

ABSTRACT
Background
Inflammation is a key driver of the transition of acute kidney injury to progressive fibrosis and chronic kidney disease (AKI-to-CKD transition). Blocking a-disintegrin-and-metalloprotease-17 (ADAM17)-dependent ectodomain shedding, in particular of epidermal growth factor receptor (EGFR) ligands and of the type 1 inflammatory cytokine tumor necrosis factor (TNF), reduces pro-inflammatory and pro-fibrotic responses after ischemic AKI or unilateral ureteral obstruction (UUO), a classical fibrosis model. Metalloprotease or EGFR inhibition show significant undesirable side effects in humans. In retrospective studies anti-TNF biologics reduce the incidence and progression of CKD in humans. Whether TNF has a role in AKI-to-CKD transition and how TNF inhibition compares to EGFR inhibition is largely unknown.
Methods
Mice were subjected to bilateral renal ischemia-reperfusion injury or unilateral ureteral obstruction. Kidneys were analyzed by histology, immunohistochemistry, qPCR, western blot, mass cytometry, scRNA sequencing, and cytokine profiling.
Results
Here we show that TNF or EGFR inhibition reduce AKI-to-CKD transition and fibrosis equally by about 25%, while combination has no additional effect. EGFR inhibition reduced kidney TNF expression by about 50% largely by reducing accumulation of TNF expressing immune cells in the kidney early after AKI, while TNF inhibition did not affect EGFR activation or immune cell accumulation. Using scRNAseq data we show that TNF is predominantly expressed by immune cells in AKI but not in proximal tubule cells (PTC), and PTC-TNF knockout did not affect AKI-to-CKD transition in UUO. Thus, the anti-inflammatory and anti-fibrotic effects of the anti-TNF biologic etanercept in AKI-to-CKD transition rely on blocking TNF that is released from immune cells recruited or accumulating in response to PTC-EGFR signals.
Conclusion
Short-term anti-TNF biologics during or after AKI could be helpful in the prevention of AKI-to-CKD transition.
Keywords: acute kidney injury, chronic kidney disease, epidermal growth factor receptor, fibrosis, TNF
Graphical Abstract
Graphical Abstract.

KEY LEARNING POINTS.
What is already known about this subject?
Epidermal growth factor receptor (EGFR) ligands and tumor necrosis factor (TNF) are key substrates of the metalloprotease a-disintegrin-and-metalloprotease-17 (ADAM17). Inhibition of ADAM17 or of EGFR activation in acute kidney injury (AKI) has anti-fibrotic effects. Whether TNF has a role in AKI-to-CKD transition and fibrosis is unresolved.
Metalloprotease and EGFR inhibition have undesirable side-effects in humans.
In retrospective studies anti-TNF biologics, such as etanercept, reduce incidence and progression of chronic kidney disease (CKD).
What this study adds?
TNF inhibition with etanercept is as efficient in preventing AKI-to-CKD transition and kidney fibrosis as is EGFR inhibition.
TNF and EGFR inhibition show a significant overlap in reducing kidney cytokines after AKI.
Mechanistically, we show that EGFR inhibition strongly blunts kidney immune cell ingress and accumulation after injury. In contrast, TNF inhibition does not, but rather exerts its effects by blocking TNF released by recruited/accumulated immune cells.
What impact this may have on practice or policy?
Anti-TNF biologics given short-term to prevent AKI-to-CKD transition could be studied prospectively.
INTRODUCTION
Inflammation is a key driver of acute kidney injury to chronic kidney disease transition (AKI-to-CKD transition), a frequent clinical diagnosis that signals the transition to kidney fibrosis with progressive loss of kidney function that carries high morbidity and mortality due to a lack of specific therapies [1–4]. In mice, an anti-inflammatory and anti-fibrotic effect of epidermal growth factor receptor (EGFR) inhibition or of EGFR-knockout in proximal-tubule-cells (PTC) in CKD models was first reported by the Friedlander laboratory and later confirmed and extended to AKI-induced fibrosis by others [5–9]. We showed that AKI-to-CKD transition in mice and likely humans is driven by a-disintegrin-and-metalloprotease-17 (ADAM17)-dependent substrate release, in particular of EGFR ligands and of the type 1 inflammatory cytokine tumor necrosis factor (TNF), causing persistent pro-fibrotic kidney inflammation after AKI. ADAM17-hypomorph or PTC-ADAM17-knockout (KO) mice subjected to bilateral kidney ischemia reperfusion injury (IRI) or unilateral ureteral obstruction (UUO) strongly reduced PTC-EGFR activation and kidney TNF expression and release, resulting in reduced pro-fibrotic kidney inflammation and reduced AKI-to-CKD transition and fibrosis [10, 11]. To date EGFR inhibition has not been studied in human AKI, AKI-to-CKD transition or in CKD, likely due to its unfavorable side-effect profile. Because EGFR signaling is important for homeostasis, proliferation and wound healing in many epithelial cells, EGFR inhibition typically leads to diarrhea, wound healing defects and skin infections. Blunting epithelial repair by EGFR inhibition has been shown to delay renal recovery after AKI in mice [12]. Thus, long-term EGFR inhibition would likely not represent a viable therapeutic strategy clinically.
The other key ADAM17 substrate identified in our AKI-to-CKD-transition studies, TNF, has also been linked to human kidney disease. CKD patients show elevated serum levels of TNF, and CKD progression correlates with increased serum levels of its soluble receptors TNFR1 and TNFR2 [13–15]. A recent large retrospective propensity-matched cohort study [16] and a second smaller study [17] suggested that anti-TNF biologics, such as etanercept (TNFR2-Fc), reduce incidence and progression of CKD in patients with rheumatoid arthritis. Whether TNF has a role in AKI-to-CKD transition and how TNF inhibition compares to EGFR inhibition in AKI-to-CKD transition is largely unknown. In rodents only limited conflicting data are available. In rats TNF inhibition by etanercept reduced kidney injury markers early after ischemic AKI, but no late fibrosis outcomes were measured [18]. TNF inhibition with soluble pegylated TNFR1 in rats reduced early fibrosis markers after UUO [19], but TNF-global-KO mice showed increased fibrosis [14]. Several studies used the human TNF inhibitor infliximab in rodent kidney injury models focusing on early injury [20–23], yet later it was shown that infliximab does not bind to rodent TNF [24], suggesting that the observed effects were independent of TNF neutralization. Thus, to date it is not fully resolved whether TNF has pro- or anti-fibrotic effects in the kidney, or whether TNF inhibition could be used to reduce AKI-to-CKD transition and fibrosis.
