

Abstract
Cysteine plays critical roles in cellular biosynthesis, enzyme catalysis, and redox metabolism. The intracellular cysteine pool can be sustained by cystine uptake or de novo synthesis from serine and homocysteine. Demand for cysteine is increased during tumorigenesis for generating glutathione to deal with oxidative stress. While cultured cells have been shown to be highly dependent on exogenous cystine for proliferation and survival, how diverse tissues obtain and use cysteine in vivo has not been characterized. We comprehensively interrogated cysteine metabolism in normal murine tissues and cancers that arise from them using stable isotope 13C1-serine and 13C6-cystine tracing. De novo cysteine synthesis was highest in normal liver and pancreas and absent in lung tissue, while cysteine synthesis was either inactive or downregulated during tumorigenesis. In contrast, cystine uptake and metabolism to downstream metabolites was a universal feature of normal tissues and tumors. However, differences in glutathione labeling from cysteine were evident across tumor types. Thus, cystine is a major contributor to the cysteine pool in tumors, and glutathione metabolism is differentially active across tumor types.
Significance:
Stable isotope 13C1-serine and 13C6-cystine tracing characterizes cysteine metabolism in normal murine tissues and its rewiring in tumors using genetically engineered mouse models of liver, pancreas, and lung cancers.
Graphical Abstract
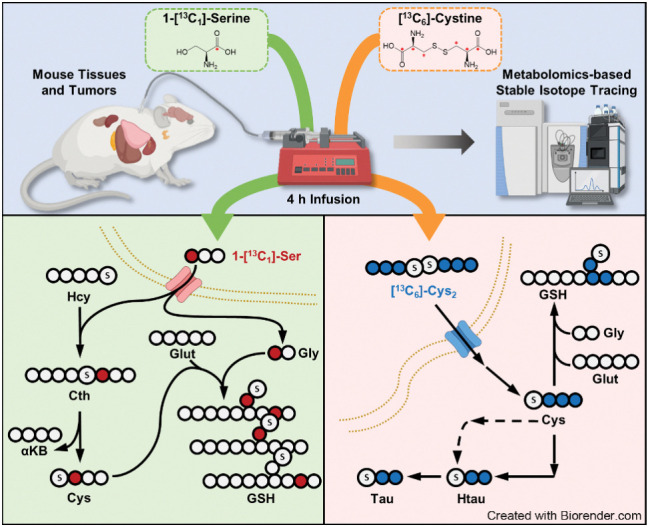
Introduction
The nonessential, thiol-containing amino acid cysteine is an essential source of sulfur for the synthesis of diverse cellular factors that play important biological functions in maintaining redox homeostasis, enzyme catalysis, and electron transfer (1). Cysteine is partitioned into various downstream metabolic pathways including glutathione and taurine synthesis. The tripeptide antioxidant glutathione is the most abundant intracellular antioxidant (2) and is synthesized from cysteine, glutamate, and glycine in two steps mediated by glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS; ref. 3). Glutathione synthesis is regulated by the rate-limiting enzyme GCL (4), which consists of a catalytic subunit (GCLC) and a modifier subunit (GCLM), which relieves the feedback inhibition of GCLC by glutathione. Glutathione synthesis is induced by oxidative stress, which stabilizes nuclear factor erythroid 2–related factor 2 (NRF2) to induce the transcription of both GCLC and GCLM and a battery of antioxidant enzymes that detoxify reactive oxygen species (5).
Tumorigenesis is accompanied by an increased demand for cysteine to deal oxidative stress (6, 7). Many cancers upregulate the system xc− cystine/glutamate antiporter (xCT) to maintain the intracellular cysteine pool and promote entry of cysteine into glutathione synthesis (8). This is achieved by mutations in oncogenes/tumor suppressors that regulate xCT expression, including Kelch-like ECH-associated protein 1 (KEAP1)/NRF2 (9, 10) and p53 (11), or regulate xCT activity (12–15). Moreover, xCT expression is induced by amino acid starvation via activating transcription factor 4 (ATF4; ref. 16), thereby ensuring adequate cysteine availability in nutrient poor conditions. There are many lines of evidence that extracellular cystine is the primary supply of the intracellular cysteine pool to support the cellular redox state. Insufficient cystine availability induces iron-dependent lipid peroxidation, leading to a form of cell death known as ferroptosis (17–19). Pharmacologic inhibition of xCT induces ferroptosis of cancer cells (17, 20), and enzyme-based cystine degradation has shown efficacy against several in vivo cancer models (20–22).
Beyond cystine uptake, the intracellular cysteine pool can be sustained by the transsulfuration pathway in the liver, although its contribution to other tissues is less clear (23). Transsulfuration is catalyzed by cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) and mediates both irreversible homocysteine removal and de novo cysteine synthesis, with serine donating the carbon backbone and homocysteine donating the sulfur to cysteine (24). While both CBS and CSE are broadly expressed (25), the contribution of transsulfuration to the cysteine pool in tumors is poorly characterized. Prior studies have found a small contribution of this pathway to the cysteine pool, which could protect against cystine starvation in Ewing sarcoma and neuroblastoma cell lines (26, 27). In contrast, we found no contribution of the transsulfuration pathway to the cysteine and glutathione pool in non–small cell lung cancer (NSCLC) cell lines, which robustly die by ferroptosis under cystine starvation (19). Moreover, the contribution of the transsulfuration pathway to the cysteine pools of non-hepatic tissues and tumors in vivo has not been investigated.
In this study, we comprehensively investigated the contribution of the both the transsulfuration pathway and exogenous cyst(e)ine to the cysteine pool and downstream metabolites in nine different healthy mouse tissues and tumors of the lung, pancreas, and liver. We found limited contribution of transsulfuration to the cysteine pool of non-hepatic tissues, in contrast to robust contribution from exogenous cyst(e)ine. Moreover, tumors from transsulfuration capable tissues downregulated this pathway, while tissues lacking transsulfuration activity generated transsulfuration deficient tumors. Finally, we characterized cysteine catabolism to glutathione and taurine across tissues and glutathione metabolism in tumors, which demonstrated complex patterns associated with enzyme expression and substrate availability.
Materials and Methods
Lead contact
Further information and requests for resources and reagents in Table 1 should be directed to and will be fulfilled by the corresponding author, Dr. Gina M. DeNicola (Gina.DeNicola@moffitt.org).
Table 1.
Materials.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ADO (Rabbit polyclonal antibody) | OriGene | Catalog no. TA322128; Lot no. D814AA091; RRID: AB_2920786 |
| CBS (D8F2P; Rabbit monoclonal antibody) | Cell Signaling Technology | Catalog no. 14782; Lot no. 1; RRID: AB_2798609 |
| CDO1 (Rabbit polyclonal antibody) | Proteintech | Catalog no. 12589-1-AP; Lot no. 57877; RRID: AB_10638145 |
| CSAD (Rabbit polyclonal antibody) | LSBio (LifeSpan) | Catalog no. LS-C375526; Lot no. 124060; RRID: AB_2801349 |
| CSE (Rabbit polyclonal antibody) | Proteintech | Catalog no. 12217-1-AP; Lot no. 00089191; RRID: AB_2087497 |
| FMO1 (Rabbit polyclonal antibody) | LSBio (LifeSpan) | Catalog no. LS-C346135-50; Lot no. 215780; RRID: AB_2920787 |
| GCLC (Mouse mAb) | Santa Cruz Biotechnology | Catalog no. sc-390811; Lot no. E1917; RRID: AB_2736837 |
| GCLM (Rabbit polyclonal antibody) | GeneTex | Catalog no. GTX114075; Lot no. 40156; RRID: AB_10619535 |
| GSS (Mouse mAb) | Novus | Catalog no. NBP2-03351; Lot no. A01; RRID: AB_2920788 |
| HSP90 (Rabbit polyclonal antibody) | Cell Signaling Technology | Catalog no. 4874; Lot no. 6; RRID: AB_2233307 |
| xCT (Rabbit polyclonal antibody) | Cell Signaling Technology | Catalog no. 98051; Lot no. 1; RRID: AB_2800296 |
| Chemicals, peptides, and recombinant proteins | ||
| Glucose | Thermo Fisher Scientific | Catalog no. D16-500 |
| Glycine | VWR | Catalog no. BP381-1 |
| L-Arginine•HCl | Sigma-Aldrich | Catalog no. A6969-25G |
| L-Aspartic acid | MP Biomedicals | Catalog no. 194633 |
| L-Asparagine | Sigma-Aldrich | Catalog no. A4159-25G |
| L-Cystine•2HCl | Sigma-Aldrich | Catalog no. C6727-25G |
| L-Glutamic acid | Sigma-Aldrich | Catalog no. G8415-100G |
| L-Glutamine | VWR | Catalog no. 02-0131-0100 |
| L-Histidine•HCl•H2O | Sigma-Aldrich | Catalog no. H5659-25G |
| Hydroxy-L-proline | TCI | Catalog no. H0296 |
| L-Isoleucine | Alfa Aesar | Catalog no. J63045 |
| L-Leucine | Sigma-Aldrich | Catalog no. L8912-25G |
| L-Lysine•HCl | Sigma-Aldrich | Catalog no. L8662-25G |
| L-Methionine | Sigma-Aldrich | Catalog no. M5308-25G |
| L-Phenylalanine | Sigma-Aldrich | Catalog no. P5482-25G |
| L-Proline | Sigma-Aldrich | Catalog no. P5607-25G |
| L-Serine | Sigma-Aldrich | Catalog no. S4311-25G |
| L-Threonine | VWR | Catalog no. E808-25G |
| L-Tryptophan | Sigma-Aldrich | Catalog no. T8941-25G |
| L-Tyrosine•2Na•2H2O | Sigma-Aldrich | Catalog no. T1145-25G |
| L-Valine | Sigma-Aldrich | Catalog no. V0513-25G |
| [13C3]-serine | Cambridge Isotope Laboratories | Catalog no. CLM-1574-H-0.1 |
| [1-13C1]-serine | Cambridge Isotope Laboratories | Catalog no. CLM-1573-0.25 |
| [13C3]-cysteine | Cambridge Isotope Laboratories | Catalog no. CLM-4320-H-PK |
| Methanol (LC/MS grade) | Thermo Fisher Scientific | Catalog no. 047192.M1 |
| H2O | Honeywell Burdick & Jackson | Catalog no. LC365-4 |
| Acetonitrile | Honeywell Burdick & Jackson | Catalog no. 34998-2.5L |
| N-ethylmaleimide (NEM) | Alfa Aesar | Catalog no. 40526-06 |
| UltraPure Distilled Water | Invitrogen | Catalog no. 10977-015 |
| Ammonium formate | Frontier Scientific | Catalog no. JK967458 |
| DL-Propargylglycine | Fisher Scientific | Catalog no. 437320010 |
| Experimental models: Cell lines | ||
| A549 | ATCC | Catalog no. CCL-185; RRID: CVCL_0023 |
