

Abstract
TGF-β plays a prominent role as an inducer of epithelial-mesenchymal transitions (EMTs) during development and wound healing and in disease conditions such as fibrosis and cancer. During these processes EMT occurs together with changes in cell proliferation, differentiation, communication, and extracellular matrix remodeling that are orchestrated by multiple signaling inputs besides TGF-β. Chief among these inputs is RAS-MAPK signaling, which is frequently required for EMT induction by TGF-β. Recent work elucidated the molecular basis for the cooperation between the TGF-β-SMAD and RAS-MAPK pathways in the induction of EMT in embryonic, adult and carcinoma epithelial cells. These studies also provided direct mechanistic links between EMT and progenitor cell differentiation during gastrulation or intra-tumoral fibrosis during cancer metastasis. These insights illuminate the nature of TGF-β driven EMTs as part of broader processes during development, fibrogenesis and metastasis.
Keywords: TGF-β, EMT, Development, Cancer, Fibrosis
1. Introduction
EMT is a key cellular process during embryogenesis, wound healing, fibrosis, and tumor progression. Epithelial cells undergoing EMT lose apicobasal polarity and adherent junctions while gaining mesenchymal traits including anteroposterior polarity, migration, and invasion of adjacent tissue. These changes additionally remodel cell contacts with neighboring cells and with basement membrane and other extracellular matrix (ECM) structures.
The ability to undergo EMT reflects the intrinsic phenotypic plasticity of epithelial stem and progenitor cells, which is essential for remodeling epithelial structures during developmental and regenerative processes. Cells that undergo an EMT frequently return to an epithelial state, regaining apicobasal polarity and adherent contact with neighboring cells through a mesenchymal-to-epithelial transition (MET). Notably, EMT is no longer thought to be a binary alternation between extreme epithelial and mesenchymal phenotypic states. Rather, EMTs represent departures from the polarized epithelial phenotype the extent of which depends on the type of cell and the signals it receives [1,2]. Distinct epithelial progenitors undergoing an EMT may manifest different degrees of departure from the epithelial state, expressing different types of mesenchymal markers and phenotypic features.
EMT is orchestrated by transcription factors (TFs) called EMT-TFs, which repress epithelial genes and stimulate expression of mesenchymal components. Combinatorial expression of the core EMT-TFs Snail (encoded by SNAI1), Slug (SNAI2), ZEB1, ZEB2, TWIST1, and TWIST2 drives the mesenchymal state and represses the epithelial state [1,2]. Other transcription factors also contribute to EMT such as GATA3 and GATA6 [3,4], SOX9 [5], PRRX1 [6], Goosecoid (GSC) [7], Brachyury [8] and LBX1 [9], EMT is additionally balanced by a network of countervailing transcription factors and microRNAs.
Expression of EMT-TFs is regulated by extracellular signals and frequently involves cooperation between various signaling pathways that shape epithelial plasticity in a context-dependent manner. Although multiple signals can modulate EMTs, transforming growth factor-β (TGF-β) frequently plays a dominant role [1,10,11]. TGF-β activates the expression of EMT-TFs and triggers EMTs is diverse developmental and regenerative processes and in the diseases that coopt these mechanisms.
EMTs do not occur as isolated cellular events. The complex biological processes that involve EMTs also involve profound changes is epithelial tissue structure. For example, EMT and migration accompany mesoderm and endoderm differentiation of pluripotent epiblast cells during embryo gastrulation, re-epithelialization and tissue remodeling during wound healing, and collective cell invasion, proliferation, and intra-tumoral fibrosis during metastasis. In these diverse contexts, epithelial cells undergo changes in proliferation, differentiation, extracellular matrix remodeling, and secretome-mediated crosstalk with fibroblast, immune, and other stromal cell types. Mechanisms likely exist that coordinate EMT with these accompanying changes for tissue formation during development and regeneration.
The diversity of EMT-TFs, modulatory signals, and associated cellular responses poses a challenge in delineating the mechanistic basis for EMT and its coupling with distinct events. Recent work on the coordinated induction of EMTs during gastrulation and tumorigenesis provided insights into the molecular basis for EMT induction by collaborative TGF-β and RAS signals and how EMT is coupled with cell differentiation, apoptosis or fibrogenesis depending on the context [11–13]. Here we review the contextual nature of EMTs and their associated programs in the light of these insights and discuss the implications for a better understanding of TGF-β regulated EMTs as fundamental events in development and disease.
2. TGF-β as a regulator of EMTs in different contexts
The existence of epithelial-mesenchymal interface regulation in developmental processes controlled by TGF-β family members was observed in Müllerian duct regression in rat [14]. Direct evidence of the ability of TGF-β to induce epithelial phenotypic transitions was first reported in embryonic heart formation [15,16], in mouse mammary epithelial cells in culture [17], and later in many other epithelial cell types in culture and during development, fibrosis, and cancer [1,18]. During development, the TGF-β related Nodal and bone morphogenetic protein (BMP) pathways establish the body axes and direct the subsequent patterning of tissues. Nodal induces expression of Snai1 and Twist1 and drives an EMT program including loss of E-cadherin expression and gain of N-cadherin, key events during gastrulation [19]. Mouse embryos deficient for Snail fail to downregulate E-cadherin and exhibit defects in cell migration and the formation of mesoderm [20]. Additionally, Nodal induces the expression of Eomesodermin (Eomes) and Mesp, TFs that contribute to EMT during gastrulation. Different levels of Nodal signaling are required to specify and pattern the mesendoderm through effects beyond EMT [21,22]. For example, formation of the endoderm and the anterior mesoderm in the mouse requires more Nodal stimulation than posterior mesoderm, in line with the observation of Nodal gradient-dependent induction of endodermal TF Foxa2 and mesodermal TFs Gsc and brachyury.
TGF-β induces Snail and Slug to drive EMT during cardiac valve formation [23]. TGF-β is associated with cells undergoing EMT at the interfaces of injured tissues and invasive tumors with the surrounding stroma [24,25]. TGF-β, and EMT, play a dual role in cancer, as clearly observed during pancreatic tumorigenesis. TGF-β acts as a tumor suppressor that eliminates pre-malignant cells harboring oncogenic KRAS mutations by triggering apoptosis, which results from a conflict between TGF-β dependent EMT and an epithelial progenitor enforcing program in these cells [12]. Cancer cell clones can bypass this pressure by decoupling TGF-β-induced EMT from apoptosis, which consequently allows EMT to support cancer cell migration, invasiveness, and metastasis, by turning TGF-β from a tumor suppressor into a tumor promoter [11,13, 26]. Moreover, cancer cells undergoing a TGF-β-induced EMT show resistance to chemotherapy and iradiation [27,28], and overexpression of Snail impairs cell cycle progression and apoptosis [29,30].
Thus, TGF-β (or Nodal signaling though the same pathway) is a potent inducer of different EMTs with different outcomes in different contexts (Fig. 1): morphogenesis and differentiation in gastrulation, organogenesis later in development, balanced epithelial regeneration in wound healing, apoptosis in pre-malignant stages, and invasive growth in tumor progression and metastasis.
Fig. 1.
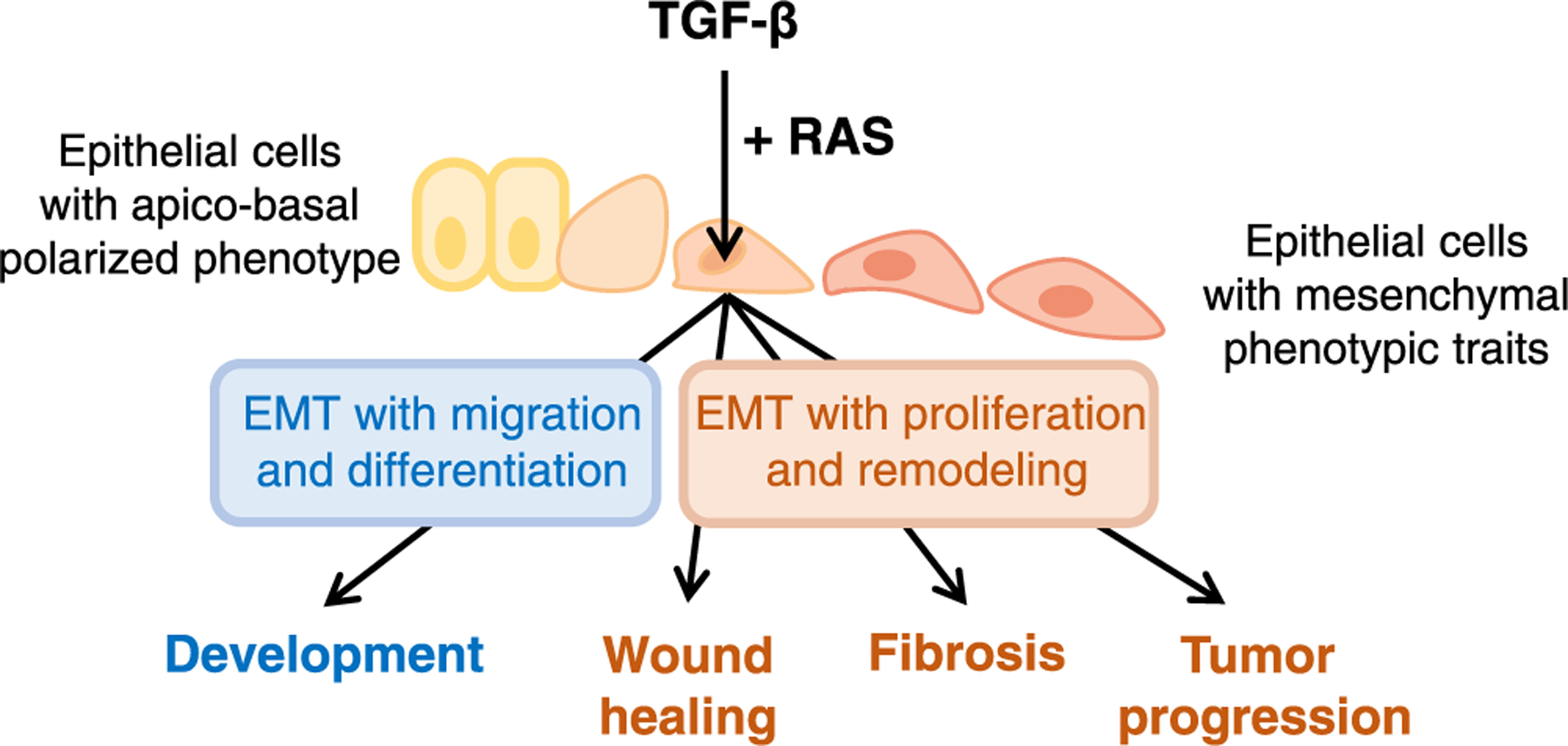
TGF-β as regulator of EMTs in different contexts. TGF-β triggers EMTs in the context of complex developmental, regenerative, and pathological processes that also involve profound changes in cell proliferation, differentiation, position, and extracellular matrix remodeling. In developmental contexts such as gastrulation, EMT driven by Nodal TGF-β signaling occurs together with mesendodermal cell differentiation. In regenerative contexts such as wound healing and their derivative pathologies –fibrosis and cancer–TGF-β driven EMT occurs alongside fibrogenic effects that remodels the extracellular matrix. Other signals converge with TGF-β on the regulation of EMT. RAS signaling is a particularly powerful collaborating signal in TGF-β induction of EMTs in diverse contexts.
