

Abstract
Although cardiac abnormalities have been observed in a growing class of human disorders caused by defective primary cilia, the function of cilia in the heart remains an underexplored area. The primary function of cilia in the heart was long thought to be restricted to left–right axis patterning during embryogenesis. However, new findings have revealed broad roles for cilia in congenital heart disease, valvulogenesis, myocardial fibrosis and regeneration, and mechanosensation. In this Review, we describe advances in our understanding of the mechanisms by which cilia function contributes to cardiac left–right axis development and discuss the latest findings that highlight a broader role for cilia in cardiac development. Specifically, we examine the growing line of evidence connecting cilia function to the pathogenesis of congenital heart disease. Furthermore, we also highlight research from the past 10 years demonstrating the role of cilia function in common cardiac valve disorders, including mitral valve prolapse and aortic valve disease, and describe findings that implicate cardiac cilia in mechanosensation potentially linking haemodynamic and contractile forces with genetic regulation of cardiac development and function. Finally, given the presence of cilia on cardiac fibroblasts, we also explore the potential role of cilia in fibrotic growth and summarize the evidence implicating cardiac cilia in heart regeneration.
Cilia are hair-like, microtubule-based cellular organelles of several micrometres in length that protrude from the surface of most animal cells, from protozoa to humans1. First described >300 years ago by van Leeuwenhoek2 and documented by Zimmerman3, cilia were initially believed to function solely for cellular propulsion. However, research from the past two decades has highlighted the central role of cilia in orchestrating numerous cellular sensory and signalling events as well as in the development of numerous diseases4–9. Of note, reports of developmental defects in the heart associated with ciliopathies have emerged in the past 10 years10–18, but a consensus on the role of cilia in cardiogenesis has not yet been reached. In this Review, we summarize the mechanisms by which cilia contribute to cardiac left–right axis development, identify a role for cilia in congenital heart disease (CHD) and common cardiac valve diseases, explore the relationship between cardiac cilia and mechanosensation, which potentially links haemodynamic and contractile forces with genetic regulation of cardiac development and function, discuss the evidence linking cilia and fibrotic growth, and explore the potential implications of cardiac cilia in heart regeneration. Together, these findings have advanced our understanding of the role of cilia in cardiac development and function, can help delineate the regulatory mechanisms guiding heart development, and provide important insights into disease pathogenesis, prognostic indicators and novel therapeutic targets to treat CHD.
Cilia.
Membrane-bound, microtubule-based, antenna-like sensory organelles that project from the surface of most animal cells to coordinate numerous signalling pathways during development and in tissue homeostasis.
Congenital heart disease.
(CHD). A disease characterized by a structural abnormality of the heart that is not an acquired condition.
Cilia organization and signalling
Cilia consist of a membrane-bound axoneme containing nine circumferentially arranged doublet microtubules that are composed of A and B subfibres1 (FIG. 1). These subfibres are held together by nexin links, which extend upwards from the basal body and out into the extracellular space. All types of cilia rely on intraflagellar transport for assembly, maintenance and signalling. Intraflagellar transport involves a specialized bidirectional trafficking system that moves axonemal precursors and cilia membrane proteins into and out of the cilium19,20. Cilia are traditionally classified into motile, nodal or primary types that follow a 9 + 0 or 9 + 2 ultra-structural arrangement of axonemal microtubules (FIG. 1), which are observable with the use of transmission electron microscopy. Motile cilia typically beat in a planar motion and have two additional central microtubules connected to each of the surrounding doublets by radial spokes that are absent in 9 + 0 axonemes21. In addition, 9 + 2 motile cilia contain accessory structures used for motility such as outer and inner dynein arms and central pair projections. Cilia that have a typical 9 + 0 axonemal arrangement usually do not have the outer and inner dynein arms and other accessory structures required for motility and are thus often immotile. These 9 + 0 cilia are frequently referred to as primary cilia and are widely distributed among tissues in vertebrates. In most cases, there is only one primary cilium per cell and it arises from the mother centriole. Although 9 + 2 structures are commonly associated with motile cilia in the setting of multiciliated cells and 9 + 0 structures are associated with immotile cilia, many exceptions to this observation exist22–27. Notably, motile cilia in the left–right organizer (LRO; also known as the ventral node in mice, Hensen’s node in chickens, Kupffer’s vesicle in zebrafish and the gastrocoel roof plate in Xenopus28–30), an embryonic structure that determines left–right body axis asymmetry, are mostly 9 + 0 in ultrastructure and have a circular beating motion rather than a planar motion31–33. Electron tomography studies have further challenged this canonical definition of ciliary subtype by demonstrating that primary cilia have the established 9 + 0 axonemal microtubule arrangement only at their proximal base, with fewer microtubule doublets near the distal tip34–37. These reports underscore the fact that the ultrastructure of cilia is far more complex than previously thought.
Fig. 1 |. Diversity of ciliary structure.
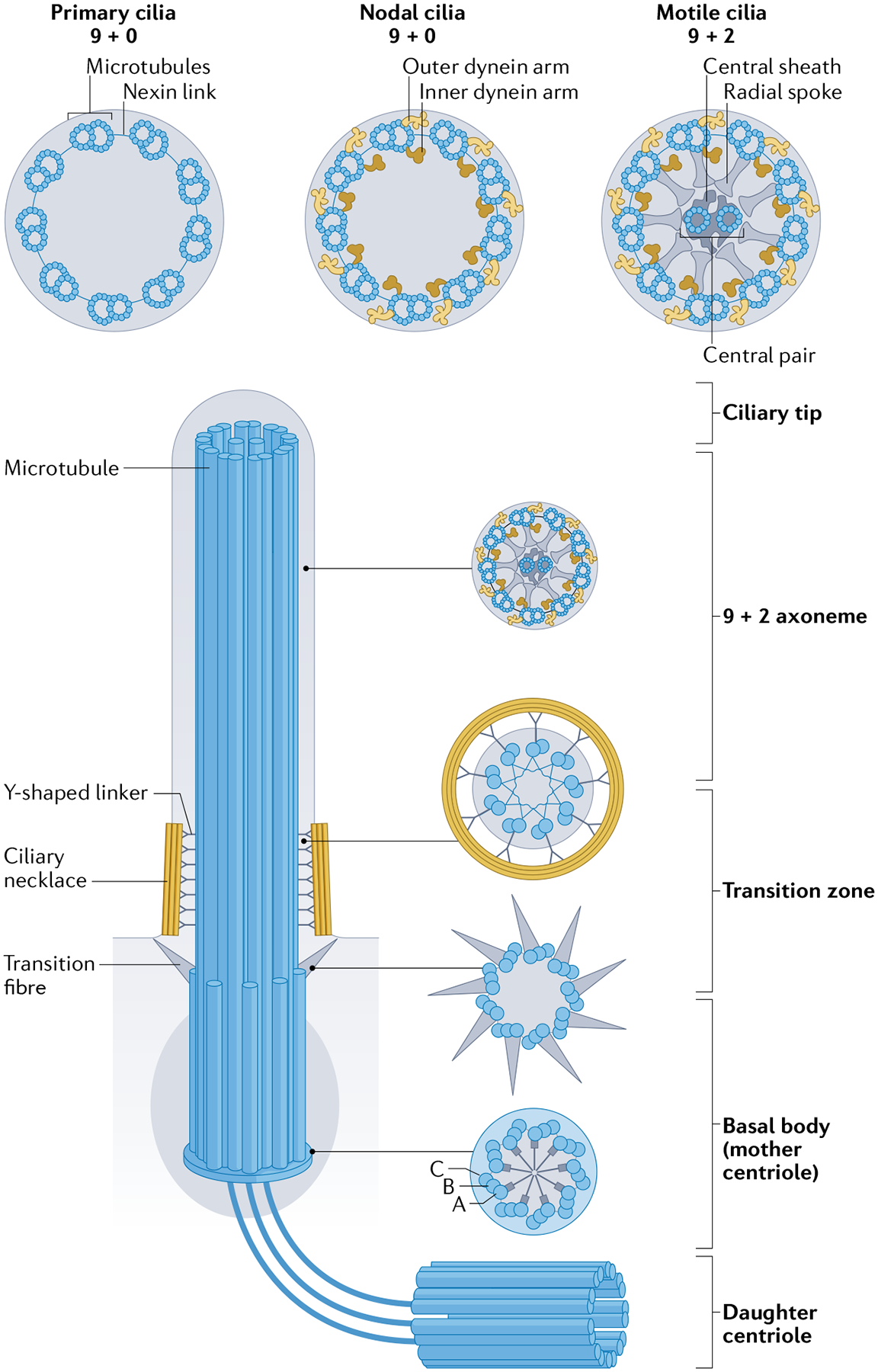
A cilium is composed of multiple pairs of microtubules (axoneme) running from the basal body. The axoneme is the structural core of a cilium. The microtubule doublets, containing an A-tubule (A) and a B-tubule (B), are continuous with the microtubules at the transition zone. The doublets are connected by nexin links and are held in place by radial spokes that extend into the axonemal centre. Depending on the type of cilium, the cilium backbone can have 9 + 0 or 9 + 2 axoneme configurations. In motile cilia (9 + 2 configuration except at the left–right organizer), inner and outer dynein arms are connected to the A-tubules and mediate cilia bending by reversibly binding to the neighbouring B-tubule. Most 9 + 0 cilia (primary cilia) do not possess inner and outer dynein arms, radial spokes or central pairs. Some 9 + 0 cilia lack the central microtubules only and are motile (nodal cilia). The basal body of each cilium is a specialized centriole that anchors the axoneme to the cell. The basal body is composed of nine triplets of microtubules (9 × 3 microtubular structure), containing an A-tubule, a B-tubule and a C-tubule (C), rooted in pericentriolar material with a proximal amorphous disc, a cartwheel structure, a middle portion that lacks appendages and transition fibres at the distal end. Proximal transition fibres connect each microtubule doublet (without dynein arms) to the membrane. The transition zone functions as a regulated gate that controls the entry and exit of ciliary proteins into the cilium. The transition zone converts the 9 × 3 microtubular structure of the basal body into the axonemal doublet structure. The distal part contains stellate fibre arrays and an amorphous disc structure and gives rise to the central microtubules in 9 + 2 axonemes.
Left–right organizer.
(LRO). Evolutionarily conserved embryonic ciliated organ of laterality in which breaking of left–right asymmetry occurs.
Primary cilia are involved in the maintenance of tissue homeostasis by regulating signalling pathways and sensing biophysical and biochemical changes in the extracellular environment. For example, primary cilia can act as flow sensors and chemosensors that respond to a plethora of extracellular signals38,39. During development and in the adult organism, primary cilia regulate multiple signalling pathways39,40, including Hedgehog41,42, WNT43,44, transforming growth factor-β (TGFβ)45 and Notch46,47 (FIG. 2a). Critically, many components of these signalling cascades localize to or near the cilium, including core Hedgehog factors such as Patched, Smoothened and glioma-associated oncogene homologue (GLI)41,42,48,49. Furthermore, primary cilia are specialized calcium signalling compartments, given that intraciliary calcium signalling is distinct from extraciliary calcium signalling and that numerous calcium channels and calcium-binding proteins localize to the cilium50–55. Of note, signals from distinct ciliary channels can be spatially distinguishable from one another within the cilium because some of these channels and binding partners occupy unique microdomains in the organelle such as the inversin compartment56,57 and linear arrays formed by polycystin 2 (PKD2; a mechanosensitive transient receptor potential channel)58. Indeed, the cilium’s compartmentalization, strategic positioning on the cell surface and unique geometry with a large surface-area-to-volume ratio are all believed to have facilitated its adoption by vertebrate cells as a master signal transduction centre. The structural integrity and performance of the cilium is critical for its function given that defects in cilia contribute to a wide array of multiorgan conditions such as airway dysfunction, anosmia, cancer, cognitive disorders, laterality defects, polycystic kidney disease, retinal degeneration, obesity and sterility as well as hepatic, pancreatic, renal and skeletal defects4–9,50,59,60 (FIG. 2b).
Fig. 2 |. Signalling pathways involved in heart development and organs involved in ciliopathies.
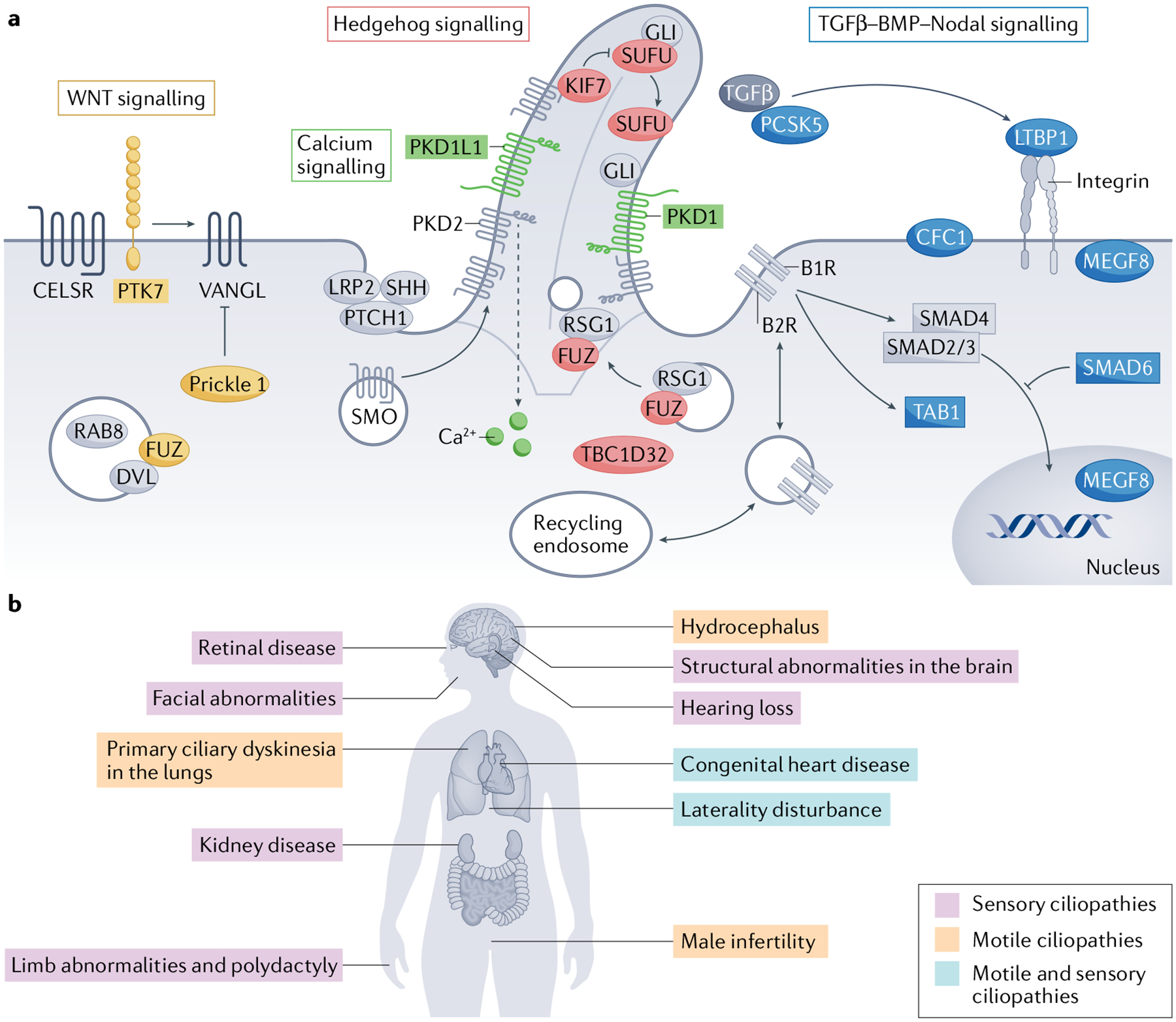
a | Diagram of ciliary signalling pathways implicated in cardiac development. The cilium is a master signal transduction centre that orchestrates numerous signalling pathways involved in heart morphogenesis. Members of these pathways identified from screens in animal models and in patients with congenital heart disease are highlighted: WNT signalling pathway proteins are shown in yellow, calcium signalling proteins are shown in green, Hedgehog signalling proteins are shown in red and the proteins in the transforming growth factor-β (TGFβ)–BMP–Nodal pathways are shown in blue. b | Diagram of organ system involvement in ciliopathies. Because motile and immotile cilia are found throughout most cells in the human body, cilia defects result in pleiotropic disorders that affect numerous organ systems such as the heart, lungs, kidney, brain, eyes, ears, craniofacial system, limbs and reproductive system. Motile ciliopathies are shown in pink, sensory ciliopathies in purple and phenotypes attributed to both are shown in blue. GLI, glioma-associated oncogene homologue; PKD, polycystin. Part a adapted from REF.237, CC BY 4.0 https://creativecommons.org/licenses/by/4.0/. Parts c, e, f, g, h, i adapted with permission from REF.53, Elsevier.