| Calu3 | Dr. John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_0609 |
| H1299 | ATCC | Catalog no. CRL-5803; RRID: CVCL_0600 |
| H1581 | Dr. John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1479 |
| H1792 | Dr. John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1495 |
| H1944 | Dr. John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1508 |
| H1975 | Dr. John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1511 |
| H1993 | ATCC | Catalog no. CRL-5909; RRID: CVCL_1512 |
| H2009 | ATCC | Catalog no. CRL-5911; RRID: CVCL_1514 |
| H2172 | Dr. John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1537 |
| H2347 | Dr. John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_1550 |
| H460 | Dr. John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_0459 |
| HCC15 | Dr. John Minna, Hamon Cancer Center Collection (University of Texas-Southwestern Medical Center) | RRID: CVCL_2057 |
| DMS79 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1178 |
| H211 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1529 |
| H526 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1569 |
| 16HC | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_X025 |
| 16HV | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_X026 |
| 86M1 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_8263 |
| H146 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1473 |
| H1838 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1499 |
| H209 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1525 |
| H2107 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1527 |
| H524 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1568 |
| H740 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1586 |
| H82 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1591 |
| H841 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1595 |
| HCC33 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_2058 |
| N417 | Moffitt Lung Cancer Center of Excellence Cell Line Bank | RRID: CVCL_1602 |
| SW210.5 | Dr. John Cleveland, Moffitt Cancer Center | RRID: CVCL_S185 |
| PATU8902 | Dr. Lewis C. Cantley, Weill Cornell Medicine | RRID: CVCL_1845 |
| PL45 | Dr. Lewis C. Cantley, Weill Cornell Medicine | RRID: CVCL_3567 |
| MiaPaca2 | Dr. Lewis C. Cantley, Weill Cornell Medicine | RRID: CVCL_0428 |
| T3M4 | Dr. Lewis C. Cantley, Weill Cornell Medicine | RRID: CVCL_4056 |
| 8988T | Dr. Lewis C. Cantley, Weill Cornell Medicine | RRID: CVCL_1847 |
| 8902 | Dr. Lewis C. Cantley, Weill Cornell Medicine | RRID: CVCL_1845 |
| BXPC3 | Dr. Lewis C. Cantley, Weill Cornell Medicine | RRID: CVCL_5I88 |
| CFPAC | Dr. Lewis C. Cantley, Weill Cornell Medicine | RRID: CVCL_1119 |
| MPANC96 | Dr. Lewis C. Cantley, Weill Cornell Medicine | RRID: CVCL_7165 |
| PANC1 | Dr. Lewis C. Cantley, Weill Cornell Medicine | RRID: CVCL_0480 |
| SUIT2 | Dr. Lewis C. Cantley, Weill Cornell Medicine | RRID: CVCL_3172 |
| SW1990 | ATCC | Cat# CRL-2172; RRID: CVCL_1723 |
| Alex | Dr. Amaia Lujambio, Icahn School of Medicine | RRID: CVCL_0485 |
| HepG2 | Dr. Amaia Lujambio, Icahn School of Medicine | RRID: CVCL_0027 |
| Hep3B | Dr. Amaia Lujambio, Icahn School of Medicine | RRID: CVCL_0326 |
| SNU398 | Dr. Amaia Lujambio, Icahn School of Medicine | RRID: CVCL_0077 |
| SNU449 | Dr. Amaia Lujambio, Icahn School of Medicine | RRID: CVCL_0454 |
| Experimental models: Organisms/strains | ||
| LSL-KrasG12D (Krastm4Tyj) | JAX | RRID:IMSR_JAX:008179 |
| Trp53flox (Trp53tm1Brn) | JAX | RRID:IMSR_JAX:008462 |
| p48-Cre (Ptf1atm1(cre)Hnak) | Dr. Karen Mann, Moffitt Cancer Center | RRID:IMSR_JAX:023329 |
| LSL-Nrf2D29H (Nfe2l2tm1Gmdn) | (28) | MGI:7327101 |
| Rb1flox/flox; Trp53flox/flox; MycLSL/LSL (Igs2tm1(CAG-Myc*T58A/luc)Wrey Trp53tm1Brn Rb1tm3Tyj) | JAX | RRID:IMSR_JAX:029971 |
| Recombinant DNA | ||
| pT3-EF1A-MYC-IRES-luc | (29) | RRID: Addgene_129775 |
| CMV-SB13 vector | (29) | |
| px330 p53 | (29) | |
| Software and algorithms | ||
| El-Maven | https://www.elucidata.io/el-maven | v 0.10.0 or 0.12.0 |
| GraphPad Prism | https://www.graphpad.com/scientific-software/prism/ | Version 9 |
| IsoCor | https://isocor.readthedocs.io/en/latest/ | Version 1.0 or 2.2.0 |
| Xcalibur | Version 4.0 | |
| Other | ||
| RPMI1640 Medium Modified w/o L-Glutamine, w/o Amino acids, Glucose (Powder) | US Biological | Catalog no. R9010-01 |
| DMEM w/ L-Glutamine and Sodium Biscarbonate, w/o Glucose, Serine, Glycine (Powder) | US Biological | Catalog no. D9800-16 |
| Dialyzed FBS (dFBS) | Sigma-Aldrich | Catalog no. F0392 |
| DMEM, 1× (Dulbecco's Modification of Eagle's Medium) with 4.5 g/L glucose, L-glutamine and sodium pyruvate | Corning | Catalog no. 10-013-CV |
| RPMI Medium 1640 (1×) [+] L-Glutamine | Gibco | Catalog no. 11875-093 |
| DPBS w/o Calcium or Magnesium | Lonza | Catalog no. 17-512F |
| FBS | Sigma-Aldrich | Catalog no. F0926 |
Material availability
All unique reagents generated in this study are available from the corresponding author without restriction.
Cell lines
Cell lines were obtained from authentic sources (ATCC; Hamon Cancer Center Collection, Moffitt Lung COE Bank) or authenticated by short tandem repeat profiling [pancreatic ductal adenocarcinoma (PDAC) cell lines]. Hepatocellular carcinoma (HCC) cell lines were not authenticated. All cell lines were immediately tested for Mycoplasma upon receipt using the MycoAlert assay (Lonza) and aliquots were frozen. Cell lines were tested for Mycoplasma monthly and used within 10–20 passages.
Generation of experimental animals
All animal experiments were approved by the University of South Florida Institutional Animal Care and Use Committee (protocols IS00003893R, IS00006358R, IS00007922R, IS00008736R, and IS00010348R). To generate Myc; p53−/− HCC tumors (29), DNA was delivered to the liver of 8-week-old C57BL/6J female via hydrodynamic tail vein injection to concomitantly integrate a Myc transposon into the mouse genome and delete Trp53. A volume of sterile saline equal to 10% of their body weight containing 10 μg of Myc-Luciferase plasmid, 10 μg of Cas9/sgp53 plasmid, and 2.5 μg of SB13 transposase plasmid was injected into the mouse tail vein. Liver tumors developed approximately 4–6 weeks later, which was monitored by bioluminescence imaging. To generate experimental PDAC mice, mice harboring LSL-KrasG12D, Trp53flox, and p48-cre alleles were intercrossed to generate LSL-KrasG12D/+; Trp53flox/+; p48-cre experimental animals. Mice developed tumors with a median survival of approximately 6 months (30), and tumor development was monitored by abdominal palpation, followed by confirmation by ultrasound. To generate experimental animals with lung adenocarcinoma (LUAD) tumors, LSL-KrasG12D/+, Trp53flox, and LSL-Nrf2D29H mice were intercrossed to generate LSL-KrasG12D/+; Trp53flox/flox and LSL-KrasG12D/+; Trp53flox/flox; LSL-Nrf2D29H mice. Mice were infected intranasally with 2 × 107 plaque-forming unit (PFU) adenovirus (Ad5CMVCre, University of Iowa, Iowa City, IA) under isofluorane anesthesia to initiate tumor development. Mice developed tumors with a median survival of approximately 4 months after infection as reported previously (28, 31), and mice were used for experiments between 3 and 3.5 months when they displayed evidence of tumor burden such as rapid respiration. To generate experimental animals with small cell lung cancer (SCLC) tumors, Rb1flox/flox; Trp53flox/flox; MycLSL/LSL mice were intercrossed to generate Rb1flox/flox; Trp53flox/flox; MycLSL/+ (RPM) and Rb1flox/flox; Trp53flox/flox; MycLSL/LSL (RPMM) experimental animals (32). Mice were infected intratracheally with 7.5×107 PFU adenovirus (Ad5CGRPCre, University of Iowa) under ketamine (100 mg/kg)/xylazine (10 mg/kg) anesthesia to initiate tumor development. Mice developed tumors around 9 to 10 weeks after infection, and tumor development was monitored by MRI.
Stable isotope animal infusions
[13C6]-cystine was generated from [13C3]-cysteine by oxidation with H2O2. A 10 mg/mL solution of [13C3]-cysteine was dissolved in sterile saline, followed by the addition of an equimolar volume of 30% H2O2 (0.94 μL/mg cysteine) and cysteine was allowed to oxidize for 30 minutes at room temperature on a rocker, during which time the resulting cystine precipitated. The H2O2 was inactivated by heating at 60°C for 5 minutes. Cystine was resolubilized by the addition of 6N HCl. For infusion of stable isotope tracers, catheters were surgically implanted into the jugular vein 2 to 7 days prior to infusion and mice were allowed to recover prior to infusion. On day of experiment, catheters were connected to a syringe pump on a tether and swivel system (SAI Technologies) to allow mice to freely move around the cage during infusions. A mouse harness with a spring prevented the tubing from disconnecting from the catheter. Syringes prefilled with saline containing 20 mg/mL of [1-13C1]-serine or 10 mg/mL of [13C6]-cystine were loaded into the pump and tracers were infused at a rate of 120 μL/hour for 4 hours. During the final minutes of the infusion, blood was collected from the cheek vein. Mice were sacrificed by cervical dislocation and organs of interest rapidly collected in cryovials and frozen in liquid nitrogen.