3. The SMAD pathway as a transcriptional activator of EMTs
Gene regulation by TGF-β is executed through SMAD transcription factors. The context-dependent determinants and operating logic of the TGF-β-SMAD signaling pathway have been reviewed [10,11,31], and are summarized here only briefly (Fig. 2A). SMAD2 and SMAD3 are directly phosphorylated by the TGF-β receptor complex (or the Nodal/Activin receptor complex). Thus activated, SMAD2 and SMAD3 form trimeric complexes with SMAD4 to transcriptionally regulate nuclear target genes increasing gene expression in most cases or repressing it in others. SMADs bind with low affinity to GC-rich DNA sequences but are directed to specific loci by interaction with context-dependent DNA-binding cofactors that recruit activated SMADs to loci genome-wide. Some of these cofactors are lineage-determining transcription factors (LDTFs) expressed in a developmentally programmed manner, while others are signal-driven transcription factors (SDTFs) activated by specific extracellular stimuli. The repertoire of specific DNA-binding SMAD partners in a cell, together with signal crosstalk inputs modulating SMAD activity and chromatin accessibility controlling the access to SMAD target loci, collectively shape the TGF-β response and determine the highly contextual and pleiotropic nature of TGF-β effects [10].
Fig. 2.
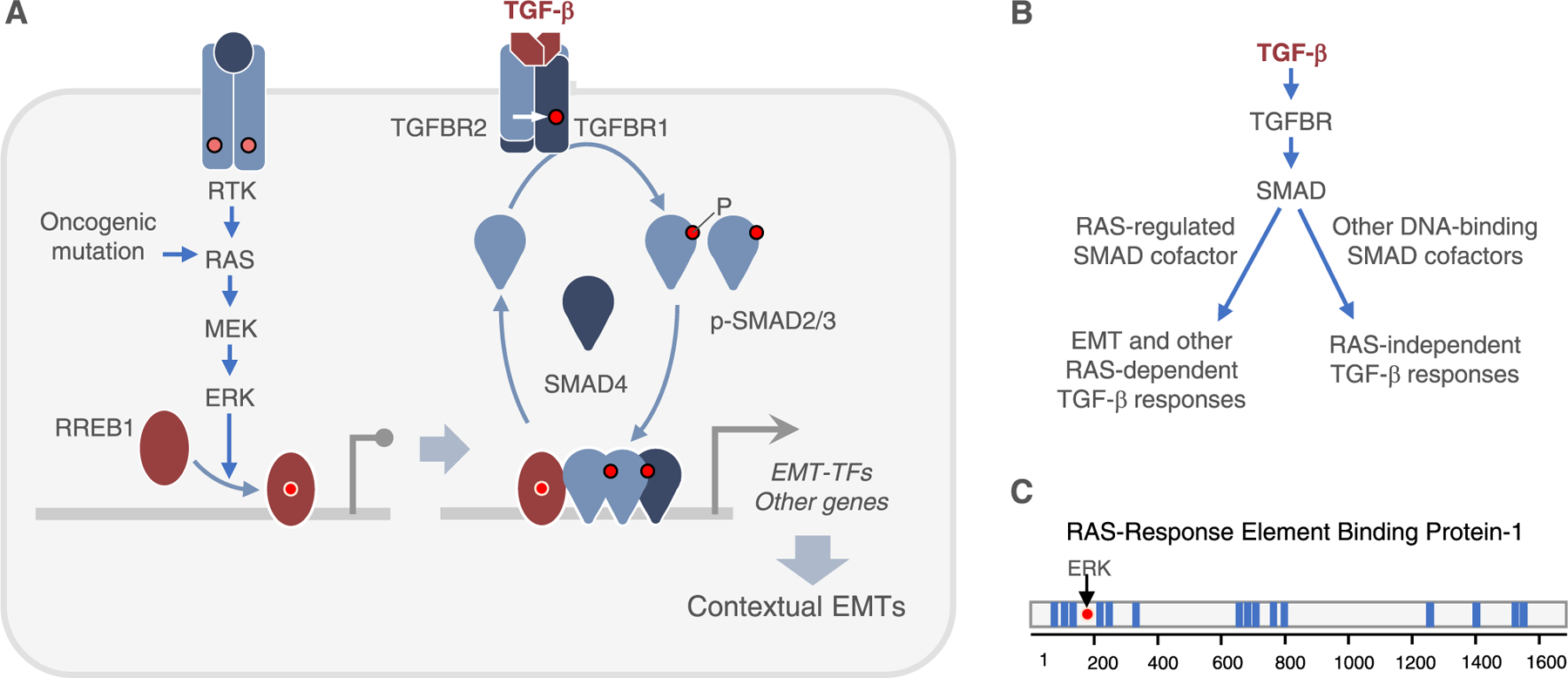
TGF-β and RAS collaboration in the induction of EMTs. A. RAS-MAPK signaling is an determines a cell’s competence to undergo EMT in response to TGF-β. RAS activation by receptor tyrosine kinases (RTK) or oncogenic mutation leads the ERK phosphorylation of RREB1, enabling its binding to cognate sites genome-wide. RREB1 binds near SMAD binding sites. TGF-β receptors activate SMAD transcription factors for activation of EMT-TFs and other genes in collaboration with RREB1, triggering different types of EMTs in different contexts. B. Only a subset of TGF-β-SMAD target genes depend on RAS-MAPK input for activation. RREB1 collaborated with SMADs in the activation of these RAS-dependent genes. C. RREB1 contains 15 zinc-finger domains (blue bars) distributed in three clusters. ERK phosphorylation of a canonical MAPK site in the N-terminal domain of RREB1 stimulates binding to DNA.
The forkhead box protein H1 (FOXH1) in mesendoderm precursors, zinc-finger protein 423 (ZFP423, also known as OAZ) in ventral mesoderm, MyoD1 in myoblast precursors, CCAAT/enhancer-binding protein-α (C/EBPα) in myeloid precursors, and GATA in erythroid precursors are classical examples of LDTFs dictating the genome binding pattern of SMADs to determine lineage commitment and cell differentiation within the lineage [32–35]. These LDTFs occupy cis-regulatory elements targeted by TGF-β-activated SMADs. FOXH1 acts as a pioneer transcription factor that binds to repressive chromatin to remodel it for subsequent recruitment and binding of activated SMADs [32,36,37]. In mesendoderm differentiation genes, FOXH1 can recruit SMAD3 in the absence of Nodal signaling, but Nodal is still required for the activation and recruitment of SMAD2 and SMAD4 to the FOXH1-SMAD3 pre-bound complex [32]. Other LDTFs may similarly act as pioneer factors that locally remodel target loci for the eventual recruitment of ligand-activated SMADs.
Alongside LDTFs, SDTFs activated by certain signals also participate in directing SMADs to specific loci. Examples include AP-1 transcription factors in the regulation of the plasminogen activator protease inhibitor SerpinE1 (also known as PAI-1) [38] and matrix metalloproteinases [39, 40], and FOXO transcription factors in the regulation of cyclin-dependent kinase (CDK) inhibitors p15INK4b (encoded by CDKN2B) and p21CIP1 (CDKN1A) [41]. Activated SMAD complexes cooperate with different co-activators, co-repressors, chromatin remodelers, and chromatin readers to shape transcriptional regulation of target genes. Early examples include SMAD interactions with the CBP and p300 histone acetyltransferases [42,43], the TGIF histone deacetylase adaptor [44], and the BRG subunit of the SWI-SNF chromatin remodeler complex [45,46].