Left–right patterning of the heart
Initiation of cardiac development.
During vertebrate embryogenesis, the early symmetrical heart tube undergoes a dynamic looping stage that results in left–right asymmetry of the developing heart. The first stages of cardiac left–right development are initiated in a transient, evolutionarily conserved, ciliated epithelium known as the LRO. Within the LRO, which is formed after development of the dorsoventral and anteroposterior axes61,62, both motile and primary cilia have a critical role in establishing the left–right asymmetry of the body axis and proper placement and patterning of the internal organs, including heart looping32,63–65. Impaired LRO ciliary signalling results in major left–right patterning defects31,66–68. Motile cilia in the LRO are oriented in a polarized fashion and rotate synchronously in a clockwise manner to produce a leftward fluid flow in the extracellular space (FIG. 3a–c). This leftward flow is essential for defining left–right asymmetry31,66–70, as shown by the demonstration that artificially applied flow to mouse embryos in vitro can determine left–right patterning68. Although approximately 300 cilia are present in the mouse LRO as few as two motile LRO cilia are enough to break the bilateral symmetry in the mouse embryo62. Accordingly, optical mapping of flow in the LRO of mouse71, zebrafish72 and Xenopus73 embryos revealed that local regional areas of flow in the LRO, rather than across the entire structure, are critical for left–right development. Indeed, LRO fluid flow is sensed by primary cilia on the cells located solely along the peripheral edges of the LRO, known as crown cells74.
Fig. 3 |. Model for intraciliary calcium signalling in left–right patterning.
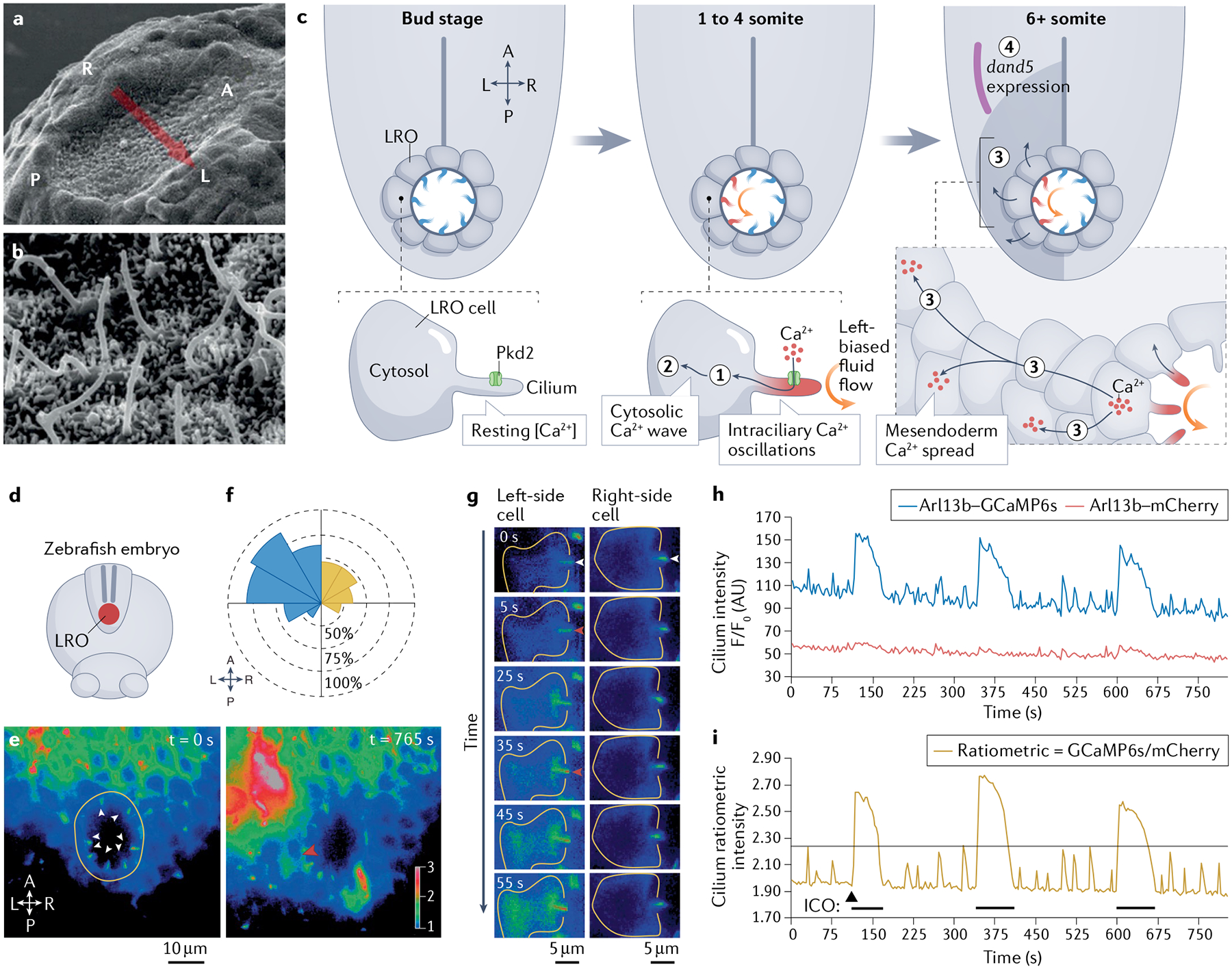
a,b | Scanning electron microscopy images of left–right organizer (LRO) cilia at the embryonic mouse LRO at 8 days post-coitum. c | Model for intraciliary calcium signalling during left–right development in the zebrafish embryo LRO. At the bud stage (~10 hours post-fertilization (hpf)), cilia (blue) in the LRO are primarily immotile and bilateral symmetry is maintained. At the 1 to 4 somite stage (~11 hpf), cilia motility initiates and drives left-biased fluid flow (orange), triggering Pkd2-dependent (green) intraciliary calcium oscillations (ICOs; red) along the left side of the LRO (1). At the 6+ somite stage (12 hpf), ciliary-to-cytosolic Ca2+ waves (2) spread to neighbouring cells to establish leftward-biased stable elevations in calcium around the mesendoderm (3), which in turn direct asymmetrical gene expression (such as dand5, shown in purple) to establish left–right patterning (4). d–i | Calcium imaging of the zebrafish LRO reveals ICOs that are mainly located on the left side of the LRO. e | Representative live images of the LRO zebrafish embryo at the 1-somite stage (~10 hpf) expressing ratiometric genetically encoded calcium indicators (arl13b–GCaMP6s/arl13b–mCherry). The ratiometric signal is shown and false-coloured with a rainbow intensometric scale. White arrowheads indicate LRO cilia at resting stage (left panel), red arrowhead highlights an ICO inducing a cytosolic calcium wave (right panel). f | Rose diagram depicting the spatial distribution and mean percentage of cells per embryo displaying ICOs in the total LRO of embryos (n = 10) spanning the entire course of LRO development (bud to 16-somite stage: ~10–17 hpf). Left-sided waves are shown in blue, right-sided waves are shown in yellow. g | Representative montage of ratiometric fluorescent images of a single LRO cell from the left side or right side of the LRO over 55 seconds of total elapsed time. White arrowheads indicate cilia at resting state and red arrowheads indicate the onset of ICO. h | Two-colour fluorescent intensity (F/F0) over time (seconds) plot from a single representative LRO cilium (left side) co-expressing arl13b–GCaMP6s and arl13b–mCherry at the 1-somite stage. Multiple ICOs are visible in the Arl13b–GCaMP6s channel (blue trace), whereas the Arl13b–mCherry signal (pink trace) is relatively quiescent. i | Ratiometric intensity (Arl13b–GCaMP6s/Arl13b–mCherry; gold trace) over time (seconds) plot. The arrowhead and black bars indicate the start of multiple ICOs. The ratiometric signal was thresholded (indicated by the grey horizontal line) to facilitate the analysis of calcium oscillation dynamics. A, anterior; L, left; P, posterior; R, right. Parts a and b reprinted from REF.238, Springer Nature Limited. Parts c and e–i modified with permission from REF.53.
The precise mechanism by which LRO flow is detected is uncertain as is the function of cilia in this process and how flow regulates Nodal signalling to establish left–right asymmetry. Several models have been proposed63,64. The ‘morphogen flow’ hypothesis suggests that LRO flow creates an asymmetrical distribution of short-lived molecules across the LRO, resulting in the accumulation of these molecules in embryonic fluid flow at the left side of the LRO31,66. A variation of this morphogen flow model proposes that these soluble molecules are encapsulated in membrane-bound vesicles (also known as nodal vesicular parcels (NVPs)) that are delivered to the left side of the LRO61,75. Although numerous studies have reported on the presence of ciliary vesicles reminiscent of NVPs in Caenorhabditis elegans, green algae and cultured cells from mice, whether these ciliary vesicles are present in the LRO and are functionally equivalent to NVPs remain uncertain76–80. An alternative model, which has become known as the ‘two-cilia’ model32,65, proposes that LRO leftward flow driven by motile cilia initiates a calcium influx in cells on the left side of the LRO via activation of the polycystin ion channels in primary cilia that function as shear flow mechanosensors32,53,54,74,81 (FIG. 3c). Indeed, calcium signalling has a critical role in left–right determination, given that one of the earliest molecular events in establishing asymmetry is an increase in cytoplasmic calcium in the left-sided cells of the LRO32,53,81 and that the cation channel PKD2 is critical for proper left–right patterning of the embryo74,82. The contribution of calcium signalling to left–right determination will be addressed in more detail below. Given the importance of proper left–right patterning for embryonic development, several mechanisms are likely to occur in concert to guarantee the integrity of this process.
After the detection of LRO flow by primary cilia, left-sided cytoplasmic mesodermal calcium transients are transmitted to surrounding tissues to induce the asymmetrical expression of the left-side determinant Nodal and its feedback inhibitor Lefty. Nodal and Lefty are highly conserved among vertebrates and some non-vertebrates and constitute a self-enhancement and lateral-inhibition system that provides robustness to the left–right determination process83–85. Asymmetrical Nodal signalling induces left-sided expression of other genes, such as Pitx2 (REF.86), which in turn induce asymmetrical morphogenesis of most visceral organs, including the heart, lungs and kidneys87.
Calcium signalling and cardiac situs.
A growing line of evidence has highlighted the critical role of calcium signalling in left–right patterning. For more than a decade, the earliest observable molecular event in the process of left–right asymmetry was an increase in cytoplasmic calcium on the left-sided mesoderm of the LRO32. However, several studies have demonstrated that before these left-sided cytoplasmic calcium events take place, intraciliary calcium oscillations occur in the left side of the LRO and initiate vertebrate left–right asymmetry53,54 (FIG. 3d–i). Intraciliary calcium oscillations were initially only observed in the zebrafish LRO but not in the mouse LRO53,88; however, a subsequent study detected intraciliary calcium oscillations in mouse embryos that were strikingly similar to those observed in zebrafish embryos54. These left-sided intraciliary calcium oscillations resist treatment by thapsigargin (a blocker of the sarco/endoplasmic reticulum calcium ATPase channel that depletes cytosolic calcium stores), thereby demonstrating that intraciliary calcium oscillations are generated independently of cytosolic calcium. These intraciliary calcium oscillations also depend on the presence of LRO flow and PKD2 in both mice54 and zebrafish53, occurring in both species at the time point and location at which bilateral symmetry is first broken. Of note, these intraciliary calcium oscillations are the earliest known left–right asymmetrical molecular signal.
Differences in the study protocols could account for the discrepancies between the findings on intraciliary calcium oscillations reported in published studies, in particular the use of different culture media. Indeed, in the study by Mizuno et al., mouse embryos were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 75% rat serum54, whereas Delling et al. analysed mouse embryos that were cultured in DMEM plus F-12 medium supplemented with 10% fetal bovine serum88. Data demonstrating that mouse embryos require culture in medium containing 50–75% rat serum for proper establishment and maintenance of left–right asymmetry54,89 might explain the failure to detect intraciliary calcium oscillations in embryos cultured without rat serum88. Another discrepancy in study protocol that might influence the capacity to detect intraciliary calcium oscillations relates to differences between the genetically encoded calcium indicators (GECIs; particularly in the dissociation constant (Kd) for calcium) used in various studies38,53,88,90–92. Our group53 and Mizuno et al.54 used the GCaMP6s and GCaMP6 sensors (~144 nM (REF.90) and ~158 nM (REF.91), respectively) in their studies, whereas Delling et al. used G-GECO1.2 (450–1,100 nM (REF.92))88. If LRO cilia contain a low concentration of calcium, the choice of GECIs and the Kd might be instrumental in dictating its capacity for detecting intraciliary calcium oscillations. Another potential factor that can account for discrepant results between studies is variation in the design of GECIs37,38,88,93,94. Our group co-expressed arl13b–GCaMP6s and arl13b–mCherry53, while Mizuno et al. conjugated GCaMP6 to mCherry54 using a p2A peptide to allow post-translational cleavage93; both reporters were therefore expressed as a ratiometric system consisting of two separate proteins (with mCherry used as an internal control to correct for motion arte-fact). However, Delling et al. used G-GECO1.2 and mCherry expressed as a direct fusion protein88, which might hamper the GECI via Förster resonance energy transfer94 and steric inhibition between the fluorophores.