Cell viability assays
Cystine-free media was prepared from RPMI1640 medium powder lacking amino acids according to instructions and amino acids were added following the RPMI1640 formulation except cystine. NSCLC cell lines (Calu3, H1944, H2009, H2347, and H1792) were plated RPMI1640 (5% FBS) in 96-well plate at density of 10,000 cells/well in a 100 μL final volume. The following day, the medium was aspirated and cells were washed with Dulbecco's phosphate buffered saline (DPBS), followed by with 100 μL of RPMI [5% dialyzed fetal bovine serum (dFBS)] containing 0 μmol/L cystine or 200 μmol/L cystine. For rescue experiments, homocysteine or cystathionine was added into media for final concentration of 1 mmol/L. For CSE inhibition experiments, propargylglycine (PPG) was added to the media for final concentration of 100 μmol/L. Three days later, cells were fixed in iced cold 4% of paraformaldehyde at 4°C for 20 minutes, then stained with 50 μL of 0.1% crystal violet in 20% methanol on orbital shaker (room temperature, 30 minutes). The plate was washed twice with dH2O and dried. Crystal violet was solubilized in 100 μL of 10% acetic and the absorbance was read at 600 nm.
Stable isotope labeling in cell culture
To prepare medium including [13C3]-serine, RPMI1640 Medium powder without glucose and amino acids (US Biological) and DMEM powder without glucose, glycine, and serine (US Biological) were reconstituted following manufacturer's instructions. Glucose and amino acids were added to match the RPMI1640 and DMEM formulation except serine. RPMI feeding media contained 300 μmol/L [13C3]-serine + 5% dFBS and DMEM contained 400 μmol/L [13C3]-serine + 10% dFBS. Both were supplemented with 1% Pen/Strep. Cell lines were plated in 6-well plates so they were 70% confluent at the time of extraction. Cells were preconditioned in medium including dFBS overnight (RPMI with 5% dFBS for NSCLC and SCLC cell lines; DMEM with 10% dFBS for PDAC and HCC cell lines). Prior to labeling, the cells were washed with 1 mL serine-free medium and then fed with medium containing [13C3]-serine for 4 hours prior to extraction.
Metabolomics sample preparation
Cells were washed with ice-cold DPBS, followed by aspiration of medium. A total of 500 μL of ice-cold extraction solvent (80% methanol and 20% water including 10 mmol/L ammonium formate and 25 mmol/L NEM, pH 7.0) was added to each well. After 30 minutes of incubation at 4°C, cells were scraped and, the supernatant moved to a 1.5 mL tube and the debris cleared by centrifugation (17,000 × g, 4°C, 20 minutes). Extracts were stored at −80°C until analysis. Cell numbers were counted by Scepter 2.0 cell counter and used to calculate intracellular metabolite concentrations. To extract metabolites from tissues, the frozen tissues were pulverized with a prechilled Bio-Pulverizer (59012MS, BioSpec). After weighing the tissues, the extraction solvent (80% methanol and 20% water including 10 mmol/L ammonium formate and 25 mmol/L NEM, pH 7.0) was added to the pulverized tissue for a final concentration of 50 mg tissue/mL extraction solvent for 30 minutes at 4°C. To extract metabolites from serum, 390 μL of extraction solvent (82% methanol and 18% water including 10 mmol/L ammonium formate and 25 mmol/L NEM, pH 7.0) was added to 10 μL of serum, followed by incubation at −80°C for 30 minutes. Debris was cleared by centrifugation (17,000 × g, 4°C, 20 minutes). Extracts were stored at −80°C until analysis.
LC/MS analysis and data processing
The instrumental conditions of LC/MS analysis were optimized on the basis of previously established methods (19). The chromatography system for separation was the Vanquish UPLC system equipped with a SeQuant ZIC-pHILIC column (150 × 4.6 mm, 5 μm, MilliporeSigma) connected to a SeQuant ZIC-pHILIC guard column (20 × 4.6 mm, 5 μm, MilliporeSigma) or an Atlantis Premier BEH Z-HILIC VanGuard FIT column (2.1 mm × 150 mm, 2.5 μm, Waters). The column was kept in a 30°C column chamber 5 μL of sample loaded via autosampler. For the gradient, mobile phase A (10 mmol/L ammonium carbonate and 0.05% ammonium hydroxide in water) and mobile phase B (100% acetonitrile) were used as follow: 0–13 minutes, 80% to 20% of B; 13–15 minutes, 20% of B; 15–20 minutes, 80% of B. For separated metabolite detection, a Q Exactive HF (QE-HF) Orbitrap mass spectrometer (Thermo Fisher Scientific) with H-ESI was used. The ions were detected by both positive and negative modes. The MS1 scan range was m/z 65–950 for both modes. The capillary temperature and voltage were 320°C and 3.5 kV, respectively. The mass resolution was 120,000 and the AGC target was 3 × 106. After data conversion from .raw to .cdf using Xcalibur (Version 4.0), further data processing for targeted metabolomics was performed by El-Maven (Version 0.10.0 or 0.12.0) and the default parameters were used for data processing except as follows: ionization mode, positive; isotopic tracer, C13; extracted ion chromatogram (EIC) extraction window (+/−), 10.00 ppm. Identification of metabolites was performed on the basis of retention time and exact precursor ion m/z in previously established authentic standard-based in-house library (19). The peak intensity of each EIC was measured as AreaTop (mean of three top points in the peak). For isotope correction, the extracted metabolite signals from El-Maven were loaded into the IsoCor (https://isocor.readthedocs.io/en/latest/; Version 1.0 or 2.2.0) as .tsv file according to the recommended format and processed with following parameters: Isotopic tracer, 13C; “Low resolution” was selected; “Correct natural abondance of the tracer element” was selected; Isotopic purity of the tracer, 12C was 0.01 and 13C was 0.99. To examine the fractional contribution of intracellular serine or cysteine to downstream metabolites, we normalized the labeled fraction of downstream metabolites to the fraction labeling of intracellular serine or cysteine as follows:
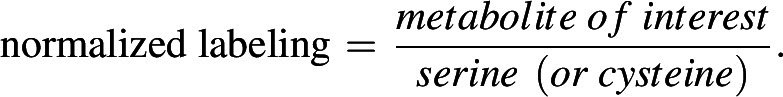 |
Immunoblotting
To prepare cell lysates, cells were washed with ice-cold DPBS, detached from 6-well plates by scraping, transferred to microcentrifuge tubes, and pelleted. Cell pellets were lysed in RIPA lysis buffer (20 mmol/L Tris-HCl, pH 7.5; 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 1% NP-40, 1% sodium deoxycholate) containing protease inhibitors and phosphatase inhibitors on ice for 30 minutes. To extract protein from tissue samples, 25 μL of RIPA containing protease inhibitors and phosphatase inhibitors was added per 1 mg of tissue. After homogenization with a dounce homogenizer, the samples were incubated on ice for 30 minutes, followed by sonication for 5 minutes on medium power (30 seconds on/ 30 seconds off). Debris was cleared by centrifugation (13,000 × g, 4°C, 15 minutes) and supernatants were stored at −20°C until analysis.
Protein quantification was conducted using DC assay (Bio-Rad) according to the manufacturer's instructions. The protein lysates were combined with 6× loading buffer containing 2-mercaptoethanol and loaded on NuPAGE 4%-12% Bis-Tris Midi gels (Invitrogen). After separation of protein by SDS-PAGE, the proteins were transferred to 0.45 μmol/L nitrocellulose blotting membrane (Cytiva). Following the blocking of membrane with 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 (TBST) for 30 minutes, the membranes were washed three times with TBST for 10 minutes each, and the membranes were incubated in primary antibodies at a 1:1,000 dilution in 2% milk overnight at 4°C. After incubation, the membranes were washed again in TBST and developed with ECL using X-ray film. When comparing cell line or tissue samples across multiple membranes, the same lysate was loaded on multiple membranes to ensure exposures were equal.
Database mining for mutation and gene expression data
The mutation status of KEAP1, KRAS, EGFR, STK11, NFE2L2, RB1, and MYC in NSCLC, SCLC, PDAC, and HCC cell lines was determined from the DepMap Portal (https://depmap.org/portal/), the Sanger Catalogue of Somatic Mutations in Cancer database (COSMIC, https://cancer.sanger.ac.uk/cosmic), and the TP53 Database (https://tp53.isb-cgc.org/). For gene expression analysis of CBS, CSE, GCLM, GSS, and SLC7A11 (xCT) in human patient with liver hepatocellular carcinoma, pancreatic adenocarcinoma, LUAD, and lung squamous cell carcinoma (LUSC) compared with healthy tissue, the GEPIA2 (gepia2.cancer-pku.cn) open access online tool was used to query The Cancer Genome Atlas (TCGA)/GTEx data.
Quantification and statistical analysis
GraphPad Prism 9 was used for all statistical analysis. The Mann–Whitney test (nonparametric t test) was conducted for statistical comparisons.
Data availability
The data generated in this study are included in the article and its supplementary figures. Raw data are available upon request without restriction from the corresponding author.
