

Abstract
Alzheimer’s disease (AD) is an age-related disease pathologically defined by the deposition of amyloid plaques and neurofibrillary tangles in the brain parenchyma. Single cell profiling reveals Alzheimer’s dementia involves the complex interplay of virtually every major brain cell type. Here, we highlight cell-type specific molecular perturbations in AD. We discuss how genomic information from single cells expand existing paradigms of AD pathogenesis and highlight new opportunities for therapeutic interventions.
Introduction
Single cell genomics have defined the complex molecular regulation of major cell types in the mouse1–4 and human brain5,6. Profiling genetic information from single cells from individuals with various stages of AD pathology (Table 1) uncovers detailed cell-type specific molecular programs in AD (Figure 1). Keeping in mind challenges related to interpreting genomic studies from single cells, such as the common necessity to profile single nuclei instead of single cells from archived brain tissue (Box 1), we argue molecular disturbances across major cell types converge on common signaling pathways such as lipid handling, immune response, and metabolic reprogramming (Figure 2). Further defining and manipulating core signaling nodes may generate new opportunities for therapeutic intervention.
Table 1.
Single-cell transcriptomic and epigenetic datasets from post-mortem AD tissue.
| Study | Data ID | Patient cohort | Brain region | Sequencing strategy | Total nuclei |
|---|---|---|---|---|---|
| ♦Mathys8 | syn18485175 | 48 | PFC (BA10) | snRNA-seq | 80,660 |
| Davila13 | N/A | 112 | Hippocampus | snRNA-seq | 489,558 |
| Entorhinal cortex | |||||
| ♦Grubman7 | GSE138852 | 12 | Entorhinal cortex | snRNA-seq | 13,214 |
| ♦Leng18 | GSE147528 | 10 | Caudal entorhinal cortex | snRNA-seq | 42,528 |
| Superior frontal gyrus | snRNA-seq | 63,608 | |||
| Zhou9 | syn21125841 | 32 | Dorsolateral prefrontal cortex | snRNA-seq | 66,311 |
| Lau14 | GSE157827 | 21 | PFC (BA6, BA8, and BA9) | snRNA-seq | 169,496 |
| Otero-Garcia12 | GSE129308 | 8 | Prefrontal cortex (BA9) | AT8 and MAP2 FACS | 63,110 |
| Alsema127 | GSE146639 | 27 | superior parietal lobe superior frontal gyrus | CD11/CD45 FACS; bc-Smart-seq2 | |
| Marinaro128 | N/A | 12 | PFC (BA9) | FACS neurons and glia; snRNA-seq | 89,325 |
| ♦Yang116 | GSE163577 | 17 | hippocampus | Vascular enriched fraction then snRNA-seq | 143,793 |
| 8 | superior frontal cortex | ||||
| Gerrits129 | GSE148822 | 18 | occipitotemporal cortex and fusiform gyrus | NEUN−/OLIG2− FACS, then snRNA-seq | 482,472 nuclei |
| Del-Aguila130 | http://ngi.pub/snuclRNA-seq/ | 3 | Parietal lobe | snRNA-seq | 26,331 |
| Olah131 | 14 | dorsolateral prefrontal cortex | CD11b+/CD45+, snRNA-seq | 16,242 | |
| 3 | TNC | ||||
| ♦Morabito77 | syn3219045 | 20 | PFC | snATAC-seq and snRNA-seq | 191,890 |
| Xu132 | GSE181279 | 5 | PBMCs | CD45 selection, then TCR-seq | 36,849 |
| Gate133 | GSE134578 | 18 | peripheral CD8+ TEMRA; CSF cells | TCR-seq | 21,267 |
| Smith134 | GSE160936 | 12 | entorhinal and somatosensory cortex | NEUN-/SOX10- | 52,706 astrocytes and 27,592 microglia |
signifies particularly noteworthy study
Figure 1: Overview of central advantages of single cell approaches for the study of AD.
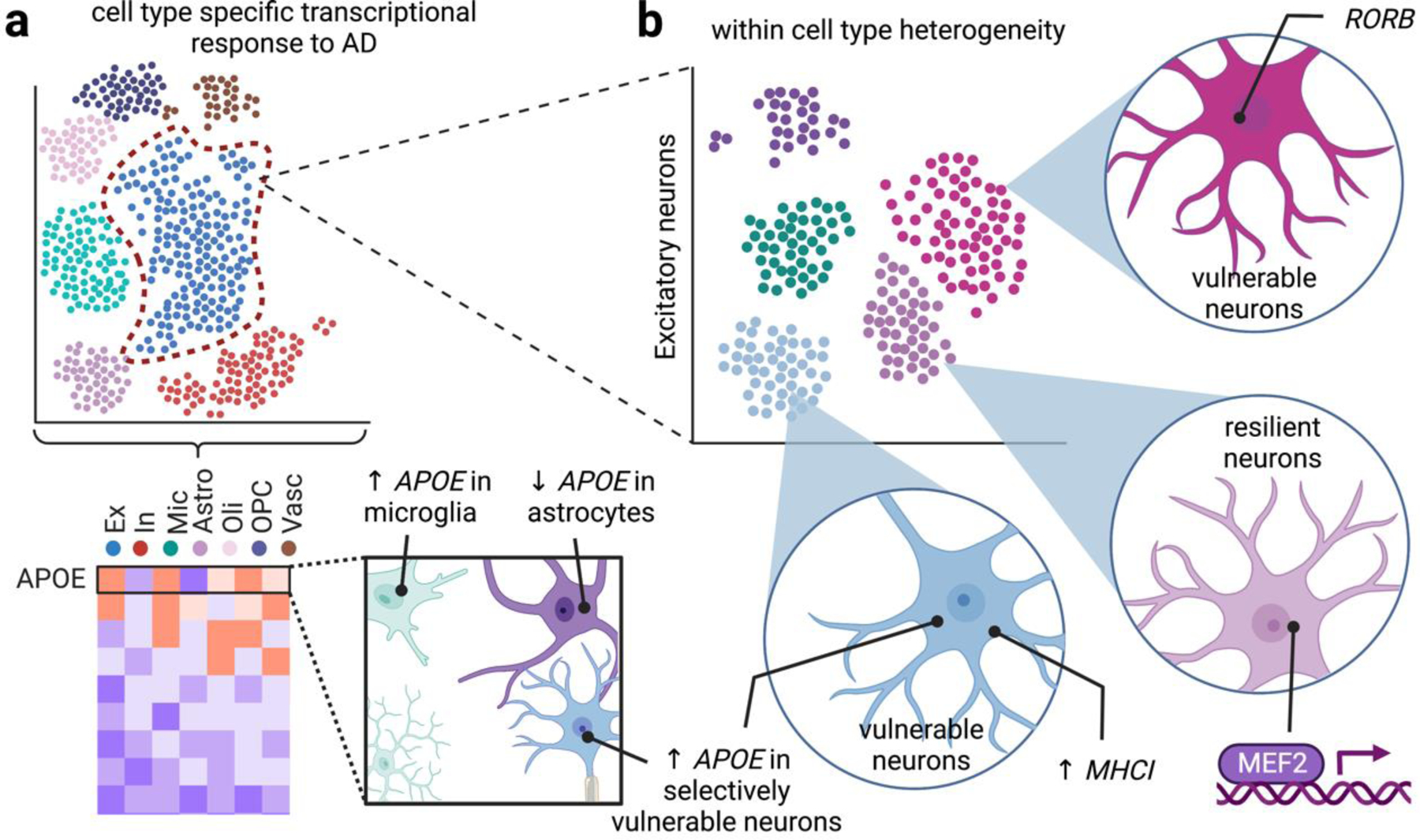
Single cell approaches highlight cell type and cell subtype specific vulnerability to disease. (a) Cell type specific responses to disease. Bulk quantifications of gene expression report population averages, which belie changes from specific cell populations that may drive distinct pathological responses. For example, snRNA-seq revealed that APOE is downregulated in AD astrocytes but upregulated in microglia7,8 and some neurons20. (b) Cell subtype responses to disease. Bulk profiling based on cell type markers might mask within-cell type heterogeneity, such as layer-specific neurons, non-myelinating oligodendrocytes. In contrast, single cell profiling unmasks differential vulnerabilities to AD within distinct subsets of major cell types. For example, neurons selectively vulnerable to AD neurodegeneration are marked by RORB18 and elevated APOE/MHC-I signaling20, and neurons resilient to AD pathology are enriched in MEF219.
Box 1. Considerations related to interpreting single cell data for the study of Alzheimer’s disease.
AD classification.
Assigning AD status is nontrivial because some individuals bearing AD pathology are cognitively normal, while some individuals clinically diagnosed with AD are found to not harbor AD pathology19. Variants in genetic risk factors such as APOE or TREM29 generate pleiotropic molecular effects. These factors underscore the importance of considering AD classification in profiling patient samples and interpreting single cell data.
Patient selection.
Sex is a critical consideration in patient selection, as sex-specific AD associations have emerged in single cell profiling8. Additionally, racial and ethnic factors may be associated with differential AD risk125,126, but these potential differences driving AD susceptibility are poorly understood at the single cell level. Many other confounds, including education, diet, sleep patterns, and exercise habits are known to generate epidemiological effects on AD risk and therefore may affect conclusions from single cell profiling—and may confer a tractable opportunity to define underpinnings of AD vulnerability.
Brain region.
The anatomical routes AD pathology progresses through the brain are incompletely understood. Single cell studies are beginning to unravel this complexity by examining multiple brain regions, and spatial transcriptomics is facilitating insight into brain-wide transcriptional modules associated with AD dysfunction.
Single cell preparations.
Archived brain tissue often suffers from compromised structural integrity, complicating efforts to purify whole cells, so most studies of human tissue rely on nuclei purification135. However, single nuclei preparations of microglia may not capture many AD-associated microglial genes that are observed in single soma preparations136, and cytoplasmic mRNA may be lost in many other cell types. Additionally, debris related to single cell preparations following nuclei purification can complicate downstream analyses; while sorting nuclei by FACS prior to loading cells may remove some debris, stress-associated artifacts from cell sorting may confound some analyses.
Tissue preparation.
Enzymatic dissociation induces stress-related transcriptional changes in microglia, and a cocktail of transcription/translation inhibitors (actinomycin D, ansiomycin, and triptolide) are thought to prevent dissociation related transcriptional responses137. Differences in single cell preparations between experimenters may further bias the enrichment of certain cell types, which may explain differences in proportions in brain cell types across datasets. Furthermore, some cell types evade certain preparations. For example, many vascular cells resist single cell dissociation protocols, which may account for their low yields in most datasets, and strainer-based methods to capture intact blood vessels helps enrich vascular segments for single cell analysis116.
Computational analysis.
Quality filtering, such as metrics of cell health (e.g., number of mitochondrial genes in nuclei datasets) and feature selection (e.g., number of genes detected, which can be a proxy for doublets), can affect downstream conclusions. Additionally, clustering resolution to define cell types can be highly dependent on individual experiments, and individual clustering algorithms can require user-defined parameters. Differential gene expression analysis is also highly variable, reinforcing the importance of biological validation. As sample sizes increase, batch correction and data integration across multiple datasets presents several statistical challenges. Thus, harmonizing single cell datasets may be essential to generate biological insight across analytical methods and tissue preparation protocols.