Although further research is required to understand the downstream molecular targets of intraciliary calcium oscillations, the likeliest ultimate target is Dand5 mRNA (also called Cerl2 in mouse, charon in zebrafish and Coco in Xenopus)95. Dand5 encodes a Nodal antagonist and is the earliest known asymmetrically expressed gene involved in left–right development96,97. Intraciliary calcium oscillations are likely to lead to the entry of calcium in the cell bodies on the left side of the LRO, triggering inositol triphosphate-receptor-dependent release of calcium from the endoplasmic reticulum, which induces the degradation of Dand5 mRNA along the left side of the LRO54,74,81 (FIG. 3c). Given that decay of Dand5 mRNA is post-transcriptionally regulated via its 3′ untranslated region98, microRNAs or microRNA-related proteins might be involved in left–right patterning99. Once Nodal signalling inhibition is lifted on the left side of the lateral plate mesoderm, left-sided expression of downstream target genes can be induced, which ultimately leads to asymmetrical morphogenesis of most visceral organs100.
Overall, this model strongly suggests that LRO cells respond to calcium signals. This concept is reinforced by the observation that many calcium signalling proteins are expressed in LRO cells and are required for proper left–right patterning such as calmodulin-dependent kinase II, which senses differential frequency of calcium spikes101, or inversin, a ciliary protein that has calmodulin-binding domains102. Nonetheless, the mechanism linking intraciliary calcium oscillations and subsequent cytoplasmic transients to Dand5 mRNA degradation remains to be determined.
Impaired LRO ciliary signalling in CHD.
As described above, cilia are essential for the development of vertebrate left–right asymmetry. Failure to establish normal left–right patterning results in severe CHD13,14,18. Numerous studies have implicated impaired signalling in cilia as one of the mechanisms underlying CHD pathogenesis10–18,103. Defects in cilia are also known to cause heterotaxy, a syndrome characterized by abnormal left–right asymmetrical body patterning that commonly affects the heart and is tightly correlated with a broad spectrum of CHDs104–106 (FIG. 4), including most known CHD subtypes, but especially in atrioventricular septal defects (AVSDs) and transposition of the great arteries104–106 (FIG. 4b). The incidence of heterotaxy is greatly increased in patients with cilia motility defects compared with the general population (6.3% versus 0.004%)104,106. Mutations affecting cilia structure and function have been identified in patients with heterotaxy and other congenital diseases107,108 (FIGS 4,5; TABLE 1). Indeed, the incidence of CHD is higher in patients with heterotaxy than in the general population (57% versus 1%), with AVSDs and transposition of the great arteries being the most prevalent CHD subtypes104 (FIG. 4b).
Fig. 4 |. Cilia defects and left–right patterning impairment leading to heterotaxy and congenital heart disease.
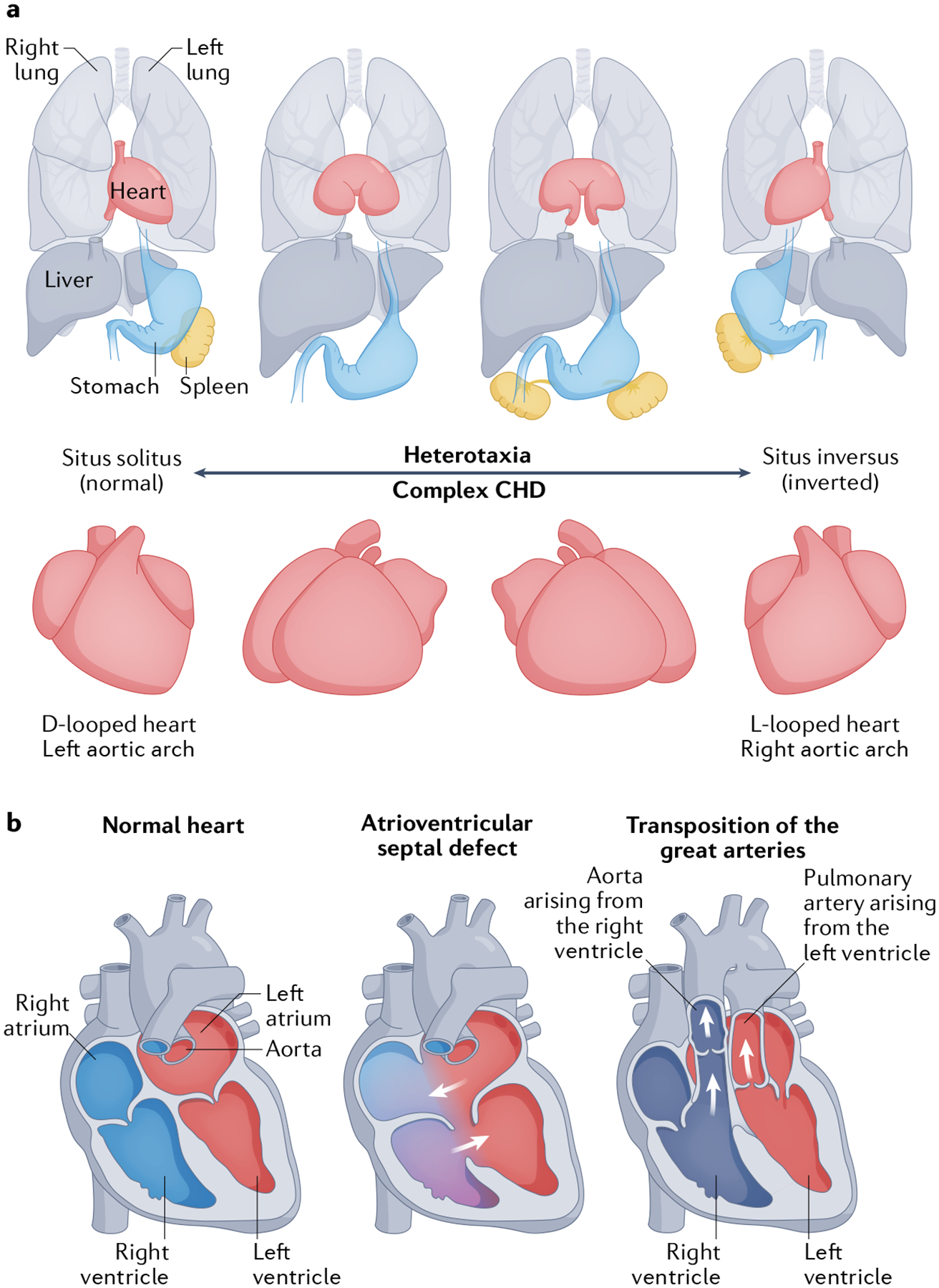
a | Anatomical spectrum of organ and heart laterality associated with heterotaxy and congenital heart disease (CHD). b | Atrioventricular septal defects and transposition of the great arteries are CHDs associated with cilia and laterality defects. Oxygenated blood is shown in red, deoxygenated blood is shown in blue, and a mixture of oxygenated and deoxygenated blood is shown in purple.
Fig. 5 |. Cilia-related genes linked to human congenital heart disease.
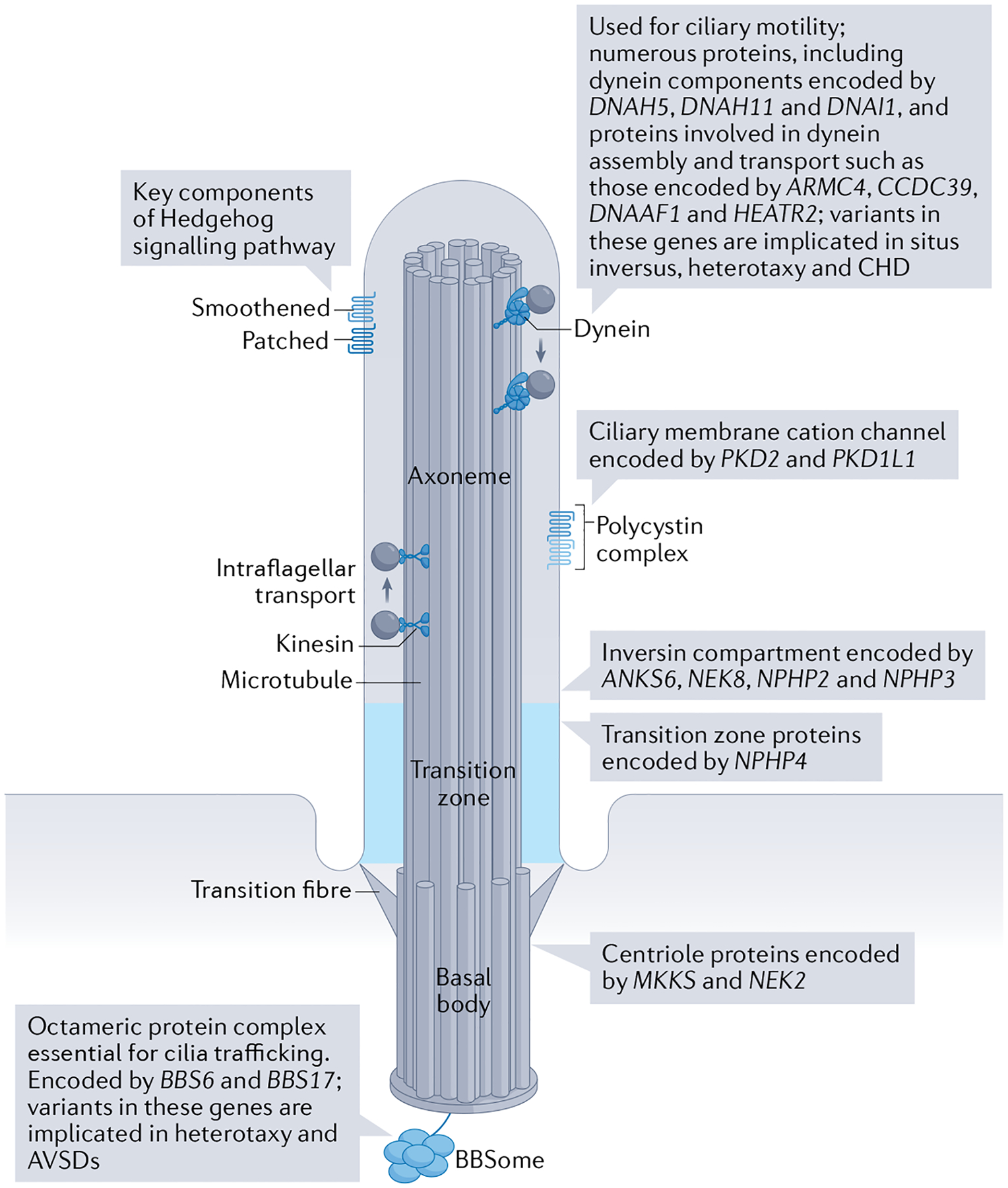
Schematic of a cilium and related components that have been associated with the development of congenital heart disease (CHD). Heterotaxy and a range of other CHDs have been associated with alterations of most ciliary components. Dynein and dynein assembly genes are required for ciliary motility, and mutations in these genes are more frequently associated with situs inversus. Mutations in genes required for Hedgehog signalling are more frequently associated with atrioventricular canal defects. See TABLE 1 for a full list of cilia-related genes linked to CHD, the ciliary function or compartment they encode, and the associated heart diseases. AVSD, atrioventricular septal defect.
Table 1 |.
Cilia-related genes linked to human congenital heart disease
| Cilia component | Gene | Molecular function | Cardiac phenotype | Refs |
|---|---|---|---|---|
| Cilia motility | DNAI1 | Dynein intermediate chain | SI, HIX, CHD | 104,193–195 |
| DNAAF3 | Dynein assembly | SI | 196 | |
| DNAH5 | Dynein outer arm | SI, HIX, CHD | 104,197 | |
| TXNDC3 | Dynein outer arm | SI, HIX | 198 | |
| DNAH11 | Dynein outer arm | SI, HIX, CHD | 121,199,200 | |
| DNAAF2 | Dynein assembly factor | SI | 201 | |
| CCDC39 | Dynein assembly | SI, HIX | 202 | |
| CCDC40 | Dynein assembly | SI | 203 | |
| DNAL1 | Dynein light chain | SI | 204 | |
| CCDC103 | Dynein assembly | SI, HIX | 205 | |
| HEATR2 | Dynein transport and assembly | SI, HIX, CHD | 206 | |
| LRRC6 | Dynein transcription | SI | 207 | |
| CCDC114 | Dynein docking | SI, HIX, CHD | 208 | |
| ZMYND10 | Dynein transcription | SI | 209,210 | |
| ARMC4 | Dynein assembly | SI | 211 | |
| DYX1C1 | Dynein assembly | SI, HIX, CHD | 212 | |
| C21ORF59 | Dynein assembly | SI, HIX | 213 | |
| SPAG1 | Dynein transport and assembly | SI | 214 | |
| CCDC151 | Dynein assembly | SI, HIX, CHD | 215 | |
| PIH1D3 | Dynein assembly | SI | 216 | |
| DNAAF1 | Dynein assembly | SI | 217–219 | |
| CCDC11 | CentrioCe | SI, HIX | 220 | |
| DNAH6 | Dynein inner arm | HIX | 221 | |
| TTC25 | Outer arm docking | SI | 222 | |
| PoCycystin complex | PKD2 | Ciliary membrane cation channel | SI | 223 |
| PKD1L1 | Ciliary membrane cation channel | HIX | 224 | |
| Transition zone or inversin compartment | NPHP2 | Inversin compartment | SI, IGA | 130,225,226 |
| NPHP3 | Inversin compartment | VSD, HIX | 227,228 | |
| ANKS6 | Inversin compartment | SI, CHD | 229 | |
| NPHP4 | Transition zone | CHD, HIX | 129 | |
| NEK8 | Inversin compartment | CHD, HIX | 195,230 | |
| Centriole | MKKS | Centriole | CHD | 231 |
| NEK2 | Centriole | HIX | 11 | |
| BBSome | BBS6 | Cilia trafficking | HIX, AVC | 124 |
| BBS17 | Cilia trafficking | HIX, AVC | 123 | |
| LRO formation | GDF1 | LRO formation | RAI, CHD | 15,232 |
| GALNT11 | LRO formation | HIX | 11,233 | |
| Others | EVC | Hedgehog signalling | AVC | 234 |
| EVC2 | Hedgehog signalling | AVC | 234 | |
| MKS1 | Hedgehog signalling | AVC | 116 | |
| DCHS1 | Not reported, possibly near ciliary base | Mitral prolapse | 235 | |
| NUP188 | Nuclear or ciliary pore | CHD | 11,236 | |
| RNF20 | Transcriptional regulation | CHD, HIX | 108 |
AVC, atrioventricular canal; CHD, congenital heart disease; HTX, heterotaxy; LRO, left–right organizer; SI, situs inversus; RAI, right atrial isomerism; TGA, transposition of the great arteries; VSD, ventricular septal defect.
