

Summary
VLCFA (Very Long-Chain Fatty Acids) are the most abundant fatty acids in myelin. Hence, during demyelination or aging, glia are exposed to higher levels of VLCFA than normal. We report that glia convert these VLCFA into Sphingosine 1-Phosphate (S1P) via a glial-specific S1P pathway. Excess S1P causes neuroinflammation, NF-κB activation, and macrophage infiltration into the CNS. Suppressing the function of S1P in fly glia or neurons, or administration of Fingolimod, an S1P receptor antagonist, strongly attenuates the phenotypes caused by excess VLCFA. In contrast, elevating the VLCFA levels in glia and immune cells exacerbates these phenotypes. Elevated VLCFA and S1P are also toxic in vertebrates based on a mouse model of Multiple Sclerosis (MS), Experimental Autoimmune Encephalomyelitis (EAE). Indeed, reducing VLCFA with Bezafibrate ameliorates the phenotypes. Moreover, simultaneous use of Bezafibrate and Fingolimod synergizes to improve EAE, suggesting that lowering VLCFA and S1P is a treatment avenue for MS.
Keywords: Ceramide, Lipid metabolism, Myelin lipid, NF-κB activation, VLCFA β-oxidation, Fingolimod, Bezafibrate, Drosophila, Mice, Multiple Sclerosis
Graphical Abstract
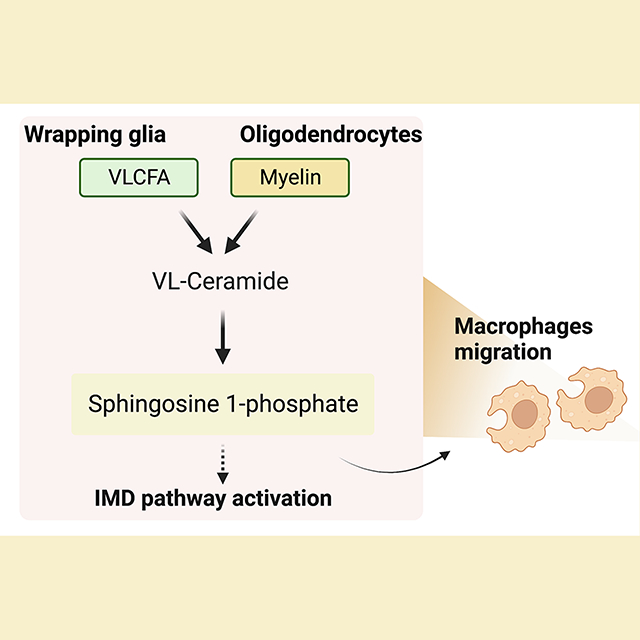
During demyelination and aging, glia are exposed to myelin components that contain high levels of VLCFAs. Here, Chung and Ye et al. document that VLCFA accumulation increases sphingosine 1-phosphate (S1P) levels in glia. Glial S1P leads to neuroinflammation by activating the immune deficiency pathway and promoting peripheral macrophage infiltration into the CNS. Therapeutically, bezafibrate and fingolimod, to reduce VLCFAs and S1P function, have additive beneficial effects in experimental autoimmune encephalomyelitis, a model of MS in mice.
Introduction
White matter accounts for approximately 50% of the total volume of the human brain and consists mostly of myelin that protects neurons and axons, improves nerve conduction, and serves as an energy source for axons1,2. Myelin is part of multilayered membranes found in oligodendrocytes and Schwann cells and consists of approximately 80% lipids and 20% protein. This is a high lipid concentration, given that the membranes of most cells contain 50% lipids and 50% protein3. Myelin is composed of 40% sphingolipids, 25% phospholipids, and 35% cholesterol3–5. Gal-Ceramide and Sphingomyelin are the major sphingolipids of myelin, and they are known to play a critical role in the regulation of membrane stiffness as well as in the interaction of myelin membranes with axonal membranes and other myelin membranes6–9. Interestingly, it has been reported that 50% of sphingolipids in myelin contain VLCFAs (C≥22)10–12. This is remarkable considering that VLCFAs are rare fatty acids that make up only 5% of the total fatty acids in the body. An increase in VLCFAs is known to increase membrane stiffness and decrease fluidity, and VLCFAs are thought to be critical for proper myelin function10,11,13,14.
In glia, Gal-Ceramide and Sphingomyelin are synthesized via a series of enzymatic reactions that include Ceramide Synthases, Ceramide galactosyltransferase, and Sphingomyelin synthase upon which they are incorporated into myelin. However, they are also synthesized and used as membrane components in other cells that are not part of the nervous system. Hence, the enzymes that synthesize these sphingolipids are typically found to be ubiquitously expressed15–18. Even though flies lack myelin, the enzymes that synthesize sphingolipids are highly conserved, and these lipids are present in wrapping glial membranes and genes encoding these enzymes are ubiquitously expressed in the fly CNS19. In contrast, the enzymes that degrade Gal-Ceramide or Sphingomyelin that contain VLCFA are not well characterized but the role of sphingolipids and their synthesis, transport and function have been studied in flies20–24.
Humans experience a slow and progressive degeneration of myelin as they age. This demise of myelin is associated with reduced nerve conduction velocity and gradually disrupts the function of neural circuits25. Moreover, MS, Huntington’s Disease, and Alzheimer’s Disease are also associated with progressive demyelination26,27. Hence, oligodendrocytes and Schwann cells get exposed to myelin components that contain high levels of VLCFAs but how these VLCFAs are metabolized in glia remains to be established.
We previously described a novel phenotype in patients harboring a novel variant in the peroxisomal enzyme ACOX128. Loss of ACOX1 leads to an elevation of VLCFA as it is the rate-limiting step in β-oxidation of the VLCFA29. We found that ACOX1 is expressed and required in glia, but not in neurons28. Here, we find that glial loss of ACOX1 leads to an elevation of VLCFA, which results in a concomitant increase in S1P. S1P is secreted by glial cells and taken up by neurons where the elevated S1P level is toxic and causes neuroinflammation as well as the invasion of macrophages into the CNS. This toxicity is very significantly suppressed by a drug that lowers VLCFA synthesis and one that interferes with S1P signaling. Importantly, lowering VLCFA levels ameliorates the clinical severity of an experimental autoimmune encephalomyelitis (EAE) mouse model of MS and additionally interfering with S1P signaling significantly improves the clinical phenotypes in the EAE model.
Results
Elevated levels of VLCFAs in glia cause lethality and neurodegeneration
We have previously shown that the proper activity of peroxisomal β-oxidation is essential for glial survival and that both loss and gain of ACOX1 severely affect glial function in flies and humans, albeit via different pathways. VLCFA levels are significantly increased when dACOX1 is lost. We showed that the loss of dACOX1 causes lethality and only a few percent of the flies eclose. These flies exhibit climbing defects and a decrease in life-span. Both defects are suppressed by reducing the level of VLCFA by inhibiting their synthesis. Indeed, knockdown of dELOVL an enzyme that produces VLCFAs, suppress the defects associated with dACOX1 loss. Hence, ACOX1 and ELOVL1 function antagonistically28.
To dissect the function of VLCFAs, specifically in glia, we either reduced the levels of dACOX1 using RNAi or elevated the levels of ELOVL128. The dACOX1 RNAi reduced the dACOX1 transcript levels by less than 50% (Figure S1A) and failed to cause any noticeable phenotype when ubiquitously expressed (Figure S1B). We therefore examined the lethality and behavioral phenotypes by increasing the production of VLCFA by expressing human ELOVL1 in glia (Repo>UAS-ELOVL1). Approximately 50% of the animals died as pupae (Figure 1A). The loss of viability associated with human ELOVL1 expression was significantly suppressed by RNAi-mediated reduction of fly dELOVL (CG31522) indicating that the induced phenotypes are sensitive to protein levels. Interestingly, Bezafibrate, a drug that inhibits ELOVL1 function30,31, significantly suppressed the decreased eclosion rate, again indicating that the induced phenotypes are highly sensitive to ELOVL1 function. The flies that eclose as adults (Repo>ELOVL1) exhibit climbing defects after 10 days but no climbing defects are observed in three-day-old flies (Figure 1B). These flies also have a short lifespan and all flies die before Day 25 (Figure 1C). Hence, elevating the levels of VLCFA in glia induces a shorter life span and decreases climbing performance.
Figure 1. Elevated levels of VLCFAs induce S1P production and neuronal dysfunction.
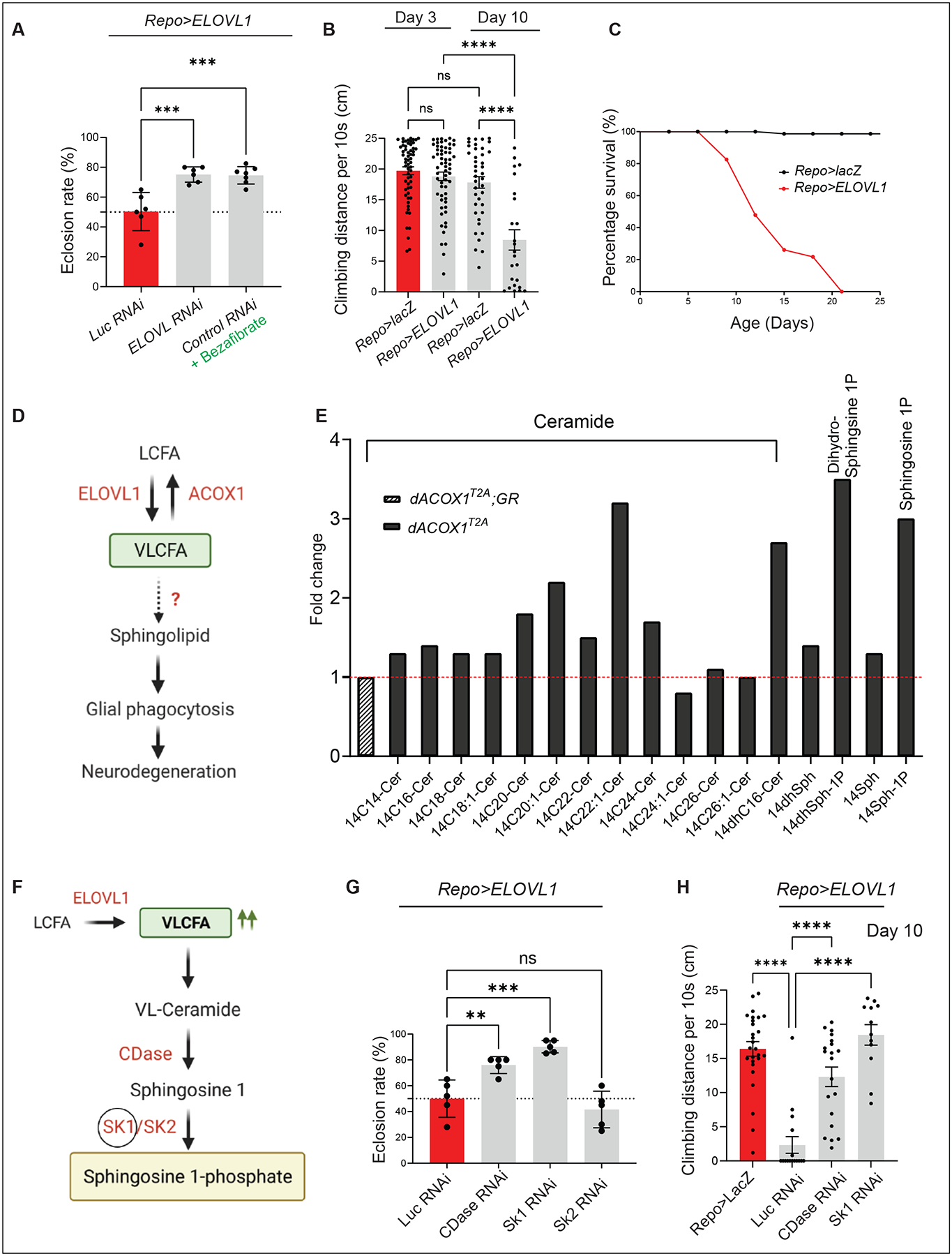
(A) Expressing ELOVL RNAi to downregulate the fly homologue of ELOVL1 or Bezafibrate supplementation suppresses the low eclosion rate observed in Repo>ELOVL1 flies. Quantification of the percentage of expected animals per cross (n>6). (B) Glial ELOVL1 expression causes progressive climbing defects (n>24) and (C) significantly decreases lifespan (n=100 for Repo>lacZ and Repo>ELOVL1). (D) Model of the mechanisms of VLCFA leading to neurodegeneration. (E) Sphingolipid profiling in heads of dACOX1T2A mutants (n=500 per each genotype). Cer: Ceramide, Sph: Sphingsoine, dhSph: dihydro Sphingosine, Sph-1P: Sphingosine 1-phosphate, dhSph-1P: dehydro sphingosine 1-phosphate. GR: Genomic Rescue construct (F) Pathway to convert VLCFAs into S1P. (G) A decrease in the levels of CDase or SK1 but not SK2 significantly suppresses the lethality observed in Repo>ELOVL1 flies. Quantification of the percentage of expected animals per cross (n=5 per each genotype) (H) A decrease in the levels of CDase or SK1 significantly suppresses the progressive climbing defects observed in Repo>ELOVL1 flies (n>11). Statistical analyses are one-way ANOVA followed by a Tukey post hoc test. Results are mean ± s.e.m. (****p < 0.0001, ***p < 0.001, **p < 0.01; n.s., not significant).
To examine if the lifespan and climbing defects are due to developmental issues or defects induced after eclosion, we assessed the phenotypes associated with ELOVL1 expression in adults only. We expressed Tub-GAL80ts in Repo-GAL4>ELOVL1 and raised the flies at 18C (no expression of ELOVL1), and shifted the animals to 29°C 1–3 days after eclosion (high expression of ELOVL1). As shown in Figure S1C, expression of ELOVL1 post-eclosion causes climbing defects at day 15, indicating that ELOVL1 expression in adult glia is sufficient to induce behavioral phenotypes.
Glial VLCFA are converted to VL-Ceramide and Sphingosine 1-Phosphate
To assess how VLCFA elevation in glia may affect lipid metabolism, we performed mass spectrometry analysis of 26 ceramides and other sphingolipid intermediates (including dihydroceramide, dihydrosphingosine, and sphingosine) in adult fly heads that lack dACOX1 (Figure 1E). These analyses revealed that the levels of VL-Ceramides C22 and C24 were increased by 2–3 fold (Figure 1E), whereas the levels of Ceramides with short- and medium-chain fatty acids remained unchanged with the exception of C20 when compared to controls. Note that VLCFAs can be promptly converted to VL-Ceramides via CerS232.
Additionally, dihydro-sphingosine 1-phosphate (Dihydro S1P) and sphingosine 1-phosphate (S1P) levels were significantly increased in dACOX1 mutant flies (Figure 1E). Given that dACOX1 is almost exclusively expressed in glia in the brain28 and given that glia correspond to ~10% of the cells in the brain33, the amount of lipids accumulated in glia should be much greater than the levels quantified in whole heads. Furthermore, the data show that the levels of VL-Ceramides, Sphingosine (Sph), and Sphingosine 1-phosphate (S1P) in the adult heads of Repo>ELOVL1 flies are also significantly increased, similar to those of dACOX1T2A flies (Figure S1D).
There are at least three pathways to produce ceramides: the de novo synthesis pathway and two salvage pathways mediated by the neutral and acidic sphingomyelinases in cytoplasm and lysosomes, respectively. Hence, it is not obvious what the source is of the ceramides that produce VL-ceramides. To assess if the de novo synthesis pathway is affected, we measured the transcript level of CG4162 (Lace) & CG4016 (Spt1) in heads of Repo>ELOVL1 flies. We found that the transcript levels of these genes are reduced, indicating that the increase of VL-Ceramide level may transcriptionally inhibit the synthesis of SPT (Serine palmitoyltransferase) controlled by Lace and Spt1 (Figure S1E). It is likely that the salvage pathways compensate for this decrease.
We further examined if a neuronal or glial expression of these enzymes (Lace or Spt1) causes defects. However, either glial or neuronal overexpression of Lace & Spt1 does not cause obvious defects, as shown in Figures S1F–G. However, it is well established that ORMDL functions as a potent negative regulator of SPT activity in the ER34. In the presence of ORMDL and elevated levels of ceramides, SPT activity is dramatically reduced35. However, overexpression of Spt1 significantly enhances lethality induced by the expression of ELOVL1 in glia (Figure S1F), suggesting that ORMDL is not inhibited by VL-Ceramides, and hence the production of Ceramides derived from the salvage pathways is not inhibited by ORMDL35. This may lead to more substrate for the production of VL-Ceramides and increased S1P production and toxicity. In summary, these data suggest that the elevated levels of VLCFA in glia lead to an increase in VL-Ceramides and S1P.
