

Abstract
Hyperfiltration (HF) is a state of high glomerular filtration rate (GFR) observed in early diabetes that damages glomeruli, resulting in an iterative process of increasing filtration load on fewer and fewer remaining functional glomeruli. To delineate underlying cellular mechanisms of damage associated with HF, transcriptional profiles of kidney biopsies from Pima Indians with type 2 diabetes with or without early-stage diabetic kidney disease (DKD) were grouped into two HF categories based on annual iothalamate GFR measurements. Twenty-six participants with a peak GFR measurement within two years of biopsy were categorized as the “HF group”, and 26 in whom biopsy preceded peak GFR by >2 years were considered “pre-HF”. The HF group had higher hemoglobin A1c, higher urine albumin-to-creatinine ratio, increased glomerular basement membrane width and lower podocyte density compared to the pre-HF group. A glomerular 1240-gene transcriptional signature identified in the HF group was enriched for endothelial stress response signaling genes, including from endothelin-1, tec-kinase and TGF-β1 pathways, with the majority of the transcripts mapped to endothelial and inflammatory cell clusters in kidney single cell transcriptional data. This analysis reveals molecular pathomechanisms associated with HF in early DKD involving putative ligand-receptor pairs with downstream intracellular targets linked to cellular crosstalk between endothelial and mesangial cells.
Introduction
Diabetic kidney disease (DKD) is a common cause of kidney failure with rising prevalence worldwide1, 2. Alterations in glomerular hemodynamic function at the onset of diabetes often lead to sustained increases in the glomerular filtration rate (GFR), commonly referred to as hyperfiltration (HF). The presence of HF is considered a key early driver of DKD as HF is associated with development and progression of DKD3–5 and with increased mortality6 and is considered a prime therapeutic target in DKD. HF may also be part of the etiology of some non-diabetic chronic kidney diseases including obesity-related glomerulopathy7, 8. The pathomechanisms triggered in HF are not fully understood. Proposed mechanisms include increased intra-renal nitric oxide signaling, tubular sodium and glucose reabsorption, and intraglomerular mechanical stress from glomerular hypertension7, 9–12. However compelling evidence on how these and other pathways are regulated in the kidneys in HF is yet to be established.
We explored pathomechanisms associated with HF in Pima Indians from the Gila River Indian Community in Arizona who have a very high prevalence of obesity, type 2 diabetes and DKD. Individuals from this community have participated in decades-long prospective studies of early DKD that included clinical data and serial measurements of GFR by iothalamate clearance. A subset of these participants also underwent research kidney biopsies. The structural lesions observed in kidney biopsies in the Pima Indians are attributable to diabetes, without evidence of obesity-related or hypertensive glomerulopathy13, which could also induce HF. Previous studies in this cohort documented the presence of HF and established a temporal link to the onset of diabetes5, 14. Although the diagnosis of HF typically relies on measures of whole kidney GFR, without accounting for individual differences in nephron numbers15, 16, a previous study of kidney donors found similar levels of single-nephron GFR across a range of whole-kidney GFR, underlining the problem with absolute whole-kidney GFR HF thresholds17. To more accurately reflect HF at a single nephron level, we defined HF as peak GFR within each individual based on observed trends during long-term follow-up. We then examined transcriptional differences in kidney tissue obtained from individuals who reached peak GFR around the time of kidney biopsy and in those who reached peak GFR well after biopsy to identify signatures associated with HF that may contribute to progression of DKD. The aim of the present study was to integrate clinical measurements, structural morphometry, and gene expression analyses of kidney tissue, including single cell RNAseq (scRNAseq) analyses, to identify pathways associated with HF that may promote progressive DKD and be amenable to targeted therapies. An overview of the analytical strategy used in this study is shown in Figure 1.
Figure 1. Explanatory figure describing the analytical approach of the study.
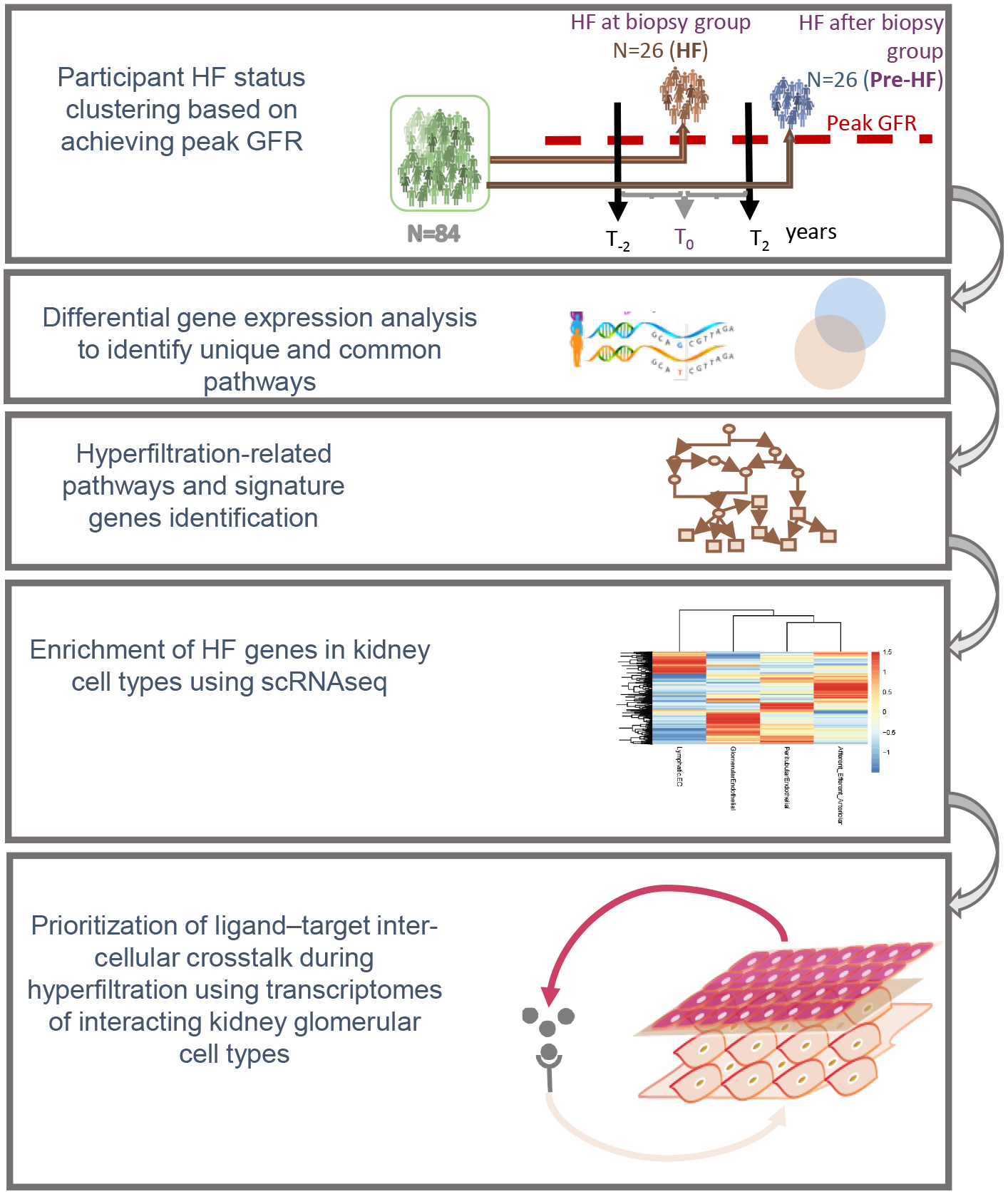
Participants were clustered based on peak measured GFR and their kidney biopsy transcriptional profiles compared to identify HF related pathways and the specific cells in the kidney involved in HF related glomerular injury pathways.
Methods
Study population
Pima Indians from the Gila River Indian Community participated in a longitudinal study of diabetes and its complications between 1965 and 2007. In 1996, 169 Pima adults with type 2 diabetes participated in a prospective, randomized, placebo-controlled, double-blinded intervention trial (Renoprotection in Early Diabetic Nephropathy in Pima Indians trial, clinicaltrials.gov, NCT00340678)18. At the end of the 6-year trial, 111 of the participants underwent a kidney biopsy. Subsequently, all trial participants were followed-up annually. In 2015, 1st degree relatives of clinical trial participants (N=42) enrolled in the follow-up study, and clinical trial participants not previously biopsied (N=2), provided kidney biopsies which were used for the scRNAseq analysis described below. Follow-up continued through 2019. All participants provided informed consent. Due to privacy protection concerns, individual-level genotype and gene expression data from this study cannot be made publicly available.
Peak GFR categorization
Individuals who had a mean GFR <60 ml/min and/or a mean urine albumin-to-creatinine ratio (ACR) >300 mg/g within 1 year of their kidney biopsy were considered to have advanced DKD and were excluded, leaving 84 participants considered for the present study. GFR measurements performed in all kidney study protocols were reviewed, and these participants were classified into 3 groups based on the date they had their highest recorded GFR measurement (peak GFR), regardless of its absolute value. The absolute GFR was used because the study involved overweight and obese participants and indexing for body surface area may significantly underestimate their actual GFR and mask HF19. Those who achieved peak GFR <2 years from the time of biopsy (N=26) were categorized as the “HF group”. The remaining 58 participants were split into two groups based on whether their peak was >2 years before (N=32) or >2 years after (N=26) the biopsy. Those whose GFR peak was >2 years before the biopsy were considered likely to have more advanced DKD because of their declining GFR and were excluded from subsequent analysis, whereas the 26 participants with a peak >2 years after biopsy were included in the analyses and were referred to as the “Pre-HF group”. A flow chart of participant selection for this study is summarized in Figure 2. Seven participants reached the identical GFR peak twice. Of these, two had their peaks within the same time interval category whereas three participants had their initial peak >2 years before biopsy and the second peak within 2 years of biopsy, and two had their first peak within 2 years of biopsy and then a second peak >2 years after biopsy. In these cases, the first GFR peak was used for the purpose of categorization. Individual time-course plots of GFR for study participants by timing of peak GFR are provided in Supplemental Figure S1. As a sensitivity analysis, peak GFR was also categorized based on creatinine-based estimated GFR computed using the CKD-EPI equation20.
