

SUMMARY
Hematopoietic stem cells (HSCs) regenerate blood cells throughout life. To preserve their fitness, HSCs are particularly dependent on maintaining protein homeostasis (proteostasis). However, how HSCs purge misfolded proteins is unknown. Here we show that in contrast to most cells that primarily utilize the proteasome to degrade misfolded proteins, HSCs preferentially traffic misfolded proteins to aggresomes in a Bag3-dependent manner, and depend on aggrephagy, a selective form of autophagy, to maintain proteostasis in vivo. When autophagy is disabled, HSCs compensate by increasing proteasome activity, but proteostasis is ultimately disrupted as protein aggregates accumulate and HSC function is impaired. Bag3-deficiency blunts aggresome formation in HSCs, resulting in protein aggregate accumulation, myeloid-biased differentiation, and diminished self-renewal activity. Furthermore, HSC aging is associated with a severe loss of aggresomes and reduced autophagic flux. Protein degradation pathways are thus specifically configured in young adult HSCs to preserve proteostasis and fitness, but become dysregulated during aging.
Keywords: stem cell, hematopoietic stem cell, autophagy, proteostasis, aggresome, proteasome, Bag3, aggrephagy, aging, protein degradation
eTOC Blurb
Inability to grow HSCs in culture is a major barrier to their expanded use in cell-based therapies. Kruta et al. demonstrate that cultured HSCs rapidly increase protein synthesis, which severely disrupts proteostasis and impairs their self-renewal. Hsf1 activation promotes HSC fitness and proteostasis in culture and during aging in vivo.
Graphical Abstract
INTRODUCTION
Maintenance of protein homeostasis (proteostasis) has emerged as fundamentally and preferentially important for stem cells1. Proteostasis disruption impairs stem cell self-renewal, which contributes to poor ex vivo expansion and is associated with degenerative disorders, cancer predisposition syndromes, and age-related pathologies in vivo2–5. To maintain proteostasis, cells employ a network of pathways to balance protein synthesis, folding, trafficking and degradation6. Despite being highly-conserved, the proteostasis network can be specifically configured to support stem cell fitness and longevity. Stem cells exhibit and depend on unusually low protein synthesis rates compared to restricted progenitors7–11. Modest increases in protein synthesis disrupt stem cell proteostasis and impair self-renewal by increasing the biogenesis of misfolded proteins, but similar changes minimally impact progenitors3. Similarly, activation of the unfolded protein response (UPR) has dichotomous effects in stem and progenitor cells. UPR activation safeguards the integrity of the stem cell pool by preferentially inducing apoptosis in stressed stem cells, whereas it typically promotes an adaptive response in progenitors12.
Protein degradation is a critical component of the proteostasis network13. Canonically, the ubiquitin proteasome system functions as the primary pathway for destruction of misfolded proteins13. In embryonic stem cells, high proteasome activity provides proteostasis buffering capacity by degrading and preventing accumulation of misfolded proteins14. In contrast, proteasome activity is low within some somatic stem cells like neural stem cells15 and HSCs3. This raises a fundamental paradox: if somatic stem cells are highly dependent on proteostasis maintenance, why do they have such limited proteasome capacity to degrade misfolded proteins? We set out to address this question in HSCs and to uncover how protein degradation is configured to support stem cell proteostasis and function.
RESULTS
HSCs have high autophagic activity at steady state
To investigate protein degradation, we quantified proteasome activity within HSCs and progenitors. Consistent with prior studies3,16, CD150+CD48−Lineage−Sca1+ckit+ (CD150+CD48−LSK)17 HSCs and CD150−CD48−LSK multipotent progenitors (MPPs)18,19 exhibited substantially less proteasome activity than common myeloid progenitors (CMPs), granulocyte macrophage progenitors (GMPs), megakaryocyte erythroid progenitors (MEPs), and common lymphoid progenitors (CLPs) (Fig. 1A, S1A). The paucity of proteasome activity prompted us to investigate whether HSCs utilize an alternative pathway to degrade misfolded proteins.
