

Abstract
Over recent years, members of the APOBEC3 family of cytosine deaminases have been implicated in increased cancer genome mutagenesis, thereby contributing to intra- and inter-tumor genomic heterogeneity and therapy resistance in, amongst others, breast cancer. Understanding the available methods for clinical detection of these enzymes, the conditions required for their (dysregulated) expression, the clinical impact they have, and the clinical implications they may offer is crucial in understanding the current impact of APOBEC3-mediated mutagenesis in breast cancer. Here, we provide a comprehensive review of recent developments in the detection of APOBEC3-mediated mutagenesis and responsible APOBEC3 enzymes, summarize the pathways that control their expression, and explore the clinical ramifications and opportunities they pose. We propose that APOBEC3-mediated mutagenesis can function as a helpful predictive biomarker in several standard-of-care breast cancer treatment plans and may be a novel target for treatment.
Keywords: APOBEC3, breast cancer, tumor evolution, therapy resistance, immunotherapy
Introduction
The genomic landscape of breast cancer is shaped by many mutagenic processes, which promote intra- and inter-tumor genomic heterogeneity and contribute to tumor evolution and thereby treatment resistance (1, 2). These mutational processes are computationally distinguishable as signatures with different etiologic causes (1). For example, a mutational signature attributable to deamination of cytosine bases in DNA catalyzed by apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-3 (APOBEC3) enzymes is evident in a large proportion of breast cancers (1). Two of the seven APOBEC3 enzymes found in humans, APOBEC3A [A3A] and APOBEC3B [A3B], have been causally linked to the observed APOBEC mutation signature in breast cancer. This review focuses firstly on how APOBEC3-positive tumors can be diagnosed, secondly on how the proteins responsible may become dysregulated in breast cancer, and finally on the clinical impact and implications of APOBEC3-mediated mutagenesis for novel and patient-specific treatment opportunities. Of note, APOBEC3 enzymes may also have roles in cancer that are independent of mutagenesis, for example estrogen dependent gene expression (3), R-loop homeostasis (bioRxiv 2021.08.30.458235v1) and RNA editing (bioRxiv 2022.06.01.494353), which are beyond the scope of this review.
The APOBEC3 ABC’s and how to detect them
Family member profiles
APOBEC3 proteins catalyze the deamination of cytosines, thereby converting them into pre-mutagenic uracils [reviewed in (4)]. Human cells can express up to seven APOBEC3 proteins, A3A, A3B, A3C, A3D, A3F, A3G, and A3H, which can be further distinguished by single amino acid variants, of which A3H is the most variable in the human population with over a dozen phylogenetically distinct haplotypes (5, 6) [Fig. 1A]. In addition, although all A3 members are structurally similar, differences in amino acid sequence and functionality allow sub-classification into different domain groupings [called Z1, Z2, and Z3, respectively color-coded green, orange and blue in Fig. 1A]. The composition of these domains is evolutionarily conserved amongst higher primates and most of these three domains are also expressed in other mammalian orders, including even- and odd-toed ungulates, bats, and afrotheres (5). In humans, the Z1 domain provides the catalytically active pocket in A3A, A3B and A3G, while the catalytically active pockets of A3C-F and A3H are encoded by Z2 and Z3 domains, respectively [Fig. 1A]. Additionally, a catalytically inactive form of Z2 is present as the N-terminal domain of A3B, A3D, A3F, and A3G, possibly serving to regulate catalytic activity, subcellular localization and the packaging into viral particles (7–9). As such, several A3 members, including A3D, A3F, A3G, and A3H, are capable of restricting HIV-1, whereas other virus types may be restricted by A3A, A3B, and A3H [(10–13), and reviewed in (4, 14)]. For instance, the DNA-based hepatitis B virus may be restricted through the editing capabilities of A3B, A3C, A3F, and A3G (15), and the large DNA herpesviruses by A3B and potentially also by A3A (16).
Figure 1. The APOBEC3 enzymes, their association with breast cancer, and the diagnostic methods available.
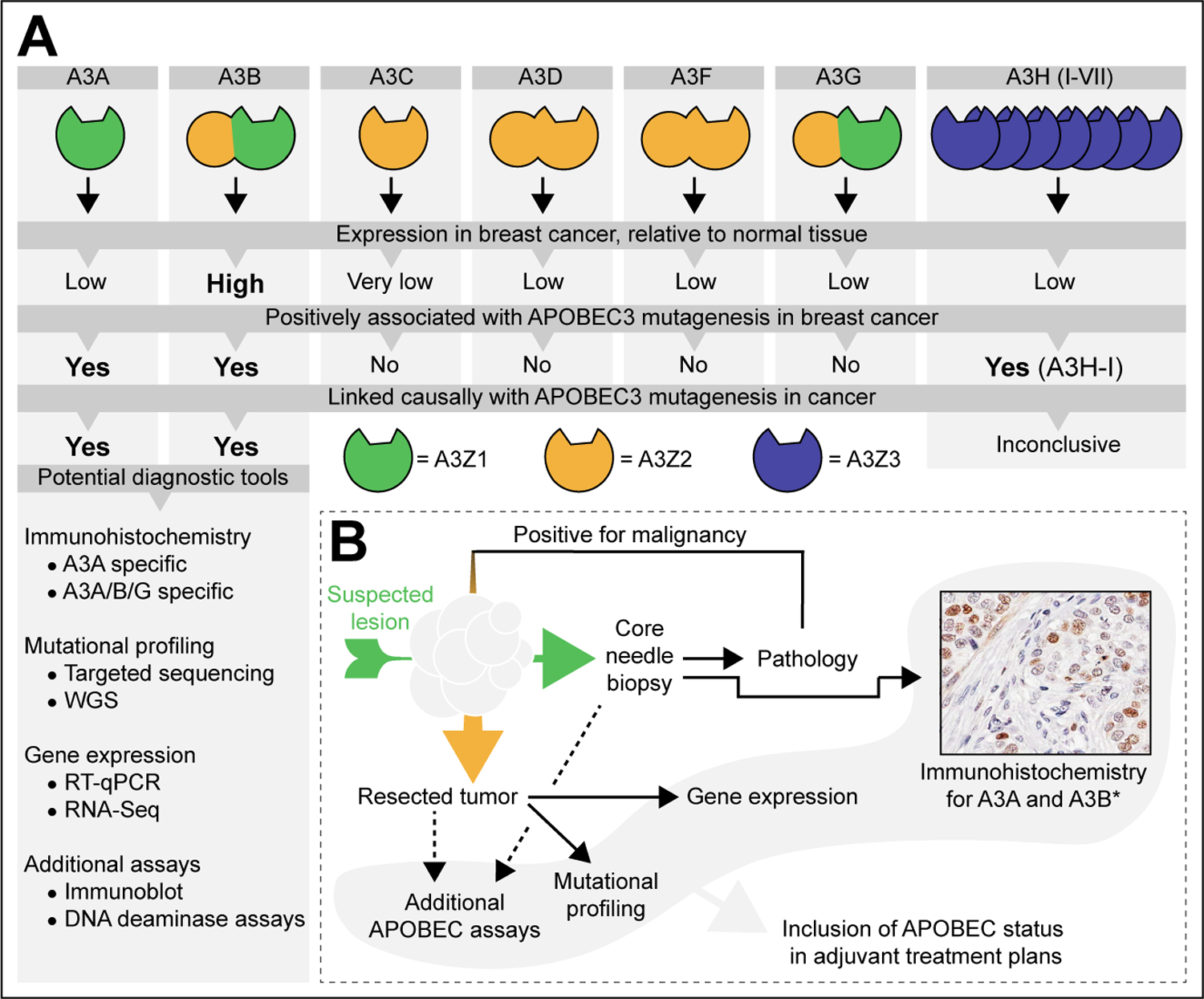
A Break-down of individual APOBEC3 family members, their conserved domain composition (5, 6), expression levels in cancer (17, 22, 30), and their causal involvement in the observed APOBEC mutagenesis pattern observed in cancer (13, 17, 22, 33). The list of potential diagnostic tools denotes published methods suitable for the detection of APOBEC3 enzymes, their deaminase activity, or the APOBEC single base substitutions [SBS] signatures (bioRxiv 2022.04.26.489523v2, (12, 17, 35, 39, 45–50).
B Proposed flow chart for the inclusion of the APOBEC status in the consideration of suitable adjuvant treatment plans. An initial core needle biopsy is taken from the suspected lesion [green arrow] and immunohistochemistry for A3A and/or A3B is performed in parallel to conventional clinical pathology. *The example shown here is considered A3B specific because of its nuclear localization. If malignant and operable, the freshly resected tumor [orange arrow] is subjected to additional assays, including mutational profiling and gene expression analyses. The resultant APOBEC status may then be included in the adjuvant treatment plans.
A3A and A3B are major contributors to cancer
Whole genome sequencing found that off-target activity of APOBEC3 to chromosomal DNA constitutes a major source of somatically acquired mutations in a variety of malignancies, including breast cancer (17–20). Mutational activity by APOBEC3 proteins can be computationally identified in sequencing data as mutations occurring at cytosines within a 5’-TCW [W = A or T] trinucleotide context (21). The mutational process within this context starts with the deamination of cytosine into uracil, which then templates for thymine during replication and base pairs with adenosine. After a round of replication, a C-to-T transition is then immortalized into the genome. Alternatively, uracil excision by DNA glycosylases and subsequent error-prone repair by translesion synthesis polymerases can generate C-to-G transversions. These two distinguishable single base substitution [SBS] mutations are included in the over 40 etiologically distinct mutation signatures found in pan-cancer datasets [referred to as SBS2 and SBS13, respectively: see (1)]. Importantly, these mutation signatures are consistent with the mutagenic characteristics of A3A, A3B, and A3H haplotype I [A3H-I]. Although previously proposed as a likely contributor, the role of A3H-I in cancer-related mutagenesis has recently been questioned (bioRxiv 2022.04.26.489523v2 (22, 23). In comparison, A3A and A3B are currently seen as major contenders to the origin of APOBEC3 deaminase activity in cancer, which is further described below.