The Glial S1P exit pathway converts VL-Ceramide to S1P
The production of S1P depends on a series of enzymes that are shown in Figure S2A. In mammals, ceramides are synthesized by a family of six ceramide synthases (CerS1–6) (Figure S2A), each of which synthesizes ceramides with distinct acyl chain lengths. However, there is only a single ceramide synthase in flies, Schlank36,37. Ceramides are hydrolyzed into Sphingosine 1 by three different types of ceramidases in human. Ceramidases are encoded by five distinct genes in vertebrates which encode enzymes with diverse subcellular localizations (Figure S2A). In flies, there is only a single CDase gene (Figure 1F and Figure S2A). Hence, studying this pathway in mice is complex38. In contrast, in flies, the absence of ceramide synthase (Schlank) or ceramidase (CDase) results in obvious phenotypes, and severe loss of function mutations in these genes cause lethality22,36.
Sphingosine is used to synthesize S1P, which is mediated by SPHK1 and SPHK2 in humans, or SK1 and SK2 in flies (Figures 1F and S2A). S1P serves both a cell-autonomous and non-autonomous function39, and its levels are regulated by the balance of its synthesis and degradation by SGPL1, which is localized to the ER and is responsible for the irreversible breakdown of S1P40. Fruit flies have a single conserved homolog of mammalian, SGPL1, Sply (Figure S2A)41. We named the pathway that converts VL-Ceramide into S1P consisting of CDase, SK, the S1P exit pathway (Figures 1F and S2A).
Elevated levels of S1P are toxic
The elevated levels of the intermediate products leading to the production of S1P or S1P itself in glia may be toxic when the VLCFA are elevated, either when ELOVL1 is overexpressed or when dACOX1 is lost. We therefore reduced the levels of CDase, SK1, and SK242–44 in glia of Repo>ELOVL1 flies. Expression of RNAi for CDase or SK1 in glia significantly alleviates the decreased viability and climbing defects caused by Repo>ELOVL1 (Figures 1G–H). However, RNAi for SK2 did not alter the survival rate (Figure 1G). These findings indicate that accumulation of S1P, but not the VLCFA or intermediate products in glia, is detrimental, and that CDase and SK1 are indeed required in glia.
S1P is produced in glia but not in neurons
We next investigated which cells express CDase and SK1 in the fly CNS. We inserted the SA-T2A-GAL4-polyA artificial exon into a coding intron of CDase (Figures 2A and S2B)45. Given that SK1 lacks a suitable intron to insert T2A-GAL4, we replaced the entire ORF with a Kozak-GAL4 allele using CRISPR-Cas9-mediated homologous recombination (Figures 2A and S2C)46. To determine where these genes are expressed, we crossed the T2A-GAL4 CDase and Kozak-GAL4 SK1 flies to flies carrying the UAS- nuclear mCherry detector (UAS-nls-mCherry) (Figures 2B) and stained them with antibodies against the nuclear Repo. The expression of both SK1 and CDase is confined to glia, approximately 70% of the glia of the larval CNS and 90% of the glia of the adult CNS (Figures 2B). We did not observe expression of either gene in neurons. These data are consistent with single-cell and single-nucleus sequencing data of the adult Drosophila brain19,47. Similarly, vertebrate expression data reported in Brain-RNA Seq for CDase and SK1 homologues are highly enriched in oligodendrocytes and microglia17,18,48. Hence, the two key enzymes involved in S1P synthesis are highly enriched or selectively expressed in subpopulations of glia in flies, mice, and humans.
Figure 2. S1P is synthesized in glia and transported to neurons.
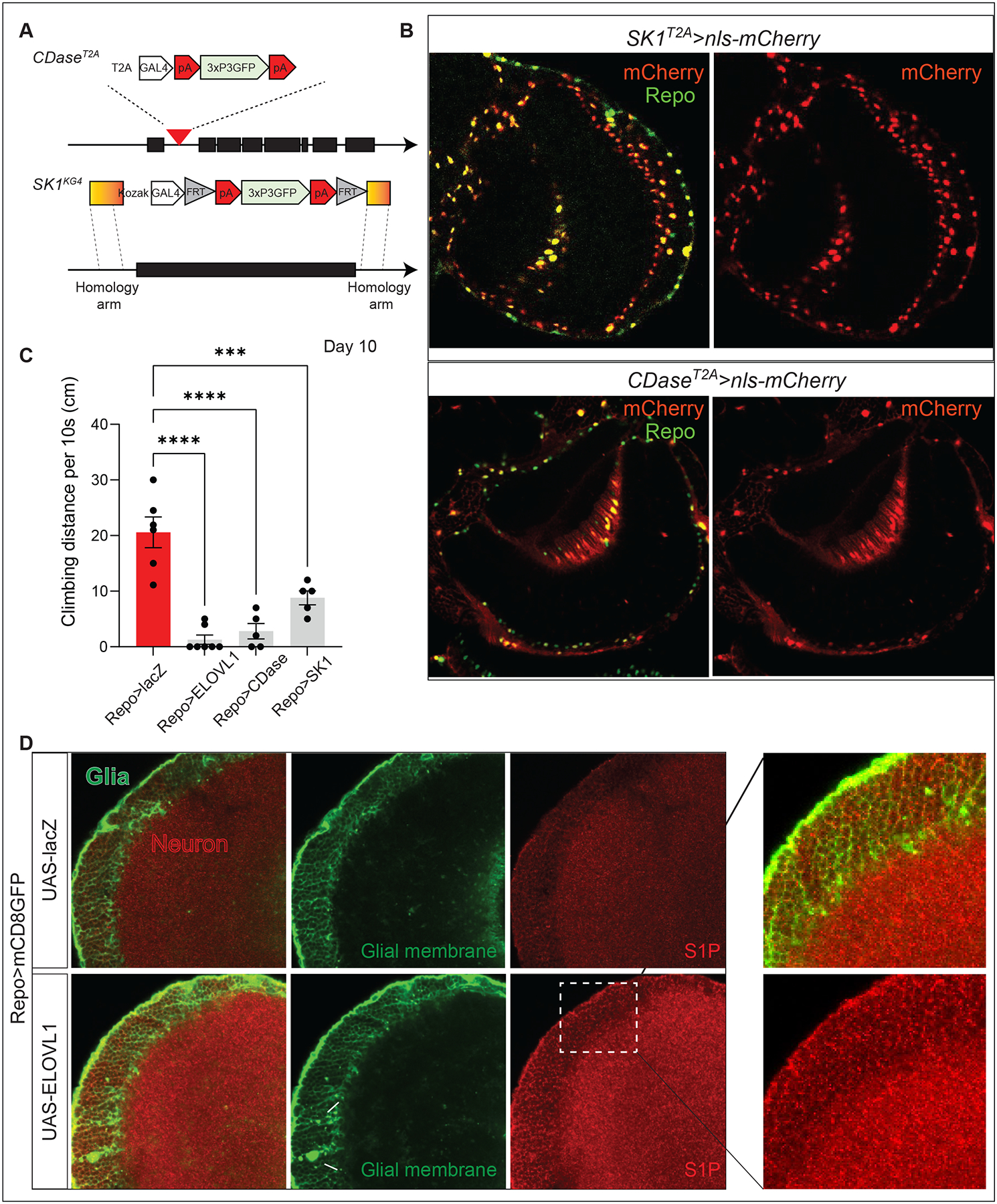
(A) Scheme of the structure of the CDaseT2A and SK1T2A alleles. (B) Both CDase and SK1 are expressed in glia of the adult CNS. Expression of CDaseT2A>nls-mCherry (red) colocalized with anti-Repo (green), marking the glia nuclei in adult CNS (Top). Expression of SK1T2A>nls-mCherry (red) colocalized with anti-Repo (green) (Bottom). Scale bar: 5 μm. (C) Increased synthesis of S1P results in severe climbing defects on Day 10 at 29°C degree (n>5). (D) Expression of ELOVL1 in glia increases the level of S1P in neurons. mCD8-GFP (green) labels the glial membrane, and anti-S1P (red) documents the expression pattern of S1P in the adult CNS. White arrows indicate abnormal glial membranes caused by glial ELOVL1 expression. The white dotted box indicates the region that is enlarged. Scale bar: 5 μm
Given that reducing the expression of these two enzymes strongly reduces the toxicity associated with elevated levels of VLCFA, we elevated glial expression of CDase or SK1 to determine if they may lead to the overproduction of S1P and cause toxicity. Indeed, overexpression of these enzymes is sufficient to cause climbing defects similar to the expression of ELOVL1 in glia (Figure 2C), again indicating that elevated production of S1P via CDase and SK1 in glia is sufficient to induce neurodegenerative phenotypes.
S1P is transported from glia to neuron
S1P is a bioactive lipid that has been implicated in synaptic transmission, differentiation of oligodendrocytes, and microglial proinflammatory responses39,49–51. It has been shown to be secreted and activates several signaling pathways through a G protein-coupled receptor signaling pathway49. However, S1P can also be taken up by recipient cells, independent of S1P receptors52.
Our data clearly indicate that glial cells produce excess S1P when VLCFA levels are elevated. This raises the following questions: Is the observed toxicity confined to glia (autocrine signaling), or do glial cells secrete S1P that is toxic to neurons and/or does it affect other cells (paracrine or endocrine signaling)? To determine where S1P is localized in wild-type adult fly brains, we immunostained brains with an anti-S1P antibody53,54 and co-expressed Repo>UAS-mCD8GFP to label the glial cell membranes (green, Figures 2D and S3A–C). S1P is not only enriched in the glial membrane but also highly enriched in the cytoplasm of neurons in a vesicular pattern (Red, Figures 2D, and S3A–C), as previously documented in vertebrates55–57.
To determine whether an elevation of VLCFA levels in glia results in an increase in S1P levels in neurons, we expressed ELOVL1 in glia and immunostained for S1P. We observed some aberrant membrane structures in glia (Figures 2D and S3A–C, white arrows). Importantly, there is an obvious increase in immunoreactivity when stained with S1P antibody compared to control animals (Repo>mCD8GFP), both in neurons and glia (Figures 2D and S3A–B). Hence, S1P produced in glia must be transported to neurons.
S1P induces the demise of neurons
S1P is irreversibly degraded by the human SGPL1 enzyme. This highly conserved ER enzyme catalyzes the irreversible degradation of endogenous and dietary S1P, the final step of sphingolipid catabolism58,59. Loss of SGPL1 (human homolog of sply) in human has been associated with a rare disease characterized by an elevation of S1P level and neurologic phenotypes60,61. Metabolic studies based on plasma/fibroblasts of these individuals have revealed elevated levels of S1P. The fly protein, Sply, is evolutionarily conserved with a DIOPT Score of 15/1562.
If the elevated levels of S1P produced in glial cells are taken up by neurons and are toxic in neurons, lowering the levels of S1P by elevating the expression of Sply in neurons may suppress the toxic effects (Figure 3A). We therefore overexpressed sply in neurons upon elevated glial VLCFA production. We used the lexA-lexop binary expression system to generate flies expressing lexop-ELOVL1 driven by Repo-lexA63. Repo>lexop-ELOVL1 flies exhibit climbing defects beginning at Day 20 (Figure 3B), two weeks later than the Repo>UAS-ELOVL1 flies, possibly because the Repo-lexA is not as potent as the Repo-Gal463. Next, we generated transgenic flies with UAS-sply or UAS-human SGPL1 to co-express these genes in neurons in the Repo>lexop-ELOVL1 background. Notably, neuronal expression of fly sply or human SGPL1 significantly improves the climbing defects caused by Repo>lexop-ELOVL1 flies (Figure 3B). However, neuronal knockdown of sply (elav>sply RNAi) in a wild-type background does not cause a decrease in lifespan or climbing defects at Day 15 (Figure S3C). These data indicate that glial transport of S1P to neurons is a major source of neuronal S1P increase and that the elevated levels of S1P cause toxicity in glia and neurons. Importantly, the observation that neuronal Sply expression can significantly suppress the toxic effects of overproduction of S1P in glia clearly shows that S1P has a paracrine function and that it can be taken up by neurons to cause neuronal dysfunction (Figure 3A).
Figure 3. Lowering S1P production or signaling improves neuronal function.
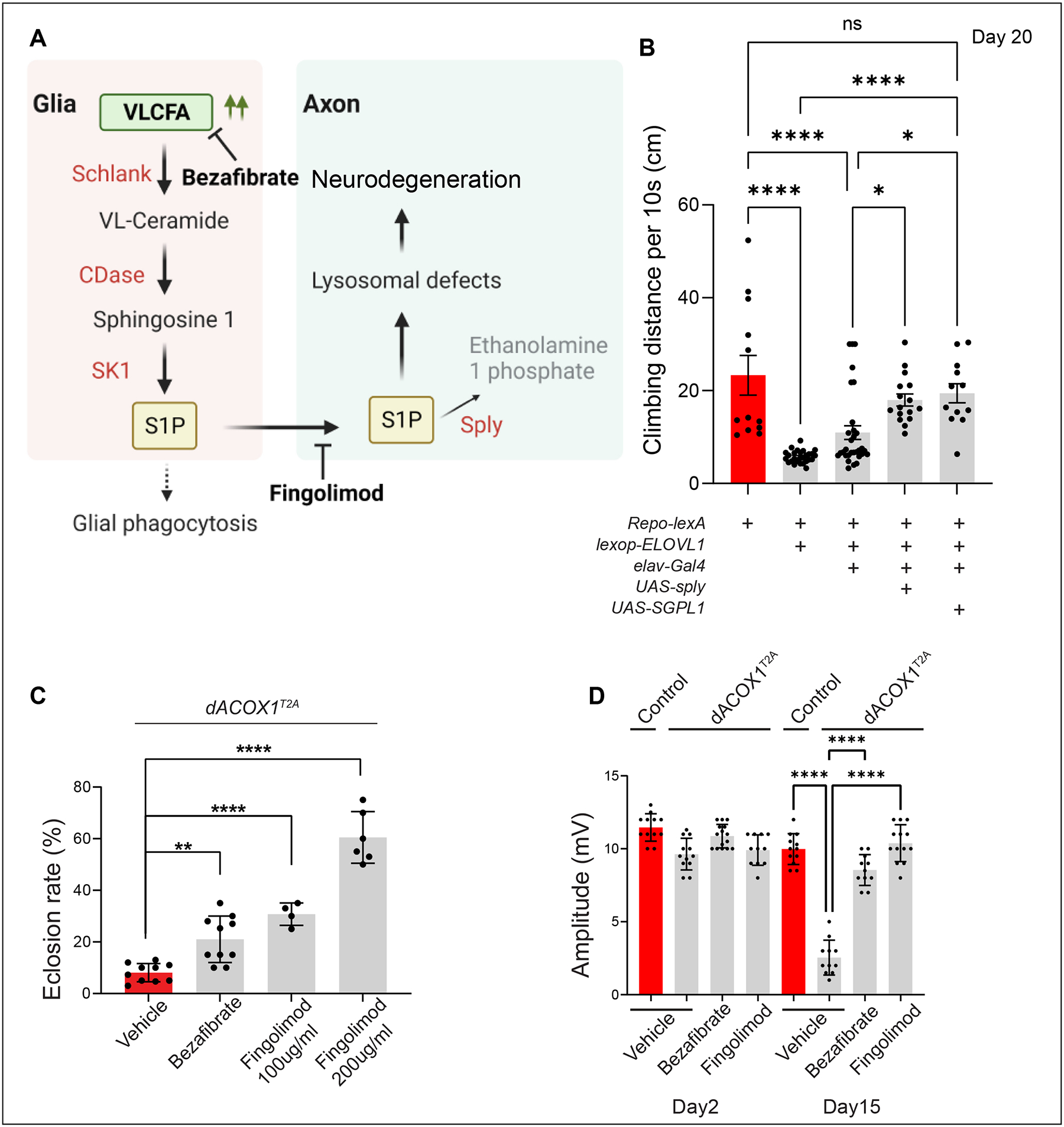
(A) Schematic illustrating how VLCFA in glia may lead to neurodegeneration. (B) Neuronal expression of either UAS-sply or UAS-SGPL1 suppresses the progressive climbing defects observed in flies (Repo-lexA/lexop-ELOVL1) at Day 20 (n>12). (C) Supplementation of either Bezafibrate or Fingolimod can significantly suppress the lethality observed in dACOX1T2A mutant flies. Quantification of the percentage of expected animals per cross (n>4). (D) Bezafibrate and Fingolimod rescued the ERG amplitude defects of homozygous dACOX1T2A flies at Day 15 (n>10 per genotype). Statistical analyses are one-way ANOVA followed by a Tukey post hoc test. Results are mean ± s.e.m. (****p < 0.0001, ***p < 0.001, **p < 0.01; n.s., not significant).