Figure 1.

HSCs exhibit high levels of autophagy in vivo.
A. Proteasome activity in HSCs/MPPs (CD48−LSK), CMPs, GMPs, and MEPs measured in relative luminescence units (n=21–22 replicates in 8 experiments).
B. Autophagic activity in HSCs, MPPs, and myeloid progenitors measured by frequency of RFP+ cells in CAG-RFP-EGFP-LC3 mice (n=9 mice from 3 experiments).
C. Representative histograms for RFP expression in bone marrow cells from wildtype mice and HSCs, MPPs, CMPs, GMPs, and MEPs from CAG-RFP-EGFP-LC3 mice (n=9 mice from 3 experiments). Mean frequency and fluorescence intensity of individual mice are shown ± standard deviation (SD).
D. Representative confocal images of FACS isolated RFP+EGFP− (high flux), RFP+EGFP+ (low flux), and RFP−EGFP− (undetectable flux) HSCs.
E-F Flow cytometry gating strategy for identifying high (RFP+EGFP−), low (RFP+EGFP+), and no flux (RFP−EGFP−) autophagy HSCs (E) or progenitors (F) in CAG-RFP-EGFP-LC3 mice.
G-I Frequency of cells undergoing high (G), low (H), and no autophagic flux (I) in CAG-RFP-EGFP-LC3 mice determined by flow cytometry (n=9 mice from 3 experiments).
Data show individual replicates and mean ± SD in A-B and G-I. Data were assessed using a student’s t-test (A) or a one-way ANOVA with Dunnett’s test relative to HSCs (B, G-I). **P≤ 0.01, ***P≤ 0.001, ****P≤ 0.0001.
Macroautophagy (autophagy hereafter) is a bulk degradative process whereby organelles and macromolecules, including proteins, are recycled in a lysosomal-dependent manner20. In most cells, autophagy is minimally active at steady state, but is activated in response to starvation or stress21. In HSCs, autophagy is readily activated in response to cytokine withdrawal ex vivo22, and promotes metabolic function, mitochondrial health and regulates cell signaling in vivo22–26. However, if/how autophagy contributes to HSC proteostasis is largely unexplored.
To measure autophagy in vivo, we used CAG-RFP-EGFP-LC3 mice27. LC3 is a cytosolic protein that when lipidated, localizes to the surface of autophagosomes20. To assess the frequency of HSCs and progenitors with autophagic activity, we quantified the fraction of RFP+ cells within each population. Strikingly, HSCs exhibited unusually high autophagic activity at steady state (Fig. 1B,C). 74% of HSCs were RFP+ as compared to 55% of MPPs, 23% of CMPs, 31 of GMPs, and 8% of MEPs. In addition, RFP fluorescence intensity was much higher in HSCs compared to all progenitor populations (Fig. 1C), suggesting that autophagic HSCs have more autophagic vesicles than progenitors.
Since LC3 can be cytoplasmic, we confirmed that RFP expression faithfully reflected the presence of autophagosomes. Cytoplasmic and autophagosomal LC3 can be distinguished using microscopy based on diffuse and punctate expression, respectively28,29. 100% of RFP+ HSCs and MPPs contained RFP+ puncta when examined by confocal microscopy (Fig. 1D), validating that RFP+ HSCs and MPPs contain autophagosomes.