Of all APOBEC3 proteins possibly involved in breast cancer mutagenesis, A3B is the only deaminase overexpressed and steadily present in the nuclear compartment (7, 17, 24, 25). Various studies have directly and indirectly connected A3B-activity with APOBEC3-mediated mutagenesis in several cancer types, including breast cancer [Fig. 1A] (bioRxiv 2022.04.26.489523v2, (17, 26). However, APOBEC3-mediated mutagenesis can still be detected in breast cancers of patients carrying loss of A3B. Loss of A3B presents as a chimeric allele, where the A3A coding region is fused to the A3B 3’UTR, which is rare in European and African populations, but present in ~37%, ~58%, and ~93% of East Asians, native American, and Oceanic populations, respectively (27, 28). This A3A-B fusion allele indicates that additional APOBEC3 members, such as A3A, also contribute to the overall level of APOBEC signature SBS mutations in breast cancer.
Like A3B, the potent deamination activity of A3A has also been causally linked to increased levels of APOBEC3-mediated mutagenesis [Fig. 1A] (bioRxiv 2022.04.26.489523v2, (23, 29–32). In fact, A3A has been proposed as the dominant deaminase over A3B in breast cancer, predominantly based on the reported computationally distinguishable mutational signatures of A3A and A3B as established in yeast, cell lines and subsequently tumors (30, 33, 34). However, although potentially useful in the detection of A3A-mediated mutagenesis specifically, more recent analyses in HAP1 cells have shown that this approach may not provide the necessary resolution between these two deaminases and highlights the appreciable contribution of A3A as well as A3B (bioRxiv 2022.04.26.489523v2). Furthermore, while A3A was recently proposed as a major contributor to SBS2 and SBS13 in breast cancer cells, A3B still has an appreciable contribution to APOBEC3-mediated mutagenesis (23). Therefore, since both A3A and A3B are directly implicated with the accumulation of APOBEC signatures, and a detailed correlation of the most relevant deaminase in relation to breast cancer development is yet to be fully established (and they may also contribute combinatorially), APOBEC3-mediated mutagenesis in this review will not specifically be attributed to either enzyme Fig. 1A]. Reproducible and clinically implementable detection methods, discussed below, can further consolidate the clinical relevance of APOBEC3 proteins and their mutagenic activity.
Options for clinical detection of APOBEC3-mediated mutagenesis
As part of the initial histopathological assessment of malignancy, expression of APOBEC3 proteins can be detected through immunohistochemistry [IHC] in, for instance, diagnostic core needle biopsies [Fig. 1B]. A rabbit monoclonal antibody has recently proven to be suitable for the specific detection of A3A protein using immunofluorescence (bioRxiv 2022.04.26.489523v2). For detection of A3B, the most frequently used antibody is 5210-87-13, a rabbit monoclonal that detects A3A, A3B, and A3G due to a shared epitope (35). As the only APOBEC3 protein with dominant nuclear localization, A3B expressed by tumor cells can be clearly distinguished from other APOBEC3 proteins, including A3A, which are present as cell-wide or cytoplasmic proteins (36). The immunohistochemical detection of A3B has been demonstrated in tumor tissue from several cancer types, including head/neck and ovarian cancer (37–39). Given the low expression of APOBEC3 proteins in most healthy cells, staining of A3A and A3B in tumor cells can be readily detected. It is currently unknown which molecular breast cancer subtype is most likely to score positive for the immunohistochemical detection of A3A and A3B protein. However, protein expression can be seen at early stages of tumor development (40) and, therefore, immunohistochemical detection of A3A and A3B may conveniently be included in histopathologic analysis and stained in parallel to molecular markers such as estrogen receptor [ER] and human epidermal growth factor receptor 2 [HER2]. We therefore recommend all samples be subjected to the A3A/B immunohistochemical analysis. Furthermore, to establish mutational contributions, the resected primary tumor may also be analyzed by DNA sequencing [Fig. 1B]. To the best of our knowledge, targeted sequencing approaches have yet to be optimized to detect APOBEC SBS signatures. Therefore, whole exome sequencing [WES] or whole genome sequencing [WGS] is necessary to gain a comprehensive overview of APOBEC3-mediated mutagenesis. Based on available sequencing data, HER2-amplified breast cancers are most likely to display pan-genomic APOBEC SBS signatures, and ER+ disease may additionally contain APOBEC-induced mutations with clinical relevance (41, 42). These samples may be prioritized in sequencing efforts. Moreover, because tumors with homologous recombination [HR] deficiencies such as loss of BRCA1 or BRCA2 rarely show APOBEC SBS signatures (43, 44), testing labs may be prudent to focus APOBEC diagnostic efforts on HR-proficient tumors.
Importantly, by combining genome sequencing with IHC, historical APOBEC3 activity [i.e., presence of APOBEC SBS signatures, but absence of A3A and A3B protein], may be distinguished from ongoing APOBEC3 activity [i.e., presence of both APOBEC SBS signatures and IHC-positivity]. Other techniques that can consolidate the expression and/or activity of A3A and A3B include quantification by RNA-based methods [i.e., RT-qPCR, RNA sequencing, and/or RNA scope], immunoblotting, A3A dependent RNA editing (45), and DNA deaminase assays [Fig. 1B] (1, 12, 17, 39, 46–50). In order to understand the biological context surrounding APOBEC3-mediated mutagenesis it is important to know how these enzymes can be expressed in breast cancer [discussed below].
(Dys)regulation of APOBEC3 enzymes in breast cancer
Due to their prominent role in the innate immune system, much of the initial data on APOBEC3 regulation stems from virology research. These observations have proven insightful in the identification of mechanisms underlying APOBEC3 (dys)regulation, even though viruses are unlikely to be directly involved in breast cancer [reviewed in (51)].
Regulation of A3A expression
One of the most prominent factors that induce A3A expression are interferons [IFNs]. The pleotropic group of IFNs, most commonly type-I and type-II IFN, are produced as first-responder inflammatory cytokines by, amongst others, tumor-resident immune cells. Type-I IFNs potently induce A3A, while type-II IFNs only activate A3A marginally (52). In breast epithelial cells A3A transcriptional activation through type-I IFNs requires the transcription factor complex STAT2 and its upstream regulator JAK [Fig. 2A] (53). Type-I IFN also induces A3A in tumor cell lines, including those from lung, bladder, and breast cancer (45, 46, 54–56).
Figure 2. Transcriptional regulation of A3A and A3B.
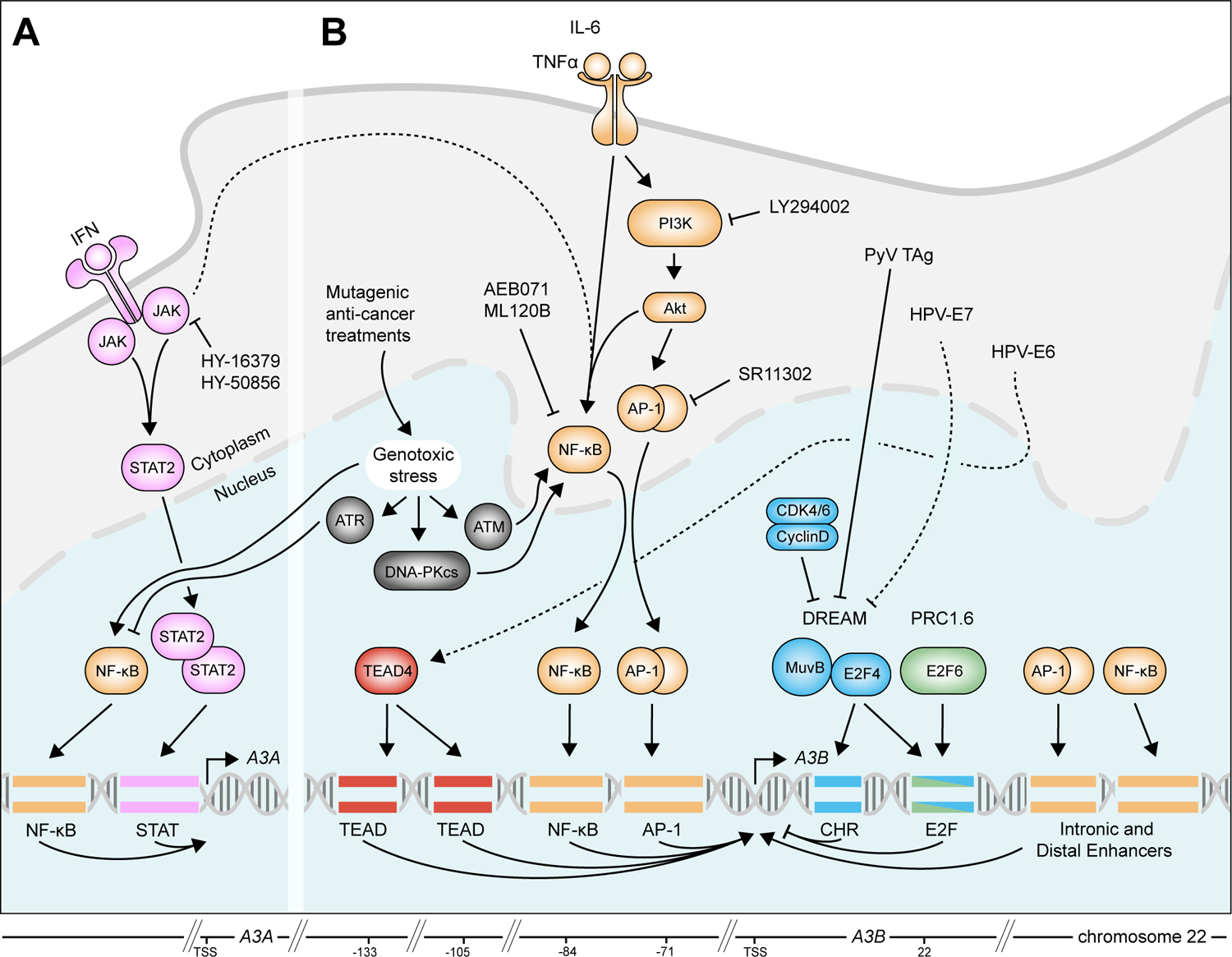
A Activation of IFN signaling facilitates the recruitment of STAT2 to the promoter of A3A, while genotoxic stress promotes the recruitment of the NF-κB transcription factor complex. Transcriptional activation of A3A through genotoxic stress is inhibited by ATR.