Figure 2. Flow chart describing participant selection.
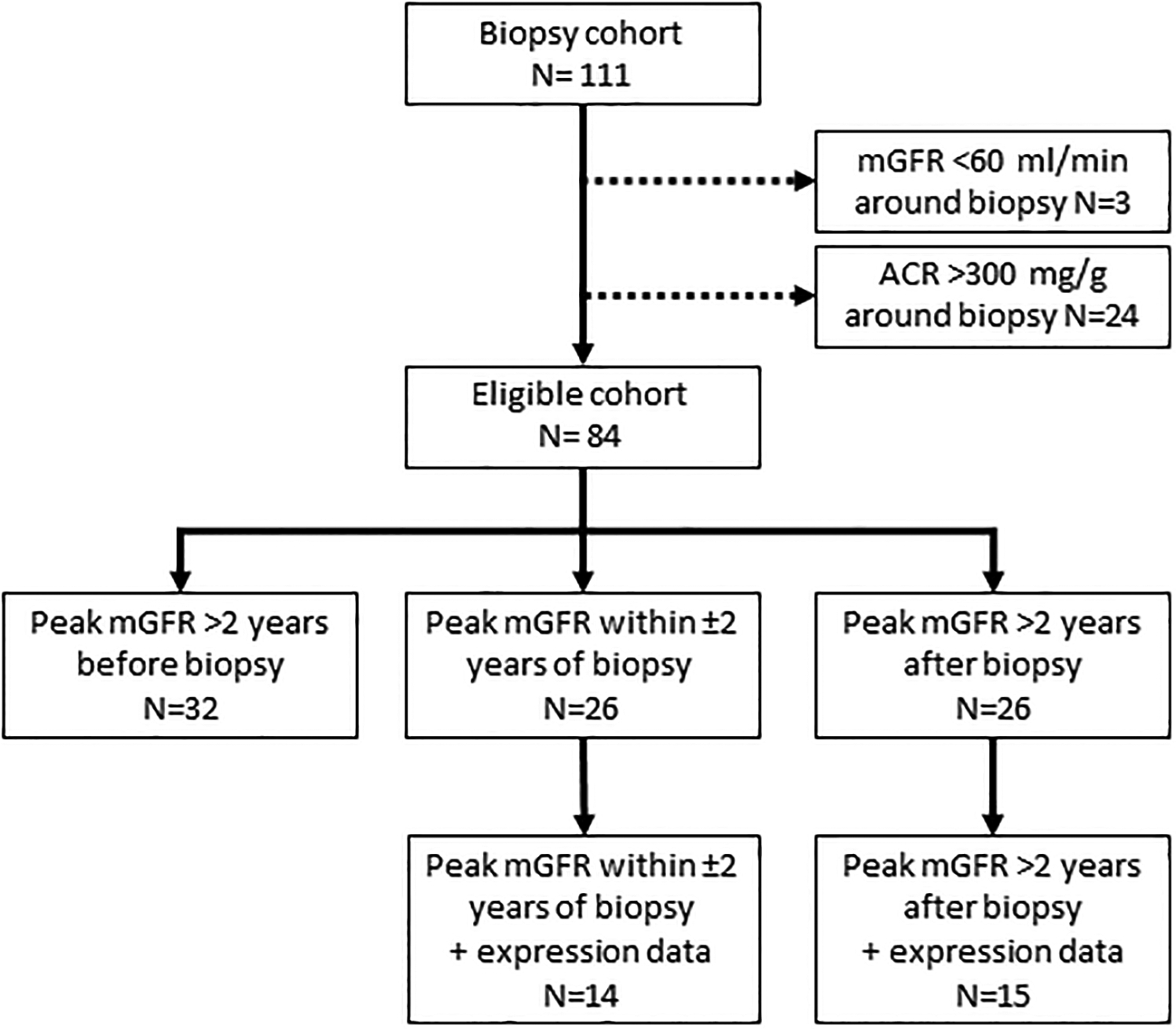
Participants, twenty-six each, in the HF group and pre-HF group were identified from those in the study cohort with a kidney biopsy (N=111), based on their peak GFR and in a subset with expression profiles.
Quantitative morphometry
Structural parameters were measured by unbiased random sampling. Biopsy tissue was processed and embedded in epoxy resin (LX112; Ladd Research Industries, Williston, VT). Measurements were made from digital micrographs, and stereological methods were used to account for 2-dimensional sampling of 3-dimensional objects18. Tissue was prepared for light and electron microscopy studies according to standard procedures21–23. The following glomerular structural parameters were measured on electron microscopy images as described elsewhere21, 22, 24: glomerular basement membrane width25, 26, mesangial fractional volume, (including mesangial cell and mesangial matrix fractional volumes)25, 26, glomerular filtration surface density25, 26, foot process width27, percentage of endothelial fenestrations27, and the glomerular podocyte fractional volume per glomerulus28. Cortical interstitial fractional volume23 and mean glomerular volume29, 30 were estimated using light microscopy.
Gene co-expression analyses
RNA sequencing data obtained from microdissected glomerular and tubulointerstitial compartments from kidney biopsy tissue were analyzed (supplemental methods). Eigengene-based weighted gene co-expression network analysis modules were constructed using Weighted Gene Co-expression Network Analysis (WGCNA)31(supplemental methods). Eigen genes were then correlated (Pearson correlation) with the HF categories and the morphometric parameters. Transcripts contained in modules with statistically significant (p ≤0.05) associations to these traits were used for downstream functional analysis. All statistical analysis were done in R statistical software (www.r-project.org) and Stata MP 15 (Stata Corp., College Station, TX; www.stata.com). Ingenuity Pathway System (IPA) (Qiagen, Redwood City, CA, USA) was used to reveal associated functional pathways. Statistical significance was set at a Bonferroni adjusted p- value of < 0.05.
Pathway Analysis
Pathway analysis was performed using the IPA software querying the HR associated transcripts. Cytoscape32 visualization platform and ggplot2 package in R statistical platform was used to render the network images from the pathway network. An in-house custom python script was used to parse the IPA output for the Cytoscape subnetwork generations. Pathways with fewer than 5 shared genes were filtered out to reduce the number of nodes in the pathway network. Major cancer pathways were also removed. The resulting network yielded 175 nodes and 5288 edges. Default parameters in the Cytoscape plugin, MCODE33 were used to construct subnetworks.
scRNAseq data generation and analysis
scRNAseq data were generated from CryoStor® (Stemcell Technologies) preserved DKD (N=44) and control (living donor, N=18) biopsies34, as previously published34, 35. Individual cell barcoding, reverse RNA transcription, library generation and single cell sequencing using Illumina were all performed using the 10X Genomics protocol34. The output from the sequencer was first processed by CellRanger (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger). CellRanger output data files were analyzed using the Seurat 3 R package (https://cran.r-project.org/web/packages/Seurat/index.html)34.
Hub genes from the HF signature modules were compared with an external scRNAseq data set obtained from protocol biopsies of patients with youth onset type 2 diabetes36. This data set included healthy controls (HC, n=6) and participants ranging from 12–21 years with type 2 diabetes (T2D, n=6) from the Renal Hemodynamics, Energetics and Insulin Resistance in Youth Onset Type 2 Diabetes Study (Renal-HEIR, NCT03584217) and the Impact of Metabolic Surgery on Pancreatic, Renal and Cardiovascular Health in Youth with Type 2 Diabetes (IMPROVE-T2D, NCT03620773). T2D were obese (BMI 38.0±4.9 kg/m2) and had elevated measured iohexol GFR (208±53 ml/min), but normal to borderline elevated ACR (10±10 mg/g). Morphometric evaluation of the kidney biopsies showed higher mesangial and glomerular volumes in T2D compared to HC, consistent with early kidney dysfunction but no features indicative of advanced DKD36. scRNAseq followed the same protocol described above.
Cell-Cell Crosstalk Analysis
NicheNetR37 (https://github.com/saeyslab/nichenetr) was used to identify ligand-receptor (LR) interactions that drive the observed expression changes in the target cell population in the single cell transcriptome. NichNetR compiles literature-based ligand-receptor interactions, signal transductions and regulatory networks to prioritize the ligand-receptor-target gene identification. Based on cell type enrichment of HF genes, the LR interactions and downstream target genes were identified from the cell-cell communication between endothelial and mesangial cells.
Results
Participant characteristics
Clinical and demographic characteristics for the HF and pre-HF groups (Table 1) showed a mean GFR in the HF group of 173±48 ml/min, 21 ml/min higher, on average, than in the pre-HF group (152±38 ml/min, p=0.08). The HF group also had higher mean HbA1c (hemoglobin A1c, p=0.03) and median ACR (p=0.007). Though not statistically significant, there was also a trend towards longer duration of diabetes. There were no statistically significant differences in the initial study GFR or the maximal GFR between groups. Comparison of histopathological structural parameters from the kidney biopsies (Table 2) showed wider glomerular basement membrane (p=0.02) and higher mesangial fractional volume (p=0.004) among those in the HF group, reflecting greater structural changes near peak GFR, compared to pre-HF. The difference in mesangial fractional volume was due predominantly to differences in the mesangial matrix fractional volume (p=0.0001) and not to differences in mesangial cell fractional volume (p=0.25). Podocyte density (p=0.03) was also slightly lower among those in the HF group, but podocyte fractional volume of the glomerulus was maintained, indicating that podocytes were fewer in number but larger. Clinical characteristics and structural parameters in the 32 participants not included in the primary analyses, because their peak GFR occurred >2 years before the kidney biopsy, compared with the 52 participants included in the study are provided in Supplemental Table S1. These patients had longer diabetes duration (p<0.001), lower GFR (p=0.007), and tissue morphometric measurements including increased cortical interstitial fractional volume and decreased glomerular podocyte fractional volume (p=0.032), reflective of their more advanced stage of DKD.
Table 1.