We previously showed that loss of dACOX1 leads to an increase in VLCFA levels and causes age-dependent Electroretinogram (ERG) defects at Day 1528. However, the ELOVL1 inhibitor, Bezafibrate, significantly improved viability and neuronal function. Another drug, Fingolimod, used to treat MS, is known to bind to the S1P receptors and to downregulate receptor levels64. However, no S1P receptor has been identified in flies, and the five known vertebrate S1PR have no homologs in flies41,62. Hence, it is not obvious if Fingolimod may suppress the action of elevated S1P in flies. We supplemented flies that lack dACOX1 (dACOX1T2A/dACOX1T2A) with Fingolimod and assessed their viability and neuronal function using ERGs in flies. To our surprise, Fingolimod supplementation caused a dramatic improvement in viability (Figure 3C) and strongly attenuated the progressive loss of ERG amplitude (Figure 3D). Note that ERGs have been used to study retinal disorders but that they are often relevant to our understanding of neurological diseases65–67. The ERG depolarization amplitude provides a read-out of the phototransduction process, whereas the on/off transient spikes at the onset and offset of light correspond to postsynaptic potential changes68. Considering the behavioral defects (climbing defects and life-span decrease) when VLCFA/S1P are elevated, the ERG data indicate that neuronal CNS functions are affected when the animals age, and an unidentified S1P receptor must be present in flies. Alternatively, Fingolimod can act through another mechanism to attenuate the toxicity associated with elevated levels of S1P. Most importantly, inhibiting S1P toxicity is critical to delaying the neurodegeneration caused by elevated VLCFA levels.
Elevated S1P induces phagocytosis through Draper
Given that elevated S1P levels in neurons cause neurodegeneration, we wondered if and how dying neurons are cleared. Wrapping glia in Drosophila ensheath peripheral axons in a manner similar to vertebrate oligodendrocytes or Schwann cells. Even though flies lack myelin, the enzymes that synthesize sphingolipids, the components of myelin, are highly conserved in flies and important components of the wrapping glial membranes which seem functionally equivalent to myelin containing membranes69 (Figure S2A). To determine whether wrapping glia is affected when S1P levels are elevated, we performed transmission electron microscopy (TEM) of peripheral wing nerves of 7-day-old Repo>ELOVL1 flies. We found that axons in these flies have a reduced diameter and exhibit abnormal morphology when compared to controls (Figures 4A–B, black arrows) and the number of axons is mildly but significantly reduced (Figure 4C). Additionally, we observe membrane expansions of the three glial cell types: wrapping glia, perineurial glia, and subperineurial glia (Figures 4A–B; marked by orange;70. However, the total number of glia remains unchanged (Figure 4D). Taken together, these findings suggest that glia expand their membrane when S1P is elevated in glia and neurons.
Figure 4. Elevated S1P induces phagocytosis through Draper.
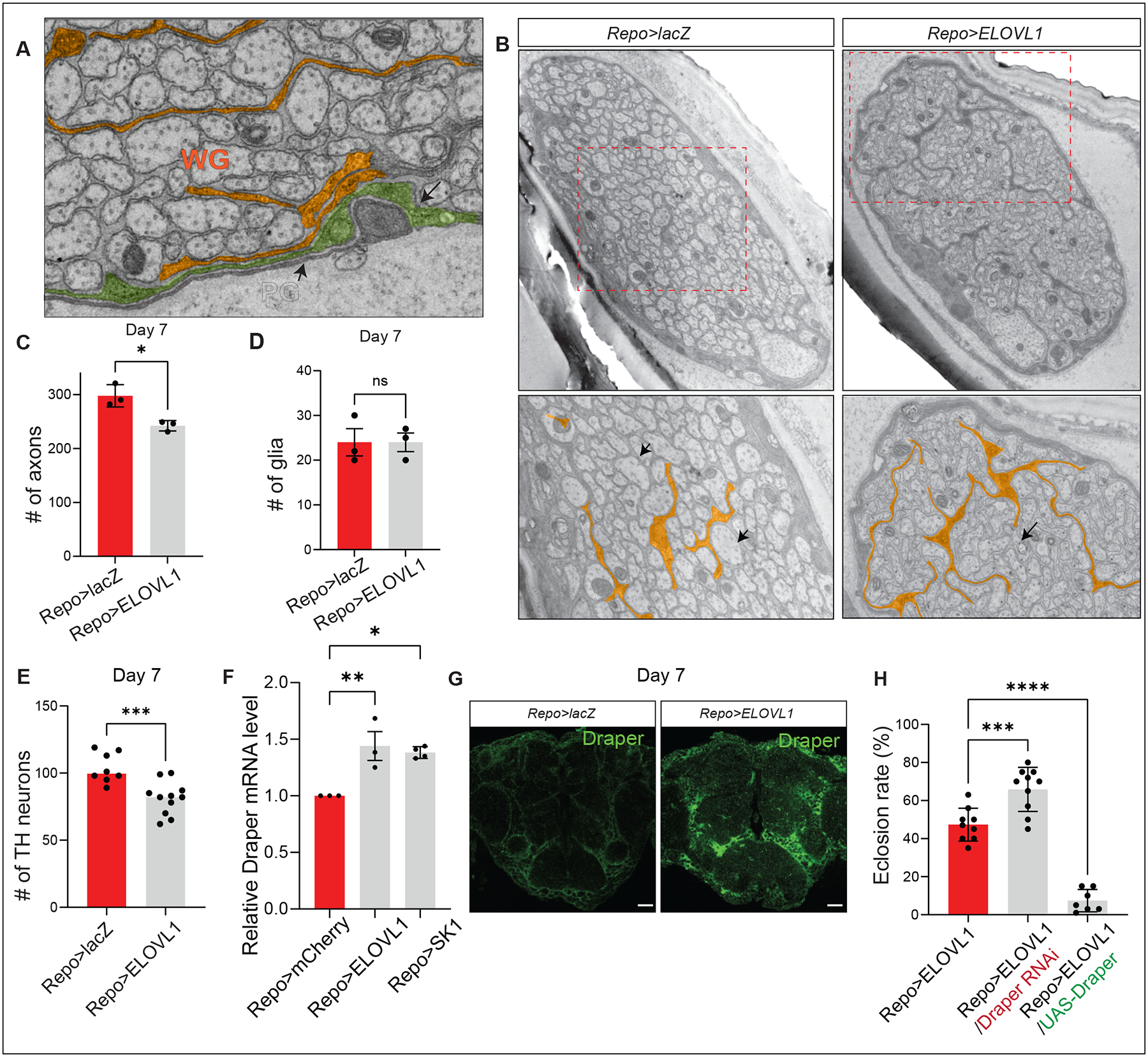
(A) Image of a section of the wing nerve showing the three types of glia: Perineurial (PG), Subperineurial (SPG), and wrapping glia (WG). (B) Glial ELOVL1 expression leads to extended and expanded glial membranes. (C) The number of axons is significantly reduced in the wing margin nerves of Repo>ELOVL1 flies at Day 7 (n=3 per each genotype), however the (D) number of glia is not altered (E) The number of TH neurons is significantly reduced in CNS of Repo>ELOVL1 flies (n=6 for Repo>lacZ, n=11 for Repo>ELOVL1). (F) The relative Draper mRNA levels are significantly increased in the CNS of Repo>ELOVL1 and Repo>SK1 flies (n=3 per each genotype). (G) Draper protein levels are increased in Repo>ELOVL1 fly heads. (H) Eclosion rates of Repo>ELOVL1 flies are modulated by Draper expression. Quantification of the percentage of expected animals per cross (n>7). Statistical analyses are one-way ANOVA followed by a Tukey post hoc test. Results are mean ± s.e.m. (****p < 0.0001, ***p < 0.001, **p < 0.01; n.s., not significant).
The observed glial membrane expansion indicates that the glia may be hyperactive when S1P is elevated71. It has been previously shown that hyperactive glial phagocytosis is sufficient to induce neuronal loss of TH (Tyrosine hydroxylase) expressing neurons and to reduce the life span of flies72. Since glial phagocytosis is mediated by Draper, a phagocytic receptor present on glia73,74, we assessed if the elevation of S1P in glia affects Draper expression in the adult CNS. We found that Draper mRNA expression, as well as protein levels, are significantly increased (Figures 4F–G), and the number of TH (Tyrosine hydroxylase) positive neurons is significantly reduced at Day 7 (Figure 4E). To assess if the Draper levels affect the phenotypes associated with the elevated levels of VLCFA/S1P, we reduced Draper levels with an established RNAi in a Repo>ELOVL1 background. This partially suppressed lethality. In contrast, overexpression of Draper and ELOVL1 severely reduced lifespan, and the flies that eclose were very short-lived (2 days) (Figure 4H). To examine if overexpression of Draper can induce similar glial phenotypes as Repo>ELOVL1, we further performed TEM of peripheral wing nerves of 3-day-old Repo>UAS-Draper flies. Interestingly, we find that overexpression of Draper enlarges the glial membrane and decreases the number of axons, similar to what we observe in Repo>ELOVL1 flies (Figures S4A–B). In contrast, decreasing the levels of Draper result in less enlarged glial membrane morphology than the control (Repo>mCherry), and the number of axons is not affected (Figures S4A–B). Since Draper transcription is increased in glia when axons are injured71, the data indicate that expression of ELOVL1 or SK1 in glia leads to an elevation of S1P and an increased level of Draper that promotes glial phagocytosis.
Loss of dACOX1 induces a strong cellular and humoral immune response
The significant suppression of neurodegenerative phenotypes observed in dACOX1T2A flies by Fingolimod, a key drug used to treat relapsing-remitting MS, an autoimmune disease64,75, prompted us to explore immune pathways in flies. We noted that escaper homozygous dACOX1T2A flies accumulate melanotic masses that are visible through the cuticle throughout their body, including eyes, heads, abdomens, and wing margins (Figures 5A and S4C). Black masses are typically associated with an immune response against foreign bodies mediated by hemocytes. Bacteria, parasitic wasp eggs, or other immunogens typically induce differentiation of plasmatocytes, the most common cells circulating in the hemolymph, into lamellocytes76. The latter cell type surrounds the target tissue and forms tight junctions to sequester the target. The lamellocytes accumulate black pigment produced by another type of immune cells, the crystal cells77–79. Given that wild type flies (dACOX1T2A/ dACOX1T2A; GR (genomic rescue construct)) do not exhibit melanotic masses, the occurrence of melanized tissues in adult flies in the absence of foreign objects or bacterial infections suggests that loss of dACOX1 induces an autoimmune response, i.e., the organism is attacking its own cells80. Furthermore, we found that the melanization phenotypes are progressive and ROS-independent, as treatment with the antioxidant NACA (AD4) does not alter the accumulation of melanotic masses (Figure S4C). This is in sharp contrast to the observed suppression of NACA (AD4) of the gain of function phenotype associated with the ACOX1 variant that causes Mitchell Syndrome28.
Figure 5. Glial VLCFA/S1P accumulation induces a robust immune response via the IMD pathway.
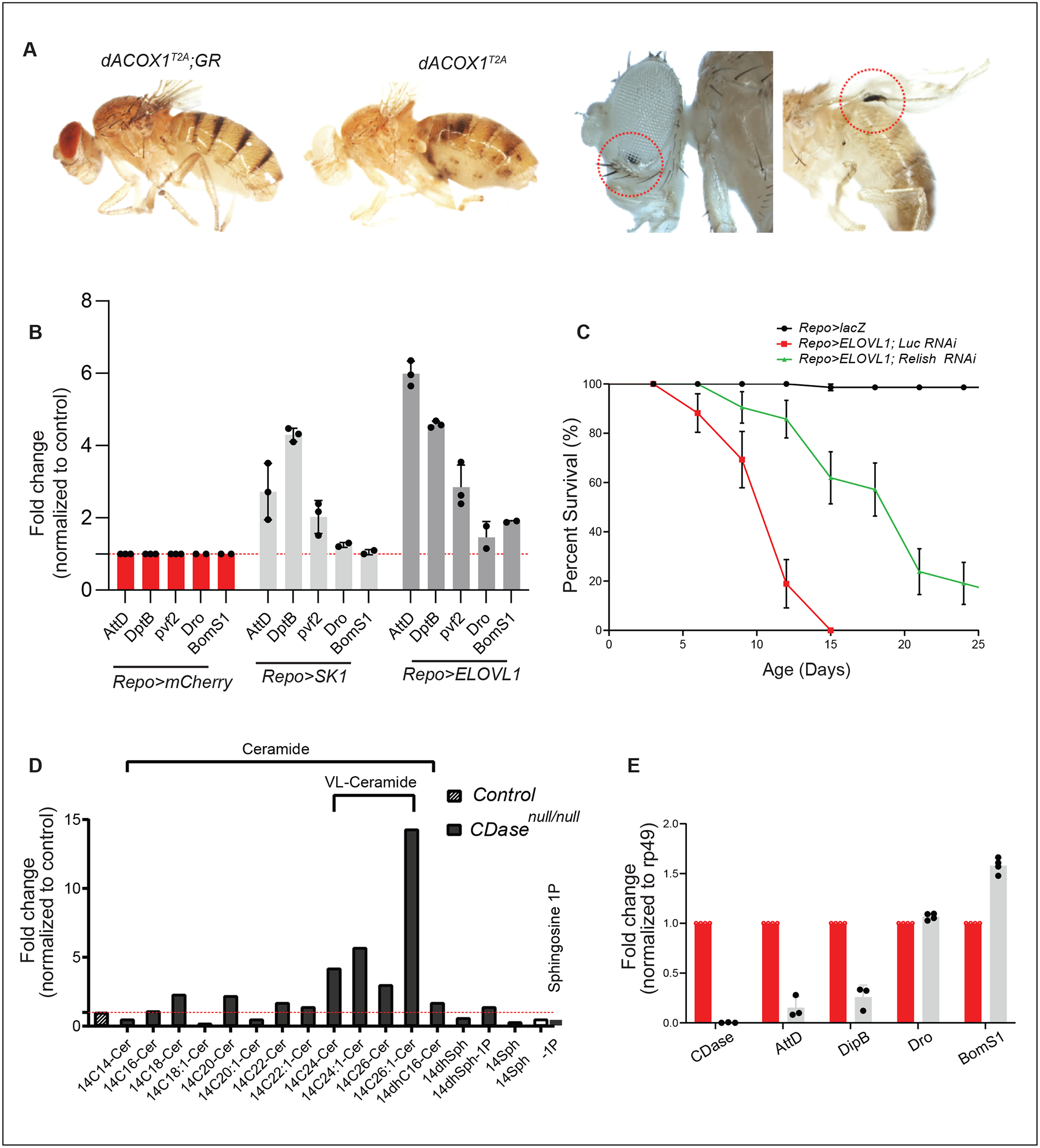
(A) Melanization is observed in numerous tissues of dACOX1 mutant flies (dACOX1T2A). These are not observed in control flies (dACOX1T2A;GR). (B) Repo>ELOVL1 and Repo>SK1 induce a robust elevation of the AntiMicrobacterial Peptides (AMPs) dependent on the IMD pathway but not the Toll pathway in adult heads (80> heads of flies were used per genotype. n=3). (C) Expression of Relish RNAi suppresses the short life span (n>50 per genotype) observed in Repo>ELOVL1 flies. (D) Sphingolipid profiling in heads of yw (control) and CDase mutants (yw;CDasenull) (n=500 for each genotype). (E) Fold changes of the levels of AMPs in control (yw), CDase null flies (yw;CDasenull) (n>30 heads of flies were used per genotype. n=3).
Given the above observations, we also explored if other aspects of the immune system are activated. Drosophila mounts a potent host defense when challenged by various microorganisms among which antimicrobial peptides are prominent. The proteins implicated in the induction of these peptides are very similar to those that play a critical role in the mammalian innate immune defenses81. Melanotic masses encapsulate tissues that are perceived as being abnormal or alternatively originate when the Toll/Cactus/dorsal axis is perturbed. Indeed, the constitutive activation of the Toll pathway during development as observed in Tl[10B] or cactus mutants82, leads to the formation of melanotic pseudo-tumors that in the most severe cases cause developmental lethality. However, the melanization phenotype in dACOX1T2A flies is not observed in larvae or pupae, nor are they observed in young flies (1–3 days old). They are first observed in 4-day-old flies and gradually increase in size and number in aging flies. (Figures 5A and S4C–D), indicating that melanization does not cause developmental lethality of dACOX1T2A animals.
To determine whether S1P is required for NF-κB activation, we generated a null mutant allele of CDase (CDasenull) (Figure S2D). The loss of CDase should lead to a block of S1P production in glia and promote an increase in VL-Ceramides. Indeed, CDase null mutant fly heads have a ~15-fold increase in VL-Ceramides, but they contain less than 40% of the S1P observed in controls (Figure 5D). Importantly, the IMD pathway activity, as assessed by AMP production (Attacin-D (AttD) and Diptericin-B (DptB)83,84), is very significantly decreased (by about 70–90%) when compared to control flies; however the level of the Toll-activated AMPs (Drosomycin (Drs) and BomS185) is not reduced in CDase mutant heads (Figure 5E), indicating that S1P produced in glia indeed induces NF-κB activation.