B The PKC/ncNF-κB pathway dominates A3B transcription and recruits transcription factor complexes to the A3B promoter, intronic, and distal enhancers. In turn, the PKC/ncNF-κB pathway is activated by genotoxic stress, TNFα, IL-6, and possibly IFN signaling. Transcriptional repression is facilitated by the DREAM complex [and the upstream RB/E2F pathway] and the PRC1.6 complex. Viral oncogenes, including HPV-E6, HPV-E7, and PyV Tag can also activate A3B, although this is unlikely to contribute to A3B expression in breast cancer.
A3A transcription can also be activated by the canonical PKC/ncNF-κB pathway, which itself is activated by a myriad of inflammatory and genotoxic stresses [Fig. 2A and discussed below] (53, 57). However, the activation of A3A upon genotoxic insults is closely guarded by the protein Ataxia Telangiectasia and Rad3-related [ATR], which generally serves as a protective protein during DNA replication stress [Fig. 2A](58). Interestingly, ATR also prevents direct induction of A3A through commonly used cancer treatments, particularly those that cause replication stress (53). However, whether anti-cancer treatments can stimulate inflammatory pathways and subsequently induce A3A expression, and whether this impacts disease trajectory, remains unclear. Moreover, the current body of knowledge on the transcriptional regulation of A3A strongly indicates that chemical inhibition of the IFN and/or PKC/ncNF-κB pathways may be a useful approach to limit A3A expression. Currently, the best-defined inhibitor that has been directly investigated within this context targets JAK2 and effectively prevents A3A induction by IFN (53) [Fig. 2].
Regulation of A3B expression
The PKC/ncNF-κB signaling pathway and its associated proteins PI3K and AKT are at the center of A3B transcriptional activation (39, 59, 60). PKC/ncNF-κB and related AP-1 complexes are recruited to sites within the A3B promoter, intronic regions, and a distant enhancer, thus activating A3B transcription [Fig. 2B] (61). Various upstream stimuli converge to activate the PKC/ncNF-κB signaling pathway, thereby eliciting an increase in A3B expression in breast cancer. For example, tumor necrosis factor alpha [TNFα], a pro-inflammatory cytokine, activates the PKC/ncNF-κB signaling pathway and stimulates A3B expression [Fig. 2B] (61). Furthermore, the pro-inflammatory cytokine IL-6, produced by both leukocytes and several solid tumor cell lines, can activate PKC/ncNF-κB [reviewed in (62)] and thereby A3B [Fig. 2B] (61, 63).
Induction of A3B by type I and type II IFNs by PKC/ncNF-κB cross-activation is also observed in (oropharangeal and lung) cancer cell lines (54, 56). However, relative to A3A, this induction of A3B by IFNs is less consistent between different tissue types, indicating the presence of currently unknown regulatory mechanisms [Fig. 2B]. Finally, DNA double strand breaks, which commonly occur in response to ionizing radiation, various chemotherapeutic drugs, or advanced genomic instability [reviewed in (64)] can increase A3B expression in, amongst others, breast cancer cell lines (55, 60, 61, 65–67). Induction of A3B through the PKC/ncNF-κB pathway is also dependent on several main responders to DNA double strand breaks, specifically DNA-PKcs and ATM [ataxia telangiectasia mutated] [Fig. 2B] (60, 61). Interestingly, and further emphasizing the central role of the PKC/ncNF-κB pathway in A3B induction, several pre-clinical and clinically approved PKC inhibitors have been shown to effectively, and dose-responsively, inhibit expression of A3B in various [breast] cancer cell lines (39, 61) [Fig. 2]. Future studies could further explore the usefulness of these compounds in restricting the mutagenic activities of A3B.
Additionally, several viral oncoproteins, including human papillomavirus [HPV] E6, E7, and polyomavirus T-antigen, are strongly implicated with A3B transcriptional dysregulation and the accumulation of APOBEC SBS signatures in several virally induced cancers [Fig. 2B] (52, 68–73). Specifically, HPV-E6 may drive expression of A3B through recruitment of the transcription factor TEAD to two distinct binding sites at the A3B promoter (68). Additionally, both HPV-E7 and polyomavirus T-antigen target the transcriptional repressor DREAM which, as an integral component of the RB/E2F pathway, facilitates the timely expression of cell cycle-associated genes during proliferation [Fig. 2B]. A3B is repressed by the DREAM and the PRC1.6 complex, which are recruited to an E2F binding site within the A3B promoter in normal-like breast epithelial cells [Fig. 2B] (26, 72). Importantly, disruption of the RB/E2F pathway is common in breast cancer and associates with increased APOBEC SBS signatures (26, 74). Moreover, given the functional implication of the RB/E2F pathway with proliferation, it is likely that A3B expression is regulated in a fashion similar to many cell cycle genes. Multiple lines of evidence have indeed classified A3B as a gene that associates strongly with cell cycle progression and proliferation in cancer cells (26, 37, 75).
Thus, in contrast to A3A, A3B is readily induced by therapeutic agents. In fact, the occurrence of treatment-induced mutations is relatively well documented and predominantly showcases the direct induction of mutations by cancer drugs, including cis-platin (44), and induction of APOBEC-mutagenesis has been documented comparing gliomas before and after irradiation (76). However, it would be insightful, although challenging to control, to investigate the contribution of treatment-induced A3B on the total APOBEC signature load of breast cancer patients. A minor contribution of treatment-induced A3B-activity is to be expected and might, on a background level, amplify the impact of A3B on disease progression [discussed below].
APOBEC3-mediated mutagenesis and disease trajectory
Recent tumor sequencing efforts have revealed the genetically heterogeneous and evolving nature of tumor cells, changing their genetic makeup during different cancer stages or when facing anti-cancer treatments (1, 77, 78). Specifically, APOBEC3-mediated mutagenesis has been shown to, in varying degrees, influence the mutational landscape of pre-malignant breast lesions, primary disease, and metastatic breast cancer.
APOBEC3 activity during pre-malignancy
Although samples sizes remain limited, APOBEC3 expression and/or -mediated mutagenesis is found in about 8% of ductal carcinoma in situ [DCIS] samples and is detectable in approximately 16% of specimens when DCIS is associated with invasive disease (50, 79–82). However, although APOBEC3-mediated mutagenesis is appreciable in DCIS, no clear evidence of APOBEC3-mediated mutagenesis towards driver genes has been found (80, 82). This indicates that although APOBEC3-mediated mutagenesis can influence the cellular genome at the precursor-stage, this frequently takes place before clonal selection overtakes the overall genomic makeup [Fig. 3]. Therefore, the overall impact on tumor evolution is yet to be fully determined.
Figure 3. APOBEC3-mediated mutagenesis and disease trajectory.
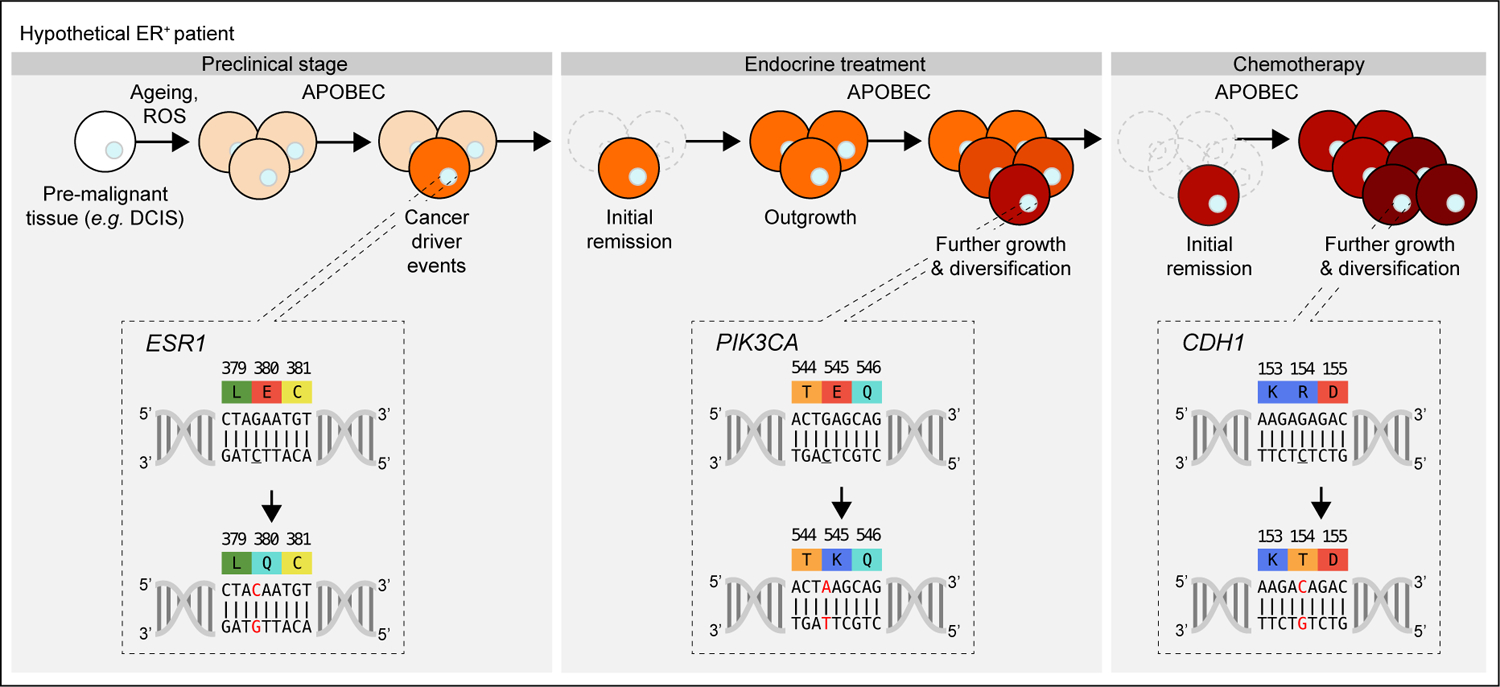
Clinical progression of a hypothetical ER+ patient from preclinical stage [initial outgrowth and initial cancer driver events], to first- and second-line anti-cancer treatments. In this example, APOBEC3-mediated mutation of ESR1 is the first driver mutation. Mutations in PIK3CA provide the tumor with resistance to adjuvant endocrine treatment. Although remission is obtained, further growth and APOBEC3-mediated diversification occurs. Second-line chemotherapeutics provided temporary remission, butfurther APOBEC3-mediated mutagenesis affected genes involved in metastatic behavior, here exemplified with CDH1, leading to treatment failure. The cytosine targeted by APOBEC3 proteins is underlined.
