carry predicted tRNA genes.
Next, as a combined solution against horizontal gene transfer and viral infections, including tRNA-expressing viruses, we have created a new type of biocontained E. coli strain, Ec_Syn61Δ3-SL, carrying an amino-acid-swapped genetic code. Ec_Syn61Δ3-SL achieves unprecedented gene-transfer resistance by the reassignment of TCR codons to leucine—an amino acid distant in physicochemical properties compared to their natural serine identity—using reprogrammed, virus-derived tRNAs. In contrast to prior studies2–4,33, we confirmed the virus resistance of this strain using a broad range of viruses. We found that viral tRNAs provide exceptional codon reassignment efficiency and are superior for establishing altered genetic codes compared to cellular tRNAs. We also showed that the swapped genetic code of Ec_Syn61Δ3-SL simultaneously prevents viral replication and the escape of synthetic genetic information into wild organisms. As a direct application, we addicted the most widely used plasmid vectors to express leucine-containing proteins with TCR codons and developed the first plasmids that cannot function in natural organisms. By demonstrating efficient serine-to-leucine reassignment at a previously unprecedented scale, we made biological containment possible for genes, operons, up to entire chromosomes. Adaptive laboratory evolution, that have significantly improved doubling times of organisms with altered genetic codes4,44, will be applied to Ec_Syn61Δ3-SL to help move toward industrial use.
Potential limitations of our work include the sampled viral diversity and the inability of our viral resistance assays to sample multi-step, long-term evolutionary processes of viruses. Although our tests of Ec_Syn61Δ3-SL’s viral resistance included twelve lytic Syn61Δ3 phages and a complex mixture of environmental samples, including fresh sewage, we cannot rule out the existence of Ec_Syn61Δ3-SL-infecting phages in Earth’s biome. Our assays provided an example of Ec_Syn61Δ3-SL culture contamination with soil, sewage, and fecal material (Extended Data Table 1a); however, lysogenic viruses or jumbo- and megaphages, with genomes of more than 200 and 500 kbp, respectively, could have remained undetected in our assays due to their frequent inability to form visible plaques9,36. We hypothesize that bacteriophages with few TCR codons in essential genes, e.g., Escherichia phage EC6098 with only 33 TCR positions in its six protein-coding genes45, have the highest potential to overcome an amino-acid-swapped code. Alternatively, viral tRNA-degrading proteins37,38 could evolve to selectively destroy mistranslating tRNAs and thus promote viral escape; however, both mechanisms would require multiple simultaneous mutations and thus unlikely to occur. Future work will explore whether such viruses can evade the amino-acid-swapped genetic code of Ec_Syn61Δ3-SL.
After the publication of our preprint46, Zürcher et al.33 confirmed some of these findings by identifying two tRNAs that could overcome Syn61Δ3’s virus resistance and by noting the utility of swapped genetic codes to obstruct HGT. The use of weak E. coli tRNAs in our hands was insufficient to produce true resistance to a broad set of viruses. The apparent success with such weak tRNAs in Zürcher et al. may be due to testing only two (related) viral strains and using efficiency-of-plating34,35 and conjugation assays (Supplementary Note). Furthermore, the lower expression level of mistranslating tRNAs used by Zürcher et al., in combination with the use of E. coli tRNAs still susceptible to viral tRNA-degrading enzymes37,38, might result in compromised resistance in the presence of complex environmental viral communities. Zürcher et al. did not implement biocontainment strategies to limit the unwanted proliferation of their potentially virus-resistant host.
These findings fundamentally impact ongoing prokaryotic and eukaryotic genome recoding projects5,47–49, including our aim to engineer a virus-resistant stop-codon-recoded human cell line and a 57-codon strain of E. coli, as some of the identified tRNAs are expected to enable viral replication in these recoded organisms. Therefore, we are now implementing the swapped genetic code of this work to ensure the virus and HGT resistance of these engineered hosts. We expect that our results will have implications for the safe use of GMOs in open environments by establishing a generalizable method for genetic code alteration that simultaneously prevents viral predation in natural ecosystems and blocks incoming and outgoing HGT with natural organisms. Follow-up works could utilize biocontained swapped genetic codes to prevent transgene release during bioremediation, offer containment for open cultures of engineered photoautotrophs for carbon sequestration, and enable safe microbiome engineering and vaccine production by addicting living treatments to use a swapped code. The combination of genome recoding and codon swap may provide a universal strategy to make any species resistant to many or all natural viruses.
Methods
Bacterial media and reagents
Lysogeny Broth Lennox (LBL) was prepared by dissolving 10 g/l tryptone, 5 g/l yeast extract, and 5 g/l sodium chloride in deionized H2O and sterilized by autoclaving. Super Optimal Broth (SOB) was prepared by dissolving 20 g/l tryptone, 5 g/l yeast extract, 0.5 g/l sodium chloride, 2.4 g/l magnesium sulfate, and 0.186 g/l potassium chloride in deionized H2O and sterilized by autoclaving. 2×YT media consisted of 16 g/l casein digest peptone, 10 g/l yeast extract, 5 g/l sodium chloride. LBL and 2×YT agar plates were prepared by supplementing LBL medium or 2×YT with agar at 1.6% w/v before autoclaving. Top agar for agar overlay assays was prepared by supplementing LBL medium with agarose at 0.7% w/v before autoclaving. SM Buffer, 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 8 mM MgSO4, 0.01% gelatin, was used for storing and diluting bacteriophage stocks (Geno Technology, Inc., St. Louis, MO, USA). L-4,4’-biphenylalanine (bipA) was obtained from PepTech Corporation (USA).
Bacteriophage isolation
Bacteriophages were isolated from environmental samples from Massachusetts, United States, collected in Q3 2021 (Samples 1–2, 4–13) and Q1 2022 (Samples 1–3, Extended Data Table 1a), and by using E. coli Syn61Δ3(ev5) (from the laboratory of Jason W. Chin (Addgene strain #174514)) as host. For aqueous samples, including sewage, we directly used 50 ml filter-sterilized filtrates, while samples with mainly solid components, like soil and animal feces, were first resuspended to release phage particles and then sterilized by centrifugation and subsequent filtration. Environmental samples and the sterilized filtrates were stored at 4 °C in the dark. This protocol avoided the inactivation of chloroform-sensitive viruses. Sterilized samples were then mixed with exponentially growing cultures of Syn61Δ3(ev5) in SOB supplemented with 10 mM CaCl2 and MgCl2. Infected cultures were grown overnight at 37 °C aerobically and then filter sterilized by centrifugation at 4000× g for 15 minutes and filtered through a 0.45 μm PVDF Steriflip™ disposable vacuum filter unit (MilliporeSigma). Next, 1 ml from each sterilized enriched culture was mixed with 10 ml exponentially growing Syn61Δ3 (OD600 = 0.2), supplemented with 10 mM CaCl2 and MgCl2, and mixed with 10 ml 0.7% LBL top agar. Top agar suspensions were then poured on top of LBL agar plates in 145×20 mm Petri dishes (Greiner Bio-One). Petri dishes were incubated overnight at 37 °C and inspected for phage plaques on the next day. Areas with visible lysis or plaques were excised, resuspended in SM buffer, and diluted to single plaques on top agar lawns containing 99% Syn61Δ3 and 1% MDS42 cells. We note that adding trace amounts of MDS42 cells increased the visibility of plaques, and clear plaques, indicating phage replication on the recoded host, could be easily picked. Dilutions and single plaque isolations were repeated four times for each plaque to purify isogenic phages. Finally, high-titer stocks were prepared by mixing sterilized suspensions from single plaques with exponentially growing MDS42 cells (OD600 = 0.3) in SOB supplemented with 10 mM CaCl2 and MgCl2. Phage-infected samples were grown at 37 °C until complete lysis (~4 hrs) and then sterilized by filtration.
Bacteriophage culturing
Bacteriophage stocks were prepared by a modified liquid lysate Phages on Tap protocol in LBL medium51. High-titer lysates were prepared from single plaques by picking well-isolated phage plaques into SM buffer and then seeding 3–50 ml early exponential phase cultures of E. coli MDS42 cells with the resulted phage suspension in SOB supplemented with 10 mM CaCl2 and MgCl2. Phage infected samples were grown at 37 °C until complete lysis and then sterilized by filtration. High-titer phage lysates were stored at 4 °C in the dark. Phages were archived as virocells and stored at −80 °C in the presence of 25% glycerol for long-term storage.
Phage replication assay
Genomic TCR-suppressor tRNA-SerUGA gene containing phages (based on Supplementary Data 1), corresponding to NCBI GenBank numbers MZ501046, MZ501058, MZ501065, MZ501066, MZ501067, MZ501074, MZ501075, MZ501089, MZ501096, MZ501098, MZ501105, MZ501106 24, were obtained from DSMZ (Germany). Exponential phase cultures (OD600 = 0.3) of MDS42 and Syn61Δ3(ev5) were grown in SOB supplemented with 10 mM CaCl2 and MgCl2 at 37 °C. Cultures were infected with phage at an MOI of approximately 0.001. Simultaneously, the same amount of each phage was added to sterile SOB supplemented with 10 mM CaCl2 and MgCl2 to act as a cell-free control for input phage calculation. Replication assays with the T6 bacteriophage in Syn61Δ3 on Figure 1d were performed by infecting exponential phase cultures at an MOI of 0.01 with T6 phage. The figure shows total T6 titer after 24 hours of incubation. Infected cultures were grown at 37 °C with shaking at 250 rpm. After 24 hours, cultures were transferred to 1 ml tubes and centrifuged at 19,000x g to remove cells and cellular debris, and the clarified supernatant was serially diluted in SM buffer to enumerate output phage concentration. 1.5 μl of the diluted supernatants were applied to LBL 0.7% top agar seeded with MDS42 cells and 10 mM CaCl2 and MgCl2 using a 96 fixed pin multi-blot replicator (VP407, V&P Scientific). Following 18 hours of incubation at 37 °C, plaques were counted, and the number of plaques was multiplied by the dilution to calculate the phage titer of the original sample.
Single-step phage growth curve
An exponential phase culture (OD600 = 0.3) of Syn61Δ3 was grown in 50 ml SOB supplemented with 10 mM CaCl2 and MgCl2 at 37 °C with shaking at 250 rpm. Cultures were then spun down and resuspended in 3 ml SOB supplemented with 10 mM CaCl2 and MgCl2, and 1 ml samples were infected with REP12 phage at an MOI of 0.01. Infected cultures were incubated at 37 °C for 10 minutes without shaking for phage attachment and then washed twice with 1 ml SOB by pelleting cells at 4000× g for 3 minutes. Infected cells were then diluted into 50 ml SOB supplemented with 10 mM CaCl2 and MgCl2 and incubated at 37 °C with shaking at 250 rpm. At every 20 minutes, 1 ml sample was measured out into a sterile Eppendorf tube containing 100 μl chloroform, immediately vortexed, and then placed on ice. Phage titers were determined by centrifuging chloroformed cultures at 6000× g for 3 minutes and then serially diluting supernatants in SM buffer and spotting 1 μl dilutions to LBL 0.7% top agar plates seeded with MDS42 cells and 10 mM CaCl2 and MgCl2. Following 18 hours of incubation at 37 °C, plaques were counted, and the number of plaques was multiplied by the dilution to calculate the phage titer of the original sample.
Bacteriophage genome sequencing, assembly, and annotation
Genomic DNA of bacteriophages was prepared from high-titer (i.e., >1010 PFU/mL) stocks after DNase treatment using the Norgen Biotek Phage DNA Isolation Kit (Cat# 46800) according to the manufacturer’s guidelines and sequenced at the Microbial Genome Sequencing Center (MiGS, Pittsburgh, PA, USA). Sequencing libraries were prepared using the Illumina DNA Prep kit and IDT 10 bp UDI indices and sequenced on an Illumina NextSeq 2000, producing 150 bp paired-end reads. Demultiplexing, quality control, and adapter trimming were performed with bcl-convert (v3.9.3). Reads were trimmed to Q28 using BBDuk from BBTools. Phage genomes were then assembled de novo using SPAdes 3.15.2 in --careful mode with an average read coverage of 10–50×. Assembled genomes were then annotated using Prokka version 1.14.652 with default parameters, except that the PHROGs HMM database53 was used as input to improve phage functional gene annotations.
Bacterial genome sequencing and annotation
Genomic DNA from overnight saturated cultures of isogenic bacterial clones was prepared using the MasterPure™ Complete DNA and RNA Purification Kit (Lucigen) according to the manufacturer’s guidelines and sequenced at the Microbial Genome Sequencing Center (MiGS, Pittsburgh, PA, USA). Sequencing libraries were prepared using the Illumina DNA Prep kit and IDT 10 bp UDI indices and sequenced on an Illumina NextSeq 2000, producing 150 bp paired-end reads. Demultiplexing, quality control, and adapter trimming were performed with bcl-convert (v3.9.3). Reads were then trimmed to Q28 using BBDuk from BBTools and aligned to their corresponding reference by using Bowtie2 2.3.054 in --sensitive-local mode. Single-nucleotide polymorphisms (SNPs) and indels were called using breseq (version 0.36.1)55. Only variants with a prevalence higher than 75% were voted as mutations. Following variant calling, mutations were also manually inspected within the aligned sequencing reads in all cases.