TGF-β triggers EMTs by inducing the expression of the EMT-TFs Snail and/or Slug, which repress the expression of the epithelial adherens junction molecule E-cadherin (encoded by CDH1) [47–49] and the epithelial progenitor transcription factor KLF5 [12]. SMAD transcription factors are central mediators of TGF-β-induced EMTs in many contexts. For example, SMADs are required for TGF-β induction of Snai1 and/or Snai2 in kidney epithelial cells [50,51], mammary epithelial and breast carcinoma cells [51–53], hepatocytes [54], lens epithelial cells [55], and human pancreatic ductal adenocarcinoma (PDAC) cells [56]. TGF-β-activated SMADs may cooperate with EMT-TFs in subsequent waves of transcriptional regulation. For example, TGF-β activated SMADs cooperate with Snail to downregulate the expression of epithelial markers such as E-cadherin, occludin, and claudin-3 in mouse mammary epithelial cells [57], and Klf5 in mouse pancreatic ductal adenocarcinoma cells [12]. In turn ZEB1 represses epithelial markers by recruiting co-repressors together with SMAD3 [58].
Various additional cofactors have been identified as important participants in SMAD-mediated induction of EMT-TF expression, including the enhancer organizing factor HMGA2 [59–61], the histone demethylases KDM1A (LSD1) [62] and KDM6B (JMJD3) [63], the COMPASS complex component RBBP5 [64], the histone methyltransferase EZH2 [65], and the SWI/SNF chromatin remodeling complex core component BRG1 [45]. ZEB expression by TGF-β additionally involves the transcription factor ETS1 [66,67].
4. TGF-β and RAS cooperate in the induction of EMTs
Notably, TGF-β induces EMT in epithelial cells under certain conditions and not in others depending on inputs from other pathways. RAS-MAPK signaling is a particularly important determinant of a cell’s competence to undergo EMT in response to TGF-β, as first observed in mouse mammary epithelial cells [68–70]. Mammary ductal morphogenesis and gland formation involves EMT [71] and mammary gland development requires EMT-TFs [72]. TGF-β induction of EMT in mammary epithelial cells requires ERK MAPK activity [73]. TGF-β cooperates with RAS-MAPK signaling to induce EMTs in normal and malignant mammary, kidney, liver, pancreatic and lung epithelial cells [12,70, 74–81]. Oncogenic RAS mutations enable TGF-β-SMAD signaling to induce EMT in cancer cells [12,13,77,82]. The cooperation of TGF-β and RAS pathways allows carcinoma cells at the tumor invasive front to undergo EMT, migration and hematogenous dissemination [83]. During gastrulation, Nodal requires FGF and WNT signals to activate mesendoderm differentiation and drive an EMT [18,84–87]. Epiblast cells in Fgfr1-null mice fail to downregulate E-cadherin and do not undergo EMT [84]. In sum, although various pathways cooperate with the TGF-β pathway during processes that involve EMTs, the RAS-MAPK pathway stands out as a potent partner of TGF-β in triggering EMTs.
In principle, the involvement of RAS signaling in TGF-β induction of EMT could be due to a role of RAS-MAPK directly downstream of TGF-β receptors. TGF-β addition to cells in culture can activate the ERK MAPK, p38 MAPK and PI3K pathways [88,89], and several reports suggested that TGF-β receptors trigger EMT by activating ERK MAPK [73], p38 MAPK [88] or PI3K pathways [90]. While the biochemical and structural basis for the coupling of TGF-β receptors to these pathways is not clear, each of these pathways is potently and directly activated by well-established receptor tyrosine kinase ligands, cell metabolism effectors, and cellular stresses. In developing tissues and tumors TGF-β operates in the presence of these potent MAPK and PI3K agonists, therefore raising questions about the significance of TGF-β as an activator of MAPKs or PI3K for the induction of EMTs in vivo.
An alternative hypothesis is that the synergy between TGF-β-SMAD and RAS-MAPK as inducers of EMTs is based on a convergence of these pathways, each activated by its corresponding agonists, to bring about strong activation of EMT-TF expression. Support for this model comes from experiments using inducible expression of an oncogenic RAS allele (KRASG12D) in mouse pancreatic organoids. In these experiments, TGF-β strongly induced Snail expression and EMT when KRASG12D was turned on, but not when it was turned off [12,13,26]. Intriguingly, other work showed that knockdown of KRAS in human PDAC cells prevented the downregulation of E-cadherin, while sparing other TGF-β gene responses intact [77], suggesting a selective participation of RAS in some but not all TGF-β gene responses.
5. TGF-β and RAS pathways converge on RREB1 for EMT induction
The preceding observations raised the possibility that a RAS-dependent transcription factor is required for SMAD-driven expression of EMT-TFs, but not for other SMAD-dependent responses (Fig. 2B). This hypothesis was confirmed with the identification of RAS-responsive element binding protein 1 (RREB1) as a RAS-MAPK-activated partner of SMADs in the transcriptional activation of Snai1 together with a specific subset of TGF-β target genes [13] (Fig. 2A). Motif analysis of SMAD binding sites in RAS-dependent and RAS-independent TGF-β target genes revealed an enrichment for RREB1 binding elements in genes requiring RAS input for activation. In an orthogonal approach, a genetic screen for transcription factors required in PDAC cells for TGF-β-dependent apoptosis identified SOX4 and RREB1 as hits. Biochemical characterization showed that N-terminal phosphorylation of RREB1 by ERK induces its binding to DNA. In cells with high levels of RAS-MPK activity, RREB1 is pre-bound to these target genes prior to stimulation of cells by TGF-β [13]. RREB1 is essential for SMAD activation of Snai1 in PDAC cells, lung adenocarcinoma (LUAD) cells, mammary epithelial cells (which harbor high ERK activity levels), and mouse embryonic stem cells. Smad4-restored PDAC cells harboring oncogenic KRAS mutant, where TGF-β-induced EMT triggers apoptosis [12], grew poorly as subcutaneous tumors in mice compared to Rreb1-knockout counterparts. In contrast, TGF-β- and RREB1-dependent EMT in KRAS-mutant LUAD is decoupled from apoptosis and instead promotes the growth of pulmonary metastases [13].
RREB1 contains 15 C2H2 zinc-fingers (ZFs) distributed in three clusters (Fig. 2C) and exists in various splice variants. The RREB1 orthologue in Drosophila, known as hindsight or pebbled, mediates collective migration of border cells [91]. Previous work on this relatively obscure RAS effector showed that RREB1 activates or represses transcription depending on context. In mammalian adipose tissues, RREB1 selectively recruits JMJD3 to brown fat-specific genes and removes H3K27me3 to activate their gene expression, which leads to development of beige adipocytes [92]. In pancreatic endocrine β cells, RREB1 occupies the promoter of insulin genes and recruits CtBP-LSD1 to stimulate gene transcription by removal of repressive histone marks [93]. In colorectal adenocarcinoma, RREB1 mediates KRAS repression of miR-143/145 expression [94].
RREB1 shows alterations in cancer, and particularly in PDAC tumors, 95 % of which harbor KRAS mutations. Heightened RAS signaling in premalignant pancreatic progenitor cells leads to TGF-β-induced EMT coupled with apoptosis. Nearly half of human PDAC show a genetic loss of SMAD4 or other components of the TGF-β pathway, thus disabling TGF-β-mediated tumor suppression. Expression of RREB1 is markedly downregulated in early stage of PDAC [95], and truncating RREB1 mutations occur in a small but significant proportion of human PDAC [96].
6. TGF-β EMTs are part of broad developmental and regenerative programs
6.1. TGF-β EMTs in gastrulation and other developmental stages
EMT is essential for gastrulation, which generates the three germ layers –endoderm, mesoderm, and ectoderm–from pluripotent epiblast cells. Epithelial epiblast cells exhibit apical-basal polarity and express E-cadherin, as these cells lose pluripotency they undergo EMT to migrate to different locations. The first stage of gastrulation is the formation of a primitive streak within the epiblast epithelial layer where EMT is induced by Nodal-activated SMAD signaling [97].
Nodal-activated SMADs integrate multiple inputs by cooperating with other transcription factors, resulting in the parallel induction of EMT-TFs and differentiation genes, which orchestrate epiblast cell migration and differentiation. Genome-wide prebound FoxH1 directs Nodal-activated SMAD2/3 to target loci and jointly binds to the regulatory elements of mesendoderm differentiation genes, including Eomes, Brachyury, Gsc, Mixl1, and Foxa2 [32]. FoxH1-occupied cis-elements are poised along with repressive histone mark H3K9me3. To resolve chromatin accessibility, Nodal-activated SMAD3 cooperates with E3 ubiquitin-protein ligase TRIM33. TRIM33 interacts with histone marks at regulatory loci and displaces the interaction of histone reader heterochromatin protein 1 (HP1) with the histone marks, resulting in the activation of mesendodermal gene expressions [98].
The neural crest is a transient group of progenitor cells characterized by multipotency and migratory ability. After specification, pre-migratory neural crest precursors undergo EMT and delaminate from the neuroepithelium to migrate within the embryo and in a context-specific manner, where they contribute to the development of a variety of organs. BMP secreted from the dorsal neural tube induces segregation of pre-migratory neural crest cells and initiates the EMT program [99]. WNT induces BMP expression and provides ground for the cooperation of the BMP-Wnt pathways to induce Snai1, Snai2, and Sox9 [100]. TGF-β also endows endothelial plasticity in heart development via endothelial-mesenchymal transition (EndMT) which leads to heart valve formation [101].