APOBEC3 activity throughout malignant disease
APOBEC3-mediated deamination is actively involved in tumor evolution in early and advanced breast cancer (2, 44, 56, 78, 81, 83, 84), adding novel “branches” to the cancer evolutionary “tree” that may unfavorably impact disease trajectory. For instance, in hypermutated breast tumors APOBEC3 activity can account for almost two-thirds of the total mutational burden (44, 85). Indeed, mutations in ~25% of cancer driver genes occur within the preferred 5’-TCW APOBEC context [see Fig. 3 and Table 1 for examples].
| Gene | Mutations | Trinucleotide context | Pathogenicity score (FATHMM) | Hotspot (Yes/No) | Clinical associations | References |
|---|---|---|---|---|---|---|
| PIK3CA | E453K | TCT > TTT | 0.99 | No | Loss of normal PIK3CA function has been recognized as a contributing factor to the acquired resistance to chemotherapy and endocrine therapy. Multi-hit mutations in PIK3CA are also associated with APOBEC activity. | (78, 86, 87, 95, 123) |
| E542K | TCA > TTA | 0.97 | Yes | |||
| E545K | TCA > TTA | 0.97 | Yes | |||
| E545Q | TCA > TGA | 0.98 | Yes | |||
| E726K | TCA > TTA | 0.99 | No | |||
| E970K | TCT > TTT | 0.99 | No | |||
| SPEN | E2151K | TCT > TTT | N/A | No | Associated with acquired resistance to endocrine treatment | (95, 96) |
| ESR1 | E380Q | TCT > TGT | 1.00 | Yes | Associated with acquired resistance to endocrine treatment | (95, 97) |
| NF1 | Q554* | TCA > TTA | 0.99 | No | Confers resistance to endocrine treatment, possibly by enabling cell cycle progression overdrive | (92, 124) |
| Q1218* | TCA > TTA | 0.98 | No | |||
| Q1399* | TCA > TTA | 0.98 | No | |||
| Q2234* | TCA > TTA | 1.00 | No | |||
| CDH1 | R154T | TCT > TGT | 0.98 | No | Associated with cellular discohesion and hyperplasia | (81, 94) |
| ARID1A | SNNN* | not reported | various | No | Associated with acquired resistance to endocrine treatment and lowered PFS | (83) |
| KMT2C | QNNN* |
Abbreviations: FATHMM, Functional Analysis Through Hidden Markov Models (125)
As driver mutations occur predominantly in the early stages of tumor evolution, this indicates that APOBEC3-mediated mutagenesis provides mutagenic fuel during the early stages of breast cancer (78, 83). However, APOBEC3-mediated alterations of driver genes can also occur as late events (44, 81, 83) [Fig. 3]. Notable APOBEC3-associated driver mutations are the E542K and E545K hot spot mutations in PIK3CA, the second most frequently altered breast cancer driver gene (2). These mutations account for ≥ 1/3rd of the PIK3CA single-point mutations in breast cancer and are thought to predominantly occur as early events (29, 42, 83). Tumors can also carry multiple APOBEC-associated PIK3CA mutations which are enriched in metastatic breast cancer as compared to primary breast cancer (83). This cis-PIK3CA mutational genotype provides enhanced downstream signaling, associates with lower progression-free survival, and has been recognized as a contributing factor to acquired treatment resistance (42, 86–88).
Other genes affected by APOBEC3-mediated mutagenesis include, amongst others, KMT2C and ARID1A (78), which exhibit widespread, non-hotspot truncating S>X and Q>X mutations in an APOBEC context (83). Importantly, loss of KMT2C is associated with resistance to endocrine therapy, while ARID1A mutations may confer resistance to both endocrine therapy and chemotherapy (89–91). Moreover, the emergence of truncating NF1 mutations bearing APOBEC SBS signatures may also occur during endocrine therapy (92). Loss of NF1 has been shown to confer resistance to endocrine treatment, possibly by enabling cell cycle progression overdrive (93). An APOBEC-associated mutation in the tumor suppressor CDH1, unique to the metastatic tumor, grew to dominance during chemotherapy (81). Pathogenic mutations in CDH1 have been associated with cellular decohesion and hyperplasia and contribute to lobular breast cancer [reviewed in (94)]. Finally, post-mortem sequencing of metastases of endocrine-resistant ER+ breast cancer revealed novel acquired APOBEC-associated mutations in SPEN and ESR1 (95), genes that have been associated with acquired resistance to endocrine treatment (96, 97). Importantly, APOBEC-associated mutations in almost all the aforementioned driver genes [PIK3CA, KMT2C, ARID1A, NF1, and CDH1] have recently been shown to be enriched in metastatic breast cancer, strongly emphasizing their relevance in cancer development (83). Other genes carrying APOBEC-associated mutations in these studies were, amongst others, the tumor suppressors MAP3K1, TP53, and ZFHX3.
Leveraging APOBEC3 activity for clinical benefit
APOBEC3 as a prognostic biomarker
Considering the active contribution of APOBEC3-mediated mutagenesis to disease trajectory it can be expected that APOBEC3 expression and/or APOBEC SBS signatures can serve as prognostic biomarkers in breast cancer. Indeed, in ER+ breast cancer high expression of A3B correlates with unfavorable clinical parameters, including disease-free survival, metastasis-free survival, and overall survival (48). Although prognostic studies are rare as these have to be performed in the absence of systemic treatment to distinguish them from predictive biomarkers, the independent nature of these findings emphasizes the suitability of A3B as a prognostic marker in ER+ breast cancer.
APOBEC3 as a predictive biomarker
The role of APOBEC3 expression and/or APOBEC SBS signatures as predictive biomarkers has become increasingly established over recent years. Increased A3B expression in breast cancer has been strongly associated with treatment failure of adjuvant endocrine drugs (49), implying that commonly used endocrine drugs [such as tamoxifen] are less suitable for APOBEC3-positive tumors [Fig. 4]. Conversely, in breast cancer as well as ovarian carcinoma and bladder cancer, APOBEC3-mediated mutagenesis was found to predict beneficial treatment response (38, 55, 98, 99). Although diverse treatment regimens were used in these studies, all applied DNA intercalating agents. These observations suggest that APOBEC3-mediated mutagenesis and DNA intercalators combine to exert synergistic levels of DNA damage during breast cancer treatment [Fig. 4]. Overall, APOBEC3-mediated mutagenesis is a predictive biomarker for response to both endocrine- and chemotherapy. Of note, recent work in lung cancer suggests A3A and A3B may also contribute to resistance to targeted therapies (bioRxiv 2021.01.20.426852v1; bioRxiv 2020.12.18.423280v2).
Figure 4. Clinical implications of APOBEC3-mediated mutagenesis.