Genomic programs and biological function.
AD-specific transcriptional differences may not reflect biological differences, partly because gene transcripts may not generate differences in protein levels owing to multiple levels of regulation. Furthermore, non-genomic programs may account for many aspects of AD dysfunction, including post-translational modifications and mRNA regulation, such as regulation of local protein synthesis in distinct cellular compartments and cell types, which may not be captured by single cell profiling.
Figure 2. Shared cellular pathways disturbed in AD as revealed by single cell genomics.
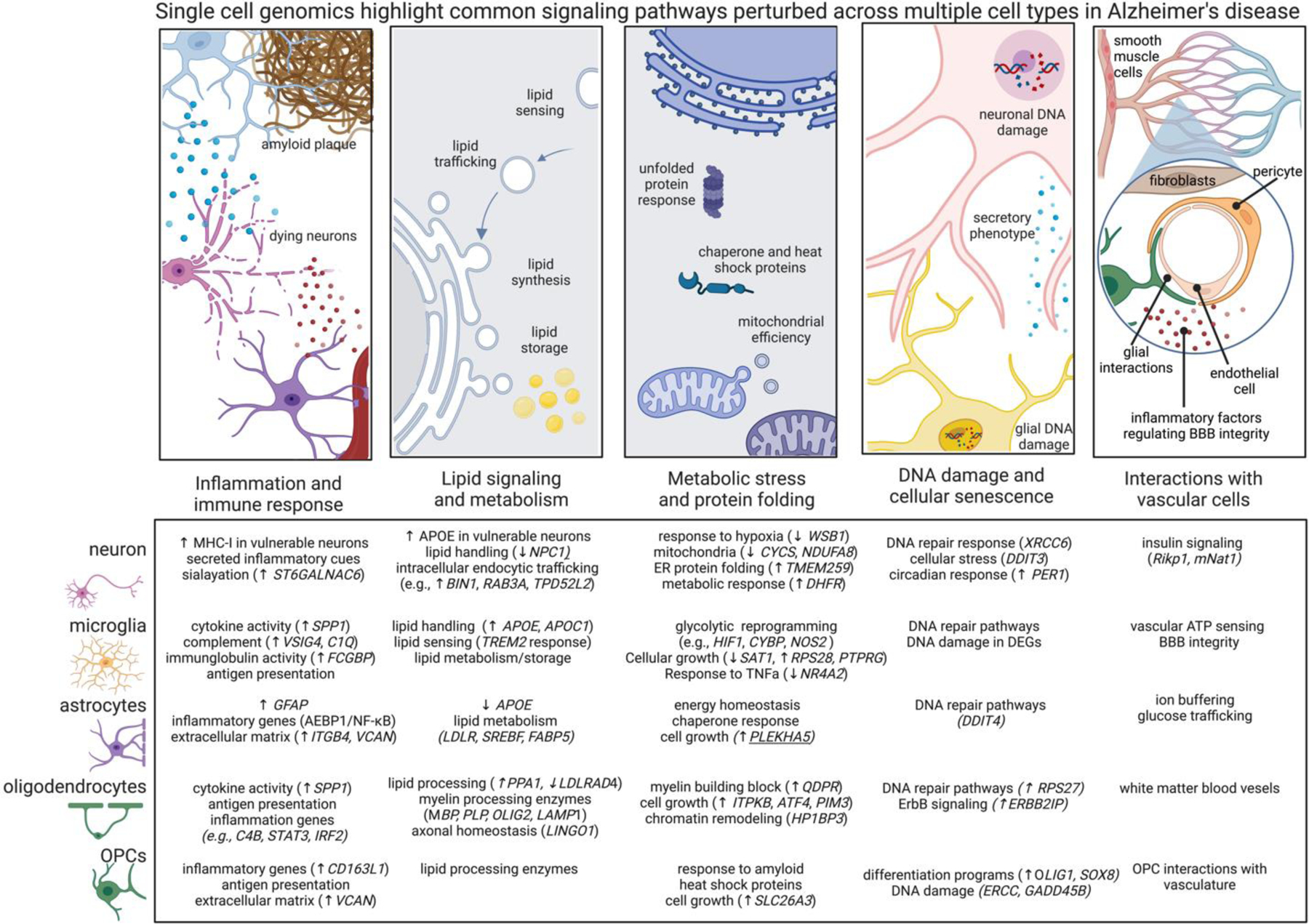
Differentially expressed genes across cell types are related to shared signaling motifs. Identifying common disrupted pathways may uncover core nodes of perturbation and lead to new therapeutic interventions related to multiple cells. Below we highlight common cellular pathways that are disrupted across multiple cell types in AD. Arrows denote transcriptional directions from prefrontal cortex8 (up arrow means up in AD compared to non-AD) and with a focus on genes showing concordant expression changes from other datasets and brain regions as highlighted in the text.
•Immune signaling. Nearly every cell type generates immune responses in AD, including transcriptional responses related to cytokine, chemokine, and MHC signaling. MHC signaling may related to synaptic plasticity and the unfolded protein response. The low-grade AD-related inflammation in every cell type associated with AD may be associated with metabolic reprogramming.
•Lipid handling. Lipid signaling is crucial for many cell functions, such as sensing and shuttling lipid species and to accommodate the dynamic remodeling of plasma membrane required for the structural plasticity of dendritic spines, microglial processes, astrocytic endfeet, and nodes of Ranvier. Perturbed lipid signaling in many brain cell types in AD, underscore the importance of lipid signaling and metabolism.
•Unfolded protein response. Nearly every major cell type modulates protein misfolding pathways and integrated stress responses, and, related, mitochondrial function, highlighting energetic disruptions in AD cells. These findings suggest the milieu of the AD brain affects unfolded protein response and cellular stress even in cells not directly burdened by pathology.
•DNA damage and cellular senescence. DNA damage in neurons is associated with aging and is elevated in neurodegeneration23, DNA damage is essential for the expression of learning related immediate early gene expression27. Many cells in AD have impaired DNA repair enzyme pathways, potentially suggesting senescent state and loss of core cellular functions.
•Vascular interactions. Recent studies are beginning to profile the complex network of vascular cells in AD. Existing datasets highlight signaling pathways perturbed across multiple brain cell types relating to neurovascular coupling and BBB dysfunction in AD, including the cell-type specific secretion of inflammatory molecules known to regulate vascular cells.
Excitatory neurons.
Synaptic alterations and neuronal loss are well-established in AD, and single cell profiling has revealed molecular programs regulating neuronal dysfunction (Figure 3). Several single nucleus RNA-sequencing (snRNA-seq) studies reveal excitatory neurons from AD patients alter genes regulating neurotransmitter release, synaptic vesicle recycling, and glutamate metabolism7–9. Histological findings indicate post-synaptic terminals are lost in AD, and several differentially expressed genes relate to post-synaptic scaffolding molecules, glutamate receptor trafficking, and calmodulin signaling8,9. Importantly, in situ hybridization confirms some of these findings, such as the reduced number of excitatory neurons8 and the downregulation of NTNG1, a gene involved in the regulation of neurite outgrowth8. Inhibitory synapses, which are highly plastic in the adult brain and are thought to enable flexible modulation of stable excitatory connections10, are also reduced in number in AD11. These findings are partly reflected in altered expression of genes critical for inhibitory synapses, such as modulation of some integrin genes7,8. The integrity of myelinated axons is critical for long-range projections, and genes related to neuronal-oligodendrocyte interactions are modulated in AD neurons, such as upregulation of LINGO17–9, a negative regulator of myelination (a finding repeated across several transcriptomic studies7–9 and by in situ hybridization8). These transcriptional changes suggest synaptic elements are not simply structurally lost in AD, but the very molecular machinery governing their integrity are dysregulated.
Figure 3. Single cell genomics reveal cell type specific perturbations in sensing and regulating neural activity in AD.
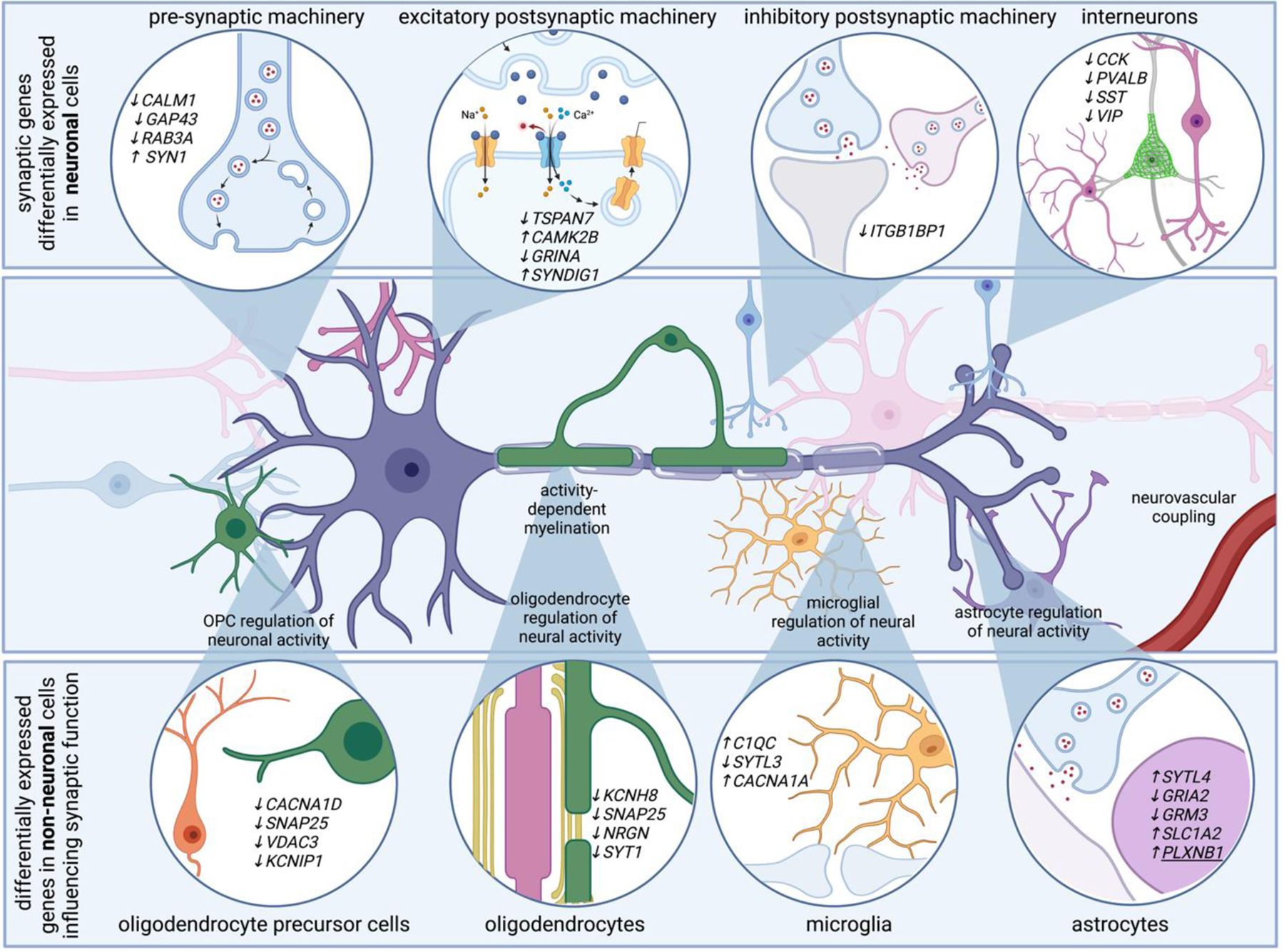
Neurons account for the vast majority of differentially expressed genes in AD. Genes related to pre-synaptic, post-synaptic, and inhibitory synaptic machinery emerge in single transcriptomes of AD neurons. For example, AD neurons upregulate SYN1, a gene that encodes synapsin 1, critical for synaptic vesicle function, and downregulate TSPAN7, which encodes a tetraspanin thought to regulate post-synaptic dendritic spine structure. Transcriptional programs associated with altered electrical properties may be associated with neuronal vulnerability to AD. Additionally, non-neuronal cells modulate genes that are involved in synaptic function. For example, genes related to synaptotagmin related genes are differentially expressed in astrocytes, oligodendrocytes, oligodendrocyte precursor cells, and microglia. Several differentially expressed genes in non-neuronal cells converge on pathways that ultimately influence neuronal function, such as genes related to synaptic pruning and activity-dependent ion channels. For example, voltage gated ion channels, which might help non-neuronal cells sense neuronal activity, are also modulated in multiple cell types. These highlight how many brain cell types are involved in sensing and regulating neural activity, and suggest neural circuit dysfunction in AD is likely the consequence of multi-cellular signaling cascades.