Results
Cultured cancer cell lines lack of de novo cysteine synthesis capacity
In our previous study, we found that NSCLC cysteine pools are not supported by transsulfuration, resulting in cumulative cell death following extracellular cystine starvation (19). To evaluate the origin of cysteine more broadly in cancer cells in culture, we examined de novo cysteine synthesis from 13C3-serine to cysteine through the transsulfuration pathway in a panel of SCLC, PDAC, and HCC cell lines representative of the genetic diversity of these cancers. Incubation with 13C3-serine as an extracellular serine source for 4 hours resulted in almost complete labeling of the intracellular serine pool (Fig. 1A), and metabolism to cystathionine (Fig. 1B). We found that SCLC and NSCLC showed higher cystathionine labeling in this time period than HCC and PDAC cell lines (Fig. 1B). However, regardless of cancer type, labeling of cysteine from serine was absent in all cell lines, suggesting a bottleneck at cysteine synthesis from cystathionine (Fig. 1C). To examine whether impaired cysteine synthesis is a consequence of lack of transsulfuration enzyme expression, we performed immunoblotting for CBS and CSE, which mediate the first and second steps of transsulfuration (Fig. 1D), respectively. Interestingly, while some PDAC cell lines lacked CBS expression and some HCC cell lines lacked CSE expression, almost all cancer cell lines investigated expressed both enzymes despite being unable to synthesize cysteine (Fig. 1D). Moreover, there was no association between mutation status and CBS or CSE expression (Supplementary Fig. S1).
Figure 1.

Cultured cancer cell lines lack de novo cysteine synthesis capacity. A–C, Analysis of de novo cysteine synthesis in cultured NSCLC, SCLC, PDAC, and HCC cell lines with 13C3-serine tracing. Cell lines were incubated with 13C3-serine containing media for 4 hours, followed by analysis of the fraction labeling in serine (A), cystathionine (B), and cysteine (C). Data are presented as mean ± SD and N = 3 biological replicates for each cell line. D, Immunoblotting for the transsulfuration enzymes CBS and CSE. HSP90 was used for the loading control and HepG2 was used for relative comparison between different membranes. Cth, cystathionine; Cys, cysteine; Ser, serine.
Prior studies have suggested that generation of homocysteine via the methionine cycle is a critical limiting factor for transsulfuration flux (26, 33). To examine whether transsulfuration may be substrate limited, we examined whether excess cystathionine or homocysteine could rescue viability under cystine starvation in a panel of NSCLC cell lines (Supplementary Fig. S2A). We observed a full rescue of viability by either substrate, which was reversed by treatment with PPG, an irreversible inhibitor of CSE, demonstrating the requirement for transsulfuration for this rescue (Supplementary Fig. S2B). To confirm that these substrates were actively contributing to the cysteine and glutathione pools, we cultured cells with 13C3-serine for 24 hours under cystine replete or starved conditions in the presence or absence of cystathionine or homocysteine. Homocysteine treatment elevated both homocysteine and cystathionine levels within cells, with cystathionine demonstrating M+3 labeling from serine (Supplementary Fig. S2C). Because cystathionine already contains serine carbons, 13C3-serine was not useful for assaying transsulfuration under cystathionine treatment conditions. Nevertheless, we observed that cystathionine treatment was more effective at elevating cystathionine and did not alter homocysteine levels as expected. Both treatments elevated cysteine levels under cysteine starved conditions, although levels were still much lower than replete conditions. Despite this, glutathione levels were completely restored, with M+2 labeling (via glycine) demonstrating active de novo synthesis despite the lack of exogenous cystine. Moreover, M+3 labeling of cystathionine, cysteine, and glutathione was evident in homocysteine-treated conditions, demonstrating active cysteine synthesis from serine. These results indicate that under these culture conditions transsulfuration enzymes are substrate limited for the synthesis of cysteine.
Contribution of de novo cysteine synthesis to the cysteine pool varies across healthy mouse tissues
Transsulfuration activity is known to be high in healthy liver (25), but the contribution of this pathway to the cysteine pool across diverse tissues is not known. To this end, we infused healthy C57BL/6J mice with 1-[13C1]-serine for 4 hours via the jugular vein to label intracellular intermediates in the transsulfuration and glutathione synthesis pathways (Fig. 2A). We analyzed their fraction labeling (Fig. 2B–G) and total levels (Supplementary Fig. S3A–S3F) in nine different tissues (liver, pancreas, kidney, heart, thymus, spleen, lung, cerebellum, and brain) and serum by mass spectrometry. The resulting total signal intensity of intermediates revealed that liver and pancreas are the most cystathionine abundant tissues, while pancreas and kidney have a larger cysteine pool than the others (Supplementary Fig. S3C and S3E). Four hours of 1-[13C1]-serine infusion labeled around 50% of circulating serine (Fig. 2B) and this time frame was sufficient to detect 13C label in cysteine and glutathione (Fig. 2E and F). Like what we observed in cultured cell lines (Fig. 1B), cystathionine labeling was detected in all tissues but not serum (Fig. 2D), demonstrating that CBS was active. Analysis of cysteine labeling across tissues revealed robust de novo cysteine synthesis in liver tissue, with low labeling detected in other tissues (Fig. 2F). Within each tissue, the cysteine fraction labeling was normalized to labeling in the serine pool to evaluate the fractional contribution of serine to cysteine. The highest labeling was observed in the liver (25%), and the second highest labeling was observed in pancreas (3%), with other tissues deriving less than 3% of their cysteine from serine (Fig. 2H). Given the significant labeling in cystathionine across tissues (Fig. 2D), these results suggest that the cleavage of the bond between sulfur and the gamma carbon by CSE is a bottle neck of de novo cysteine synthesis.
Figure 2.
![Figure 2. Contribution of de novo cysteine synthesis to the cysteine pool varies across healthy mouse tissues. A, Schematic depicting 1-[13C1]-serine infusion and its metabolism via the transsulfuration and glutathione synthesis pathways. B–G, Healthy C57BL/6J mice were infused with 1-[13C1]-serine, followed by analysis of the fraction labeling in serine (B), glycine (C), cystathionine (D), glutathione (E), cysteine (F), and γ-glutamylcysteine (G). For B–G, data are presented as mean ± SD and N = 10 mice (5 male, 5 female). N.D., not detected. H, Fractional contribution of serine to intracellular cysteine synthesis in each tissue from B–G. Cysteine labeling was normalized to the fraction labeling of serine in each tissue. I, Immunoblots of CBS and CSE for each tissue. HSP90 was used for the loading control. αKB, α-ketobutyrate; Cth, cystathionine; Cys, cysteine; Gly, glycine; Glut, glutamate; GSH, glutathione; γ-Glu-Cys, γ-glutamylcysteine; Ser, serine. (A, Created with BioRender.com.)](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10152234_1426fig2.jpg)
Contribution of de novo cysteine synthesis to the cysteine pool varies across healthy mouse tissues. A, Schematic depicting 1-[13C1]-serine infusion and its metabolism via the transsulfuration and glutathione synthesis pathways. B–G, Healthy C57BL/6J mice were infused with 1-[13C1]-serine, followed by analysis of the fraction labeling in serine (B), glycine (C), cystathionine (D), glutathione (E), cysteine (F), and γ-glutamylcysteine (G). For B–G, data are presented as mean ± SD and N = 10 mice (5 male, 5 female). N.D., not detected. H, Fractional contribution of serine to intracellular cysteine synthesis in each tissue from B–G. Cysteine labeling was normalized to the fraction labeling of serine in each tissue. I, Immunoblots of CBS and CSE for each tissue. HSP90 was used for the loading control. αKB, α-ketobutyrate; Cth, cystathionine; Cys, cysteine; Gly, glycine; Glut, glutamate; GSH, glutathione; γ-Glu-Cys, γ-glutamylcysteine; Ser, serine. (A, Created with BioRender.com.)
Next, we examined CBS and CSE expression levels across tissues to examine their association with cysteine synthesis (Fig. 2I). We found that liver and pancreas both have high expression of CBS, with the liver demonstrating higher CSE expression than pancreas (Fig. 2I). Even when accounting for cystathionine labeling, liver had much higher synthesis of cysteine compared with pancreas, suggesting that second step of transsulfuration mediated by CSE was more active (Fig. 2H), and the expression of CSE and additional factors may be key regulators of cysteine synthesis (Fig. 2I). Tissues lacking cysteine labeling generally had low expression of both CBS and CSE, including lung, cerebellum, thymus, and spleen (Fig. 2F, H and I). Kidney had high expression of CSE, but lower CBS expression than liver and pancreas (Fig. 2I), which likely explained its lower cysteine labeling (1.5%; Fig. 2H). Collectively, these results demonstrate that transsulfuration of serine to cysteine is a minor contributor to the cysteine pool in most non-hepatic tissues and the bottleneck is the CSE step.
1-[13C1]-serine infusion for 4 hours was also sufficient to evaluate the glutathione synthesis pathway downstream of cysteine metabolism. Evaluation of total metabolite pools revealed that liver had the highest total signal of glutathione (Supplementary Fig. S3D), consistent with its established role in glutathione synthesis to supply the circulating pool (34). Liver, pancreas, and kidney and spleen demonstrated detectable labeling from serine (Fig. 2E), with kidney demonstrating the highest labeling, followed by liver, pancreas, and spleen. However, because 1-[13C1]-serine can label glutathione via both cysteine and glycine, with both resulting in M+1 labeling, this labeling overestimates the contribution of cysteine (Fig. 2F). To directly examine the entry of serine-derived cysteine into the glutathione synthesis pathway, we examined labeling in γ-glutamylcysteine (Fig. 2G). In agreement with cysteine labeling, only liver showed a substantial fraction of γ-glutamylcysteine labeling (5%). In addition, M+2 labeling of glutathione was absent across all tissues (Fig. 2E), further supporting that serine-derived cysteine comprises a minor proportion of glutathione.