The de novo sequencing and genome assembly of Syn61Δ3(ev5) (from a single-colony isolate of Addgene strain #174514) was performed by generating 84,136 Oxford Nanopore (ONT) long-reads by PCR-free library generation (Oxford Nanopore, UK) on a MinION Flow Cell (R9.4.1) and 4.5 × 106 150 bp paired-end reads on an Illumina NextSeq 2000. Quality control and adapter trimming were performed with bcl2fastq 2.20.0.445 and porechop 0.2.3_seqan2.1.1 for Illumina and ONT sequencing, respectively. Next, we performed hybrid assembly with Illumina and ONT reads by using Unicycler 0.4.8 by using the default parameters. Finally, the resulted single, circular contig representing the entire genome was manually inspected for errors in Geneious Prime® 2022.1.1. and annotated based on sequence homology by using the BLAST function (version 2.11.0) implemented in Geneious Prime® 2022.1.1. based on E. coli K-12 MG1655 (NCBI ID: U00096.3) as reference. Gene essentiality was determined based on Ref56.
Transcriptome analysis of phage-infected cells
We explored transcriptomic changes and mRNA production in phage-infected Syn61Δ3 cells by performing a modified single-step growth experiment and collected samples at 20 minutes intervals. 50 ml of early-exponential (OD600 = 0.15) Syn61Δ3 cells (corresponding to 2×1010 CFU) growing at 37 °C, 250 rpm in SOB containing 10 mM CaCl2 and MgCl2 were spun down at room temperature and resuspended in 1 ml of SOB. 50 μl of this uninfected sample was immediately frozen in liquid N2 and stored at −80 °C until RNA extraction. Next, 900 μl of this cell suspension was mixed with 10 ml prewarmed REP12 phage stock (i.e., ~7×1010 PFU to achieve a MOI of ~4) in SOB containing 10 mM CaCl2 and MgCl2, and then incubated at 37 °C for 10 minutes without shaking for phage absorption. Following phage attachment, samples were spun down, washed with 1 ml SOB twice to remove unabsorbed phages, and then resuspended in 10 ml SOB containing 10 mM CaCl2 and MgCl2. Samples were then incubated at 37 °C, 250 rpm. After 20- and 40-minutes post-infection, we spun down 1 ml cell suspension from each sample, and the cell pellets were frozen in liquid nitrogen and stored at −80 °C until RNA extraction. As expected, after 60 minutes post-infection, no cell pellet was visible. Phage infections were performed in three independent replicates. Total RNA from frozen samples was extracted by using the RNeasy Mini Kit (Qiagen, USA) according to the manufacturer’s instructions and the extracted RNA was DNAse treated with Invitrogen RNase-free DNAse (Thermo Fisher Scientific, USA). Sequencing library preparation was then performed using Stranded Total RNA Prep Ligation kit with Ribo-Zero Plus for rRNA depletion and by using 10 bp IDT for Illumina indices (all from Illumina, USA). Sequencing was done on a NextSeq2000 instrument in 2×50 bp paired-end mode. Demultiplexing, quality control, and adapter trimming were performed with bcl-convert (v3.9.3). cDNA reads were aligned to their corresponding reference by using Bowtie2 2.3.054 in --sensitive-local mode, and read count and expression metrics were determined by using Geneious Prime® 2022.1.1. (Biomatters Ltd.). Finally, differential expression analysis was performed by using DESeq250 (version 1.38.3) with default settings.
tRNA sequencing sample preparation
We explored tRNA expression levels and changes in phage-infected Syn61Δ3 cells by performing a modified single-step growth experiment with high MOI and cell mass. An early-exponential phase culture (OD600 = 0.2) of Syn61Δ3 cells (corresponding to approximately 5×1010 CFU) growing at 37 °C, 250 rpm in SOB containing 10 mM CaCl2 and MgCl2 were spun down at room temperature and resuspended in 1.1 ml of SOB. 100 μl of this uninfected sample was immediately frozen in liquid N2 and stored at −80 °C until tRNA extraction. Next, 1000 μl of this cell suspension was mixed with 20 ml prewarmed REP12 phage stock (i.e., ~1012 PFU to achieve a MOI of ~20) in SOB containing 10 mM CaCl2 and MgCl2, and then incubated at 37 °C for 10 minutes without shaking for phage absorption. Following phage attachment, samples were spun down, the supernatant containing unabsorbed phages was removed, and the cell pellet was then resuspended in 7 ml SOB containing 10 mM CaCl2 and MgCl2. Samples were then incubated at 37 °C, 250 rpm. Immediately after phage attachment and after 20- and 40-minutes post-infection, 1 ml cell suspensions from each sample were spun down, and cell pellets were frozen in liquid N2 and stored at −80 °C until total RNA extraction. Phage infections were performed in two independent replicates.
We analyzed the total tRNA content of Ec_Syn61Δ3-SL cells expressing KP869110.1 viral tRNA24-LeuUGA and tRNA24-LeuCGA by pelleting cells from 5 ml mid-exponential (OD600 = 0.3) culture at 4000× g and flash-freezing the cell pellet in liquid nitrogen.
We extracted tRNAs by lysing samples at room temperature (RT) for 30 mins in 150 μl lysis buffer containing 8 mg/mL lysozyme (from chicken egg white, #76346–678, VWR, USA), 10 mM Tris HCl pH 7.5, and 1 μl murine RNase inhibitor (New England Biolabs). Samples were then mixed with 700 μl Qiazol reagent (#79306, Qiagen) and incubated for 5 minutes at RT. Next, 150 μl chloroform was added, vortexed, and incubated until phase separation. Samples were then spun at 15,000x g for 15 min at in a cooled centrifuge. The supernatant was transferred into a new Eppendorf tube and mixed with 350 μl 70% ethanol. Larger RNA molecules were then bound to an RNeasy MinElute spin column (#74204, Qiagen), and the flow-through was mixed with 450 μl of 100% ethanol, and tRNAs were bound to a new RNeasy MinElute spin column. The tRNA fraction was then washed first with 500 μl wash buffer (#74204, Qiagen), next with 80% ethanol, and then eluted in RNase-free water. The eluted tRNAs were deacylated in 60 mM pH 9.5 borate buffer (J62154-AK, Alfa Aesar, Thermo Fisher Scientific) for 30 minutes and then purified using a Micro Bio-Spin P-30 Gel Column (7326251, from Bio-Rad).
tRNA sequencing library preparation, sequencing, and data analysis
We prepared tRNA cDNA libraries by reverse-transcribing tRNAs using the TGIRT™-III template-switching reverse-transcriptase (TGIRT50, InGex, USA) according to the manufacturer’s instructions. In brief, we prepared reaction mixtures containing 1 μl (~100 ng) of the deacylated tRNAs, 2 μl of 1 μM TGIRT DNA/RNA heteroduplex (prepared by hybridizing equimolar amounts of rCrUrUrUrGrArGrCrCrUrArArUrGrCrCrUrGrArArArGrArUrCrGrGrArArGrArGrCrArCrArCrGrUrCrUrArGrUrUrCrUrArCrArGrUrCrCrGrArCrGrArU/3SpC3/ and ATCGTCGGACTGTAGAACTAGACGTGTGCTCTTCCGATCTTTCAGGCATTAGGCTCAAAGN oligos), 4 μl 5× TGIRT™ reaction buffer (2.25 M NaCl, 25 mM MgCl2, 100 mM Tris-HCl, pH 7.5), 2 μl of 100 mM DTT, 9 μl RNase-free water, and 1 μl TGIRT-III, and incubated at room temperature for 30 minutes to initiate template-switching. Next, 1 μl of 25 mM dNTPs (Thermo Fisher Scientific, USA) was added to the reaction mixture, and samples were incubated at 60 °C for 30 minutes to perform reverse transcription. RNA was then hydrolyzed by NaOH, neutralized by HCl, and the cDNA library was purified using MinElute PCR purification kit. cDNAs were then ligated to a preadenylated DNA adapter/5Phos/GATCNNNAGATCGGAAGAGCGTCGTGT/3SpC3/, in which NNN denotes an N, NN, NNN spacer to increase library diversity during sequencing (preadenylated oligos were prepared by 5’ DNA adenylation kit (E2610L) using thermostable 5’ App DNA/RNA ligase (M0319L, both from New England Biolabs) following the manufacturer’s protocol. The cDNA library was purified using the MinElute PCR purification kit (Qiagen) and amplified using Q5 Host-Start High-Fidelity 2x Master Mix (New England Biolabs). PCR products were then size selected to remove adaptor-dimers below 200 bp using three subsequent size-selection rounds with a Select-a-Size DNA Clean & Concentrator Kit (D4080, Zymo Research). Finally, amplicon libraries were barcoded using the IDT 10 bp UDI indices (Illumina) and sequenced on an Illumina MiSeq to produce 250 bp paired-end reads. Read-demultiplexing was performed with bcl-convert (v3.9.3). Paired-end reads were then aligned to their reference sequences by using Geneious assembler, implemented in Geneious Prime® 2022.1.1., allowing a maximum of ten SNPs within tRNA reads compared to their reference. These settings allowed us to map lower-fidelity TGIRT-III-transcribed cDNA reads to their corresponding reference sequence without cross-mapping to tRNAs sharing sequence homology. tRNA reads from Ec_Syn61Δ3-SL cells expressing KP869110.1 viral tRNA24-LeuUGA and tRNA24-LeuCGA were mapped without allowing the presence of SNPs in sequencing reads to distinguish tRNA24-LeuUGA and tRNA24-LeuCGA that differs by only a single SNP within the anticodon region.
Genome editing and biocontainment of Syn61Δ3
We first generated a deficient recombination variant of Syn61Δ3(ev5) by eliminating the expression of the genomic recA gene using Cas9-assisted recombineering. RecA deletion experiments were performed by first transforming Syn61Δ3(ev5) cells with a plasmid carrying a pSC101 origin-of-replication, a constitutively expressed chloramphenicol resistance marker, SpCas9 and tracrRNA (from pCas957,58, Addgene #42876), and the λRed operon, consisting of gam, exo, and bet (from pORTMAGE311B59, Addgene #120418). Next, cells were made electrocompetent using a standard protocol57,58 for Cas9-assisted recombineering and transformed with 2 μl of 100 μM 90 nucleotide-long ssDNA oligonucleotide inserting a stop codon and a frameshift mutation into recA (Supplementary Data 3). Successful edits were selected by cotransforming 1 μg from a variant of the pCRISPR plasmid57,58 carrying a 5’-AGTTGATACCTTCGCCGTAG guide sequence to cleave the genomic recA sequence in unedited cells. All plasmids were recoded to lack TCR and TAG codons in protein-coding genes, and synthesized by GenScript USA Inc. The resulted Syn61Δ3(ev5) ΔrecA strain was validated by whole genome sequencing and then evolved for increased fitness (available in section “Adaptive laboratory evolution of Syn61Δ3”). Finally, the replacement of the genomic adk gene of Syn61Δ3(ev5) ΔrecA (ev1) with the bipA-dependent adk.d6 variant10 was performed by first transforming cells with a plasmid carrying a constitutively expressed MjTyrRS-derived bipA aaRS (variant 10, based on Ref60) together with its associated tRNA under the control of a proK tRNA promoter and an aminoglycoside-(3)-N-acetyltransferase gene, conferring gentamycin resistance, all on a plasmid containing a p15A origin-of-replication (Supplementary Data 3). Next, we integrated the adk.d6 variant by Cas9-assisted recombineering as described above, but instead of oligonucleotide-mediated recombineering, we transformed 4 μg of a dsDNA cassette carrying the full-length adk.d6 variant with 400 bp flanking genomic homology (constructed by GenScript USA Inc., Supplementary Data 3). Cells were grown in the presence of 200 μM bipA in 2×YT media throughout the entire procedure. Successful edits were selected using a dual-targeting crRNA expression construct, carrying 5’-GCAATGCGTATCATTCTGCT and 5’- GCCGTCAACTTTCGCGTATT guide sequences (from GenScript USA Inc.). Positive colonies were selected by screening colonies with allele-specific PCR (Supplementary Data 3) and validated by whole genome sequencing. Finally, the escape rate of the resulted Syn61Δ3(ev5) ΔrecA (ev1) adk.d6 strain was determined as described earlier10, but instead of chloramphenicol, cells were grown in the presence of 10 μg/ml gentamycin in 2×YT. Plates were incubated for seven days at 37 °C. Escape rate measurements were performed in triplicates; ± indicates standard deviation.