Several studies highlight how AD neurons modulate stress related genes8,12, particularly genes related to chaperone mediated protein folding7–9,13,14 (e.g., DNAJA1). Altered genes in AD neurons also relate to mitochondrial translocase, glucose sensing, and glycolysis (e.g., SLC2A3, which encodes a glucose transporter enriched at synaptic terminals)8,9. Synaptic mitochondria are critical for sustained synaptic efficacy15,16, and transcriptional profiles provide insight into metabolic programs disrupted in AD neurons.
Defining genetic signatures of neurons selectively vulnerable to dysfunction may reveal the molecular logic governing AD degeneration (Box 2). Tau aggregates form neurofibrillary tangles within some neurons, a canonical pathological hallmark of AD closely associated with neuronal loss. Fluorescence activated cell sorting (FACS) based on neurons with neurofibrillary tangles reveal synaptic genes are dysregulated in tangle-bearing neurons compared to non-tangle-bearing neurons from AD individuals12. A separate snRNA-seq study revealed BAG3, a master regulator for proteotoxicity-induced signaling, regulates tau homeostasis17. Reducing BAG3 in primary cortical neurons led to tau accumulation, and BAG3 overexpression attenuated tau pathology17, suggesting selective vulnerability related to tau metabolism may be governed in part by BAG3. Similarly, the transcription factor RORB is thought to mark a population of tangle-burdened neurons that modulate genes related to synaptic proteins and neurotransmitter receptors18. Thus, RORB may be a marker for selectively vulnerable neurons18. Conversely, some neurons may be preferentially resilient to AD: a subset of individuals harbor extensive amyloid and tau pathology but do not exhibit dementia19 and therefore offer a tractable opportunity to interrogate the molecular basis of resilience to cognitive decline. MEF2C is upregulated in excitatory neurons in individuals with high AD pathology and normal cognition compared to age-matched individuals with high pathology and low cognition19. Impairing Mef2 in mice causes neuronal hyperexcitability, and Mef2 overexpression in Tau P301S mice rescued tauopathy-induced hyerpexcitability19. These findings suggest properties related to neuronal firing may explain individual neuronal susceptibility or resilience to degeneration.
Box 2. Cellular identity, cell states, and disease-associated molecular programs.
Classifying cell types requires multiple levels of characterization to correlate transcriptional and epigenomic profiles of cellular identity with functional and developmental state138. In the study of AD, single cell genomics have provided a great deal of insight into transcriptional alterations in major brain cell types, but how these transcriptional profiles correlate with functional state in AD progression are incompletely understood. Sub-clustering analysis within major cell types often reveals transcriptionally distinct subtypes of cells within the AD brain that may be associated with AD pathology and cognitive dysfunction. Given the functional plasticity of cells, these sub-clusters may represent functionally distinct cell states that emerge in disease. Nevertheless, even neurons not burdened by tau pathology18 and microglia not directly phagocytosing amyloid plaque50 modulate genes related to cellular stress and protein folding in AD. These findings highlight the heterogeneity of disease-related molecular programs across and within cell types.
Disease-related molecular signatures may reflect cells sampling a distinct space of their cell identity, and the plasticity of these cell states may reflect the capacity for disease-modifying treatments to return cells to a homeostatic equilibrium. For example, the transcription factor PU.1 is thought to govern aspects of microglial state in AD77,79, and modulating PU.1 can alter microglial gene expression139 in a potentially therapeutically useful manner. Similarly, genes related to neuronal hyperactivity may relate to vulnerability or resilience to degeneration, and targeting neuronal hyperactivity using levetiracetam, an anti-epileptic agent which reduces hyperactivity and improves memory in APP/PS1 mice140, may provide benefit for some AD patients141. Other efforts have identified molecular regulators of disease-associated states in astrocytes77,86 and oligodendrocytes77. Further defining the molecular regulators of cellular states—and interrogating how therapeutic intervention might target state-related pathways—may advance the treatment of AD.
Single cell transcriptomics reveal downregulation of cell-type specific marker genes for some cell types. For example, microglia, interneurons, and endothelial cells downregulate canonical markers in AD. Impairment of signaling pathways directly encoded by these marker genes may influence neuronal circuits (such as reduced fractalkine and purine signaling in microglia67,68, or reduced neuropeptide signaling in interneurons46). More broadly, loss of marker genes may indicate the very transcriptional programs governing the identity of distinct cell types is lost in AD, potentially leading to cellular senescence. Accordingly, senolytic treatment, which ameliorates pathology and cognitive impairment in APP/PS1 mice108 may represent a therapeutic strategy for the treatment of AD142.
Risk variants from genome wide-association studies (GWAS) further highlight differential risk to AD (Box 3). The ɛ4 allele of the apolipoprotein E gene (APOE) is considered the most highly validated genetic risk factor for sporadic late-onset AD. While APOE is predominantly expressed by astrocytes and microglia8, variable expression of APOE among individual neurons suggests some neurons express relatively higher levels of APOE20. Increased APOE levels were associated with cellular stress and death20, suggesting neuronal APOE and MHC-I signaling might be an important factor driving differential vulnerability to AD neurodegeneration. Apolipoproteins are critical for lipid transport, and other lipid related genes modulated in AD neurons include those related to cholesterol transport (e.g., NPC18,9, which encodes a lysosomal cholesterol transporter whose loss of function is associated with neuronal death21) and lipid signaling (e.g., LAMTOR5, related to endosomal/lysosomal transport7–9,14). The findings highlight complex lipid-related signaling networks disrupted in AD neurons.
Box 3. Single cell genetics, GWAS risk variants, and AD biomarkers.
Genome-wide association studies (GWAS) have highlighted genetic variants associated with sporadic, late-onset Alzheimer’s disease47,143,144. Genomic information from single cells facilitates biological insight captured from GWAS studies in several ways:
Single cell approaches help define which cell types highly express AD risk genes. For example, compared to other brain cell types, microglia are thought to express relatively higher levels of the risk variants TREM2 and CD3358,145. The observation that expression of AD risk genes are enriched in immune cells has contributed to the hypothesis that immune mechanisms may play causal roles in AD47.
Distinct cell types differentially modulate the expression of AD risk genes144. For example, the risk gene CNTNAP2, a neuroxin family gene involved in cell adhesion, is upregulated in late-pathology AD neurons8 but downregulated in astrocytes7,8; BIN1 is upregulated only in AD excitatory neurons7,8; APOE is upregulated in microglia7–9 and vulnerable populations of neurons20 but downregulated in astrocytes7–9. The gene products of these risk variants likely affect many cellular processes in AD. Manipulations of risk variants within certain cells types, such as selective removal of neuronal APOE4146 or astrocytic APOE488 in mice, highlight how cell type specific expression of risk gene variants governs signaling alterations in AD.
Mutations in one gene can modify the expression and function of many other genes across many cell types. For example, individuals harboring APOE4123 and TREM2 R47H9 mutations carry differential expression for many genes across many cell types. Single genetic variants, such as 5XFAD mice lacking Ccr7147 or Trem29, and APOE4-knock in mice88,148, generate widespread transcriptional variations in many cell types. Collectively, these findings underscore how single genetic perturbations vastly alter many cell types. Recent in vitro studies enable the perturbation of AD risk factors in distinct cell types, such as APOE genotype in distinct vascular123 and glial149 cell types, which further illustrate how risk variants affect distinct cell types. Single cell studies also suggest highlight expression of genes upstream of GWAS genes. For example, AD astrocytes upregulate of TFEB, which is upstream of ten GWAS loci, and which might control astrocytic disease-state transition7.
Single cell genomics also generate insight into cell-type specific contributions to AD biomarkers. For example, CHI3L1, which encodes chitinase-like protein, a candidate cerebrospinal fluid (CSF) biomarker for preclinical AD, is upregulated in AD microglia9. Similarly, AD microglia upregulate SORL19 and A2M9, which encode CSF biomarkers, and downregulate FTH19, a serum marker. These findings highlight the potentially causal roles of immune mechanisms in AD. AD neurons downregulate NEFL and BDNF7,8, which encode plasma biomarkers. Glial fibrillary acidic protein (GFAP), another biomarker, is upregulated in astrocytes from AD patients7,8 and 5XFAD mice50. These findings showcase how single cell approaches define cell states potentially responsible for AD biomarkers, which may facilitate efforts to define cellular substrates driving distinct clinically distinct subtypes and pathological stages of AD.
Collectively, the overlap between risk variants and single cell genomics provide insight into cell types and signaling pathways that may govern AD progression. Further defining how genetic risk interface with non-genetic factors my further reveal signaling nodes governing AD pathogenesis, potentially informing new therapeutic approaches.
Neuronal DNA damage.
DNA damage is well known to occur in AD neurons22, and snRNA-seq reveals AD neurons modulate genes related to DNA repair enzymes7–9,14 (e.g., XRCC6, which is involved in DNA repair initiation, is downregulated in AD neurons8). Moreover, single cell whole genome amplification sequencing suggests DNA damage in neurons is elevated in neurodegeneration23. High DNA damage in human neurons is enriched in differentially expressed genes of AD individuals24, potentially suggesting dysregulated gene expression in AD neurons may be related to impaired DNA repair. DNA damage may be part of a switch in cellular states associated with metabolic stress (e.g., upregulation of the DNA damage inducible transcript DDIT38,9 may be associated with broader program in cellular stress). Neurons burned with DNA damage also active inflammatory signaling in neurodegeneration25. DNA damage is known to occur at neuronal enhancers and promoters26, particularly during learning27, and is required for learning-related immediate early gene expression27. Disentangling learning-related DNA damage and aging-related DNA damage in AD may lead to new insights into the progression of neural circuit disruption in AD.
Neurogenesis.