Cyst(e)ine supplies the cysteine pool in all tissues
To directly assay cysteine metabolism to downstream metabolites, we infused healthy C57BL/6J mice with 13C6-cystine for 4 hours (Fig. 3A). Serum and tissues were collected and analyzed by LC/MS-based metabolomics to examine the total signal (Supplementary Fig. S4A–S4F) and labeled fraction (Fig. 3B–G) of intermediates within the glutathione and taurine synthesis pathways. Interestingly, despite infusion with pure 13C6-cystine, cystine formed mixed disulfides within the serum and tissues to form a substantial fraction of M+3 cystine (Fig. 3B). Importantly, M+3 cysteine was also detected in the serum, precluding our ability to determine uptake as cystine versus cysteine (Fig. 3D; Supplementary Fig. S4C). Cysteine synthesis low tissues including heart, thymus, and lung demonstrated the highest fraction labeling of cysteine from 13C6-cystine, while the lowest fraction labeling was observed in cysteine synthesis high pancreas and kidney (Fig. 3D). Immunoblotting for the expression of the cystine/glutamate antiporter revealed highest expression in pancreas (Fig. 3H), which also had the highest total cysteine levels (Supplementary Fig. S4C). However, xCT expression largely did not correlate with cysteine levels or labeling across tissues, suggesting other transporters for cystine or cysteine may mediate import, or additional mechanisms of xCT regulation may play a role. Indeed, we examined glutamate levels across tissues and found that brain and cerebellum levels were expectedly high (Supplementary Fig. S4G), consistent with the neurotransmitter function of this metabolite, which likely inhibits the ability of the xCT in cerebellum to import cystine (Fig. 3B), which is very low in this tissue (Supplementary Fig. S4A). Moreover, these results support our finding that liver, pancreas, and kidney contribute to the cysteine pool through de novo cysteine synthesis (Figs. 2H and 3D).
Figure 3.

Cyst(e)ine supplies the cysteine pool in all tissues. A, Schematic depicting 13C6-cystine infusion and its metabolism to glutathione and taurine. B–G, Healthy C57BL/6J mice were infused with 13C6-cystine, followed by analysis of the fraction labeling in cysteine (B), γ-glutamylcysteine (C), cysteine (D), glutathione (E), hypotaurine (F) and taurine (G). For B–G, data are presented as mean ± SD and N = 5 mice. N.D., not detected. H, Immunoblots of xCT, CDO1, CSAD, ADO, FMO1, GCLC, GCLM, and GSS for each tissue. HSP90 was used for the loading control. αKB, α-ketobutyrate; Cth, cystathionine; Cys, cysteine; Cys2, cystine; γ-Glu-Cys, γ-glutamylcysteine; Glut, glutamate; Gly, glycine; GSH, glutathione; Hcy, homocysteine; Htau, hypotaurine; Tau, taurine. (A, Created with BioRender.com.)
Because all tissues demonstrated substantial labeling in the cysteine pool from 13C6-cystine, we were able to examine differential metabolism of cysteine to downstream metabolites across tissues. We found that kidney and pancreas demonstrated significantly higher glutathione labeling compared with the other tissues, with liver also having high labeling when accounting for the relatively lower labeling in the cysteine pool (Fig. 3D and E). In contrast, brain and cerebellum demonstrated the lowest glutathione labeling, suggesting glutathione synthesis may be very slow in these tissues (Fig. 3E; Supplementary Fig. S4D). The rate-limiting step of glutathione synthesis depends on regulation of GCL (GCLC and GCLM) activity, which is positively regulated cysteine availability and negatively regulated by glutathione (4). High glutathione labeling in liver and kidney was correlated with strong expression of both subunits of GCL (Fig. 3H), with the lower level of cysteine and higher level of glutathione potentially contributing to the higher labeling in the kidney (Fig. 3E; Supplementary S4C and S3D). While pancreas had low expression of GCLC and GCLM, its high level of xCT expression and cysteine levels and low levels of glutathione may contribute to high glutathione labeling (Fig. 3E and H; Supplementary S4C and S4D). For lung, cerebellum, spleen, and thymus, lower expression of GCLC and GCLM combined with relatively low cysteine and high glutathione likely result in lower levels of de novo glutathione synthesis (Fig. 3E and H; Supplementary S4C and S4D). Interestingly, the heart has higher expression of GSS compared with other tissues (Fig. 3H), but has low glutathione labeling (Fig. 3E), raising the possibility that GSS has other functions. These results demonstrate diverse metabolism of cysteine to glutathione across murine tissues.
Hypotaurine and taurine are maintained by cross-talk between cysteine catabolism and transport
Next, we examined cysteine metabolism to taurine to determine if there are differences in cysteine entry into downstream pathways across tissues. Cysteine is metabolized to hypotaurine via the cysteine sulfinic acid pathway, mediated by cysteine dioxygenase type 1 (CDO1), or the cysteamine pathway downstream of coenzyme A breakdown, followed by oxidation to taurine. A total of 4 hours of infusion with 13C6-cystine was sufficient to label hypotaurine in the serum and all tissues but was not sufficient to label taurine (Fig. 3F and G). Interestingly, brain and cerebellum hypotaurine labeling exceeded cysteine and glutathione labeling, suggesting contribution from the circulation (Fig. 3F). Moreover, kidney and lung hypotaurine labeling mirrored serum hypotaurine labeling, suggesting hypotaurine transport may be a major contributor to these tissues (Fig. 3F). Consistent with prior reports (24, 35), kidney and liver expressed CDO1 and cysteine sulfinic acid decarboxylase (CSAD; Fig. 3H). In addition to these tissues, we also observe strong CDO1 expression in pancreas, while cerebellum and thymus had low expression. CDO1 expression was undetectable in spleen and heart, which matched the lowest hypotaurine labeling in these two organs (Fig. 3F and H). CSAD and 2-aminoethanethiol (cysteamine) dioxygenase (ADO) expression were more uniform across the tissues, except for heart, which lacked expression of both enzymes, and cerebellum, which had high ADO expression (Fig. 3H). These results demonstrate that cysteine contributes to the hypotaurine pool across tissues both directly and indirectly via the circulation.
While the lack of labeling in taurine from 13C6-cystine precluded our ability to look directly at its synthesis, there were interesting differences in hypotaurine and taurine levels across tissues that prompted us to look at the final step in taurine synthesis. Hypotaurine is enzymatically oxidized to generate taurine, although the enzyme responsible this reaction is not well defined. NAD-dependent hypotaurine dehydrogenase has been suggested as the responsible enzyme, but direct evidence is lacking (36). Recently, flavin-containing monooxygenase 1 (FMO1) was shown to mediate taurine biosynthesis from hypotaurine in vivo (37). Immunoblotting revealed that liver, heart, and lung had the highest FMO1 expression, while expression in pancreas was undetectable (Fig. 3H). These patterns match taurine levels across tissues, with levels highest in liver and heart, with the levels in the pancreas almost an order of magnitude lower than heart (Supplementary Fig. S4F). These findings suggest FMO1-expressing tissues may have a greater capacity to metabolize hypotaurine to taurine.
Tumorigenesis of liver and pancreas induces downregulation of de novo cysteine synthesis
Given that cysteine is a crucial biomolecule that contains a sulfur moiety that facilitates redox homeostasis and energy transfer, cancers have been proposed to maintain their cysteine pool by rewiring de novo synthesis or cystine uptake (8). To interrogate the source of cysteine in tumors in vivo, we first selected two genetically engineered mouse (GEM) models of liver and pancreatic cancer, because these two tissues demonstrated cysteine synthesis capacity, and examined whether cysteine synthesis capacity is maintained in tumors. A hydrodynamic tail vein injection model was used to generate Myc; Trp53−/− liver tumors for a HCC model and LSL-KrasG12D/+; Trp53flox/+; p48-Cre (KPC) mice were used for a PDAC model (Fig. 4A). 1-[13C1]-serine tracing was performed as described for healthy mice. HCC demonstrated similar labeling in the serine pool compared with normal liver, and similar labeling in downstream metabolites cystathionine and glycine (Fig. 4B). Labeling in the cysteine, γ-glutamylcysteine, and glutathione pools were lower in HCC compared with normal liver, although these differences were not significant (Fig. 4B). PDAC demonstrated higher labeling in the serine pool, with similar labeling in downstream glycine and cystathionine pools (Fig. 4C). However, labeling in both cysteine and γ-glutamylcysteine was absent. Although labeling was detected in glutathione, this was likely coming from glycine due to the absence of cysteine labeling (Fig. 4C). When normalized to serine labeling within each tissue, we found that de novo cysteine synthesis decreased in both HCC and PDAC tumors compared with each control healthy tissue (Fig. 4D). For HCC, the distribution appeared binary, with tumors either maintaining the labeling fraction of the parental tissue or having a substantially reduced fraction. For PDAC, cysteine labeling was completely absent (Fig. 4D). Immunoblotting for CBS and CSE revealed a slight reduction in these proteins in HCC and a dramatic reduction in their expression in PDAC consistent with the cysteine labeling patterns (Fig. 4E). Consistently, CBS mRNA expression was significantly downregulated in human HCC and dramatically downregulated in human PDAC in TCGA/GTEx dataset (Supplementary Fig. S5A). Despite this, the total cysteine pool in murine HCC was dramatically increased, suggesting HCC facilitates the accumulation of cysteine by other mechanisms (Supplementary Fig. S5B). In contrast, the total cysteine pool of PDAC was decreased (Supplementary Fig. S5C). These results demonstrate that transsulfuration capable tissues may maintain this capacity upon transformation, or may lose this capacity entirely.
Figure 4.