6.2. TGF-β EMTs in wound healing and tissue fibrosis
Wound repair is a fundamental property of multicellular organisms to restore tissue integrity and homeostasis. Postnatal wound repair results in a non-functioning mass of fibrotic scar conceivably due to inflammatory responses. Fibrosis, defined by the excessive accumulation of ECM components, is an outcome of dysregulated regenerative responses, such as pathological activation of repair with sustained inflammation and impaired termination. TGF-β-induced EMT plays a prominent role in the regenerative processes, as a high level of TGF-β is linked to EMT-like changes in tissue fibrosis [50,102,103]. Both in wound healing and tissue fibrosis, EMT is associated with a fibrogenic program to restore tissue architecture, and TGF-β signaling drives pro-fibrotic events in wound healing and tissue fibrosis, such as inflammation, epithelial cell migration, fibroblast recruitment, and ECM deposition.
During wound healing, re-epithelialization, characterized by the migration and proliferation of epithelial cells at the edge of damaged tissue serves to replenish tissue integrity and structure, and involves EMTs [1,104]. Single-cell transcriptome analysis shows induction of EMT gene signatures at wound sites in mice [105]. A loss of epithelial features and increased cell motility provided by a reversible EMT supports tissue homeostasis [106–108]. Injured tissue is exposed to multiple growth factors and inflammatory cytokines. Platelets, neutrophils, and monocyte-derived macrophages are key initial sources of TGF-β, which induces EMT [109]. EGF, FGF, and HGF stimulate corresponding receptors and activate downstream signaling cascades, such as MAPK pathways [110]. Activated MAPK upregulates EMT-TFs such as Snail, Slug, and ZEB [1,111], and accelerates cutaneous wound healing [112], likely in combination with TGF-β signaling.
In cutaneous wounds, TGF-β is not only an inducer of keratinocyte migration but also a potent chemoattractant for endothelial cells and fibroblasts. The dermis is restored by invading and proliferating fibroblasts that generate ECM components, mainly collagen, which ultimately forms the bulk of the mature scar. Myofibroblasts, the differentiated form of fibroblasts and the primary cell type supporting matrix-preservation, aid in the synthesis of ECM proteins and wound closure. A dominant pro-fibrotic role of TGF-β is the differentiation of fibroblasts into myofibroblasts [113], and pro-fibrotic factors such as tenascin-C, which is induced by TGF-β, support the recruitment and differentiation of fibroblasts at the wound site [114].
The pro-fibrotic actions of TGF-β are linked, in part, to their effects on epithelial cells during wound healing in which pro-fibrotic factors are secreted in response to TGF-β. TGF-β activates a matrix-preserving program including production of interstitial collagens [115] and fibronectin [116]. Scar formation after a stab wound to the cerebral cortex is reduced in Smad3-null mice [117]. Recent studies on the contribution of EMT to renal fibrosis suggest that epithelial cells undergoing EMT activated other gene programs that resulted in tissue fibrosis [118,119]. Snail-induced partial EMT in kidney epithelial cells gave rise to inflammation during fibrosis. Partial EMT in tubular epithelial cells is not coupled to their differentiation into myofibroblasts, rather, it is coupled to inflammatory gene activation. A parallel regulation of EMT and fibrogenesis by may be beneficial in acute tissue injury, while chronically it may lead to reduced organ function in tissue fibrosis or tumor progression in malignancy
6.3. TGF-β EMTs in tumor suppression
The tumor-suppressive function of TGF-β is manifest through genetic loss-of-function in TGF-β pathway components. Prominent examples include SMAD4 mutations in PDAC [96,120] and TGFBR2 mutations in colorectal cancers [121]. Genetic inactivation of TGF-β signaling components allows pre-malignant cells to avoid apoptosis induced by TGF-β. TGF-β induces expression of cyclin-dependent kinase inhibitors, which slow down the cell cycle, in normal epithelial cells [122–124], whereas carcinoma cells bypass the effectiveness of these CDK inhibitors by harboring CDK activating signals. However, TGF-β signaling induces apoptosis in pancreatic premalignant progenitor cells that harbor KRAS oncogenic mutations [125]. Recent work delineated how TGF-β-induced EMT is coupled to apoptosis (Fig. 3A). TGF-β induces Snail, which then suppresses the expression of transcription factor KLF5. SOX4 is a genome-wide binding partner of KLF5 in pancreatic epithelial progenitors. Downregulation of KLF5 by Snail deprives SOX4 from this cofactor and leads SOX4 to activate expression of proapoptotic factors including BIM and BMF, a process termed lethal EMT [12].
Fig. 3.

TGF-β EMTs in tumor suppression and tumor progression. TGF-β induces EMTs in malignant cells in collaboration with RAS signaling, a frequently hyperactivated pathway in carcinoma cells. A. In pre-malignant pancreatic progenitors, strong activation of EMT by collaboration of TGF-β with oncogenic, hyperactive RAS signaling enter into a conflict with a SOX4-dependent epithelial program and this conflict triggers apoptosis. A pro-apoptotic or lethal EMT thus explains the tumor suppressive action of TGF-β in pancreatic cancer. B. Carcinoma cells decouple EMT from apoptosis by various mechanisms and use TGF-β induced EMT and associated fibrogenic gene response for invasion and metastatic outgrowth, as observed in lung adenocarcinoma models.
6.4. TGF-β EMTs in tumor progression and metastasis
TGF-β-induced EMT is implicated in tumor progression and metastasis [1,126]. Carcinoma cells that progress by decoupling TGF-β from apoptosis can undergo TGF-β dependent EMT and adopt a highly plastic phenotype that facilitates tumor growth and metastasis (Fig. 3B), in addition to immune suppressive protection and other supportive effects of TGF-β in the tumor microenvironment. Carcinoma cells undergoing EMT at the primary site gain invasive and migratory activity for intravasation into blood and lymphatic vessels becoming circulating tumor cells (CTCs) [127], and may further benefit from this EMT during extravasation after lodging in distant organ capillaries minutes later. However, carcinoma cells can also depart from tumors as small epithelial clusters which are highly metastatic [128,129]. CTC clusters become coated with platelets in the blood circulation [130]. It has been shown that platelet-derived TGF-β induces EMT in associated CTCs to stimulate cancer cell extravasation [131]. After CTCs infiltrate distant organs, TGF-β controls dormancy of the disseminated cells [132,133]. TGF-β induces the entry of disseminated carcinoma cells into a non-proliferative dormant state [134]. Dormancy is an immune evasive state that protects cancer cells from elimination by the immune system [135]. It is not known whether TGF-β induced dormancy is accompanied by EMT. However, the outgrowth of metastasis initiating cells requires downregulation of Twist, Prrx1 and other EMT-TFs and return to an epithelial phenotype through an MET [6,136,137].
Cancer cell populations are heterogeneous in their EMT response to TGF-β. Although EMT is a common mechanism for metastatic dissemination, the cells that are most competent at forming metastases are not necessarily those which adopt the most mesenchymal-like phenotype as they undergo EMT [138]. The basis for this heterogeneity of EMT responses to TGF-β, and which forms of EMT are most effective at mediating specific steps in the metastatic cascade [139,140] are currently unknown.
TGF-β-induced EMTs are accompanied by fibrogenic activation of stromal fibroblasts not only during wound healing and fibrotic diseases, but also in primary and metastatic tumors. Cancer cells undergoing an EMT in response to TGF-β secrete pro-fibrotic factors [13], suggesting that EMT and fibrogenesis are parts of an orchestrated program (Fig. 3B). Intra-tumoral fibrosis promotes tumor growth by supporting collective cancer cell migration and cancer cell proliferation on stiff extracellular matrices. An association between fibrosis and cancer has been noted [141]. Studies have shown a strong association between fibrotic microenvironment and tumor progression [142–144]. Cancer cells in primary tumors orchestrate the recruitment and activation of stromal cells that build up desmoplasia. A fibrotic environment with increased deposition of fibrillar collagen, hyaluronic acid, fibronectin, and tenascin-C is conducive to multiple steps of metastasis. A TGFβ-driven, LRRC15+ cancer-associated fibroblasts (CAFs) lineage portends poor outcome in immunotherapy trials in different types of carcinoma [145].
7. RREB1 coordinates EMTs with developmental and fibrogenic gene expression programs
The preceding sections highlight the fact that TGF-β pathway drives cells into EMTs as part of broader developmental or regenerative programs which are closely associated with other changes that TGF-β is also known to induce, notably the differentiation of germ layers, and fibrogenesis in the context of wound healing, fibrosis, and cancer. If TGF-β signaling drives EMT together with fibrogenesis or differentiation in a context-dependent fashion, might the activation of EMT be directly and specifically coupled with the activation of these accompanying programs and an essential feature of the developmental or regenerative processes?
The identification of RREB1 as a common nexus for the TGF-β-SMAD and RAS-MARK pathways in the induction of Snail expression and EMT unexpectedly shed light on this question [13]. The subset of TGF-β responsive genes that require RAS activity and RREB1 in PDAC, LUAD and mammary epithelial cells includes not only Snai1, but also a set of genes encoding secreted fibrogenic factors and matrix remodeling molecules. These factors include interleukin-11, PDGFB, WISP1, SerpinE1 and HAS2. RREB1 binds to the regulatory regions of these genes in a RAS-MAPK-dependent manner, as it does in the regulatory region of Snai1 (Fig. 4A). Moreover, inhibiting EMT by knockout or knockdown of Snai1 and Zeb1 in LUAD cells did not interfere with the induction of these fibrogenic factors by TGF-β [13]. Thus, RREB1 connects EMT and fibrogenic effects of TGF-β as important yet distinct facets of a common program in adult epithelial cells, providing a mechanism for the coordination of EMT and fibrogenesis during normal tissue remodeling and in cancer. In similar but contrasting fashion, the set of Nodal responsive genes that RREB1 activates in mouse embryo mesendoderm precursor cells include not only Snai1 and Snai2, but also the key mesendoderm differentiation genes including Eomes, Gsc, Mixl1 and Brachyury [13]. The coupled induction of EMT-TFs and mesendoderm differentiation genes provides a mechanism for the coordination of EMT and differentiation during gastrulation (Fig. 4B).