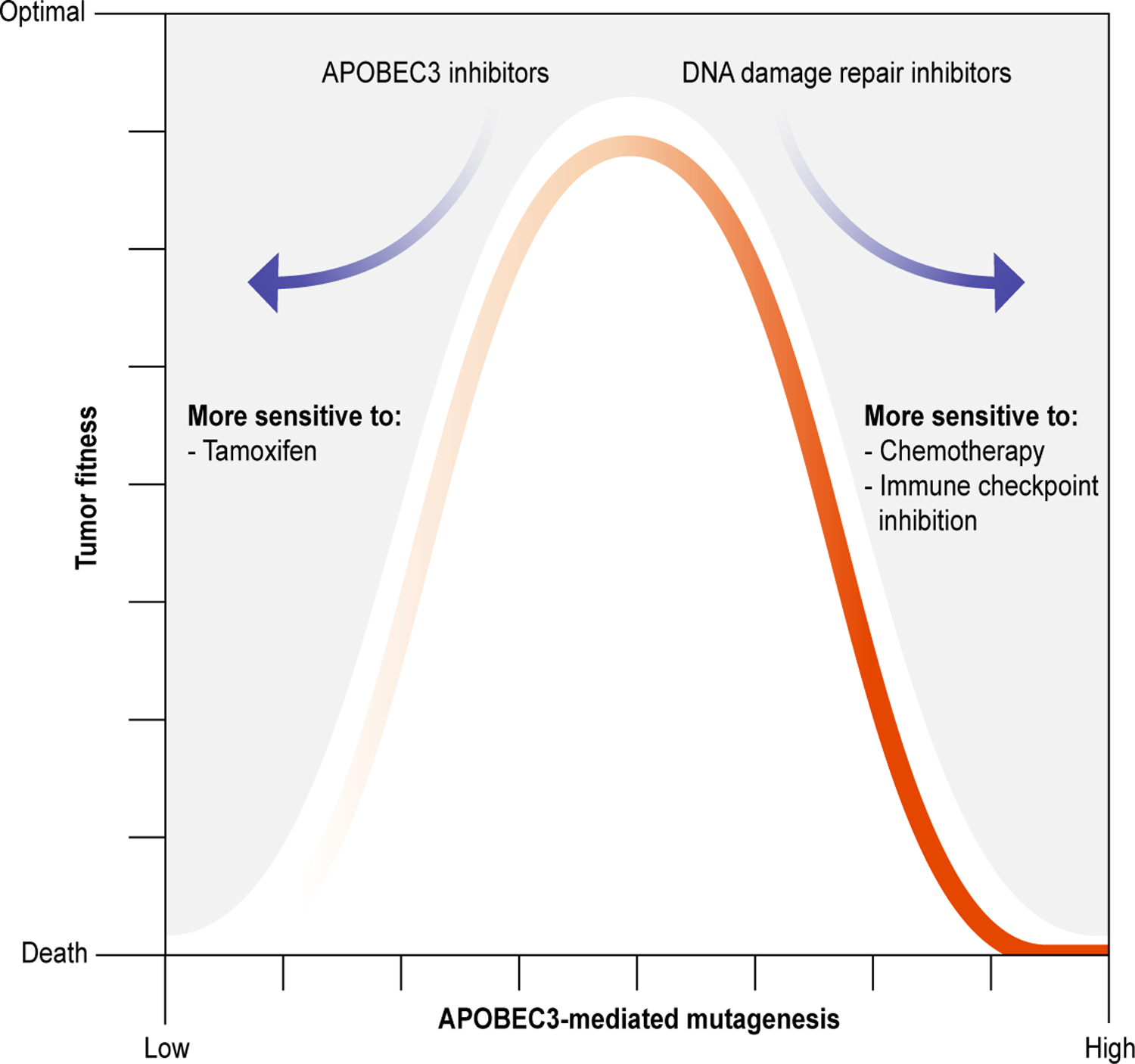
Overview depicting the relationship between APOBEC3-mediated mutagenesis [X-axis] and tumor fitness [Y-axis] as a function of suitable anti-cancer treatments. Standard-of-care therapies, such as tamoxifen and radiotherapy, are suitable when APOBEC3 activity is low, driving down tumor fitness. On the other hand, immune checkpoint inhibition, synergistic drugs [such as DNA intercalating agents], and synthetically lethal combinations [such as ATRi] can exploit the weaknesses brought about by APOBEC3-mediated mutagenesis. Inhibitors of A3A and/or A3B, currently in development, may also be used to limit APOBEC3-mediated genomic diversification and sensitize to other treatments.
APOBEC3 as a predictor for immune checkpoint inhibition response
Cancer growth relies on a disturbance in the balance between detection and subsequent elimination of cancer cells by immune cells and, conversely, the escape of cancer cells from immune cells. At the core of this interaction are antigen-presenting dendritic cells [DCs] and CD4+ or CD8+ T cells. DCs are innate immune cells specialized in recognizing neoantigens, which are proteins released by tumor cells that contain non-autologous antigens as a result from somatic mutations (100). Upon recognition, these neoantigens are used to prime naïve CD4+, and CD8+ T cells, which infiltrate tumor tissue and eliminate tumor cells displaying these neoantigens [reviewed in (101)]. Due to the highly plastic nature of tumors, cancer cells need to tip the scale in their favor in order to avoid cytotoxic elimination. Proteins expressed by tumor cells, such as PD-1, can interact with inhibitory ligands expressed by T cells, such as PD-L1, initiating a shift towards immune tolerance. This interaction, called an immune checkpoint, forms the basis for immune checkpoint inhibition [ICI], which seeks to enhance immunogenic tumor cell killing by using antibodies against key immune checkpoint proteins [Fig. 4, and reviewed in (102, 103)].
Given the involvement of APOBEC activity with shaping the tumor genome, and thereby the antigen repertoire, its suitability to predict ICI response in breast cancer has become subject of investigation. In a recent study (104), murine breast cancer cells that normally do not possess the A3B gene, were engineered to express A3B and orthotopically injected. Interestingly, expression of A3B alone already significantly slowed tumor growth as compared to cells devoid of A3B. This partial inhibition of tumor growth was dependent on CD4+ and CD8+ immune cells, suggesting that at least some cytotoxic tumor cell killing was achieved. A3B expression also promoted tumor infiltration by T cells that were likely primed with APOBEC3-induced neoantigens. Strikingly, when combined with ICI, potent and sustained growth inhibition was achieved in A3B-expressing cells, but not control cells.
Increased APOBEC3-mediated mutagenesis has also been associated with immunoactivation in human breast cancer. For instance, firstly, indicators of APOBEC3-mediated mutagenesis such as increased APOBEC SBS signatures and expression of A3A or A3B often correlate positively with infiltration of tumor tissue by immune cells, including DCs and CD8+ T cells (105–107). Secondly, the same indicators of APOBEC3-mediated mutagenesis correlate positively with expression of PD-1 and PD-L1 (108). Thirdly, in several studies -although with a limited number of breast cancer patients- the presence of APOBEC SBS signatures significantly improved the chance of ICI response (85, 108–110). Combined, these findings suggest that APOBEC3 activity can function as a powerful predictor of ICI responsiveness [Fig. 4], which merits further investigation with larger cohorts of breast cancer patients.
Synthetically lethal interactions with APOBEC3-mediated mutagenesis
In addition to somatic C-to-T and C-to-G mutations the activity of APOBEC3 enzymes can also induce DNA double-strand breaks and replication stress (17, 41, 111–113). In order to counteract these pressures, and to ensure survival, cancer cells that display APOBEC3 activity increasingly rely on DNA damage repair (111–113). This vulnerability has led to a number of synthetic lethality approaches that target DNA damage repair in A3A or A3B expressing cancer cells [Fig. 4]. Cells with high APOBEC3 activity are exceptionally vulnerable to inhibition of ATR, an important DNA damage checkpoint (29, 113). There are currently multiple ATR inhibitors being evaluated for clinical use (114). Furthermore, inhibition of at least three major repair factors involved in the resolution or neutralization of deaminated lesions, including UNG, HMCES, and APE1, show similar synthetic lethal phenotypes (111, 115–117). Altogether, these synthetic lethality models represent a rational design to systematically attack the vulnerabilities of cancers that show ongoing APOBEC3-mutagenesis and warrant further research into their suitability in breast cancer.
Dampening APOBEC3 activity using inhibitors
Efforts to inhibit A3A and A3B enzymes have chiefly relied on the design of chemical inhibitors and, so far, revolve around two molecular classes [Fig. 5]. One such approach exploits the trinucleotide preference of the Z1 domains of A3A and A3B, and features a chemically modified cytosine, called dZ, in an oligo-based substrate. These efforts yielded promising substrate-like inhibitors within the low micromolar range in in vitro assays (118–121). Additionally, another recent approach identified several candidate small molecules [as opposed to substrate-like inhibitors], with comparable in vitro effectiveness within the low micromolar range (122). After further characterization in vitro, APOBEC3 inhibitors can subsequently be investigated in clinically relevant pre-clinical platforms, as relevant genetically engineered mouse models for A3A and A3B have become available recently (31, 104). Future studies should aim to determine possible systemic toxicity of candidate inhibitors and their ability to lower the accumulation of APOBEC3 SBS signatures in murine cancers. Moreover, considering the established role of both these enzymes in the development of [breast] cancer, further research into APOBEC3 inhibition should stay focused on the dual-inhibition of both A3A and A3B. Ultimately, APOBEC3 inhibitors should be investigated as as synergistic treatments in conjunction with existing anti-breast cancer therapies, including surgery and targeted treatments based on genetic markers [Fig. 4].
Figure 5. APOBEC3A and APOBEC3B inhibitors.