To test if the Toll pathway is activated upon VLCFA accumulation, we measured the level of mRNAs of the AMP genes, Drs and BomS, which are typically elevated by the Toll pathway. We also tested AttD and DptB which are dependent on the IMD pathway. Both were assayed in heads of Repo>ELOVL1 flies. We also assessed the levels of Pvf2, a PDGF- and VEGF-related factor 2, which encodes a ligand for the receptor tyrosine kinase, Pvr, required for macrophage invasion in fly brains86. As shown in Figure 5B, glial expression of ELOVL1 in adult brains results in a sixfold increase in the expression of IMD-specific AMPs and a threefold increase in the expression of Pvf2. Additionally, glial SK1 overexpression results in a fourfold increase in IMD-specific AMPs and a twofold increase in Pvf2 expression in adult brains (Figure 5B). However, transcription of the AMPs dependent on the Toll pathway (Dro and BomS1) were not significantly increased in heads of Repo>ELOVL1 (1.3–1.7 fold) Repo>SK1 (1.1–1.3 fold) (Figure 5B). To determine whether the IMD pathway activation is critical for neurodegenerative phenotypes, we co-expressed a Relish RNAi in Repo>ELOVL1 flies. This partially but significantly suppressed the life span reduction associated with Repo>ELOVL1 (Figure 5C). These data suggest that an increase in S1P is sufficient to activate the IMD pathway in the CNS.
Finally, to assess possible contributions of microorganisms in Repo>ELOVL1 flies, we raised Repo>ELOVL1 under axenic conditions87. We did not observe changes in lethality, indicating that a contribution of microorganisms to the phenotypes associated with Repo>ELOVL1 flies is unlikely (Figure S4E). In summary, elevation of VLCFA in glia induces at least two forms of autoimmunity: a cellular immune responses (melanization) and activation of the IMD pathway.
Concurrent expression of ELOVL1 in glia and immune cells induces neurodegeneration
Intriguingly, dACOX1, which encodes a peroxisomal enzyme, is not only expressed in glial cells but is also highly expressed in most immune cells, including PPO-expressing hemocytes (Figure 6A). To assess the contribution of immune cells versus glial cells in neurodegeneration, we expressed UAS-ELOVL1 in glial cells (Repo-GAL4), hemocytes (Hml-GAL4, expressed in 70–80% of hemocytes)88,89, as well as both tissues (Repo-GAL4 and Hml-GAL4). Combined expression of ELOVL1 in glia and hemocytes induces a climbing defect in 2-day-old flies. This defect is not observed in flies in which the gene is expressed in either cell type alone (Figure 6B). Moreover, the climbing defect is significantly worse at day 15, although expression of ELOVL1 in glia is already sufficient to induce a mild climbing defect at day 15 (Figures 6B–C). In summary, increasing the production of VLCFA in immune cells and glial cells exacerbates the demise of the nervous system.
Figure 6. An elevation of S1P in glia or immune cells promotes neuroinflammation.
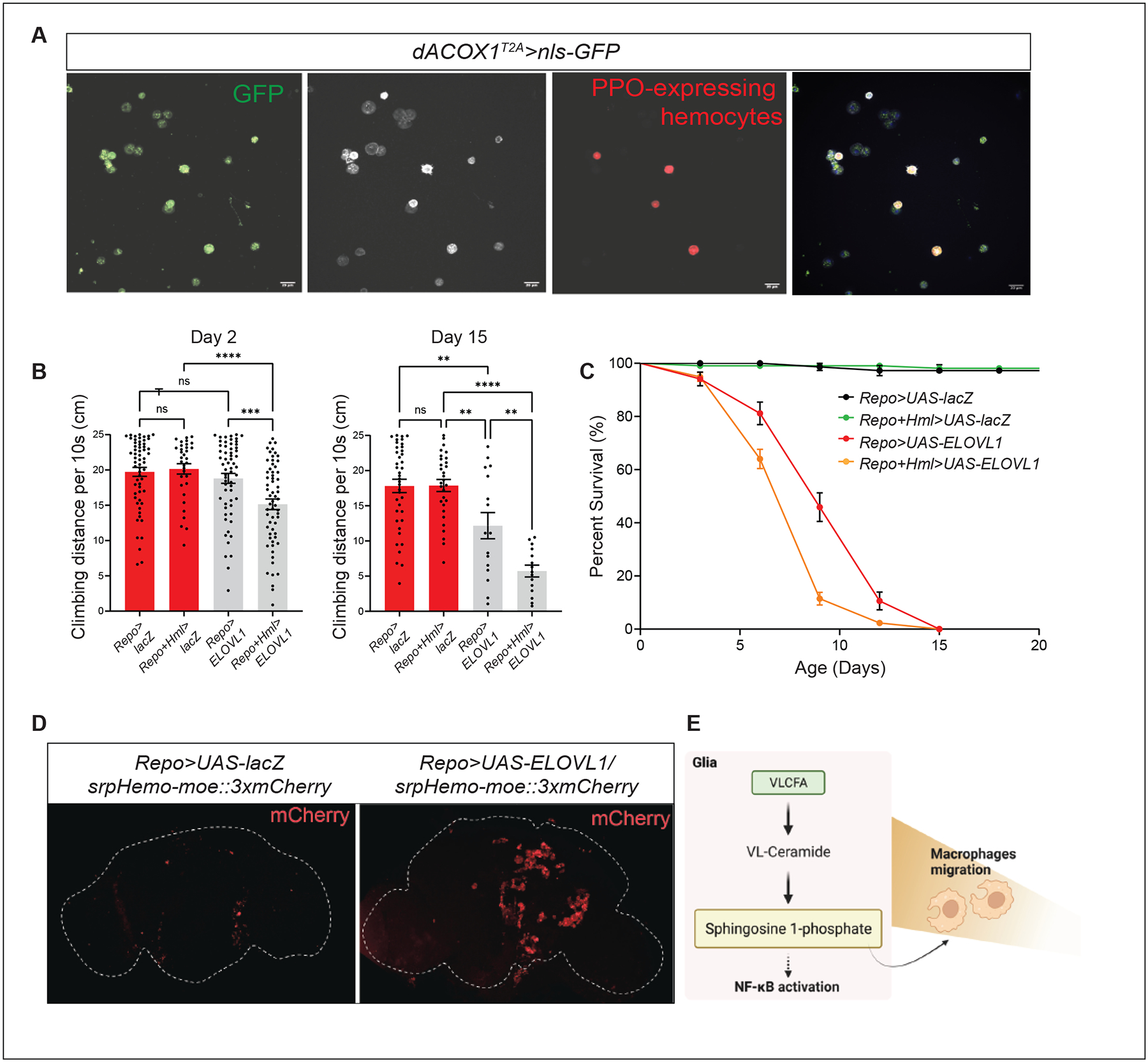
(A) dACOX1 is expressed in immune cells. Green (mCD8GFP) marks the dACOX1-expressing cells. White labels all immune cells (Hemese+). Red (BcF6-mCherry) indicates crystal cells (PPO1+). Scale bar: 20 μm. (B-C) Concurrent expression of ELOVL1 in both glia and immune cells (Repo+Hml>ELOVL1) leads to significantly enhanced progressive climbing defects on both Day 2 and 15 (n>35 per genotype) (B) and a life-span decrease (C) when compared to controls (n>50 per genotype). (D) Expression of ELOVL1 in glia (Repo>ELOVL1) induces hemocyte infiltration into the CNS when compared to control brains (Repo>UAS-LacZ). (E) Schematic showing that VLCFA accumulation leads to hemocytes recruitment into the CNS. Statistical analyses are one-way ANOVA followed by a Tukey post hoc test. Results are mean ± s.e.m. (****p < 0.0001, ***p < 0.001, **p < 0.01; n.s., not significant).
Glial S1P facilitates macrophage invasion via the NF-κB pathway
One of the primary features of neuroinflammation in MS is the peripheral immune cell recruitment to CNS from lymphoid organs64,90. We therefore, wondered if S1P secretion from glia promotes macrophage infiltration into the CNS. We labeled hemocytes in animals expressing ELOVL1 in glia (Repo>ELOVL1) and found a very significant accumulation of macrophages around as well as within the neuropil of the CNS (Figure 6D) and no accumulation was observed in control brains (Repo>lacZ) (Figure 6D). These data indicate that elevated levels of S1P induce neuroinflammation by recruiting peripheral immune cells (Figure 6E).
Finally, to assess if the perineural glia that form the Blood Brain Barrier (BBB) play a role in Repo>ELOVL1 flies, we expressed GAL80 in perineural glia in the presence of Repo>ELOVL1 to suppresses the expression of ELOVL1. It did not affect the climbing defects (Figure S4F), indicating that the behavioral defects observed in Repo>ELOVL1 flies are not likely due to a problem related to the BBB. This is consistent with a previous study that macrophage infiltration in the pupal stage is not associated with BBB integrity86. Taken together, our findings indicate that S1P can activate the IMD pathway in glia and recruit immune cells to the CNS.
The above data indicate that there are striking phenotypic parallels between loss of dACOX1 in flies, ACOX1 deficiency in human, and MS. Indeed, loss of ACOX1 in human causes a disease characterized by the accumulation of VLCFA91 (Figure S4G). The ACOX1 deficiency is often associated with seizures, failure to thrive, visual system failure, impaired hearing and vision, loss of motor achievements, demyelination and neuroinflammation.
Prophylactic bezafibrate treatment ameliorates EAE progression
Given that our data in flies indicate that an increase in VLCFA and S1P is at the root of many glia and immunological issues and given the phenotypic similarities with MS (Figure S4G), we explored the role of VLCFA and S1P in a mouse model for MS. The most commonly used animal model to study MS is the experimental autoimmune encephalomyelitis (EAE) mouse model92. In this model, mice are injected with the Myelin oligodendrocyte glycoprotein (MOG) peptide (35–55) (day 0), and their clinical phenotype is monitored over a 30-day period using a standard scoring system. Injection of myelin triggers an autoimmune reaction that targets the myelin sheath, resulting in several pathological features including neuroinflammation, demyelination, axonal loss, severe uncoordination, hind leg paralysis, and occasionally death.
Given the abundance of VLCFA in myelin and its central role in the pathogenesis of MS, we assessed if inhibition of VLCFA synthesis ameliorates the clinical progression in EAE mice. Starting from the day of immunization (day 0), we treated the mice daily with Bezafibrate, which inhibits VLCFA synthesis (Figures 7A and S5A). This prophylactic treatment of Bezafibrate (100mg/kg) significantly ameliorates the behavioral dysfunction of EAE, as assessed by the clinical score, at the peak and throughout the chronic phase of the disease (Figures 7A–B, Table 1). Demyelination and axon loss were evaluated by luxol fast blue (LFB) staining (Figures S5B–C) and immunostaining for neurofilament (NF, Figures S5D–E) in the thoracic spinal cords. Prophylactic treatment of Bezafibrate significantly reduced the demyelinating area (Figures S5B–C and I) and ameliorated axonal loss (Figures S5D–E and I) in the white matter tracts at the chronic stage of EAE. Since infiltration of immune cells and reactive gliosis play a critical role in MS93, we further examined the effect of the treatment on the infiltration of macrophages, activation of microglia, infiltration of cytotoxic T-cells, and reactive astrocytes in the demyelinated lesions at the late stage of EAE. We observed a significant inhibition of the number of macrophage and microglia (Iba1) in the spinal cord of Bezafibrate-treated EAE mice (Figures S5F–G and J), indicating a suppression of the immune response by Bezafibrate. Of note, we did not find altered responses of reactive astrocytes (GFAP) nor cytotoxic T cell infiltration (CD8) after Bezafibrate treatment (Figures S5L–P). Overall, these results suggest that prophylactic treatment of Bezafibrate alleviated demyelination, neuronal damage, and immune cell infiltration associated with EAE progression.
Figure 7. Co-treatment of EAE mice with Bezafibrate and Fingolimod synergistically improves behavior and cellular pathology.
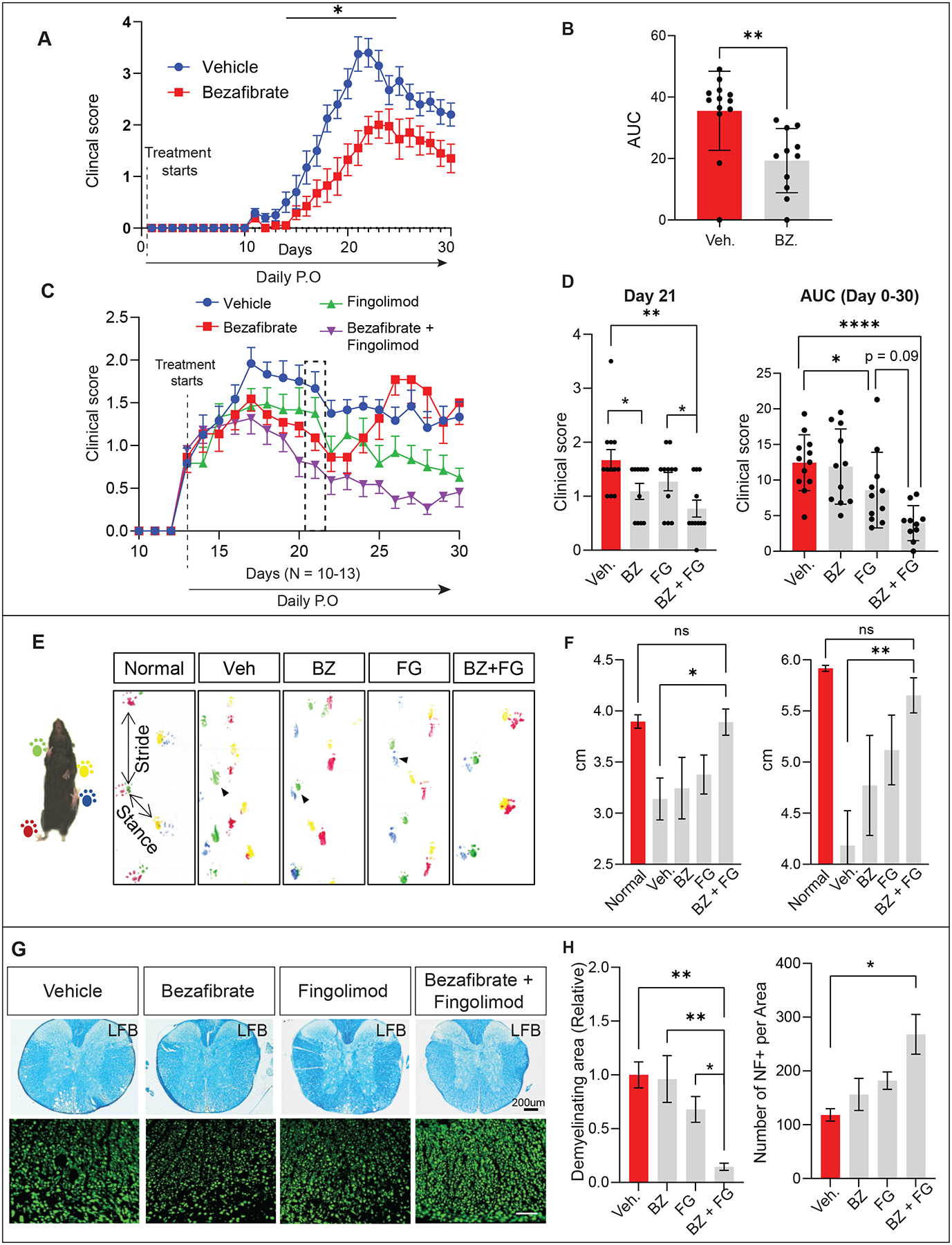
(A) The clinical score during EAE progression was recorded and statistically analyzed using two-way ANOVA, followed by Sidak post-hoc analysis; n = 10–13 per group. (B) Overall disease severity of EAE mice was compared by calculating the area under the curve (AUC) between day 14–30, followed by unpaired Student’s t-test; n = 10–13 per group (C-D) EAE mice were randomly divided into 4 groups based on the clinical score at the onset of EAE (Day 13) and treated with Bezafibrate (100 mg/kg), Fingolimod (3mg/kg), a combination of Bezafibrate (100 mg/kg) and Fingolimod (3mg/kg) or vehicle by daily oral gavage. Clinical scores were compared with vehicle control (n = 11–12 per group). Overall disease severity of EAE mice was compared by calculating the area under the curve (AUC) between day 18–30, followed by unpaired Student’s t-test; n = 11–12 per group. Data were statistically analyzed using two-way ANOVA, followed by Sidak post-hoc analysis,; (E-F) EAE mice were subjected to footprint analysis on Day 30. For normal non-EAE wt mice, green and red mark the right feet, and yellow and blue mark the left feet. For the experimental mice (Veh, BZ, FG, and BZ+FG), red and yellow mark the right feet, and green and blue mark the left feet. Stride and stance distance were measured to indicate fine motor function and were statistically analyzed using two-way ANOVA, followed by Sidak post-hoc analysis. (G) Spinal cord sections from the EAE mice were stained by Luxol fast blue (LFB) to assess demyelination and stained for neurofilament (NF) to assess neuronal loss (n =4–6 per group). (H) Quantifications of LFB and NF staining. Values were normalized to vehicle-treated control and statistically analyzed by using two-way ANOVA, followed by Sidak post-hoc analysis, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Data in all figures are represented as mean ± SEM.
Table 1.