Fig. 4.

RREB1 coordinates EMTs with developmental and fibrogenic gene expression programs. A. In various types of adult epithelial cells and carcinoma cells, TGF-β induces a fibrogenic EMT coordinated through SMAD and RREB1, which activate the expression of EMT-TFs and the cell disjunction enzyme hyaluronan synthase 2 (Has2) as well as the expression of various fibrogenic factors. Together, these two arms coordinated by SMAD and RREB1 drive a fribogenic EMT. B. Similarly, in embryonic mesendoderm progenitors Nodal receptor signaling acting through SMADs and RREB1 activates the expression of EMT-TFs together with the expression of mesendoderm differentiation TFs, two key events in gastrulation. C. The chromatin status at relevant loci determines the accessibility of SMAD and RREB1 to the EMT-TF Snai1, the lineage differentiation TF Gsc and the fibrogenic factor Wisp1 in lung adenocarcinoma cells and mesendoderm progenitors.
Insight into why different sets of genes are co-regulated by SMAD and RREB1 together with EMT-TFs in different contexts was provided by chromatin accessibility analysis in mouse mesendoderm embryoid bodies and in PDAC cells [13]. The chromatin at SMAD2/3 and RREB1 binding sites in the Snai1 promoter was accessible both in mesendoderm progenitors and PDAC cells. In contrast, the chromatin at SMAD2/3 and RREB1 binding sites in Gsc and Mixl1 was accessible in mesendoderm progenitors, but not in PDAC cells, and the contrary was true of chromatin accessibility in Wisp1 and Serpine1 (Fig. 4C). Thus, different chromatin patterns enable signal-activated SMAD and RREB1 to access either fibrogenic or mesendoderm programs in these two distinct cellular contexts, and access Snai1 in both contexts.
8. Perspectives
In this review, we have highlighted how cells utilize TGF-β signals to trigger EMTs in different contexts and for different purposes. The regulation of EMT and its associated programs provides a clear example of convergence of three proposed classes of determinants shaping the contextual nature of TGF-β effects [10], namely, SMAD regulatory signals (RAS-MAPK in this case), SMAD transcriptional partners (RREB1), and the chromatin status of target loci. Combined, these three classes of determinants shape TGF-β induced EMTs as part of broader programs in different physiological and pathological contexts [13].
Despite the recent progress in understanding the basic principles of TGF-β signaling and biology, we still lack a complete understanding of transcriptional regulation by TGF-β and its diverse roles under different conditions. For example, we do not know why certain SMAD target genes require RREB1 for activation and others do not. Although RREB1 is required for transcriptional activation of all RAS-dependent TGF-β gene responses, it is required for SMAD binding to the regulatory regions of only some of these, and more so in PDAC cells than in mammary epithelial cells [13]. If the main function of RREB1 is not recruiting SMADs to target genes (FOXH1 serves this function in mesendoderm progenitors [32,98]), what biochemical functions then does RREB1 bring to its collaboration with SMADs in the transcriptional activation of these genes?
In the context of fibrosis, if TGF-β is a direct and potent activator of stromal fibroblasts, what is the role of the TGF-β-induced production of fibrogenic factors by epithelial cells undergoing EMT? Do the epithelial derived fibrogenic factors serve to complement the direct effects of TGF-β on stromal fibroblasts? How important is this contribution, and when in the course of a wound healing or a fibrotic process?
The simplicity of the scheme in Fig. 2A belies hidden complexities that our current knowledge cannot explain. For example, in mouse embryonic stem cells which form embryoid bodies recapitulating signaling and lineage specification events of gastrulation, absence of RREB1 causes a complete loss of EMT and mesendoderm differentiation responses to Nodal [13]. However, when these Rreb1−/− ESCs were injected into wild-type mouse blastocysts and allowed to develop in utero, the resulting chimeric embryos showed gastrulation defects, but the Rreb1−/− cells in these embryos did not exhibit an absolute EMT block [13]. Moreover, germline Rreb1-null mice showed altered organization of actin filaments and adherens junctions within the epiblast, perturbed epithelial architecture, and ectopic migration through the underlying basement membrane, but many of the cells still underwent EMTs [146]. These observations suggest that Nodal- and RREB1-dependent EMTs are amalgamated with other EMT programs in embryos, where the absence of RREB1 activity may be partly buffered by other transcriptional cofactors.
To address some of these questions, it will be important to identify other critical components of SMAD-RREB1 complexes. This would provide a more complete understanding of the biochemical function of RREB1 in transcriptional regulation and reveal factors that modulate the function of this complex and partly buffer the loss of RREB1 in the precise execution of TGF-β dependent EMT programs. Defining the distinct chromatin features of TGF-β target genes in different EMT-associated contexts will also further clarify the requirement for a collaboration between TGF-β-SMAD and RAS-MAPK pathways through RREB1. Addressing these and other related questions will be important towards advancing our understanding of TGF-β EMTs in development, regeneration, fibrosis, and cancer.
Acknowledgements
We thank Anna-Katerina Hadjantonakis for input on developmental aspects of EMT, and Gabriella Johnson for help with the manuscript. The authors’ work in the subject area of this review is supported by U.S. National Cancer Institute grants R35-CA252978 (JM) and P30-CA008748 (MSKCC), and a postdoctoral fellowship (JHL) and grant support (JM) from the Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center at MSKCC.
Footnotes
Conflict of Interest statement
JM owns company stock in Scholar Rock.
Data availability
No data was used for the research described in the article.
References
- [1].Nieto MA, Huang RY, Jackson RA, Thiery JP, EMT: 2016, Cell 166 (1) (2016) 21–45. [DOI] [PubMed] [Google Scholar]
- [2].Dongre A, Weinberg RA, New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer, Nat. Rev. Mol. Cell Biol 20 (2) (2019) 69–84. [DOI] [PubMed] [Google Scholar]
- [3].Yan W, Cao QJ, Arenas RB, Bentley B, Shao R, GATA3 inhibits breast cancer metastasis through the reversal of epithelial-mesenchymal transition, J. Biol. Chem 285 (18) (2010) 14042–14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Martinelli P, Carrillo-de Santa Pau E, Cox T, Sainz B Jr., Dusetti N, Greenhalf W, Rinaldi L, Costello E, Ghaneh P, Malats N, Büchler M, Pajic M, Biankin AV, Iovanna J, Neoptolemos J, Real FX, GATA6 regulates EMT and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer, Gut 66 (9) (2017) 1665–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Akiyama H, Chaboissier MC, Behringer RR, Rowitch DH, Schedl A, Epstein JA, de Crombrugghe B, Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa, Proc. Natl. Acad. Sci. U. S. A 101 (17) (2004) 6502–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ocaña OH, Córcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA, Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1, Cancer Cell 22 (6) (2012) 709–724. [DOI] [PubMed] [Google Scholar]
- [7].Hartwell KA, Muir B, Reinhardt F, Carpenter AE, Sgroi DC, Weinberg RA, The Spemann organizer gene, Goosecoid, promotes tumor metastasis, Proc. Natl. Acad. Sci. U. S. A 103 (50) (2006) 18969–18974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fernando RI, Litzinger M, Trono P, Hamilton DH, Schlom J, Palena C, The T-box transcription factor Brachyury promotes epithelial-mesenchymal transition in human tumor cells, J. Clin. Invest 120 (2) (2010) 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yu M, Smolen GA, Zhang J, Wittner B, Schott BJ, Brachtel E, Ramaswamy S, Maheswaran S, Haber DA, A developmentally regulated inducer of EMT, LBX1, contributes to breast cancer progression, Genes Dev. 23 (15) (2009) 1737–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Massagué J, TGFβ signalling in context, Nat. Rev. Mol. Cell Biol 13 (10) (2012) 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].David CJ, Massagué J, Contextual determinants of TGFβ action in development, immunity and cancer, Nat. Rev. Mol. Cell Biol 19 (7) (2018) 419–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, Massagué J, TGF-β Tumor Suppression through a Lethal EMT, Cell 164 (5) (2016) 1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Su J, Morgani SM, David CJ, Wang Q, Er EE, Huang YH, Basnet H, Zou Y, Shu W, Soni RK, Hendrickson RC, Hadjantonakis AK, Massagué J, TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1, Nature 577 (7791) (2020) 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Trelstad RL, Hayashi A, Hayashi K, Donahoe PK, The epithelial-mesenchymal interface of the male rate Mullerian duct: loss of basement membrane integrity and ductal regression, Dev. Biol 92 (1) (1982) 27–40. [DOI] [PubMed] [Google Scholar]
- [15].Potts JD, Runyan RB, Epithelial-mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor beta, Dev. Biol 134 (2) (1989) 392–401. [DOI] [PubMed] [Google Scholar]