Heterotaxy.
(HTX). Any arrangement of the organs across the left–right axis differing from situs solitus and situs inversus.
Atrioventricular septal defects.
(AVsDs). Conditions characterized by improper atrial and ventricular septa and adjoining valve development.
Transposition of the great arteries.
(TgA). A congenital heart defect characterized by a switch in the position of the pulmonary artery and the aorta.
Cilia and cardiac morphogenesis.
The mechanisms involved in the translation of global embryonic asymmetry to asymmetrical cardiac morphogenesis remain unclear. Cells at the LRO do not directly contribute to the heart; cilia at the LRO break bilateral symmetry and provide the initial asymmetrical molecular signal, which is propagated through the lateral plate mesoderm to cardiac primordia84–86. The signal is initiated by left-sided degradation of Dand5, which then leads to amplification of Nodal signalling on the left side of the embryo, culminating in the expression of transcription factor Pitx2 in the anterior lateral plate mesoderm and the primitive left atrium85. The most pronounced cardiac asymmetry is the direction of the heart loop but striking asymmetrical development can also be observed in the venous and arterial vessels, atrial primordia and cardiac valves. The shape and direction of the heart loop have been suggested to result from a combination of ‘buckling’ of the heart tube as it elongates while its endpoints are fixed and amplification of small left–right asymmetries programmed into the poles of the heart by cilia-generated signals arising from the LRO109. In chick embryos, cardiac cells have inherent chirality that can be oriented by the NODAL protein, providing a potential mechanism linking LRO signalling to cardiac cells110.
Laterality phenotypes can be classified as situs solitus (normal orientation), situs inversus (mirror image orientation of all organs) and heterotaxy (any orientation of organs across the left–right axis diverging from situs solitus or situs inversus) (FIG. 4a). The most severe laterality phenotypes are right and left isomerism resulting from complete failure to break bilateral symmetry111. How cilia and LRO asymmetry can affect the final cardiac phenotype is not known but certain indicators can help predict the resulting CHD. When the LRO signal is ‘random’ owing to absent ciliary motility but preserved ciliary sensing, cardiac loop direction and global organ laterality are concordant and random, resulting in the most common phenotype being either situs solitus or situs inversus and normal intracardiac anatomy59. Lack of control over loop direction is hypothesized to result in some loops that are abnormally shaped and interfere with normal intracardiac interactions, leading to structural heart disease, which has been seen in mice and patients harbouring mutations that affect cilia motility104,112. Cardiac morphogenesis is most notably affected by mutations that alter ciliogenesis and signalling. These mutations can lead to bilateral right-sided molecular signatures, such as that observed in the absence of PKD2-mediated cilia sensing113, or bilateral left-sided molecular signatures resulting from loss of TMEM107 (transmembrane protein 107)114, which is a component of the cilia transition zone that is required for regulation of ciliary protein composition. Of note, cilia are found in the developing heart, and ciliary sensing has a role in cardiac development, including in valve development (discussed below). The presence of defective intracardiac cilia is predictive of more complex CHD owing to defective heart loop formation that results from abnormal left–right asymmetry in addition to aberrations in subsequent heart development115,116. Although CHD that is linked to abnormal laterality is complex (FIG. 4b), left isomerism is thought to be more frequently associated with obstruction to systemic outflow and right isomerism with obstruction to pulmonary outflow and pulmonary venous return117. Furthermore, Hedgehog signalling (which requires cilia for normal function) at the dorsal mesenchymal protrusion is necessary for cardiac septation118,119. Even pulmonary venous return might be directed by cilia function, because mice with a conditional mutation in Ift88 (encoding an intraflagellar transport component) had total anomalous pulmonary venous drainage, a form of CHD103. Given that cilia function is required for directing heart asymmetry, looping, cardiac septation and valve development, complex forms of AVSD are a hallmark of ciliopathy and laterality-associated CHD120.
Together, these studies show that mutations affecting the structure and function of LRO cilia can lead to left–right asymmetry defects associated with CHD. Given that these mutations might also have an effect on the assembly and function of primary cilia formed within the developing heart, establishing whether a heart defect results from impaired LRO cilia signalling, defective cardiac primary cilia or both remains a challenge.
Variants affecting cilia gene function in CHD
Defective cilia have been linked to a broad range of CHDs with and without abnormal laterality in both mice and humans15,18 (FIGS 4b,5). Targeted sequencing of cilia-related genes in patients with CHD in association with other features of ciliopathies has led to the identification of variants affecting genes required for cilia biogenesis, signalling and motility, all of which contribute to CHD. The link between defective cilia motility and CHD has been well established. In this setting, the resulting CHD is most often laterality associated but also includes many other CHD subtypes such as tetralogy of Fallot, AVSDs and single ventricle defects15. The variable presentation of the congenital heart defect resulting from mutation of a single cilia dynein gene was first observed in mice homozygous for a mutation in Dnah11 (REF.112). Subsequently, targeted sequencing of 37 genes linked to primary cilia dyskinesia in 42 patients with CHD associated with heterotaxy syndrome resulted in the identification of possible disease-causing heterozygous variants in 45% of patients, with the DNAH11 gene accounting for 14 of 84 alleles121. Abnormal thoracoabdominal situs and cardiac looping with or without intracardiac defects are associated with mutations that affect most of the components and processes required for cilia motility, including dynein inner and outer arms, dynein assembly, dynein docking and regulation of dynein transcription104 (FIGS 1,5). Interestingly, defects in the radial spoke proteins connecting the central pair microtubules to the peripheral A microtubules are associated with respiratory cilia dysmotility but not CHD, which suggests that radial spokes and the central pair microtubules do not have a fundamental role in cilia motility at the LRO122.
In addition to cilia motility, ciliary sensing is also required for normal heart development, and mutations affecting primary immotile cilia can likewise contribute to CHD115,116. The BBSome is an octameric complex required for the trafficking of cilia membrane proteins123–125. Variants in genes encoding BBSome components are responsible for Bardet–Biedl syndrome and are also associated with CHD, especially AVSDs124,125. The transition zone and inversin compartment are the proximal components of the cilia axoneme involved in the regulation of cilia signalling, including Hedgehog signalling126. Variants in the genes encoding these proximal components result in ciliopathies such as nephronophthisis, Joubert syndrome and Meckel–Gruber syndrome. Patients with these disorders present most commonly with polycystic kidney disease and cerebellar and neural tube defects but several of these gene variants have also been linked to abnormal heart development126. Mice with mutations in Jbts17 have pulmonary valve defects127, whereas mice with mutations in Mks1 have AVSDs116. Furthermore, variants in TMEM237 (REF.128), NPHP4 (REF.129) and NPHP2 (REF.130) have also been identified in patients with CHD. In addition to evidence linking gene variants that affect cilia function to complex CHD, either isolated or in the setting of ciliopathies or abnormal laterality, these genetic variants have been implicated in heart valve development and disease (discussed extensively below).
The majority of cilia-related genes implicated in cardiac development cause disease in a recessive manner and encode proteins that make up the structure of the cilia or the ciliary signalling machinery15,131,132. However, monoallelic variants affecting the RNF20 complex (which works in a dosage-dependent manner as a transcriptional regulator of motile cilia function) and transmitted heterozygous variants in genes related to signalling downstream of cilia (such as ROCK2 and SMAD2) have also been linked to CHD108,133.
The findings from numerous targeted-sequencing studies implicating genes that encode various ciliary components in CHD suggest that cilia have a major role in the pathogenesis of CHD. Given that an increasing number of genes and proteins have been linked to cilia function, one of the major challenges in improving our understanding of the genetics of ciliopathies is the large size of the ‘ciliome’ (the genes and proteins required for cilia function). Estimates of the size and composition of the ciliome in the literature are wide-ranging. The SYSCILIA consortium developed a manually curated list of 428 cilia-associated proteins, of which 187 have been linked to ciliopathies134. In 2019, Bayesian integration of 9 independent cilium datasets predicted a total of 956 cilia-related genes135. Genome-wide analysis of the contribution of cilia to CHD has now been performed in both mice and humans. A forward recessive mutagenesis screen for severe CHD in mice resulted in the identification of 61 CHD-related genes, 34 of which have been linked to cilia18. Subsequently, whole-exome sequencing conducted to analyse 2,781 patients with CHD demonstrated that cilia-related gene variants were disproportionately found in patients with laterality disturbances but contribute broadly to CHD, including non-syndromic CHD15. Further analysis of the same patient cohort revealed that cilia-related genes are enriched for damaging rare, recessive genotypes131, similar to the observations made in mice. The substantial contribution of cilia-related genetic variation to CHD suggests that some CHDs can be classified as a ciliopathy. However, as some cilia-related proteins might have secondary or additional functions beyond the cilium, ciliopathic defects in the heart might also be caused by cilia-independent mechanisms.
Cilia in valve development and disease
Heart valve defects in ciliopathies.
Patients with ciliopathies often have heart valve defects17. In particular, patients with nephronophthisis136, Ellis–van Creveld (EvC) syndrome137, Joubert syndrome138, Bardet–Biedl syndrome139 or autosomal dominant polycystic kidney disease140 have an increased risk of mitral valve prolapse, bicuspid aortic valve disease, and tricuspid or pulmonary valve defects141,142. Variants in ciliary genes associated with Meckel–Gruber syndrome, Bardet–Biedl syndrome and EvC syndrome can cause cardiac valve defects irrespective of left–right patterning defects104,116. A genome-wide association study revealed a strong link between bicuspid aortic valve disease and genetic variation in human primary cilia, whereby single nucleotide polymorphisms were identified in or near genes related to the regulation of ciliogenesis through the exocyst142, a protein complex that chaperones cilia cargo to the membrane. Similarly, numerous cilia proteins, such as Smoothened and EvC Ciliary Complex Subunits 1 and 2, are heavily expressed in the endocardial cushion; approximately 60% of patients with a ciliopathy and variants in these genes have endocardial cushion defects143,144. Improper tissue formation from endocardial cushions as well as other structures, such as the dorsal mesenchymal protrusion, can contribute to the development of AVSDs145. Interestingly, the first gene implicated in AVSDs in patient studies was CRELD1, which encodes a component of cilia146. Subsequently, more than 25 known mouse ciliary genes and 7 human ciliary genes have been found to cause AVSDs116. A study that examined how cilia can influence AVSD prevalence revealed that mutations in genes related to motile cilia only contributed to AVSDs if heterotaxy was also present, whereas mutations in genes involved in ciliary signalling contributed to AVSDs irrespective of left–right patterning defects116. These findings suggest that ciliary signalling, not cilia motility, is a driving force behind AVSDs and imply a role for primary cilia in cushion formation beyond establishing left–right asymmetry16,116,120.
Endocardial cushion.
(ECC). A small pocket of cardiac jelly between the endocardial lining and the myocardium in the atrioventricular canal and outflow tract.
Cilia localization in the developing valve.
The first step in valve formation is the secretion and accumulation of cardiac jelly between the endocardial lining and the myocardium in the atrioventricular canal and outflow tract147 (FIG. 6a). This process creates small pockets known as endocardial cushions that must be cellularized through endothelial-to-mesenchymal transition (EndoMT), wherein the endocardial lining differentiates and migrates into the cardiac jelly, populating the cushions with mesenchyme147. EndoMT spans from E9.0 to E10.5 in mice, Hamburger–Hamilton (HH; stages of chick development) 16 to HH22 in chicks and 72 hours post-fertilization (hpf) to 96 hpf in zebrafish. The cushion mesenchyme will then proliferate, increasing cushion size until cushion remodelling begins. Remodelling is a coordinated effort involving differentiation, migration and condensation of the mesenchymal cells, and spans from E12.5 to E16.5 in mice, HH22 to HH28 in chicks and 96 hpf to 166 hpf in zebrafish147. Endocardial primary cilia are found both upstream and downstream of the developing atrioventricular and outflow tract cushions in chick hearts at HH17 (REF.148). These cilia become increasingly scarce over time at the narrowest section of the atrioventricular canal and outflow tract where EndoMT is occurring owing to higher shear stress. Given that the atrioventricular canal is subjected to higher shear stress between two contractile regions, the outflow tract retains its cilia for a longer period of time. Mesenchymal primary cilia, which are not subject to shear stress, are present throughout valve development. In mice, cilia are similarly found on both the endocardium and mesenchyme of the atrioventricular and outflow tract cushions from E8.5 to E12.5, which encompasses the major stages of cushion development after the onset of contraction18,115,141,149. In zebrafish, endocardial primary cilia can be found protruding into the lumen throughout the heart from 24 hpf to 96 hpf (REFS150,151). Just as in chicks, mice and zebrafish show a regionalized decrease in endocardial cilia in areas of higher shear stress where EndoMT occurs115,141,150 (FIG. 6a).
Fig. 6 |. Spatiotemporal distribution of primary cilia during valve development.