Here we show that TNF inhibition is equally effective as EGFR inhibition in reducing AKI-to-CKD transition and fibrosis in mice.
MATERIALS AND METHODS
Animal studies
Eight- to 12-week-old male mice were used in accordance with the animal care and use protocol approved by the Institutional Animal Care and Use Committee of Washington University School of Medicine, in adherence to standards set in the Guide for the Care and Use of Laboratory Animals. Kidney ischemia for 21 min at 37°C was induced bilaterally using the flank approach [25]. Sham operations exposed kidneys without induction of ischemia.
Animal treatments
Erlotinib hydrochloride (LC Laboratories, Woburn, MA, USA) in 0.5% methyl cellulose + 1% tween 80 in water was stored at –20°C at a concentration of 8 mg/mL. Animals received 80 mg/kg/day via gavage [7]. Murine etanercept (Pfizer) 12.5 mg/mL in PBS was injected into animals at 10 mg/kg i.p. twice/week. Mice were randomized into groups and received treatments for 2 or 28 days after AKI as indicated. A group of mice received fluorescently labeled etanercept (VRDye™ 549 kit from LI-COR) to confirm drug delivery into the kidney interstitium.
Renal function
Serum creatinine was measured by LC-mass spectrometry at the O'Brien Core Center for AKI Research (University of Alabama School of Medicine) and blood urea nitrogen (BUN) levels by DiUR100 kit (Thermo Scientific). Glomerular filtration rate (GFR) was measured by FITC-sinistrin (75 mg/kg i.v.) clearance (Fresenius-Kabi, Linz, Austria) using a transdermal monitoring system (MediBeacon GmbH, Mannheim, Germany) as per manufacturer’s instructions.
Immunofluorescence staining and kidney histology
Kidney histology was examined in formalin-fixed sections. Ten images/kidney were collected for blinded quantification. OCT tissue cryo-sections were washed with PBST [0.05% (v/v) Tween-20 in PBS] for 5 min at room temperature (RT) and permeabilized with 1% SDS in PBS for 5 min, followed by PBST washes (3 × 5 min). After 30 min incubation in blocking buffer (5% BSA and 5% goat serum in PBS), sections were incubated with primary antibodies overnight at 4°C, followed by PBST washes (3 × 5 min), 1 h incubation with secondary antibodies (where appropriate), followed by PBST washes (3 × 5 min) and mounting in DAPI-containing medium (Vector Laboratories). Primary antibodies: αSMA (FITC-conjugated, Sigma-Aldrich, #F3777), fibronectin (Abcam, #ab2413) and CD31 (BD Biosciences #550274). Fibrosis was quantified in kidney cortex by Picrosirius red-stained area using ImageJ software [26].
Kidney whole lysate or cortical lysate preparation and western blot were performed as previously described [10]. Primary antibodies: anti-p-EGFR Y1068 (Cell Signaling Technology #2234), anti-fibronectin (Abcam, #ab2413) or anti-GAPDH (Abcam #ab181602).
Cytokine profiling was done with proteome profiler array Mouse XL Cytokine Array Kit (#ARY028, R&D Systems) according to the manufacturer's instructions. Images were captured using the ChemiDoc Imager (BioRad) and analyzed using the ImageLab software (BioRad). Gene Ontology (GO) enrichment analyses for Biological Processes and transcriptional regulatory networks (TRRUST) were performed using Metascape [27]. Venn diagrams were generated using BioVenn [28]. Heatmaps were produced using GraphPad Prism 9.0 (GraphPad Software Inc.).
Bulk mRNA sequencing
Total RNA was isolated from mouse kidneys using TRIzol (Invitrogen) and sequenced by Novogene Corporation Inc.
Single cell RNA sequencing
Single-cell RNA sequencing (scRNAseq) analysis of four pooled sham or four pooled AKI kidney samples, was performed as described previously [29]. Briefly, cells were stained with propidium iodide, and live cells were sorted using FACSAria III (BD Biosciences). Libraries were prepared using the Chromium Single Cell 5′ Library Kit v2 and Chromium instrument (10× Genomics, Pleasanton, CA, USA). Full-length cDNA was amplified, and libraries were submitted to Genome Technology Access Center of Washington University in St Louis for sequencing at a depth of 50 000 reads. All processing steps were performed in R (v4.1.0) using Seurat v3 [30]. Quality control was first performed on each library to find appropriate filtering thresholds for each. Expression matrices for each sample were loaded into R as Seurat objects, retaining only cells that have >200 and <3200 genes. Poor quality cells with >10% mitochondrial genes were removed. Any gene not expressed in at least three cells was removed. SCTransform was used for normalization, scaling and variance stabilization (https://github.com/ChristophH/sctransform). This was done to reduce bias introduced by technical variation, sequencing depth and capture efficiency. Integration of kidney single-cell data was done using Harmony to control for batch effects [31]. After quality control and integration, 13 882 kidney cells were further analyzed. We identified 15 clusters by applying a K-nearest neighbor graph and clustering with a resolution of 0.2 on the result of principal component analysis (PCA). We visualized the clustering using uniform manifold approximation and projection (UMAP). To assign cluster identities, we first compiled a list of kidney cell types and their currently established markers [32, 33] and performed manual annotation by those markers and additional differentially expressed genes (DEGs) between clusters identified by FindAllMarkers() function in Seurat. The expression levels of Tnf and Adam17 in each cluster were visualized using the ‘plot1cell’ package [34].
qPCR
Total RNA was isolated from mouse kidneys (whole kidney or cortex) using the Trizol (Invitrogen) following the manufacturer’s instructions. Total RNA was reverse transcribed using the QuantiTect RT Kit (QIAGEN) and real-time polymerase chain reaction (PCR) was performed with Fast SYBR Green (QIAGEN). Primer sequences were Gapdh: F: 5′-ACCACAGTCCATGCCATCAC-3′, R: 5′-TCCACCACCCTGTTGCTGTA-3′ and Tgfb1: F: 5′-CTGCTGACCCCCACTGATAC-3′, R: 5′-AGCCCTGTATTCCGTCTCCT-3′. Gapdh was used as the housekeeping gene. Data were analyzed using the ΔΔCt method.