Adaptive laboratory evolution of Syn61Δ3
We performed standard adaptive laboratory evolution in rich bacterial media for 30 days (~270 cell generations) on Syn61Δ3(ev5) ΔrecA cells to increase fitness. At each transfer step, 109-1010 bacterial cells were transferred into 500 ml LBL medium containing 1.5 g/l Tris/Tris-HCl and incubated aerobically for 24 hours at 37 °C, 250 rpm in a 2000 ml Erlenmeyer flask with a vented cap. Following 30 transfers, bacterial cells were spread onto LBL agar plates, and an individual colony was isolated and subjected to whole-genome sequencing. The identified mutations in the resulted evolved variant, Syn61Δ3(ev5) ΔrecA (ev1), are available in Supplementary Data 3.
Doubling time measurements
To determine growth parameters under standard laboratory conditions, saturated overnight cultures of E. coli Syn61Δ3(ev5), Syn61Δ3(ev5) ΔrecA, and its evolved variant, Syn61Δ3(ev5) ΔrecA (ev1) were diluted 1:200 into 50 ml of 2×YT and LBL in a 300 ml Erlenmeyer flask with vented cap and incubated aerobically at 37 °C, 250 rpm. Ec_Syn61Δ3-SL cells were characterized similarly, but by using 2×YT containing 50 μg/ml kanamycin. All growth measurements were performed in triplicates. Optical density at 600 nm (OD600) measurements were taken every 20 minutes for 8 hours or until stationary phase was reached on a CO8000 Cell Density Meter, WPA. The doubling time was calculated for each independent replicate by log2-transforming OD600 values and calculating the doubling time based on every six consecutive data points during the exponential growth phase. We calculated the doubling time (1/slope) from a linear fit to log2-derivatives of the six data points within this window and reported the shortest doubling time for each independent culture. Curve fitting, linear regression, and doubling time calculations were performed with Prism9 (GraphPad). Error bars show ± standard deviation.
tRNA annotation
We detected tRNA genes in the Viral genomic NCBI Reference Sequence Database (Accessed: January 2, 2022) and in individual phage isolates’ genomes by using tRNAscan-SE 2.0.9 in bacterial (-B), archaeal (-A), or eukaryotic (-E) maximum sensitivity mode (-I --max)61. tRNAscan-SE detection parameters were chosen according to the predicted host of the corresponding viral strain.
Mobile tRNAome tRNA library generation and selection
We generated our mobile tRNAome expression library by synthesizing tRNAscan-SE predicted tRNAs from diverse sources (Supplementary Data 1), driven by a strong bacterial proK tRNA promoter and followed by two transcriptional terminators as 10 pmol ssDNA oligonucleotide libraries (10 pmol oPool, from Integrated DNA Technologies, USA). Oligonucleotides were resuspended in 1× TE buffer and then amplified using 5’ phosphorylated primers. Amplicons were then blunt-end ligated into pCR4Blunt-TOPO (Invitrogen, Zero Blunt™ TOPO™ PCR Cloning Kit) for 18 hrs at 16 °C and then purified by using the Thermo Scientific GeneJET PCR Purification Kit. We then electroporated 50 ng purified plasmid in five parallel electroporations into 5 × 40 μl freshly made electrocompetent cells of MDS42 and Syn61Δ3(ev5). Prior to electrotransformation, bacterial cells were made electrocompetent by growing cells after a 1:100 dilution in SOB until mid-log phase (OD=0.3) at 32 °C and then washing cells three times using ice-cold water. Electroporated cultures were allowed to recover overnight at 37 °C and then plated to LBL agar plates containing 50 μg/ml kanamycin in 145×20 mm Petri dishes (Greiner Bio-One). Plates were incubated at 37 °C until colony formation. Approximately 1000–5000 colonies were then washed off from selection plates, and plasmids were extracted by using the Monarch® Plasmid Miniprep Kit (New England Biolabs). The tRNA insert from isolated plasmids was then amplified with primers bearing the standard Nextera Illumina Read 1 and Read 2 primer binding sites, barcoded using the IDT 10 bp UDI indices, and sequenced on an Illumina NextSeq 2000, producing 150 bp paired-end reads. Demultiplexing was performed with bcl-convert (v3.9.3). Paired-end reads were then trimmed using BBDuk from BBTools (in Geneious Prime® 2022.1.1., Biomatters Ltd.), merged, and aligned to their reference sequences by using Geneious assembler, implemented in Geneious Prime® 2022.1.1., allowing maximum a single SNV within the tRNA read.
tRNA-LeuYGA library generation and selection
We identified leucine tRNAs that can translate TCR codons as leucine by performing two consecutive screens with plasmid libraries expressing an anticodon loop mutagenized 65,536-member library of leuU tRNA variants and a smaller, 13-member tRNA-LeuYGA expression library consisting of bacteriophage derived Leu tRNA variants, both bearing two tRNAs under the control of a proK promoter and with anticodons swapped to UGA and CGA. To construct a 65,536-member leuU tRNA library, we synthesized an expression construct consisting of a proK promoter-leuUUGA-spacer-leuUCGA-proK terminator sequence, in which the anticodon loop of both leuU tRNAs has been fully randomized, as an oPool library (Supplementary Data 3) (Integrated DNA Technologies, USA). Next, we amplified these leuU variants by using Q5 Hot Start High-Fidelity Master mix using 5’ phosphorylated primers and then ligated the library into a plasmid backbone containing a high copy-number pUC origin-of-replication and an APH(3’)-I aminoglycoside O-phosphotransferase (aph3Ia29×Leu→TCR) gene in which all 29 instances of leucine coding codons were replaced with TCR serine codons (synthesized as a gBlock dsDNA fragment by Integrated DNA Technologies, USA). The ligation was performed at a 3:1 insert-to-vector ratio and by using T4 DNA ligase (New England Biolabs, USA) for 16 hours at 16 °C according to the manufacturer’s instructions. Finally, the ligation product was purified using the GeneJet PCR purification kit (Thermo Fischer Scientific, USA). We constructed the second, 13-member tRNA-LeuYGA expression library (Supplementary Data 3) consisting of bacteriophage-derived Leu tRNA variants bearing a UGA and CGA anticodon by using the same method as for our leuUYGA library. Following library generation, 100 ng from each library was electroporated into freshly made electrocompetent cells of Syn61Δ3 (ev5) ΔrecA (ev1) and recovered in SOB at 37 °C for 16 hours, 250 rpm. After recovery, the cells were plated to 2×YT agar plates containing kanamycin at 200 μg/ml concentration, and selection plates were incubated at 37 °C until colony formation. Finally, plasmids from clones were purified using a Monarch plasmid miniprep kit (New England Biolabs, USA) and subjected to whole plasmid sequencing (SNPsaurus, Eugene, Oregon, US).
Virus resistance analysis of Ec_Syn61Δ3-SL cells
An exponential phase culture (OD600 = 0.3) of the corresponding strain was grown in 3 ml SOB supplemented with 10 mM CaCl2 and MgCl2 and 75 μg/ml kanamycin at 37 °C with shaking. Cultures were then spun down and resuspended in 1 ml SOB supplemented with 10 mM CaCl2 and MgCl2 and infected with a 1:1 mixture of all 12 Syn61Δ3-lytic phage isolates from this study (Extended Data Table 1b) at an MOI of 0.1. Infected cultures were incubated at 37 °C without shaking for 10 minutes for phage attachment and then washed three times with 1 ml SOB supplemented with 10 mM CaCl2 and MgCl2 by pelleting cells at 4000× g for 3 minutes. Infected cells were then diluted into 4 ml SOB supplemented with 10 mM CaCl2 and MgCl2 and 75 μg/ml kanamycin and incubated at 37 °C with shaking at 250 rpm. After 24 hours of incubation, 500 μl samples were measured out into a sterile Eppendorf tube containing 50 μl chloroform, immediately vortexed, and then placed on ice. Phage infection experiments were performed in three independent replicates. Phage titers were determined by centrifuging chloroformed cultures at 6000× g for 3 minutes and then plating 5 μl of the supernatant directly or its appropriate dilutions mixed with 300 μl MDS42 cells in LBL 0.7% top agar with 10 mM CaCl2 and MgCl2. Following 18 hours of incubation at 37 °C, plaques were counted, and the number of plaques was multiplied by the dilution to calculate the phage titer of the original sample.
Phage enrichment experiments were performed by mixing 50 ml early exponential phase cultures (OD600 = 0.2) of bipA-biocontained Ec_Syn61Δ3-SL carrying pLS1 and pLS2 plasmids with 10 ml environmental sample mix, containing the mixture of Sample 2–13 from our study (Extended Data Table 1b). We maximized phage diversity in the environmental sample mix by using freshly collected and filter-sterilized sewage. Infected Ec_Syn61Δ3-SL cells with the corresponding plasmid were grown overnight in SOB supplemented with 200 mM bipA, 10 mM CaCl2 and MgCl2, and 75 μg/ml kanamycin at 37 °C with shaking at 250 rpm. On the next day, cells were removed by centrifugation at 4000× g for 20 minutes, and the supernatant was filter-sterilized using a 0.45 μm filter. Next, 5 ml of the sterilized sample was mixed again with 50 ml early exponential phase cultures (OD600 = 0.2) of the corresponding strain, incubated for 20 minutes at 37 °C for phage absorption, pelleted by centrifugation at 4000× g for 15 minutes, and then resuspended in 50 ml SOB supplemented with 200 mM bipA, 10 mM CaCl2 and MgCl2, and 75 μg/ml kanamycin. Infected cultures were then incubated at 37 °C with shaking at 250 rpm. Cultures were grown overnight and then sterilized by centrifugation at 4000× g for 15 minutes and filtered through a 0.45 μm PVDF Steriflip™ disposable vacuum filter unit (MilliporeSigma). Finally, phage titers were determined by using MDS42 cells as above. Phage enrichment experiments were performed in two independent replicates. The lytic phage titer of the unenriched sample mix was determined by diluting 100 μl of the input environmental sample mix, containing a mixture of Sample 2–13 (Extended Data Table 1), in SM buffer and 10 μl samples from each dilution steps were mixed with late-exponential phase MDS42 cells and 4 ml 0.7% top agar, and then poured on top of LBL agar plates. Plates were incubated until plaque formation at 37 °C.
Construction of pLS plasmids
All pLS plasmids listed in Supplementary Data 3 were synthesized as gBlocks by IDT and circularized either by ligation with T4 DNA ligase (New England Biolabs, USA), or, in the case of pSC101 and RK2 plasmid-derived variants, by isothermal assembly using the HiFi DNA Assembly Master Mix (New England Biolabs, USA). Following assembly, purified plasmid assemblies were electroporated into Ec_Syn61Δ3-SL cells carrying pLS1. pLS1 and pLS2 were designed to express two distinct combinations of the previously identified phage tRNA-LeuYGA tRNAs in antiparallel orientation to avoid repeat-mediated instability62, driven by the strong bacterial proK and SLP2018–2-101 promoters, together with a high copy-number pUC origin-of-replication, the aph3Ia29×Leu→TCR and aminoglycoside-(3)-N-acetyltransferase18×Leu→TCR marker genes. Transformants carrying either pLS1 or pLS2 were identified by transforming assemblies into Syn61Δ3(ev5) ΔrecA (ev1) and selecting for kanamycin resistance. Finally, plasmids from antibiotic-resistant clones were purified using a Monarch plasmid miniprep kit (New England Biolabs, USA) and subjected to whole plasmid sequencing (SNPsaurus, Eugene, Oregon, US).
Escape rate analysis of viral Leu-tRNAYGAs and pLS plasmids
We analyzed the ability of pLS plasmids to function outside Ec_Syn61Δ3-SL cells by transforming extracted plasmids into E. coli K-12 MG1655. Plasmids were purified from biocontained Ec_Syn61Δ3-SL cells, carrying either pLS1 or pLS2 to express tRNA-LeuYGA, or pLS1 together with pLS3–5, by using the PureLink™ Fast Low-Endotoxin Midi Plasmid Purification Kit (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. Next, we electroporated 1 μg from each plasmid prep into freshly made electrocompetent cells of E. coli K-12 MG1655. Cells were made electrocompetent by diluting an overnight SOB culture of MG1655 1:100 into 500 ml SOB in a 2000 ml flask and growing cells aerobically at 32 °C with shaking at 250 rpm. At OD600 = 0.3, cells were cooled on ice and then pelleted by centrifugation and resuspended in 10% glycerol-in-water. Cells were washed 4-times with 10% glycerol-in-water and then resuspended in 400 μl 20% glycerol-in-water. 1000 ng from each plasmid sample was then mixed with 80 μl electrocompetent cells and electroporated by using standard settings in two 1-mm electroporation cuvettes by using standard electroporator settings (1.8 kV, 200 Ohm, 25 μF). Electroporations were performed in three independent replicates. Electroporated cells were then resuspended in 1 ml SOB, and the culture was allowed to recover overnight at 37 °C with shaking at 250 rpm. Finally, 500 μl from each recovery culture was plated to LBL agar plates containing antibiotics corresponding to the given pLS plasmid’s resistance marker (15 μg/ml gentamycin plus 50 μg/ml kanamycin in the case of pLS1 and 2; 100 μg/ml carbenicillin for pLS4, 30 μg/ml chloramphenicol for pLS3 and pLS6, and 20 μg/ml gentamycin for pLS5) in 145×20 mm Petri dishes (Greiner Bio-One). Plates were incubated at 37 °C for seven days and inspected for growth. Electroporation efficiency measurements were performed by electroporating a plasmid carrying a pUC origin-of-replication and kanamycin resistance (pUC-KanR) into MG1655 electrocompetent cells under identical conditions.