CAG-RFP-EGFP-LC3 mice enable quantification of autophagic flux27. EGFP fluorescence is pH sensitive and quenched within acidic lysosomes27. Consequently, RFP+EGFP+ cells are enriched with early autophagic vesicles (autophagosomes), while RFP+EGFP− cells contain late autophagic vesicles (autolysosomes), and RFP−EGFP− cells contain no detectable autophagic vesicles (Fig. 1E). Based on these parameters, RFP+EGFP+ cells have low autophagic flux, RFP+EGFP− cells have high autophagic flux, and RFP−EGFP− cells have no detectable autophagic flux27. We implemented a conservative gating strategy that was validated by confocal microscopy to resolve RFP+EGFP+, RFP+EGFP−, and RFP−EGFP− populations by flow cytometry (Fig. 1E,F, S1C,D). We confirmed that based on this gating, sorted RFP+EGFP+, RFP+EGFP−, and RFP−EGFP− HSCs and MPPs exclusively contained RFP+EGFP+ puncta, RFP+EGFP− puncta, and no fluorescence, respectively (Fig. 1D). We then adopted this gating strategy to quantify autophagic flux within HSCs and progenitors in vivo by flow cytometry (Fig. 1E,F, S1A,B).
The majority of autophagic RFP+ HSCs, and 45% of HSCs overall, exhibited high autophagic flux at steady state (Fig. 1G). In contrast, only 27% of MPPs, 9% of CMPs, 7% of GMPs, 3% of MEPs, and 6% of CLPs exhibited high autophagic flux (Fig. 1G, S1B). There also tended to be a greater fraction of HSCs with low autophagic flux compared to progenitors (Fig. 1H). Only 25% of HSCs exhibited undetectable autophagic flux, which was significantly less than all progenitor populations (Fig. 1I, S1B). We validated this quantification by sorting unfractionated HSCs from CAG-RFP-EGFP-LC3 mice and assessing RFP and EGFP expression by confocal microscopy. Using this unbiased approach, we determined that 41% of HSCs contained RFP+EGFP− puncta, 25% contained RFP+EGFP+ puncta, and 34% contained no puncta (Fig. S1E) which was similar to flow cytometric analysis.
To further confirm that HSCs exhibited more autophagy than progenitors in vivo, we assessed autophagic activity by quantifying p62 (Sqstm130). p62 is an autophagosomal cargo protein degraded by autophagy, and thus lower p62 is indicative of elevated autophagy29. Consistent, with CAG-RFP-EGFP-LC3 mice, HSCs contained significantly less p62 than progenitors (Fig. S1F). To confirm that lower p62 was due to elevated autophagy and not to baseline differences in p62 expression, we conditionally deleted the essential autophagy gene Atg531 in the hematopoietic system of young adult Mx1-Cre;Atg5fl/fl (Atg5−/−) mice. Atg5−/− HSCs and MPPs, but not restricted progenitors, exhibited a significant increase in p62 abundance compared to controls (Fig. S1G), indicating that autophagy is readily ongoing within HSCs and MPPs at steady state. Atg5−/− HSCs exhibited similar p62 abundance compared to most restricted progenitors (Fig. S1G), indicating that baseline p62 expression is similar amongst HSCs and progenitors. Overall, data from orthogonal approaches indicate that most HSCs exhibit high autophagic activity at steady state in vivo, whereas autophagy declines in MPPs and is mostly absent in restricted myeloid progenitors.
Since autophagy is often associated with nutrient deprivation and quiescence, we wondered if activating HSCs would suppress autophagy and enhance proteasome activity. We treated young adult mice with cyclophosphamide and granulocyte colony-stimulating factor (GCSF). This treatment drives HSCs to modestly increase protein synthesis10 and undergo rapid self-renewing divisions32. Strikingly, this proliferative stress further increased autophagy in HSCs, but did not significantly affect proteasome activity (Fig. S1H,I). These data support the conclusion that autophagy is the primary protein degradation pathway utilized by young adult HSCs under steady state and stress conditions in vivo.
HSCs transit between autophagic states
Deletion of key autophagy genes impairs HSC function22,23,33,34. In agreement with these studies, conditional deletion of Atg5 in the hematopoietic system of young adult mice impaired HSC reconstituting activity and self-renewal potential (Fig. S2A-N). However, together with our quantitative assessment of autophagic flux, these data raised a key question: if autophagy promotes HSC function, do differences in steady state autophagic activity reflect functional heterogeneity amongst CD150+CD48−LSK cells?