Clinical characteristics at time of kidney biopsy by peak measured GFR group
| Characteristic | Pre-HF (n=26) | HF (n=26) | p-value |
|---|---|---|---|
| Male sex (%) | 7 (26.9) | 3 (11.5) | 0.16 |
| Age (years) | 44.8 ± 8.6 | 44.6 ± 12.0 | 0.95 |
| Diabetes duration (years) | 12.4 ± 3.4 | 14.4 ± 4.3 | 0.07 |
| BMI (kg/m2) | 37.0 ± 8.0 | 37.2 ± 9.2 | 0.91 |
| Systolic blood pressure (mmHg) | 120 ± 8 | 121 ± 8 | 0.70 |
| Diastolic blood pressure (mmHg) | 75 ± 5 | 77 ± 5 | 0.41 |
| HbA1c (%) | 8.7 ± 2.0 | 9.9 ± 1.8 | 0.03 |
| mGFR (ml/min) | 152 ± 38 | 173 ± 48 | 0.08 |
| Mean mGFR (ml/min) | 154 ± 30 | 149 ± 38 | 0.57 |
| Peak mGFR (ml/min) | 221 ± 48 | 236 ± 47 | 0.25 |
| ACR (mg/g) | 18 (13, 31) | 58 (34, 74) | 0.007 |
| RAS use (%) | 21 (80.8%) | 17 (65.4%) | 0.21 |
Values are means ± standard deviation, n and (frequency), or median (interquartile range).
Abbreviations: ACR = albumin-to-creatinine ratio, BMI = body mass index, HbA1c = hemoglobin A1c, mGFR = measured glomerular filtration rate; RAS = renin angiotensin system blockers; Mean mGFR is the mean of all available measured glomerular filtration rates; Peak mGFR = highest measured glomerular filtration rate.
p-value for male sex is from a chi-square test; p-values for continuous variables are based on t-tests; ACR was log transformed prior to analysis.
Table 2.
Morphometric measures by peak measured GFR group
| Structural parameter | Pre-HF (n=26) | HF (n=26) | p-value |
|---|---|---|---|
| Mean glomerular volume (106 μm3) | 2.25 ± 0.68 * | 2.51 ± 0.75 ‡ | 0.22 |
| Glomerular basement membrane width (nm) | 428 ± 75 | 484 ± 88 | =0.02 |
| Mesangial fractional volume per glomerulus | 0.23 ± 0.05 | 0.27 ± 0.05 | 0.004 |
| Mesangial cell fractional volume per glomerulus | 0.08 ± 0.02 | 0.09 ± 0.02 | 0.25 |
| Mesangial matrix fractional volume per glomerulus | 0.11 ± 0.03 | 0.14 ± 0.04 | 0.0001 |
| Cortical interstitial fractional volume | 0.17 (0.03) † | 0.19 (0.04) ‡ | 0.06 |
| Glomerular filtration surface density (μm2/μm3) | 0.12 ± 0.04 | 0.10 ± 0.04 | 0.21 |
| Foot process width (nm) | 726 (637, 952) | 821 (631, 1175) | 0.53 |
| Glomerular podocyte fractional volume | 0.18 ± 0.06 | 0.16 ± 0.05 | 0.19 |
| Podocyte number density per glomerulus (106 μm3) | 161 ± 81 | 116 ± 54 | 0.03 |
| Fenestrated endothelium (%) | 44.5 ± 16.4 | 43.5 ± 21.8 | 0.85 |
Values are means ± standard deviation or median (interquartile range).
n=22,
n=25,
n=24
p-values are based on t-tests; foot process width was log transformed prior to analysis.
To determine whether HF categorization based on measured GFR could be replicated using estimated GFR (eGFR), we repeated the analyses using the serum creatinine based CKD-EPI equation20 (Supplemental Table S2). eGFR trajectories resulted in significant misclassification with the vast majority (71/84; 85%) of study participants being classified as reaching peak eGFR >2 years before biopsy. Only 4 participants (5%) had a peak eGFR within 2 years of biopsy with the remaining 9 participants (11%) reaching peak eGFR >2 years after biopsy. Thus, eGFR was not a useful proxy for measured GFR in determining the timing of peak GFR. Accordingly, all gene expression analyses described below were based on measured GFR trajectories.
Twenty-nine of the 52 participants had sufficient tissue available from kidney biopsies for Affymetrix-based glomerular gene expression analyses (14 in the peak HF group and 15 in the HF after biopsy group). The clinical and morphometric characteristics of this subset with expression data were similar to those without expression data except that those with expression data were younger (p=0.005), had shorter diabetes duration (p=0.02), lower glomerular filtration surface density (p=0.02), and more fenestrated endothelium (p=0.005) (Supplemental Table S3). Just as in the full cohort, the HF group in the subset of participants with expression data had higher HbA1c (p=0.006) and ACR (p=0.05) than those in the pre-HF group. Unlike the full cohort there was significantly lower use of renin-angiotensin-system (RAS) inhibitors among the HF group compared to the pre-HF group (57.1% vs. 93.3%, p=0.04). Glomerular basement membrane width and mesangial fractional volume, again driven by mesangial matrix fractional volume, remained higher in the HF group, although only the difference in mesangial matrix fractional volume was statistically significant, and podocyte density per glomerulus was significantly lower. As with the full cohort, statistically significant differences in the other podocyte parameters were not observed in the subset of participants with expression data (Supplemental Table S4).
Glomerular compartment gene expression associations and HF gene modules
Gene expression profiles were first analyzed using data driven WGCNA resulting in 22 co-expression gene clusters (modules) from the glomerular compartment (Figure 3A). Three of the glomerular modules were significantly associated with HF. Of the 1240 differentially expressed genes represented in these modules, 95% were upregulated in the HF compared to pre-HF group (Figure 3B, Supplemental Table S5). These modules correlated (Figure 3B) positively with peak GFR at time of biopsy and negatively with mesangial cell volume, consistent with observed associations at the cohort level. However, none of these 3 modules showed strong association with GFR and ACR when analyzed as continuous variables. Similar analysis of the tubulointerstitial compartment from 22 participants, with 7 reaching peak GFR at the time of biopsy, resulted in 18 modules. None of these modules showed any association with HF status.
Figure 3. HF associated modules and their eigengenes’ association with duration of diabetic kidney disease.
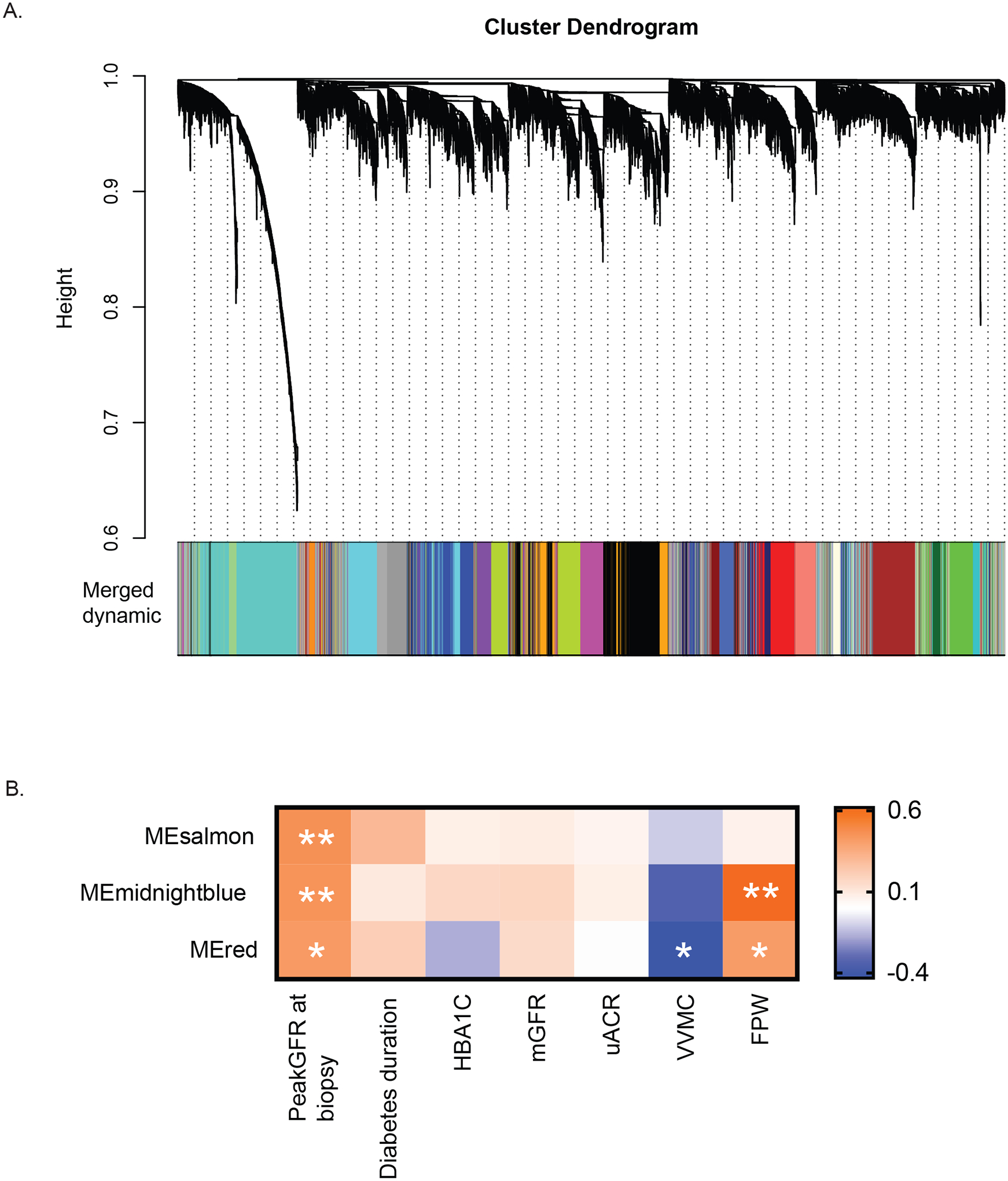
A. Cluster dendrogram of WGCNA based gene modules from glomerular transcription data from 29 participants. B. Three modules had ≥95% genes over-expressed in the HF group compared to pre-HF group. These modules were also significantly (*p<0.05, **p≤0.01) positively correlated with peak GFR at time of biopsy and negatively with mesangial cell volume (VVMC).