Clinical features of EAE after prophylactic treatment of Bezafibrate
| Group | # Sick/total (incidence) | Maximum clinical score | Cumulative disease index | Mean weight on peak day |
|---|---|---|---|---|
| Vehicle | 13/13 (100%) | 3.9 ± 0.2 | 39.5 ± 2.1 | 21.0 ± 0.7 |
| Bezafibrate | 10/10 (100%) | 2.6 ± 0.2 *** | 22.0 ± 2.8**** | 21.1 ± 0.7 |
Values shown are mean ± SEM; n =13 or 10 per group.
p< 0.001,
p< 0.0001 vs. vehicle group
Cumulative disease index: sum of clinical score of the entire period.
Therapeutic treatment of Bezafibrate and Fingolimod synergistically improve EAE behavioral dysfunctions and pathology
S1P is known to act via G-protein-coupled S1P receptors. In the oligodendrocyte lineage, activation of S1P receptors stimulates oligodendrocyte differentiation94,95, whereas activation of S1P receptors of lymphocytes mediates immune activation in MS96. To lower the levels of VLCFA and reduce the effects of S1P in immune cells, we treated EAE mice with Bezafibrate and Fingolimod at the onset of symptoms (Day 13) to reflect the therapeutic, rather than preventative effects (Figure 7C).
Treatment of the EAE symptoms was started at day 13 by daily oral gavage. Unlike prophylactic treatment with Bezafibrate, Bezafibrate only ameliorates the clinical score at the peak but not in the chronic phase of EAE development (Figure 7C). As reported75,97,98, therapeutical treatment of Fingolimod improves the clinical score throughout the EAE progression (Figure 7C, Table 2). Strikingly, a combination of Bezafibrate (100 mg/kg) and Fingolimod (3mg/kg) treatment showed synergistic effects on improving EAE-induced paralysis throughout EAE development (Figure 7D, Table 2). Given that the clinical score is a relative coarse measure of motor deficits, we also evaluated the fine motor function using a footprint assay (Figure 7E)99. Bezafibrate and Fingolimod single treatments did not rescue the ‘dragging’ footprint phenotype caused by EAE. However, a combination of both drugs restored the normal footprint and stance distance in EAE mice (Figures 7E–F). Histological analysis also reveals that Bezafibrate and Fingolimod synergistically improved the demyelination (LFB) and neuronal loss (NF) (Figures 7G–H). The treatment also showed a significant Fingolimod-induced immune suppressive effect as indicated by reduced microglia (Iba1, Figures S6A–D) and cytotoxic T-cell infiltration (CD8, Figures S6E–H). We also observed CD4+ cells at Day 19 in spinal cords as previously reported100. However, we found very few CD4+ positive T cells in the chronic stage (Figure S6P). Importantly, CD8+ T cells have been shown to play a role in the progression of both mouse EAE and human MS101–103. Fingolimod but not bezafibrate has a minor effect on reducing reactive astrocytes (GFAP, Figures S6I–L). Together, these data show that a combination of Bezafibrate and Fingolimod has additive beneficial effects on behavior dysfunction, demyelination, and neuronal loss in EAE progression.
Table 2.
Clinical features of EAE after therapeutical treatment of Bezafibrate and Fingolimod
| Group | # Sick/total (incidence) | Maximum clinical score | Cumulative disease index | Mean weight on peak day |
|---|---|---|---|---|
| Vehicle | 12/12 (100%) | 2.4 ± 0.2 | 26.0 ± 1.8 | 19.8 ± 0.5 |
| Bezafibrate | 11/11 (100%) | 2.2 ± 0.2 | 23.1 ± 2.1 | 20.0 ± 0.5 |
| Fingolimod | 11/11 (100%) | 1.8 ± 0.2 | 17.1 ± 1.9** | 20.1 ± 0.4 |
| Bezafibrate + Fingolimod | 11/11 (100%) | 1.6 ± 0.1* | 12.7 ± 1.8*** | 20.0 ± 0.5 |
Values shown are mean ± SEM; n =12 or 11 per group.
p< 0.05,
p< 0.01,
p< 0.001 vs. vehicle group.
Cumulative disease index: sum of clinical score of the entire period
Discussion
VLCFA are the most common fatty acid species in CNS myelin104. Although VLCFA accumulation has been linked to a variety of demyelination diseases in humans105, the mechanism by which VLCFAs are metabolized in glia remains unknown. We demonstrate that the S1P exit pathway, which converts VLCFA to S1P, is required in glia, not in neurons. Our findings indicate that glial cells supply S1P to the nervous system upon elevation of VLCFAs. This may occur when myelin is degraded, ACOX1 function is lost, or VLCFA synthesis is elevated. The increase in VLCFA production in glia results in neurodegeneration, which can be suppressed by Bezafibrate, a drug that lowers the production of VLCFA or Fingolimod, an S1P antagonist. These data provide compelling evidence that this enzymatic pathway in glia can drive neurodegeneration when it is too active and too much S1P is produced. The data show that S1P is secreted by glia and taken up by neurons. In addition, the elevated levels of S1P synergize with immune cells when their VLCFAs are also elevated. Finally, this pathway is at least in part evolutionarily conserved as lowering VLCFA levels with Bezafibrate improves the clinical phenotypes in the EAE model in mice, and the combination of Bezafibrate and Fingolimod improves the clinical phenotypes in a very robust additive and possibly synergistic manner. Taken together, our findings in flies and mice indicate that the VL-Ceramide pathway is critical in the nervous system and may represent a novel therapeutic target for neurodegenerative diseases associated with sphingolipid metabolism, including MS.
VLCFA toxicity in glia
A recent study documented that reactive astrocytes can eliminate neurons and mature oligodendrocytes by secreting saturated VLCFA106. Our findings reveal that S1P is a primary downstream effector of VLCFAs that causes toxicity. Intriguingly, astrocytes do not express the enzymes involved in S1P exit pathway or express them at very low levels, whereas microglia and oligodendrocytes express the corresponding genes abundantly17,18. These data support the idea that glial subtypes cooperate to metabolize lipids107 and that astrocytes may transfer VLCFAs to oligodendrocytes or microglia. It is unclear whether VLCFAs can be converted to other lipids, including phospholipids or cholesterol, which are critical lipids in myelin. However, our data show that S1P is a major downstream effector of VLCFAs in CNS (Figures 1D–E).
A potential S1P transport mechanism
Our data show that when S1P production is increased, Fingolimod or sply expression in neurons can suppress neurotoxicity. Similarly, Fingolimod and sply expression have been shown previously to block S1P function and mitigate dystrophic muscle phenotypes in Drosophila58. Secretion of S1P in vertebrate cells is mediated by SPNS2 (Sphingolipid transporter)108, and loss of the fly ortholog, Spinster, leads to neuronal demise109,110. However, it remains to be shown that Spinster is involved in the secretion of S1P. Once secreted, S1P acts through two possible pathways: a) via S1P-S1PR (S1P receptor) binding111 or b) via direct uptake by recipient cells, independent of S1PR-S1P binding52. Fingolimod primarily binds to S1P receptors50,64,75, suggesting that there may be an S1PR receptor in fruit flies.
Immune activation and neuroinflammation
We document that ELOVL1 overexpression in the glia leads to the activation of the IMD pathway in the CNS, which in turn leads to the recruitment of hemocytes. This suggests that glial-ELOVL1 expression is at the root of neuroinflammation through IMD activation. Moreover, glial knockdown of Relish, a downstream effector of the IMD pathway, significantly suppress the life-span decrease in glial-ELOVL1 expressing flies, providing further evidence that glial IMD activation is the cause of neuroinflammation. Interestingly, infiltration of macrophages in nervous system tissue in acute cases of MS is well established112. Moreover, it is also well-established that inflammatory responses in the CNS cause axonal damage and demyelination during MS113. However, the role of elevated VLCFAs in glial and immune cells in the pathophysiology of MS is not well established. Hence, our findings that increased S1P levels but not VLCFA trigger neuroinflammation are relevant with regards to immune cell accumulation and immune activation in the CNS.
Targeting VLCFA metabolism in demyelinating diseases
It has been documented that the levels of VL-Ceramides are up-regulated, but Long-chain Ceramides are down-regulated in MS patients when compared to healthy controls. In addition, the level of S1P is upregulated 3 fold in the cerebrospinal fluid (CSF) in MS patients114. These observations are in agreement with our data that elevation of VL-Ceramides leads to an increase in S1P level. Hence, supplementation of Bezafibrate, to lower VLCFA and S1P production, as well as Fingolimod, to suppress the action of S1P, improve the behavioral dysfunction and pathology associated with EAE mice significantly better than each drug separately. Bezafibrate is known to slowly cross the blood-brain barrier115, whereas Fingolimod crosses the BBB quickly116. As a result, Bezafibrate’s effect on VLCFA and S1P reduction may be limited in the brain; hence, supplementing both drugs is more effective.
The implications of our observations and therapeutic implications are not restricted to MS. We argue that myelin, which is very rich in VLCFA-ceramides (10-fold higher than in other cells), is a very important source of S1P. Hence, myelin breakdown also leads to elevated levels of VLCFA and S1P. The breakdown of myelin is observed in several other neurodegenerative diseases and is often followed by neuroinflammation. For example, Huntington’s chorea and Alzheimer’s Disease (AD) are also associated with progressive demyelination26,27. Interestingly, S1P levels in the CSF of patients with Mild Cognitive Impairment-AD are significantly elevated117, and a recent report suggests that genes implicated in the S1P exit pathway are associated with AD118. In summary, we argue that neurological diseases in which myelin is broken down may cause inflammation by elevating S1P.
Limitations of Study
In this study, we show that VLCFAs in glia are metabolized into Sphingosine 1-Phoshpate (S1P), that it is secreted by glia and causes the demise of neurons when it is elevated. We identify the pathway involved in these metabolic changes and named it the “S1P exit pathway”. The uptake of S1P by neurons causes neuroinflammation, activation of the IMD pathway as well as peripheral macrophage invasion into the CNS. However, we do not know how S1P is transported from glia to neuron, given that S1P receptor and transporter have not yet been identified in flies. Even though the fly does not have an obvious S1P receptors (S1PRs), the protein that transports S1P, SPNS2 (Spinster Homolog 2), was first identified in flies (spinster)119,120, it is highly expressed in CNS, and its loss causes lysosomal defects and neurodegeneration109. Yet, it has not been determined whether Spinster can transport S1P. In vertebrates, S1P binds to apolipoprotein M (ApoM) for its transport, which is not conserved in flies. However, we have previously shown that ApoD (glial lazarillo) in flies is required for transferring fatty acids from neuron to glia,66. Therefore, Glaz may also facilitate S1P transport to neurons. In summary, further studies focused on how S1P is transported from glia to neurons will be required.
STAR METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hyunglok Chung (hchung2@houstonmethodist.org).
MATERIALS AVAILABILITY
The Lead contact can provide Drosophila strains, plasmids, or other materials used in this study upon request.
DATA AND CODE AVAILABILITY
All data points used to create the graphs can be found in Data S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fly strains and genetics
Generation of SK1 Kozak Gal4 (SK1T2A) and CDase CRIMIC Gal4 (CDaseT2A) flies
SK1 Kozak Gal4/CDase CRIMIC alleles were generated as described by46 respectively. Briefly, sgRNAs that target the SK1 locus GTTGGCCGTCATATCTTCTTTGG for 5’ end and AATATTTCACTACTGCCCACTGG for 3’end were cloned in pCFD5 vector (Port and Bullock, 2016). 200 bps of homology arms were synthesized in pUC57-Kan_gw_OK1 vector by Genewiz (South Plainfield, NJ). KozakGAL4-polyA-FRT-3XP3EGFP-FRT fragment is subcloned from pM37_KozakGAL4 vector into the synthesis product in between the homology arms. For CDase CRIMIC allele sgRNA targeting the CDase locus (AAGGTAAGGTAGTTATAGCCAGG) were cloned in pCFD3 vector127 and 200 bps of homolog tarns were synthesized in pUC57-Kan_gw_OK1 vector by Genewiz. attP-FRT-SA-T2AGAL4-polyA-3XP3EGFP-polyA-FRT-attP cassette were subcloned from pM37_p0 vector to the synthesis product in between the homology arms. Homology donor plasmids and the sgRNA encoding plasmids were injected into nos::Cas9 embryos as described in45.
Generation of CDasenull flies by CRISPR/Cas9 deletion
CRISPR-mediated mutagenesis was performed by WellGenetics Inc. using modified methods of128. In brief, the upstream gRNA sequence CAGGAAACTTACGGCCAGAA[AGG] and the downstream gRNA sequence CTAGTCCTCCTTAACGGTGA[AGG] were cloned into U6 promoter plasmid(s) separately. Cassette RMCE-3xP3-RFP, which contains two attP site, a floxed 3xP3-RFP, and two homology arms were cloned into pUC57-Kan as donor template for repair. CDase/CG1471-targeting gRNAs and hs-Cas9 were supplied in DNA plasmids, together with donor plasmid for microinjection into embryos of control strain w[1118]. F1 flies carrying selection marker of 3xP3-RFP were further validated by genomic PCR and sequencing. CRISPR generated a 2,729-bp deletion allele of CDase/CG1471, deleting the entire CDS of CDase/CG1471 gene which was replaced by the cassette RMCE-3xP3-RFP.
Mice
C57BL/6 mice (purchased from Center for Comparative Medicine at Baylor College of Medicine) were housed in a temperature-controlled environment (21 ± 1°C) with 14h light/ 10h cycles and fed standard rodent chow (Pico Lab Standard Diet, Cat # 5053) ad libitum. Mice were routinely checked for health status three times a day. All mice were maintained and studied according to protocols approved by the Institutional Animal Care and Use Committee of Baylor college of Medicine. Experimental details for the EAE model are provided in the Method Details section.
METHOD DETAILS
Transmission electron microscopy for wing margin
Drosophila wing margins were imaged following standard electron microscopy procedures using a Ted Pella Bio Wave processing microwave with vacuum attachments. The flies were covered in 2% paraformaldehyde, 2.5% glutaraldehyde, in 0.1 M sodium cacodylate buffer at pH 7.2. Briefly, the thorax was dissected under the fixative away from the head and abdomen leaving the wings on the thorax. After dissection the thorax was incubated overnight up to 3 days in the fixative on a rotator. The pre-fixed thorax with wings was then fixed again, rinsed by 3x with Millipore water, post-fixed with 1% aqueous osmium tetroxide, and rinsed again 3x with Millipore water. Concentrations from 25–100% of ethanol were used for the initial dehydration series, followed with propylene oxide as the final dehydrant. Samples were gradually infiltrated with 3 ratios of propylene oxide and Embed 812, finally going into 3 changes of pure resin under vacuum. Samples were allowed to infiltrate in pure resin overnight on a rotator. The samples were embedded into flat silicone molds arranged so that the sample could be cross-sectioned in the wing margin area. The samples were then cured in the oven at 62°C for three days. Thin-sections of the polymerized samples were cut at 48–50 nm and stained with 1% uranyl acetate for 10 minutes followed by 2.5% lead citrate for 2 minutes before TEM examination. Grids were viewed in a JEOL 1400+ transmission electron microscope at 80kV. Images were captured using an AMT XR-16 mid-mount 16 mega-pixel digital camera. Given the difficulties in quantifying the size of the glial membrane, a subjective observation method was used.
Drosophila assays
Climbing was performed as previously described129. Briefly, for all assays, newly eclosed flies were collected and kept at 25°C until used. To test climbing ability, an acrylic climbing tube was used129. The climbing tube was gently tapped and then recorded for 10 s. This was repeated ten times for each vial. The recorded data were analyzed by Tracker software (https://tracker.physlets.org/). SEM error bars were used in the analysis for climbing assay For the longevity assay, the flies are separated into vials (10 per vial), and incubated at 25°C. These flies are transferred to fresh vials every 3 days, and the number of dead flies is counted. Survival rates are calculated for the total population.. To quantify the eclosion rate, we counted the # of eclosed progenies divided by # of expected progenies based on Mendelian ratio. The melanization index was evaluated by counting the # of melanized spots (black spots) in the body.
ERG Recording of Fly Eye
ERG recordings were performed as described in. In brief, flies were glued to a slide with Elmer’s Glue. A recording electrode filled with 100 mM NaCl was placed on the eye, and a reference electrode was placed on the fly head. During the recording, a 1 s pulse of light stimulation was given, and the ERG traces of ten flies for each genotype were recorded and analyzed with WinWCP v.5.3.3 software.
Drug administration in fly food
Fingolimod (Sigma, STAR Methods) was added freshly to regular fly food at the indicated concentrations: 100μg/ml and 200 μg/ml dissolved in distilled water. Bezafibrate (Sigma, STAR Methods) was added freshly to regular fly food at 0.4 μM dissolved in DMSO. For ERG analysis, flies were transferred to freshly prepared food supplemented with either Fingolimod or Bezafibrate every 2 days. We followed the protocol described by Koyle et al. (2016) for axenic flies.