To test this, we competitively transplanted 25 RFP+EGFP+, RFP+EGFP− or RFP−EGFP− CD150+CD48−LSK cells with 3×105 wildtype competitor bone marrow cells into lethally irradiated mice (Fig. 2A). There was no significant difference in peripheral blood or bone marrow cell chimerism in recipients of the three HSC subsets (Fig. 2B-E, S2O-R). Similarly, there was no significant difference in the frequency of donor-derived HSCs in recipient bone marrow 16 weeks after transplant (Fig. 2F). These data indicate that all autophagic subsets of CD150+CD48−LSK cells contain bona fide HSCs, and that autophagic state is not associated with differences in long-term multilineage reconstituting potential.
Figure 2.

HSCs dynamically transition between autophagic flux states.
A Schematic for competitive transplantation of RFP+EGFP− high flux, RFP+EGFP+ low flux, and RFP−EGFP− no flux HSCs into irradiated recipients.
B-E Donor hematopoietic (B), B (C), T (D), and myeloid (E) cell engraftment in peripheral blood of mice receiving 25 RFP+EGFP− high flux, RFP+EGFP+ low flux, or RFP−EGFP− no flux HSCs with 3×105 bone marrow cells (n=10–13 recipients/HSC subset in 3 experiments).
F-I HSC engraftment (F) and autophagic activity (G-I) in the bone marrow of mice from transplants in B-E 16 weeks post-transplantation (n=8–9 recipients/HSC subset in 3 experiments).
J Heat map comparing differentially expressed transcripts between RFP+EGFP−, RFP+EGFP+, and RFP−EGFP− HSCs (≥1.5-fold change, Padj<0.05, n=3).
K Normalized enrichment scores from GSEA of most upregulated and downregulated pathways in autophagic versus non-autophagic HSCs.
L Frequency of Ki67+ RFP−EGFP− or RFP+ CD48−LSK HSC/MPPs (n=3).
Data show individual mice (F-I, L) and/or mean (B-I, L) ± SEM (B-E) or SD (F-I, L). Data were assessed by a one-way repeated measures (B-E) or ordinary (F-I) ANOVA followed by Tukey’s test, or student’s t-test (L). *P≤ 0.05, **P≤ 0.01, ***P≤ 0.001, ****P≤ 0.0001.
At this point, we sought to reconcile an apparent contradiction; if autophagy is required for HSC function, how can RFP−EGFP− HSCs with no detectable autophagy have normal long-term multilineage reconstituting activity? To answer this, we assessed autophagic activity within donor-derived HSCs. Interestingly, HSCs were not fixed in their autophagic potential. Regardless of which HSC subset was transplanted, recipient mice exhibiting long-term multilineage reconstitution all contained a similar distribution of donor-derived low-flux RFP+EGFP+, high-flux RFP+EGFP− and undetectable-flux RFP−EGFP− HSCs in their bone marrow (Fig. 2G-I). These data suggest that autophagic activity does not distinguish functionally distinct or hierarchically related HSCs. Rather, the HSC autophagic state is plastic, and HSCs can dynamically transition between no-flux, low-flux, and high-flux autophagic states.
Autophagic HSCs exhibit increased quiescence
Plasticity between autophagic states could mask distinctions between low-flux RFP+EGFP+, high-flux RFP+EGFP− or no-flux RFP−EGFP− HSCs. To capture potential static differences between these HSC subsets, we performed RNA-sequencing. The transcriptomes of low-flux RFP+EGFP+ and high-flux RFP+EGFP− HSCs were very similar, as only 20 genes were differentially expressed (Fig. 2J; >1.5-fold; Padj<0.05). In contrast, RFP−EGFP− HSCs with no detectable autophagic flux were transcriptionally distinct from both subsets of RFP+ autophagic HSCs, marked by differential expression (>1.5-fold; Padj<0.05) of nearly 300 genes (Fig. 2J). Gene set enrichment analysis revealed RFP−EGFP− HSCs exhibited significantly increased expression of metabolic and biosynthetic pathways (Fig. 2K). Consistent with this, RFP−EGFP− HSCs were twice as likely to be in the cell cycle, as marked by expression of Ki67, compared to autophagic RFP+ HSCs (Fig. 2L). There were no differences in apoptosis between RFP+ and RFP−EGFP− HSCs (Fig. S2S). These data indicate that autophagic RFP+ HSCs exhibit increased quiescence, and RFP− HSCs without detectable autophagy are significantly more likely to be cycling.