Cloning of REP12 tRNA operon
We analyzed the incorporated amino acid in Syn61Δ3 cells bearing the tRNA operon and its native promoter from the REP12 phage by subcloning the genomic tRNA operon into a low-copy plasmid containing an RK2 origin-of-replication and a chloramphenicol-acetyltransferase marker, both recoded to contain no TCR or TAG codons. The genomic tRNA operon of REP12 was PCR amplified using the Q5 Hot Start Master Mix (New England Biolabs, USA) from extracted phage gDNA and purified using the GeneJet PCR purification kit (Thermo Fischer Scientific, USA). Next, 100 ng of the amplified tRNA operon was assembled into the linearized pRK2-cat backbone using the HiFi DNA Assembly Master Mix (New England Biolabs, USA). After incubation for 60 mins at 50 °C, the assembly was purified using the DNA Concentrator & Clean kit (Zymo Research, USA) and transformed into Syn61Δ3(ev5) ΔrecA (ev1) cells expressing an MSKGPGKVPGAGVPGxGVPGVGKGGGT- elastin peptide fused to sfGFP with a terminal 6×His tag (in which x denotes the analyzed codon, TCA or TCG) on a plasmid containing a kanamycin resistance gene and a pUC origin of replication63. Following an overnight recovery at 32 °C, cells were plated to 2×YT agar plates containing kanamycin and chloramphenicol. Finally, plasmid sequences in outgrowing colonies were validated by whole-plasmid sequencing. Elastin16TCR-sfGFP-6×HIS expression measurements have been performed as described below.
Elastin16TCA-sfGFP-6×HIS expression measurements
We assayed the amino acid identity of the serine tRNAUGA tRNAs of MZ501075 and REP12 (see Supplementary Data 3 for sequence information) by coexpressing selected tRNAs and a constitutively expressed MSKGPGKVPGAGVPGxGVPGVGKGGGT- elastin peptide fused to sfGFP with a terminal 6×His tag (in which x denotes the analyzed codon, TCA or TCG)63 on a plasmid containing a kanamycin resistance gene and a pUC origin-of-replication in Syn61Δ3(ev5). Similarly, the pRK2-REP12 plasmid carrying the REP12 phage tRNA operon under the control of its native promoter was coexpressed with the same pUC plasmid carrying no tRNA genes. As a control, we utilized the same elastin-sfGFP-6×HIS expression construct in which position x has been replaced with an alanine GCA codon. For fluorescence and MS/MS measurements, we diluted cultures 1:100 from overnight starters into 50 ml 2×YT in 300 ml shake flasks containing the corresponding antibiotics and cultivated for 48 hours at 37 °C, 200 rpm aerobically. We then determined sfGFP expression levels in samples by pelleting and washing 1 ml of the culture with PBS and resuspending cell pellets in 110 μL BugBuster Protein Extraction Reagent (MilliporeSigma). Reactions were incubated for 5 minutes and then spun down at 13,000× rpm for 10 minutes. The fluorescence of the BugBuster-treated supernatants and the OD600 of the original culture was measured using the Synergy H1 Hybrid Reader (BioTek) plate reader using the bottom mode analysis with an excitation at 480 nm and emission measurement at 515 nm, with the gain set to 50. Fluorescence values were normalized based on OD600 data. The remaining 49 mL culture was spun down, and the cell pellet was resuspended in 2 ml BugBuster Protein Extraction Reagent (MilliporeSigma) and incubated at room temperature for 5 minutes. The lysed cell mixture was spun down at 13,000× rpm for 10 minutes, and the supernatant was mixed in a 1:1 ratio with HIS-Binding/Wash Buffer (G-Biosciences, USA) and 50 μl HisTag Dynabeads (Thermo Fischer Scientific, USA). Following an incubation period of 5 minutes, the beads were separated on a magnetic rack and washed with 300 μl HIS-Binding/Wash Buffer and PBS (Phosphate Buffered Saline) three times. After the last wash step, the bead pellets containing the bound elastin-sfGFP-6×HIS protein samples were frozen at −80 °C until MS/MS sample preparation. Protein production experiments were performed in three independent replicates.
Tandem liquid chromatography and mass spectrometry (LC/MS-MS) analysis of tryptic elastin-sfGFP-6×HIS
Samples from elastin-sfGFP-6×HIS expression experiments were digested directly on HisTag Dynabeads according to the FASP digest procedure64. In brief, samples were washed with 50 mM TEAB (triethylammonium bicarbonate buffer) and then rehydrated with 50 mM TEAB-trypsin solution, followed by a three-hour digest at 50 °C. Digested peptides were then separated from HisTag Dynabeads and concentrated by spinning and drying samples at 3.000× rpm using a SpeedVac concentrator. Samples were then solubilized in 0.1% formic acid-in-water for subsequent analysis by tandem mass spectrometry. LC-MS/MS analysis of digested samples was performed on a Lumos Tribrid Orbitrap Mass Spectrometer equipped with an Ultimate 3000 nano-HPLC (both from Thermo Fisher Scientific, USA). Peptides were separated on a 150 μm inner diameter microcapillary trapping column packed first with 2 cm of C18 Reprosil resin (5 μm, 100 Å, from Dr. Maisch GmbH, Germany) followed by a 50 cm analytical column (PharmaFluidics, Belgium). Separation was achieved by applying a gradient from 4% to 30% acetonitrile in 0.1% formic acid over 60 mins at 200 nl/min. Electrospray ionization was performed by applying a voltage of 2 kV using a custom electrode junction at the end of the microcapillary column and sprayed from metal tips (PepSep, Denmark). The mass spectrometry survey scan was performed in the Orbitrap in the range of 400–1,800 m/z at a resolution of 6×104, followed by the selection of the twenty most intense ions for fragmentation using Collision Induced Dissociation in the second MS step (CID-MS2 fragmentation) in the Ion trap using a precursor isolation width window of 2 m/z, AGC (automatic gain control) setting of 10,000 and a maximum ion accumulation of 100 ms. Singly charged ion species were excluded from CID fragmentation. The normalized collision energy was set to 35 V and an activation time of 10 ms. Ions in a 10-ppm m/z window around ions selected for MS-MS were excluded from further selection for fragmentation for 60 seconds.
The raw data were analyzed using Proteome Discoverer 2.4 (Thermo Fisher Scientific, USA). Assignment of MS/MS spectra was performed using the Sequest HT algorithm by searching the data against a protein sequence database, including all protein entries from E. coli K-12 MG1655, all proteins sequences of interest (including the elastin-sfGFP fusion protein), as well as other known contaminants such as human keratins and common lab contaminants. Quantitative analysis between samples was performed by LFQ (label free quantitation) between different samples. Sequest HT searches were performed using a 10-ppm precursor ion tolerance and requiring each peptides N-/C termini to adhere with trypsin protease specificity while allowing up to two missed cleavages. Methionine oxidation (+15.99492 Da), deamidation (+0.98402 Da) of asparagine and glutamine amino acids, phosphorylation at serine, threonine, and tyrosine amino acids (+79.96633 Da) and N-terminus acetylation (+42.01057 Da) was set as variable modifications. We then determined the amino acid incorporated at position x in our elastin-sfGFP-6×His construct by analyzing changes compared to Phe. To cover all 20 possible amino acid exchange cases at the x position, we performed five separate searches with four different amino acids as possible variable modifications in each search. All cysteines were set to permanent no modification due to no alkylation procedure. An overall false discovery rate of 1% on both protein and peptide level was achieved by performing target-decoy database search using Percolator65.
Total proteome analysis and the detection of serine-to-leucine mistranslation events
We analyzed the translation of viral proteins in Ec_Syn61Δ3-SL cells (Syn61Δ3(ev5) ΔrecA (ev1) expressing a proK promoter-driven Leu9-tRNAYGA construct from Escherichia phage OSYSP (GenBank ID MF402939.1) and APH(3’)-I aminoglycoside O-phosphotransferase (aph3Ia29×Leu→TCR), on a high copy-number pUC plasmid), by performing a modified single-step growth experiment and subsequent time-course tandem mass spectrometry-based proteome analysis. An early-exponential phase culture (OD600 = 0.2) of Ec_Syn61Δ3-SL cells (corresponding to approximately 4×1010 CFU) growing at 37 °C, 250 rpm in SOB containing 10 mM CaCl2, MgCl2, and 75 μg/ml kanamycin were spun down at room temperature and resuspended in 1.1 ml SOB containing 10 mM CaCl2, MgCl2, and 75 μg/ml kanamycin. 100 μl of this uninfected sample was immediately frozen in liquid N2 and stored at −80 °C until proteome analysis. Next, 1000 μl of this cell suspension was mixed with 10 ml prewarmed REP12 phage stock (i.e., ~5×1011 PFU to achieve a MOI of ~12) in SOB containing 10 mM CaCl2, MgCl2, and 75 μg/ml kanamycin, and then incubated at 37 °C for 10 minutes without shaking for phage absorption. Following phage attachment, samples were spun down, the supernatant containing unabsorbed phages was removed, and the cell pellet was resuspended in 5 ml SOB containing 10 mM CaCl2 and MgCl2. Samples were then incubated at 37 °C, 250 rpm. After 20- and 40-minutes post-infection, 1 ml cell suspensions were spun down, and cell pellets were frozen in liquid N2 and stored at −80 °C until total protein extraction. Samples from control and phage-infected Syn61Δ3 cells were then digested by using the FASP digest procedure64. In brief, samples were washed with 50 mM TEAB buffer on a 10 kDa cutoff filter (Pall Corp, CA) and then rehydrated with 50 mM TEAB-trypsin solution, followed by a three-hour digest at 37 °C. Digested peptides were then extracted and separated into 10 fractions by using the Pierce™ High pH Reversed-Phase Peptide Fractionation Kit according to the manufacturer’s protocol (Thermo Fisher Scientific, USA). Following fractionation, peptides were concentrated and dried by spinning samples at 3.000× rpm using a SpeedVac concentrator. Samples were then solubilized in 0.1% formic acid-in-water for subsequent analysis by tandem mass spectrometry. LC-MS/MS analysis of digested samples was performed on a Lumos Tribrid Orbitrap Mass Spectrometer equipped with an Ultimate 3000 nano-HPLC (both from Thermo Fisher Scientific, USA). Peptides were separated on a 150 μm inner diameter microcapillary trapping column packed first with 2 cm of C18 Reprosil resin (5 μm, 100 Å, from Dr. Maisch GmbH, Germany) followed by a 50 cm analytical column (PharmaFluidics, Belgium). Separation was achieved by applying a gradient from 5% to 27% acetonitrile in 0.1% formic acid over 90 mins at 200 nl/min. Electrospray ionization was performed by applying a voltage of 2 kV using a custom electrode junction at the end of the microcapillary column and sprayed from metal tips (PepSep, Denmark). The mass spectrometry survey scan was performed in the Orbitrap in the range of 400–1,800 m/z at a resolution of 6×104, followed by the selection of the twenty most intense ions for fragmentation using Collision Induced Dissociation in the second MS step (CID-MS2 fragmentation) in the Ion trap using a precursor isolation width window of 2 m/z, AGC (automatic gain control) setting of 10,000 and a maximum ion accumulation of 100 ms. Singly charged ion species were excluded from CID fragmentation. The normalized collision energy was set to 35 V and an activation time of 10 ms. Ions in a 10-ppm m/z window around ions selected for MS-MS were excluded from further selection for fragmentation for 60 seconds.
The raw data was analyzed using Proteome Discoverer 2.4 (Thermo Fisher Scientific, USA). Assignment of MS/MS spectra was performed using the Sequest HT algorithm by searching the data against a protein sequence database, including all protein entries from E. coli K-12 MG1655, all protein sequences of the corresponding REP12 bacteriophage and the Aph3Ia APH(3’)-I aminoglycoside O-phosphotransferase, as well as other known contaminants such as human keratins and common lab contaminants. Quantitative analysis between samples was performed by LFQ (label free quantitation) between different samples. Sequest HT searches were performed using a 10-ppm precursor ion tolerance and requiring each peptides N-/C termini to adhere with trypsin protease specificity while allowing up to two missed cleavages. Methionine oxidation (+15.99492 Da), deamidation (+0.98402 Da) of asparagine and glutamine amino acids, phosphorylation at serine, threonine, and tyrosine amino acids (+79.96633 Da) and N-terminus acetylation (+42.01057 Da) was set as variable modifications. Special modification of serine to leucine amino acid exchange (+26.052036 Da) on all serine amino acid positions was used as variable modification. All cysteines were set to permanent no modification due to no alkylation procedure. An overall false discovery rate of 1% on both protein and peptide levels was achieved by performing target-decoy database search using Percolator65.
Extended Data
Extended Data Figure 1. Viral serine tRNAs decode TCA codons as serine, but Syn61Δ3 obstructs the replication of viruses containing genomic tRNA-SerUGA.