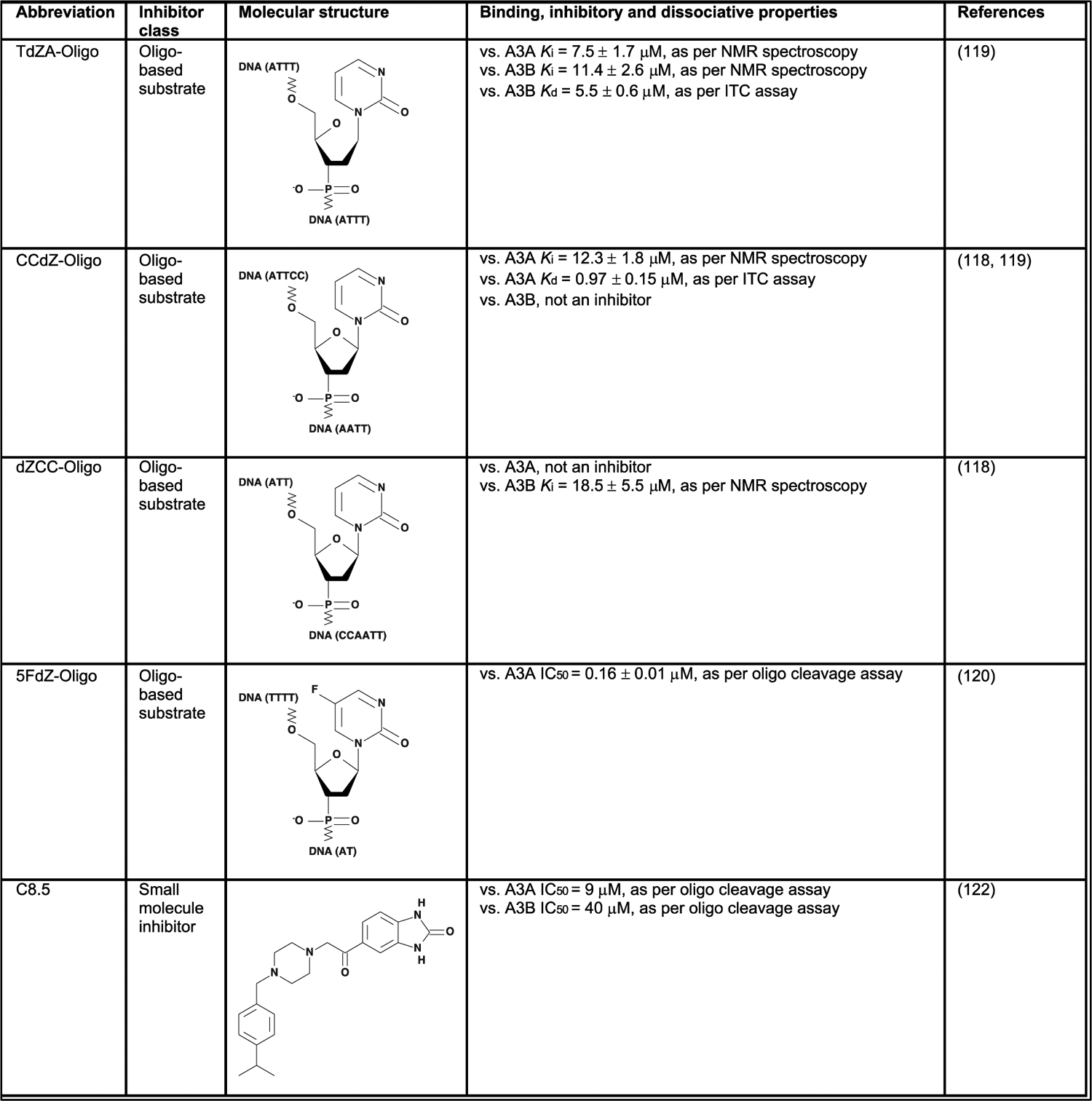
Published inhibitors of APOBEC3A and APOBEC3B, their molecular structures and pharmacological properties.
Conclusions
Over the past decade the perspective on APOBEC3 enzymes, specifically A3A and A3B, has drastically shifted from beneficial members of the innate system to direct influencers of cancer development and disease trajectory most notably in breast cancer. A collection of cellular pathways, including the PKC/ncNF-κB, the RB/E2F pathway, and IFN signaling relay proliferative and inflammatory signals that stimulate expression of A3A and/or A3B. The mutagenic activities of APOBEC3 proteins can now be traced using high-throughput sequencing approaches, implicating them with genomic alterations that stand in direct association with treatment response. They also provide prognostic and predictive value and reveal potential cancer weak spots. Furthermore, the promising characteristics of potential APOBEC3 inhibitors merit further investigation and may be instrumental in restricting the mutagenic arsenal of cancer cells.
Acknowledgements
We want to extend our gratitude to Wilke Castelijns, MSc., for helpful suggestions during the early conceptualization phase of this review. We also thank Bojana Stefanovska, PhD., and Anya Normandeau, B.A., for helpful feedback.
Funding
This work was supported, in part, by the KWF Dutch Cancer Society (KWF10270, to JWMM, PNS, and RSH) and by the National Cancer Institute (P01-CA234228, to RSH). RSH is the Ewing Halsell President’s Council Distinguished Chair, a CPRIT Scholar, and an Investigator of the Howard Hughes Medical Institute at University of Texas Health San Antonio. None of the funding agencies had any role in conceptualization, study design, data collection, interpretation of results, or the decision to submit this work for publication.
Footnotes
Author’s Disclosures
The authors declare no competing or financial interests.
References
- 1.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534(7605):47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Periyasamy M, Patel H, Lai CF, Nguyen VTM, Nevedomskaya E, Harrod A, et al. APOBEC3B-Mediated Cytidine Deamination Is Required for Estrogen Receptor Action in Breast Cancer. Cell Rep. 2015;13(1):108–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris RS, Dudley JP. APOBECs and virus restriction. Virology. 2015;479–480:131–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ito J, Gifford RJ, Sato K. Retroviruses drive the rapid evolution of mammalian APOBEC3 genes. Proc Natl Acad Sci U S A. 2020;117(1):610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Abudu A, Son S, Dang Y, Venta PJ, Zheng YH. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J Virol. 2011;85(7):3142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salamango DJ, McCann JL, Demir O, Brown WL, Amaro RE, Harris RS. APOBEC3B Nuclear Localization Requires Two Distinct N-Terminal Domain Surfaces. J Mol Biol. 2018;430(17):2695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navarro F, Bollman B, Chen H, Konig R, Yu Q, Chiles K, et al. Complementary function of the two catalytic domains of APOBEC3G. Virology. 2005;333(2):374–86. [DOI] [PubMed] [Google Scholar]
- 9.Shi K, Demir O, Carpenter MA, Wagner J, Kurahashi K, Harris RS, et al. Conformational Switch Regulates the DNA Cytosine Deaminase Activity of Human APOBEC3B. Sci Rep. 2017;7(1):17415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson BD, Ikeda T, Moghadasi SA, Martin AS, Brown WL, Harris RS. Natural APOBEC3C variants can elicit differential HIV-1 restriction activity. Retrovirology. 2018;15(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weisblum Y, Oiknine-Djian E, Zakay-Rones Z, Vorontsov O, Haimov-Kochman R, Nevo Y, et al. APOBEC3A Is Upregulated by Human Cytomegalovirus (HCMV) in the Maternal-Fetal Interface, Acting as an Innate Anti-HCMV Effector. J Virol. 2017;91(23):e01296–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010;38(13):4274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14(15):1392–6. [DOI] [PubMed] [Google Scholar]
- 14.Hultquist JF, Harris RS. Leveraging APOBEC3 proteins to alter the HIV mutation rate and combat AIDS. Future Virology. 2009;4(6):605–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suspene R, Guetard D, Henry M, Sommer P, Wain-Hobson S, Vartanian JP. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc Natl Acad Sci U S A. 2005;102(23):8321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng AZ, Yockteng-Melgar J, Jarvis MC, Malik-Soni N, Borozan I, Carpenter MA, et al. Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat Microbiol. 2019;4(1):78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494(7437):366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149(5):979–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45(9):970–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petljak M, Alexandrov LB, Brammeld JS, Price S, Wedge DC, Grossmann S, et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell. 2019;176(6):1282–94 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nik-Zainal S, Morganella S. Mutational Signatures in Breast Cancer: The Problem at the DNA Level. Clin Cancer Res. 2017;23(11):2617–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Starrett GJ, Luengas EM, McCann JL, Ebrahimi D, Temiz NA, Love RP, et al. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat Commun. 2016;7:12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petljak M, Dananberg A, Chu K, Bergstrom EN, Striepen J, von Morgen P, et al. Mechanisms of APOBEC3 mutagenesis in human cancer cells. Nature. 2022;607(7920):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lackey L, Law EK, Brown WL, Harris RS. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle. 2013;12(5):762–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCann JL, Klein MM, Leland EM, Law EK, Brown WL, Salamango DJ, et al. The DNA deaminase APOBEC3B interacts with the cell-cycle protein CDK4 and disrupts CDK4-mediated nuclear import of Cyclin D1. J Biol Chem. 2019;294(32):12099–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roelofs PA, Goh CY, Chua BH, Jarvis MC, Stewart TA, McCann JL, et al. Characterization of the mechanism by which the RB/E2F pathway controls expression of the cancer genomic DNA deaminase APOBEC3B. Elife. 2020;9:e61287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 2007;3(4):e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nik-Zainal S, Wedge DC, Alexandrov LB, Petljak M, Butler AP, Bolli N, et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat Genet. 2014;46(5):487–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buisson R, Langenbucher A, Bowen D, Kwan EE, Benes CH, Zou L, et al. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science. 2019;364(6447):eaaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cortez LM, Brown AL, Dennis MA, Collins CD, Brown AJ, Mitchell D, et al. APOBEC3A is a prominent cytidine deaminase in breast cancer. PLoS Genet. 2019;15(12):e1008545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Law EK, Levin-Klein R, Jarvis MC, Kim H, Argyris PP, Carpenter MA, et al. APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. J Exp Med. 2020;217(12):e20200261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, Roberts SA. APOBEC3A and APOBEC3B Preferentially Deaminate the Lagging Strand Template during DNA Replication. Cell Rep. 2016;14(6):1273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, Malc EP, et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet. 2015;47(9):1067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeWeerd RA, Nemeth E, Poti A, Petryk N, Chen CL, Hyrien O, et al. Prospectively defined patterns of APOBEC3A mutagenesis are prevalent in human cancers. Cell Rep. 2022;38(12):110555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown WL, Law EK, Argyris PP, Carpenter MA, Levin-Klein R, Ranum AN, et al. A Rabbit Monoclonal Antibody against the Antiviral and Cancer Genomic DNA Mutating Enzyme APOBEC3B. Antibodies (Basel). 2019;8(3):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hultquist JF, Harris RS. Leveraging APOBEC3 proteins to alter the HIV mutation rate and combat AIDS. Future Virol. 2009;4(6):605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Argyris PP, Wilkinson PE, Jarvis MC, Magliocca KR, Patel MR, Vogel RI, et al. Endogenous APOBEC3B overexpression characterizes HPV-positive and HPV-negative oral epithelial dysplasias and head and neck cancers. Mod Pathol. 2021;34(2):280–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serebrenik AA, Argyris PP, Jarvis MC, Brown WL, Bazzaro M, Vogel RI, et al. The DNA Cytosine Deaminase APOBEC3B is a Molecular Determinant of Platinum Responsiveness in Clear Cell Ovarian Cancer. Clin Cancer Res. 2020;26(13):3397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leonard B, McCann JL, Starrett GJ, Kosyakovsky L, Luengas EM, Molan AM, et al. The PKC/NF-kappaB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res. 2015;75(21):4538–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JY, Schizas M, Geyer FC, Selenica P, Piscuoglio S, Sakr RA, et al. Lobular Carcinomas In Situ Display Intralesion Genetic Heterogeneity and Clonal Evolution in the Progression to Invasive Lobular Carcinoma. Clin Cancer Res. 2019;25(2):674–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venkatesan S, Angelova M, Puttick C, Zhai H, Caswell DR, Lu W-T, et al. Induction of APOBEC3 Exacerbates DNA Replication Stress and Chromosomal Instability in Early Breast and Lung Cancer Evolution. Cancer Discovery. 