Crosstalk between proteasome and autophagy maintains HSC proteostasis
In most cells, proteasomes serve as the primary site for destruction of misfolded proteins, while autophagy typically contributes to proteostasis maintenance in response to stress35,36. However, given the low proteasome and high autophagic activity in HSCs, we wondered what the relative contributions of these pathways were to proteostasis maintenance. We previously established assays to quantify misfolded and unfolded protein in HSCs and progenitors by quantifying polyubiquitinated protein and tetraphenylethene maleimide (TMI)37 fluorescence by flow cytometry, respectively3. Using these tools, we evaluated misfolded and unfolded protein abundance in HSCs and progenitors from mice treated with the proteasome inhibitor bortezomib38 as well as autophagy-deficient Atg5−/− mice (Fig. 3A). We confirmed that Atg5 deletion disrupted autophagy and that bortezomib inhibited proteasome activity in Atg5−/− cells (Fig. S1G, S3A). Surprisingly, neither proteasome inhibition nor disabling autophagy significantly increased the abundance of either polyubiquitinated (misfolded) or unfolded proteins in HSCs or progenitors (Fig. 3B,C, S3B,C). However, when proteasome and autophagy were simultaneously inhibited by administering bortezomib to Atg5−/− mice, misfolded and unfolded protein abundance increased significantly in HSCs (Fig. 3B,C).
Figure 3.

Proteasome and autophagy collaborate to clear unfolded/misfolded proteins.
A Strategy for inhibition of proteasome and simultaneous inhibition of proteasome and autophagy in vivo.
B-C Relative misfolded (ubiquitinated) and unfolded (TMI) protein in HSCs from vehicle (DMSO)-treated wildtype (WT), vehicle-treated Atg5−/−, WT bortezomib-treated, and Atg5−/− bortezomib-treated mice (n=5 from 5 experiments).
D-F Gene set enrichment plots showing that a UPR gene set is not increased in HSCs when proteasome (D) or autophagy (E) alone are inhibited but significantly upregulated when both pathways are inhibited (F) in vivo (n=3).
G Frequency of RFP+ autophagic cells after bortezomib treatment in CAG-RFP-EGFP-LC3 mice (n=6 from 3 experiments).
H Representative histograms and mean fluorescence intensity ± SD of RFP in WT bone marrow cells and bortezomib-treated or control CAG-RFP-EGFP-LC3 HSCs.
I-J Relative proteasome activity in WT or Atg5−/− CD48−LSK HSC/MPPs (I) and restricted progenitors (J) (n=6 from 6 experiments).
K Frequency of cells with high autophagic flux (RFP+EGFP−) in young (3 month) and old (22–24 month) CAG-RFP-EGFP-LC3 mice (n=5–9).
L Relative proteasome activity in young (3 month) and old (22–24 month) CD48−LSK HSC/MPPs (n=6 from 6 experiments).
Data show individual mice and means ± SD (B-C, G, I-L). Data were assessed by ordinary one-way ANOVA with Dunnett’s test relative to WT (B-C) or student’s t-test (G, I-L). *P≤ 0.05, **P≤ 0.01, ***P≤ 0.001, ****P≤ 0.0001.
Disruption of HSC proteostasis by combined proteasome and autophagy inhibition was also transcriptionally evident. RNA-sequencing revealed that combined proteasome and autophagy inhibition induced significant upregulation of gene sets associated with UPR activation, but neither proteasome inhibition nor Atg5-deficiency had that effect on its own (Fig. 3D-F, Table S1).