![Figure 4. Tumorigenesis of liver and pancreas induces downregulation of de novo cysteine synthesis. A, Schematic for the generation of Myc; p53−/− HCC and KrasG12D; p53+/− PDAC GEMM tumors. B, Analysis of the fraction labeling in serine, glycine, cystathionine, glutathione, cysteine, and γ-glutamylcysteine in liver tissues (N = 8) compared with HCC tumors (N = 8) and their matched serum normal (N = 8) and HCC (N = 5) following infusion with 1-[13C1]-serine. C, Analysis of the fraction labeling in serine, glycine, cystathionine, glutathione, cysteine, and γ-glutamylcysteine in pancreas tissues (N = 5) compared with PDAC tumors (N = 5) and their matched serum from normal (N = 5) and PDAC (N = 5) following infusion with 1-[13C1]-serine. D, Fractional contribution of serine to intracellular cysteine synthesis in HCC and PDAC. Cysteine labeling was normalized to the fraction labeling of serine in each tissue. One healthy pancreas sample was excluded because of a division error. For B–D, data are presented as mean ± SD. N.D., not detected. E, Immunoblots of CBS and CSE for each tissue. HSP90 was used for the loading control. *, P < 0.05; **, P < 0.01. Cth, cystathionine; Cys, cysteine; Gly, glycine; GSH, glutathione; γ-Glu-Cys, γ-glutamylcysteine; Ser, serine. (A, Created with BioRender.com.)](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10152234_1426fig4.jpg)
Tumorigenesis of liver and pancreas induces downregulation of de novo cysteine synthesis. A, Schematic for the generation of Myc; p53−/− HCC and KrasG12D; p53+/− PDAC GEMM tumors. B, Analysis of the fraction labeling in serine, glycine, cystathionine, glutathione, cysteine, and γ-glutamylcysteine in liver tissues (N = 8) compared with HCC tumors (N = 8) and their matched serum normal (N = 8) and HCC (N = 5) following infusion with 1-[13C1]-serine. C, Analysis of the fraction labeling in serine, glycine, cystathionine, glutathione, cysteine, and γ-glutamylcysteine in pancreas tissues (N = 5) compared with PDAC tumors (N = 5) and their matched serum from normal (N = 5) and PDAC (N = 5) following infusion with 1-[13C1]-serine. D, Fractional contribution of serine to intracellular cysteine synthesis in HCC and PDAC. Cysteine labeling was normalized to the fraction labeling of serine in each tissue. One healthy pancreas sample was excluded because of a division error. For B–D, data are presented as mean ± SD. N.D., not detected. E, Immunoblots of CBS and CSE for each tissue. HSP90 was used for the loading control. *, P < 0.05; **, P < 0.01. Cth, cystathionine; Cys, cysteine; Gly, glycine; GSH, glutathione; γ-Glu-Cys, γ-glutamylcysteine; Ser, serine. (A, Created with BioRender.com.)
De novo cysteine synthesis does not contribute to the cysteine pool of lung tumors
We next wanted to explore whether a transsulfuration incapable tissue could gain the use of this pathway upon transformation. To this end, we explored the transsulfuration capacity of lung tumors. Thus, we employed 1-[13C1]-serine tracing using three different GEM models (GEMM). Given the role of NRF2 in cysteine metabolism, LSL-KrasG12D/+; Trp53flox/flox (KP) mice and LSL-KrasG12D/+; Trp53flox/flox; LSL-Nfe2l2D29H (KPN) mice (both C57BL/6J background) were used for LUAD (Fig. 5A). We also examined the activity of the transsulfuration pathway in SCLC, given the very different cell of origin of this lung cancer cell type from NSCLC. For the SCLC model, Rb1flox/flox; Trp53flox/fox; MycLSL/+ or Rb1flox/flox; Trp53flox/fox; MycLSL/LSL [RPM(M)] mice (mixed C57BL/6/FVB/129 background) were used (Fig. 5A). In LUAD, the cystathionine labeling was similar to control lung, which was not influenced by Nrf2 mutation (Fig. 5B). In contrast, SCLC demonstrated a significant reduction of cystathionine labeling compared with control (Fig. 5C). Most importantly, none of the GEM tumors demonstrated cysteine labeling from serine, indicating a lack of transsulfuration of serine to cysteine (Fig. 5B–D). Despite a lack of de novo cysteine synthesis, all tumor models accumulated tumoral cysteine, suggesting other mechanisms of cysteine accumulation (Fig. 5D; Supplementary Fig. S6A and S6B). Immunoblotting for CBS and CSE revealed that both LUAD models (KP and KPN models) had downregulated CBS compared with normal lung tissue, but CSE was overexpressed (Fig. 5E). In contrast, the patterns in SCLC tumors were reversed, with a modest increase in CBS and downregulation of CSE (Fig. 5E). Consistently, the mRNA expression of CBS in the human LUAD and LUSC tumors was nonsignificantly reduced, while CSE was nonsignificantly elevated in LUAD, but reduced in LUSC (Supplementary Fig. S6C). These results demonstrate that lung tumors do not acquire de novo cysteine synthesis capacity and accumulate cysteine via other mechanisms.
Figure 5.
![Figure 5. De novo cysteine synthesis does not contribute to the cysteine pool of lung tumors. A, Schematic for the generation of KrasG12D; p53−/− and KrasG12D; p53−/−; Nrf2D29H LUAD, and Rb1−/−; p53−/−; MycT58A/+ or Rb1−/−; p53−/−; MycT58A/T58A SCLC GEMM tumors. B, Analysis of the fraction labeling in serine, glycine, cystathionine, glutathione, cysteine, and γ-glutamylcysteine in normal lung tissues (N = 8) compared with Nrf2WT LUAD and (N = 10), Nrf2D29H LUAD tumors (N = 10) and their matched serum from normal (N = 8), Nrf2WT (N = 5), and Nrf2D29H (N = 5) following infusion with 1-[13C1]-serine. C, Analysis of the fraction labeling in serine, glycine, cystathionine, glutathione, cysteine, and γ-glutamylcysteine in normal lung tissues (N = 8) compared with SCLC tumors (N = 9), and their matched serum normal (N = 8) and SCLC (N = 9) following infusion with 1-[13C1]-serine. The control lung samples in C are the same as in B. D, Fractional contribution of serine to intracellular cysteine synthesis in LUAD and SCLC. Cysteine labeling was normalized to the fraction labeling of serine in each tissue. For B–D, data are presented as mean ± SD. N.D., not detected. E, Immunoblots of CBS and CSE for each tissue. HSP90 was used for the loading control. *, P < 0.05; ****, P < 0.0001. Cth, cystathionine; Cys, cysteine; Gly, glycine; GSH, glutathione; γ-Glu-Cys, γ-glutamylcysteine; Ser, serine. (A, Created with BioRender.com.)](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10152234_1426fig5.jpg)
De novo cysteine synthesis does not contribute to the cysteine pool of lung tumors. A, Schematic for the generation of KrasG12D; p53−/− and KrasG12D; p53−/−; Nrf2D29H LUAD, and Rb1−/−; p53−/−; MycT58A/+ or Rb1−/−; p53−/−; MycT58A/T58A SCLC GEMM tumors. B, Analysis of the fraction labeling in serine, glycine, cystathionine, glutathione, cysteine, and γ-glutamylcysteine in normal lung tissues (N = 8) compared with Nrf2WT LUAD and (N = 10), Nrf2D29H LUAD tumors (N = 10) and their matched serum from normal (N = 8), Nrf2WT (N = 5), and Nrf2D29H (N = 5) following infusion with 1-[13C1]-serine. C, Analysis of the fraction labeling in serine, glycine, cystathionine, glutathione, cysteine, and γ-glutamylcysteine in normal lung tissues (N = 8) compared with SCLC tumors (N = 9), and their matched serum normal (N = 8) and SCLC (N = 9) following infusion with 1-[13C1]-serine. The control lung samples in C are the same as in B. D, Fractional contribution of serine to intracellular cysteine synthesis in LUAD and SCLC. Cysteine labeling was normalized to the fraction labeling of serine in each tissue. For B–D, data are presented as mean ± SD. N.D., not detected. E, Immunoblots of CBS and CSE for each tissue. HSP90 was used for the loading control. *, P < 0.05; ****, P < 0.0001. Cth, cystathionine; Cys, cysteine; Gly, glycine; GSH, glutathione; γ-Glu-Cys, γ-glutamylcysteine; Ser, serine. (A, Created with BioRender.com.)
Cystine is a major contributor to the cysteine pool in tumors
To interrogate the contribution of cystine to the cysteine pool and downstream metabolites, we performed 13C6-cystine tracing in the HCC, PDAC, and LUAD GEMMs and the respective normal controls. We observed significant labeling in the cysteine pool in tumors, with higher labeling in cysteine synthesis–eficient tumors (50%) compared with cysteine synthesis competent HCC (19%; Fig. 6A). HCC cysteine labeling was not significantly different from normal liver, while PDAC labeling was higher than pancreas and LUAD labeling was lower than normal lung (Fig. 6A). Moreover, Nrf2D29H increased cysteine labeling in tumors (Fig. 6A), which is consistent with the know role of NRF2 in promoting the uptake of cystine via xCT and its reduction to cysteine via the glutathione/thioredoxin systems (38, 39). γ-glutamylcysteine labeling patterns mirrored cysteine labeling, apart from in lung tumors, where labeling was dramatically lower. Despite this, lung tumors had higher glutathione levels and labeling, with Nrf2D29H tumors demonstrating significantly higher levels and labeling than Nrf2WT tumors (Fig. 6A and B). The incongruence between γ-glutamylcysteine labeling and glutathione labeling in the lung tumors suggests potential dilution of the γ-glutamylcysteine pool by another source or cell type. HCC demonstrated similar glutathione labeling compared with normal liver, while PDAC demonstrated significantly lower labeling (Fig. 6A). Despite this, HCC demonstrated lower glutathione levels, and PDAC demonstrated higher glutathione levels (Fig. 6B), with the inverse observed in cysteine levels (Fig. 6C). Immunoblotting revealed very low xCT expression in HCC, like normal liver, with similar expression of GCLC, GCLM, and GSS between HCC and normal liver (Fig. 6D). Normal pancreas and PDAC had high expression of xCT, with PDAC upregulating GCLC and GCLM, despite a reduction in glutathione labeling (Fig. 6A and D). LUAD upregulated xCT and GCLC, with Nrf2 promoting a further increase in xCT, GCLC, and GCLM (Fig. 6D). In addition, the gene expression profile of xCT, GCLC, and GCLM in the TCGA/GTEx was consistent with immunoblotting in our GEM models (Supplementary Fig. S7A). These findings reveal that cystine is a major contributor to the cysteine pool in tumors, but glutathione metabolism displays complex regulation across diverse tumor types.
Figure 6.

Cystine is a major contributor to the cysteine pool in tumors. A, Analysis of the fraction labeling in cysteine, γ-glutamylcysteine, and glutathione in liver tissues (N = 9), HCC tumors (N = 9), lung tissues (N = 10), Nrf2WT LUAD tumors (N = 16), Nrf2D29H LUAD tumors (N = 10), pancreas tissues (N = 3), PDAC tumors (N = 12), and their matched serum from normal control mice for HCC (N = 6), HCC (N = 7), normal control mice for PDAC (N = 3), PDAC (N = 6), normal control mice for LUAD (N = 5), Nrf2WT LUAD (N = 8), and Nrf2D29H LUAD serum (N = 5) following infusion with 13C6-cystine. B, Total signal of glutathione in the tissues from A. C, Total signal of cysteine in the tissues from A. For A–C, data are presented as mean ± SD. N.D., not detected. D, Immunoblots of xCT, GCLC, GCLM, and GSS for each tissue. HSP90 was used for the loading control. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Cys, cysteine; GSH, glutathione; γ-Glu-Cys, γ-glutamylcysteine.