Mass cytometry CyTOF
Single-cell preparations were analyzed by mass cytometry as previously described [35]. Briefly, cells were labeled using a previously validated and titrated antibody cocktail for surface markers (all antibodies conjugated by the manufacturer; Fluidigm) diluted in Fluidigm MaxPar Cell Staining Buffer (CSB) (1 h at 4°C). After two washes in CSB, cells were fixed in 2% paraformaldehyde for 20 min at room temperature, washed, stained with MaxPar Intercalator-IR (Fluidigm) and filtered into cell strainer cap tubes. Data were then acquired on a CyTOF2/Helios instrument (Fluidigm) and analyzed with the CytoBank software using our recently described gating strategy [35].
Statistics
All results are reported as the mean ± SEM. Comparison of two groups was performed using an unpaired, two-tailed t-test or a Pearson correlation analysis where appropriate. Comparison of three or more groups was performed via ANOVA and Tukey's post hoc test. Statistical analyses were performed using GraphPad Prism 9.0 (GraphPad Software Inc.). A P-value of <.05 was considered significant.
RESULTS
Inhibition of TNF by etanercept reduces AKI-to-CKD transition and kidney fibrosis after AKI equally effectively as EGFR inhibition
We used an AKI-to-CKD severe bilateral renal IRI model for our studies. To conclusively establish that this model leads to significant progressive loss of GFR, we evaluated mice after bilateral IRI or sham operation for a period of 6 months (180 days) (Supplementary data, Fig. S1A). Bilateral IRI caused significant AKI with serum BUN and creatinine elevations on Day 1 after AKI, which returned to baseline after 3–4 weeks (Supplementary data, Fig. S1B and C). GFR, assessed trans-dermally by detecting the excretion of injected FITC-sinistrin [36], also returned to normal by 1 month after injury (Supplementary data, Fig. S1D). This occurred due to renal compensatory mechanisms and despite the fact that severe fibrosis is observed at this time point in AKI-injured animals, as we and others previously documented [7, 9–11], and we also demonstrate later in Fig. 1. To assess whether established fibrosis at 1 month would lead to progressive renal failure as expected, we measured GFR over 6 months. GFR declined progressively in AKI-injured animals but not sham animals starting 3–4 months after injury, leading to a total loss of 40% of baseline GFR by 6 months after AKI as compared with sham (Supplementary data, Fig. S1D). Serum BUN and creatinine measured over the same time period were normal (Supplementary data, Fig. S1B and C), highlighting known insensitivity of these markers for determination of early kidney failure. Thus, our rodent AKI model behaves as predicted from studies in patients with AKI that progress to CKD (AKI-to-CKD transition) [1, 2].
Figure 1:

TNF inhibition is equally effective as EGFR inhibition in reducing AKI-to-CKD transition and fibrosis. (A) Experimental scheme of mice subjected to AKI and treated with vehicle (V/V), TNF inhibition (ET, etanercept) or EGFR inhibition (ER, erlotinib) or their combination (COMB) after AKI. (B) Serum BUN values. (C) Serum creatinine Day 1 or Day 28 after AKI. (D) Glomerular filtration rate Day 28. (E) Immunofluorescence staining of cortical area at Day 28 after AKI for fibrosis markers fibronectin (green) and αSMA (red). Nuclei were stained with DAPI (blue). (F) Picrosirius red staining of cortical area at Day 28 after AKI. (G) Fibronectin representative western blot in total kidney lysates and quantification. (H) TGF-β qPCR in total kidney lysates. n = 6–10 mice/group; except n = 3 for fibronectin western blot and TGF-β qPCR. We used ANOVA with Tukey's multiple comparisons test to compare samples. *P < .05; **P < .01; ****P < .0001; NS: non-significant.
To test whether TNF has a role in AKI-to-CKD transition and the development of fibrosis after AKI, and to compare it to EGFR inhibition, we treated mice with TNF inhibition (etanercept, ET), EGFR (kinase) inhibition (erlotinib, ER) or their combination (COMB) for 28 days after inducing ischemic AKI by severe bilateral IRI [10, 11] (Fig. 1A). ET is a TNFR2-Fc conjugate that acts as a decoy receptor for soluble TNF. We used murine Tnfr2-Fc (Pfizer) [30, 31] to ensure specificity for mouse TNF [37, 38]. Fluorescently labeled ET was effectively delivered into the interstitial compartment of the kidney (Supplementary data, Fig. S2). ET, similar to ER, did not blunt initial injury of the kidney, as determined by serum BUN (Fig. 1B) and serum creatinine (Fig. 1C). GFR at Day 28 after AKI, as expected due to compensatory mechanisms (see above), did not differ between sham, vehicle or inhibitor treatments and was normal (Fig. 1D). However, at Day 28 after AKI, TNF inhibition (ET) prevented AKI-induced fibrosis equally as well as EGFR inhibition (ER), with combination treatment (COMB) providing no additional benefit. Fibrosis was assessed by the expression of pro-fibrotic markers α-smooth muscle actin (α-SMA) and fibronectin (Fig. 1E) and by picrosirius red staining of collagen fibers (Fig. 1F). Finally, we confirmed the anti-fibrotic effect of all treatments by western blot for kidney fibronectin (Fig. 1G) and by qPCR for kidney expression of transforming growth factor-beta (TGF-β), a key pro-fibrotic cytokine (Fig. 1H). Both are strongly reduced by all treatments. There was no significant difference between the treatments.