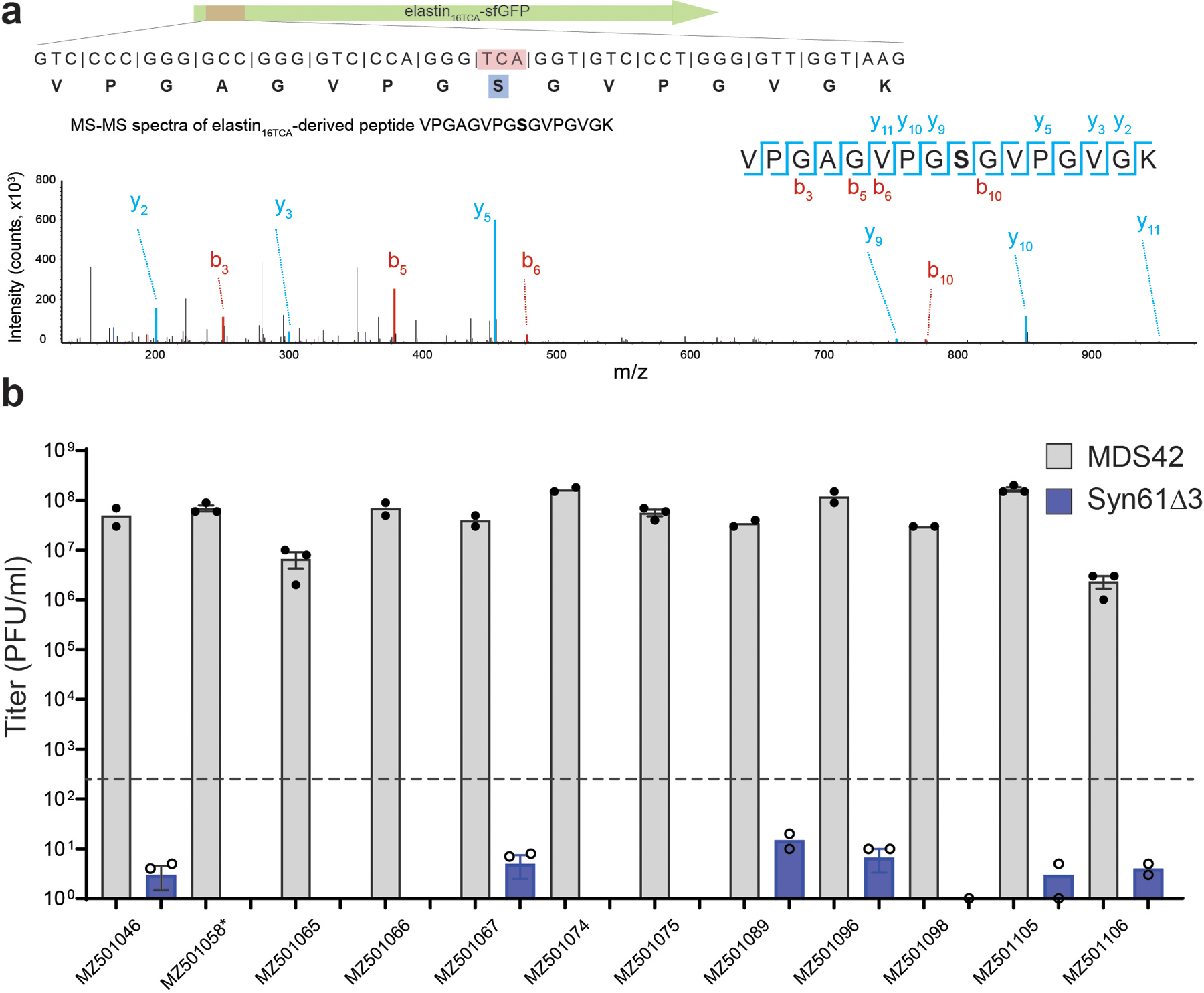
a) Viral TCR suppressor tRNAs decode TCA codons as serine. The amino acid identity of the translated TCR codon within elastin16 TCA-sfGFP-His6 was confirmed by tandem mass spectrometry from Syn61Δ3 expressing the tRNA-SerUGA of Escherichia phage IrisVonRoten (GenBank ID MZ501075)24. The figure shows the amino acid sequence and MS/MS spectrum of the analyzed elastin16 TCA peptide. MS/MS data was collected once.
b) Syn61Δ3 obstructs the replication of viruses containing genomic tRNA-SerUGA. Figure shows the titer of twelve tRNA gene-containing coliphages24, after 24 hours of growth on MDS42 and Syn61Δ3. All analyzed bacteriophages, except MZ501058, contain a genomic tRNA-SerUGA tRNA that provides TCR suppressor activity based on our screen (Figure 1b, Supplementary Data 1). Early exponential phase cultures of MDS42 and Syn61Δ3 were infected at an MOI of 0.001 with the corresponding phages, and free phage titers were determined after 24 hours of incubation. Measurements were performed in n=3 independent experiments (i.e., MDS42 + MZ501058, MZ501065, MZ501075, MZ501105, MZ501106; Syn61Δ3 + MZ501046, MZ501067, MZ501066, MZ501096, MZ501074, MZ501098) or in n=2 independent experiments (i.e., MDS42 + MZ501046, MZ501066, MZ501067, MZ501074, MZ501089, MZ501096, MZ501098; Syn61Δ3 + MZ501058, MZ501065, MZ501075, MZ501089, MZ501105, MZ501106); dashed line represents input phage titer without bacterial cells (i.e., a titer of 420 PFU/ml); dots represent data from independent experiments; bar graphs represent the mean; error bars represent the SEM based on n=3 independent experiments.
Extended Data Figure 2. Viruses overcome genetic-code-based resistance by rapidly complementing the cellular tRNA pool with virus-encoded tRNAs.
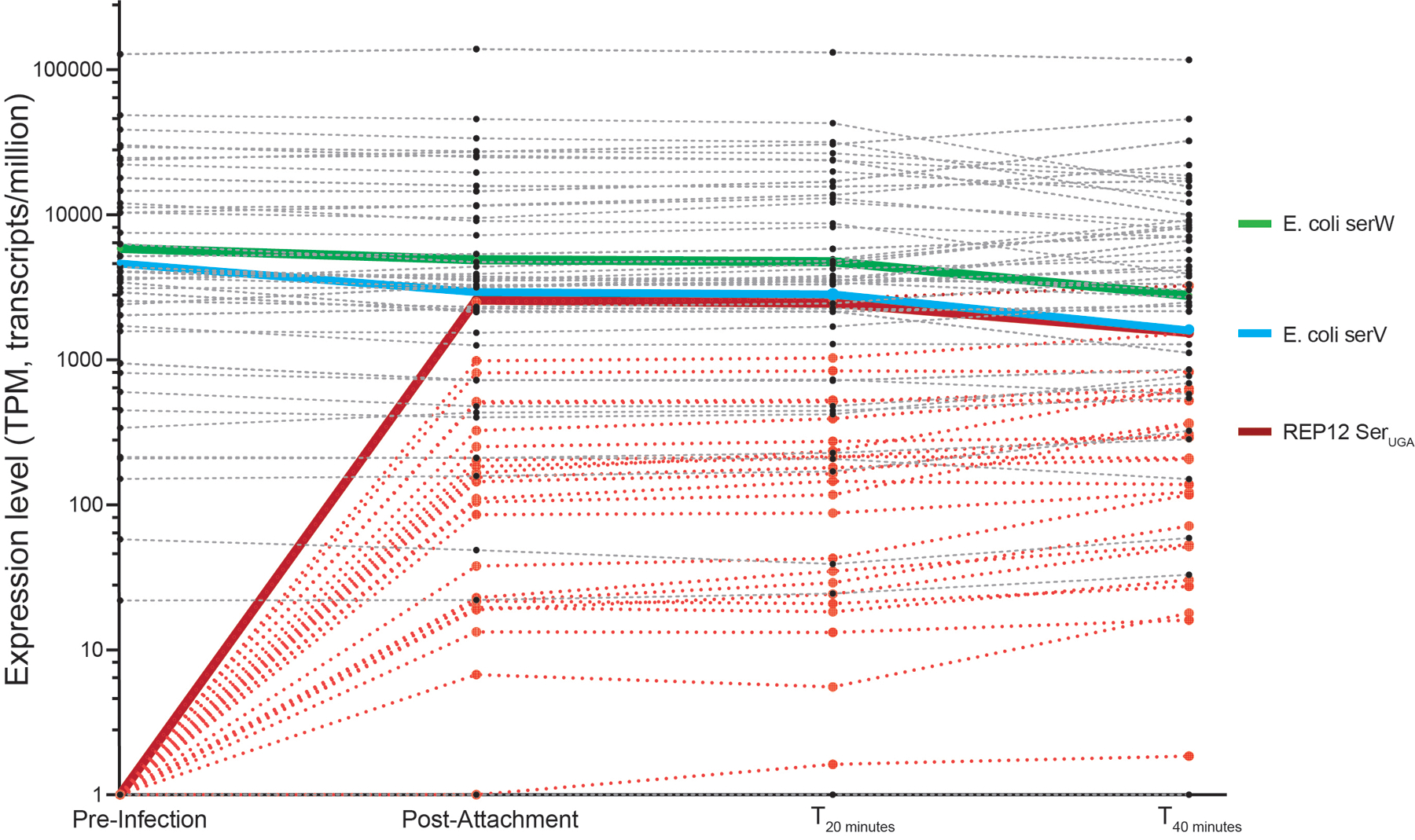
The time-course kinetics of host and viral tRNA expression in Syn61Δ3 cells following REP12 phage infection was quantified using tRNAseq (Methods). The endogenous serV and serW tRNAs of the host Syn61Δ3 are highlighted in green and blue, respectively, while the tRNA-SerUGA of the REP12 virus is highlighted in red. REP12 viral tRNAs are shown in light red; endogenous tRNAs of Syn61Δ3 are shown in gray. Data represent mean TPM (transcript/million). Source data is available within this paper.
Extended Data Figure 3. tRNAseq-based detection of CCA tRNA tail addition to REP12 tRNA-SerUGA.
The tRNA tail is modified into CCA even if the phage-encoded tRNA-SerUGA does not encode a CCA end. The sequence of genomic phage-encoded tRNA-SerUGA is highlighted in magenta. Black letters indicate example tRNAseq sequencing reads from REP12 infected Syn61Δ3 cells directly after phage attachment based on a single experiment.
Extended Data Figure 4. Transcriptomic changes in Syn61Δ3 following phage infection.
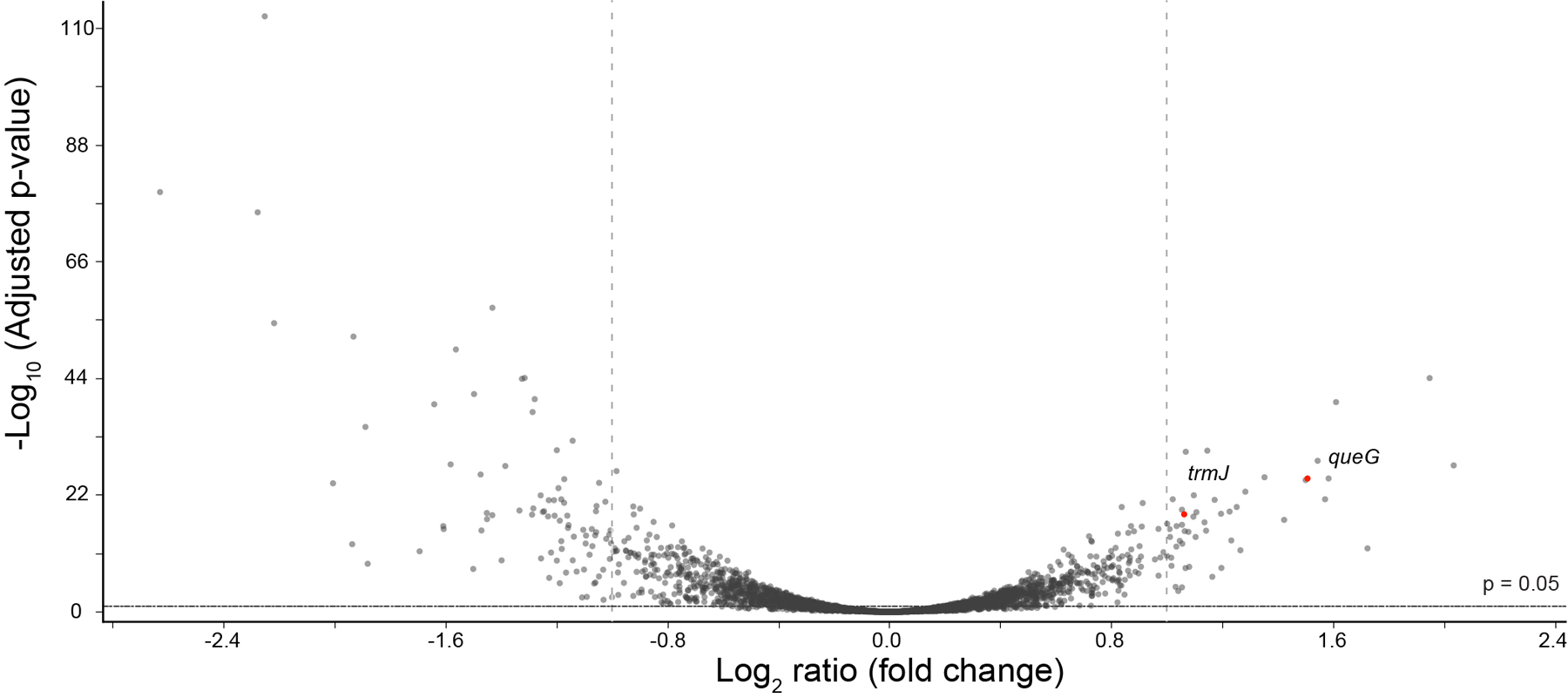
Volcano plot shows the differential expression between uninfected and REP12-infected Syn61Δ3 cells, 40 minutes post-infection, based on n=3 independent experiments (Methods). QueG encodes epoxyqueuosine reductase that catalyzes the final step in the de novo synthesis of queuosine in tRNAs. TrmJ encodes tRNA Cm32/Um32 methyltransferase that introduces methyl groups at the 2’-O position of U32 of several tRNAs, including tRNA-SerUGA. Differential expression and −Log10 adjusted p-values were calculated using the DESeq2 algorithm50. Source data is available within this paper.