Real-Time PCR
Flies were incubated at a 25°C incubator before eclosion. For experiments shown in Figure 6A, flies were transferred to a 29 °C incubator right after the eclosion, and only heads (n > 80 per genotype) were collected on day 3. For experiments shown in Figure 6E, flies were always kept in a 25 °C incubator, and 1–3-day old adult fly heads (n > 30) were dissected and collected. All the collection and dissection steps were done on the ice to prevent RNA degradation. Total RNA was extracted using TRIzol (Invitrogen, 15596026) following the manufacturer’s instructions. EtOH washing step were performed twice for higher RNA purity. Only RNAs with high purity (the ratio of A260/A230 >2.0) were used for further applications. The cDNA synthesis and the genomic DNA (gDNA) removal steps were carried out using All-In-One 5X RT MasterMix (abm, G592). Real-time PCR experiments were conducted in triplicates and analyzed using a CFX96 Real-Time system (Bio-Rad, USA) with iTaq Universal SYBR Green Supermix (Bio-Rad, 1725121). Detailed Real-Time PCR steps were as follows; PCR reactions were initially incubated at 95 °C for 3 min for polymerase activation and DNA denaturation. After the pre-treatment, reactions were subjected to the following thermal cycling conditions: 40 cycles of denaturation at 95 °C for 5 s and annealing/extension at 60 °C for 30 s. After cycling, the melting curve was analyzed to check the existence of non-specific amplification or inefficient reaction. Experiments were repeated three times. All primers were synthesized (GENEWIZ, USA) and purified with HPLC. Following primers with high primer efficiency (>90%) were used for amplification:
Rp49 F: TACAGGCCCAAGATCGTGAA (Tm:60)
Rp49 R: TCTCCTTGCGCTTCTTGGA (Tm:60)
pvf2 F: CAGGGCGACGACAATCATCT (Tm:60)
pvf2 R: TGGATAGTCATCGCTCCCATC (Tm:60)
Dip-B F: TTCTCGAGTGCCTGGGCTTA (Tm:60)
Dip-B R: ATTGGGAGCATATGCCAGTG (Tm:60)
AttD F: GTATTCGCCTCCACTCCAGG (Tm:60)
AttD R: TGCATGACCATTGGCGTTGA (Tm:60)
Drosomycin F: CTGGGACAACGAGACCTGTC (Tm: 60)
Drosomycin R: ATCCTTCGCACCAGCACTTC (Tm: 60)
BomS1 F: CTGGGACAACGAGACCTGTC (Tm: 60)
BomS1 R: ATCCTTCGCACCAGCACTTC (Tm: 60)
Draper F: CTGGATGGACCCAATATCTGC (Tm: 60)
Draper R: GTTTAATGCGATAGGTGGAGCA (Tm: 60)
SK1 F: ACACATCCGCAGTATGCCAA (Tm: 60)
SK1 R: CACAATGCCCGAATAGCGTG (Tm: 60)
Lace F: TTTCCAAGATTGGCGCTGTG (Tm: 60)
Lace R: AGAATCGGATGCGTCCTTCC (Tm: 60)
Spt F: TCGGCGGATATTTCACGCAT (Tm: 60)
Spt R: GTTCCGTGCCCTTTCACAAA (Tm:60)
CDase F: CCTTTCTGGCCGTAAGTTTCC (Tm:60)
CDase R: CCACTTGCTTGATGTTGGCATAG (Tm:60).
Primer-BLAST program (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) was used for the primer design of pvf2, Dip-B, and AttD. FlyPrimerBank tool was utilized for the CDase primer design130.
Immunocytochemistry
In brief, immunostaining was performed as previously described129,131. Tissues were dissected in PBS and fixed in 4% paraformaldehyde (PFA) in PBS at 4°C on a nutating platform, then transferred to 0.2% Triton X-100 in PBS (0.2% PBST) at 4°C on a nutating platform for overnight incubation. For immunostaining, the samples were blocked in 5% BSA/2% PBST and incubated with the primary antibodies: Rat anti-Elav (1:500, 7E8A10, DSHB132), Mouse anti-Repo (1:50, 8D12, DSHB,) and Mouse anti-S1P (1:100, Z-P300. Echelon Biosciences) diluted in 5% BSA/0.2% PBST at 4°C for 48 hrs with nutation, then washed 3x with 0.2% PBST for 5 min. The secondary antibodies Donkey anti-rabbit (Cy3) and Donkey anti-mouse (Alexa-647) (Jackson ImmunoResearch) were diluted 1:250 in 5% BSA/0.2% PBST and incubated with the samples at 4°C for 48 hrs on a rotating platform. Samples were cleared and mounted in RapiClear (SunJin Lab Co.) and imaged with a Leica SP8 Confocal Microscope under a 20x objective lens and analyzed using Fiji133. Pupal brain staining was performed as described in86.
Immune cell staining:
Immunostaining of Drosophila larval hemocytes (immune cells) was performed as described previously134. Briefly, 20 late 3rd instar larvae (LL3) of the genotype yw;dACOX1T2A, mCD8GFP; BcF6-mCherry were vortexed and bled in 300 μl of Schneider’s media. The immune cell suspension was then transferred to a chambered coverglass slide (VWR, cat# 62407–056) and cells were allowed to settle down at room temperature (RT) for 30 min and fixed in 4% PFA for 20 min. Next, the cells were washed three times using 1x PBS, permeabilized using PBS with 0.1% Triton-X (PBST) for 10 min and blocked with 5% BSA in PBST for 20 min. Lastly, the cells were incubated with 1:100 anti-Hemese (H2) antibody [1:100 dilution] overnight at 4°C. The next day, cells were washed and incubated with anti-mouse Alexa Fluor 633 (far red) secondary antibody (Invitrogen, cat# A-21052) for 1 hr at RT. Finally, cells were washed three times with 1x PBS and mounting media with DAPI (Vector Laboratories Inc., cat# H-1200) was added to cells. Imaging was performed using Nikon Spinning disk confocal microscope.
EAE mouse model
While there are some variations in the doses and schedule of EAE induction135–138, we performed pilot study and optimized our protocol to ensure a consistent >90% successful rate of EAE symptoms. Briefly, EAE was induced in female C57BL/6 mice (purchased from Center for Comparative Medicine) at 8 –10-weeks-old by subcutaneous flank administration of 200 μg of myelin oligodendrocyte glycoprotein (MOG) peptide (amino acid 35–55; CSBio) in Complete Freund’s Adjuvant (CFA, Millipore-Sigma) containing 1g non-viable Mycobacterium tuberculosis (H37Ra, ATCC 25177). 300 ng pertussis toxin (Fisher Scientific) was administered intraperitoneally on day 0, 2 and 7, along with the MOG administration on day 0 and 7 (Figure S5A). EAE mice were treated daily with Bezafibrate (100mg/kg) and/or Fingolimod (3mg/kg) by oral gavage. All drugs were dissolved in 0.5% Sodium carboxymethyl cellulose (CMC) solution with 0.5% Tween80. Control mice were administrated with vehicle. Mice were scored daily as follows: 0 = no signs; 0.5 = distal limp tail; 1 = limp tail; 1.5 = wobbly walking; 2 = crossed hindlegs; 2.5 = one hind limb paresis; 3 = bilateral hind limb paresis; 3.5 = severe bilateral hind limb paralysis; 4 = beginning forelimb paresis;4.5 = moribund; (animals were euthanized); 5 = dead.”
For footprint test, mice with clinical scores up to 2 were habituated to the walkway so that they learned to perform the test with a reliable walking pattern. The footprint analysis was performed on day 29 in EAE mice with or without drug treatment. The forepaws and hindpaws of the mice were painted with different non-toxic paint using cotton swap, and the mice were allowed to run on a 50 cm × 10 cm paper-lined tunnel to a hiding box to create consecutive footprints with colors. Three independent measurements were made from each mouse every 20 minutes to avoid stress. Stride and stance length was measured and averaged from all three independent measurements.
Immunostaining for mouse tissues
Mice were anesthetized and intracardially perfused with phosphate-buffered saline (PBS), followed by 4% paraformaldehyde (PFA). The spinal cords were fixed by 4% PFA for overnight and then dehydrated using serial sucrose-gradient osmosis. The spinal cord was embedded in OCT compound (Tissue TEK) and stored at −80 °C until further use. Tissue sections were prepared with cryostat (Leica) at a thickness of 15 um, followed by immunostaining as described before139,140. Following primary antibodies were used: rabbit anti-GFAP (1:500; Agilent Dako; SIS), chicken anti-Neurofilament H (1:500; EnCor Biotechnology, CPCA-NF-H), rabbit anti-Iba1 (1:500, Wako, 019–19741), rabbit Anti-CD8 alpha (1:500, Abcam, ab251596). Secondary antibodies included the following: AlexaFluor-488- and AlexaFluor-568-conjugated secondary antibodies to rabbit or chicken (1:500; Thermo Scientific). Cell nuclei were stained with DAPI (Vector Labs). Images were obtained by Zeiss Imager.M2m equipped with ApoTome.2, Axiocam 506 mono.
QUANTIFICATION AND STATISTICAL ANALYSIS
Results are presented as dot or bar plots, in which the mean +/− standard error of the mean (SEM) are depicted. All statistical analysis was performed using Graphpad Prism (GraphPad Software, Inc., Ca, US). When the means of 2 groups were compared, a two-tailed unpaired t-test was used, and when the means of 2 variables of more than 2 groups were compared, a two-way ANOVA with Tukey’s multiple comparisons post-test was used. Results were designated significant when the P-value (p)<0.05: *= p<0.05, **= p<0.01, ***= p<0.001, n.s. = non-significant. For mice works, GraphPad Prism v9 was used for generating graphs and statistical analysis. Data were reported as mean +/− SEM. Two-tailed, unpaired Student’s t-test was used for two-group comparisons. Two-way ANOVA was used for multiple group comparisons. Differences were considered significant at P < 0.05. The sample size for each experiment is indicated in the corresponding figure legend.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-elav | DSHB Cat# 7E8A10 | RRID:AB_528218 |
| anti-Repo | DSHB Cat# 8D12 | RRID:AB_528448 |
| Anti-Hemese | 121 | N/A |
| anti-mouse-647 | Jackson ImmunoResearch Labs Cat# 715-605-151 | RRID:AB_2340863 |
| anti-rabbit-Cy3 | Jackson ImmunoResearch Labs Cat# 712-165-153 | RRID:AB_2340667 |
| anti-GFP | Abcam Cat# ab6662 | RRID:AB_305635 |
| anti-HA | BioLegend Cat# 902301 | RRID:AB_2565018 |
| anti-actin | Millipore Cat# MAB1501 | RRID:AB_2223041 |
| anti-Sphingosine 1-phosphate | Echelon Biosciences Cat# Z-P300 | |
| anti-Relish | DSHB Cat# 21F3 | RRID:AB_1553772 |
| anti-RFP | Invitrogen Cat# R10367 | RRID:AB_10563941 |
| anti-Draper | DSHB Cat# 8A1 | RRID:AB_2618106 |
| anti-Neurofilament-H | BioLegend Cat# 835601 | RRID:AB_2565349 |
| anti-Iba-1 | FUJIFILM Wako Shibayagi Cat# 013-26471 | RRID:AB_2687911 |
| anti-ACOX1 | Sigma-Aldrich Cat# HPA021195 | RRID:AB_1844528 |
| anti-CD8 | Abcam Cat# ab236230 | RRID:AB_2756381 |
| anti-GFAP | Agilent Cat# N1506 | RRID:AB_10013482 |
| Bacterial and Virus Strains | ||
| NEB® 5-alpha Competent E. coli | NEB | Cat#C2987 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CHAPS hydrate | Sigma-Aldrich | Cat# C9426 |
| cOmplete™ Protease Inhibitor Cocktail | Roche | Cat# 11697498001 |
| PhosSTOP™ | Sigma-Aldrich | Cat# 4906845001 |
| 10x Tris/Glycine Buffer | Biorad | Cat# 1610734 |
| Bezafibrate | Sigma-Aldrich | Cat# BP755 |
| N-acetylcystein amide | Sigma-Aldrich | Cat# A0737 |
| 4%−15% Mini-PROTEAN TGX precast native gel | Biorad | Cat# 4561086 |
| Fingolimod (FTY720) | Millipore sigma | Cat# 162359-56-0 |
| Experimental Models: Organisms/Strains | ||
| UAS-2xEGFP, hs-Cre,vas-dϕC31 | 122 | N/A |
| Repo-Gal4 | BDSC_7415 | RRID: BDSC_7415 |
| Hml-Delta-Gal4 | 88 | N/A |
| UAS-dELOVL RNAi | BDSC_50710 | RRID: BDSC_50710 |
| UAS-hELOVL1 | BDSC_77932 | RRID: BDSC_77932 |
| dACOX1 T2A | This paper | N/A |
| UAS-nls. mCherry | BDSC_38424 | RRID: BDSC_38424 |
| UAS-lacZ | Bellen lab stock | N/A |
| UAS-Draper | BDSC_67035 | RRID: BDSC_67035 |
| UAS-Draper RNAi | BDSC_67034 | RRID: BDSC_67034 |
| UAS-Luciferase RNAi | BDSC_35785 | RRID: BDSC_35785 |
| UAS-CDase RNAi | BDSC_36764 | RRID: BDSC_36764 |
| UAS-SK1 RNAi | BDSC_36747 | RRID: BDSC_36747 |
| UAS-SK2 RNAi | BDSC_35570 | RRID: BDSC_35570 |
| UAS-CDase | 123 | N/A |
| UAS-SK1 | 123 | N/A |
| UAS-mCD8GFP | BDSC_32184 | RRID: BDSC_32184 |
| Repo-LexA-GAD | BDSC_67096 | RRID: BDSC_67096 |
| LexAop-ELOVL1 | This paper | N/A |
| Sply RNAi | VDRC_105752 | RRID: FlyBase_FBst0324722 |
| UAS-sply | This paper | N/A |
| UAS-SGPL1 | This paper | N/A |
| UAS-Relish RNAi | BDSC_33661 | RRID: BDSC_33661 |
| dACOX1ywg | N/A | |
| BcF6-mCherry | 124 | N/A |
| P{w[+mC]=srpHemo-Moe.3XmCherry}3 | 125 | N/A |
| UAS-lace | DGGR_109638 | RRID: DGGR_109638 |
| UAS-spt | 126 | N/A |
| Tubulin Gal80 | 86 | N/A |
| Perineurial glia Gal80 | 86 | N/A |
| TubGal80ts;Repo Gal4 | S. Schirmeir & C. Klambt | N/A |
| Software and Algorithms | ||
| ImageJ | Version 2.0 | https://www.imagej.nih.gov |
| Prism7 | Graph Pad | https://www.graphpad.com |
| Zen Blue | Zeiss | https://www.zeiss.com |
| Zen Black | Zeiss | https://www.zeiss.com |
| Snapgene | Snapgene | www.snapgene.com |
| Imagelab | Bio-rad | https://www.bio-rad.com/en-us/product/image-lab-software |
| Other | ||
Highlights.
Glia convert VLCFA into Sphingosine 1-Phosphate (S1P) via a glial-specific S1P pathway
Excess S1P causes neuroinflammation, NF-kB activation, and macrophage infiltration
Suppressing S1P function attenuates the pathological phenotypes caused by excess VLCFAs
Bezafibrate and fingolimod synergize to ameliorate symptoms in the EAE model of MS
Acknowledgments:
We thank Dr. Christian Klämbt, Dr. Usha Acharya, Dr. Marc Freeman, Dr. Bruno Lemaitre, and Dr. Sean Sweeney for generously sharing fly reagents. We thank Hongling Pan for creating transgenic flies and Dinghui Yu for assistance and maintenance of microscopes. We thank Prof. István Andó for the generous gift of anti-Hemese (H2) antibody. We also thank Dr. Paula Montero Llopis of the Microscopy Resources on the North Quad (MicRoN) core facility at Harvard Medical School. Drosophila stocks were obtained from the Bloomington Stock Center (NIH P40OD018537) at Indiana University. H.K.L. is supported by NIH/NINDS (R01NS110859) and NMSS (RG-1907-34551). H.J.B. is supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director (U54NS093793), NIH/ORIP (R24OD022005; R24OD031447), and the Chair in Neuroscience of the Neurological Research Institute of TCH. Confocal microscopy was performed in the Neurovisualization core of the BCM IDDRC (NICHD U54HD083092). H.C. is supported by the Warren Alpert Foundation. J-W.M. is supported by NRF-2021R1A6A3A14044510 from the National Research Foundation of Korea. NP is an investigator of the Howard Hughes Medical Institute (HHMI). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing financial interests.