Abbreviations: ME- modules; PeakGFRatBx – Peak GFR at kidney biopsy/HF group; HbA1c- Hemoglobin A1c; mGFR-measured glomerular filtration rate; uACR- urine albumin-to-creatinine ratio; VVMC - mesangial fractional volume per glomerulus; FPW- foot process width
HF Pathways
Pathway analyses of the genes in these three modules yielded 194 significantly associated pathways (p≤0.05) (Supplemental Table S6). Figure 4 lists the top 100 pathways associated with the HF genes. Pathways included in this list were selected based on the adjusted p-value and number of HF genes in each pathway, presented alphabetically. Among these key enriched pathways representing the HF milieu were known growth factor signaling pathways such as platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF) as well as endhothelin-1, TGFB1, integrin, angiogenesis and epithelial-mesenchymal transition pathways. The network maps of the top 30 interconnected HF genes (Supplemental Figure S2) and Cytoscape pathway analysis (Supplemental Figure S3) identified many of the same key pathways.
Figure 4. Top 100 significantly enriched pathways represented in the HF gene set arranged alphabetically.
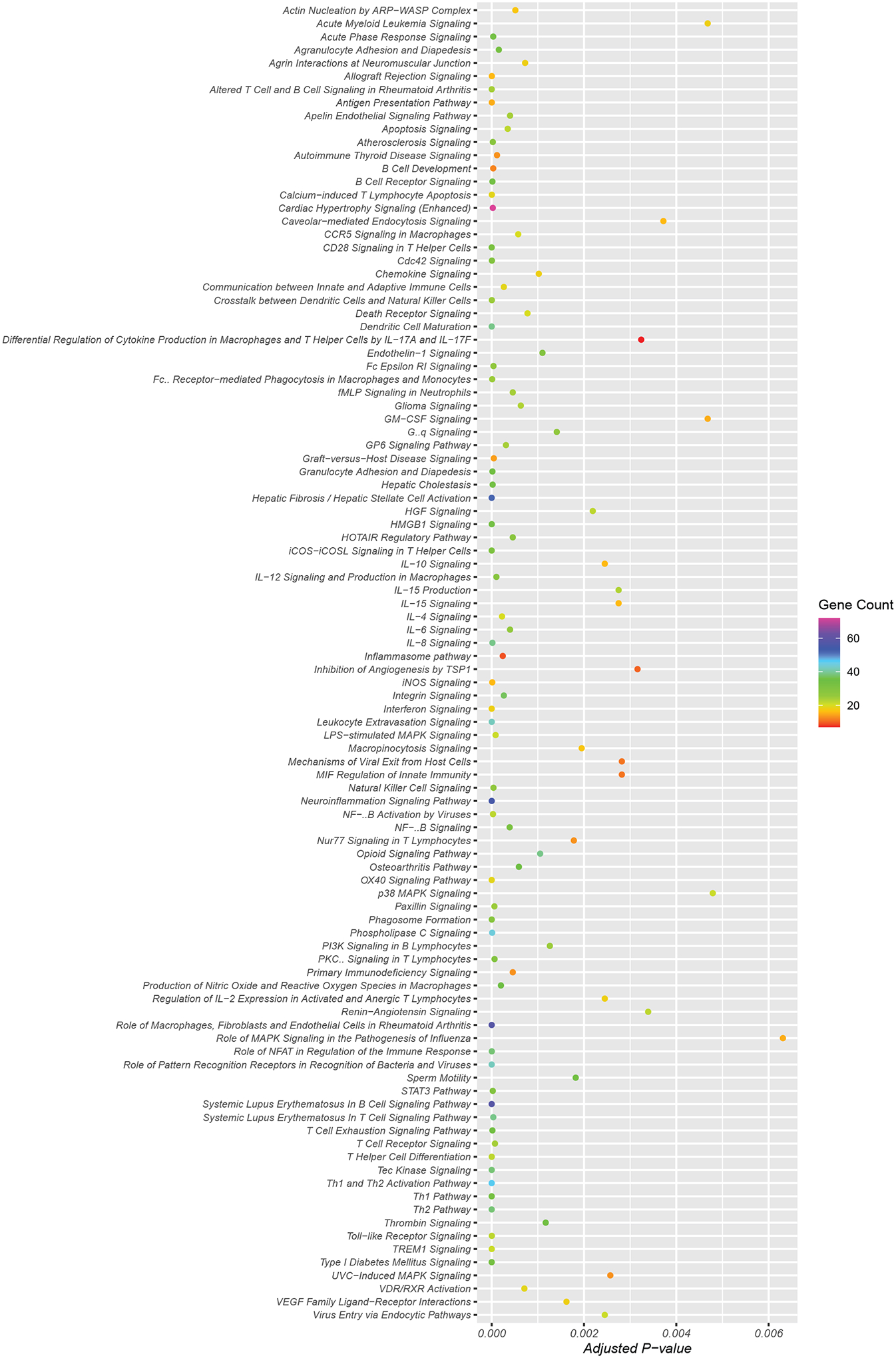
The top100 pathways encompassed by the HF genes were sorted based on adjusted p-values and plotted using the R package ggplot. The pathways in the figure are ordered alphabetically. The dots (Gene counts) are colored based on the number of pathway genes (purple >70; red <10) represented in the HF gene set and the x-axis describes membership of these genes in the corresponding pathway (adjusted p-values for all the genes in a dot)
Kidney localization of HF activation signals using scRNAseq analyses
To better elucidate the regulation of these pathways within the kidney, the cellular localization of active HF associated pathways were determined using scRNAseq data. Kidney biopsy samples from an independent group of 44 Pima Indians representing the same population pool with type 2 diabetes and 18 non-Pima controls were analyzed34. Clinical and morphometric measures for this group are shown in Supplemental Table S7. The 1240 genes from the 3 HF associated modules (ME salmon, ME midnight blue, ME red) identified in the primary Pima cohort were mapped onto the 20 cell clusters identified from this cohort (Figure 5A). Mapping these modules associated with HF to the single cell signature enabled us to identify the representative cell types responsible for the association. The highest enrichment (28%) was to the endothelial clusters indicating more than 350 of the HF genes were enriched (higher expression) in endothelial cells followed by fibroblasts and myeloid cells (Figure 5B). Further subclustering of the endothelial cell cluster and mapping of HF gene expression (Figure 5C) revealed a clear pattern of enriched activation of HF genes in intrinsic glomerular endothelial cells, where 30% of the HF genes showed maximal expression compared to all other cell clusters. The HF signature was evaluated in an independent protocol biopsy cohort of HC (n=6) and T2D (n=6). Even with the limited sample size available, a subset (296/1240, 24%) of these HF genes also showed differential regulation with significant enrichment (adjusted p-value <=0.05) in the endothelial cluster of this hyperfiltrating cohort compared to HC. The hub genes from the 3 modules also displayed endothelial cell-specific expression differences (Supplemental Figure S4).
Figure 5. Single cell RNAseq (scRNAseq) analyses.
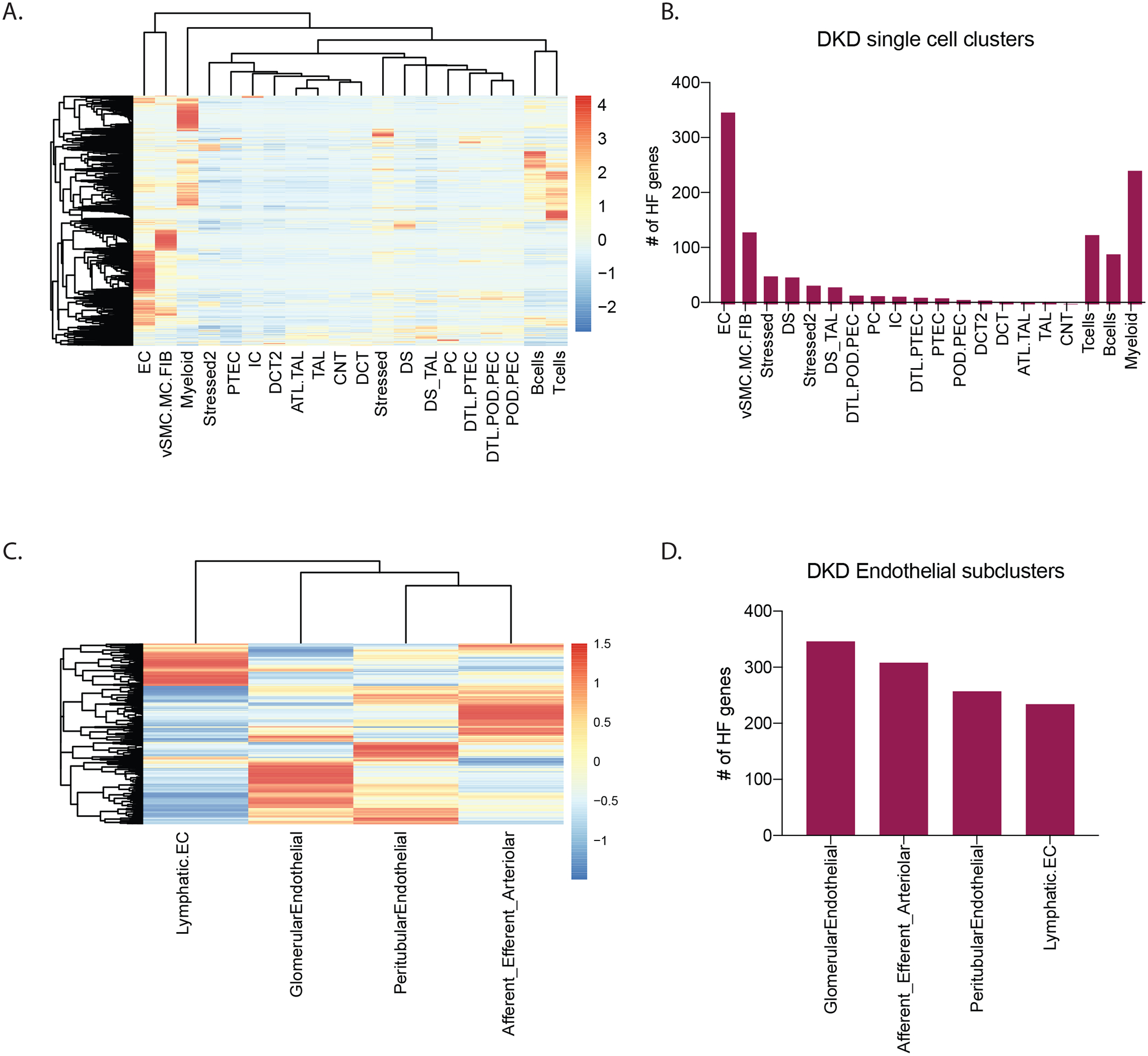
A. Heatmap shows relative expression of HF genes in each of the cell type clusters derived from scRNAseq of kidney biopsies, with higher expression (red) of HF genes visualized in the endothelial cell (EC) cluster. B. Plot of the number of expressed HF genes in each scRNAseq cell type cluster also shows presence of more HF genes in the EC cluster. C. Heatmap shows expression profiles of HF genes and D. number of HF genes expressed in each EC subcluster. Number of genes in panels B and D were defined by their maximum average expression in each of the clusters. Each gene was assigned to only one cluster based on its maximum expression.