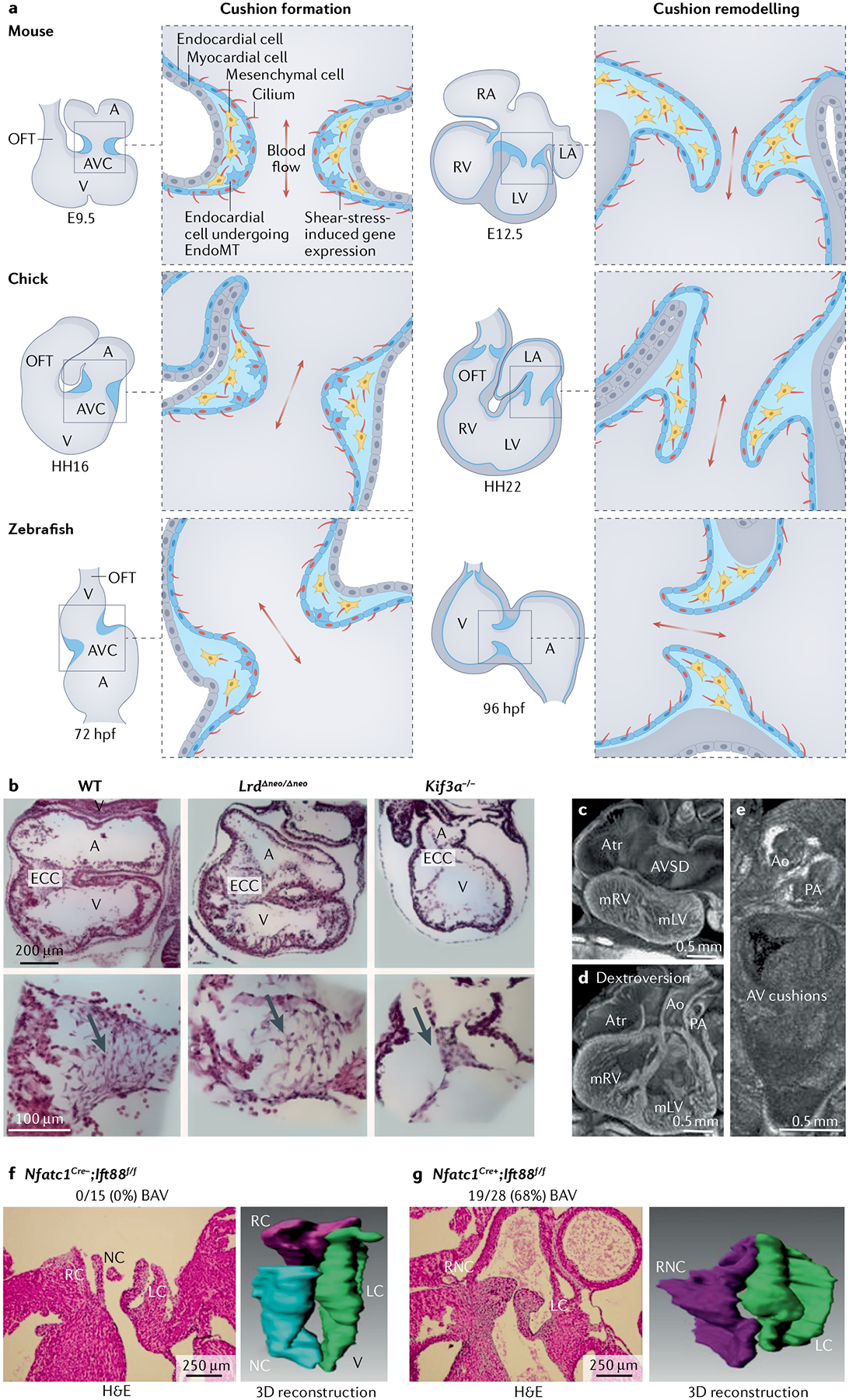
The initial critical steps of cushion formation and remodelling in three vertebrate model organisms are presented (part a). The cellular processes are conserved across mice, chicks and zebrafish, and the figure indicates the spatial context of the forming valves within the heart at relevant times during development in each model. Haematoxylin and eosin (H&E)-stained sections of mouse hearts at E12.5 (part b). Left panels show hearts from wild-type (WT) mice, middle panels from mice with mutant Lrd (encoding left–right dynein), which have left–right patterning defects, and right panels from mice with deletion of Kif3a (encoding a motor subunit of the heterotrimeric kinesin complex). The arrows pinpoint the lack of cushion cellularization in cilia mutants irrespective of left–right patterning defects. Histopathological images of mouse hearts with deletion of Cc2d2a (which encodes a basal body component) at E15.5, which have normal outflow tract cushions but atrioventricular (AV) cushions with dextrocardia (part c) and AV septal defects (AVSDs) (part d). Mutant AV cushions are improperly formed (part e). Mice with conditional deletion of Ift88 (Nfatc1Cre;Ift88f/f), which encodes an intraflagellar transport component, have cushion remodelling phenotypes that result in bicuspid aortic valve (BAV) defects compared with mice with WT Ift88 (Nfatc1Cre−;Ift88f/f) (parts f, g). This phenotype was highly penetrant, being observed in 19 of 28 mice with the genetic deletion. The left panel shows H&E-stained sections and the right panel shows 3D reconstruction images depicting the penetrance of this phenotype. A, atrium; Ao, aorta; Atr, atrium; AVC, atrioventricular canal; ECC, endocardial cushion; EndoMT, endothelial-to-mesenchymal transition; HH, Hamburger–Hamilton; hpf, hours post-fertilization; LA, left atrium; LC, left coronary; LV, left ventricle; mLV, morphological left ventricle; mRV, morphological right ventricle; NC, non-coronary; OFT, outflow tract; PA, pulmonary artery; RA, right atrium; RC, right coronary; RNC, right non-coronary; RV, right ventricle; V, ventricle. Part b reprinted with permission from REF.115, Wiley. Parts c–e reprinted from REF.18, Springer Nature Limited. Parts f and g reprinted with permission from REF.141, Wiley.
Deciliation and reduction in cilia length in response to strong fluid flow have been previously reported in human umbilical vein endothelial cells152. Interestingly, modulation of cilia length, as well as the presence of cilia, can alter genetic signalling in the tissues that these cilia occupy153,154. Markers of mechanical stress, such as KLF2 (encoding Krüppel-like factor 2 (KLF2)), are expressed in regions with deciliation and reduced cilia length148,155, suggesting that regional deciliation is involved in the modulation of mechanosensitive signalling in the endocardium.
Atrioventricular canal.
(AVC). The channel connecting the developing atrium and ventricles in embryonic development.
Outflow tract.
(OFT). The channel connecting the developing ventricles and dorsal aorta in embryonic development.
Endothelial-to-mesenchymal transition.
(EndoMT). The differentiation of endothelial cells to mesenchyme during cardiac cushion development.
Primary cilia in heart cushion formation.
Loss of primary cilia has been found to prime the endothelium for EndoMT, a critical process for cardiac cushion development156. Mutant mouse endothelium that lacks cilia undergoes EndoMT in response to shear stress. However, when wild-type endothelium is exposed to sufficient shear stress, resulting in deciliation, the endothelium also undergoes EndoMT156. Interestingly, while the mechanosensitive Klf2 and Klf4 genes are increased in response to shear stress in wild-type endothelium in mice, removal of cilia inhibits the induction of Klf2 expression and significantly downregulates KLF4 at the mRNA and protein levels in response to shear stress150,157. This observation contradicts earlier findings of Van der Heiden et al., who reported that only regions of endocardium with few or no primary cilia expressed KLF2 in chick148. Whether this discrepancy is related to differences between endothelial populations (kidney versus heart) or distinct regulatory mechanisms is not known. This finding suggests that primary cilia are required in regions of higher shear stress (such as the atrioventricular canal) to induce Klf2 expression before being lost, allowing Klf2 expression to persist while Klf4 (which encodes an inhibitor of EndoMT) is downregulated. This theory can help explain how modulation of gene expression and the resulting EndoMT and cushion formation can be controlled by gradual, regional deciliation.
Mutant mice lacking Kif3a or Kif3b (which encode motor subunits of the heterotrimeric kinesin complex) are unable to assemble cilia and are non-viable by E10.5 owing to heart failure69. These mouse mutants have a complete loss of cushion cellularity not seen in other mutants with defective left–right patterning and irrespective of heart looping, indicating a severe EndoMT defect independent of its left–right randomization69,115,158 (FIG. 6b). A less severe defect in cushion cellularization can be seen in mice lacking Pkd2 (which encodes a mechanosensitive transient receptor potential cation channel), suggesting that abnormal cilia calcium signalling can contribute to inhibition of cushion cellularity115,159,160. Similarly, targeted insertional Ift88−/− mice, which lack a subunit of the intraflagellar transport complex B, have residual, but few, cilia present with reduced cushion cellularity and are non-viable by E11.5 (REFS158,161). Therefore, primary cilia might act as mechanosensors in cushion formation just as they do in the endothelium. Overall, these findings support a role for cilia, deciliation and shear stress in initiating EndoMT during cushion formation in the heart.
Primary cilia in valve leaflet remodelling.
In addition to their role in early valve formation, primary cilia also contribute to later stages of valve development during leaflet remodelling141,142,162,163. In mice at E12.5, selective loss of mesenchymal primary cilia in the atrioventricular cushions through loss of the basal body component CC2D2A resulted in remodelling defects and AVSDs, whereas the outflow tract cushion remained unaffected18 (FIG. 6c–e). To discriminate between ciliary signalling in valvulogenesis and left–right patterning, study investigators assessed mutant mice with a hypomorphic allele of Ift88 (Ift88cbbs) established from chemical mutagenesis that leads to reduced cilia numbers but normal left–right development149. These mutants live longer than Ift88−/− mice and also have impaired cushion remodelling and AVSDs. Other mutant mouse models that have reduced cilia numbers through conditionally deleted intraflagellar transport components also evade left–right patterning defects. Conditional knockout of Ift88 in the endocardium using a Nfatc1–Cre driver results in a phenotype that is reminiscent of myxomatous bicuspid aortic valve disease141 (FIG. 6f,g). The increased cusp size was secondary to increased extracellular matrix (ECM) production, suggesting that primary cilia can repress collagen deposition and maturation to control leaflet size. Subsequently, RNA sequencing confirmed that the conditional mutation leads to the upregulation of ECM-related gene pathways in the anterior mitral leaflets162. Interestingly, conditional knockout of Klf2 in the endocardium using the same Nfatc1–Cre driver resulted in a similar cushion remodelling phenotype, further suggesting a role for cilia in modulating the expression of Klf2 in vivo164. Similar to the conditional Ift88 knockout, Ift88orpk mutant mice (also referred to as polaris), which is a hypomorphic allele established by insertional mutagenesis, also have increased ECM deposition in the heart at E11.5 (REF.156). Furthermore, Ift88orpk mice have renal cysts, polydactyly and hydrocephalus but survive past birth and do not have left–right patterning defects165. Mutations affecting the exocyst, a shuttling complex necessary for ciliogenesis and ciliary signalling, result in aortic valve defects in both mice and zebrafish142. Endocardial expression of Desert Hedgehog is also necessary for cilia-induced activation of the TIAM1–RAC1 axis, which in turn stimulates ECM remodelling necessary for proper valve remodelling163. Perturbations in this paracrine system lead to adult myxomatous mitral valve disease, which is related to the mitral valve prolapse observed in many patients with ciliopathies163. Although disease pathogenesis in patients with mitral valve prolapse was believed to be through gradual valve degeneration, the latest evidence points to two major disease phases, wherein secondary effects such as inflammation can aggravate a pre-existing congenital valve defect162,163. These findings demonstrate how defects in cilia-dependent leaflet remodelling during development can prime conditions such as mitral valve prolapse.
In contrast to the findings by Slough et al., who reported that mutant mice lacking cilia had no cushion cellularization115, mice with NFatc1–Cre conditional Ift88 mutations have normal cushion cellularization, suggesting that endocardial primary cilia are not necessary for EndoMT141. However, given that some cilia components are long-lived, cilia priming of EndoMT should already be under way after induction of Nfatc1 expression166 (which in mice occurs at E9.5 at the earliest167). Removing Exoc5 (encoding a subunit of the exocyst complex) with the use of the same Nfatc1–Cre driver mediates cilia loss by E13.5 (REF.142), well past the stage at which EndoMT occurs (E9–E10.5). The product of Nfatc1 has also been identified as an inhibitor of EndoMT; therefore, endocardial cells expressing Nfatc1 will not undergo EndoMT regardless of the presence of primary cilia168. This finding might explain the discrepancy observed between the conditional Ift88 mutant mice and the Kif3a-knockout mice used by Slough et al.115, which have severe EndoMT defects irrespective of left–right randomization but still indicate a role for primary cilia in remodelling.
Cilia as mechanosensors in valve development
Primary cilia as endothelial mechanosensors.
Primary cilia are known to act as mechanosensors in endothelial tissue of the vasculature169,170 and kidney171. Mechanical stimulation of cilia on Madin–Darby Canine Kidney (MDCK) cells resulted in an influx of cytosolic calcium172. The same response was elicited by fluid flow over the endothelium and removal of cilia by treatment with chloral hydrate or by genetic knockdown approaches ablated this mechanosensitive response173,174. Mouse endothelial cells with knockdown or knockout of Pkd1 or Pkd2 have the same loss of calcium influx as endothelium lacking cilia, suggesting that cilia-localized PKD1 and PKD2 are important for the transduction of mechanical signals175. This finding also mirrors the observation that cilia localization of PKD2 in the mouse LRO is required for sensing LRO flow and normal left–right development74.
Increased fluid flow in zebrafish vasculature leads to increased cilia deflection and calcium influx in a dose-dependent manner in vivo35. Furthermore, a reduction in blood viscosity or fluid shear stress as well as a loss of contractility, cilia or pkd2 expression can all individually result in loss of endothelial calcium influx in zebrafish35. Similar to findings in the heart, endothelial cilia gradually deplete as flow increases. Intriguingly, vascular endothelial cilia have incomplete microtubules towards the end of the axoneme, which potentially allows for greater flexibility and sensitivity. This observation lends support to the hypothesis that vascular endothelial cilia are necessary for mechanosensation in low-flow settings and are subsequently lost when no longer required.
Primary cilia as endocardial mechanosensors.
The endocardium of chicken arterial vasculature has a higher sensitivity to mechanical stimuli than endothelium in vivo155,176. Furthermore, shear stress-induced gene expression is higher in chicken ciliated endocardium than in non-ciliated endocardium177. The removal of endocardial cilia in chicken by the administration of chloral hydrate diminished shear stress-induced gene expression to the level of expression in the arterial endothelium. These findings suggest that the developing heart is more sensitive to mechanical cues than the vasculature, wherein smaller stress increments can affect resulting genetic programmes. Furthermore, these observations indicate that specialized mechanosensors are present in the developing heart that are sensitive to these smaller stress increments.
The role of primary cilia as endocardial mechanosensors in both the setting of the cardiovascular system and cilia biology has been a topic of debate178. The main argument against primary cilia as mediators of mechanical stress during valve development was formed on the basis of the observation that cilia are absent from the endocardium at seemingly critical stages of valve development owing to deciliation. Even before deciliation, not all cells are ciliated at one time, leading to the question of how mechanosensitive gene expression could be initiated ubiquitously without the mechanosensor present on every cell179. However, a two-phase hypothesis might address this issue. First, primary cilia can influence the endocardial response to flow via cilia signalling. Second, the cilia can amplify mechanical signals on the cytoskeleton to allow instantaneous and lasting gene expression. In this model, cilia are transient by design to enable spatiotemporal regulation of mechanosensation35,148,156,177,180. Cytoplasmic calcium can also be subsequently induced through cell–cell junctions or communication, removing the need for complete ciliation174. However, given that deciliation has been found to influence gene expression, prior resorption of cilia on the endocardium might be a critical priming step in valve development156.
Numerous lines of evidence have implicated primary cilia as endocardial mechanosensors through the same pathways used in vascular endothelium (FIG. 7). In zebrafish, mutations in pkd2 result in both the loss of klf2a expression and cushion cellularization to the same degree as loss of contractility with lidocaine treatment, thereby demonstrating the importance of cilia calcium influx for endocardial mechanosensation and EndoMT160,181. Zebrafish with an ift88 knockdown have reduced endocardial notch1b expression in a flow-dependent manner but notch1b loss only occurred within a narrow timeframe when cilia were pharmacologically removed, indicating that primary cilia are needed for EndoMT-dependent Notch1b activation at the onset of flow150. As in vascular endothelium, primary cilia in the endocardium are hypothesized to detect flow through activation of calcium transient receptor potential channels localized in cilia, such as Pkd2, that are stretched in response to low laminar flow150,182. In zebrafish, endocardial Notch signalling is also involved in the regeneration of the heart, a process that requires blood flow and subsequent klf2a expression183. Knockdown of ift88 prevented the upregulation of the expression of both klf2a and notch1b in the atrioventricular canal during ventricular regeneration151,182. Whether these mechanisms are dependent on cilia calcium influx or whether primary cilia similarly act as endocardial valve mechanosensors outside of ventricular regeneration remains unclear. Together, these data implicate primary cilia as critical mediators between laminar flow and genetic regulation of pathways responsible for complex cellular processes in the endocardium.