- [16].Potts JD, Dagle JM, Walder JA, Weeks DL, Runyan RB, Epithelial-mesenchymal transformation of embryonic cardiac endothelial cells is inhibited by a modified antisense oligodeoxynucleotide to transforming growth factor beta 3, Proc. Natl. Acad. Sci. U. S. A 88 (4) (1991) 1516–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Miettinen PJ, Ebner R, Lopez AR, Derynck R, TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors, J. Cell Biol 127 (6 Pt 2) (1994) 2021–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Katsuno Y, Derynck R, Epithelial plasticity, epithelial-mesenchymal transition, and the TGF-β family, Dev. Cell 56 (6) (2021) 726–746. [DOI] [PubMed] [Google Scholar]
- [19].Schäfer G, Narasimha M, Vogelsang E, Leptin M, Cadherin switching during the formation and differentiation of the Drosophila mesoderm - implications for epithelial-to-mesenchymal transitions, J. Cell Sci 127 (Pt 7) (2014) 1511–1522. [DOI] [PubMed] [Google Scholar]
- [20].Carver EA, Jiang R, Lan Y, Oram KF, Gridley T, The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition, Mol. Cell Biol 21 (23) (2001) 8184–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Economou AD, Hill CS, Temporal dynamics in the formation and interpretation of Nodal and BMP morphogen gradients, Curr. Top. Dev. Biol 137 (2020) 363–389. [DOI] [PubMed] [Google Scholar]
- [22].Vincent SD, Dunn NR, Hayashi S, Norris DP, Robertson EJ, Cell fate decisions within the mouse organizer are governed by graded Nodal signals, Genes Dev. 17 (13) (2003) 1646–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Romano LA, Runyan RB, Slug is an essential target of TGFbeta2 signaling in the developing chicken heart, Dev. Biol 223 (1) (2000) 91–102. [DOI] [PubMed] [Google Scholar]
- [24].Oshimori N, Oristian D, Fuchs E, TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma, Cell 160 (5) (2015) 963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, Deschler DG, Varvares MA, Mylvaganam R, Rozenblatt-Rosen O, Rocco JW, Faquin WC, Lin DT, Regev A, Bernstein BE, Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer, Cell 171 (7) (2017) 1611–1624, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Huang YH, Hu J, Chen F, Lecomte N, Basnet H, David CJ, Witkin MD, Allen PJ, Leach SD, Hollmann TJ, Iacobuzio-Donahue CA, Massagué J, ID1 mediates escape from TGFβ tumor suppression in pancreatic cancer, Cancer Disco 10 (1) (2020) 142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, Schwabe RF, Vahdat LT, Altorki NK, Mittal V, Gao D, Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance, Nature 527 (7579) (2015) 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R, Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer, Nature 527 (7579) (2015) 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Vega S, Morales AV, Ocaña OH, Valdés F, Fabregat I, Nieto MA, Snail blocks the cell cycle and confers resistance to cell death, Genes Dev. 18 (10) (2004) 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY, Bapat SA, Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells, Stem Cells 27 (9) (2009) 2059–2068. [DOI] [PubMed] [Google Scholar]
- [31].Batlle E, Massagué J, Transforming growth factor-β signaling in immunity and cancer, Immunity 50 (4) (2019) 924–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Aragón E, Wang Q, Zou Y, Morgani SM, Ruiz L, Kaczmarska Z, Su J, Torner C, Tian L, Hu J, Shu W, Agrawal S, Gomes T, Márquez JA, Hadjantonakis AK, Macias MJ, Massagué J, Structural basis for distinct roles of SMAD2 and SMAD3 in FOXH1 pioneer-directed TGF-β signaling, Genes Dev. 33 (21–22) (2019) 1506–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hata A, Seoane J, Lagna G, Montalvo E, Hemmati-Brivanlou A, Massagué J, OAZ uses distinct DNA- and protein-binding zinc fingers in separate BMP-Smad and Olf signaling pathways, Cell 100 (2) (2000) 229–240. [DOI] [PubMed] [Google Scholar]
- [34].Trompouki E, Bowman TV, Lawton LN, Fan ZP, Wu DC, DiBiase A, Martin CS, Cech JN, Sessa AK, Leblanc JL, Li P, Durand EM, Mosimann C, Heffner GC, Daley GQ, Paulson RF, Young RA, Zon LI, Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration, Cell 147 (3) (2011) 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mullen AC, Orlando DA, Newman JJ, Lovén J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, DeKoter RP, Young RA, Master transcription factors determine cell-type-specific responses to TGF-β signaling, Cell 147 (3) (2011) 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M, Smad4 and FAST-1 in the assembly of activin-responsive factor, Nature 389 (6646) (1997) 85–89. [DOI] [PubMed] [Google Scholar]
- [37].Chen X, Rubock MJ, Whitman M, A transcriptional partner for MAD proteins in TGF-beta signalling, Nature 383 (6602) (1996) 691–696. [DOI] [PubMed] [Google Scholar]
- [38].Qing J, Zhang Y, Derynck R, Structural and functional characterization of the transforming growth factor-beta -induced Smad3/c-Jun transcriptional cooperativity, J. Biol. Chem 275 (49) (2000) 38802–38812. [DOI] [PubMed] [Google Scholar]
- [39].Sundqvist A, Zieba A, Vasilaki E, Herrera Hidalgo C, Söderberg O, Koinuma D, Miyazono K, Heldin CH, Landegren U, Ten Dijke P, van Dam H, Specific interactions between Smad proteins and AP-1 components determine TGFβ-induced breast cancer cell invasion, Oncogene 32 (31) (2013) 3606–3615. [DOI] [PubMed] [Google Scholar]
- [40].Sundqvist A, Morikawa M, Ren J, Vasilaki E, Kawasaki N, Kobayashi M, Koinuma D, Aburatani H, Miyazono K, Heldin CH, van Dam H, Ten Dijke P, JUNB governs a feed-forward network of TGFβ signaling that aggravates breast cancer invasion, Nucleic Acids Res. 46 (3) (2018) 1180–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Seoane J, Le HV, Shen L, Anderson SA, Massagué J, Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation, Cell 117 (2) (2004) 211–223. [DOI] [PubMed] [Google Scholar]
- [42].Pouponnot C, Jayaraman L, Massagué J, Physical and functional interaction of SMADs and p300/CBP, J. Biol. Chem 273 (36) (1998) 22865–22868. [DOI] [PubMed] [Google Scholar]
- [43].Janknecht R, Wells NJ, Hunter T, TGF-beta-stimulated cooperation of smad proteins with the coactivators CBP/p300, Genes Dev. 12 (14) (1998) 2114–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wotton D, Lo RS, Swaby LA, Massagué J, Multiple modes of repression by the Smad transcriptional corepressor TGIF, J. Biol. Chem 274 (52) (1999) 37105–37110. [DOI] [PubMed] [Google Scholar]
- [45].Xi Q, He W, Zhang XH, Le HV, Massagué J, Genome-wide impact of the BRG1 SWI/SNF chromatin remodeler on the transforming growth factor beta transcriptional program, J. Biol. Chem 283 (2) (2008) 1146–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ross S, Cheung E, Petrakis TG, Howell M, Kraus WL, Hill CS, Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription, Embo J. 25 (19) (2006) 4490–4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A, The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells, Nat. Cell Biol 2 (2) (2000) 84–89. [DOI] [PubMed] [Google Scholar]
- [48].Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA, The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression, Nat. Cell Biol 2 (2) (2000) 76–83. [DOI] [PubMed] [Google Scholar]
- [49].Bolós V, Peinado H, Pérez-Moreno MA, Fraga MF, Esteller M, Cano A, The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors, J. Cell Sci 116 (Pt 3) (2003) 499–511. [DOI] [PubMed] [Google Scholar]
- [50].Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A, Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction, J. Clin. Invest 112 (10) (2003) 1486–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Morita T, Mayanagi T, Sobue K, Dual roles of myocardin-related transcription factors in epithelial–mesenchymal transition via slug induction and actin remodeling, J. Cell Biol 179 (5) (2007) 1027–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Du D, Katsuno Y, Meyer D, Budi EH, Chen SH, Koeppen H, Wang H, Akhurst RJ, Derynck R, Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial-mesenchymal transition, EMBO Rep. 19 (1) (2018) 135–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Deckers M, van Dinther M, Buijs J, Que I, Löwik C, van der Pluijm G, ten Dijke P, The tumor suppressor Smad4 is required for transforming growth factor beta-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells, Cancer Res. 66 (4) (2006) 2202–2209. [DOI] [PubMed] [Google Scholar]
- [54].Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A, Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro, J. Biol. Chem 282 (30) (2007) 22089–22101. [DOI] [PubMed] [Google Scholar]
- [55].Saika S, Kono-Saika S, Ohnishi Y, Sato M, Muragaki Y, Ooshima A, Flanders KC, Yoo J, Anzano M, Liu CY, Kao WW, Roberts AB, Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury, Am. J. Pathol 164 (2) (2004) 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Takano S, Kanai F, Jazag A, Ijichi H, Yao J, Ogawa H, Enomoto N, Omata M, Nakao A, Smad4 is essential for down-regulation of E-cadherin induced by TGF-beta in pancreatic cancer cell line PANC-1, J. Biochem 141 (3) (2007) 345–351. [DOI] [PubMed] [Google Scholar]