Discussion
In this study, we comprehensively evaluated the contribution of transsulfuration and exogenous cyst(e)ine to the cysteine pool and its downstream intermediates using 13C-based stable isotope tracers in vivo and in vitro. Prior work has established that flux through the methionine cycle to convert S-adenosylmethionine to S-adenosylhomocysteine, and subsequently homocysteine, is a critical limiting factor for transsulfuration. It was suggested methylation of lipids by phosphatidylethanolamine N-methyltransferase in the liver drives the production of homocysteine for transsulfuration, but non-hepatic tissues lack this activity (33). Using 13C1-serine tracing, we directly assayed activity of the transsulfuration pathway downstream of the methionine cycle, where homocysteine condenses with serine to produce cystathionine. Indeed, we find that cystathionine demonstrated the highest abundance in the liver compared with other tissues, which is likely explained by both methionine cycle flux and expression of transsulfuration enzymes in this tissue. Interestingly, across both cultured cancer cell lines and diverse tissues in vivo, we find that cystathionine is robustly labeled from serine, while cysteine labeling is low or absent, even when exogenous cystine is removed from the culture system. CBS is considered the rate-limiting enzyme of transsulfuration, and its activity is positively regulated by S-adenosylmethionine availability to promote entry of homocysteine into the transsulfuration pathway (40). However, our results demonstrate that the second step of transsulfuration mediated by CSE is likely to also restrict de novo cysteine synthesis in vitro as well as in vivo. Recently, interleukin-1 receptor accessory protein (IL1RAP) was identified as a novel positive regulator of cysteine availability that functions through both the regulation of xCT and transsulfuration (27). Interestingly, IL1RAP promotes transsulfuration through the transcriptional regulation of CSE, suggesting CSE is the limiting component. Moreover, ATF4 transcriptionally regulates CSE (41), suggesting amino acid stress promotes cysteine synthesis at the CSE step. CSE is a PLP-dependent enzyme, and its activity is modified by calcium, nitric oxide, and carbon dioxide (42), which are not captured in our metabolomics. These modifiers may display different abundance across these tissues. Therefore, it is possible that the regulation of CBS is important to avoid the toxicity associated with homocysteine accumulation, while regulation of CSE is important to control cysteine synthesis.
Given the importance of cysteine, why is the transsulfuration pathway not a major contributor to the cysteine pool in tumors? We find that de novo cystine synthesis is either inactive (lung cancer) or downregulated (HCC and PDAC) during tumorigenesis. Downregulation of transsulfuration is associated with a decreased expression of both CBS and CSE. Prior studies have reported the downregulation of CBS in both HepG2 cells and patients with HCC, which is associated with poor prognosis (43, 44). Downregulation of CSE is also observed in HCC and associated with poor prognosis (45). The analysis of CBS and CSE expression in PDAC is complicated by the dense stroma typical of these tumors (46), which is comprised of fibroblasts, immune cells, and other cell types. While this stroma can account for half of the tumor cellularity, it is unlikely to completely account for the complete absence of cysteine synthesis and CBS/CSE expression we observe in the GEMMs. In contrast to HCC, low expression of CBS in PDAC is associated with better outcomes (40). In addition to their role in cysteine synthesis, CBS and CSE play a role in hydrogen sulfide (H2S) generation, which can be toxic in high concentrations (40). Thus, tumors in which adequate cysteine is supplied from other pathways may downregulate these enzymes to limit the other metabolic consequences of these enzymes. However, colorectal carcinomas are reported to increase CBS expression, which is associated with worse outcomes (40), and CBS heterozygous knockout mice are protected from azoxymethane-induced aberrant colonic crypt formation (47). Moreover, CBS or CSE, respectively, promote xenograft growth of neuroblastoma and Ewing sarcoma cell lines (26, 27). Additional studies are needed to evaluate the contribution of CBS and CSE to the synthesis of cysteine and other products of the transsulfuration pathway in these other models in vivo.
We find that in normal tissues, cyst(e)ine readily labels the cysteine pool. However, xCT expression alone is not a good predictor of cysteine labeling. The uptake of cystine and its reduction to cysteine via xCT is influenced by other factors, including both intracellular and extracellular glutamate availability, cellular reducing potential, and xCT posttranslational modification and subcellular localization of xCT (15, 23). Indeed, we find that concomitant with high xCT expression the brain tissues also have high glutamate levels, which likely limit cystine/glutamate exchange. Moreover, xCT knockout mice are viable (48), indicating that other cystine and/or cysteine transporters can support normal cysteine homeostasis. Cysteine transporters are poorly characterized, particularly in the context of cancer. Interestingly, we find that despite an increase in the total cysteine pool in HCC, this cannot be accounted for by an increase in transsulfuration or cystine uptake, suggesting that HCC tumors have an alternative source of cysteine. Glutathione degradation via gamma-glutamyl transpeptidase may locally generate available cysteine (49), or tumors may recycle micropinocytosis-derived protein to contribute to the cysteine pool as has been shown in HCC cell lines (50). Additional work is needed to understand the reliance of HCC tumors on other cysteine sources. Given this potential metabolic flexibility, it may be challenging to target cysteine availability in HCC.
We examined the metabolism of cysteine to downstream metabolites in tumors. Cancer cells are generally thought to have an increased demand for antioxidant protection, particularly via glutathione synthesis (7). We find that LUAD tumors show increased contribution of cysteine to the glutathione pool, with NRF2 activation further promoting glutathione synthesis as expected. Interestingly, PDAC decreased the contribution of cysteine to glutathione despite having a very high total glutathione content. PDAC has high macropinocytic activity (51), which facilitates the uptake of protein from the protein-rich extracellular environment for degradation to supply the intracellular amino acid pools. Glutathione is about seven times higher in PDAC interstitial fluid compared with plasma (184 vs. 26 μmol/L) in a GEMM (52), raising the interesting possibility that PDAC can acquire glutathione via micropinocytosis to supply the intracellular pool. In addition to glutathione synthesis, we examined taurine synthesis as another downstream cysteine catabolic pathway. Taurine is thought to be predominantly synthesized in the liver, and to a lesser extent in other tissues, then released into circulation for uptake via the ubiquitously expressed taurine transporter (TAUT, SLC6A6; ref. 35). However, our results suggest that kidney, liver, and brain (including cerebellum) label their hypotaurine pool from circulating hypotaurine, suggesting uptake of hypotaurine itself in these tissues. Hypotaurine is transported by γ-aminobutyric acid transporter type 2 (GAT2, SLC6A13) and TAUT (53). GAT2 is primarily expressed in not only brain including cerebellum but also liver and kidney (54). Moreover, pancreas expresses high levels of CDO1, but lacks the expression of FMO1, raising the possibility that pancreas synthesizes high levels of hypotaurine for export. Because we could not distinguish hypotaurine labeled directly in tissues from cysteine from label coming from circulating hypotaurine, we could not evaluate cysteine catabolism to hypotaurine in tumors. Taurine synthesis is frequently downregulated in many cancers by CDO1 promoter methylation (55), raising the possibility that cancers supply their taurine pool by hypotaurine and/or taurine transport. Additional studies are needed to examine this possibility.
Limitations of the study
There are several limitations of our study. First, we examined cysteine labeling and downstream metabolism at a single timepoint after a 4-hour infusion with 13C1-serine or 13C6-cystine, which allowed us to examine the contribution of exogenous cyst(e)ine and the transulfuration pathway to the cysteine pool and downstream metabolism. We were unable to evaluate the contribution of other sources of cysteine, including glutathione, protein, and even circulating cysteine precursors like cystathionine, with this approach. We analyzed steady state labeling of metabolites in cysteine metabolic pathways but did not assay flux over time like what was recently reported for tricarboxylic acid cycle flux (56), which would provide additional information. Our analyses were also limited to macrodissected tissues that are comprised of multiple cell types that, together with metabolic and microenvironmental heterogeneity, may influence cysteine metabolism that may be masked by bulk tissue analysis. Combining labeling with spatial metabolomics will be critical to deconvoluting the impact of these factors on cysteine metabolism (57). Finally, our studies are limited to mice and while sulfur metabolism is highly conserved, there may be microbiome, diet, and environmental differences that influence the translation of our studies to humans.
Supplementary Material
Figure S1 reports the status of common mutations in the cell lines used in this study.
Figure S2 demonstrates that transsulfuration intermediates are limiting for activity in NSCLC cells
Figure S3 contains the total metabolite levels in healthy mouse tissues related to Figure 2
Figure S4 contains the total metabolite levels in healthy mouse tissues related to Figure 3
Figure S5 contains the total metabolite levels in liver and pancreas cancer models related to Figure 4
Figure S6 contains total metabolite levels in lung cancer models related to Figure 5
Figure S7 contains mRNA expression of cysteine metabolic enzymes in patient liver, pancreas and lung tumors
Acknowledgments
The authors would like to thank all members of the DeNicola laboratory and Isaac Harris for their very helpful discussions. This work was supported by grants from the NIH/NCI R37CA230042, which supported J. Combs, A. Falzone, N. Prieto-Farigua, and G. DeNicola, and P01CA250984, which supported S.J. Yoon, S. Caldwell, H. Ackerman, G. DeNicola, and E. Flores. This work was also supported by Miles for Moffitt funds awarded to the Lung Cancer Metabolism Working Group and the Proteomics/Metabolomics Core, which is funded in part by Moffitt's Cancer Center Support Grant (NCI, P30-CA076292).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
S. Caldwell reports grants from Moffitt Cancer Center during the conduct of the study. G.M. DeNicola reports grants from NIH/NCI during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
S.J. Yoon: Conceptualization, data curation, formal analysis, investigation, visualization, writing–original draft, project administration, writing–review and editing. J.A. Combs: Data curation, formal analysis, methodology, writing–review and editing. A. Falzone: Resources, investigation, methodology, writing–review and editing. N. Prieto-Farigua: Formal analysis, investigation, writing–review and editing. S. Caldwell: Resources, investigation, writing–review and editing. H.D. Ackerman: Resources, writing–review and editing. E.R. Flores: Resources, funding acquisition, writing–review and editing. G.M. DeNicola: Conceptualization, data curation, supervision, funding acquisition, investigation, visualization, methodology, project administration, writing–review and editing.