Extended Data Figure 5. Multiple sequence alignment of leuUYGA and phage-derived tRNA-LeuYGA variants.
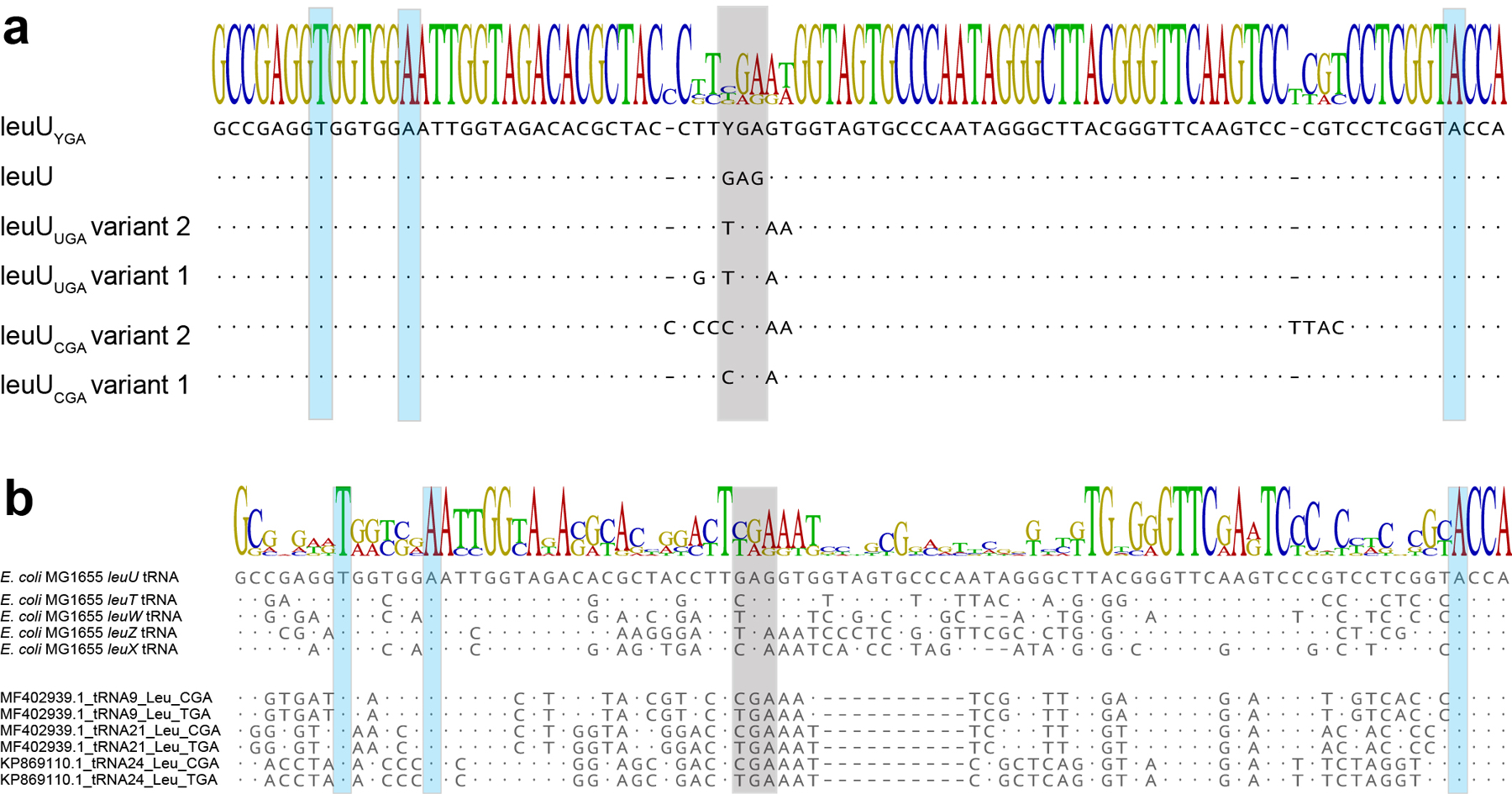
a) Multiple sequence alignment of leuUYGA variants selected in aminoglycoside O-phosphotransferase expression screen, compared to E. coli leuU and the YGA anticodon-swapped E. coli leuU tRNA variant. Grey shading indicates the anticodon region, and the host’s LeuS leucine-tRNA-ligase identity elements31 are shown in blue. Sequence information of the leuUYGA variants is available in Supplementary Data 3.
b) Multiple sequence alignment of phage-derived tRNA-LeuYGA variants selected in the aph3Ia29×Leu→TCR aminoglycoside O-phosphotransferase expression screen, compared to endogenous E. coli leucine tRNAs. Grey shading indicates the anticodon region, while the host’s LeuS leucine-tRNA-ligase identity elements31 are shown in blue. Sequence information of the phage-derived tRNA-LeuYGA variants is available in Supplementary Data 3.
Extended Data Figure 6. tRNAseq-based quantification of viral Leu-tRNAUGA and Leu-tRNACGA in the tRNAome of Ec_Syn61Δ3-SL.
tRNA levels of Ec_Syn61Δ3-SL were quantified using tRNAseq (Methods). Viral Leu-tRNAUGA and Leu-tRNACGA are highlighted in red, and the host’s endogenous E. coli serV tRNA is highlighted in orange. tRNAseq data was collected once. Data represent TPM (transcript/million). Source data is available within this paper.
Extended Data Figure 7. Serine-to-leucine mistranslation of TCT codons in Ec_Syn61Δ3-SL cells.
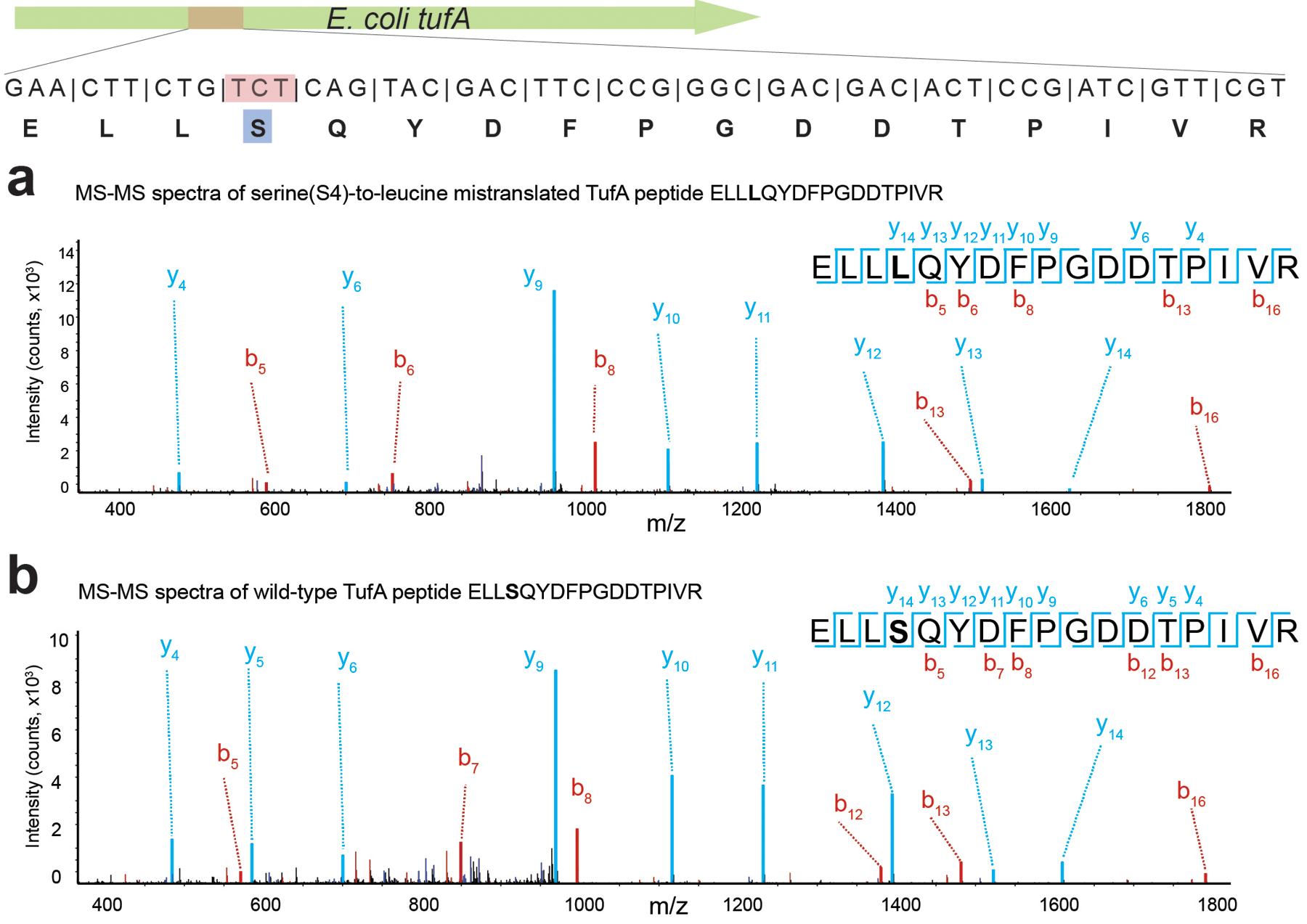
a) MS/MS spectrum of the serine-to-leucine mistranslated TufA peptide. b) MS/MS spectrum of the wild-type TufA peptide. The figure shows the amino acid sequence and MS/MS spectrum of the Ec_Syn61Δ3-SL-expressed TufA protein fragment, together with its genomic sequence, in which the serine-coding TCT codon (as shown in panel b) is partially mistranslated as leucine (as shown in panel a). The experiment was performed by analyzing the total proteome of Ec_Syn61Δ3-SL cells, expressing Leu9-tRNAYGA from Escherichia phage OSYSP (GenBank ID MF402939.1) by tandem mass spectrometry (Methods). MS/MS data was collected once.
Extended Data Figure 8. Doubling time and growth curves of Syn61Δ3(ev5), Syn61Δ3(ev5)ΔrecA, Syn61Δ3(ev5)ΔrecA(ev1), and Ec_Syn61Δ3-SL.