- [57].Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, Crystal RG, de Herreros AG, Moustakas A, Pettersson RF, Fuxe J, A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition, Nat. Cell Biol 11 (8) (2009) 943–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Postigo AA, Depp JL, Taylor JJ, Kroll KL, Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins, Embo J. 22 (10) (2003) 2453–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tan EJ, Thuault S, Caja L, Carletti T, Heldin CH, Moustakas A, Regulation of transcription factor Twist expression by the DNA architectural protein high mobility group A2 during epithelial-to-mesenchymal transition, J. Biol. Chem 287 (10) (2012) 7134–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Thuault S, Tan EJ, Peinado H, Cano A, Heldin CH, Moustakas A, HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition, J. Biol. Chem 283 (48) (2008) 33437–33446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A, Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition, J. Cell Biol 174 (2) (2006) 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lin T, Ponn A, Hu X, Law BK, Lu J, Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition, Oncogene 29 (35) (2010) 4896–4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ramadoss S, Chen X, Wang CY, Histone demethylase KDM6B promotes epithelial-mesenchymal transition, J. Biol. Chem 287 (53) (2012) 44508–44517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Li D, Sun H, Sun WJ, Bao HB, Si SH, Fan JL, Lin P, Cui RJ, Pan YJ, Wen SM, Zheng XL, Yu XG, Role of RbBP5 and H3K4me3 in the vicinity of Snail transcription start site during epithelial-mesenchymal transition in prostate cancer cell, Oncotarget 7 (40) (2016) 65553–65567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cardenas H, Zhao J, Vieth E, Nephew KP, Matei D, EZH2 inhibition promotes epithelial-to-mesenchymal transition in ovarian cancer cells, Oncotarget 7 (51) (2016) 84453–84467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Shirakihara T, Saitoh M, Miyazono K, Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta, Mol. Biol. Cell 18 (9) (2007) 3533–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nishimura G, Manabe I, Tsushima K, Fujiu K, Oishi Y, Imai Y, Maemura K, Miyagishi M, Higashi Y, Kondoh H, Nagai R, DeltaEF1 mediates TGF-beta signaling in vascular smooth muscle cell differentiation, Dev. Cell 11 (1) (2006) 93–104. [DOI] [PubMed] [Google Scholar]
- [68].Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E, TGF-beta1 and Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells, Genes Dev. 10 (19) (1996) 2462–2477. [DOI] [PubMed] [Google Scholar]
- [69].Gotzmann J, Huber H, Thallinger C, Wolschek M, Jansen B, Schulte-Hermann R, Beug H, Mikulits W, Hepatocytes convert to a fibroblastoid phenotype through the cooperation of TGF-beta1 and Ha-Ras: steps towards invasiveness, J. Cell Sci 115 (Pt 6) (2002) 1189–1202. [DOI] [PubMed] [Google Scholar]
- [70].Oft M, Akhurst RJ, Balmain A, Metastasis is driven by sequential elevation H-ras and Smad2 levels, Nat. Cell Biol 4 (7) (2002) 487–494. [DOI] [PubMed] [Google Scholar]
- [71].Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, Weinberg RA, Distinct EMT programs control normal mammary stem cells and tumour-initiating cells, Nature 525 (7568) (2015) 256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chakrabarti R, Hwang J, Andres Blanco M, Wei Y, Lukačišin M, Romano RA, Smalley K, Liu S, Yang Q, Ibrahim T, Mercatali L, Amadori D, Haffty BG, Sinha S, Kang Y, Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2, Nat. Cell Biol 14 (11) (2012) 1212–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL, Activation the Erk pathway is required for TGF-beta1-induced EMT in vitro, Neoplasia 6 (2004) 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lehmann K, Janda E, Pierreux CE, Rytömaa M, Schulze A, McMahon M, Hill CS, Beug H, Downward J, Raf induces TGFbeta production while blocking apoptotic but not invasive responses: a mechanism leading to increased malignancy in epithelial cells, Genes Dev. 14 (20) (2000) 2610–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H, Grünert S, Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways, J. Cell Biol 156 (2) (2002) 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Fischer AN, Herrera B, Mikula M, Proell V, Fuchs E, Gotzmann J, Schulte-Hermann R, Beug H, Mikulits W, Integration of Ras subeffector signaling in TGF-beta mediated late stage hepatocarcinogenesis, Carcinogenesis 26 (5) (2005) 931–942. [DOI] [PubMed] [Google Scholar]
- [77].Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M, Role of Ras signaling in the induction of snail by transforming growth factor-beta, J. Biol. Chem 284 (1) (2009) 245–253. [DOI] [PubMed] [Google Scholar]
- [78].Meno C, Gritsman K, Ohishi S, Ohfuji Y, Heckscher E, Mochida K, Shimono A, Kondoh H, Talbot WS, Robertson EJ, Schier AF, Hamada H, Mouse Lefty2 and zebrafish antivin are feedback inhibitors of nodal signaling during vertebrate gastrulation, Mol. Cell 4 (3) (1999) 287–298. [DOI] [PubMed] [Google Scholar]
- [79].Sun X, Meyers EN, Lewandoski M, Martin GR, Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo, Genes Dev. (14) (1999) 1834–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J, fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation, Genes Dev. 8 (24) (1994) 3032–3044. [DOI] [PubMed] [Google Scholar]
- [81].Zhou X, Sasaki H, Lowe L, Hogan BL, Kuehn MR, Nodal is a novel TGF-beta-like gene expressed in the mouse node during gastrulation, Nature 361 (6412) (1993) 543–547. [DOI] [PubMed] [Google Scholar]
- [82].Peinado H, Quintanilla M, Cano A, Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions, J. Biol. Chem 278 (23) (2003) 21113–21123. [DOI] [PubMed] [Google Scholar]
- [83].Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E, Localized reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility, Nat. Cell Biol 11 (11) (2009) 1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ciruna B, Rossant J, FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak, Dev. Cell 1 (1) (2001) 37–49. [DOI] [PubMed] [Google Scholar]
- [85].Kemler R, Hierholzer A, Kanzler B, Kuppig S, Hansen K, Taketo MM, Vries WN, Knowles BB, Solter D, Stabilization of beta-catenin in the mouse zygote leads to premature epithelial-mesenchymal transition in the epiblast, Development 131 (23) (2004) 5817–5824. [DOI] [PubMed] [Google Scholar]
- [86].Ben-Haim N, Lu C, Guzman-Ayala M, Pescatore L, Mesnard D, Bischofberger M, Naef F, Robertson EJ, Constam DB, The nodal precursor acting via activin receptors induces mesoderm by maintaining a source of its convertases and BMP4, Dev. Cell 11 (3) (2006) 313–323. [DOI] [PubMed] [Google Scholar]
- [87].Wang Q, Zou Y, Nowotschin S, Kim SY, Li QV, Soh CL, Su J, Zhang C, Shu W, Xi Q, Huangfu D, Hadjantonakis AK, Massagué J, The p53 Family Coordinates Wnt and Nodal Inputs in Mesendodermal Differentiation of Embryonic Stem Cells, Cell Stem Cell 20 (1) (2017) 70–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Yu L, Hébert MC, Zhang YE, TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses, Embo J. 21 (14) (2002) 3749–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lamouille S, Derynck R, Emergence of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin axis in transforming growth factor-β-induced epithelial-mesenchymal transition, Cells Tissues Organs 193 (1–2) (2011) 8–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R, TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion, J. Cell Sci 125 (Pt 5) (2012) 1259–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Melani M, Simpson KJ, Brugge JS, Montell D, Regulation of cell adhesion and collective cell migration by hindsight and its human homolog RREB1, Curr. Biol 18 (7) (2008) 532–537. [DOI] [PubMed] [Google Scholar]
- [92].Pan D, Huang L, Zhu LJ, Zou T, Ou J, Zhou W, Wang YX, Jmjd3-Mediated H3K27me3 dynamics orchestrate brown fat development and regulate white fat plasticity, Dev. Cell 35 (5) (2015) 568–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ray SK, Li HJ, Metzger E, Schüle R, Leiter AB, CtBP and associated LSD1 are required for transcriptional activation by NeuroD1 in gastrointestinal endocrine cells, Mol. Cell Biol 34 (12) (2014) 2308–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kent OA, Fox-Talbot K, Halushka MK, RREB1 repressed miR-143/145 modulates KRAS signaling through downregulation of multiple targets, Oncogene 32 (20) (2013) 2576–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Costello LC, Zou J, Desouki MM, Franklin RB, Evidence for changes in RREB-1, ZIP3, and Zinc in the early development of pancreatic adenocarcinoma, J. Gastrointest. Cancer 43 (4) (2012) 570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma, Cancer Cell 32(2) (2017) 185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lim J, Thiery JP, Epithelial-mesenchymal transitions: insights from development, Development 139 (19) (2012) 3471–3486. [DOI] [PubMed] [Google Scholar]
- [98].Xi Q, Wang Z, Zaromytidou AI, Zhang XH, Chow-Tsang LF, Liu JX, Kim H, Barlas A, Manova-Todorova K, Kaartinen V, Studer L, Mark W, Patel DJ, Massagué J, A poised chromatin platform for TGF-β access to master regulators, Cell 147 (7) (2011) 1511–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Correia AC, Costa M, Moraes F, Bom J, Nóvoa A, Mallo M, Bmp2 is required for migration but not for induction of neural crest cells in the mouse, Dev. Dyn 236 (9) (2007) 2493–2501. [DOI] [PubMed] [Google Scholar]
- [100].Betancur P, Bronner-Fraser M, Sauka-Spengler T, Assembling neural crest regulatory circuits into a gene regulatory network, Annu Rev. Cell Dev. Biol 26 (2010) 581–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].van Meeteren LA, ten Dijke P, Regulation of endothelial cell plasticity by TGF-β, Cell Tissue Res. 347 (1) (2012) 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Rygiel KA, Robertson H, Marshall HL, Pekalski M, Zhao L, Booth TA, Jones DE, Burt AD, Kirby JA, Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease, Lab Invest. 88 (2) (2008) 112–123. [DOI] [PubMed] [Google Scholar]