Reference
- 1.Nave KA (2010). Myelination and support of axonal integrity by glia. Nature 468, 244–252. 10.1038/nature09614. [DOI] [PubMed] [Google Scholar]
- 2.Chrast R, Saher G, Nave KA, and Verheijen MH (2011). Lipid metabolism in myelinating glial cells: lessons from human inherited disorders and mouse models. J Lipid Res 52, 419–434. 10.1194/jlr.R009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams KA, and Deber CM (1993). The structure and function of central nervous system myelin. Crit Rev Clin Lab Sci 30, 29–64. 10.3109/10408369309084665. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien JS (1965). Stability of the Myelin Membrane. Science 147, 1099–1107. 10.1126/science.147.3662.1099. [DOI] [PubMed] [Google Scholar]
- 5.Norton WT, and Poduslo SE (1973). Myelination in rat brain: changes in myelin composition during brain maturation. J Neurochem 21, 759–773. 10.1111/j.1471-4159.1973.tb07520.x. [DOI] [PubMed] [Google Scholar]
- 6.Garbay B, Heape AM, Sargueil F, and Cassagne C (2000). Myelin synthesis in the peripheral nervous system. Prog Neurobiol 61, 267–304. 10.1016/s0301-0082(99)00049-0. [DOI] [PubMed] [Google Scholar]
- 7.Aggarwal S, Yurlova L, and Simons M (2011). Central nervous system myelin: structure, synthesis and assembly. Trends Cell Biol 21, 585–593. 10.1016/j.tcb.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Weinstock NI, Shin D, Dhimal N, Hong X, Irons EE, Silvestri NJ, Reed CB, Nguyen D, Sampson O, Cheng YC, et al. (2020). Macrophages Expressing GALC Improve Peripheral Krabbe Disease by a Mechanism Independent of Cross-Correction. Neuron 107, 65–81 e69. 10.1016/j.neuron.2020.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinstock NI, Kreher C, Favret J, Nguyen D, Bongarzone ER, Wrabetz L, Feltri ML, and Shin D (2020). Brainstem development requires galactosylceramidase and is critical for pathogenesis in a model of Krabbe disease. Nat Commun 11, 5356. 10.1038/s41467-020-19179-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sassa T, Suto S, Okayasu Y, and Kihara A (2012). A shift in sphingolipid composition from C24 to C16 increases susceptibility to apoptosis in HeLa cells. Biochim Biophys Acta 1821, 1031–1037. 10.1016/j.bbalip.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 11.Sassa T, and Kihara A (2014). Metabolism of very long-chain Fatty acids: genes and pathophysiology. Biomol Ther (Seoul) 22, 83–92. 10.4062/biomolther.2014.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imgrund S, Hartmann D, Farwanah H, Eckhardt M, Sandhoff R, Degen J, Gieselmann V, Sandhoff K, and Willecke K (2009). Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J Biol Chem 284, 33549–33560. 10.1074/jbc.M109.031971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simons K, and Ikonen E (1997). Functional rafts in cell membranes. Nature 387, 569–572. 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 14.Strachan LR, Stevenson TJ, Freshner B, Keefe MD, Miranda Bowles D, and Bonkowsky JL (2017). A zebrafish model of X-linked adrenoleukodystrophy recapitulates key disease features and demonstrates a developmental requirement for abcd1 in oligodendrocyte patterning and myelination. Hum Mol Genet 26, 3600–3614. 10.1093/hmg/ddx249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milhas D, Clarke CJ, and Hannun YA (2010). Sphingomyelin metabolism at the plasma membrane: implications for bioactive sphingolipids. FEBS Lett 584, 1887–1894. 10.1016/j.febslet.2009.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slotte JP (2013). Biological functions of sphingomyelins. Prog Lipid Res 52, 424–437. 10.1016/j.plipres.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, et al. (2016). New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 113, E1738–1746. 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, and Barres BA (2018). Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A 115, E1896–E1905. 10.1073/pnas.1800165115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Janssens J, De Waegeneer M, Kolluru SS, Davie K, Gardeux V, Saelens W, David FPA, Brbic M, Spanier K, et al. (2022). Fly Cell Atlas: A single-nucleus transcriptomic atlas of the adult fruit fly. Science 375, eabk2432. 10.1126/science.abk2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin G, Lee PT, Chen K, Mao D, Tan KL, Zuo Z, Lin WW, Wang L, and Bellen HJ (2018). Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to alpha-Synuclein Gain. Cell Metab 28, 605–618 e606. 10.1016/j.cmet.2018.05.019. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Lin G, Zuo Z, Li Y, Byeon SK, Pandey A, and Bellen HJ (2022). Neuronal activity induces glucosylceramide that is secreted via exosomes for lysosomal degradation in glia. Sci Adv 8, eabn3326. 10.1126/sciadv.abn3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan C, Rao RP, Jesmin N, Bamba T, Nagashima K, Pascual A, Preat T, Fukusaki E, Acharya U, and Acharya JK (2011). CDase is a pan-ceramidase in Drosophila. Mol Biol Cell 22, 33–43. 10.1091/mbc.E10-05-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acharya JK, Dasgupta U, Rawat SS, Yuan C, Sanxaridis PD, Yonamine I, Karim P, Nagashima K, Brodsky MH, Tsunoda S, and Acharya U (2008). Cell-nonautonomous function of ceramidase in photoreceptor homeostasis. Neuron 57, 69–79. 10.1016/j.neuron.2007.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh A, Kling T, Snaidero N, Sampaio JL, Shevchenko A, Gras H, Geurten B, Gopfert MC, Schulz JB, Voigt A, and Simons M (2013). A global in vivo Drosophila RNAi screen identifies a key role of ceramide phosphoethanolamine for glial ensheathment of axons. PLoS Genet 9, e1003980. 10.1371/journal.pgen.1003980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peters R (2006). Ageing and the brain. Postgrad Med J 82, 84–88. 10.1136/pgmj.2005.036665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nasrabady SE, Rizvi B, Goldman JE, and Brickman AM (2018). White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun 6, 22. 10.1186/s40478-018-0515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bae HG, Kim TK, Suk HY, Jung S, and Jo DG (2020). White matter and neurological disorders. Arch Pharm Res 43, 920–931. 10.1007/s12272-020-01270-x. [DOI] [PubMed] [Google Scholar]
- 28.Chung HL, Wangler MF, Marcogliese PC, Jo J, Ravenscroft TA, Zuo Z, Duraine L, Sadeghzadeh S, Li-Kroeger D, Schmidt RE, et al. (2020). Loss- or Gain-of-Function Mutations in ACOX1 Cause Axonal Loss via Different Mechanisms. Neuron. 10.1016/j.neuron.2020.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watkins PA, McGuinness MC, Raymond GV, Hicks BA, Sisk JM, Moser AB, and Moser HW (1995). Distinction between peroxisomal bifunctional enzyme and acyl-CoA oxidase deficiencies. Ann Neurol 38, 472–477. 10.1002/ana.410380322. [DOI] [PubMed] [Google Scholar]
- 30.Engelen M, Schackmann MJ, Ofman R, Sanders RJ, Dijkstra IM, Houten SM, Fourcade S, Pujol A, Poll-The BT, Wanders RJ, and Kemp S (2012). Bezafibrate lowers very long-chain fatty acids in X-linked adrenoleukodystrophy fibroblasts by inhibiting fatty acid elongation. J Inherit Metab Dis 35, 1137–1145. 10.1007/s10545-012-9471-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engelen M, Tran L, Ofman R, Brennecke J, Moser AB, Dijkstra IM, Wanders RJ, Poll-The BT, and Kemp S (2012). Bezafibrate for X-linked adrenoleukodystrophy. PLoS One 7, e41013. 10.1371/journal.pone.0041013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohno Y, Suto S, Yamanaka M, Mizutani Y, Mitsutake S, Igarashi Y, Sassa T, and Kihara A (2010). ELOVL1 production of C24 acyl-CoAs is linked to C24 sphingolipid synthesis. Proc Natl Acad Sci U S A 107, 18439–18444. 10.1073/pnas.1005572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kremer MC, Jung C, Batelli S, Rubin GM, and Gaul U (2017). The glia of the adult Drosophila nervous system. Glia 65, 606–638. 10.1002/glia.23115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siow DL, and Wattenberg BW (2012). Mammalian ORMDL proteins mediate the feedback response in ceramide biosynthesis. J Biol Chem 287, 40198–40204. 10.1074/jbc.C112.404012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis DL, Gable K, Suemitsu J, Dunn TM, and Wattenberg BW (2019). The ORM-DL/Orm-serine palmitoyltransferase (SPT) complex is directly regulated by ceramide: Reconstitution of SPT regulation in isolated membranes. J Biol Chem 294, 5146–5156. 10.1074/jbc.RA118.007291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acharya U, and Acharya JK (2005). Enzymes of sphingolipid metabolism in Drosophila melanogaster. Cell Mol Life Sci 62, 128–142. 10.1007/s00018-004-4254-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bauer R, Voelzmann A, Breiden B, Schepers U, Farwanah H, Hahn I, Eckardt F, Sandhoff K, and Hoch M (2009). Schlank, a member of the ceramide synthase family controls growth and body fat in Drosophila. EMBO J 28, 3706–3716. 10.1038/emboj.2009.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao C, and Obeid LM (2008). Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim Biophys Acta 1781, 424–434. 10.1016/j.bbalip.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosen H, and Goetzl EJ (2005). Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol 5, 560–570. 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- 40.Aguilar A, and Saba JD (2012). Truth and consequences of sphingosine-1-phosphate lyase. Adv Biol Regul 52, 17–30. 10.1016/j.advenzreg.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Al-Ouran R, Hu Y, Kim SY, Wan YW, Wangler MF, Yamamoto S, Chao HT, Comjean A, Mohr SE, et al. (2017). MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am J Hum Genet 100, 843–853. 10.1016/j.ajhg.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu T, Sims D, and Baum B (2009). Parallel RNAi screens across different cell lines identify generic and cell type-specific regulators of actin organization and cell morphology. Genome Biol 10, R26. 10.1186/gb-2009-10-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schnorrer F, Schonbauer C, Langer CC, Dietzl G, Novatchkova M, Schernhuber K, Fellner M, Azaryan A, Radolf M, Stark A, et al. (2010). Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 464, 287–291. 10.1038/nature08799. [DOI] [PubMed] [Google Scholar]
- 44.Wicker-Thomas C, Garrido D, Bontonou G, Napal L, Mazuras N, Denis B, Rubin T, Parvy JP, and Montagne J (2015). Flexible origin of hydrocarbon/pheromone precursors in Drosophila melanogaster. J Lipid Res 56, 2094–2101. 10.1194/jlr.M060368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanca O, Zirin J, Garcia-Marques J, Knight SM, Yang-Zhou D, Amador G, Chung H, Zuo Z, Ma L, He Y, et al. (2019). An efficient CRISPR-based strategy to insert small and large fragments of DNA using short homology arms. Elife 8. 10.7554/eLife.51539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanca O, Zirin J, Hu Y, Tepe B, Dutta D, Lin WW, Ma L, Ge M, Zuo Z, Liu LP, et al. (2022). An expanded toolkit for Drosophila gene tagging using synthesized homology donor constructs for CRISPR-mediated homologous recombination. Elife 11. 10.7554/eLife.76077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davie K, Janssens J, Koldere D, De Waegeneer M, Pech U, Kreft L, Aibar S, Makhzami S, Christiaens V, Bravo Gonzalez-Blas C, et al. (2018). A Single-Cell Transcriptome Atlas of the Aging Drosophila Brain. Cell 174, 982–998 e920. 10.1016/j.cell.2018.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE, et al. (2019). Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 101, 207–223 e210. 10.1016/j.neuron.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spiegel S, and Milstien S (2003). Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4, 397–407. 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 50.Taha TA, Argraves KM, and Obeid LM (2004). Sphingosine-1-phosphate receptors: receptor specificity versus functional redundancy. Biochim Biophys Acta 1682, 48–55. 10.1016/j.bbalip.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 51.Maceyka M, Harikumar KB, Milstien S, and Spiegel S (2012). Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol 22, 50–60. 10.1016/j.tcb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goto H, Miyamoto M, and Kihara A (2021). Direct uptake of sphingosine-1-phosphate independent of phospholipid phosphatases. J Biol Chem 296, 100605. 10.1016/j.jbc.2021.100605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Pardo A, Pepe G, Castaldo S, Marracino F, Capocci L, Amico E, Madonna M, Giova S, Jeong SK, Park BM, et al. (2019). Stimulation of Sphingosine Kinase 1 (SPHK1) Is Beneficial in a Huntington’s Disease Pre-clinical Model. Front Mol Neurosci 12, 100. 10.3389/fnmol.2019.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Pardo A, and Maglione V (2018). Sphingolipid Metabolism: A New Therapeutic Opportunity for Brain Degenerative Disorders. Front Neurosci 12, 249. 10.3389/fnins.2018.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grassi S, Mauri L, Prioni S, Cabitta L, Sonnino S, Prinetti A, and Giussani P (2019). Sphingosine 1-Phosphate Receptors and Metabolic Enzymes as Druggable Targets for Brain Diseases. Front Pharmacol 10, 807. 10.3389/fphar.2019.00807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karaca I, Tamboli IY, Glebov K, Richter J, Fell LH, Grimm MO, Haupenthal VJ, Hartmann T, Graler MH, van Echten-Deckert G, and Walter J (2014). Deficiency of sphingosine-1-phosphate lyase impairs lysosomal metabolism of the amyloid precursor protein. J Biol Chem 289, 16761–16772. 10.1074/jbc.M113.535500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Proia RL, and Hla T (2015). Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest 125, 1379–1387. 10.1172/JCI76369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pantoja M, Fischer KA, Ieronimakis N, Reyes M, and Ruohola-Baker H (2013). Genetic elevation of sphingosine 1-phosphate suppresses dystrophic muscle phenotypes in Drosophila. Development 140, 136–146. 10.1242/dev.087791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lovric S, Goncalves S, Gee HY, Oskouian B, Srinivas H, Choi WI, Shril S, Ashraf S, Tan W, Rao J, et al. (2017). Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J Clin Invest 127, 912–928. 10.1172/JCI89626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Janecke AR, Xu R, Steichen-Gersdorf E, Waldegger S, Entenmann A, Giner T, Krainer I, Huber LA, Hess MW, Frishberg Y, et al. (2017). Deficiency of the sphingosine-1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications. Hum Mutat 38, 365–372. 10.1002/humu.23192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prasad R, Hadjidemetriou I, Maharaj A, Meimaridou E, Buonocore F, Saleem M, Hurcombe J, Bierzynska A, Barbagelata E, Bergada I, et al. (2017). Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J Clin Invest 127, 942–953. 10.1172/JCI90171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, and Mohr SE (2011). An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12, 357. 10.1186/1471-2105-12-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lai SL, and Lee T (2006). Genetic mosaic with dual binary transcriptional systems in Drosophila. Nat Neurosci 9, 703–709. 10.1038/nn1681. [DOI] [PubMed] [Google Scholar]
- 64.Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, and Burtin P (2010). Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov 9, 883–897. 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 65.Yamamoto S, Jaiswal M, Charng WL, Gambin T, Karaca E, Mirzaa G, Wiszniewski W, Sandoval H, Haelterman NA, Xiong B, et al. (2014). A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 159, 200–214. 10.1016/j.cell.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu L, MacKenzie KR, Putluri N, Maletic-Savatic M, and Bellen HJ (2017). The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab 26, 719–737 e716. 10.1016/j.cmet.2017.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moulton MJ, Barish S, Ralhan I, Chang J, Goodman LD, Harland JG, Marcogliese PC, Johansson JO, Ioannou MS, and Bellen HJ (2021). Neuronal ROS-induced glial lipid droplet formation is altered by loss of Alzheimer’s disease-associated genes. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2112095118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hardie RC, and Raghu P (2001). Visual transduction in Drosophila. Nature 413, 186–193. 10.1038/35093002. [DOI] [PubMed] [Google Scholar]
- 69.Vacaru AM, van den Dikkenberg J, Ternes P, and Holthuis JC (2013). Ceramide phosphoethanolamine biosynthesis in Drosophila is mediated by a unique ethanolamine phosphotransferase in the Golgi lumen. J Biol Chem 288, 11520–11530. 10.1074/jbc.M113.460972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Freeman MR (2015). Drosophila Central Nervous System Glia. Cold Spring Harb Perspect Biol 7. 10.1101/cshperspect.a020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, and Freeman MR (2006). The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 50, 869–881. 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 72.Hakim-Mishnaevski K, Flint-Brodsly N, Shklyar B, Levy-Adam F, and Kurant E (2019). Glial Phagocytic Receptors Promote Neuronal Loss in Adult Drosophila Brain. Cell Rep 29, 1438–1448 e1433. 10.1016/j.celrep.2019.09.086. [DOI] [PubMed] [Google Scholar]