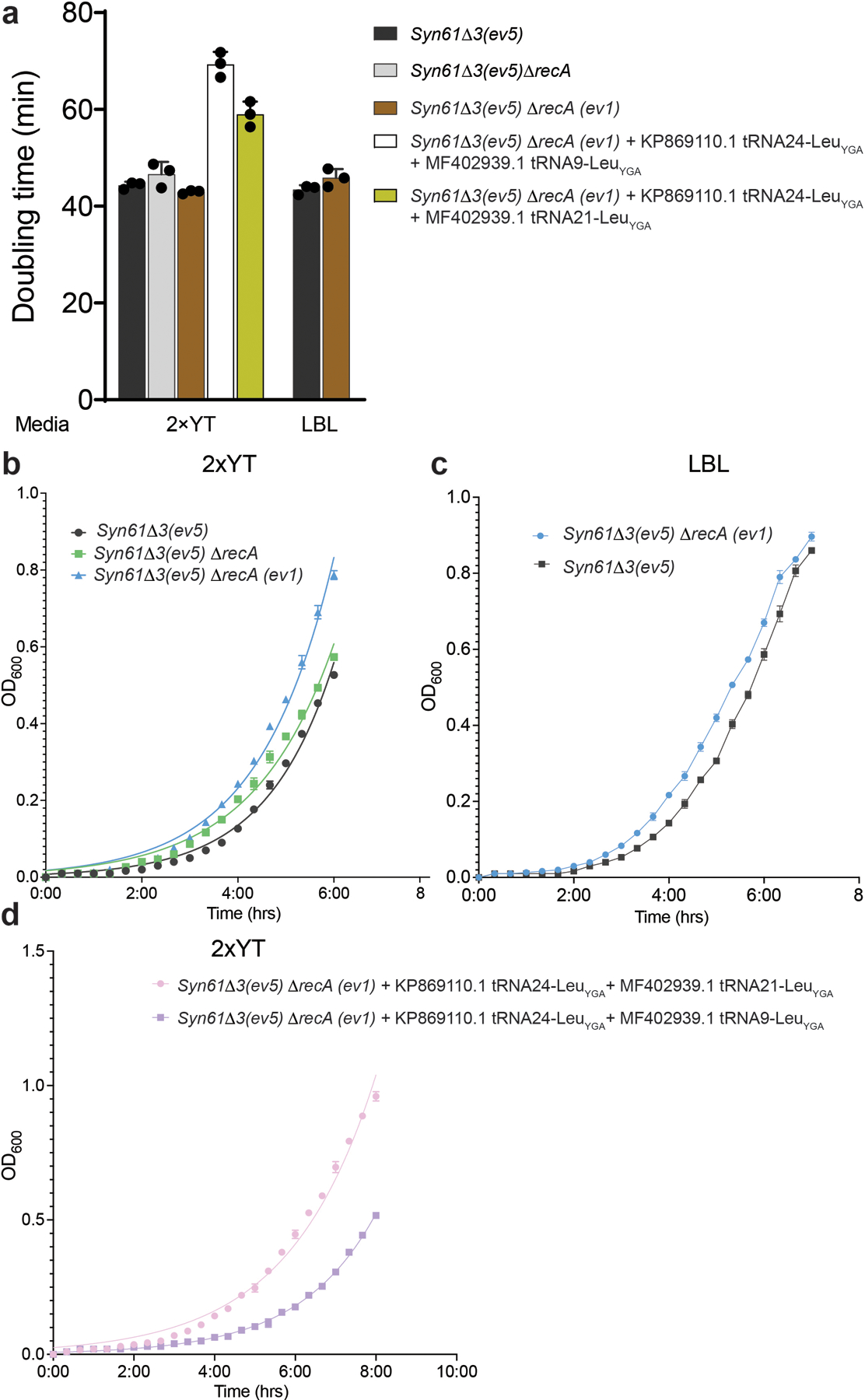
a) Doubling times of Syn61Δ3(ev5), Syn61Δ3(ev5)ΔrecA, Syn61Δ3(ev5)ΔrecA(ev1), and Ec_Syn61Δ3-SL, calculated based on growth curves (shown in panels b, c, d) in rich bacterial media under standard laboratory conditions. b) Growth curves of Syn61Δ3(ev5), Syn61Δ3(ev5)ΔrecA, and Syn61Δ3(ev5)ΔrecA(ev1) in LBL broth. c) Growth curves of Syn61Δ3(ev5) and Syn61Δ3(ev5)ΔrecA(ev1) in 2×YT broth. d) Growth curves of Ec_Syn61Δ3-SL in 2×YT broth containing 50 μg/ml kanamycin. Three independent cultures were grown aerobically in vented shake flasks at 37 °C, and OD600 measurements were taken during exponential growth (Methods). Data curves and bars represent the mean. Error bars show standard deviation based on n=3 independent experiments.
Extended Data Figure 9. Ec_Syn61Δ3-SL resists viruses in environmental samples.
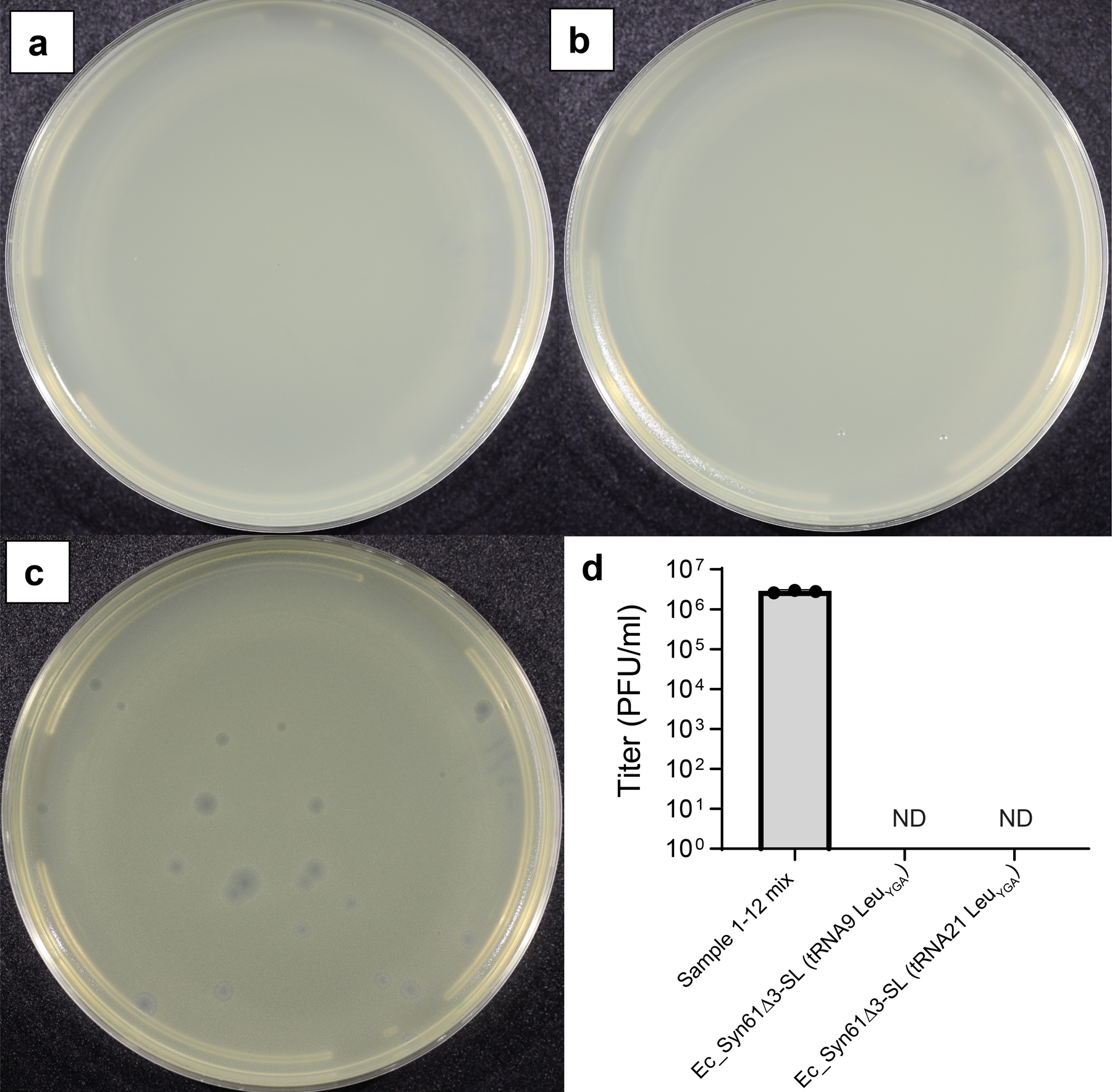
Phage enrichment experiment using Ec_Syn61Δ3-SL, expressing KP869110.1 tRNA24 LeuYGA and MF402939.1 tRNA9 LeuYGA, as host. (b) Phage enrichment experiment using Ec_Syn61Δ3-SL, expressing KP869110.1 tRNA24 LeuYGA and MF402939.1 tRNA21 LeuYGA, as host. Phage enrichment experiments were performed by mixing early exponential cultures of Ec_Syn61Δ3-SL with 10 ml environmental sample mix containing the mixture of Sample 2–13 from our study (Extended Data Table 1a). After two enrichment cycles (Methods, Supplementary Note), filter-sterilized culture supernatants were mixed with phage-susceptible E. coli MDS42 cells in top agar and plated on LBL agar plates to determine viral titer. Enrichment experiments were performed in n=2 independent replicates with the same result. (c) Lytic E. coli MDS42 phage plaques after 103-fold dilution of the environmental sample mix. d) Lytic phage titer of the environmental sample mix, before and after enrichment on Ec_Syn61Δ3-SL. Dots represent the viral titer of the unenriched sample based on three independent experiments, measured on E. coli MDS42 cells. ND represents no plaques detected. Bar represents the mean. Error bar shows standard deviation based on n=3 independent experiments.
Extended Data Table 1. Isolation of lytic viruses infecting Syn61Δ3.
a) Environmental samples analyzed in this study for the presence of lytic viruses of Syn61Δ3. Samples containing lytic phages of Syn61Δ3 are highlighted in gray.
b) Genome size and taxonomy of lytic viruses infecting Syn61Δ3. Annotated genome sequences of all REP phage isolates are available in the Supplementary Data of this paper. REP is an abbreviation of Recoded E. coli Phage. The annotated genomes of REP phages have been deposited to NCBI GenBank under Accession numbers OQ174500, OQ174501, OQ174502, OQ174503, OQ174504, OQ174505, OQ174506, OQ174507, OQ174508, OQ174509, OQ174510, and OQ174511.
| a | |||
|---|---|---|---|
| Sample ID | Sample description | Sample treatment | Sample source location |
| 1 | Wastewater (primary effluent) | Chloroform treated | Massachusetts, USA |
| 2 | Wastewater (primary effluent) | 0.22 μm filtered | Massachusetts, USA |
| 3 | River water | 0.22 μm filtered | Massachusetts, USA |
| 4 | Soil from horse and alpaca enclosure | 0.22 μm filtered | Massachusetts, USA |
| 5 | Water from porta-potty overflow | 0.22 μm filtered | Massachusetts, USA |
| 6 | Irrigation water near wild rat nest | 0.22 μm filtered | Massachusetts, USA |
| 7 | Soil from pasture-raised chicken enclosure | 0.22 μm filtered | Massachusetts, USA |
| 8 | Soil from egg-laying chicken shed | 0.22 μm filtered | Massachusetts, USA |
| 9 | Fresh pig feces | 0.22 μm filtered | Massachusetts, USA |
| 10 | Mixed fresh horse and cow feces | 0.22 μm filtered | Massachusetts, USA |
| 11 | Farm compost pile | 0.22 μm filtered | Massachusetts, USA |
| 12 | Farm compost pile | 0.22 μm filtered | Massachusetts, USA |
| 13 | Water from pig and goose water trough | 0.22 μm filtered | Massachusetts, USA |
| b | ||
|---|---|---|
| Isolate | Genome size (kb) | Taxonomic classification |
| REP1 | 167.4 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Tevenvirinae; Tequatrovirus |
| REP2 | 167.9 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Tevenvirinae; Tequatrovirus |
| REP3 | 167.9 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Tevenvirinae; Tequatrovirus |
| REP4 | 166.8 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Tevenvirinae; Tequatrovirus |
| REP5 | 88.9 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Ounavirinae; Felixounavirus |
| REP6 | 87.2 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Ounavirinae; Felixounavirus |
| REP7 | 88.9 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Ounavirinae; Felixounavirus |
| REP8 | 85.8 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Ounavirinae; Felixounavirus |
| REP9 | 85.8 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Ounavirinae; Felixounavirus |
| REP10 | 85.7 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Ounavirinae; Felixounavirus |
| REP11 | 85.7 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Ounavirinae; Felixounavirus |
| REP12 | 85.8 | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Caudovirales; Myoviridae; Ounavirinae; Felixounavirus |
Supplementary Material
Acknowledgments
We thank György Pósfai (Biological Research Centre, Hungary) for sharing MDS42 and Jason W. Chin’s team (Medical Research Council Laboratory of Molecular Biology, UK) for sharing Syn61Δ3 via Addgene. Funding for this research was provided by the US Department of Energy (DOE) under grant DE-FG02–02ER63445 and by the National Science Foundation (NSF) Award number: 2123243 (both to G.M.C.). M.B. acknowledges support from the NIGMS of the National Institutes of Health (R35GM133700), the David and Lucile Packard Foundation, the Pew Charitable Trusts, and the Alfred P. Sloan Foundation. A.N. was supported by the EMBO LTF 160–2019 Long-Term fellowship. The authors thank Andrew Millard’s laboratory for making the PHROG HMM database available for bacteriophage annotation, GenScript USA Inc. for their DNA synthesis support, and Dan Snyder, Katrina Harris, and all members of the Microbial Genome Sequencing Center (MiGS), Pittsburgh, PA for their support with DNA and RNA sequencing. We are thankful to Ting Wu for her support, Yue Shen and Shirui Yan (Institute of Biochemistry, Beijing Genomics Institute) for our collaboration on genome recoding, and Behnoush Hajian for graphical design and her help with illustrations.