Abbreviations: ATL-Ascending thin loop of Henle; CNT-Connecting tubule; DCT-Distal connecting tubule; DTL- Descending loop of Henle; EC- endothelial cells; IC- intercalated cells; MC-Mesangial Cell; PC- principal cells; PEC- Parietal epithelial cell: POD- Podocyte; PTEC- proximal tubular cells; TAL- thick ascending loop; vSMC-Vascular smooth muscle cells; Stressed/ Stressed 2-Clusters showing ribosomal/stress genes/ injury markers as top marker genes (Stressed cluster were akin to stressed proximal or descending loop of Henle cells whereas cells in the Stressed 2 cluster shared similarities with distal nephron cells.)
Ligand-Receptor interactions during HF
Pathway and single cell enrichment analysis of HF associated genes all indicate activation of transcription pathways in endothelial cells (Figure 5, Supplemental Table S6, Supplemental Figure S2). This activation was further explored using a ligand-receptor-target gene network approach37. In addition to establishing the link between the ligand and receptors in the sender/receiver cells, NicheNetR also connects the ligand-receptor networks with upregulated downstream intracellular signaling target transcripts in the receiver cells as a surrogate for pathway activation, with significance defined by NicheNetR. The top 15 ligand-receptor pairs based on the NicheNetR predicted ligand activity in the endothelial cell cluster and corresponding receptor activity in mesangial cells (Supplemental Figure S5). Top ligands in endothelial cells include endothelin-1 (EDN1), transforming growth factor β1 (TGFB1), vascular endothelial growth factor A (VEGFA), C-X-C motif chemokine ligand 10 (CXCL10). The corresponding receptors in mesangial cells include endothelin receptors A and B (EDNRA/B), transforming growth factor β receptor 2/ Erb-B2 receptor tyrosine kinase 2 (TGFBR2/ERBB2) and protein tyrosine phosphatase receptor type B/ neuropilin 1/ platelet derived growth factor receptor β (PTPRB/NRP1/PDGFRB). Figure 6A shows the relative enrichment of these ligands in endothelial cells versus other kidney cell types, while Figure 6B shows an increase in relative expression of ligands in endothelial cells in DKD versus living donor (LD) controls34. By mapping the main ligands from endothelial cells to the corresponding receptors in mesangial cells, we were able to identify primary activation cascades in the endothelial-mesangial crosstalk in early DKD and by inference in HF (Figure 6C and Supplemental Figure S5). These networks were enriched for genes in the signaling pathways of Rho GTPase small molecules, axon guidance, actin skeleton, VEGFA-NRP1 induced angiogenesis and the endothelin-1 pathway.
Figure 6. NicheNet Analysis of ligand receptor crosstalk.
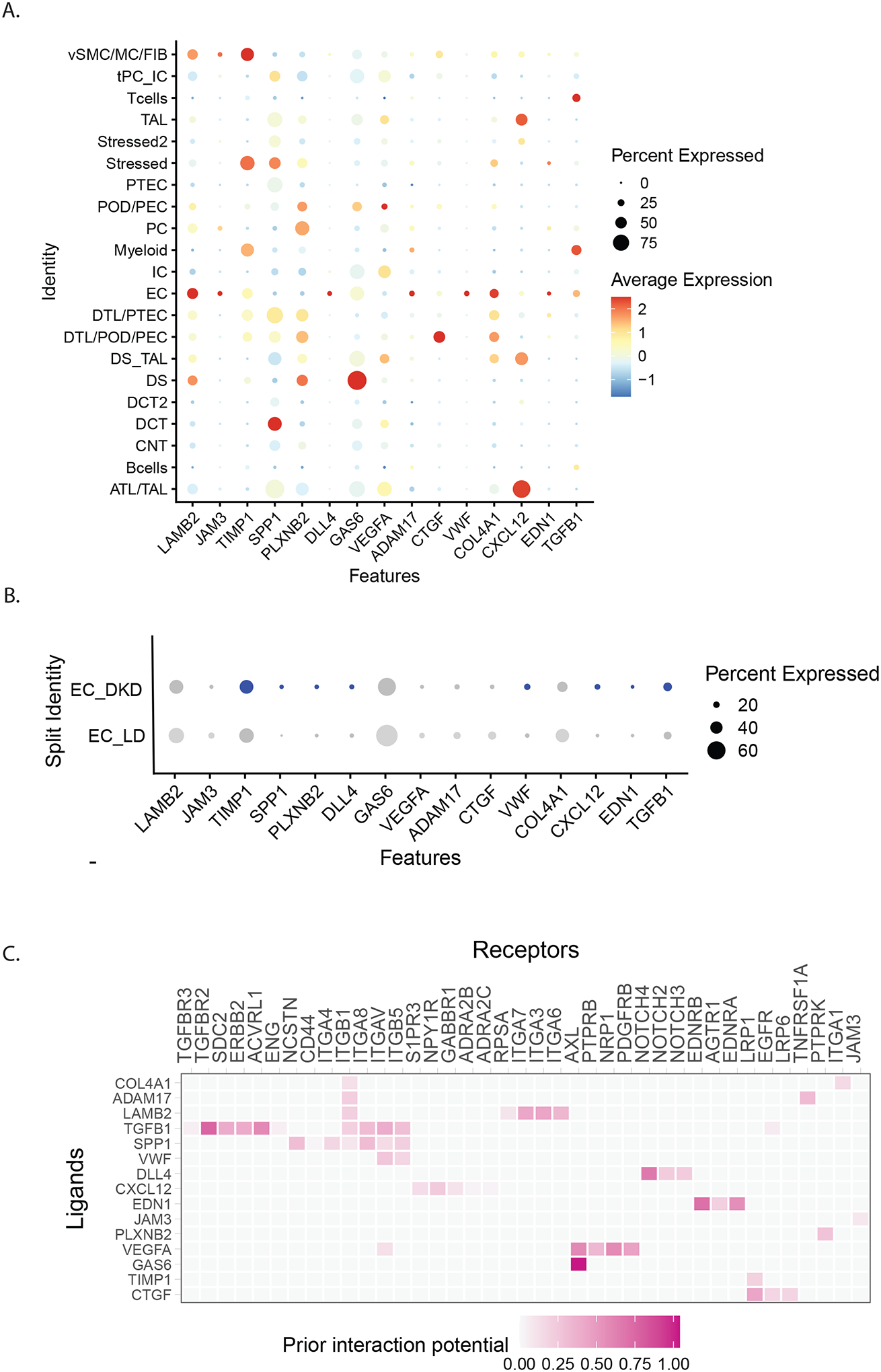
NicheNetR (https://github.com/saeyslab/nichenetr) was used to identify ligand-receptor (LR) interactions that drive the observed expression changes in the single cell transcriptome. A. Expression of the top 15 nichenetR predicted ligands were compared across cell types. B. Several of these ligands are upregulated in DKD endothelial cells (EC_DKD) compared to endothelial cell of controls (EC_LD). The dot size represents the percentage of cells expressing the gene in the respective clusters and the color represent the intensity of the expression level from grey(low) to blue (high). C.Predicted ligand-receptor interaction for the top 15 ligands in the endothelial and mesangial cell. The interaction pairs are prioritized based on the weights derived from the ligand-receptor network from nichenet data sources.
Abbreviations: ATL-Ascending thin loop of Henle; CNT-Connecting tubule; DCT-Distal connecting tubule; DTL- Descending loop of Henle; EC- endothelial cells; IC- intercalated cells; MC-Mesangial Cell; PC- principal cells; PEC- Parietal epithelial cell: POD- Podocyte; PTEC- proximal tubular cells; TAL- thick ascending loop; vSMC-Vascular smooth muscle cells; Stressed/ Stressed 2-Clusters showing ribosomal/stress genes/ injury markers as top marker genes (Stressed cluster were akin to stressed proximal or descending loop of Henle cells whereas cells in the Stressed 2 cluster shared similarities with distal nephron cells.)
Discussion
To our knowledge, this study is the first to report differences in gene expression associated with HF in humans and to link these differences to contemporaneous glomerular structural lesions characteristic of HF8, 17, 38. These associations were identified through the use of an individual’s peak measured GFR to classify HF rather than an arbitrary, absolute HF threshold not allowing for individual variations, e.g. in nephron numbers17 and in the capacity of nephrons to handle prolonged states of HF. Gene expression differences between the HF groups suggest mechanisms associated with the zenith of GFR in individuals with type 2 diabetes as potential therapeutic targets. Mapping the differentially regulated HF genes onto specific kidney cell populations allowed us to identify key cellular mediators of HF and demonstrate that HF genes are significantly enriched in endothelial cells, macrophages, and fibroblasts relative to other kidney cell types. These observations provide unbiased evidence of endothelial stress in the core regulation unit of glomerular hemodynamics.