2021;11(10):2456–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kingston B, Cutts RJ, Bye H, Beaney M, Walsh-Crestani G, Hrebien S, et al. Genomic profile of advanced breast cancer in circulating tumour DNA. Nat Commun. 2021;12(1):2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pitt JJ, Riester M, Zheng Y, Yoshimatsu TF, Sanni A, Oluwasola O, et al. Characterization of Nigerian breast cancer reveals prevalent homologous recombination deficiency and aggressive molecular features. Nat Commun. 2018;9(1):4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Angus L, Smid M, Wilting SM, van Riet J, Van Hoeck A, Nguyen L, et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat Genet. 2019;51(10):1450–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jalili P, Bowen D, Langenbucher A, Park S, Aguirre K, Corcoran RB, et al. Quantification of ongoing APOBEC3A activity in tumor cells by monitoring RNA editing at hotspots. Nat Commun. 2020;11(1):2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carpenter MA, Li M, Rathore A, Lackey L, Law EK, Land AM, et al. Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A. J Biol Chem. 2012;287(41):34801–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sieuwerts AM, Schrijver WA, Dalm SU, de Weerd V, Moelans CB, Ter Hoeve N, et al. Progressive APOBEC3B mRNA expression in distant breast cancer metastases. PLoS One. 2017;12(1):e0171343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sieuwerts AM, Willis S, Burns MB, Look MP, Meijer-Van Gelder ME, Schlicker A, et al. Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Horm Cancer. 2014;5(6):405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Law EK, Sieuwerts AM, LaPara K, Leonard B, Starrett GJ, Molan AM, et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci Adv. 2016;2(10):e1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sieuwerts AM, Doebar SC, de Weerd V, Verhoef EI, Beauford CM, Agahozo MC, et al. APOBEC3B Gene Expression in Ductal Carcinoma In Situ and Synchronous Invasive Breast Cancer. Cancers (Basel). 2019;11(8):1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lawson JS, Glenn WK. Catching viral breast cancer. Infect Agent Cancer. 2021;16(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baker SC, Mason AS, Slip RG, Skinner KT, Macdonald A, Masood O, et al. Induction of APOBEC3-mediated genomic damage in urothelium implicates BK polyomavirus (BKPyV) as a hit-and-run driver for bladder cancer. Oncogene. 2022;41(15):2139–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oh S, Bournique E, Bowen D, Jalili P, Sanchez A, Ward I, et al. Genotoxic stress and viral infection induce transient expression of APOBEC3A and pro-inflammatory genes through two distinct pathways. Nat Commun. 2021;12(1):4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kondo S, Wakae K, Wakisaka N, Nakanishi Y, Ishikawa K, Komori T, et al. APOBEC3A associates with human papillomavirus genome integration in oropharyngeal cancers. Oncogene. 2017;36(12):1687–97. [DOI] [PubMed] [Google Scholar]
- 55.Middlebrooks CD, Banday AR, Matsuda K, Udquim KI, Onabajo OO, Paquin A, et al. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat Genet. 2016;48(11):1330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roper N, Gao S, Maity TK, Banday AR, Zhang X, Venugopalan A, et al. APOBEC Mutagenesis and Copy-Number Alterations Are Drivers of Proteogenomic Tumor Evolution and Heterogeneity in Metastatic Thoracic Tumors. Cell Rep. 2019;26(10):2651–66 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Siriwardena SU, Perera MLW, Senevirathne V, Stewart J, Bhagwat AS. A Tumor-Promoting Phorbol Ester Causes a Large Increase in APOBEC3A Expression and a Moderate Increase in APOBEC3B Expression in a Normal Human Keratinocyte Cell Line without Increasing Genomic Uracils. Mol Cell Biol. 2019;39(1):e00238–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 2017;18(10):622–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Madsen P, Anant S, Rasmussen HH, Gromov P, Vorum H, Dumanski JP, et al. Psoriasis upregulated phorbolin-1 shares structural but not functional similarity to the mRNA-editing protein apobec-1. J Invest Dermatol. 1999;113(2):162–9. [DOI] [PubMed] [Google Scholar]
- 60.Periyasamy M, Singh AK, Gemma C, Farzan R, Allsopp RC, Shaw JA, et al. Induction of APOBEC3B expression by chemotherapy drugs is mediated by DNA-PK-directed activation of NF-kappaB. Oncogene. 2021;40(6):1077–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin L, Holmes B, Shen MW, Kammeron D, Geijsen N, Gifford DK, et al. Comprehensive Mapping of Key Regulatory Networks that Drive Oncogene Expression. Cell Rep. 2020;33(8):108426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu W, Wu J, Yang F, Ma L, Ni C, Hou X, et al. Genetic Polymorphisms Predisposing the Interleukin 6-Induced APOBEC3B-UNG Imbalance Increase HCC Risk via Promoting the Generation of APOBEC-Signature HBV Mutations. Clin Cancer Res. 2019;25(18):5525–36. [DOI] [PubMed] [Google Scholar]
- 64.Habraken Y, Piette J. NF-kappaB activation by double-strand breaks. Biochem Pharmacol. 2006;72(9):1132–41. [DOI] [PubMed] [Google Scholar]
- 65.Kanu N, Cerone MA, Goh G, Zalmas LP, Bartkova J, Dietzen M, et al. DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer. Genome Biol. 2016;17(1):185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saito Y, Miura H, Takahashi N, Kuwahara Y, Yamamoto Y, Fukumoto M, et al. Involvement of APOBEC3B in mutation induction by irradiation. J Radiat Res. 2020;61(6):819–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamazaki H, Shirakawa K, Matsumoto T, Kazuma Y, Matsui H, Horisawa Y, et al. APOBEC3B reporter myeloma cell lines identify DNA damage response pathways leading to APOBEC3B expression. PLoS One. 2020;15(1):e0223463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mori S, Takeuchi T, Ishii Y, Yugawa T, Kiyono T, Nishina H, et al. Human Papillomavirus 16 E6 Upregulates APOBEC3B via the TEAD Transcription Factor. J Virol. 2017;91(6):e02413–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Periyasamy M, Singh AK, Gemma C, Kranjec C, Farzan R, Leach DA, et al. p53 controls expression of the DNA deaminase APOBEC3B to limit its potential mutagenic activity in cancer cells. Nucleic Acids Res. 2017;45(19):11056–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vieira VC, Leonard B, White EA, Starrett GJ, Temiz NA, Lorenz LD, et al. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. mBio. 2014;5(6):e02234–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Warren CJ, Van Doorslaer K, Pandey A, Espinosa JM, Pyeon D. Role of the host restriction factor APOBEC3 on papillomavirus evolution. Virus Evol. 2015;1(1):vev015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Starrett GJ, Serebrenik AA, Roelofs PA, McCann JL, Verhalen B, Jarvis MC, et al. Polyomavirus T Antigen Induces APOBEC3B Expression Using an LXCXE-Dependent and TP53-Independent Mechanism. mBio. 2019;10(1):e02690–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zapatka M, Borozan I, Brewer DS, Iskar M, Grundhoff A, Alawi M, et al. The landscape of viral associations in human cancers. Nat Genet. 2020;52(3):320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hirabayashi S, Shirakawa K, Horisawa Y, Matsumoto T, Matsui H, Yamazaki H, et al. APOBEC3B is preferentially expressed at the G2/M phase of cell cycle. Biochem Biophys Res Commun. 2021;546:178–84. [DOI] [PubMed] [Google Scholar]
- 76.Kocakavuk E, Anderson KJ, Varn FS, Johnson KC, Amin SB, Sulman EP, et al. Radiotherapy is associated with a deletion signature that contributes to poor outcomes in patients with cancer. Nat Genet. 2021;53(7):1088–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brady SW, McQuerry JA, Qiao Y, Piccolo SR, Shrestha G, Jenkins DF, et al. Combating subclonal evolution of resistant cancer phenotypes. Nature Communications. 2017;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7(283):283ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nachmanson D, Officer A, Mori H, Gordon J, Evans MF, Steward J, et al. The breast pre-cancer atlas illustrates the molecular and micro-environmental diversity of ductal carcinoma in situ. NPJ Breast Cancer. 2022;8(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pareja F, Brown DN, Lee JY, Da Cruz Paula A, Selenica P, Bi R, et al. Whole-Exome Sequencing Analysis of the Progression from Non-Low-Grade Ductal Carcinoma In Situ to Invasive Ductal Carcinoma. Clin Cancer Res. 2020;26(14):3682–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brady SW, McQuerry JA, Qiao Y, Piccolo SR, Shrestha G, Jenkins DF, et al. Combating subclonal evolution of resistant cancer phenotypes. Nat Commun. 2017;8(1):1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pang JB, Savas P, Fellowes AP, Mir Arnau G, Kader T, Vedururu R, et al. Breast ductal carcinoma in situ carry mutational driver events representative of invasive breast cancer. Mod Pathol. 2017;30(7):952–63. [DOI] [PubMed] [Google Scholar]