Footnotes
Competing Interests statement
The authors declare competing financial interests. Harvard Medical School has filed a provisional patent application related to this work on which A.N., S.V., and G.M.C. are listed as inventors. M.L., K.C., and F.H. are employed by GenScript USA Inc., but the company had no role in designing or executing experiments. G.M.C. is a founder of the following companies in which he has related financial interests: GRO Biosciences, EnEvolv (Ginkgo Bioworks), and 64x Bio. Other potentially relevant financial interests of G.M.C. are listed at http://arep.med.harvard.edu/gmc/tech.html.
Supplementary Information is available for this paper.
Data availability Statement
Raw data from whole-genome sequencing, transcriptome, and tRNA sequencing experiments have been deposited to Sequence Read Archive (SRA) under BioProject ID PRJNA856259. tRNA data and the generated sequences in this study are included in the Supplementary Data files. Mass spectra and proteome measurements have been deposited to MassIVE (MSV000089854; doi:10.25345/C5FF3M41W)66. The Syn61Δ3(ev5) ΔrecA (ev1) strain is available from Addgene (Bacterial strain #189857). We cannot deposit Ec_Syn61Δ3-SL and Ec_Syn61Δ3 adk.d6 at Addgene due to the incompatibility of Addgene’s methods of strain distribution and growth media requirements, but all materials used in this study are freely available from the corresponding authors for academic research use upon request. The PHROG HMM database is available at https://phrogs.lmge.uca.fr/ and from reference53. The assembled annotated genome of Escherichia coli Syn61 substr. Syn61Δ3(ev5) is available in Supplementary Data 4, and the annotated genome of REP phages have been deposited to NCBI GenBank under accession numbers OQ174500, OQ174501, OQ174502, OQ174503, OQ174504, OQ174505, OQ174506, OQ174507, OQ174508, OQ174509, OQ174510, andOQ174511. Source data is provided with this paper.
References
- 1.Church GM & Regis E Regenesis: How Synthetic Biology Will Reinvent Nature and Ourselves. (Basic Books, 2014). [Google Scholar]
- 2.Lajoie MJ et al. Genomically Recoded Organisms Expand Biological Functions. Science 342, 357–360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma NJ & Isaacs FJ Genomic Recoding Broadly Obstructs the Propagation of Horizontally Transferred Genetic Elements. cels 3, 199–207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robertson WE et al. Sense codon reassignment enables viral resistance and encoded polymer synthesis. Science 372, 1057–1062 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostrov N et al. Design, synthesis, and testing toward a 57-codon genome. Science 353, 819–822 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Fujino T, Tozaki M & Murakami H An Amino Acid-Swapped Genetic Code. ACS Synth. Biol 9, 2703–2713 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Abrahão J et al. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nature Communications 9, 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgado S & Vicente AC Global In-Silico Scenario of tRNA Genes and Their Organization in Virus Genomes. Viruses 11, 180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Shayeb B et al. Clades of huge phages from across Earth’s ecosystems. Nature 578, 425–431 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mandell DJ et al. Biocontainment of genetically modified organisms by synthetic protein design. Nature 518, 55–60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zou X et al. Systematic strategies for developing phage resistant Escherichia coli strains. Nat Commun 13, 4491 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barone PW et al. Viral contamination in biologic manufacture and implications for emerging therapies. Nature Biotechnology 1–10 (2020) doi: 10.1038/s41587-020-0507-2. [DOI] [PubMed] [Google Scholar]
- 13.Baltz RH Bacteriophage-resistant industrial fermentation strains: from the cradle to CRISPR/Cas9. Journal of Industrial Microbiology and Biotechnology 45, 1003–1006 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Ostrov N et al. Synthetic genomes with altered genetic codes. Current Opinion in Systems Biology (2020) doi: 10.1016/j.coisb.2020.09.007. [DOI] [Google Scholar]
- 15.Fredens J et al. Total synthesis of Escherichia coli with a recoded genome. Nature 1 (2019) doi: 10.1038/s41586-019-1192-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang JY et al. Degradation of host translational machinery drives tRNA acquisition in viruses. Cell Systems 12, 771–779.e5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peters S et al. Validation that human microbiome phages use alternative genetic coding with TAG stop read as Q. http://biorxiv.org/lookup/doi/10.1101/2022.01.06.475225 (2022) doi: 10.1101/2022.01.06.475225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borges AL et al. Stop codon recoding is widespread in diverse phage lineages and has the potential to regulate translation of late stage and lytic genes. BioRxiv (2021) doi: 10.1101/2021.08.26.457843. [DOI] [Google Scholar]
- 19.Abe T et al. tRNADB-CE 2011: tRNA gene database curated manually by experts. Nucleic Acids Research 39, D210–D213 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alamos P et al. Functionality of tRNAs encoded in a mobile genetic element from an acidophilic bacterium. RNA Biology 15, 518–527 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santamaría-Gómez J et al. Role of a cryptic tRNA gene operon in survival under translational stress. Nucleic Acids Research (2021) doi: 10.1093/nar/gkab661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bustamante P et al. ICEAfe1, an Actively Excising Genetic Element from the Biomining Bacterium Acidithiobacillus ferrooxidans. MMB 22, 399–407 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Bowden RJ, Simas JP, Davis AJ & Efstathiou S 1997. Murine gammaherpesvirus 68 encodes tRNA-like sequences which are expressed during latency. Journal of General Virology 78, 1675–1687. [DOI] [PubMed] [Google Scholar]
- 24.Maffei E et al. Systematic exploration of Escherichia coli phage–host interactions with the BASEL phage collection. PLOS Biology 19, e3001424 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brok-Volchanskaya VS et al. Phage T4 SegB protein is a homing endonuclease required for the preferred inheritance of T4 tRNA gene region occurring in co-infection with a related phage. Nucleic Acids Res 36, 2094–2105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miles ZD, McCarty RM, Molnar G & Bandarian V Discovery of epoxyqueuosine (oQ) reductase reveals parallels between halorespiration and tRNA modification. Proceedings of the National Academy of Sciences 108, 7368–7372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu R-J, Long T, Zhou M, Zhou X-L & Wang E-D tRNA recognition by a bacterial tRNA Xm32 modification enzyme from the SPOUT methyltransferase superfamily. Nucleic Acids Research 43, 7489–7503 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt M & Kubyshkin V How To Quantify a Genetic Firewall? A Polarity-Based Metric for Genetic Code Engineering. ChemBioChem 22, 1268–1284 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Two conformations of a crystalline human tRNA synthetase–tRNA complex: implications for protein synthesis. The EMBO Journal 25, 2919–2929 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobayashi T et al. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat Struct Mol Biol 10, 425–432 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Giege R, Sissler M & Florentz C Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Research 26, 5017–5035 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Church G, Baynes B & Pitcher E Hierarchical Assembly Methods for Genome Engineering. (2007).
- 33.Zürcher JF et al. Refactored genetic codes enable bidirectional genetic isolation. Science doi: 10.1126/science.add8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Łoś JM, Golec P, Węgrzyn G, Węgrzyn A & Łoś M Simple Method for Plating Escherichia coli Bacteriophages Forming Very Small Plaques or No Plaques under Standard Conditions. Appl Environ Microbiol 74, 5113–5120 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abedon ST & Yin J Bacteriophage Plaques: Theory and Analysis. in Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions (eds. Clokie MRJ & Kropinski AM) 161–174 (Humana Press, 2009). doi: 10.1007/978-1-60327-164-6_17. [DOI] [PubMed] [Google Scholar]
- 36.Serwer P, Hayes SJ, Thomas JA & Hardies SC Propagating the missing bacteriophages: a large bacteriophage in a new class. Virology Journal 4, 21 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Yashiro Y, Sakaguchi Y, Suzuki T & Tomita K Mechanistic insights into tRNA cleavage by a contact-dependent growth inhibitor protein and translation factors. Nucleic Acids Research 50, 4713–4731 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomita K, Ogawa T, Uozumi T, Watanabe K & Masaki H A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proceedings of the National Academy of Sciences 97, 8278–8283 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takai K, Takaku H & Yokoyama S In Vitro Codon-Reading Specificities of Unmodified tRNA Molecules with Different Anticodons on the Sequence Background of Escherichia coli tRNASer1. Biochemical and Biophysical Research Communications 257, 662–667 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Takai K, Okumura S, Hosono K, Yokoyama S & Takaku H A single uridine modification at the wobble position of an artificial tRNA enhances wobbling in an Escherichia coli cell-free translation system. FEBS Letters 447, 1–4 (1999). [DOI] [PubMed] [Google Scholar]
- 41.Kunjapur AM et al. Synthetic auxotrophy remains stable after continuous evolution and in coculture with mammalian cells. Science Advances 7, eabf5851 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rovner AJ et al. Recoded organisms engineered to depend on synthetic amino acids. Nature 518, 89–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nyerges Á et al. A highly precise and portable genome engineering method allows comparison of mutational effects across bacterial species. PNAS 201520040 (2016) doi: 10.1073/pnas.1520040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wannier TM et al. Adaptive evolution of genomically recoded Escherichia coli. PNAS 201715530 (2018) doi: 10.1073/pnas.1715530115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirchberger PC & Ochman H Resurrection of a global, metagenomically defined gokushovirus. eLife 9, e51599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nyerges A et al. Swapped genetic code blocks viral infections and gene transfer. 2022.07.08.499367 Preprint at 10.1101/2022.07.08.499367 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y et al. Multiplex base editing to convert TAG into TAA codons in the human genome. 2021.07.13.452007 Preprint at 10.1101/2021.07.13.452007 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boeke JD et al. The Genome Project–Write. Science aaf6850 (2016) doi: 10.1126/science.aaf6850. [DOI] [PubMed] [Google Scholar]
- 49.Dai J, Boeke JD, Luo Z, Jiang S & Cai Y Sc3.0: revamping and minimizing the yeast genome. Genome Biol 21, 205 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonilla N et al. Phage on tap–a quick and efficient protocol for the preparation of bacteriophage laboratory stocks. PeerJ 4, e2261 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seemann T Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Terzian P et al. PHROG: families of prokaryotic virus proteins clustered using remote homology. NAR Genomics and Bioinformatics 3, lqab067 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deatherage DE & Barrick JE Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq.Methods Mol. Biol 1151, 165–188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goodall ECA et al. The Essential Genome of Escherichia coli K-12. mBio 9, e02096–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang W, Bikard D, Cox D, Zhang F & Marraffini LA RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotech 31, 233–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Umenhoffer K et al. Genome-Wide Abolishment of Mobile Genetic Elements Using Genome Shuffling and CRISPR/Cas-Assisted MAGE Allows the Efficient Stabilization of a Bacterial Chassis. ACS Synth. Biol 6, 1471–1483 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Szili P et al. Rapid evolution of reduced susceptibility against a balanced dual-targeting antibiotic through stepping-stone mutations. Antimicrobial Agents and Chemotherapy AAC.00207–19 (2019) doi: 10.1128/AAC.00207-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kunjapur AM et al. Engineering posttranslational proofreading to discriminate nonstandard amino acids. PNAS 115, 619–624 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan PP, Lin BY, Mak AJ & Lowe TM tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Research 49, 9077–9096 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hossain A et al. Automated design of thousands of nonrepetitive parts for engineering stable genetic systems. Nat Biotechnol 38, 1466–1475 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Mohler K et al. MS-READ: Quantitative measurement of amino acid incorporation. Biochimica et Biophysica Acta (BBA) - General Subjects 1861, 3081–3088 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wiśniewski JR, Zougman A, Nagaraj N & Mann M Universal sample preparation method for proteome analysis. Nat Methods 6, 359–362 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Käll L, Storey JD & Noble WS Non-parametric estimation of posterior error probabilities associated with peptides identified by tandem mass spectrometry. Bioinformatics 24, i42–i48 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nyerges A et al. MassIVE Dataset: A swapped genetic code prevents viral infections and gene transfer (MSV000089854). (2023) doi: 10.25345/C5FF3M41W. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data from whole-genome sequencing, transcriptome, and tRNA sequencing experiments have been deposited to Sequence Read Archive (SRA) under BioProject ID PRJNA856259. tRNA data and the generated sequences in this study are included in the Supplementary Data files. Mass spectra and proteome measurements have been deposited to MassIVE (MSV000089854; doi:10.25345/C5FF3M41W)66. The Syn61Δ3(ev5) ΔrecA (ev1) strain is available from Addgene (Bacterial strain #189857). We cannot deposit Ec_Syn61Δ3-SL and Ec_Syn61Δ3 adk.d6 at Addgene due to the incompatibility of Addgene’s methods of strain distribution and growth media requirements, but all materials used in this study are freely available from the corresponding authors for academic research use upon request. The PHROG HMM database is available at https://phrogs.lmge.uca.fr/ and from reference53. The assembled annotated genome of Escherichia coli Syn61 substr. Syn61Δ3(ev5) is available in Supplementary Data 4, and the annotated genome of REP phages have been deposited to NCBI GenBank under accession numbers OQ174500, OQ174501, OQ174502, OQ174503, OQ174504, OQ174505, OQ174506, OQ174507, OQ174508, OQ174509, OQ174510, andOQ174511. Source data is provided with this paper.