Single cell genomics have generated molecular insight into the progression of new neurons in the adult brain, including the molecular definition of stem cells and their terminal fates28–33, although common markers for neural progenitors may complicate efforts to define human neurogenesis34. Mutations in PSEN1 may alter the stem cell niche35 and some neural stem cells may be particularly affected by amyloid toxicity36, so AD risk genes could potentially alter neurogenesis in the context of AD.
Interneurons.
Interneurons critically sculpt synchronized patterns of neural activity, and single cell transcriptomics provide molecular definitions of interneuron subtypes from mouse3,37,38 and human39,40 brain, which reflect their functional repertoire41. Interneurons share transcriptional changes with excitatory neurons in AD, including genes related to metabolic stress, ion transport, DNA damage, perineuronal net assembly, and ErbB signaling7,8,13. Despite the well-documented functional perturbations of interneuron-dependent neuronal rhythms in amyloid mouse models42 and mice with targeted replacement of mouse Apoe with human APOE443, transcriptional alterations within distinct interneuron subtypes have evaded comprehensive characterization in AD7–9,13,14. Downregulated genes in AD interneurons include marker genes for canonical interneuron subtypes, including SST, PVALB, and VIP7–9. Interneuron neuropeptides are thought to regulate inhibitory circuits44,45 and neurovascular coupling46, so impaired interneuron peptide signaling in AD may broadly impair neuronal signaling in dementia. The loss of canonical markers might also suggest interneurons lose core aspects of their transcriptional identity, which may reflect global loss of cellular function (Box 2).
Microglia.
GWAS findings highlight AD-associated variants in immune related genes (e.g., TREM2, CD33, HLA-DR)47; accordingly, microglia have received a great deal of attention by single cell profiling48 (Figure 4).
Figure 4: Molecular programs adopted in AD microglia revealed by single cell genomics.
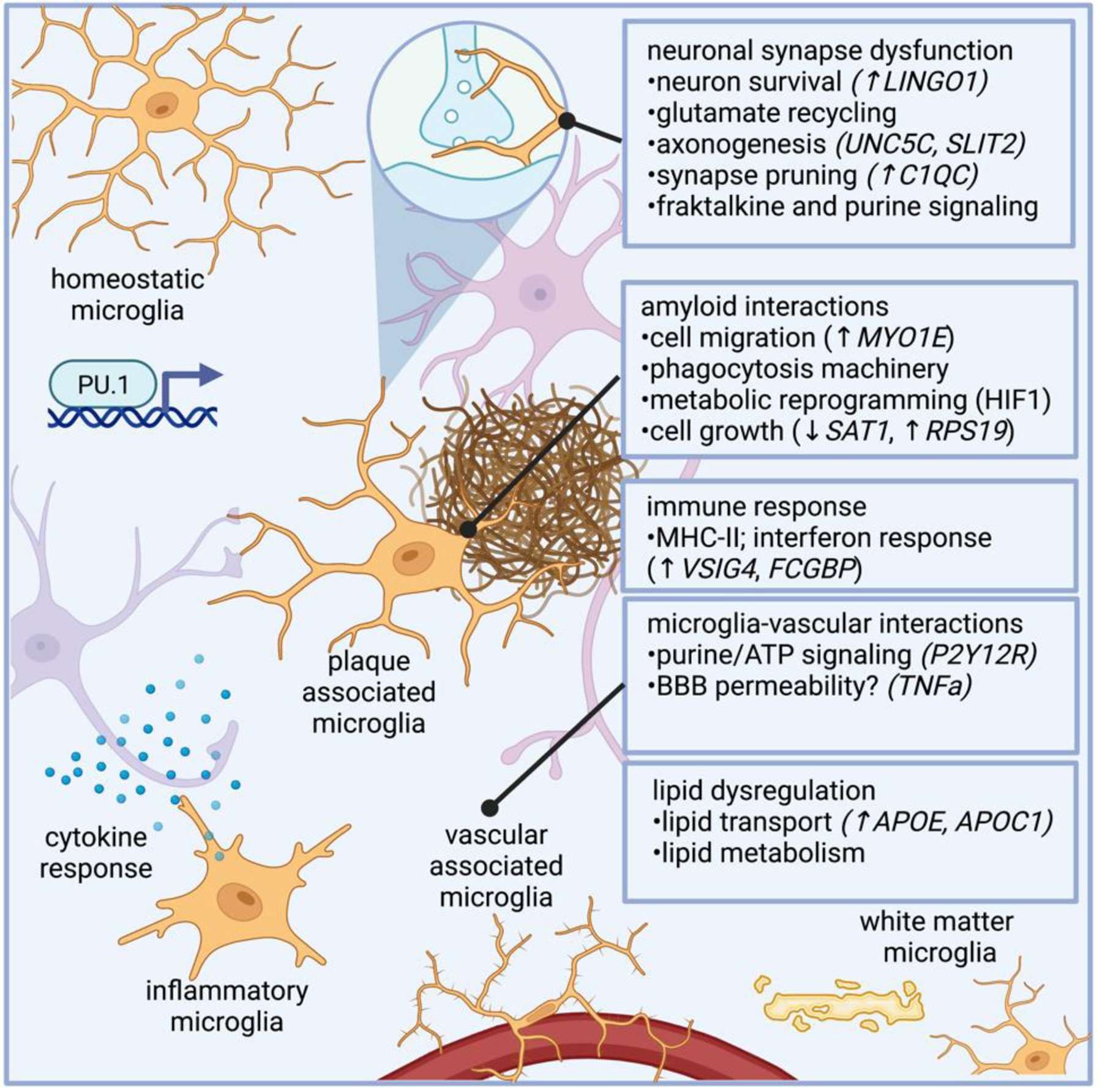
Microglia dysfunction in AD modulate genes related to synapse function, phagocytosis, and immune response. Microglia regulate genes involved in myelination, such as LINGO1, a negative regulator of myelination, as well as genes involved in axonogenesis (e.g., UNC5C and SLTI2). AD microglia also modulate complement related genes, such as C1QC, which regulate synaptic pruning. Microglial harbor properties associated with phagocytosis, and microglial response to amyloid has been well characterized, and genes in AD microglia potentially related to amyloid response include those related to microglial plaque clustering phenotypes, such as cell migration (e.g., MYO1E, which encodes a gene related to myosin), as well as genes involved in metabolic reprogramming and cell growth (e.g., SAT1, which encodes an acetyltransferase, and RPS19, a ribosomal subunit). As the innate immune cells of the brain, microglia are intimately involved in immune response, and several differentially expressed genes in AD microglia are involved in immune response, such as VSIG4 and FCGBP, genes involved in immunoglobulin response. TREM2 is a lipid receptor that is thought to govern microglia transitions to disease-associated states9,49. Subtypes of microglia that regulate plaques are marked by Hif1a in 5XFAD mice, which is associated with metabolic reprogramming in human AD microglia.
Plaque-associated microglia.
One of the first single cell transcriptomic studies profiling microglia revealed a distinct subtype in 5XFAD mice, a popular amyloid model harboring five mutations associated with familial AD. Microglia from the hippocampi of 5XFAD mice were dubbed “disease-associated microglia” (DAM)49. Microglia harboring DAM transcriptional signatures were revealed to localize to amyloid plaques. One study elegantly sorted microglia from 5XFAD mice according to their levels of plaque labeled with methoxy-X04, a fluorescent probe that binds to fibrillar β-sheets of amyloid50. Interestingly, methoxy-labeled microglia increased expression of hypoxia-inducible factor Hif1a50, which may be associated with a broad switch in metabolic programs in human AD microglia associated with HIF1 signaling51. Studies in 5XFAD mice indicate plaque-associated microglia upregulate genes related to cell surface receptors (Trem2, Tyrobp, Clec7a), integrins (Itgax), and immune-related pathways (Csf1, Ccl6)9,49,50,52. This transcriptional signature is partly recapitulated in CKp2552, APP-PS153, APPNL-G-F 54, Tau P301S, and P301L mice53 (although genetic background may drive substantial microglial diversity55). While additional studies are needed to clarify the extent to which mice recapitulate human AD microglia, several studies indicate human AD microglia modulate genes related to cell migration and phagocytosis7–9,14. Collectively, these findings highlight the transcriptional basis for microglia to cluster around plaques and phagocytose debris7–9,14.
Lipid metabolism in AD microglia.
TREM2 acts in microglia as a sensor for a wide array of lipids, and the R47H variant of TREM2 is associated with increased risk of AD and enhanced Akt signaling in microglia56. In adipose tissue of non-AD individuals, TREM2 drives a gene expression program involved in phagocytosis, lipid catabolism, and energy metabolism57, suggesting TREM2 may broadly regulate lipid-related metabolic programs in macrophages. In accord with experimental studies highlighting the importance of lipid sensing and metabolism in microglia, microglial lipid metabolism pathways are broadly disrupted in AD7–9. Interestingly, the DAM phenotype in 5XFAD mice is dependent on Trem29,49. Several findings in mice suggest manipulating microglial lipid pathways regulate pathology. For example, knocking down mouse Apoe conferred neuroprotection in APP/PS1 mice58, and overexpressing low-density lipoprotein receptor, which mediates lipid clearance, alleviates pathology when overexpressed in Tau P301S mice and shifts microglia transcriptional signatures to a homeostatic state59. Lipid metabolism is critical for microglia to rapidly remodel plasma membrane for local brain surveillance and regulate neural activity, and these findings suggest targeting microglial lipid-related pathways may alleviate AD pathology.
Microglial perturbations in neuronal support.
It is increasingly relevant how microglia regulate neuronal circuits60,61. Microglia regulate neural computations in part by complement-mediated synapse elimination62, fractalkine signaling63, and purine sensing60, and, accordingly, several studies indicate AD microglia modulate genes related to complement (C1Q7,8), fractalkine receptors (e.g., CX3CR18,14), and purine receptors (e.g., P2Y12R7). Thus, neuronal dysfunction in AD may arise in part due in part to alterations in microglial capacity to sense and control neuronal activity. Perturbations in microglial metabolic state, related in part to cellular stress and glycolytic shifts, may converge on signaling pathways that impair microglial capacity to regulate neuronal activity (for example, a model using CRISPR-edited induced pluripotent stem cells found lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal network activity64). Microglia state may also be associated with distinct GABAergic circuits65 and pyramidal neuron subtypes66, so future work defining microglial-neuronal crosstalk may reveal the molecular logic of microglia governing neuronal dysfunction in AD.
Vascular function of microglia and macrophages.
Capillary-associated microglia are thought to regulate blood flow via purinergic signaling67,68. Microglia and perivascular macrophages are thought to harbor distinct ontogeny69, and subpopulation of cells marked by high Mrc1, a marker of peripheral macrophages70, may represent a transcriptionally distinct population of vascular-associated macrophages71,72. Signaling between vascular-associated microglia/macrophages and vascular cells may influence neurovascular function in AD. For example, secreted factors from AD microglial might regulate the integrity of endothelial tight junctions. Chemokines secreted by microglia may influence the inflammatory state of endothelial walls (which in turn can lead to neutrophil adhesions and capillary stall-induced blood flow reductions73). Future studies may further define how vascular-associated macrophages influence vascular permeability and neurovascular coupling in AD.
White matter associated microglia.