- 83.Bos MK, Smid M, Sleijfer S, Martens JWM. Apolipoprotein B mRNA-Editing Catalytic Polypeptide-Like-Induced Protein Changes in Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer Throughout Disease Progression. JCO Precis Oncol. 2022;6:e2100190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell. 2017;32(2):169–84 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barroso-Sousa R, Jain E, Cohen O, Kim D, Buendia-Buendia J, Winer E, et al. Prevalence and mutational determinants of high tumor mutation burden in breast cancer. Ann Oncol. 2020;31(3):387–94. [DOI] [PubMed] [Google Scholar]
- 86.Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol. 2011;29(33):4452–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mosele F, Stefanovska B, Lusque A, Tran Dien A, Garberis I, Droin N, et al. Outcome and molecular landscape of patients with PIK3CA-mutated metastatic breast cancer. Ann Oncol. 2020;31(3):377–86. [DOI] [PubMed] [Google Scholar]
- 88.Vasan N, Razavi P, Johnson JL, Shao H, Shah H, Antoine A, et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kalpha inhibitors. Science. 2019;366(6466):714–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gala K, Li Q, Sinha A, Razavi P, Dorso M, Sanchez-Vega F, et al. KMT2C mediates the estrogen dependence of breast cancer through regulation of ERalpha enhancer function. Oncogene. 2018;37(34):4692–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hanker AB, Sudhan DR, Arteaga CL. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell. 2020;37(4):496–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu G, Chhangawala S, Cocco E, Razavi P, Cai Y, Otto JE, et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat Genet. 2020;52(2):198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sokol ES, Feng YX, Jin DX, Basudan A, Lee AV, Atkinson JM, et al. Loss of function of NF1 is a mechanism of acquired resistance to endocrine therapy in lobular breast cancer. Ann Oncol. 2019;30(1):115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pearson A, Proszek P, Pascual J, Fribbens C, Shamsher MK, Kingston B, et al. Inactivating NF1 Mutations Are Enriched in Advanced Breast Cancer and Contribute to Endocrine Therapy Resistance. Clin Cancer Res. 2020;26(3):608–22. [DOI] [PubMed] [Google Scholar]
- 94.Bruner HC, Derksen PWB. Loss of E-Cadherin-Dependent Cell-Cell Adhesion and the Development and Progression of Cancer. Cold Spring Harb Perspect Biol. 2018;10(3):a029330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Savas P, Teo ZL, Lefevre C, Flensburg C, Caramia F, Alsop K, et al. The Subclonal Architecture of Metastatic Breast Cancer: Results from a Prospective Community-Based Rapid Autopsy Program “CASCADE”. PLoS Med. 2016;13(12):e1002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Legare S, Cavallone L, Mamo A, Chabot C, Sirois I, Magliocco A, et al. The Estrogen Receptor Cofactor SPEN Functions as a Tumor Suppressor and Candidate Biomarker of Drug Responsiveness in Hormone-Dependent Breast Cancers. Cancer Res. 2015;75(20):4351–63. [DOI] [PubMed] [Google Scholar]
- 97.Zundelevich A, Dadiani M, Kahana-Edwin S, Itay A, Sella T, Gadot M, et al. ESR1 mutations are frequent in newly diagnosed metastatic and loco-regional recurrence of endocrine-treated breast cancer and carry worse prognosis. Breast Cancer Res. 2020;22(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mullane SA, Werner L, Rosenberg J, Signoretti S, Callea M, Choueiri TK, et al. Correlation of Apobec Mrna Expression with overall Survival and pd-l1 Expression in Urothelial Carcinoma. Sci Rep. 2016;6:27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Denkert C, Untch M, Benz S, Schneeweiss A, Weber KE, Schmatloch S, et al. Reconstructing tumor history in breast cancer: signatures of mutational processes and response to neoadjuvant chemotherapy(small star, filled). Ann Oncol. 2021;32(4):500–11. [DOI] [PubMed] [Google Scholar]
- 100.Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. 2021;18(4):215–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dougan M, Pietropaolo M. Time to dissect the autoimmune etiology of cancer antibody immunotherapy. J Clin Invest. 2020;130(1):51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Blank CU, Enk A. Therapeutic use of anti-CTLA-4 antibodies. Int Immunol. 2015;27(1):3–10. [DOI] [PubMed] [Google Scholar]
- 103.Philips GK, Atkins M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int Immunol. 2015;27(1):39–46. [DOI] [PubMed] [Google Scholar]
- 104.DiMarco AV, Qin X, McKinney BJ, Garcia NMG, Van Alsten SC, Mendes EA, et al. APOBEC Mutagenesis Inhibits Breast Cancer Growth through Induction of T cell-Mediated Antitumor Immune Responses. Cancer Immunol Res. 2022;10(1):70–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cescon DW, Haibe-Kains B, Mak TW. APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc Natl Acad Sci U S A. 2015;112(9):2841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen Z, Wen W, Bao J, Kuhs KL, Cai Q, Long J, et al. Integrative genomic analyses of APOBEC-mutational signature, expression and germline deletion of APOBEC3 genes, and immunogenicity in multiple cancer types. BMC Med Genomics. 2019;12(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Smid M, Rodriguez-Gonzalez FG, Sieuwerts AM, Salgado R, Prager-Van der Smissen WJ, Vlugt-Daane MV, et al. Breast cancer genome and transcriptome integration implicates specific mutational signatures with immune cell infiltration. Nat Commun. 2016;7:12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Boichard A, Pham TV, Yeerna H, Goodman A, Tamayo P, Lippman S, et al. APOBEC-related mutagenesis and neo-peptide hydrophobicity: implications for response to immunotherapy. Oncoimmunology. 2019;8(3):1550341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell. 2021;184(3):596–614 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Guo H, Zhu L, Huang L, Sun Z, Zhang H, Nong B, et al. APOBEC Alteration Contributes to Tumor Growth and Immune Escape in Pan-Cancer. Cancers (Basel). 2022;14(12):2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Buisson R, Lawrence MS, Benes CH, Zou L. APOBEC3A and APOBEC3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res. 2017;77(17):4567–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Green AM, Landry S, Budagyan K, Avgousti DC, Shalhout S, Bhagwat AS, et al. APOBEC3A damages the cellular genome during DNA replication. Cell Cycle. 2016;15(7):998–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Green AM, Budagyan K, Hayer KE, Reed MA, Savani MR, Wertheim GB, et al. Cytosine Deaminase APOBEC3A Sensitizes Leukemia Cells to Inhibition of the DNA Replication Checkpoint. Cancer Res. 2017;77(17):4579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Barnieh FM, Loadman PM, Falconer RA. Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr Res Pharmacol Drug Discov. 2021;2:100017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Biayna J, Garcia-Cao I, Alvarez MM, Salvadores M, Espinosa-Carrasco J, McCullough M, et al. Loss of the abasic site sensor HMCES is synthetic lethal with the activity of the APOBEC3A cytosine deaminase in cancer cells. PLoS Biol. 2021;19(3):e3001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Serebrenik AA, Starrett GJ, Leenen S, Jarvis MC, Shaban NM, Salamango DJ, et al. The deaminase APOBEC3B triggers the death of cells lacking uracil DNA glycosylase. Proc Natl Acad Sci U S A. 2019;116(44):22158–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mehta KPM, Lovejoy CA, Zhao R, Heintzman DR, Cortez D. HMCES Maintains Replication Fork Progression and Prevents Double-Strand Breaks in Response to APOBEC Deamination and Abasic Site Formation. Cell Rep. 2020;31(9):107705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Barzak FM, Harjes S, Kvach MV, Kurup HM, Jameson GB, Filichev VV, et al. Selective inhibition of APOBEC3 enzymes by single-stranded DNAs containing 2’-deoxyzebularine. Org Biomol Chem. 2019;17(43):9435–41. [DOI] [PubMed] [Google Scholar]
- 119.Kvach MV, Barzak FM, Harjes S, Schares HAM, Jameson GB, Ayoub AM, et al. Inhibiting APOBEC3 Activity with Single-Stranded DNA Containing 2’-Deoxyzebularine Analogues. Biochemistry. 2019;58(5):391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kvach MV, Barzak FM, Harjes S, Schares HAM, Kurup HM, Jones KF, et al. Differential Inhibition of APOBEC3 DNA-Mutator Isozymes by Fluoro- and Non-Fluoro-Substituted 2’-Deoxyzebularine Embedded in Single-Stranded DNA. Chembiochem. 2020;21(7):1028–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Barzak FM, Ryan TM, Kvach MV, Kurup HM, Aihara H, Harris RS, et al. Small-Angle X-ray Scattering Models of APOBEC3B Catalytic Domain in a Complex with a Single-Stranded DNA Inhibitor. Viruses. 2021;13(2):290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.King JJ, Borzooee F, Im J, Asgharpour M, Ghorbani A, Diamond CP, et al. Structure-Based Design of First-Generation Small Molecule Inhibitors Targeting the Catalytic Pockets of AID, APOBEC3A, and APOBEC3B. ACS Pharmacol Transl Sci. 2021;4(4):1390–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.De Mattos-Arruda L. PIK3CA mutation inhibition in hormone receptor-positive breast cancer: time has come. ESMO Open. 2020;5(4):e000890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Su J, Ruan S, Dai S, Mi J, Chen W, Jiang S. NF1 regulates apoptosis in ovarian cancer cells by targeting MCL1 via miR-142–5p. Pharmacogenomics. 2019;20(3):155–65. [DOI] [PubMed] [Google Scholar]
- 125.Rogers MF, Shihab HA, Mort M, Cooper DN, Gaunt TR, Campbell C. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics. 2018;34(3):511–3. [DOI] [PMC free article] [PubMed] [Google Scholar]