TNF and EGFR inhibition overlap in reduction of kidney cytokines after AKI, but TNF inhibition blocks leukocyte migration signals much less efficiently than EGFR inhibition
To gain a better understanding of the effects of TNF inhibition on early AKI-induced inflammation and how it compared with EGFR inhibition or combination treatment, we performed proteomic analysis of kidney tissue lysates for the expression of 98 proinflammatory/profibrotic cytokines, chemokines and related molecules (in the following simply referred to as cytokines). We compared IRI-injured mice treated with vehicle (V/V), etanercept (ET), erlotinib (ER) or combination (COMB) on Day 2 after AKI (Fig. 2A). Initial kidney injury was unaffected by any treatment, as shown by serum BUN elevations at Day 1 after AKI (Fig. 2B). TNF inhibition (ET) partially overlapped in suppression of cytokines with EGFR inhibition (ER) or combination (COMB) treatment (Fig. 2C). GO term analysis of all affected cytokines revealed an even more significant overlap between all treatments on the functional level (Fig. 2D). Notably, treatments overlapped most strongly in downregulating immune response, tissue remodeling and inflammatory cell signaling events. Early in injury, TNF inhibition had only small negative effects on leukocyte migration and cell–cell adhesion signals as compared with EGFR inhibition or combination treatment (Fig. 2E). A complete list of affected cytokines and their functional GO terms stratified by treatment condition is shown in Supplementary data, Table S1. Six cytokines were strongly downregulated by all three treatments (Fig. 2F), suggesting they might be particularly central to shared beneficial effects in reducing profibrotic inflammation and fibrosis after AKI: TNF and interferon gamma (IFNG; early response cytokine that orchestrates activation of the innate immune system), complement-factor-5 (C5; a strong chemoattractant for neutrophils and monocytes), p-Selectin (a mediator of the adhesion of immune cells to endothelial cells) and Serpin E1 (also called plasminogen activator inhibitor 1; innate immune system activator and regulator of cell migration). This suggests that the severity of type 1 inflammatory response and of innate immune cell activation, as well as the commensurate degree of neutrophil and immune cell attraction into the kidney, play important roles in AKI-to-CKD-transition. Interleukin-10 (IL10), a key anti-inflammatory cytokine produced by activated immune cells, was moderately reduced by all treatments, likely as a result of dampened inflammation overall, one key stimulus for IL-10 secretion. GO term analysis of the six commonly downregulated cytokines is shown in Fig. 2G and a complete list of functional GO terms for common downregulated cytokines is available in Supplementary data, Table S2. To test for additional regulatory overlap of treatments on the transcriptional level, we performed an enrichment analysis of transcription factors known to regulate the expression of any of the cytokines downregulated by ET, ER or COMB treatment. The top enriched transcription factor family was the nuclear factor κB (NFκB) family and the transcription factor c-Jun, important immediate early genes which are induced upon tissue injury in early inflammation (Fig. 2H). TNF itself, which was among the top two strongly downregulated cytokines by all treatments (Fig. 2F), indeed represents a major regulator as well as target of the NFκB transcription factor family [39], providing further evidence that TNF plays an important role in inflammation after AKI. A summary of our transcription factor enrichment analysis is available in Supplementary data, Table S3.
Figure 2:
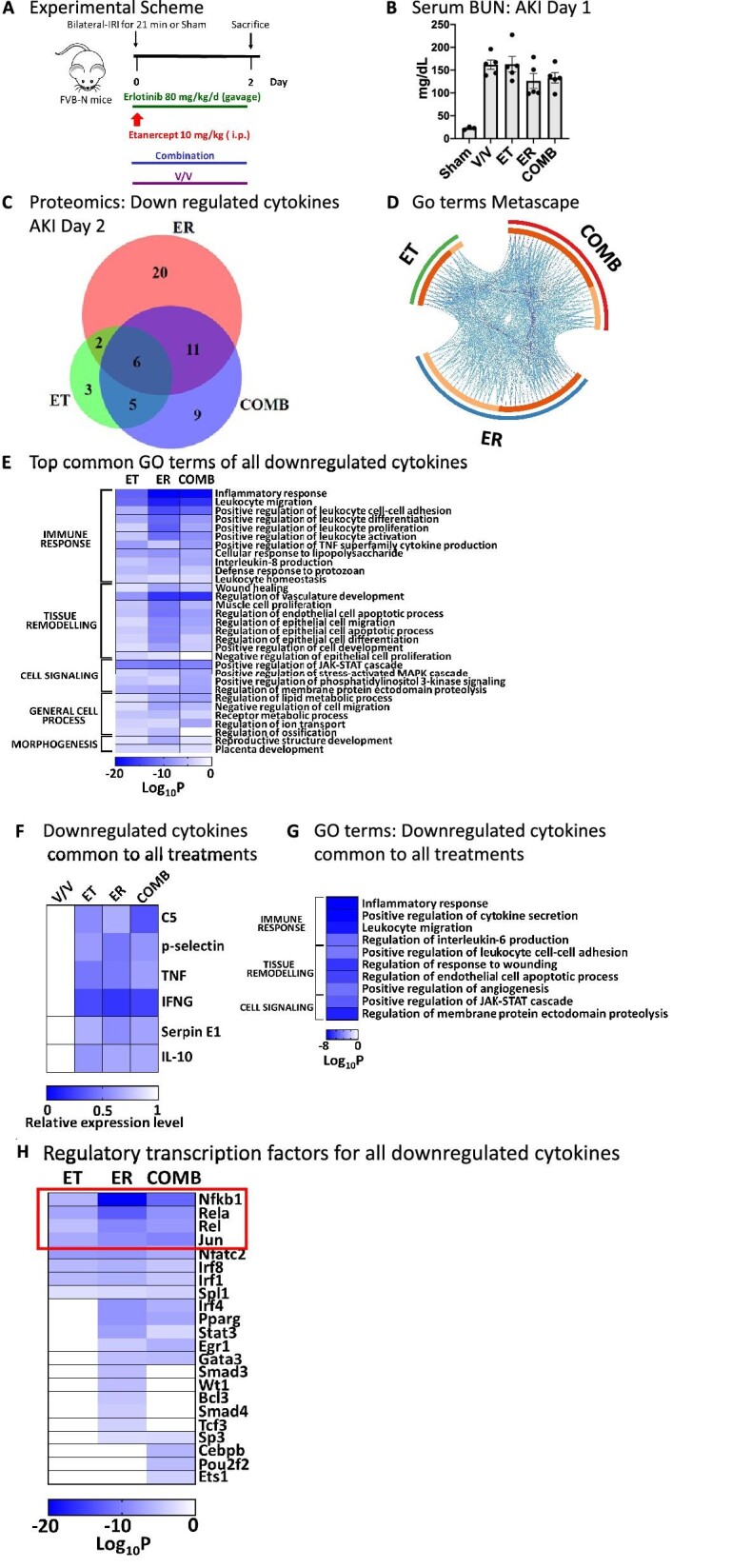
TNF and EGFR inhibition overlap in reduction of kidney cytokines after AKI, but TNF inhibition blocks leukocyte migration signals early after AKI much less efficiently than EGFR inhibition. (A) Experimental scheme: mice were subjected to bilateral IRI and treated with vehicle (V/V), etanercept (ET), erlotinib (ER) or their combination (COMB) for 2 days after AKI. (B) Serum BUN. (C) Proteomics: Venn diagram of cytokines downregulated by each treatment as compared with vehicle-treated AKI animals (n = 3/group). (D) GO term analysis of all downregulated cytokines. Outer circle denotes treatments; inner circle denotes downregulated cytokines: common between treatments (dark orange), unique (light orange). Purple curves connect identical cytokines and blue curves connect cytokines that belong to the same enriched GO term. (E) Top common GO terms of all downregulated cytokines. (F) Downregulated cytokines common to all treatments. (G) Top common GO terms of downregulated cytokines common to all treatments. (H) Enrichment analysis of transcription factors known to regulate any of the cytokines downregulated by ET, ER or COMB. NFκB family members are highlighted in red rectangle.