Microglia in white matter have a distinct transcriptional state compared to grey matter microglia74 and share genes associated with disease-associated microglia (e.g., upregulation of APOE, complement, and lipid metabolism related genes, and downregulation of homeostatic markers)75. Similar transcriptional signatures of white matter microglia are present in 5XFAD and APPNL-G-F mice, and TREM2 knockout reduces the presence of white matter microglia75. The dysfunction of microglia in white matter may relate to repairing and phagocytosing myelin76. Further defining white matter associated microglia may reveal interventions to promote the health of myelinated axons.
State transitions of microglia inflammation.
Morabito et al. performed snATAC-seq and snRNA-seq to define AD-associated gene-regulatory programs at the epigenomic and transcriptomic levels77. Some microglia in AD had more open binding sites for SPI1, which encodes PU.1, a master regulator of myeloid cell differentiation. PU.1 may act as a transcriptional repressor in late-stage AD microglia77, and a complex network of transcription factors in AD microglia (e.g., ELF5, ETS1, ETV5, SPIC)77 may be involved in microglia state transitions in AD77,78. Indeed, PU.1 expression levels79 and PU.1-dependent transcriptional control53,80 are thought to critical regulate microglial function, such as microglial clearance of amyloid. Single cell transcriptomics and CRISPRi/a screens might further reveal gene programs modulating microglial state81.
Astrocytes.
Astrocytes are involved in neuronal trophic support, extracellular ion homeostasis, and brain fluid balance. Single cell profiling reveals the molecular heterogeneity of astrocytes82–85 and astrocytic perturbations in AD (Figure 5).
Figure 5. Molecular programs adopted in AD astrocytes revealed by single cell genomics.
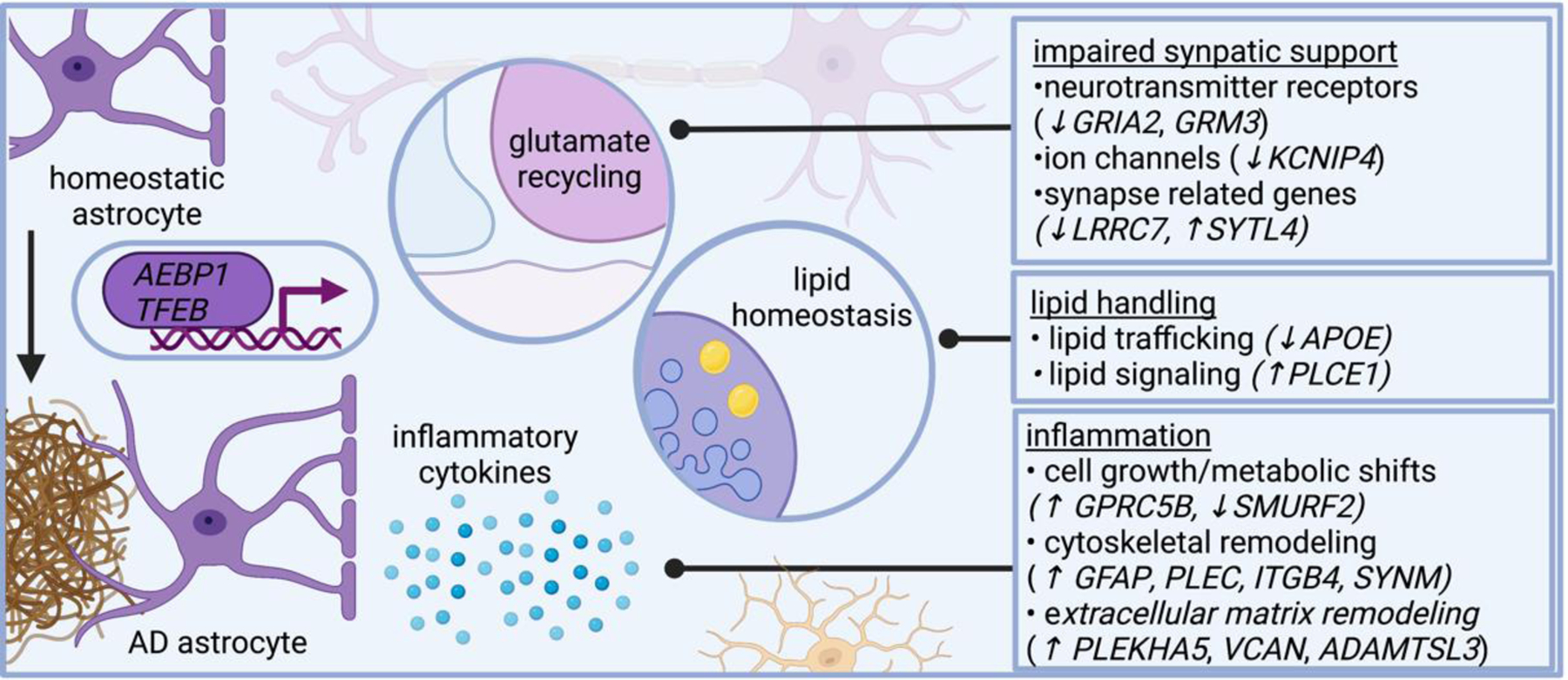
Several lines of evidence suggest astrocytes in AD become inflammatory and impair neural circuit function, including plaque-associated barriers, and modulating lipid-related signaling networks. Single cell genomics shed additional insight on these pathways and reveals astrocytes in AD modulate genes related to neurotransmitter recycling, inflammatory response, and lipid metabolism. AD dysregulates astrocytic genes involved in neurotransmitter receptors (such as GRIA2 and GRM3, which encode subunits of glutamate receptors), ion channels (such as KCNIP4, which encodes a protein that interacts with voltage-gated potassium channels), and even genes involved in synapses (such as LRRC7, which encodes a component of the post synaptic density of excitatory synapses, and SYTL4, which encodes a synaptotagmin). AD astrocytes also modulate genes involved in lipid metabolism, including APOE and PLCE1, which encodes a phospholipase. Several astrocytic genes differentially expressed in AD relate to cytoskeletal remodeling, including GFAP (which encodes an intermediate filament), PLEC (which encodes plectin, a protein that interacts with intermediate filaments), SYNM (which encodes another intermediate filament), and ITGB4 (which encodes an integrin). AD astrocytes modify genes involved in cell growth, such as SMURF2 (a member of the SMAD family important for cell growth). Collectively, these transcriptional changes highlight signaling pathways altered in AD astrocytes.
Astrocyte metabolism.
Astrocytes are a central driver of energy homeostasis in the brain. Several snRNA-seq from human AD cortex reveal AD astrocytes alter genes related to cellular stress and metabolic reprogramming genes related to cell stress (e.g., CIRBP, CABLES, CSRP1)7–9, as well as many genes related to the structural remodeling of the astrocytic cytoskeleton (e.g., GFAP)7–9 and extracellular matrix (e.g., the versican gene VCAN and integrin genes ITGB8 and ITGB4)7–9. Upregulation of GFAP in AD astrocytes is also observed cross several snRNA-seq datasets from AD patients7–9 and 5XFAD mouse hippocampus86, which may contribute to clinically relevant biomarkers (Box 3). Given that astrocytes critically respond to and regulate the inflammatory state of the brain following injury and neurodegeneration, potentially in a region-specific manner84, these findings highlight transcriptional underpinnings of astrocytic metabolic reprogramming in AD.
Many dysregulated pathways in AD astrocytes converge on lipid signaling (e.g., PLCE1, which encodes a phospholipase, and apolipoprotein family genes)7–9. Genes related to lipid synthesis and transport (e.g., LDLR)8 are perturbed in AD astrocytes7–9. Metabolic reprogramming in AD astrocytes may contribute to impaired capacity to regulate neuronal circuits. One study showed that astrocytes break down cytotoxic fatty acids secreted by hyperactive neurons87. Lipid associated pathways are dysregulated in disease-associated astrocytes from 5XFAD mouse hippocampus, including those in cholesterol pathways86, recapitulating aspects of AD astrocyte dysfunction in humans related to transport and storage in lipid droplets, and detoxification of reactive oxygen species (e.g., SOD2)9. Removing astrocytic APOE4 in mice decreases disease-associated transcriptional signatures across multiple cell types and protects against tau-mediated neurodegeneration88. These findings suggest lipid-related signaling networks in astrocytes may represent a core perturbation in AD.
Dysfunctional synaptic communication in AD astrocytes.
Astrocytes facilitate neurotransmitter shuttling and synaptic trophic support. Several snRNA-seq from human patients reveal AD astrocytes alter genes related to glutamate receptor subunits (e.g., GRIA2, GRM3, GRID2)7,8,13,86. Gap junctions and potassium handling in astrocytes critically support neural function, and several snRNA-seq studies from human AD patients reveal genes related to gap junctions (e.g., upregulation of GJA1, which encodes the major astrocytic gap junction component connexin 43)8 and ion transporters (e.g., downregulation of KCNIP4)7–9 are dysregulated in AD astrocytes. Collectively, these transcriptional changes highlight molecular pathways dysregulated in astrocytes governing extracellular ion homeostasis and neuronal function.
To gain insight into the molecular basis driving transitions between astrocytic cell states, several groups have analyzed transcription factor expression profiles. The transcription factor AEBP1, a coactivator of the master immune signaling regulator NFκB, may regulate AD-related state transitions in astrocytes7. The master lysosomal regulator TFEB was upregulated in astrocytes in AD patients7,8, and given the importance of TFEB to lysosomal pathways89, dysfunction in intracellular lipid related processes may represent a key driver of AD related dysfunction7. Several genes overlap between AD microglia and astrocytes (e.g., CTSB, CTSD, and APOE)90, potentially suggesting a common glial inflammatory milieu may emerge in AD including weakened metabolic coordination with neurons9. The complex, bidirectional inflammatory milieu generated by astrocytes and microglia (and shared in part likely is shared by oligodendrocyte related cells), collectively may regulate neuronal function. Dysfunction in secreted cytokines and lipid related trafficking may represent a functionally redundant glial response to AD pathology. For example, the metabolic shift in microglia highlighted above may be shared in part by AD astrocytes, which modulate genes involved in cell growth and signal transduction. These changes collectively highlight common lipid- and immune-related signaling pathways shared across major cell types in AD.
Oligodendrocytes.
Single cell transcriptomics from mouse91 and human brain92,93 reflect the highly heterogeneous nature of oligodendrocytes. Molecular programs perturbed in AD oligodendrocytes reflect alterations in the diverse functional repertoire of these cells, including myelination, sensing neural activity, and immune function7–9,13,14,18 (Figure 6).
Figure 6. Molecular programs adopted in AD oligodendroglia revealed by single cell genomics.