Integration of kidney tissue level and single cell transcriptional data revealed the main ligands EDN1, TGFB1, VEGFA, growth arrest-specific gene 6 (Gas6) and delta like canonical notch ligand 4 (DLL4) were primarily produced by endothelial cells in the HF state and enabled identification of predicted downstream cellular targets with corresponding over-expressed receptors in the mesangial cells using NicheNet37, 39, 40 (Figure 7). Hyperglycemic conditions, AngII, and reactive oxygen species all activate TGFB1 signaling which then leads to inflammation and fibrosis in progressive DKD41. The receptors included multiple integrins in the cell matrix signaling mechanisms. Mesangial cells targeted by this receptor-ligand interaction showed enrichment of genes in the signaling pathways of Rho GTPase small molecules, axon guidance, actin skeleton, VEGFA-NRP1 induced angiogenesis, and the endothelin-1 pathway. Identification of the endothlin-142 and TGFB1 pathways in our study is supported by prior literature on the role of these pathways in DKD susceptibility43–54. The Gas6-AKT pathway was shown to be involved in mesangial hypertrophy in rat models of DKD55, 56. DLL4 induced Notch signaling influences nephron number and segmentation during kidney development but in DKD, promotes glomerulopathy, tubulointerstitial fibrosis and possibly arteriopathy and inflammation, likely through VEGFA mediated signalling57–60. Several of these pathways were identified and effectively targeted in established DKD44. Our results extend the activation of these pathways into the earliest stages of DKD, arguing for potential beneficial effects of these treatment modalities over the entire course of DKD. Further, early detection of maximum GFR could provide an opportunity for mitigating loss of kidney function. For example, treatment with sodium-glucose co-transporter 2 (SGLT2) inhibitors in early DKD attenuated GFR while reducing serum glucose concentrations, likely through a reduction in HF11, among other mechanisms.
Figure 7. Endothelial signaling activates intracellular targets in mesangial cells.
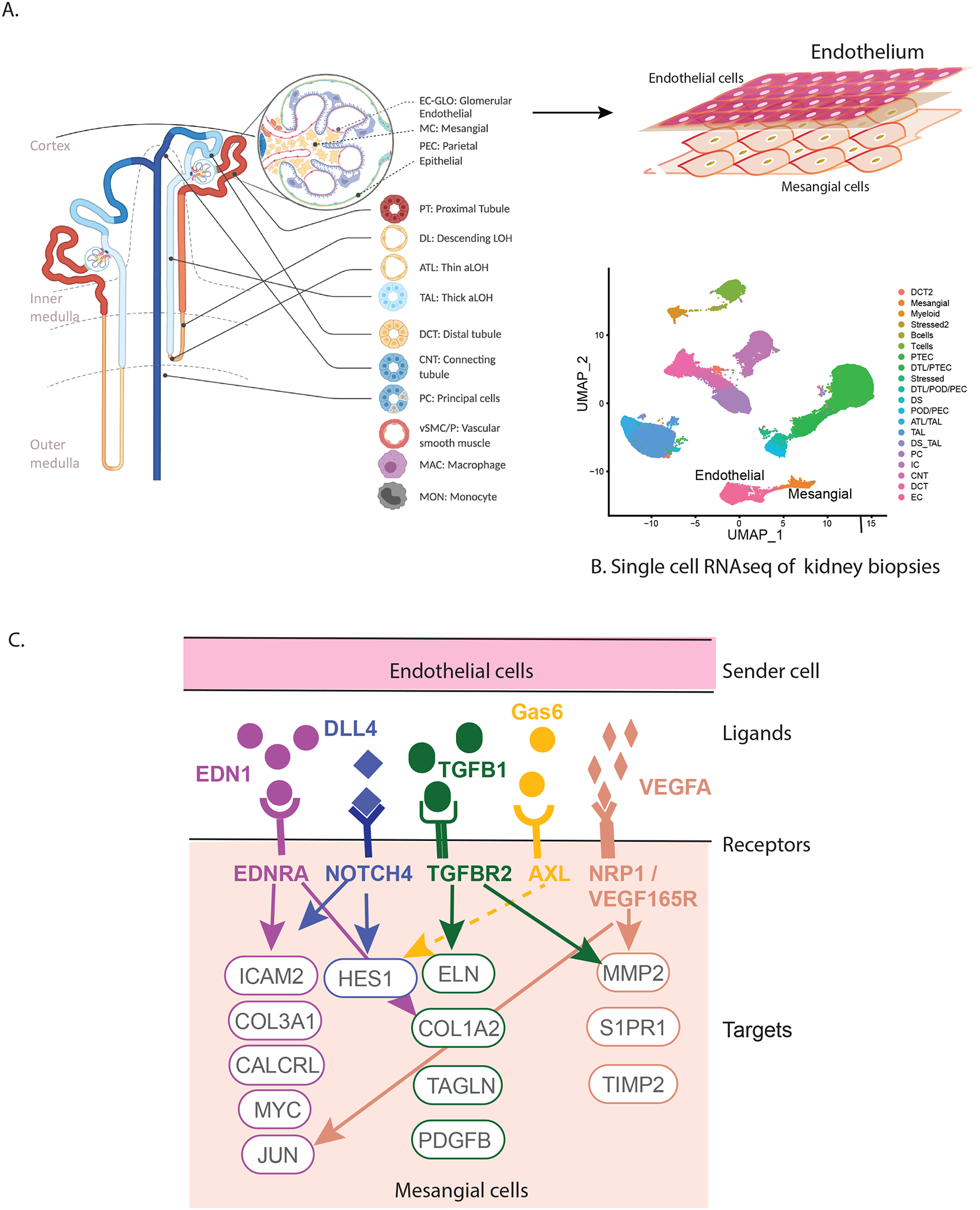
HF genes in the kidney are involved in the crosstalk between endothelial and mesangial cells in the glomerulus (A). The transcriptional profiles of these cell types in DKD could be distinguished using scRNAseq (B) leading to the identification of intracellular targets in mesangial cells (C) providing molecular insights into HF associated with early DKD.
The presence of an inflammatory signal derived from an intraglomerular macrophage cell population suggests that HF is linked to immigrating immune cells in the first steps of glomerular volume expansion61–64. Indeed, increased macrophage infiltration in glomeruli and increased expression of several genes related to inflammation, including MCP-1, were observed in hyperfiltering mice with 5/6 nephrectomy65. Furthermore, increased excretion of cytokines in the urine was previously reported during diabetes-related HF of mGFR >135 ml/min/1.73m2 61.
Nitric oxide signaling pathways and tumor necrosis factor-alpha receptor 2 (TNFR2) signaling pathways which are associated with HF in rats10, are implicated in decline in eGFR, incident CKD and ESRD in diabetes66–68. Indeed, associations were found with early glomerular lesions and ESRD in a Pima Indian cohort69, 70. However, a study of the non-diabetic general population using mGFR found an opposite relationship between TNFR2 and GFR decline, perhaps indicating a disease-specific role of these pathways in diabetes, or alternatively an association of TNFR2 with early HF (longitudinal increase in GFR)71. More recently, the mineralocorticoid receptor antagonist finerenone was found to reduce CKD progression (eGFR decline) and cardiovascular events in patients with diabetes and CKD64. Mineralocorticoid receptor blocking counters several overexpressed pathways in the HF group in the present study, including the profibrotic TGF-β signaling, the vasoconstrictive endothelin-1 signaling and proinflammatory IL-6 signaling. Meanwhile, the GLP-1 receptor antagonist, Exedin-4, ameliorates HF, glomerular hypertrophy and albuminuria in rats, likely through its anti-inflammatory action72.
The main strengths of this study are the repeated GFR measurements using iothalamate clearance providing GFR trajectories for each participant through a substantial portion of their adult life, and the research kidney biopsies not performed for a clinical indication. The GFR measurements preceded and followed research kidney biopsies in the same individuals, allowing an approach where the structural and gene expression findings could be placed in the context of each individual’s historical GFR trajectory. Some participants may have already been past their active HF stage before their GFR measurements began or their kidney biopsy was performed, which is why we excluded participants who had a low GFR and/or a high ACR at the time of biopsy, and those who had their measured GFR peak more than 2 years prior to their biopsy. To our knowledge, no other study population has combined frequently repeated GFR measurements in early diabetes with a kidney biopsy when participants could still plausibly be in the HF stage.
Some of the study’s strengths are also its limitations, in that replicating the study findings in other populations has not been possible as there are no other studies collecting these comprehensive invasive measures across decades. A protocol biopsy study in participants with youth onset T2D with significantly elevated iGFR and histological features of early DKD, however, allowed the replication of key molecular findings of endothelial cell activation during the HF stage of T2D. The rates and severity of obesity and DKD in Pima Indians are higher at a younger age than in most Caucasian or ethnically diverse research populations, and they also have a lower rate of concurrent cardiovascular disease. However, this does not necessarily mean the mechanisms of HF in this population are different from those in other populations, and previous mechanistic insights from gene expression findings in this population have indeed been replicated in unrelated populations73. Other limitations may include the effect of spontaneous variability of GFR measurements on our definition of peak GFR, although the misclassification associated with estimated GFR appears to be much greater. Moreover, the study population was reduced from 52 to 29 for gene expression analyses due to unavailability of tissue for these studies. This might have adversely affected statistical power, and the small clinical and morphometric differences we reported when comparing those with and without expression data could reduce the representativeness of the subgroup with expression data. However, the results from analyses were significant through WGCNA based dimensionality reduction. The scRNAseq data were generated from tissue samples provided by a distinct set of study participants who did not undergo long-term serial GFR measurements, but they were part of the same population. Finally, effect of RAS inhibitors on GFR trajectories could not be determined.
In conclusion, the integration of long-term GFR trajectories with morphometric and molecular analyses of research kidney biopsies enabled the identification of an endothelial stress response with concomitant activation of downstream mesangial cell pathways at peak HF. As these changes occur very early in the course of DKD, they may present an opportunity to prevent the serious complications that may follow. Taken together, this study, provides a potential molecular link between HF in humans and molecular pathways in the kidneys that may lead to structural injury and progressive GFR decline, providing a framework to explore existing and new therapeutic targets for HF in DKD.