TNF inhibition does not affect EGFR activation after AKI in vivo and PTC-derived TNF does not contribute to injury-induced fibrosis
The overlapping effects of TNF and EGFR inhibition on kidney cytokines could indicate that TNF action feeds back into the EGFR pathway. To test this, we examined EGFR phosphorylation at early time points in ET- or ER-treated mice, using cortical kidney lysates, which are highly enriched for proximal tubule. TNF inhibition (ET) did not significantly affect kidney tubule EGFR phosphorylation in vivo, whereas EGFR inhibition (ER) blocked it completely (Fig. 3A), suggesting that TNF does not feedback to EGFR early after kidney injury in vivo. Consistent with this, TNF inhibition also did not affect kidney expression of several EGFR ligands (Fig. 3B). Based on our previous observations implicating ADAM17 PTC-released EGFR ligands and TNF in AKI-to-CKD transition and fibrosis after ischemic AKI or after UUO [10, 11], we tested whether PTC-released TNF was involved in the development of pro-fibrotic inflammation after kidney injury. We generated PTC-TNF-KO mice using SLC34A1Cre-ERT2 and TNFflox/flox mice [40] and confirmed TNF KO by qPCR of cortical lysates (Fig. 3C). TNF-PTC-KO mice subjected to either sham or UUO did not show any significant differences in the development of fibrosis as determined by staining for the profibrotic markers fibronectin and αSMA (Fig. 3D), suggesting that PTC-derived TNF has no significant role in injury-induced fibrosis, and pointing to other cellular TNF sources in the injured kidney in order to explain the effects of TNF inhibition. To better understand cellular sources of kidney TNF expression after AKI, we analyzed our recently published scRNAseq dataset of sham and AKI kidneys [29]. TNF was significantly upregulated on Day 1 after AKI predominantly in immune cells, in particular in neutrophils, macrophages and some dendritic cells, which also express the metalloprotease ADAM17 which controls TNF release (Fig. 3E). Consistent with the lack of effect of PTC-TNF-KO in UUO, TNF expression in PTC was nearly absent in sham and showed only very small upregulation in a very small percentage of PTCs after kidney injury. Taken together, this suggests that immune cells represent the main kidney TNF source after AKI.
Figure 3:

TNF inhibition does not affect EGFR activation after AKI in vivo and PTC-derived TNF does not contribute to injury-induced fibrosis. (A) Representative western blot of cortical PTC-enriched kidney lysates using anti-phospho-EGFR (Y1068) antibody and quantification (n = 4). (B) Expression levels (bulk RNAseq of total kidney mRNA) of EGFR ligands amphiregulin (AREG), heparin-binding-EGF (HB-EGF) and transforming growth factor α (TGFA) at Day 2 after AKI in vehicle- or erlotinib-treated animals (n = 3). (C) qPCR of cortical lysates for TNF in control versus proximal tubule TNF knockout mice (PTC-TNF-KO) subjected to unilateral ureteral obstruction (UUO) at Day 7 after UUO (ND: not dectected). (D) Immunofluorescence staining of kidney tissue for fibrosis markers fibronectin and αSMA in sham versus UUO PTC-TNF-KO mice at Day 28 after UUO (n = 3). (E) scRNAseq analysis: TNF and ADAM17 expression in sham versus AKI kidneys at Day 1 after AKI. *P < .05; **P < .01. We used one-way ANOVA for statistical calculations in A. and two-way ANOVA in (C).
TNF inhibition, unlike EGFR inhibition, does not reduce kidney immune cell recruitment or accumulation
We thus tested the hypothesis that TNF or EGFR inhibition might affect recruitment and/or accumulation of TNF-expressing immune cells in the kidney early after AKI. Using mass cytometry (CYTOF), we show that TNF inhibition (ET) shows no significant difference from vehicle treatment in the number of macrophages that accumulate in the kidney, whereas EGFR inhibition (ER) or combination treatment (COMB) strongly reduces macrophage accumulation by about 50%, while the number of accumulating kidney neutrophils are equal between vehicle control and all treatments (Fig. 4A). Most pro-fibrotic macrophages that accumulate in the kidney after injury are derived from recruited circulating chemokine-receptor-2 positive (CCR2+) monocytes, the receptor for the monocyte-chemoattractant cytokine Ccl2 (also called monocyte-chemoattractant-protein-1, MCP-1), and blockade of CCR2 reduces kidney macrophage accumulation and ameliorates kidney fibrosis after injury [41–43]. Consistent with our CYTOF data, bulk mRNA sequencing analysis showed that TNF inhibition (ET), but not EGFR inhibition (ER), significantly reduced kidney expression of the global immune cell marker CD45, of the macrophage/dendritic cell marker F4/80, of CCR2 (chemokine-receptor-2) and of the macrophage marker Lyz2 (Lysozyme 2), but not the dendritic cell marker Itgae (Integrin subunit alpha E) (Fig. 4B). These findings collectively suggest that the anti-fibrotic function of TNF inhibition, in contrast to EGFR inhibition, does not rely on a reduction in immune cells but rather on neutralization of TNF released from pro-fibrotic immune cells.
Figure 4:

TNF inhibition, unlike EGFR inhibition, does not reduce kidney immune cell recruitment or accumulation. (A) Mass cytometry (CYTOF) analysis of kidney immune cells AKI Day 2. (B) Bulk mRNA sequencing analysis of kidney gene expression at Day 2 after AKI: CD45 (general immune cell marker), F4/80 (macrophage/dendritic cell marker), CCR2 (monocyte marker and receptor for monocyte recruitment), Lyz2 (macrophage marker), Itgae (dendritic cell marker) (n = 3). *P < .05; **P < .01. We used t-test for statistical analysis.