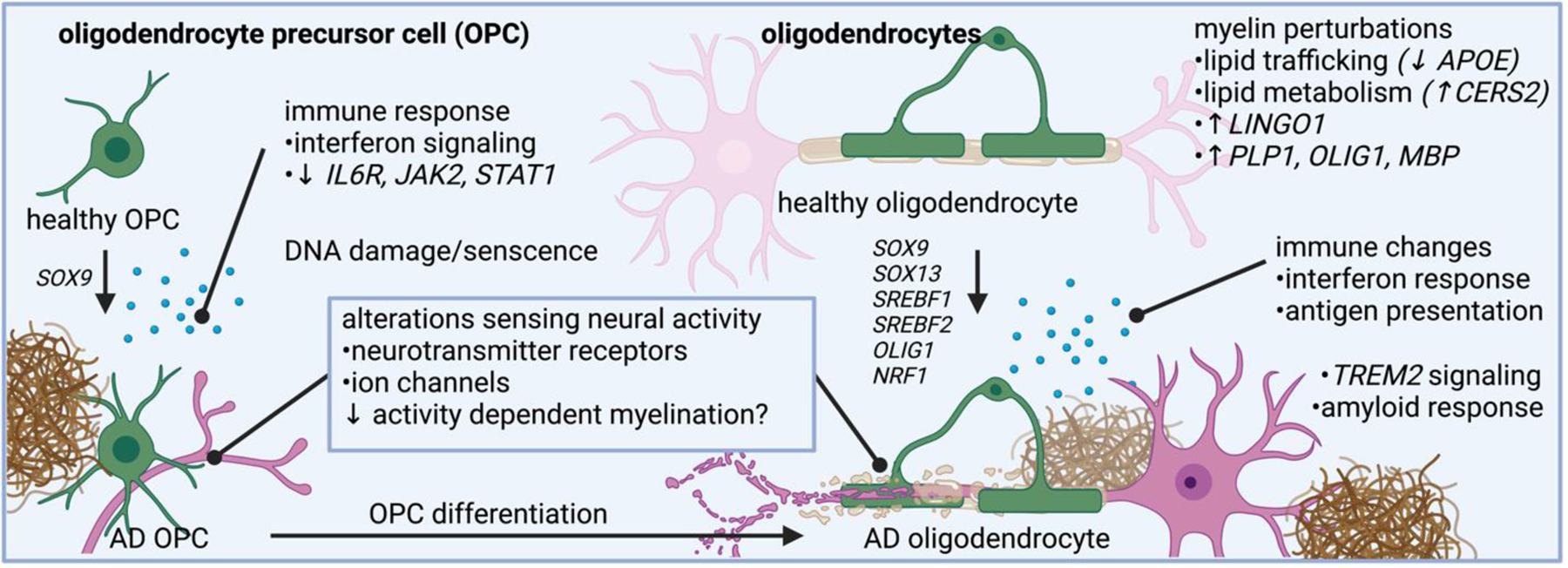
Oligodendrocytes in AD have altered pathways related to myelin synthesis, lipid trafficking, lipid metabolism, and immune related changes. Oligodendrocyte precursor cells also changes expression of genes related to neurotransmitter sensing and immune response.
Deficits in white matter volume and myelination rates are well appreciated in AD94. Many myelin related genes are perturbed in AD oligodendrocytes, such as upregulation of LINGO17,8 (a negative regulator of myelination, which is also upregulated in AD excitatory neurons) and downregulation of CNTN2 and OPALIN (genes thought to regulate axon homeostasis)14. Genes involved in myelin production are altered in oligodendrocytes early in AD, including genes for enzymes related to fundamental cellular building blocks (e.g., CERS28, CARNS17–9, and QDPR7–9, which encode genes important for the processing of molecules important for white matter integrity) and myelination itself (such as PLP1, OLIG1, and MBP)8. Several studies from human brain also highlight alterations in genes for lipid transport, including genes related to apolipoproteins and lipid receptors (e.g., ABCA6, LRP1, LDLRAD4)7–9, lysosome function (e.g., LAMP1)8, and solute carriers (e.g., SLC38A2, SLC5A11)7–9. These findings highlight cellular pathways related to biosynthetic processes and lipid handling converge on myelin synthesis, and may explain in part how oligodendrocyte dysfunction contributes to deficits in the structure and plasticity of myelination in AD.
Myelination is a highly regulated and dynamic process that may be specific for anatomically distinct neural circuits95,96. The plasticity of myelin may relate to the capacity of oligodendrocytes to sense neural activity, such as the sensory experience-dependent myelination remodeling on parvalbumin inhibitory interneurons but not on excitatory callosal projection neurons97. snRNA-seq suggests AD oligodendrocytes modulate genes related to ion channels (e.g., KCNH8)7–9 and glutamate receptor subunits (e.g., GRM3, GRID2)7,8,13,14. These findings suggest AD oligodendrocytes may exhibit impaired capacity to sense and regulate neural activity. Memory preservation is thought to require new myelin formation98, so impaired oligodendrocyte capacity to adaptively monitor neural activity and facilitate myelin remodeling may govern cognitive decline in AD.
Oligodendroglial immune and vascular functions.
Studies in mice reveal antigen processing and presentation capabilities of oligodendrocytes92,99, and AD oligodendrocytes modulate genes related to MHC-I and MHC-II8. Many other immune pathways are perturbed in AD, including interferon response and inflammation (e.g., CD63 and IRF2, which are involved in the activation of immune cells)13. Oligodendrocytes display major transcriptomic alterations in 5XFAD mice100. Reactive oligodendrocytes marked by Serpina3n+ C4b+ in plaque bearing regions of 5XFAD mice are thought to emerge in a TREM2-dependent manner9. Oligodendrocytes may also respond to101 and participate in102 blood-brain barrier integrity by secreting growth factors that regulate vascular cell function (e.g., PDGF signaling, which is thought to regulate vascular function, is dysregulated AD oligodendrocytes8). Oligodendrocytes also support extracellular matrix remodeling, a critical factor in remyelination, and AD oligodendrocytes modulate genes related to collagens, laminins, and chondrotins8. Some of these changes may reflect injury related to white matter injury, a common clinical finding in AD. Collectively, these findings highlight heterogeneous molecular programs adopted by AD oligodendrocytes.
Oligodendrocyte precursor cells (OPCs).
OPCs are distributed throughout grey and white matter. snRNA-seq reveals heterogeneous subtypes of OPCs in mouse91,103,104 and in the human cortex105,106. OPCs regulate neural activity and harbor immune and vascular related function, and snRNA-seq reflects alterations in OPC state perturbations in these processes. For example, several snRNA-seq studies from human subjects indicate AD OPCs downregulate genes for ion channels (e.g., KCNIP1, CACNA1D)7,8, and may alter genes encoding neurotransmitter receptors (e.g., GALR1)7,8, glutamate receptors (e.g., GRIA2)9, and synaptic genes (e.g., SNAP25)8. Like oligodendrocytes, OPCs are thought to dynamically sense and modulate neural activity107. Transcriptional findings highlighting OPC modulation of genes related to voltage gated ion channels suggest OPCs may harbor altered capacity to sense neural networks, which may explain in part dysfunction in adaptive myelination and neuronal integrity in AD. OPCs modulate immune related genes (e.g., IFIT1)13, highlighting a potential role for inflammation and immune mechanisms in OPC mediated dysfunction in AD. Oligodendrocyte differentiation is partly dependent on OLIG1, which is upregulated in AD OPCs8, supporting a role for alterations in the dynamic reprogramming of OPC fate that may be responsible for oligodendrocyte alterations in AD. OPCs with DNA damage may alter their differentiation programs, and, notably, amyloid itself is thought to induce senescence in OPCs, which can be reversed by senolytic treatment108.
Vascular cells.
Alterations in vascular function critical contribute to brain homeostasis, and reduced blood flow may emerge early in AD109. snRNA-seq has provided molecular definitions of functionally distinct vascular cells in mouse71,110–113 and human brain114–117. These studies suggest vascular cells in AD have altered immune signaling, neurovascular coupling, and permeability14,116.
Brain endothelial cells control the movement of ions, molecules, and cells between the blood and the parenchyma, a constellation of properties collectively referred to as the blood brain barrier (BBB)118. snRNA-seq from AD patients reveal transcriptional perturbations in many BBB processes, including alterations in tight junctions (e.g., CLDN5), solute transporters (e.g., SLC2A1), and adhesion molecules14,116. In endothelial cells of APP/PS1 mice, mNat1, a regulator of insulin sensitivity, was found to govern endothelial cell necroptosis119.
Pericytes reside in the basement membrane and wrap around capillaries. Pericytes express genes involved in actomyosin contraction71, consistent with functional evidence suggesting pericyte contractility controls vascular dynamics (e.g., by controlling blood flow at capillary junctions120 and regulating basal capillary diameters121), which may contribute to AD related hypoperfusion122. Human hippocampal pericytes from APOE4 carriers elevate expression of NFAT, and modulating NFAT through calcineurin signaling reduces APOE expression and ameliorates amyloid deposition NFAT123. Mice with targeted expression of APOE4 in vascular mural cells modulate the transcriptome of many cells, particularly astrocytes124. Collectively, these studies provide transcriptomic evidence for observations related to pericyte dysfunction in vascular dysfunction in AD related to neurovascular coupling and BBB integrity.
Future Directions
Single cell profiling reveals cell-type specific alterations in AD, and highlights core signaling pathways that are dysfunctional across cell types. Combining genetic information with other metrics of cellular functions will enhance our understanding of AD alterations in distinct cell types (Figure 7). For example, preserving anatomical information using spatial transcriptomics may generate insight into the anatomical progression of AD and define distinct neuronal projections susceptible to AD dysfunction. Expanding patient samples to resolve contributions from sex, race/ethnicity125,126, genetic risk variants, and factors such as education, sleep, and exercise habits, will further enhance our understanding of the cellular phenotypes driving memory decline in AD patients. Existing datasets must be integrated and made easily shareable to expand the heterogeneity of AD response and to define concordant gene expression changes across studies. Further defining how human molecular signatures are recapitulated in mouse models and cell culture preparations will lead to new experimental opportunities to dissect disease mechanisms.
Figure 7. Emerging methods to interrogate single cell profiles in AD.