References
- 1. Ward NP, DeNicola GM. Sulfur metabolism and its contribution to malignancy. Int Rev Cell Mol Biol 2019;347:39–103. [DOI] [PubMed] [Google Scholar]
- 2. Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radical Biol Med 2008;45:549–61. [DOI] [PubMed] [Google Scholar]
- 3. Anderson ME. Glutathione: an overview of biosynthesis and modulation. Chem Biol Interact 1998;111–112:1–14. [DOI] [PubMed] [Google Scholar]
- 4. Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J 1999;13:1169–83. [PubMed] [Google Scholar]
- 5. Harris IS, DeNicola GM. The complex interplay between antioxidants and ROS in cancer. Trends Cell Biol 2020;30:440–51. [DOI] [PubMed] [Google Scholar]
- 6. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab 2016;23:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pavlova NN, Zhu J, Thompson CB. The hallmarks of cancer metabolism: still emerging. Cell Metab 2022;34:355–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang HF, Klein Geltink RI, Parker SJ, Sorensen PH. Transsulfuration, minor player or crucial for cysteine homeostasis in cancer. Trends Cell Biol 2022;32:800–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, et al. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem 2002;277:44765–71. [DOI] [PubMed] [Google Scholar]
- 10. Kang YP, Torrente L, Falzone A, Elkins CM, Liu M, Asara JM, et al. Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. Elife 2019;8:e45572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015;520:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gu Y, Albuquerque CP, Braas D, Zhang W, Villa GR, Bi J, et al. mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Mol Cell 2017;67:128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsuchihashi K, Okazaki S, Ohmura M, Ishikawa M, Sampetrean O, Onishi N, et al. The EGF receptor promotes the malignant potential of glioma by regulating amino acid transport system xc(-). Cancer Res 2016;76:2954–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lien EC, Ghisolfi L, Geck RC, Asara JM, Toker A. Oncogenic PI3K promotes methionine dependency in breast cancer cells through the cystine-glutamate antiporter xCT. Sci Signal 2017;10:eaao6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mukhopadhyay S, Biancur DE, Parker SJ, Yamamoto K, Banh RS, Paulo JA, et al. Autophagy is required for proper cysteine homeostasis in pancreatic cancer through regulation of SLC7A11. Proc Natl Acad Sci U S A 2021;118:e2021475118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sato H, Nomura S, Maebara K, Sato K, Tamba M, Bannai S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun 2004;325:109–16. [DOI] [PubMed] [Google Scholar]
- 17. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang T, Bauer C, Newman AC, Uribe AH, Athineos D, Blyth K, et al. Polyamine pathway activity promotes cysteine essentiality in cancer cells. Nat Metab 2020;2:1062–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab 2021;33:174–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol 2019;26:623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020;368:85–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med 2017;23:120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Combs JA, DeNicola GM. The non-essential amino acid cysteine becomes essential for tumor proliferation and survival. Cancers 2019;11:678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stipanuk MH. Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu Rev Nutr 2004;24:539–77. [DOI] [PubMed] [Google Scholar]
- 25. Mudd SH, Finkelstein JD, Irreverre F, Laster L. Transsulfuration in mammals. Microassays and tissue distributions of three enzymes of the pathway. J Biol Chem 1965;240:4382–92. [PubMed] [Google Scholar]
- 26. Zhu J, Berisa M, Schwörer S, Qin W, Cross JR, Thompson CB. Transsulfuration activity can support cell growth upon extracellular cysteine limitation. Cell Metab 2019;30:865–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang HF, Hughes CS, Li W, He JZ, Surdez D, El-Naggar AM, et al. Proteomic screens for suppressors of anoikis identify IL1RAP as a promising surface target in Ewing sarcoma. Cancer Discov 2021;11:2884–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeBlasi JM, Falzone A, Caldwell S, Kang YP, Prieto-Farigua N, Prigge JR, et al. Distinct Nrf2 signaling thresholds mediate lung tumor initiation and progression. biorxiv 2022. [DOI] [PMC free article] [PubMed]
- 29. Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, Lindblad KE, Maier B, Sia D, et al. β-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov 2019;9:1124–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A 2010;107:246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res 2005;65:10280–8. [DOI] [PubMed] [Google Scholar]
- 32. Mollaoglu G, Guthrie MR, Böhm S, Brägelmann J, Can I, Ballieu PM, et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell 2017;31:270–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ye C, Sutter BM, Wang Y, Kuang Z, Tu BP. A metabolic function for phospholipid and histone methylation. Mol Cell 2017;66:180–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ookhtens M, Kaplowitz N. Role of the liver in interorgan homeostasis of glutathione and Cyst(e)ine. Semin Liver Dis 1998;18:313–29. [DOI] [PubMed] [Google Scholar]
- 35. Stipanuk MH. Role of the liver in regulation of body cysteine and taurine levels: a brief review. Neurochem Res 2004;29:105–10. [DOI] [PubMed] [Google Scholar]
- 36. Sumizu K. Oxidation of hypotaurine in rat liver. Biochim Biophys Acta 1962;63:210–2. [DOI] [PubMed] [Google Scholar]
- 37. Veeravalli S, Phillips IR, Freire RT, Varshavi D, Everett JR, Shephard EA. Flavin-containing monooxygenase 1 catalyzes the production of taurine from hypotaurine. Drug Metab Dispos 2020;48:378–85. [DOI] [PubMed] [Google Scholar]
- 38. Jaganjac M, Milkovic L, Sunjic SB, Zarkovic N. The NRF2, thioredoxin, and glutathione system in tumorigenesis and anticancer therapies. Antioxidants 2020;9:1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Habib E, Linher-Melville K, Lin HX, Singh G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol 2015;5:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ascenção K, Szabo C. Emerging roles of cystathionine β-synthase in various forms of cancer. Redox Biol 2022;53:102331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bai X, Ni J, Beretov J, Wasinger VC, Wang S, Zhu Y, et al. Activation of the eIF2α/ATF4 axis drives triple-negative breast cancer radioresistance by promoting glutathione biosynthesis. Redox Biol 2021;43:101993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao K, Li H, Li S, Yang G. Regulation of cystathionine gamma-lyase/H₂S system and its pathological implication. Front Biosci 2014;19:1355–69. [DOI] [PubMed] [Google Scholar]
- 43. Wang L, Han H, Liu Y, Zhang X, Shi X, Wang T. Cystathionine β-synthase induces multidrug resistance and metastasis in hepatocellular carcinoma. Curr Mol Med 2018;18:496–506. [DOI] [PubMed] [Google Scholar]
- 44. Kim J, Hong SJ, Park JH, Park SY, Kim SW, Cho EY, et al. Expression of cystathionine beta-synthase is downregulated in hepatocellular carcinoma and associated with poor prognosis. Oncol Rep 2009;21:1449–54. [DOI] [PubMed] [Google Scholar]
- 45. Lin Z, Huang W, He Q, Li D, Wang Z, Feng Y, et al. FOXC1 promotes HCC proliferation and metastasis by upregulating DNMT3B to induce DNA hypermethylation of CTH promoter. J Exp Clin Cancer Res 2021;40:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Helms E, Onate MK, Sherman MH. Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer Discov 2020;10:648–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Phillips CM, Zatarain JR, Nicholls ME, Porter C, Widen SG, Thanki K, et al. Upregulation of cystathionine-β-synthase in colonic epithelia reprograms metabolism and promotes carcinogenesis. Cancer Res 2017;77:5741–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem 2005;280:37423–9. [DOI] [PubMed] [Google Scholar]
- 49. Asantewaa G, Harris IS. Glutathione and its precursors in cancer. Curr Opin Biotechnol 2021;68:292–9. [DOI] [PubMed] [Google Scholar]
- 50. Byun JK, Lee S, Kang GW, Lee YR, Park SY, Song IS, et al. Macropinocytosis is an alternative pathway of cysteine acquisition and mitigates sorafenib-induced ferroptosis in hepatocellular carcinoma. J Exp Clin Cancer Res 2022;41:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013;497:633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 2019;8:e44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nishimura T, Higuchi K, Yoshida Y, Sugita-Fujisawa Y, Kojima K, Sugimoto M, et al. Hypotaurine is a substrate of GABA transporter family members GAT2/Slc6a13 and TAUT/Slc6a6. Biol Pharm Bull 2018;41:1523–9. [DOI] [PubMed] [Google Scholar]
- 54. Zhou Y, Danbolt NC. GABA and glutamate transporters in brain. Front Endocrinol 2013;4:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brait M, Ling S, Nagpal JK, Chang X, Park HL, Lee J, et al. Cysteine dioxygenase 1 is a tumor suppressor gene silenced by promoter methylation in multiple human cancers. PLoS One 2012;7:e44951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bartman CR, Shen Y, Lee WD, TeSlaa T, Jankowski CSR, Wang L, et al. Slow TCA flux implies low ATP production in tumors. biorxiv 2021.
- 57. Wang L, Xing X, Zeng X, Jackson SR, TeSlaa T, Al-Dalahmah O, et al. Spatially resolved isotope tracing reveals tissue metabolic activity. Nat Methods 2022;19:223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 reports the status of common mutations in the cell lines used in this study.
Figure S2 demonstrates that transsulfuration intermediates are limiting for activity in NSCLC cells
Figure S3 contains the total metabolite levels in healthy mouse tissues related to Figure 2
Figure S4 contains the total metabolite levels in healthy mouse tissues related to Figure 3
Figure S5 contains the total metabolite levels in liver and pancreas cancer models related to Figure 4
Figure S6 contains total metabolite levels in lung cancer models related to Figure 5
Figure S7 contains mRNA expression of cysteine metabolic enzymes in patient liver, pancreas and lung tumors
Data Availability Statement
The data generated in this study are included in the article and its supplementary figures. Raw data are available upon request without restriction from the corresponding author.