DISCUSSION
We show that TNF inhibition prevents chronic profibrotic inflammation in AKI-to-CKD transition equally effectively as EGFR activation. Both TNF inhibition and EGFR inhibition limit profibrotic inflammation. While there is significant overlap between cytokines reduced by TNF or EGFR inhibition, including of TNF itself, both treatments also reduce the expression of separate, yet functionally overlapping sets of cytokines; both effects together explain their similar anti-inflammatory and anti-fibrotic effects after AKI. Mechanistically, TNF inhibition acts on soluble TNF released by profibrotic immune cells, including neutrophils, monocytes, macrophages and dendritic cells, and blunts profibrotic kidney inflammation. In contrast, EGFR inhibition acts by reducing recruitment and/or accumulation of TNF-producing immune cells after AKI (Fig. 5).
Figure 5:
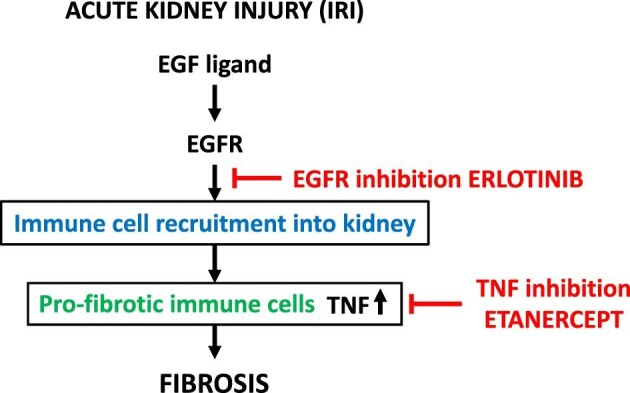
Summary scheme.
Macrophages are known key players in the development of kidney fibrosis [44]. Dendritic cells also represent a major source of TNF early after renal IRI (24 h) [45], consistent with our AKI kidney scRNAseq data. However, the role of dendritic cells in kidney fibrosis has not been studied to our knowledge. Yet, dendritic cells and dendritic cell-derived TNF have been associated with fibrosis in the liver [46] and the lung [47, 48], suggesting this may also be the case in the kidney. While PTCs are a key cell type in which activation of EGFR mediates profibrotic effects [6, 7, 49], we found that PTCs are not a main source of profibrotic TNF after AKI, based on our scRNAseq data. In addition, TNF-PTC-KO did not affect the development of injury-induced fibrosis.
As many immune cells are known to respond to soluble TNF and express its main receptor TNFR1, one main target of TNF released from immune cells may be immune cells themselves with TNF acting in a positive forward loop. However, TNF could theoretically also act on a number of non-immune cells based on the fact that TNFR1 is very broadly expressed [6, 7, 49]. It is possible that PTCs are a target of TNF signals in AKI-to-CKD transition, as it was recently demonstrated that a “failed repair” Vcam1+/Ccl2+ PTC cluster after AKI shows significant activation of NFκB, TNF and AP-1 signaling pathways [50]. It is thus possible that immune cell-derived TNF affects kidney disease progression in part through its action on “failed repair” epithelial cells. It is also possible that TNF may act directly on kidney fibroblasts/myofibroblasts.
Complicating any analysis of TNF-targeting agents is the fact that they not only bind to soluble TNF, which preferentially activates TNFR1, but also to uncleaved transmembrane pro-TNF, thus blocking its activation of TNFR2. TNFR2 is expressed in immune cells and requires cell–cell contact for activation, with the ligand and receptor expressed in opposing cells [51]. While TNFR1 activation mediates the classic proinflammatory injurious actions of soluble TNF, TNFR2 activation by pro-TNF can have either proinflammatory or anti-inflammatory/pro-repair effects, depending on the cellular context [52]. Thus, some effects of etanercept in our studies could be related to blocking TNFR2 signaling in immune cells.
Consistent with our data, TNF inhibition has also been shown to decrease glomerular inflammation and glomerulosclerosis (glomerular fibrosis) in diabetic nephropathy. In diabetic Akita mice, anti-TNF antibody reduced glomerular macrophages, glomerulosclerosis and albuminuria, and prevented reduction in glomerular filtration rate [53], consistent with an anti-fibrotic effect of TNF inhibition in inflamed glomeruli. This study also highlighted the importance of TNF expression in macrophages for induction of fibrosis in the injured kidney, as suggested by our findings. In the streptozosin (STZ) diabetic model, TNF-macrophage-KO reduced BUN and creatinine elevations, glomerular macrophage accumulation, albuminuria and TNF mRNA/protein in the kidney [53].
Our current studies raise the possibility that TNF inhibition, instead of EGFR inhibition, could be tested and potentially used clinically to prevent AKI-to-CKD transition after AKI. TNF inhibition has been used in various autoimmune diseases for long periods of time with significant benefits that are weighed against relatively rare serious adverse effects and risk of morbidity/mortality due to autoimmune disease progression. Typical adverse effects are mild or minimal, and include injection site reactions, infusion reactions (both usually manageable), neutropenia (usually mild), and in some instances skin lesions and infections; autoimmune diseases develop only rarely [54–56]. The risk of heart failure remains unclear, as combined analysis of the two relevant clinical trials showed no effect on death or new heart failure, with only few patients possibly experiencing heart failure exacerbations [57]. Thus, in our view, the opportunity for TNF inhibition in AKI patients could center on short- or medium-term treatment starting early after onset of AKI when AKI-to-CKD transition could be prevented or blunted. TNF inhibition would likely be preferrable to EGFR inhibition based on potential adverse effects. Given the high yearly mortality of CKD patients, this could be a justifiable approach at reasonable risk.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Pfizer for the provision of murine etanercept and Dr Nedospasov and Dr Grivennikov for the provision of the TNF floxed mice.
Contributor Information
Mai M Abdelmageed, Washington University School of Medicine in Saint Louis, Department of Medicine, St. Louis, MO, USA; Division of Nephrology.
Eirini Kefaloyianni, Washington University School of Medicine in Saint Louis, Department of Medicine, St. Louis, MO, USA; Division of Nephrology.
Akshayakeerthi Arthanarisami, Washington University School of Medicine in Saint Louis, Department of Medicine, St. Louis, MO, USA; Division of Nephrology.