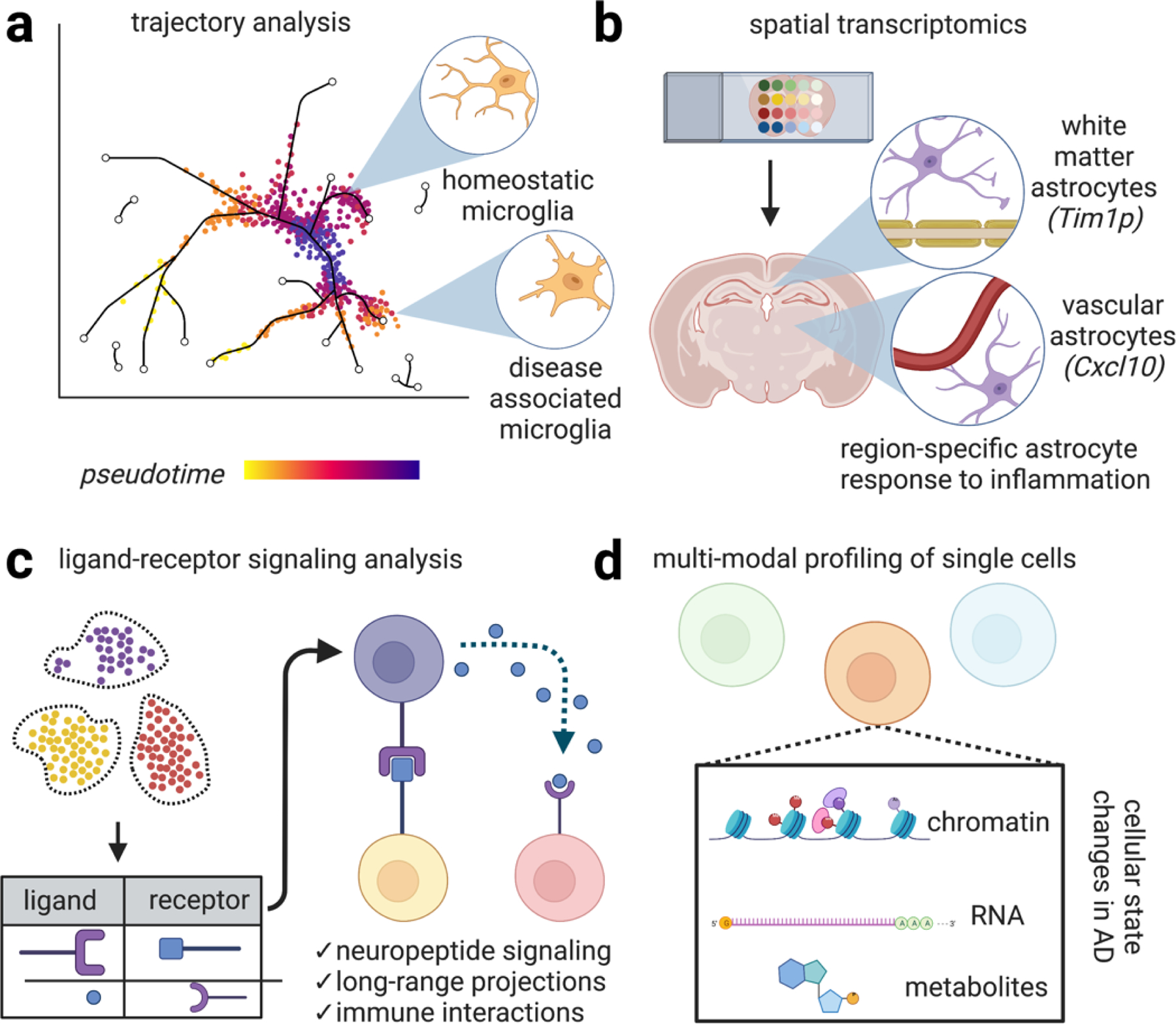
Emerging methods in single cell profiling will enhance our understanding of the distinct cellular signaling networks perturbed in AD. (a) Genetic analysis of single cells potentially enables the construction of dynamical cellular models according to “pseudotime,” a quantitative measure of biological progression through a cellular process. Applying these models to AD potentially enables the trajectory of distinct cell types adopting new transcriptional states relating to disease progression, progression of microglia from homeostatic states to disease-associated states52. (b) Spatial transcriptomics is an umbrella term referring to techniques that combine mRNA readouts with spatial information. For example, one study revealed reactive subsets of astrocytes occupy distinct anatomical locations, such one astrocyte population lining white matter tracts that express the matrix metalloprotease inhibitor Timp1, which has been shown to be involved in amyloid response and has been shown to drive oligodendrocyte production and myelination and another astrocyte population marked by the cytokine Cxcl10+ adjacent to blood vessels84. Preserving spatial information while performing single cell profiling will likely further enhance our understanding of the molecular mechanisms driving AD. (c) Ligand-receptor signaling involves predicting signaling interactions based on ligand/receptor databases. This analysis has revealed, for example, dense peptidergic intracortical signaling networks44. Expanding our understanding of cellular physical and signaling interactions between brain cells with enhanced methods of examining these interaction networks will undoubtedly yield important insight into the progression of signaling networks in AD. (d) Multi-modal profiling of single cells to simultaneously chromatin, RNA, and potentially metabolites are emerging methods. These studies have revealed additional levels of regulation in distinct cell types in AD77. Combined with enhanced sequencing depth, these methods will generate a richer portrait of cellular function in neurodegeneration.
Further characterizing distinct neuronal microcircuits and cell types that become dysfunctional in AD—and defining which cell states contribute to CSF and plasma biomarkers—may lead to new frameworks to define cellular substrates of AD progression. By identifying vulnerable cell types and the molecular programs that give rise to them, therapeutic interventions might reverse aberrant cellular trajectories. While many transcriptional alterations cell type specific, these changes ultimately might converge on shared signaling pathways across cell types that might represent targets for new therapeutic strategies.
Conclusion
Single cell profiling facilitates a nuanced portrait of the diverse cellular processes perturbed in the AD brain. These varied molecular programs help explain the divergence between healthy aging and cognitive decline, and highlight cell-type specific molecular programs involved in AD. Core signaling modules are disrupted across multiple cell types, and manipulating disrupted cellular states will pave the way for new therapeutic opportunities.
Acknowledgements
We are grateful to Hansruedi Mathys, Manolis Kellis and all members of his lab, and all members of the Tsai lab for insightful discussions. We thank the following individuals for valuable discussions and helpful feedback on this manuscript: Matheus Victor, Jay Penney, Emily Niederst, Leyla Akay, Djuna von Maydell, Ping-Chieh Pao, Lorenzo Bozzelli, Adele Bubnys, Gwyneth Welch, Dong-Shin Park, and Julia Maeve Bonner. L-H.T. acknowledges NIH R01AT011460-01 and NIH R37-NS051874-2. We thank the JPB Foundation, the Belfer Neurodegeneration Consortium, the Glenn Foundation for Medical Research, the Cure Alzheimer’s Fund and the Alana Foundation. We gratefully acknowledge generous support from the following individuals and institutions: Robert A. and Renee Belfer, the Ko Hahn family, the Carol and Gene Ludwig Family Foundation, the Halis Family Foundation, Lester A. Gimpelson, the Dolby family, Jay L. and Carroll D. Miller, David B. Emmes, the Marc Haas Foundation.
All figures were created with BioRender.
Footnotes
Competing interests
The authors declare no competing interests related to this project.
References:
- 1.Yao Z et al. A transcriptomic and epigenomic cell atlas of the mouse primary motor cortex. Nature 598, 103–110 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeisel A et al. Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tasic B et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saunders A et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lake BB et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong S et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555, 524–528 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Grubman A et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci 22, 2087–2097 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Mathys H et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26, 131–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villa KL et al. Inhibitory synapses are repeatedly assembled and removed at persistent sites in vivo. Neuron 89, 756–769 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurucu H et al. Inhibitory synapse loss and accumulation of amyloid beta in inhibitory presynaptic terminals in Alzheimer’s disease. European Journal of Neurology n/a,. [DOI] [PubMed] [Google Scholar]
- 12.Otero-Garcia M et al. Molecular signatures underlying neurofibrillary tangle susceptibility in Alzheimer’s disease. Neuron 0, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davila-Velderrain J et al. Single-cell anatomical analysis of human hippocampus and entorhinal cortex uncovers early-stage molecular pathology in Alzheimer’s disease 2021.07.01.450715 10.1101/2021.07.01.450715v1 (2021) doi:. [DOI] [Google Scholar]
- 14.Lau S-F, Cao H, Fu AKY & Ip NY Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. PNAS 117, 25800–25809 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li S & Sheng Z-H Energy matters: presynaptic metabolism and the maintenance of synaptic transmission. Nat Rev Neurosci 1–19 (2021) doi: 10.1038/s41583-021-00535-8. [DOI] [PubMed] [Google Scholar]
- 16.Cheng X-T, Huang N & Sheng Z-H Programming axonal mitochondrial maintenance and bioenergetics in neurodegeneration and regeneration. Neuron 110, 1899–1923 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu H et al. A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat Neurosci 22, 47–56 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leng K et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat Neurosci 24, 276–287 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barker SJ et al. MEF2 is a key regulator of cognitive potential and confers resilience to neurodegeneration. Science Translational Medicine 13, eabd7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zalocusky KA et al. Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer’s disease. Nat Neurosci 24, 786–798 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tiscione SA et al. IP3R-driven increases in mitochondrial Ca2+ promote neuronal death in NPC disease. PNAS 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welch G & Tsai L-H Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep 23, e54217 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lodato MA et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Q, Niu Y, Gundry M & Zong C Single-cell damagenome profiling unveils vulnerable genes and functional pathways in human genome toward DNA damage. Science Advances 7, eabf3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welch GM et al. Neurons burdened by DNA double-strand breaks incite microglia activation through antiviral-like signaling in neurodegeneration. Sci Adv 8, eabo4662 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu W et al. Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 593, 440–444 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madabhushi R et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 161, 1592–1605 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Habib N et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 353, 925–928 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hochgerner H, Zeisel A, Lönnerberg P & Linnarsson S Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nat Neurosci 21, 290–299 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Durante MA et al. Single-cell analysis of olfactory neurogenesis and differentiation in adult humans. Nat Neurosci 23, 323–326 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rohrback S et al. Submegabase copy number variations arise during cerebral cortical neurogenesis as revealed by single-cell whole-genome sequencing. PNAS 115, 10804–10809 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayhan F et al. Resolving cellular and molecular diversity along the hippocampal anterior-to-posterior axis in humans. Neuron 109, 2091–2105.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Y et al. Molecular landscapes of human hippocampal immature neurons across lifespan. Nature 607, 527–533 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franjic D et al. Transcriptomic taxonomy and neurogenic trajectories of adult human, macaque, and pig hippocampal and entorhinal cells. Neuron (2021) doi: 10.1016/j.neuron.2021.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arber C et al. Familial Alzheimer’s Disease Mutations in PSEN1 Lead to Premature Human Stem Cell Neurogenesis. Cell Reports 34, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cosacak MI et al. Single-Cell Transcriptomics Analyses of Neural Stem Cell Heterogeneity and Contextual Plasticity in a Zebrafish Brain Model of Amyloid Toxicity. Cell Reports 27, 1307–1318.e3 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Mi D et al. Early emergence of cortical interneuron diversity in the mouse embryo. Science 360, 81–85 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tasic B et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci 19, 335–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krienen FM et al. Innovations present in the primate interneuron repertoire. Nature 586, 262–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu Y et al. Interneuron origin and molecular diversity in the human fetal brain. Nat Neurosci 24, 1745–1756 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Bugeon S et al. A transcriptomic axis predicts state modulation of cortical interneurons. Nature 607, 330–338 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez-Losa M et al. Nav1.1-Overexpressing Interneuron Transplants Restore Brain Rhythms and Cognition in a Mouse Model of Alzheimer’s Disease. Neuron 98, 75–89.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nuriel T et al. Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat Commun 8, 1464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith SJ et al. Single-cell transcriptomic evidence for dense intracortical neuropeptide networks. eLife 8, e47889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong W et al. The neuropeptide landscape of human prefrontal cortex. Proceedings of the National Academy of Sciences 119, e2123146119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cauli B et al. Cortical GABA Interneurons in Neurovascular Coupling: Relays for Subcortical Vasoactive Pathways. J. Neurosci 24, 8940–8949 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jansen IE et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51, 404–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y & Colonna M Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? Journal of Experimental Medicine 218, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keren-Shaul H et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Grubman A et al. Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat Commun 12, 3015 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.March-Diaz R et al. Hypoxia compromises the mitochondrial metabolism of Alzheimer’s disease microglia via HIF1. Nat Aging 1, 385–399 (2021). [DOI] [PubMed] [Google Scholar]
- 52.Mathys H et al. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep 21, 366–380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Friedman BA et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Reports 22, 832–847 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Sala Frigerio C et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Reports 27, 1293–1306.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang HS et al. Natural genetic variation determines microglia heterogeneity in wild-derived mouse models of Alzheimer’s disease. Cell Reports 34, 108739 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sayed FA et al. AD-linked R47H-TREM2 mutation induces disease-enhancing microglial states via AKT hyperactivation. Science Translational Medicine (2021) doi: 10.1126/scitranslmed.abe3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jaitin DA et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178, 686–698.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krasemann S et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi Y et al. Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron 109, 2413–2426.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Badimon A et al. Negative feedback control of neuronal activity by microglia. Nature 586, 417–423 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Merlini M et al. Microglial Gi-dependent dynamics regulate brain network hyperexcitability. Nat Neurosci 24, 19–23 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hong S et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gunner G et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat Neurosci 22, 1075–1088 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Victor MB et al. Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell 29, 1197–1212.e8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Favuzzi E et al. GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell 184, 4048–4063.e32 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stogsdill JA et al. Pyramidal neuron subtype diversity governs microglia states in the neocortex. Nature 1–7 (2022) doi: 10.1038/s41586-022-05056-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bisht K et al. Capillary-associated microglia regulate vascular structure and function through PANX1-P2RY12 coupling in mice. Nat Commun 12, 5289 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Császár E et al. Microglia modulate blood flow, neurovascular coupling, and hypoperfusion via purinergic actions. Journal of Experimental Medicine 219, e20211071 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Masuda T et al. Specification of CNS macrophage subsets occurs postnatally in defined niches. Nature 604, 740–748 (2022). [DOI] [PubMed] [Google Scholar]