Fig. 7 |. Primary cilia as endocardial mechanosensors.
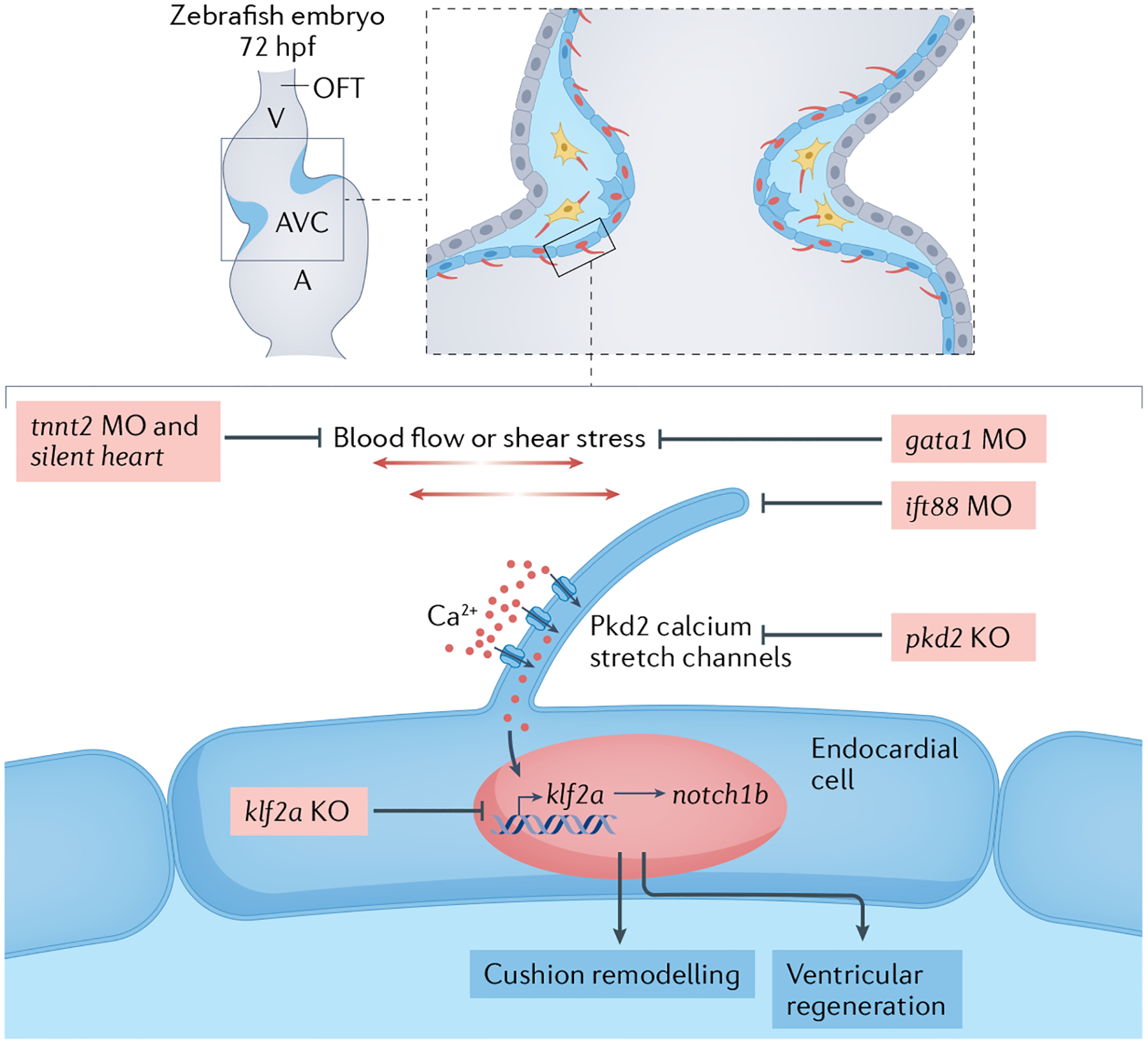
Schematic depicting the hypothesis that primary cilia serve as endocardial mechanosensors through cilia-dependent calcium influx during low blood flow in a zebrafish embryo at 72 hours post-fertilization (hpf). Polycystin 2 (Pkd2) must be localized to primary cilia to initiate cytoplasmic calcium influx in the endothelium. Perturbations at every level of the mechanosensory pathway would result in the same loss of target genes downstream of this pathway: cessation of blood flow or contraction (induced by tnnt2a morpholinos (MO; nucleotide analogues that recognize and bind short nucleotide sequences to modify or block mRNA transcription) or the silent heart mutation), reduction in blood viscosity and resulting shear stress during blood flow (induced by gata1 MO or gata2 MO), blocking of cilia calcium influx (in pkd2 knockout (KO)), removal of cilia (with ift88 MO) or removal of the downstream mechanosensitive transcription factor Krüppel-like factor 2a (in klf2a KO). A, atrium; AVC, atrioventricular canal; OFT, outflow tract; V, ventricle.
Cilia in cardiac fibrosis and regeneration
Studies within the past 5 years have implicated primary cilia and polycystin signalling in the process of heart regeneration. Although primary cilia have been located in the embryonic and adult heart of several species184, including humans185,186, the specific cell location and function of cilia remain unclear. A 2019 study reported that, in mouse, rat and human hearts, primary cilia were specifically found in cardiac fibroblasts not cardiomyocytes187. Of note, the study investigators found that, after heart injury, ciliated fibroblasts accumulated at the sites of injured myocardium. In these cells, cilia and PKD1 participate in the fibrogenic response observed after injury by inducing TGFβ1–SMAD3 activation, the production of ECM proteins and cell contractility, processes that are all compromised in fibroblasts depleted of primary cilia. These results suggest that signalling in primary cilia after cardiac injury in mammals leads to a maladaptive fibrogenic response that impairs cardiac regeneration. This notion is further reinforced by findings that, after myocardial infarction, the induction of cilia disassembly in epicardial cells via short hairpin RNA-mediated silencing of Ift88 between postnatal days 7 and 28 in mice stimulates cardiac regeneration by promoting epithelial-to-mesenchymal transition and reducing scar formation188. Modulating cilia response after cardiac injury in mammals might therefore help to promote regenerative responses, and uncovering the mechanisms involving cilia in cardiac regeneration in adult animals might provide valuable insights. For example, in zebrafish, cardiac injury induces cardiomyocyte proliferation189, with Notch signalling activation in the endocardium being essential for regeneration of the myocardium190–192. This activation was mediated by haemodynamic changes that occur after injury183. Interestingly, mechanical haemodynamic activation of Notch signalling after ventricular ablation was mediated by endocardial primary cilia via upregulation of klf2 expression151. How primary cilia transduce mechanical shear stress to regulate Notch signalling to ultimately promote heart regeneration remains to be determined.
Conclusions
Although cilia were once meekly described as ‘eyelashes’ on cells by early microscopists, this tiny but mighty organelle’s essential function in heart development and disease is now firmly in the limelight. Advances in research into intraciliary calcium signalling have addressed major long-standing gaps in our understanding of cardiac left–right development. Genomic and patient studies have critically linked cilia function and biogenesis to several cardiovascular disorders, namely CHD and mitral valve prolapse. Pioneering work in animal models has unravelled surprising roles for intracardiac cilia in haemodynamic mechanosensation, valvulogenesis and myocardial regeneration. Despite this immense progress, more research will be necessary to uncover how primary cilia integrate the different signalling pathways they are associated with during heart formation and how these affect different stages of heart development. To do so, discriminating between ciliary signalling in the LRO and embryonic and adult cardiac tissues will be crucial. Generating conditional knockout models of primary cilia from different heart tissues at distinct time points could help to establish the contribution of cardiac primary cilia to the various steps of cardiac morphogenesis and maturation independently of their involvement in the LRO during early left–right specification. The strong association between ciliopathies and congenital defects in the heart irrespective of heterotaxy indicates that primary cilia act as important genetic regulators of cardiac morphology and function throughout development. Ultimately, a deeper understanding of the mechanisms and function of cilia in the heart might lead to the development of novel diagnostic and therapeutic strategies to manage the increasing number of ciliopathic cardiovascular disorders.
Key points.
Cilia are antenna-like organelles that extend from most eukaryotic cells to obtain and interpret information from the extracellular environment; impaired ciliary signalling has been linked with congenital heart disease.
Intraciliary calcium signalling in the embryonic left–right organizer initiates vertebrate left–right patterning; abnormal left–right asymmetry is associated with major congenital heart disease, especially in heterotaxy syndrome.
Impaired cilia function and signalling are associated with heterotaxy, congenital heart disease, mitral valve prolapse and numerous other cilia-related disorders with cardiac abnormalities.
Primary cilia have a role in heart development beyond establishing left–right asymmetry.
Endocardial primary cilia might translate mechanical signals to gene expression during heart valve development.
Fibroblast cilia have been implicated in the regulation of cardiac regeneration.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Satir P & Christensen ST Overview of structure and function of mammalian cilia. Annu. Rev. Physiol 69, 377–400 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Leeuwenhoek VA Concerning little animals. Philos. Trans. R. Soc 12, 821–831 (1677). [Google Scholar]
- 3.Zimmermann KW Beiträge zur kenntniss einiger drüsen und epithelien [German]. Arch. Mikrosk. Anat 52, 552–706 (1898). [Google Scholar]
- 4.Ibanez-Tallon I, Heintz N & Omran H To beat or not to beat: roles of cilia in development and disease. Hum. Mol. Genet 12, R27–35 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Eggenschwiler JT & Anderson KV Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol 23, 345–373 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waters AM & Beales PL Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol 26, 1039–1056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hildebrandt F, Benzing T & Katsanis N Ciliopathies. N. Engl. J. Med 364, 1533–1543 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kathem SH, Mohieldin AM & Nauli SM The roles of primary cilia in polycystic kidney disease. AIMS Mol. Sci 1, 27–46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitchison HM & Valente EM Motile and non-motile cilia in human pathology: from function to phenotypes. J. Pathol 241, 294–309 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Yuan S, Zaidi S & Brueckner M Congenital heart disease: emerging themes linking genetics and development. Curr. Opin. Genet. Dev 23, 352–359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fakhro KA et al. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc. Natl Acad. Sci. USA 108, 2915–2920 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaidi S et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabriel GC & Lo CW Left-right patterning in congenital heart disease beyond heterotaxy. Am. J. Med. Genet. C Semin. Med. Genet 184, 90–96 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabriel GC, Young CB & Lo CW Role of cilia in the pathogenesis of congenital heart disease. Semin. Cell Dev. Biol 110, 2–10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin SC et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet 49, 1593–1601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klena N et al. in Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology (eds Nakanishi T et al. ) 67–79 (Springer, 2016). [PubMed] [Google Scholar]
- 17.Klena NT, Gibbs BC & Lo CW Cilia and ciliopathies in congenital heart disease. Cold Spring Harb. Perspect. Biol 9, a028266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 521, 520–524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozminski KG, Forscher P & Rosenbaum JL Three flagellar motilities in Chlamydomonas unrelated to flagellar beating. Video supplement. Cell Motil. Cytoskeleton 39, 347–348 (1998). [PubMed] [Google Scholar]
- 20.Rosenbaum JL & Witman GB Intraflagellar transport. Nat. Rev. Mol. Cell Biol 3, 813–82 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Lin J & Nicastro D Asymmetric distribution and spatial switching of dynein activity generates ciliary motility. Science 360, eaar1968 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McEwen DP, Jenkins PM & Martens JR Olfactory cilia: our direct neuronal connection to the external world. Curr. Top. Dev. Biol 85, 333–370 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Flock A & Duvall AJ 3rd The ultrastructure of the kinocilium of the sensory cells in the inner ear and lateral line organs. J. Cell Biol 25, 1–8 (1965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boo KS & Richards AG Fine structure of scolopidia in Johnston’s organ of female Aedes aegypti compared with that of the male. J. Insect Physiol 21, 1129–1139 (1975). [DOI] [PubMed] [Google Scholar]
- 25.Corbiere-Tichane G Fine structure of the sensory apparatus of the mandible of the Speophyes lucidulus larva. (Cavernicolous coleoptera of the subfamily Bathysciinae) [French]. Z. Zellforsch. Mikrosk. Anat 112, 129–138 (1971). [PubMed] [Google Scholar]
- 26.Gibbons BH, Gibbons IR & Baccetti B Structure and motility of the 9 + 0 flagellum of eel spermatozoa. J. Submicrosc. Cytol 15, 15–20 (1983). [PubMed] [Google Scholar]
- 27.Feistel K & Blum M Three types of cilia including a novel 9+4 axoneme on the notochordal plate of the rabbit embryo. Dev. Dyn 235, 3348–3358 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Kramer-Zucker AG et al. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Development 132, 1907–1921 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Schweickert A et al. Cilia-driven leftward flow determines laterality in Xenopus. Curr. Biol 17, 60–66 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Nakamura T & Hamada H Left-right patterning: conserved and divergent mechanisms. Development 139, 3257–3262 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Nonaka S et al. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 95, 829–837 (1998). [DOI] [PubMed] [Google Scholar]
- 32.McGrath J, Somlo S, Makova S, Tian X & Brueckner M Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell 114, 61–73 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Caspary T, Larkins CE & Anderson KV The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell 12, 767–778 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Gluenz E et al. Beyond 9+0: noncanonical axoneme structures characterize sensory cilia from protists to humans. FASEB J. 24, 3117–3121 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goetz JG et al. Endothelial cilia mediate low flow sensing during zebrafish vascular development. Cell Rep. 6, 799–808 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Sun S, Fisher RL, Bowser SS, Pentecost BT & Sui H Three-dimensional architecture of epithelial primary cilia. Proc. Natl Acad. Sci. USA 116, 9370–9379 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiesel P et al. The molecular structure of mammalian primary cilia revealed by cryo-electron tomography. Nat. Struct. Mol. Biol 27, 1115–1124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singla V & Reiter JF The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science 313, 629–633 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB & Christensen ST Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol 15, 199–219 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wheway G, Nazlamova L & Hancock JT Signaling through the primary cilium. Front. Cell Dev. Biol 6, 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huangfu D & Anderson KV Cilia and Hedgehog responsiveness in the mouse. Proc. Natl Acad. Sci. USA 102, 11325–11330 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corbit KC et al. Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Lienkamp S, Ganner A & Walz G Inversin, Wnt signaling and primary cilia. Differentiation 83, S49–55 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Oh EC & Katsanis N Context-dependent regulation of Wnt signaling through the primary cilium. J. Am. Soc. Nephrol 24, 10–18 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Clement CA et al. TGF-beta signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 3, 1806–1814 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Ezratty EJ et al. A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell 145, 1129–1141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Z et al. Primary cilia regulate hematopoietic stem and progenitor cell specification through Notch signaling in zebrafish. Nat. Commun 10, 1839 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rohatgi R, Milenkovic L & Scott MP Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372–376 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Haycraft CJ et al. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 1, e53 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nauli SM et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet 33, 129–137 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Delling M, DeCaen PG, Doerner JF, Febvay S & Clapham DE Primary cilia are specialized calcium signalling organelles. Nature 504, 311–314 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeCaen PG, Delling M, Vien TN & Clapham DE Direct recording and molecular identification of the calcium channel of primary cilia. Nature 504, 315–318 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan S, Zhao L, Brueckner M & Sun Z Intraciliary calcium oscillations initiate vertebrate left-right asymmetry. Curr. Biol 25, 556–567 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mizuno K et al. Role of Ca2+ transients at the node of the mouse embryo in breaking of left-right symmetry. Sci. Adv 6, eaba1195 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Su S et al. Genetically encoded calcium indicator illuminates calcium dynamics in primary cilia. Nat. Methods 10, 1105–1107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shiba D et al. Localization of Inv in a distinctive intraciliary compartment requires the C-terminal ninein-homolog-containing region. J. Cell Sci 122, 44–54 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Bennett HW et al. Novel fibrillar structure in the inversin compartment of primary cilia revealed by 3D single-molecule superresolution microscopy. Mol. Biol. Cell 31, 619–639 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu P et al. Chlamydomonas PKD2 organizes mastigonemes, hair-like glycoprotein polymers on cilia. J. Cell Biol 219, e202001122 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Afzelius BA A human syndrome caused by immotile cilia. Science 193, 317–319 (1976). [DOI] [PubMed] [Google Scholar]