- [103].Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z, Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis, Am. J. Pathol 166 (5) (2005) 1321–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Coulombe PA, Wound epithelialization: accelerating the pace of discovery, J. Invest Dermatol 121 (2) (2003) 219–230. [DOI] [PubMed] [Google Scholar]
- [105].Haensel D, Jin S, Sun P, Cinco R, Dragan M, Nguyen Q, Cang Z, Gong Y, Vu R, MacLean AL, Kessenbrock K, Gratton E, Nie Q, Dai X, Defining epidermal basal cell states during skin homeostasis and wound healing using single-cell transcriptomics, Cell Rep. 30 (11) (2020) 3932–3947, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Gilles C, Polette M, Zahm JM, Tournier JM, Volders L, Foidart JM, Birembaut P, Vimentin contributes to human mammary epithelial cell migration, J. Cell Sci 112 (Pt 24) (1999) 4615–4625. [DOI] [PubMed] [Google Scholar]
- [107].Park S, Gonzalez DG, Guirao B, Boucher JD, Cockburn K, Marsh ED, Mesa KR, Brown S, Rompolas P, Haberman AM, Bellaïche Y, Greco V, Tissue-scale coordination of cellular behaviour promotes epidermal wound repair in live mice, Nat. Cell Biol 19 (2) (2017) 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Aragona M, Dekoninck S, Rulands S, Lenglez S, Mascŕe G, Simons BD, Blanpain C, Defining stem cell dynamics and migration during wound healing in mouse skin epidermis, Nat. Commun 8 (2017) 14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Shaw TJ, Martin P, Wound repair at a glance, J. Cell Sci 122 (Pt 18) (2009) 3209–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Lemmon MA, Schlessinger J, Cell signaling by receptor tyrosine kinases, Cell 141 (7) (2010) 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Tsai JH, Yang J, Epithelial-mesenchymal plasticity in carcinoma metastasis, Genes Dev. 27 (20) (2013) 2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Escuin-Ordinas H, Li S, Xie MW, Sun L, Hugo W, Huang RR, Jiao J, de-Faria FM, Realegeno S, Krystofinski P, Azhdam A, Komenan SM, Atefi M, Comin-Anduix B, Pellegrini M, Cochran AJ, Modlin RL, Herschman HR, Lo RS, McBride WH, Segura T, Ribas A, Cutaneous wound healing through paradoxical MAPK activation by BRAF inhibitors, Nat. Commun 7 (2016) 12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Desmoulìere A, Geinoz A, Gabbiani F, Gabbiani G, Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts, J. Cell Biol 122 (1) (1993) 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Tamaoki M, Imanaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, Sakakura T, Yoshida T, Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury, Am. J. Pathol 167 (1) (2005) 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lee CH, Shah B, Moioli EK, Mao JJ, CTGF directs fibroblast differentiation from human mesenchymal stem/stromal cells and defines connective tissue healing in a rodent injury model, J. Clin. Invest 120 (9) (2010) 3340–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Romberger DJ, Beckmann JD, Claassen L, Ertl RF, Rennard SI, Modulation of fibronectin production of bovine bronchial epithelial cells by transforming growth factor-beta, Am. J. Respir. Cell Mol. Biol 7 (2) (1992) 149–155. [DOI] [PubMed] [Google Scholar]
- [117].Wang Y, Moges H, Bharucha Y, Symes A, Smad3 null mice display more rapid wound closure and reduced scar formation after a stab wound to the cerebral cortex, Exp. Neurol 203 (1) (2007) 168–184. [DOI] [PubMed] [Google Scholar]
- [118].Grande MT, Sánchez-Laorden B, López-Blau C, De Frutos CA, Boutet A, Aŕevalo M, Rowe RG, Weiss SJ, López-Novoa JM, Nieto MA, Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease, Nat. Med 21 (9) (2015) 989–997. [DOI] [PubMed] [Google Scholar]
- [119].Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC, Pentcheva-Hoang T, Nischal H, Allison JP, Zeisberg M, Kalluri R, Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis, Nat. Med 21 (9) (2015) 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Shi Y, Hata A, Lo RS, Massagué J, Pavletich NP, A structural basis for mutational inactivation of the tumour suppressor Smad4, Nature 388 (6637) (1997) 87–93. [DOI] [PubMed] [Google Scholar]
- [121].Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, et al. , Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability, Science 268 (5215) (1995) 1336–1338. [DOI] [PubMed] [Google Scholar]
- [122].Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massagué J, Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals, Cell 78 (1) (1994) 59–66. [DOI] [PubMed] [Google Scholar]
- [123].Reynisdóttir I, Polyak K, Iavarone A, Massagué J, Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta, Genes Dev. 9 (15) (1995) 1831–1845. [DOI] [PubMed] [Google Scholar]
- [124].Hannon GJ, Beach D, p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest, Nature 371 (6494) (1994) 257–261. [DOI] [PubMed] [Google Scholar]
- [125].Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, DePinho RA, Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer, Genes Dev. 20 (22) (2006) 3130–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Aiello NM, Kang Y, Context-dependent EMT programs in cancer metastasis, J. Exp. Med 216 (5) (2019) 1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Thiery JP, Acloque H, Huang RY, Nieto MA, Epithelial-mesenchymal transitions in development and disease, Cell 139 (5) (2009) 871–890. [DOI] [PubMed] [Google Scholar]
- [128].Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, Brannigan BW, Kapur R, Stott SL, Shioda T, Ramaswamy S, Ting DT, Lin CP, Toner M, Haber DA, Maheswaran S, Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis, Cell 158 (5) (2014) 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Aceto N, Toner M, Maheswaran S, Haber DA, En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition, Trends Cancer 1 (1) (2015) 44–52. [DOI] [PubMed] [Google Scholar]
- [130].Lim M, Park S, Jeong HO, Park SH, Kumar S, Jang A, Lee S, Kim DU, Cho YK, Circulating tumor cell clusters are cloaked with platelets and correlate with poor prognosis in unresectable pancreatic cancer, Cancers (Basel) 13 (21) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Labelle M, Begum S, Hynes RO, Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis, Cancer Cell 20 (5) (2011) 576–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Brown JA, Yonekubo Y, Hanson N, Sastre-Perona A, Basin A, Rytlewski JA, Dolgalev I, Meehan S, Tsirigos A, Beronja S, Schober M, TGF-β-Induced quiescence mediates chemoresistance of tumor-propagating cells in squamous cell carcinoma, Cell Stem Cell 21 (5) (2017) 650–664, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Yu-Lee LY, Yu G, Lee YC, Lin SC, Pan J, Pan T, Yu KJ, Liu B, Creighton CJ, Rodriguez-Canales J, Villalobos PA, Wistuba II E, de Nadal F, Posas GE, Gallick Lin SH, Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFβRIII-p38MAPK-pS249/T252RB pathway, Cancer Res. 78 (11) (2018) 2911–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DY, Chen EI, Lyden D, Bissell MJ, The perivascular niche regulates breast tumour dormancy, Nat. Cell Biol 15 (7) (2013) 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E, Massagué J, Metastatic latency and immune evasion through autocrine inhibition of WNT, Cell 165 (1) (2016) 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J, Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis, Cancer Cell 22 (6) (2012) 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, Massagué J, Benezra R, TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition, Cell Rep. 5 (5) (2013) 1228–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D, Moers V, Lemaire S, De Clercq S, Minguijón E, Balsat C, Sokolow Y, Dubois C, De Cock F, Scozzaro S, Sopena F, Lanas A, D’Haene N, Salmon I, Marine JC, Voet T, Sotiropoulou PA, Blanpain C, Identification of the tumour transition states occurring during EMT, Nature 556 (7702) (2018) 463–468. [DOI] [PubMed] [Google Scholar]
- [139].Ganesh K, Massagué J, Targeting metastatic cancer, Nat. Med 27 (1) (2021) 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Massagué J, Ganesh K, Metastasis-initiating cells and ecosystems, Cancer Disco 11 (4) (2021) 971–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Schäfer M, Werner S, Cancer as an overhealing wound: an old hypothesis revisited, Nat. Rev. Mol. Cell Biol 9 (8) (2008) 628–638. [DOI] [PubMed] [Google Scholar]
- [142].Jacobs TW, Byrne C, Colditz G, Connolly JL, Schnitt SJ, Radial scars in benign breast-biopsy specimens and the risk of breast cancer, N. Engl. J. Med 340 (6) (1999) 430–436. [DOI] [PubMed] [Google Scholar]
- [143].Yu Y-Y, Pinsky PF, Caporaso NE, Chatterjee N, Baumgarten M, Langenberg P, Furuno JP, Lan Q, Engels EA, Lung cancer risk following detection of pulmonary scarring by chest radiography in the prostate, lung, colorectal, and ovarian cancer screening trial, Arch. Intern Med 168 (21) (2008) 2326–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Saluja A, Maitra A, Pancreatitis and pancreatic cancer, Gastroenterology 156 (7) (2019) 1937–1940. [DOI] [PubMed] [Google Scholar]
- [145].Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, Breart B, Foreman O, Bainbridge TW, Castiglioni A, Senbabaoglu Y, Modrusan Z, Liang Y, Junttila MR, Klijn C, Bourgon R, Turley SJ, Single-cell RNA sequencing reveals stromal evolution into LRRC15(+) myofibroblasts as a determinant of patient response to cancer immunotherapy, Cancer Disco 10 (2) (2020) 232–253. [DOI] [PubMed] [Google Scholar]
- [146].Morgani SM, Su J, Nichols J, Massagué J, Hadjantonakis AK, The transcription factor Rreb1 regulates epithelial architecture, invasiveness, and vasculogenesis in early mouse embryos, Elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.