Yohei Komaru, Washington University School of Medicine in Saint Louis, Department of Medicine, St. Louis, MO, USA; Division of Nephrology.
Jeffrey J Atkinson, Washington University School of Medicine in Saint Louis, Department of Medicine, St. Louis, MO, USA; Division of Pulmonary and Critical Care Medicine.
Andreas Herrlich, Washington University School of Medicine in Saint Louis, Department of Medicine, St. Louis, MO, USA; Division of Nephrology.
FUNDING
A.H. was supported by National Institute of Diabetes and Digestive and Kidney diseases (R01DK108947 and R01DK121200) and E.K. was supported by the American Heart Association (Career Development Award 20CDA35320006) and the American Society of Nephrology (KidneyCure Carl W. Gottschalk Research Scholar Grant). Y.K. was supported by Mochida Memorial Foundation and The Uehara Memorial Foundation.
AUTHORS’ CONTRIBUTIONS
M.M.A., E.K. and A.A. did in vivo and in vitro experiments and prepared figures; Y.K. performed the scRNAseq analysis; J.J.A. provided material and expertise for the CyTOF analysis; A.H. designed the study and wrote the manuscript; E.K. contributed to manuscript writing.
DATA AVAILABILITY STATEMENT
The data underlying this article are available in this article and in supplemental material online, as well as in a previously published manuscript [29].
CONFLICT OF INTEREST STATEMENT
The authors have no financial interests to disclose. The results presented in this paper have not been published previously in whole or part, except in abstract format.
REFERENCES
- 1. Thakar CV, Christianson A, Himmelfarb Jet al. Acute kidney injury episodes and chronic kidney disease risk in diabetes mellitus. Clin J Am Soc Nephrol 2011;6:2567–72. 10.2215/cjn.01120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coca SG, Singanamala S, Parikh CR.. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int 2012;81:442–8. 10.1038/ki.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McWilliam SJ, Wright RD, Welsh GIet al. The complex interplay between kidney injury and inflammation. Clin Kidney J 2021;14:780–8. 10.1093/ckj/sfaa164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rayego-Mateos S, Marquez-Expósito L, Rodrigues-Diez Ret al. Molecular mechanisms of kidney injury and repair. Int J Mol Sci 2022;23:1542. 10.3390/ijms23031542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Terzi F, Burtin M, Hekmati Met al. Targeted expression of a dominant-negative EGF-R in the kidney reduces tubulo-interstitial lesions after renal injury. J Clin Invest 2000;106:225–34. 10.1172/jci8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lautrette A, Li S, Alili Ret al. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: a new therapeutic approach. Nat Med 2005;11:867–74. 10.1038/nm1275. [DOI] [PubMed] [Google Scholar]
- 7. Chen J, Chen J-K, Nagai Ket al. EGFR signaling promotes TGFβ-dependent renal fibrosis. J Am Soc Nephrol 2012;23:215–24. 10.1681/asn.2011070645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang J, Liu N, Zhuang S.. Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int 2013;83:804–10. 10.1038/ki.2012.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu N, Guo J-K, Pang Met al. Genetic or pharmacologic blockade of EGFR inhibits renal fibrosis. J Am Soc Nephrol 2012;23:854–67. 10.1681/asn.2011050493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kefaloyianni E, Muthu ML, Kaeppler Jet al. ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI Insight 2016;1:e87023. 10.1172/jci.insight.87023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kefaloyianni E, Raja MRK, Schumacher Jet al. Proximal tubule-derived amphiregulin amplifies and integrates profibrotic EGF receptor signals in kidney fibrosis. J Am Soc Nephrol 2019;30:2370. 10.1681/asn.2019030321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen J, Chen J-K, Harris RC.. Deletion of the epidermal growth factor receptor in renal proximal tubule epithelial cells delays recovery from acute kidney injury. Kidney Int 2012;82:45–52. 10.1038/ki.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Niewczas MA, Gohda T, Skupien Jet al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J Am Soc Nephrol 2012;23:507–15. 10.1681/asn.2011060627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gupta J, Mitra N, Kanetsky PAet al. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin J Am Soc Nephrol 2012;7:1938–46. 10.2215/cjn.03500412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gohda T, Niewczas MA, Ficociello LHet al. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J Am Soc Nephrol 2012;23:516–24. 10.1681/asn.2011060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sumida K, Molnar MZ, Potukuchi PKet al. Treatment of rheumatoid arthritis with biologic agents lowers the risk of incident chronic kidney disease. Kidney Int 2018;93:1207–16. 10.1016/j.kint.2017.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim HW, Lee C-K, Cha H-Set al. Effect of anti-tumor necrosis factor alpha treatment of rheumatoid arthritis and chronic kidney disease. Rheumatol Int 2015;35:727–34. 10.1007/s00296-014-3146-4. [DOI] [PubMed] [Google Scholar]
- 18. Choi DE, Jeong JY, Lim BJet al. Pretreatment with the tumor nerosis factor-alpha blocker etanercept attenuated ischemia-reperfusion renal injury. Transplant Proc 2009;41:3590–6. 10.1016/j.transproceed.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 19. Meldrum KK, Misseri R, Metcalfe Pet al. TNF-α neutralization ameliorates obstruction-induced renal fibrosis and dysfunction. Am J Physiol Regul Integr Comp Physiol 2007;292:R1456–64. 10.1152/ajpregu.00620.2005. [DOI] [PubMed] [Google Scholar]
- 20. Paola RD, Genovese T, Impellizzeri Det al. The renal injury and inflammation caused by ischemia-reperfusion are reduced by genetic inhibition of TNF-αR1: a comparison with infliximab treatment. Eur J Pharmacol 2013;700:134–46. 10.1016/j.ejphar.2012.11.066. [DOI] [PubMed] [Google Scholar]
- 21. Homsi E, Andreazzi DD, de Faria JBLet al. TNF-α-mediated cardiorenal injury after rhabdomyolysis in rats. Am J Physiol Ren Physiol 2015;308:F1259–67. 10.1152/ajprenal.00311.2014. [DOI] [PubMed] [Google Scholar]
- 22. Saritemur M, Un H, Cadirci Eet al. Tnf-α inhibition by infliximab as a new target for the prevention of glycerol-contrast-induced nephropathy. Environ Toxicol Pharmacol 2015;39:577–88. 10.1016/j.etap.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 23. Nagata Y, Fujimoto M, Nakamura Ket al. Anti-TNF-α agent infliximab and splenectomy are protective against renal ischemia-reperfusion injury. Transplantation 2016;100:1675–82. 10.1097/tp.0000000000001222. [DOI] [PubMed] [Google Scholar]