- 70.Munro DAD, Movahedi K & Priller J Macrophage compartmentalization in the brain and cerebrospinal fluid system. Science Immunology 7, eabk0391 (2022). [DOI] [PubMed] [Google Scholar]
- 71.Vanlandewijck M et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Van Hove H et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22, 1021–1035 (2019). [DOI] [PubMed] [Google Scholar]
- 73.Hernández JCC et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nature Neuroscience 22, 413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sankowski R et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat Neurosci 22, 2098–2110 (2019). [DOI] [PubMed] [Google Scholar]
- 75.Safaiyan S et al. White matter aging drives microglial diversity. Neuron 109, 1100–1117.e10 (2021). [DOI] [PubMed] [Google Scholar]
- 76.Hughes AN & Appel B Microglia phagocytose myelin sheaths to modify developmental myelination. Nat Neurosci 23, 1055–1066 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morabito S et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat Genet 53, 1143–1155 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Novikova G et al. Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat Commun 12, 1610 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang K et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci 20, 1052–1061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cao H et al. Association of SPI1 Haplotypes with Altered SPI1 Gene Expression and Alzheimer’s Disease Risk. J Alzheimers Dis 86, 1861–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dräger NM et al. A CRISPRi/a platform in human iPSC-derived microglia uncovers regulators of disease states. Nat Neurosci 1–14 (2022) doi: 10.1038/s41593-022-01131-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Batiuk MY et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat Commun 11, 1220 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bayraktar OA et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat Neurosci 23, 500–509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hasel P, Rose IVL, Sadick JS, Kim RD & Liddelow SA Neuroinflammatory astrocyte subtypes in the mouse brain. Nat Neurosci 24, 1475–1487 (2021). [DOI] [PubMed] [Google Scholar]
- 85.Escartin C et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 24, 312–325 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Habib N et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci 23, 701–706 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ioannou MS et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 177, 1522–1535.e14 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Wang C et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 109, 1657–1674.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ballabio A & Bonifacino JS Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol 21, 101–118 (2020). [DOI] [PubMed] [Google Scholar]
- 90.Xu Y, Kong J & Hu P Computational Drug Repurposing for Alzheimer’s Disease Using Risk Genes From GWAS and Single-Cell RNA Sequencing Studies. Frontiers in Pharmacology 12, 1614 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marques S et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352, 1326–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Falcão AM et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med 24, 1837–1844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jäkel S et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543–547 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen J-F et al. Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer’s disease. Neuron 109, 2292–2307.e5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bonetto G, Belin D & Káradóttir RT Myelin: A gatekeeper of activity-dependent circuit plasticity? Science 374, eaba6905. [DOI] [PubMed] [Google Scholar]
- 96.de Faria O et al. Periods of synchronized myelin changes shape brain function and plasticity. Nat Neurosci 24, 1508–1521 (2021). [DOI] [PubMed] [Google Scholar]
- 97.Yang SM, Michel K, Jokhi V, Nedivi E & Arlotta P Neuron class–specific responses govern adaptive myelin remodeling in the neocortex. Science (2020) doi: 10.1126/science.abd2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pan S, Mayoral SR, Choi HS, Chan JR & Kheirbek MA Preservation of a remote fear memory requires new myelin formation. Nat Neurosci 23, 487–499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kirby L et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat Commun 10, 3887 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kenigsbuch M et al. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat Neurosci 25, 876–886 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Arai K & Lo EH An Oligovascular Niche: Cerebral Endothelial Cells Promote the Survival and Proliferation of Oligodendrocyte Precursor Cells. J Neurosci 29, 4351–4355 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pham L-DD et al. Crosstalk between oligodendrocytes and cerebral endothelium contributes to vascular remodeling after white matter injury. Glia 60, 875–881 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marques S et al. Transcriptional Convergence of Oligodendrocyte Lineage Progenitors during Development. Developmental Cell 46, 504–517.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Beiter RM et al. Evidence for oligodendrocyte progenitor cell heterogeneity in the adult mouse brain. Sci Rep 12, 12921 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huang W et al. Origins and Proliferative States of Human Oligodendrocyte Precursor Cells. Cell 182, 594–608.e11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fu Y et al. Heterogeneity of glial progenitor cells during the neurogenesis-to-gliogenesis switch in the developing human cerebral cortex. Cell Reports 34, 108788 (2021). [DOI] [PubMed] [Google Scholar]
- 107.Káradóttir R, Hamilton NB, Bakiri Y & Attwell D Spiking and nonspiking classes of oligodendrocyte precursor glia in CNS white matter. Nat Neurosci 11, 450–456 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang P et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci 22, 719–728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang H et al. Cerebral blood flow in mild cognitive impairment and Alzheimer’s disease: A systematic review and meta-analysis. Ageing Research Reviews 71, 101450 (2021). [DOI] [PubMed] [Google Scholar]
- 110.Chen MB et al. Brain Endothelial Cells Are Exquisite Sensors of Age-Related Circulatory Cues. Cell Reports 30, 4418–4432.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kalucka J et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 180, 764–779.e20 (2020). [DOI] [PubMed] [Google Scholar]
- 112.Yousef H et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat Med 25, 988–1000 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhao L et al. Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain. Nat Commun 11, 4413 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wälchli T et al. Molecular atlas of the human brain vasculature at the single-cell level 2021.10.18.464715 10.1101/2021.10.18.464715v1 (2021) doi:. [DOI] [Google Scholar]
- 115.Sun N et al. Single-cell multi-region dissection of brain vasculature in Alzheimer′s Disease 2022.02.09.479797 Preprint at 10.1101/2022.02.09.479797 (2022). [DOI] [Google Scholar]
- 116.Yang AC et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 1–8 (2022) doi: 10.1038/s41586-021-04369-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Winkler EA et al. A single-cell atlas of the normal and malformed human brain vasculature. Science 375, eabi7377 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Profaci CP, Munji RN, Pulido RS & Daneman R The blood–brain barrier in health and disease: Important unanswered questions. J Exp Med 217, e20190062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zou C et al. Reduction of mNAT1/hNAT2 Contributes to Cerebral Endothelial Necroptosis and Aβ Accumulation in Alzheimer’s Disease. Cell Reports 33, 108447 (2020). [DOI] [PubMed] [Google Scholar]
- 120.Gonzales AL et al. Contractile pericytes determine the direction of blood flow at capillary junctions. PNAS 117, 27022–27033 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hartmann DA et al. Brain capillary pericytes exert a substantial but slow influence on blood flow. Nat Neurosci 24, 633–645 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nortley R et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 365, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Blanchard JW et al. Reconstruction of the human blood–brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat Med 26, 952–963 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yamazaki Y et al. Vascular ApoE4 Impairs Behavior by Modulating Gliovascular Function. Neuron 109, 438–447.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barnes LL Alzheimer disease in African American individuals: increased incidence or not enough data? Nat Rev Neurol 18, 56–62 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vila-Castelar C, Fox-Fuller JT, Guzmán-Vélez E, Schoemaker D & Quiroz YT A cultural approach to dementia — insights from US Latino and other minoritized groups. Nat Rev Neurol 18, 307–314 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Alsema AM et al. Profiling Microglia From Alzheimer’s Disease Donors and Non-demented Elderly in Acute Human Postmortem Cortical Tissue. Frontiers in Molecular Neuroscience 13, 134 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Marinaro F et al. Molecular and cellular pathology of monogenic Alzheimer’s disease at single cell resolution 2020.07.14.202317 10.1101/2020.07.14.202317v1 (2020) doi:. [DOI] [Google Scholar]
- 129.Gerrits E et al. Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol 141, 681–696 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Del-Aguila JL et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimer’s Research & Therapy 11, 71 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Olah M et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun 11, 6129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xu H & Jia J Single-Cell RNA Sequencing of Peripheral Blood Reveals Immune Cell Signatures in Alzheimer’s Disease. Frontiers in Immunology 12, 2727 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gate D et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Smith AM et al. Diverse human astrocyte and microglial transcriptional responses to Alzheimer’s pathology. Acta Neuropathol 143, 75–91 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Krishnaswami SR et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat Protoc 11, 499–524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Thrupp N et al. Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep 32, 108189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Marsh SE et al. Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nat Neurosci 25, 306–316 (2022). [DOI] [PubMed] [Google Scholar]
- 138.Zeng H What is a cell type and how to define it? Cell 185, 2739–2755 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rustenhoven J et al. PU.1 regulates Alzheimer’s disease-associated genes in primary human microglia. Molecular Neurodegeneration 13, 44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sanchez PE et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proceedings of the National Academy of Sciences 109, E2895–E2903 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vossel K et al. Effect of Levetiracetam on Cognition in Patients With Alzheimer Disease With and Without Epileptiform Activity: A Randomized Clinical Trial. JAMA Neurology 78, 1345–1354 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gonzales MM et al. Senolytic Therapy to Modulate the Progression of Alzheimer’s Disease (SToMP-AD): A Pilot Clinical Trial. J Prev Alzheimers Dis 9, 22–29 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wightman DP et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet 53, 1276–1282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bellenguez C et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 54, 412–436 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hammond TR et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50, 253–271.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Grone BP et al. Early and lifelong effects of APOE4 on neuronal gene expression networks relevant to Alzheimer’s disease 2022.06.16.496371 Preprint at 10.1101/2022.06.16.496371 (2022). [DOI] [Google Scholar]
- 147.Da Mesquita S et al. Aging-associated deficit in CCR7 is linked to worsened glymphatic function, cognition, neuroinflammation, and β-amyloid pathology. Science Advances 7, eabe4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Taubes A et al. Experimental and real-world evidence supporting the computational repurposing of bumetanide for APOE4-related Alzheimer’s disease. Nat Aging 1, 932–947 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lin Y-T et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 98, 1141–1154.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]