- 73.Doherty J, Logan MA, Tasdemir OE, and Freeman MR (2009). Ensheathing glia function as phagocytes in the adult Drosophila brain. J Neurosci 29, 4768–4781. 10.1523/JNEUROSCI.5951-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ziegenfuss JS, Biswas R, Avery MA, Hong K, Sheehan AE, Yeung YG, Stanley ER, and Freeman MR (2008). Draper-dependent glial phagocytic activity is mediated by Src and Syk family kinase signalling. Nature 453, 935–939. 10.1038/nature06901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Choi JW, Gardell SE, Herr DR, Rivera R, Lee CW, Noguchi K, Teo ST, Yung YC, Lu M, Kennedy G, and Chun J (2011). FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A 108, 751–756. 10.1073/pnas.1014154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stofanko M, Kwon SY, and Badenhorst P (2010). Lineage tracing of lamellocytes demonstrates Drosophila macrophage plasticity. PLoS One 5, e14051. 10.1371/journal.pone.0014051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Galko MJ, and Krasnow MA (2004). Cellular and genetic analysis of wound healing in Drosophila larvae. PLoS Biol 2, E239. 10.1371/journal.pbio.0020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Gregorio E, Han SJ, Lee WJ, Baek MJ, Osaki T, Kawabata S, Lee BL, Iwanaga S, Lemaitre B, and Brey PT (2002). An immune-responsive Serpin regulates the melanization cascade in Drosophila. Dev Cell 3, 581–592. 10.1016/s1534-5807(02)00267-8. [DOI] [PubMed] [Google Scholar]
- 79.Tang H (2009). Regulation and function of the melanization reaction in Drosophila. Fly (Austin) 3, 105–111. 10.4161/fly.3.1.7747. [DOI] [PubMed] [Google Scholar]
- 80.Shaukat Z, Liu D, and Gregory S (2015). Sterile inflammation in Drosophila. Mediators Inflamm 2015, 369286. 10.1155/2015/369286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hoffmann JA (2003). The immune response of Drosophila. Nature 426, 33–38. 10.1038/nature02021. [DOI] [PubMed] [Google Scholar]
- 82.Roth S, Hiromi Y, Godt D, and Nusslein-Volhard C (1991). cactus, a maternal gene required for proper formation of the dorsoventral morphogen gradient in Drosophila embryos. Development 112, 371–388. 10.1242/dev.112.2.371. [DOI] [PubMed] [Google Scholar]
- 83.Kaneko T, Yano T, Aggarwal K, Lim JH, Ueda K, Oshima Y, Peach C, Erturk-Hasdemir D, Goldman WE, Oh BH, et al. (2006). PGRP-LC and PGRP-LE have essential yet distinct functions in the drosophila immune response to monomeric DAP-type peptidoglycan. Nat Immunol 7, 715–723. 10.1038/ni1356. [DOI] [PubMed] [Google Scholar]
- 84.Gottar M, Gobert V, Michel T, Belvin M, Duyk G, Hoffmann JA, Ferrandon D, and Royet J (2002). The Drosophila immune response against Gram-negative bacteria is mediated by a peptidoglycan recognition protein. Nature 416, 640–644. 10.1038/nature734. [DOI] [PubMed] [Google Scholar]
- 85.Busse MS, Arnold CP, Towb P, Katrivesis J, and Wasserman SA (2007). A kappaB sequence code for pathway-specific innate immune responses. EMBO J 26, 3826–3835. 10.1038/sj.emboj.7601798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Winkler B, Funke D, Benmimoun B, Speder P, Rey S, Logan MA, and Klambt C (2021). Brain inflammation triggers macrophage invasion across the blood-brain barrier in Drosophila during pupal stages. Sci Adv 7, eabh0050. 10.1126/sciadv.abh0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Koyle ML, Veloz M, Judd AM, Wong AC, Newell PD, Douglas AE, and Chaston JM (2016). Rearing the Fruit Fly Drosophila melanogaster Under Axenic and Gnotobiotic Conditions. J Vis Exp. 10.3791/54219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sinenko SA, and Mathey-Prevot B (2004). Increased expression of Drosophila tetraspanin, Tsp68C, suppresses the abnormal proliferation of ytr-deficient and Ras/Raf-activated hemocytes. Oncogene 23, 9120–9128. 10.1038/sj.onc.1208156. [DOI] [PubMed] [Google Scholar]
- 89.Boulet M, Renaud Y, Lapraz F, Benmimoun B, Vandel L, and Waltzer L (2021). Characterization of the Drosophila Adult Hematopoietic System Reveals a Rare Cell Population With Differentiation and Proliferation Potential. Front Cell Dev Biol 9, 739357. 10.3389/fcell.2021.739357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prinz M, and Priller J (2017). The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci 20, 136–144. 10.1038/nn.4475. [DOI] [PubMed] [Google Scholar]
- 91.Poll-The BT, Roels F, Ogier H, Scotto J, Vamecq J, Schutgens RB, Wanders RJ, van Roermund CW, van Wijland MJ, Schram AW, and et al. (1988). A new peroxisomal disorder with enlarged peroxisomes and a specific deficiency of acyl-CoA oxidase (pseudo-neonatal adrenoleukodystrophy). Am J Hum Genet 42, 422–434. [PMC free article] [PubMed] [Google Scholar]
- 92.Constantinescu CS, Farooqi N, O’Brien K, and Gran B (2011). Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol 164, 1079–1106. 10.1111/j.1476-5381.2011.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baecher-Allan C, Kaskow BJ, and Weiner HL (2018). Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 97, 742–768. 10.1016/j.neuron.2018.01.021. [DOI] [PubMed] [Google Scholar]
- 94.Roggeri A, Schepers M, Tiane A, Rombaut B, van Veggel L, Hellings N, Prickaerts J, Pittaluga A, and Vanmierlo T (2020). Sphingosine-1-Phosphate Receptor Modulators and Oligodendroglial Cells: Beyond Immunomodulation. Int J Mol Sci 21. 10.3390/ijms21207537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dukala DE, and Soliven B (2016). S1P1 deletion in oligodendroglial lineage cells: Effect on differentiation and myelination. Glia 64, 570–582. 10.1002/glia.22949. [DOI] [PubMed] [Google Scholar]
- 96.Subei AM, and Cohen JA (2015). Sphingosine 1-phosphate receptor modulators in multiple sclerosis. CNS Drugs 29, 565–575. 10.1007/s40263-015-0261-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Volpi C, Orabona C, Macchiarulo A, Bianchi R, Puccetti P, and Grohmann U (2019). Preclinical discovery and development of fingolimod for the treatment of multiple sclerosis. Expert Opin Drug Discov 14, 1199–1212. 10.1080/17460441.2019.1646244. [DOI] [PubMed] [Google Scholar]
- 98.Zhang Y, Li X, Ciric B, Ma CG, Gran B, Rostami A, and Zhang GX (2017). Effect of Fingolimod on Neural Stem Cells: A Novel Mechanism and Broadened Application for Neural Repair. Mol Ther 25, 401–415. 10.1016/j.ymthe.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vogelaar CF, Mandal S, Lerch S, Birkner K, Birkenstock J, Buhler U, Schnatz A, Raine CS, Bittner S, Vogt J, et al. (2018). Fast direct neuronal signaling via the IL-4 receptor as therapeutic target in neuroinflammation. Sci Transl Med 10. 10.1126/scitranslmed.aao2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang H, Newton G, Wu L, Lin LL, Miracco AS, Natesan S, and Luscinskas FW (2021). CD47 antibody blockade suppresses microglia-dependent phagocytosis and monocyte transition to macrophages, impairing recovery in EAE. JCI Insight 6. 10.1172/jci.insight.148719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wagner CA, Roque PJ, Mileur TR, Liggitt D, and Goverman JM (2020). Myelin-specific CD8+ T cells exacerbate brain inflammation in CNS autoimmunity. J Clin Invest 130, 203–213. 10.1172/JCI132531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S, et al. (2000). Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med 192, 393–404. 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jacobsen M, Cepok S, Quak E, Happel M, Gaber R, Ziegler A, Schock S, Oertel WH, Sommer N, and Hemmer B (2002). Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain 125, 538–550. 10.1093/brain/awf059. [DOI] [PubMed] [Google Scholar]
- 104.MacBrinn MC, and O’Brien JS (1969). Lipid composition of optic nerve myelin. J Neurochem 16, 7–12. 10.1111/j.1471-4159.1969.tb10337.x. [DOI] [PubMed] [Google Scholar]
- 105.Manor J, Chung H, Bhagwat PK, and Wangler MF (2021). ABCD1 and X-linked adrenoleukodystrophy: A disease with a markedly variable phenotype showing conserved neurobiology in animal models. J Neurosci Res 99, 3170–3181. 10.1002/jnr.24953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guttenplan KA, Weigel MK, Prakash P, Wijewardhane PR, Hasel P, Rufen-Blanchette U, Munch AE, Blum JA, Fine J, Neal MC, et al. (2021). Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 599, 102–107. 10.1038/s41586-021-03960-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Robinson RR, Dietz AK, Maroof AM, Asmis R, and Forsthuber TG (2019). The role of glial-neuronal metabolic cooperation in modulating progression of multiple sclerosis and neuropathic pain. Immunotherapy 11, 129–147. 10.2217/imt-2018-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fukuhara S, Simmons S, Kawamura S, Inoue A, Orba Y, Tokudome T, Sunden Y, Arai Y, Moriwaki K, Ishida J, et al. (2012). The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Invest 122, 1416–1426. 10.1172/JCI60746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dermaut B, Norga KK, Kania A, Verstreken P, Pan H, Zhou Y, Callaerts P, and Bellen HJ (2005). Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J Cell Biol 170, 127–139. 10.1083/jcb.200412001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yuva-Aydemir Y, Bauke AC, and Klambt C (2011). Spinster controls Dpp signaling during glial migration in the Drosophila eye. J Neurosci 31, 7005–7015. 10.1523/JNEUROSCI.0459-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Blaho VA, and Hla T (2014). An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res 55, 1596–1608. 10.1194/jlr.R046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bruck W, Sommermeier N, Bergmann M, Zettl U, Goebel HH, Kretzschmar HA, and Lassmann H (1996). Macrophages in multiple sclerosis. Immunobiology 195, 588–600. 10.1016/S0171-2985(96)80024-6. [DOI] [PubMed] [Google Scholar]
- 113.Chitnis T, and Weiner HL (2017). CNS inflammation and neurodegeneration. J Clin Invest 127, 3577–3587. 10.1172/JCI90609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kulakowska A, Zendzian-Piotrowska M, Baranowski M, Kononczuk T, Drozdowski W, Gorski J, and Bucki R (2010). Intrathecal increase of sphingosine 1-phosphate at early stage multiple sclerosis. Neurosci Lett 477, 149–152. 10.1016/j.neulet.2010.04.052. [DOI] [PubMed] [Google Scholar]
- 115.Deplanque D, Gele P, Petrault O, Six I, Furman C, Bouly M, Nion S, Dupuis B, Leys D, Fruchart JC, et al. (2003). Peroxisome proliferator-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J Neurosci 23, 6264–6271. 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Foster CA, Howard LM, Schweitzer A, Persohn E, Hiestand PC, Balatoni B, Reuschel R, Beerli C, Schwartz M, and Billich A (2007). Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther 323, 469–475. 10.1124/jpet.107.127183. [DOI] [PubMed] [Google Scholar]
- 117.Ibanez C, Simo C, Barupal DK, Fiehn O, Kivipelto M, Cedazo-Minguez A, and Cifuentes A (2013). A new metabolomic workflow for early detection of Alzheimer’s disease. J Chromatogr A 1302, 65–71. 10.1016/j.chroma.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 118.Baloni P, Arnold M, Moreno H, Nho K, Buitrago L, Huynh K, Brauner B, Louie G, Kueider-Paisley A, Suhre K, et al. (2021). Multi-Omic Analyses Characterize the Ceramide/Sphingomyelin Pathway as a Therapeutic Target in Alzheimer’s Disease. 2021.2007.2016.21260601. 10.1101/2021.07.16.21260601 %J medRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nakano Y, Fujitani K, Kurihara J, Ragan J, Usui-Aoki K, Shimoda L, Lukacsovich T, Suzuki K, Sezaki M, Sano Y, et al. (2001). Mutations in the novel membrane protein spinster interfere with programmed cell death and cause neural degeneration in Drosophila melanogaster. Mol Cell Biol 21, 3775–3788. 10.1128/MCB.21.11.3775-3788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sweeney ST, and Davis GW (2002). Unrestricted synaptic growth in spinster-a late endosomal protein implicated in TGF-beta-mediated synaptic growth regulation. Neuron 36, 403–416. 10.1016/s0896-6273(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 121.Kurucz E, Vaczi B, Markus R, Laurinyecz B, Vilmos P, Zsamboki J, Csorba K, Gateff E, Hultmark D, and Ando I (2007). Definition of Drosophila hemocyte subsets by cell-type specific antigens. Acta Biol Hung 58 Suppl, 95–111. 10.1556/ABiol.58.2007.Suppl.8. [DOI] [PubMed] [Google Scholar]
- 122.Diao F, Ironfield H, Luan H, Diao F, Shropshire WC, Ewer J, Marr E, Potter CJ, Landgraf M, and White BH (2015). Plug-and-play genetic access to drosophila cell types using exchangeable exon cassettes. Cell Rep 10, 1410–1421. 10.1016/j.celrep.2015.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yonamine I, Bamba T, Nirala NK, Jesmin N, Kosakowska-Cholody T, Nagashima K, Fukusaki E, Acharya JK, and Acharya U (2011). Sphingosine kinases and their metabolites modulate endolysosomal trafficking in photoreceptors. J Cell Biol 192, 557–567. 10.1083/jcb.201004098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tokusumi T, Tokusumi Y, Brahier MS, Lam V, Stoller-Conrad JR, Kroeger PT, and Schulz RA (2017). Screening and Analysis of Janelia FlyLight Project Enhancer-Gal4 Strains Identifies Multiple Gene Enhancers Active During Hematopoiesis in Normal and Wasp-Challenged Drosophila Larvae. G3 (Bethesda) 7, 437–448. 10.1534/g3.116.034439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gyoergy A, Roblek M, Ratheesh A, Valoskova K, Belyaeva V, Wachner S, Matsubayashi Y, Sanchez-Sanchez BJ, Stramer B, and Siekhaus DE (2018). Tools Allowing Independent Visualization and Genetic Manipulation of Drosophila melanogaster Macrophages and Surrounding Tissues. G3 (Bethesda) 8, 845–857. 10.1534/g3.117.300452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Oswald MC, West RJ, Lloyd-Evans E, and Sweeney ST (2015). Identification of dietary alanine toxicity and trafficking dysfunction in a Drosophila model of hereditary sensory and autonomic neuropathy type 1. Hum Mol Genet 24, 6899–6909. 10.1093/hmg/ddv390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Port F, Chen HM, Lee T, and Bullock SL (2014). Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc Natl Acad Sci U S A 111, E2967–2976. 10.1073/pnas.1405500111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kondo S, and Ueda R (2013). Highly improved gene targeting by germline-specific Cas9 expression in Drosophila. Genetics 195, 715–721. 10.1534/genetics.113.156737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Park YJ, Kim S, Shim HP, Park JH, Lee G, Kim TY, Jo MC, Kwon AY, Lee M, Lee S, et al. (2021). Phosphatidylserine synthase plays an essential role in glia and affects development, as well as the maintenance of neuronal function. iScience 24, 102899. 10.1016/j.isci.2021.102899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hu Y, Sopko R, Foos M, Kelley C, Flockhart I, Ammeux N, Wang X, Perkins L, Perrimon N, and Mohr SE (2013). FlyPrimerBank: an online database for Drosophila melanogaster gene expression analysis and knockdown evaluation of RNAi reagents. G3 (Bethesda) 3, 1607–1616. 10.1534/g3.113.007021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chung HL, Augustine GJ, and Choi KW (2016). Drosophila Schip1 Links Expanded and Tao-1 to Regulate Hippo Signaling. Dev Cell 36, 511–524. 10.1016/j.devcel.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 132.O’Neill EM, Rebay I, Tjian R, and Rubin GM (1994). The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell 78, 137–147. [DOI] [PubMed] [Google Scholar]
- 133.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tattikota SG, Cho B, Liu Y, Hu Y, Barrera V, Steinbaugh MJ, Yoon SH, Comjean A, Li F, Dervis F, et al. (2020). A single-cell survey of Drosophila blood. Elife 9. 10.7554/eLife.54818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Arima Y, Kamimura D, Atsumi T, Harada M, Kawamoto T, Nishikawa N, Stofkova A, Ohki T, Higuchi K, Morimoto Y, et al. (2015). A pain-mediated neural signal induces relapse in murine autoimmune encephalomyelitis, a multiple sclerosis model. Elife 4. 10.7554/eLife.08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bittner S, Afzali AM, Wiendl H, and Meuth SG (2014). Myelin oligodendrocyte glycoprotein (MOG35–55) induced experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice. J Vis Exp. 10.3791/51275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McCarthy DP, Richards MH, and Miller SD (2012). Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods Mol Biol 900, 381–401. 10.1007/978-1-60761-720-4_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Stromnes IM, and Goverman JM (2006). Active induction of experimental allergic encephalomyelitis. Nat Protoc 1, 1810–1819. 10.1038/nprot.2006.285. [DOI] [PubMed] [Google Scholar]
- 139.Jo J, Woo J, Cristobal CD, Choi JM, Wang CY, Ye Q, Smith JA, Ung K, Liu G, Cortes D, et al. (2021). Regional heterogeneity of astrocyte morphogenesis dictated by the formin protein, Daam2, modifies circuit function. EMBO Rep 22, e53200. 10.15252/embr.202153200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ding X, Jo J, Wang CY, Cristobal CD, Zuo Z, Ye Q, Wirianto M, Lindeke-Myers A, Choi JM, Mohila CA, et al. (2020). The Daam2-VHL-Nedd4 axis governs developmental and regenerative oligodendrocyte differentiation. Genes Dev 34, 1177–1189. 10.1101/gad.338046.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data points used to create the graphs can be found in Data S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.