- 60.Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL & Witman GB Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr. Biol 12, R378–380 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Hirokawa N, Tanaka Y, Okada Y & Takeda S Nodal flow and the generation of left-right asymmetry. Cell 125, 33–45 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Shinohara K et al. Two rotating cilia in the node cavity are sufficient to break left-right symmetry in the mouse embryo. Nat. Commun 3, 622 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Norris DP Cilia, calcium and the basis of left-right asymmetry. BMC Biol. 10, 102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hirokawa N, Tanaka Y & Okada Y Cilia, KIF3 molecular motor and nodal flow. Curr. Opin. Cell Biol 24, 31–39 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Tabin CJ & Vogan KJ A two-cilia model for vertebrate left-right axis specification. Genes. Dev 17, 1–6 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Okada Y et al. Abnormal nodal flow precedes situs inversus in iv and inv mice. Mol. Cell 4, 459–468 (1999). [DOI] [PubMed] [Google Scholar]
- 67.Supp DM et al. Targeted deletion of the ATP binding domain of left-right dynein confirms its role in specifying development of left-right asymmetries. Development 126, 5495–5504 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nonaka S, Shiratori H, Saijoh Y & Hamada H Determination of left-right patterning of the mouse embryo by artificial nodal flow. Nature 418, 96–99 (2002). [DOI] [PubMed] [Google Scholar]
- 69.Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR & Goldstein LS Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc. Natl Acad. Sci. USA 96, 5043–5048 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tisler M, Thumberger T, Schneider I, Schweickert A & Blum M Leftward flow determines laterality in conjoined twins. Curr. Biol 27, 543–548 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Hashimoto M et al. Planar polarization of node cells determines the rotational axis of node cilia. Nat. Cell Biol 12, 170–176 (2010). [DOI] [PubMed] [Google Scholar]
- 72.Sampaio P et al. Left-right organizer flow dynamics: how much cilia activity reliably yields laterality? Dev. Cell 29, 716–728 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Vick P et al. Flow on the right side of the gastrocoel roof plate is dispensable for symmetry breakage in the frog Xenopus laevis. Dev. Biol 331, 281–291 (2009). [DOI] [PubMed] [Google Scholar]
- 74.Yoshiba S et al. Cilia at the node of mouse embryos sense fluid flow for left-right determination via Pkd2. Science 338, 226–231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tanaka Y, Okada Y & Hirokawa N FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 435, 172–177 (2005). [DOI] [PubMed] [Google Scholar]
- 76.Wang J et al. C. elegans ciliated sensory neurons release extracellular vesicles that function in animal communication. Curr. Biol 24, 519–525 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wood CR & Rosenbaum JL Ciliary ectosomes: transmissions from the cell’s antenna. Trends Cell Biol. 25, 276–285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nager AR et al. An actin network dispatches ciliary GPCRs into extracellular vesicles to modulate signaling. Cell 168, 252–263.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Phua SC et al. Dynamic remodeling of membrane composition drives cell cycle through primary cilia excision. Cell 168, 264–279.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wood CR, Huang K, Diener DR & Rosenbaum JL The cilium secretes bioactive ectosomes. Curr. Biol 23, 906–911 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takao D et al. Asymmetric distribution of dynamic calcium signals in the node of mouse embryo during left-right axis formation. Dev. Biol 376, 23–30 (2013). [DOI] [PubMed] [Google Scholar]
- 82.Pennekamp P et al. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr. Biol 12, 938–943 (2002). [DOI] [PubMed] [Google Scholar]
- 83.Meno C et al. Mouse Lefty2 and zebrafish antivin are feedback inhibitors of nodal signaling during vertebrate gastrulation. Mol. Cell 4, 287–298 (1999). [DOI] [PubMed] [Google Scholar]
- 84.Saijoh Y et al. Two nodal-responsive enhancers control left-right asymmetric expression of Nodal. Dev. Dyn 232, 1031–1036 (2005). [DOI] [PubMed] [Google Scholar]
- 85.Levin M, Johnson RL, Stern CD, Kuehn M & Tabin C A molecular pathway determining left-right asymmetry in chick embryogenesis. Cell 82, 803–814 (1995). [DOI] [PubMed] [Google Scholar]
- 86.Shiratori H, Yashiro K, Shen MM & Hamada H Conserved regulation and role of Pitx2 in situs-specific morphogenesis of visceral organs. Development 133, 3015–3025 (2006). [DOI] [PubMed] [Google Scholar]
- 87.Little RB & Norris DP Right, left and cilia: how asymmetry is established. Semin. Cell Dev. Biol 110, 11–18 (2021). [DOI] [PubMed] [Google Scholar]
- 88.Delling M et al. Primary cilia are not calcium-responsive mechanosensors. Nature 531, 656–660 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Behringer RGM, Vintersten Nagy K & Nagy A Manipulating the Mouse Embryo: A Laboratory Manual. 4th edn, 814 (Cold Spring Harbor Laboratory Press, 2014). [Google Scholar]
- 90.Chen TW et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ohkura M et al. Genetically encoded green fluorescent Ca2+ indicators with improved detectability for neuronal Ca2+ signals. PLoS ONE 7, e51286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhao Y et al. An expanded palette of genetically encoded Ca2+ indicators. Science 333, 1888–1891 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim JH et al. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE 6, e18556 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cho JH et al. The GCaMP-R family of genetically encoded ratiometric calcium indicators. ACS Chem. Biol 12, 1066–1074 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schweickert A et al. The nodal inhibitor Coco is a critical target of leftward flow in Xenopus. Curr. Biol 20, 738–743 (2010). [DOI] [PubMed] [Google Scholar]
- 96.Marques S et al. The activity of the Nodal antagonist Cerl-2 in the mouse node is required for correct L/R body axis. Genes Dev. 18, 2342–2347 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kawasumi A et al. Left-right asymmetry in the level of active Nodal protein produced in the node is translated into left-right asymmetry in the lateral plate of mouse embryos. Dev. Biol 353, 321–330 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nakamura T et al. Fluid flow and interlinked feedback loops establish left-right asymmetric decay of Cerl2 mRNA. Nat. Commun 3, 1322 (2012). [DOI] [PubMed] [Google Scholar]
- 99.Minegishi K et al. Fluid flow-induced left-right asymmetric decay of Dand5 mRNA in the mouse embryo requires a Bicc1-Ccr4 RNA degradation complex. Nat. Commun 12, 4071 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hamada H Molecular and cellular basis of left-right asymmetry in vertebrates. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci 96, 273–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Francescatto L, Rothschild SC, Myers AL & Tombes RM The activation of membrane targeted CaMK-II in the zebrafish Kupffer’s vesicle is required for left-right asymmetry. Development 137, 2753–2762 (2010). [DOI] [PubMed] [Google Scholar]
- 102.Mochizuki T et al. Cloning of inv, a gene that controls left/right asymmetry and kidney development. Nature 395, 177–181 (1998). [DOI] [PubMed] [Google Scholar]
- 103.Burns TA et al. A novel mouse model for cilia-associated cardiovascular anomalies with a high penetrance of total anomalous pulmonary venous return. Anat. Rec 302, 136–145 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kennedy MP et al. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation 115, 2814–2821 (2007). [DOI] [PubMed] [Google Scholar]
- 105.Lin AE et al. Laterality defects in the national birth defects prevention study (1998–2007): birth prevalence and descriptive epidemiology. Am. J. Med. Genet. A 164A, 2581–2591 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nakhleh N et al. High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation 125, 2232–2242 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Burkhalter MD et al. Imbalanced mitochondrial function provokes heterotaxy via aberrant ciliogenesis. J. Clin. Invest 129, 2841–2855 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Robson A et al. Histone H2B monoubiquitination regulates heart development via epigenetic control of cilia motility. Proc. Natl Acad. Sci. USA 116, 14049–14054 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Desgrange A, Le Garrec JF & Meilhac SM Left-right asymmetry in heart development and disease: forming the right loop. Development 145, dev162776 (2018). [DOI] [PubMed] [Google Scholar]
- 110.Ray P et al. Intrinsic cellular chirality regulates left-right symmetry breaking during cardiac looping. Proc. Natl Acad. Sci. USA 115, E11568–E11577 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jacobs JP et al. The nomenclature, definition and classification of cardiac structures in the setting of heterotaxy. Cardiol. Young 17 (Suppl. 2), 1–28 (2007). [DOI] [PubMed] [Google Scholar]
- 112.Icardo JM & Sanchez de Vega MJ Spectrum of heart malformations in mice with situs solitus, situs inversus, and associated visceral heterotaxy. Circulation 84, 2547–2558 (1991). [DOI] [PubMed] [Google Scholar]
- 113.Field S et al. Pkd1l1 establishes left-right asymmetry and physically interacts with Pkd2. Development 138, 1131–1142 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shylo NA, Emmanouil E, Ramrattan D & Weatherbee SD Loss of ciliary transition zone protein TMEM107 leads to heterotaxy in mice. Dev. Biol 460, 187–199 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Slough J, Cooney L & Brueckner M Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis. Dev. Dyn 237, 2304–2314 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Burnicka-Turek O et al. Cilia gene mutations cause atrioventricular septal defects by multiple mechanisms. Hum. Mol. Genet 25, 3011–3028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Frescura C, Ho SY, Giordano M & Thiene G Isomerism of the atrial appendages: morphology and terminology. Cardiovasc. Pathol 47, 107205 (2020). [DOI] [PubMed] [Google Scholar]
- 118.Briggs LE et al. Wnt/beta-catenin and sonic hedgehog pathways interact in the regulation of the development of the dorsal mesenchymal protrusion. Dev. Dyn 245, 103–113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hoffmann AD, Peterson MA, Friedland-Little JM, Anderson SA & Moskowitz IP Sonic hedgehog is required in pulmonary endoderm for atrial septation. Development 136, 1761–1770 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Digilio MC et al. Atrioventricular canal defect and genetic syndromes: the unifying role of sonic hedgehog. Clin. Genet 95, 268–276 (2019). [DOI] [PubMed] [Google Scholar]
- 121.Liu S et al. DNAH11 variants and its association with congenital heart disease and heterotaxy syndrome. Sci. Rep 9, 6683 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Frommer A et al. Immunofluorescence analysis and diagnosis of primary ciliary dyskinesia with radial spoke defects. Am. J. Respir. Cell Mol. Biol 53, 563–573 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pugnaloni F, Versacci P, Marino B & Digilio MC Atrioventricular canal defect is the classic congenital heart disease in Bardet-Biedl syndrome. Ann. Hum. Genet 85, 101–102 (2021). [DOI] [PubMed] [Google Scholar]
- 124.Scott CA et al. Nuclear/cytoplasmic transport defects in BBS6 underlie congenital heart disease through perturbation of a chromatin remodeling protein. PLoS Genet. 13, e1006936 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Olson AJ, Krentz AD, Finta KM, Okorie UC & Haws RM Thoraco-abdominal abnormalities in Bardet-Biedl syndrome: situs inversus and heterotaxy. J. Pediatr 204, 31–37 (2019). [DOI] [PubMed] [Google Scholar]
- 126.Gabriel GC, Pazour GJ & Lo CW Congenital heart defects and ciliopathies associated with renal phenotypes. Front. Pediatr 6, 175 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Damerla RR et al. Novel Jbts17 mutant mouse model of Joubert syndrome with cilia transition zone defects and cerebellar and other ciliopathy related anomalies. Hum. Mol. Genet 24, 3994–4005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang L et al. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am. J. Hum. Genet 89, 713–730 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.French VM et al. NPHP4 variants are associated with pleiotropic heart malformations. Circ. Res 110, 1564–1574 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.van Bon BW et al. Transposition of the great vessels in a patient with a 2.9 Mb interstitial deletion of 9q31.1 encompassing the inversin gene: clinical report and review. Am. J. Med. Genet. Part. A 146A, 1225–1229 (2008). [DOI] [PubMed] [Google Scholar]
- 131.Watkins WS et al. De novo and recessive forms of congenital heart disease have distinct genetic and phenotypic landscapes. Nat. Commun 10, 4722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Knowles MR, Zariwala M & Leigh M Primary ciliary dyskinesia. Clin. Chest Med 37, 449–461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li AH et al. Genetic architecture of laterality defects revealed by whole exome sequencing. Eur. J. Hum. Genet 27, 563–573 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.van Dam TJ et al. The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia 2, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.van Dam TJP et al. CiliaCarta: an integrated and validated compendium of ciliary genes. PLoS ONE 14, e0216705 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tory K et al. Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int. 75, 839–847 (2009). [DOI] [PubMed] [Google Scholar]
- 137.Baujat G & Le Merrer M Ellis-van Creveld syndrome. Orphanet J. Rare Dis 2, 27 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Karp N, Grosse-Wortmann L & Bowdin S Severe aortic stenosis, bicuspid aortic valve and atrial septal defect in a child with Joubert syndrome and related disorders (JSRD) — a case report and review of congenital heart defects reported in the human ciliopathies. Eur. J. Med. Genet 55, 605–610 (2012). [DOI] [PubMed] [Google Scholar]
- 139.Deveault C et al. BBS genotype-phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum. Mutat 32, 610–619 (2011). [DOI] [PubMed] [Google Scholar]
- 140.Lumiaho A et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am. J. Kidney Dis 38, 1208–1216 (2001). [DOI] [PubMed] [Google Scholar]
- 141.Toomer KA et al. A role for primary cilia in aortic valve development and disease. Dev. Dyn 246, 625–634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fulmer D et al. Defects in the exocyst-cilia machinery cause bicuspid aortic valve disease and aortic stenosis. Circulation 140, 1331–1341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Koefoed K, Veland IR, Pedersen LB, Larsen LA & Christensen ST Cilia and coordination of signaling networks during heart development. Organogenesis 10, 108–125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sund KL, Roelker S, Ramachandran V, Durbin L & Benson DW Analysis of Ellis van Creveld syndrome gene products: implications for cardiovascular development and disease. Hum. Mol. Genet 18, 1813–1824 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pierpont ME, Markwald RR & Lin AE Genetic aspects of atrioventricular septal defects. Am. J. Med. Genet 97, 289–296 (2000). [PubMed] [Google Scholar]