Supplementary Material
Supplemental Methods
Supplemental References
Supplemental Tables
Supplemental Table S1: Clinical and morphometric measurements comparing the participants included in the study (HF and pre-HF) and those who were excluded because their peak GFR occurred >2 years before their kidney biopsy
Supplemental Table S2: Timing of peak measured GFR compared to timing of peak eGFR in relation to kidney biopsy
Supplemental Table S3: Clinical and morphometric measures at the time of biopsy for the subset of participants with gene expression data compared to those without expression data
Supplemental Table S4: Clinical and morphometric measures at the time of biopsy by peak measured GFR group for the subset of participants with gene expression data
Supplemental Table S5: Complete list of genes in the three WGCNA modules where 95% genes (1240 genes) were upregulated in the HF compared to pre-HF group
Supplemental Table S6: List of 194 significantly associated pathways (p<0.05) from genes in the three HF modules
Supplemental Table S7: Clinical and morphometric measures for the participants who contributed single-cell expression data from kidney biopsies
Supplemental Figures
Supplemental Figure S1: Individual time-course plots of measured glomerular filtration rate (GFR) for study participants by time from kidney biopsy A) where GFR peak was within 2 years of kidney biopsy and B) where GFR peak was greater than 2 years after kidney biopsy
Supplemental Figure S2: Top 30 interconnected genes in HF associated modules
Supplemental Figure S3: HF gene networks and pathways using cytoscape visualization
Supplemental Figure S4: Hub genes T2D (n=6) and HC (n=6). A dot plot showing significantly enriched hubgenes selected from the 3 HF modules in T2D endothelial cells (T2D) compared to endothelial cell in HC (HC). The dot size represents the percentage of cells expressing the gene in the respective clusters and the color represent the intensity of the expression level from grey(low) to blue (high)
Supplemental Figure S5. NicheNet predicted intracellular targets of endothelial cell activated ligands in crosstalk with mesangial cells
Acknowledgements:
We thank Ms. Lois Jones, RN, Mr. Enrique Diaz, RN, Ms. Bernadine Waseta, and Ms. Camille Waseta for performing the studies in the diabetes cohort. We thank Dr. Abhijit Naik for his scientific insights. This work was performed in partial fulfillment of the requirements for the doctoral work of V.N. from the Medical Faculty of Ludwig-Maximilians-University Munich, Germany. This work was supported in part by the Intramural Research Program at the National Institute of Diabetes and Digestive and Kidney Diseases (DK069062) to HCL and RGN, the American Diabetes Association (Clinical Science Award 1-08-CR-42) to RGN, and (DK083912, DK082841, DK020572, DK092926) to RGN, by the extramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases R24 DK082841 ‘Integrated Systems Biology Approach to Diabetic Microvascular Complications’ and P30 DK081943 ‘University of Michigan O’Brien Kidney Translational Core Center’ to MK, via the Chan Zuckerberg Initiative ‘Human Cell Atlas Kidney Seed Network’ to MK and by JDRF 5-COE-2019-861-S-B ‘JDRF and M-Diabetes Center of Excellence at the University of Michigan’ to MK. The Renal-HEIR/IMPROVE-T2D and CROCODILE studies were supported by NIDDK (K23 DK116720, R01 DK132399, UC2 DK114886, P30 DK116073), JDRF (2-SRA-2019-845-S-B, 3-SRA-2022-1097-M-B), Boettcher Foundation and in part by the Intramural Research Program at NIDDK and the Centers for Disease Control and Prevention (CKD Initiative) under inter-Agency Agreement #21FED2100157DPG. P.B. receives salary and research support from NIDDK (R01 DK129211, R01 DK132399, R21 DK129720, K23 DK116720, UC2 DK114886), JDRF (3-SRA-2022-1097-M-B, 3-SRA-2022-1243-M-B, 3-SRA-2022-1230-M-B), Boettcher Foundation, American Heart Association (20IPA35260142), Ludeman Family Center for Women’s Health Research at the University of Colorado, the Department of Pediatrics, Section of Endocrinology and Barbara Davis Center for Diabetes at University of Colorado School of Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure statement
Dr. Kretzler reports grants from NIH, non-financial support from University of Michigan, during the conduct of the study; grants from JDRF, Astra-Zeneca, NovoNordisc, Eli Lilly, Gilead, Goldfinch Bio, Merck, Chan Zuckerberg Initiative, Janssen, Boehringer-Ingelheim, Moderna, Chinook, amfAR, Angion, RenalytixAI, Retrophin, European Union Innovative Medicine Initiative and Certa outside the submitted work; In addition, Dr. Kretzler has a patent PCT/EP2014/073413 “Biomarkers and methods for progression prediction for chronic kidney disease” licensed. Dr. L. H. Mariani has received unrelated research funding from NIH-NIDDK, NephCure Kidney International, NCATS, unrelated consulting fees from Reata Pharmaceuticals, Calliditas Therapeutics, Travere Therapeutics, support from Reata Pharmaceuticals for travel to meetings. Dr. J.B. Hodgin reports unrelated grant funding from NIDDK and was Chair of the Renal Pathology Society Research Committee from 2019–2020. Dr. P. Bjornstad reports serving as a consultant for AstraZeneca, Bayer, Bristol-Myers Squibb, Boehringer Ingelheim, Eli-Lilly, LG Chemistry, Sanofi, Novo Nordisk, and Horizon Pharma; and also serves on the advisory boards and/or steering committees of AstraZeneca, Bayer, Boehringer Ingelheim, Novo Nordisk, and XORTX.
Footnotes
Study approval
The study was approved by the Institutional Review Board of the National Institute of Diabetes and Digestive and Kidney Diseases.
References
- 1.System USRD. 2020. USRDS Annual Data Report: Epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD. [Google Scholar]
- 2.Ng M, Fleming T, Robinson M, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014; 384: 766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brenner BM, Hostetter TH, Olson JL, et al. The role of glomerular hyperfiltration in the initiation and progression of diabetic nephropathy. Acta Endocrinol Suppl (Copenh) 1981; 242: 7–10. [PubMed] [Google Scholar]
- 4.Ruggenenti P, Porrini EL, Gaspari F, et al. Glomerular hyperfiltration and renal disease progression in type 2 diabetes. Diabetes Care 2012; 35: 2061–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson RG, Bennett PH, Beck GJ, et al. Development and progression of renal disease in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetic Renal Disease Study Group. N Engl J Med 1996; 335: 1636–1642. [DOI] [PubMed] [Google Scholar]
- 6.Park M, Yoon E, Lim YH, et al. Renal hyperfiltration as a novel marker of all-cause mortality. J Am Soc Nephrol 2015; 26: 1426–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helal I, Fick-Brosnahan GM, Reed-Gitomer B, et al. Glomerular hyperfiltration: definitions, mechanisms and clinical implications. Nat Rev Nephrol 2012; 8: 293–300. [DOI] [PubMed] [Google Scholar]
- 8.D’Agati VD, Chagnac A, de Vries AP, et al. Obesity-related glomerulopathy: clinical and pathologic characteristics and pathogenesis. Nat Rev Nephrol 2016; 12: 453–471. [DOI] [PubMed] [Google Scholar]
- 9.Lewko B, Stepinski J. Hyperglycemia and mechanical stress: targeting the renal podocyte. J Cell Physiol 2009; 221: 288–295. [DOI] [PubMed] [Google Scholar]
- 10.Veelken R, Hilgers KF, Hartner A, et al. Nitric oxide synthase isoforms and glomerular hyperfiltration in early diabetic nephropathy. J Am Soc Nephrol 2000; 11: 71–79. [DOI] [PubMed] [Google Scholar]
- 11.Fioretto P, Zambon A, Rossato M, et al. SGLT2 Inhibitors and the Diabetic Kidney. Diabetes Care 2016; 39 Suppl 2: S165–171. [DOI] [PubMed] [Google Scholar]
- 12.Kriz W, Lemley KV. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol 2015; 26: 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fufaa GD, Weil EJ, Lemley KV, et al. Structural Predictors of Loss of Renal Function in American Indians with Type 2 Diabetes. Clin J Am Soc Nephrol 2016; 11: 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson RG, Tan M, Beck GJ, et al. Changing glomerular filtration with progression from impaired glucose tolerance to Type II diabetes mellitus. Diabetologia 1999; 42: 90–93. [DOI] [PubMed] [Google Scholar]
- 15.Pagtalunan ME, Miller PL, Jumping-Eagle S, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest 1997; 99: 342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Looker HC, Mauer M, Saulnier P-J, et al. Changes in Albuminuria But Not GFR are Associated with Early Changes in Kidney Structure in Type 2 Diabetes. J Am Soc Nephrol 2019; 30: 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denic A, Mathew J, Lerman LO, et al. Single-Nephron Glomerular Filtration Rate in Healthy Adults. N Engl J Med 2017; 376: 2349–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weil EJ, Fufaa G, Jones LI, et al. Effect of losartan on prevention and progression of early diabetic nephropathy in American Indians with type 2 diabetes. Diabetes 2013; 62: 3224–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delanaye P, Radermecker RP, Rorive M, et al. Indexing glomerular filtration rate for body surface area in obese patients is misleading: concept and example. Nephrology Dialysis Transplantation 2005; 20: 2024–2028. [DOI] [PubMed] [Google Scholar]
- 20.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009; 150: 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fioretto P, Kim Y, Mauer M. Diabetic nephropathy as a model of reversibility of established renal lesions. Curr Opin Nephrol Hypertens 1998; 7: 489–494. [DOI] [PubMed] [Google Scholar]
- 22.Mauer M, Zinman B, Gardiner R, et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med 2009; 361: 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ibrahim HN, Jackson S, Connaire J, et al. Angiotensin II blockade in kidney transplant recipients. J Am Soc Nephrol 2013; 24: 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mauer M, Caramori ML, Fioretto P, et al. Glomerular structural-functional relationship models of diabetic nephropathy are robust in type 1 diabetic patients. Nephrol Dial Transplant 2015; 30: 918–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caramori ML, Kim Y, Huang C, et al. Cellular basis of diabetic nephropathy: 1. Study design and renal structural-functional relationships in patients with long-standing type 1 diabetes. Diabetes 2002; 51: 506–513. [DOI] [PubMed] [Google Scholar]
- 26.Klein R, Zinman B, Gardiner R, et al. The relationship of diabetic retinopathy to preclinical diabetic glomerulopathy lesions in type 1 diabetic patients: the Renin-Angiotensin System Study. Diabetes 2005; 54: 527–533. [DOI] [PubMed] [Google Scholar]