- 24. Assas BM, Levison SE, Little Met al. Anti-inflammatory effects of infliximab in mice are independent of tumour necrosis factor α neutralization. Clin Exp Immunol 2017;187:225–33. 10.1111/cei.12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang L, Besschetnova TY, Brooks CRet al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 2010;16:535–43, 1p following 143. 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schneider CA, Rasband WS, Eliceiri KW.. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou Y, Zhou B, Pache Let al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 2019;10:1523. 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hulsen T, de Vlieg J, Alkema W. BioVenn – a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 2008;9:488. 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khamissi FZ, Ning L, Kefaloyianni Eet al. Identification of kidney injury released circulating osteopontin as causal agent of respiratory failure. Sci Adv 2022;8:eabm5900. 10.1126/sciadv.abm5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stuart T, Butler A, Hoffman Pet al. Comprehensive integration of single-cell data. Cell 2019;177:1888–902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Korsunsky I, Millard N, Fan Jet al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat Methods 2019;16:1289–96. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kirita Y, Wu H, Uchimura Ket al. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci USA 2020;117:15874–83. 10.1073/pnas.2005477117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Angelidis I, Simon LM, Fernandez IEet al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun 2019;10:963. 10.1038/s41467-019-08831-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu H, Villalobos RG, Yao Xet al. Mapping the single-cell transcriptomic response of murine diabetic kidney disease to therapies. Cell Metab 2022;34:1064–78.e6. 10.1016/j.cmet.2022.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brody SL, Gunsten SP, Luehmann HPet al. Chemokine receptor 2–targeted molecular imaging in pulmonary fibrosis. A clinical trial. Am J Respir Crit Care Med 2021;203:78–89. 10.1164/rccm.202004-1132oc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pérez ZH, Weinfurter S, Gretz N.. Transcutaneous assessment of renal function in conscious rodents. J Vis Exp 2016;e53767. 10.3791/53767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim HY, Renshaw-Gegg LW, Balciunas AMet al. Construction and purification of the murine p75-murine IgG1 fusion protein. J Investig Dermatol Symp Proc 2007;12:48–9. 10.1038/sj.jidsymp.5650035. [DOI] [PubMed] [Google Scholar]
- 38. Hu Y-L, Kim HY, Kohno Tet al. Pharmacodynamic effects of the murine p75-Fc fusion protein in mice. J Investig Dermatol Symp Proc 2007;12:50–1. 10.1038/sj.jidsymp.5650030. [DOI] [PubMed] [Google Scholar]
- 39. Hayden MS, Ghosh S.. Regulation of NF-κB by TNF family cytokines. Semin Immunol 2014;26:253–66. 10.1016/j.smim.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grivennikov SI, Tumanov AV, Liepinsh DJet al. Distinct and nonredundant in vivo functions of TNF produced by T cells and macrophages/neutrophils protective and deleterious effects. Immunity 2005;22:93–104. 10.1016/j.immuni.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 41. Furuichi K, Wada T, Iwata Yet al. CCR2 signaling contributes to ischemia-reperfusion injury in kidney. J Am Soc Nephrol 2003;14:2503–15. 10.1097/01.asn.0000089563.63641.a8. [DOI] [PubMed] [Google Scholar]
- 42. Kitagawa K, Wada T, Furuichi Ket al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol 2004;165:237–46. 10.1016/s0002-9440(10)63292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li L, Huang L, Sung S-SJet al. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int 2008;74:1526–37. 10.1038/ki.2008.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang X, Chen J, Xu Jet al. The role of macrophages in kidney fibrosis. Front Physiol 2021;12:705838. 10.3389/fphys.2021.705838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dong X, Swaminathan S, Bachman LAet al. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int 2007;71:619–28. 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- 46. Connolly MK, Bedrosian AS, JM Clairet al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-α. J Clin Invest 2009;119:3213–25. 10.1172/jci37581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shin J-S. Unexpected role of dendritic cells in pulmonary fibrosis. Thorax 2019;74:925. 10.1136/thoraxjnl-2019-213510. [DOI] [PubMed] [Google Scholar]
- 48. Bocchino M, Zanotta S, Capitelli Let al. Dendritic cells are the intriguing players in the puzzle of idiopathic pulmonary fibrosis pathogenesis. Front Immunol 2021;12:664109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Overstreet JM, Wang Y, Wang Xet al. Selective activation of epidermal growth factor receptor in renal proximal tubule induces tubulointerstitial fibrosis. FASEB J 2017;31:4407–21. 10.1096/fj.201601359rr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gerhardt LMS, Liu J, Koppitch Ket al. Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proc Natl Acad Sci USA 2021;118:e2026684118. 10.1073/pnas.2026684118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wajant H, Siegmund D.. TNFR1 and TNFR2 in the control of the life and death balance of macrophages. Front Cell Dev Biol 2019;7:91. 10.3389/fcell.2019.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang S, Wang J, Brand DDet al. Role of TNF–TNF receptor 2 signal in regulatory T cells and its therapeutic implications. Front Immunol 2018;9:784. 10.3389/fimmu.2018.00784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Awad AS, You H, Gao Tet al. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int 2015;88:722–33. 10.1038/ki.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stokes MB, Foster K, Markowitz GSet al. Development of glomerulonephritis during anti-TNF-α therapy for rheumatoid arthritis. Nephrol Dial Transplant 2005;20:1400–6. 10.1093/ndt/gfh832. [DOI] [PubMed] [Google Scholar]
- 55. Kaushik P, Rahmani M, Ellison W.. Membranous glomerulonephritis with the use of etanercept in ankylosing spondylitis. Ann Pharmacother 2011;45:1585. 10.1345/aph.1q492. [DOI] [PubMed] [Google Scholar]
- 56. Ammar A, Mahmood HZA, Shahid Zet al. Etanercept-associated nephropathy. Cureus 2019;11:e5419. 10.7759/cureus.5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mann DL, McMurray JJV, Packer Met al. Targeted anticytokine therapy in patients with chronic heart failure. Circulation 2004;109:1594–602. 10.1161/01.cir.0000124490.27666.b2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in this article and in supplemental material online, as well as in a previously published manuscript [29].