- 146.Maslen CL et al. CRELD1 mutations contribute to the occurrence of cardiac atrioventricular septal defects in Down syndrome. Am. J. Med. Genet. A 140, 2501–2505 (2006). [DOI] [PubMed] [Google Scholar]
- 147.Ahuja N, Ostwald P, Bark D & Garrity D Biomechanical cues direct valvulogenesis. J. Cardiovasc. Dev. Dis 7, 18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Van der Heiden K et al. Monocilia on chicken embryonic endocardium in low shear stress areas. Dev. Dyn 235, 19–28 (2006). [DOI] [PubMed] [Google Scholar]
- 149.Willaredt MA, Gorgas K, Gardner HA & Tucker KL Multiple essential roles for primary cilia in heart development. Cilia 1, 23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Samsa LA et al. Cardiac contraction activates endocardial Notch signaling to modulate chamber maturation in zebrafish. Development 142, 4080–4091 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li X et al. Primary cilia mediate Klf2-dependant Notch activation in regenerating heart. Protein Cell 11, 433–445 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Iomini C, Tejada K, Mo W, Vaananen H & Piperno G Primary cilia of human endothelial cells disassemble under laminar shear stress. J. Cell Biol 164, 811–817 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rydholm S et al. Mechanical properties of primary cilia regulate the response to fluid flow. Am. J. Physiol. Ren. Physiol 298, F1096–1102 (2010). [DOI] [PubMed] [Google Scholar]
- 154.Ritter A, Louwen F & Yuan J Deficient primary cilia in obese adipose-derived mesenchymal stem cells: obesity, a secondary ciliopathy? Obes. Rev 19, 1317–1328 (2018). [DOI] [PubMed] [Google Scholar]
- 155.Groenendijk BC, Hierck BP, Gittenberger-De Groot AC & Poelmann RE Development-related changes in the expression of shear stress responsive genes KLF-2, ET-1, and NOS-3 in the developing cardiovascular system of chicken embryos. Dev. Dyn 230, 57–68 (2004). [DOI] [PubMed] [Google Scholar]
- 156.Egorova AD et al. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ. Res 108, 1093–1101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ten Dijke P, Egorova AD, Goumans MJ, Poelmann RE & Hierck BP TGF-beta signaling in endothelial-to-mesenchymal transition: the role of shear stress and primary cilia. Sci. Signal 5, pt2 (2012). [DOI] [PubMed] [Google Scholar]
- 158.Clement CA et al. The primary cilium coordinates early cardiogenesis and hedgehog signaling in cardiomyocyte differentiation. J. Cell Sci 122, 3070–3082 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wu G et al. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat. Genet 24, 75–78 (2000). [DOI] [PubMed] [Google Scholar]
- 160.Paavola J et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J. Mol. Cell Cardiol 58, 199–208 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Peralta M et al. Intraflagellar transport complex B proteins regulate the Hippo effector Yap1 during cardiogenesis. Cell Rep. 32, 107932 (2020). [DOI] [PubMed] [Google Scholar]
- 162.Toomer KA et al. Primary cilia defects causing mitral valve prolapse. Sci. Transl. Med 11, eaax0290 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fulmer D et al. Desert hedgehog-primary cilia cross talk shapes mitral valve tissue by organizing smooth muscle actin. Dev. Biol 463, 26–38 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Goddard LM et al. Hemodynamic forces sculpt developing heart valves through a KLF2-WNT9B paracrine signaling axis. Dev. Cell 43, 274–289.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lehman JM et al. The Oak Ridge polycystic kidney mouse: modeling ciliopathies of mice and men. Dev. Dyn 237, 1960–1971 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lambrechts D & Carmeliet P Sculpting heart valves with NFATc and VEGF. Cell 118, 532–534 (2004). [DOI] [PubMed] [Google Scholar]
- 167.Wu B, Baldwin HS & Zhou B Nfatc1 directs the endocardial progenitor cells to make heart valve primordium. Trends Cardiovasc. Med 23, 294–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wu B et al. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ. Res 109, 183–192 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Luu VZ, Chowdhury B, Al-Omran M, Hess DA & Verma S Role of endothelial primary cilia as fluid mechanosensors on vascular health. Atherosclerosis 275, 196–204 (2018). [DOI] [PubMed] [Google Scholar]
- 170.Van der Heiden K, Egorova AD, Poelmann RE, Wentzel JJ & Hierck BP Role for primary cilia as flow detectors in the cardiovascular system. Int. Rev. Cell Mol. Biol 290, 87–119 (2011). [DOI] [PubMed] [Google Scholar]
- 171.Praetorius HA & Spring KR The renal cell primary cilium functions as a flow sensor. Curr. Opin. Nephrol. Hypertens 12, 517–520 (2003). [DOI] [PubMed] [Google Scholar]
- 172.Praetorius HA & Spring KR Bending the MDCK cell primary cilium increases intracellular calcium. J. Membr. Biol 184, 71–79 (2001). [DOI] [PubMed] [Google Scholar]
- 173.Praetorius HA & Spring KR Removal of the MDCK cell primary cilium abolishes flow sensing. J. Membr. Biol 191, 69–76 (2003). [DOI] [PubMed] [Google Scholar]
- 174.Nauli SM et al. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 117, 1161–1171 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.AbouAlaiwi WA et al. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ. Res 104, 860–869 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Groenendijk BC et al. Changes in shear stress-related gene expression after experimentally altered venous return in the chicken embryo. Circ. Res 96, 1291–1298 (2005). [DOI] [PubMed] [Google Scholar]
- 177.Hierck BP et al. Primary cilia sensitize endothelial cells for fluid shear stress. Dev. Dyn 237, 725–735 (2008). [DOI] [PubMed] [Google Scholar]
- 178.Ferreira RR, Fukui H, Chow R, Vilfan A & Vermot J The cilium as a force sensor-myth versus reality. J. Cell Sci 132, jcs213496 (2019). [DOI] [PubMed] [Google Scholar]
- 179.Poelmann RE, Van der Heiden K, Gittenberger-de Groot A & Hierck BP Deciphering the endothelial shear stress sensor. Circulation 117, 1124–1126 (2008). [DOI] [PubMed] [Google Scholar]
- 180.Pala R, Jamal M, Alshammari Q & Nauli SM The roles of primary cilia in cardiovascular diseases. Cells 7, 233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Heckel E et al. Oscillatory flow modulates mechanosensitive klf2a expression through trpv4 and trpp2 during heart valve development. Curr. Biol 25, 1354–1361 (2015). [DOI] [PubMed] [Google Scholar]
- 182.Li H, Chang C, Li X & Zhang R The roles and activation of endocardial Notch signaling in heart regeneration. Cell Regen. 10, 3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Galvez-Santisteban M et al. Hemodynamic-mediated endocardial signaling controls in vivo myocardial reprogramming. eLife 8, e44816 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Rash JE, Shay JW & Biesele JJ Cilia in cardiac differentiation. J. Ultrastruct. Res 29, 470–484 (1969). [DOI] [PubMed] [Google Scholar]
- 185.Myklebust R, Engedal H, Saetersdal TS & Ulstein M Primary 9 + 0 cilia in the embryonic and the adult human heart. Anat. Embryol 151, 127–139 (1977). [DOI] [PubMed] [Google Scholar]
- 186.Diguet N, Le Garrec JF, Lucchesi T & Meilhac SM Imaging and analyzing primary cilia in cardiac cells. Methods Cell Biol. 127, 55–73 (2015). [DOI] [PubMed] [Google Scholar]
- 187.Villalobos E et al. Fibroblast primary cilia are required for cardiac fibrosis. Circulation 139, 2342–2357 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Blom JN, Lu X, Kim MY & Feng Q Primary cilia disassembly promotes mammalian cardiac regeneration [abstr.]. FASEB J. 30 (Suppl. 1), 1207.7 (2016).26644352 [Google Scholar]
- 189.Poss KD, Wilson LG & Keating MT Heart regeneration in zebrafish. Science 298, 2188–2190 (2002). [DOI] [PubMed] [Google Scholar]
- 190.Raya A et al. Activation of Notch signaling pathway precedes heart regeneration in zebrafish. Proc. Natl Acad. Sci. USA 100 (Suppl. 1), 11889–11895 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhao L et al. Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc. Natl Acad. Sci. USA 111, 1403–1408 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhao L, Ben-Yair R, Burns CE & Burns CG Endocardial notch signaling promotes cardiomyocyte proliferation in the regenerating zebrafish heart through wnt pathway antagonism. Cell Rep. 26, 546–554.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Guichard C et al. Axonemal dynein intermediate-chain gene (DNAI1) mutations result in situs inversus and primary ciliary dyskinesia (Kartagener syndrome). Am. J. Hum. Genet 68, 1030–1035 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zariwala MA et al. Mutations of DNAI1 in primary ciliary dyskinesia: evidence of founder effect in a common mutation. Am. J. Respir. Crit. Care Med 174, 858–866 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liu H et al. Next-generation sequencing in a series of 80 fetuses with complex cardiac malformations and/or heterotaxy. Hum. Mutat 41, 2167–2178 (2020). [DOI] [PubMed] [Google Scholar]
- 196.Mitchison HM et al. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet 44, 381–389 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hornef N et al. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am. J. Respir. Crit. Care Med 174, 120–126 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Duriez B et al. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc. Natl Acad. Sci. USA 104, 3336–3341 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Knowles MR et al. Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax 67, 433–441 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bartoloni L et al. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc. Natl Acad. Sci. USA 99, 10282–10286 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Omran H et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature 456, 611–616 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Merveille AC et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet 43, 72–78 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Becker-Heck A et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet 43, 79–84 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mazor M et al. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am. J. Hum. Genet 88, 599–607 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Panizzi JR et al. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat. Genet 44, 714–719 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Horani A et al. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet 91, 685–693 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kott E et al. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet 91, 958–964 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Onoufriadis A et al. Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am. J. Hum. Genet 92, 88–98 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zariwala MA et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am. J. Hum. Genet 93, 336–345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Moore DJ et al. Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am. J. Hum. Genet 93, 346–356 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hjeij R et al. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet 93, 357–367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tarkar A et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet 45, 995–1003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Austin-Tse C et al. Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. Am. J. Hum. Genet 93, 672–686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Knowles MR et al. Mutations in SPAG1 cause primary ciliary dyskinesia associated with defective outer and inner dynein arms. Am. J. Hum. Genet 93, 711–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hjeij R et al. CCDC151 mutations cause primary ciliary dyskinesia by disruption of the outer dynein arm docking complex formation. Am. J. Hum. Genet 95, 257–274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Paff T et al. Mutations in PIH1D3 cause X-linked primary ciliary dyskinesia with outer and inner dynein arm defects. Am. J. Hum. Genet 100, 160–168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Duquesnoy P et al. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet 85, 890–896 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Loges NT et al. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet 85, 883–889 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hartill VL et al. DNAAF1 links heart laterality with the AAA+ ATPase RUVBL1 and ciliary intraflagellar transport. Hum. Mol. Genet 27, 529–545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Perles Z et al. A human laterality disorder associated with recessive CCDC11 mutation. J. Med. Genet 49, 386–390 (2012). [DOI] [PubMed] [Google Scholar]
- 221.Li Y et al. DNAH6 and its interactions with PCD genes in heterotaxy and primary ciliary dyskinesia. PLoS Genet. 12, e1005821 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wallmeier J et al. TTC25 deficiency results in defects of the outer dynein arm docking machinery and primary ciliary dyskinesia with left-right body asymmetry randomization. Am. J. Hum. Genet 99, 460–469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bataille S et al. Association of PKD2 (polycystin 2) mutations with left-right laterality defects. Am. J. Kidney Dis 58, 456–460 (2011). [DOI] [PubMed] [Google Scholar]
- 224.Vetrini F et al. Bi-allelic mutations in PKD1L1 are associated with laterality defects in humans. Am. J. Hum. Genet 99, 886–893 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Otto EA et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet 34, 413–420 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Okada M et al. Association of INVS (NPHP2) mutation in an adolescent exhibiting nephronophthisis (NPH) and complete situs inversus. Clin. Nephrol 69, 135–141 (2008). [DOI] [PubMed] [Google Scholar]
- 227.Chaki M et al. Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int. 80, 1239–1245 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Bergmann C et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am. J. Hum. Genet 82, 959–970 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Hoff S et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat. Genet 45, 951–956 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Frank V et al. Mutations in NEK8 link multiple organ dysplasia with altered Hippo signalling and increased c-MYC expression. Hum. Mol. Genet 22, 2177–2185 (2013). [DOI] [PubMed] [Google Scholar]
- 231.Stone DL et al. Mutation of a gene encoding a putative chaperonin causes McKusick–Kaufman syndrome. Nat. Genet 25, 79–82 (2000). [DOI] [PubMed] [Google Scholar]
- 232.Kaasinen E et al. Recessively inherited right atrial isomerism caused by mutations in growth/differentiation factor 1 (GDF1). Hum. Mol. Genet 19, 2747–2753 (2010). [DOI] [PubMed] [Google Scholar]
- 233.Boskovski MT et al. The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature 504, 456–459 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ruiz-Perez VL et al. Mutations in a new gene in Ellis–van Creveld syndrome and Weyers acrodental dysostosis. Nat. Genet 24, 283–286 (2000). [DOI] [PubMed] [Google Scholar]
- 235.Durst R et al. Mutations in DCHS1 cause mitral valve prolapse. Nature 525, 109–113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Del Viso F et al. Congenital heart disease genetics uncovers context-dependent organization and function of nucleoporins at cilia. Dev. Cell 38, 478–492 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Williams K, Carson J & Lo C Genetics of congenital heart disease. Biomolecules 9, 879 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hamada H in Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology (eds Nakanishi T et al. ) 57–65 (Springer, 2016). [PubMed] [Google Scholar]