- 27.Najafian B, Mauer M. Quantitating glomerular endothelial fenestration: an unbiased stereological approach. Am J Nephrol 2011; 33 Suppl 1: 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Najafian B, Tondel C, Svarstad E, et al. One Year of Enzyme Replacement Therapy Reduces Globotriaosylceramide Inclusions in Podocytes in Male Adult Patients with Fabry Disease. PLoS One 2016; 11: e0152812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weibel E Stereological Methods. Practical methods for biological morphometry. Academic Press: London, 1979. [Google Scholar]
- 30.Lane PH, Steffes MW, Mauer SM. Estimation of glomerular volume: a comparison of four methods. Kidney Int 1992; 41: 1085–1089. [DOI] [PubMed] [Google Scholar]
- 31.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics 2008; 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bader GD, Hogue CWV. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003; 4: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menon R, Otto EA, Sealfon R, et al. SARS-CoV-2 receptor networks in diabetic and COVID-19-associated kidney disease. Kidney Int 2020; 98: 1502–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menon R, Otto EA, Hoover P, et al. Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI insight 2020; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaub JA, AlAkwaa FM, McCown PJ, et al. SGLT2 inhibition mitigates perturbations in nephron segment-specific metabolic transcripts and mTOR pathway activity in kidneys of young persons with type 2 diabetes. MedRxiv 2022. [Google Scholar]
- 37.Browaeys R, Saelens W, Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods 2020; 17: 159–162. [DOI] [PubMed] [Google Scholar]
- 38.Elsherbiny HE, Alexander MP, Kremers WK, et al. Nephron hypertrophy and glomerulosclerosis and their association with kidney function and risk factors among living kidney donors. Clin J Am Soc Nephrol 2014; 9: 1892–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gladka MM. Cellular communication in a ‘virtual lab’: going beyond the classical ligand-receptor interaction. Cardiovasc Res 2020; 116: e67–e69. [DOI] [PubMed] [Google Scholar]
- 40.Matsui I, Matsumoto A, Inoue K, et al. Single cell RNA sequencing uncovers cellular developmental sequences and novel potential intercellular communications in embryonic kidney. Sci Rep 2021; 11: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao L, Zou Y, Liu F. Transforming Growth Factor-Beta1 in Diabetic Kidney Disease. Front Cell Dev Biol 2020; 8: 187–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dagamajalu SA-O, Rex DA-O, Gopalakrishnan L, et al. A network map of endothelin mediated signaling pathway. LID - 10.1007/s12079-020-00581-4 [doi] FAU - Dagamajalu, Shobha. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barton M, Yanagisawa M. Endothelin: 30 Years From Discovery to Therapy. Hypertension 2019; 74: 1232–1265. [DOI] [PubMed] [Google Scholar]
- 44.Cherney DZI, Bakris GL. Novel therapies for diabetic kidney disease. Kidney Int Suppl (2011) 2018; 8: 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu J, Lee K, Chuang PY, et al. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am J Physiol Renal Physiol 2015; 308: F287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kohan DE, Pollock DM. Endothelin antagonists for diabetic and non-diabetic chronic kidney disease. Br J Clin Pharmacol 2013; 76: 573–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li YH, Sheu WH, Lee WJ, et al. Synergistic effect of renalase and chronic kidney disease on endothelin-1 in patients with coronary artery disease ‒ a cross-sectional study. Sci Rep 2018; 8: 7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi H, Casalena G, Shi S, et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2017; 66: 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zanatta CM, Veronese FV, Loreto Mda S, et al. Endothelin-1 and endothelin a receptor immunoreactivity is increased in patients with diabetic nephropathy. Ren Fail 2012; 34: 308–315. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L, Chen L, Gao C, et al. Loss of Histone H3 K79 Methyltransferase Dot1l Facilitates Kidney Fibrosis by Upregulating Endothelin 1 through Histone Deacetylase 2. J Am Soc Nephrol 2020; 31: 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong Q, Zhang L, Fu J, et al. LRG1 Promotes Diabetic Kidney Disease Progression by Enhancing TGF-beta-Induced Angiogenesis. J Am Soc Nephrol 2019; 30: 546–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jung KY, Chen K, Kretzler M, et al. TGF-beta1 regulates the PINCH-1-integrin-linked kinase-alpha-parvin complex in glomerular cells. J Am Soc Nephrol 2007; 18: 66–73. [DOI] [PubMed] [Google Scholar]
- 53.Loeffler I, Hopfer U, Koczan D, et al. Type VIII Collagen Modulates TGF-β1-induced Proliferation of Mesangial Cells. J Am Soc Nephrol 2011; 22: 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mariani LH, Martini S, Barisoni L, et al. Interstitial fibrosis scored on whole-slide digital imaging of kidney biopsies is a predictor of outcome in proteinuric glomerulopathies. Nephrol Dial Transplant 2018; 33: 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arai H, Nagai K, Doi T. Role of growth arrest-specific gene 6 in diabetic nephropathy. Vitam Horm 2008; 78: 375–392. [DOI] [PubMed] [Google Scholar]
- 56.Nagai K, Matsubara T, Mima A, et al. Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int 2005; 68: 552–561. [DOI] [PubMed] [Google Scholar]
- 57.Bonegio R, Susztak K. Notch signaling in diabetic nephropathy. Exp Cell Res 2012; 318: 986–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kretzler M, Allred L. Notch inhibition reverses kidney failure. Nat Med 2008; 14: 246–247. [DOI] [PubMed] [Google Scholar]
- 59.Lin CL, Wang FS, Hsu YC, et al. Modulation of notch-1 signaling alleviates vascular endothelial growth factor-mediated diabetic nephropathy. Diabetes 2010; 59: 1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walsh DW, Roxburgh SA, McGettigan P, et al. Co-regulation of Gremlin and Notch signalling in diabetic nephropathy. Biochim Biophys Acta 2008; 1782: 10–21. [DOI] [PubMed] [Google Scholar]
- 61.Har R, Scholey JW, Daneman D, et al. The effect of renal hyperfiltration on urinary inflammatory cytokines/chemokines in patients with uncomplicated type 1 diabetes mellitus. Diabetologia 2013; 56: 1166–1173. [DOI] [PubMed] [Google Scholar]
- 62.Tonneijck L, Muskiet MHA, Smits MM, et al. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J Am Soc Nephrol 2017; 28: 1023–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heerspink HJL, Perco P, Mulder S, et al. Canagliflozin reduces inflammation and fibrosis biomarkers: a potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019; 62: 1154–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bakris GL, Agarwal R, Anker SD, et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N Engl J Med 2020. [DOI] [PubMed] [Google Scholar]
- 65.Sasaki M, Shikata K, Okada S, et al. The macrophage is a key factor in renal injuries caused by glomerular hyperfiltration. Acta Med Okayama 2011; 65: 81–89. [DOI] [PubMed] [Google Scholar]
- 66.Gohda T, Niewczas MA, Ficociello LH, et al. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J Am Soc Nephrol 2012; 23: 516–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Niewczas MA, Gohda T, Skupien J, et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J Am Soc Nephrol 2012; 23: 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shankar A, Sun L, Klein BE, et al. Markers of inflammation predict the long-term risk of developing chronic kidney disease: a population-based cohort study. Kidney Int 2011; 80: 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pavkov ME, Weil EJ, Fufaa GD, et al. Tumor necrosis factor receptors 1 and 2 are associated with early glomerular lesions in type 2 diabetes. Kidney Int 2016; 89: 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pavkov ME, Nelson RG, Knowler WC, et al. Elevation of circulating TNF receptors 1 and 2 increases the risk of end-stage renal disease in American Indians with type 2 diabetes. Kidney Int 2015; 87: 812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schei J, Stefansson VT, Eriksen BO, et al. Association of TNF Receptor 2 and CRP with GFR Decline in the General Nondiabetic Population. Clin J Am Soc Nephrol 2017; 12: 624–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kodera R, Shikata K, Kataoka HU, et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia 2011; 54: 965–978. [DOI] [PubMed] [Google Scholar]
- 73.Nair V, Komorowsky CV, Weil EJ, et al. A molecular morphometric approach to diabetic kidney disease can link structure to function and outcome. Kidney Int 2018; 93: 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Methods
Supplemental References
Supplemental Tables
Supplemental Table S1: Clinical and morphometric measurements comparing the participants included in the study (HF and pre-HF) and those who were excluded because their peak GFR occurred >2 years before their kidney biopsy
Supplemental Table S2: Timing of peak measured GFR compared to timing of peak eGFR in relation to kidney biopsy
Supplemental Table S3: Clinical and morphometric measures at the time of biopsy for the subset of participants with gene expression data compared to those without expression data
Supplemental Table S4: Clinical and morphometric measures at the time of biopsy by peak measured GFR group for the subset of participants with gene expression data
Supplemental Table S5: Complete list of genes in the three WGCNA modules where 95% genes (1240 genes) were upregulated in the HF compared to pre-HF group
Supplemental Table S6: List of 194 significantly associated pathways (p<0.05) from genes in the three HF modules
Supplemental Table S7: Clinical and morphometric measures for the participants who contributed single-cell expression data from kidney biopsies
Supplemental Figures
Supplemental Figure S1: Individual time-course plots of measured glomerular filtration rate (GFR) for study participants by time from kidney biopsy A) where GFR peak was within 2 years of kidney biopsy and B) where GFR peak was greater than 2 years after kidney biopsy
Supplemental Figure S2: Top 30 interconnected genes in HF associated modules
Supplemental Figure S3: HF gene networks and pathways using cytoscape visualization
Supplemental Figure S4: Hub genes T2D (n=6) and HC (n=6). A dot plot showing significantly enriched hubgenes selected from the 3 HF modules in T2D endothelial cells (T2D) compared to endothelial cell in HC (HC). The dot size represents the percentage of cells expressing the gene in the respective clusters and the color represent the intensity of the expression level from grey(low) to blue (high)
Supplemental Figure S5. NicheNet predicted intracellular targets of endothelial cell activated ligands in crosstalk with mesangial cells