Next, we investigated potential crosstalk between the proteasome and autophagy pathways. We administered bortezomib to CAG-RFP-EGFP-LC3 mice and found compensatory activation of autophagy. The frequencies of RFP+ HSCs and progenitors were significantly increased in response to proteasome inhibition (Fig. 3G,H). To confirm this was not due to decreased LC3 turnover by the proteasome39, we determined that p62 abundance decreased in HSCs after bortezomib treatment (Fig. S3D), supporting the finding that autophagic activity is enhanced upon proteasome inhibition. We also quantified proteasome activity in HSCs and progenitors in Atg5−/− and control mice. Proteasome activity was significantly increased in Atg5−/− CD48−LSK HSCs/MPPs compared to controls (Fig. 3I). In contrast, restricted progenitors, which have low autophagic activity at steady state, exhibited no compensatory increase in proteasome activity when autophagy was disabled (Fig. 3J). These data indicate that similar to other cells40,41, HSCs and progenitors can activate autophagy in response to proteasome inhibition. However, only HSCs and MPPs that have high steady state autophagic activity, trigger compensatory increases in proteasome activity in response to autophagy disruption. Overall, these data demonstrate that HSCs can utilize bidirectional compensatory crosstalk between proteasome and autophagy to prevent the accumulation of misfolded and unfolded proteins.
HSC aging is associated with reduced autophagic flux and increased proteasome activity
The finding that young adult HSCs exhibit high autophagic activity at steady state, along with previous reports that autophagy becomes disrupted in HSCs during aging24, prompted us to examine protein degradation activity within old adult (22–24 month-old) HSCs. Although the frequency of cells undergoing autophagy was unchanged between young and old HSCs (Fig. S3F), there was a significant age-associated decrease in the frequency of high flux RFP+EGFP− HSCs from 45% to 19% and a concomitant increase in low flux RFP+EGFP+ HSCs from 13% to 29% (Fig. 3K, S3E). These data indicate that autophagic flux declines in HSCs during aging. Interestingly, the reduction in autophagic flux was opposed by increased proteasome activity (Fig. 3L), mirroring the crosstalk observed within young adult HSCs when autophagy was disrupted (Fig. 3G,I). These data suggest that crosstalk between protein degradation systems is physiologically operative during HSC aging, and that HSC aging is associated with a shift in protein degradation characterized by diminished autophagic flux and heightened proteasome activity.
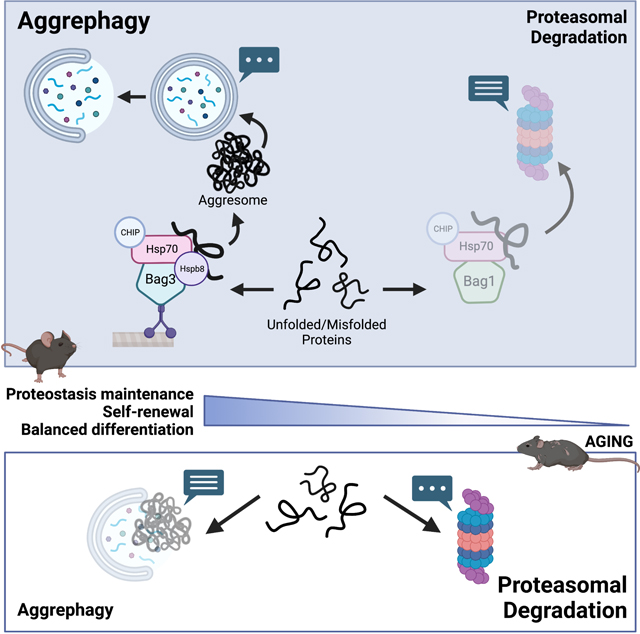
HSCs preferentially traffic misfolded proteins to aggresomes in a Bag3-dependent manner
We next simultaneously inhibited proteasome activity and autophagy by administering bortezomib to Atg5−/− mice and performed RNA-sequencing on HSCs. Combined proteasome and autophagy inhibition induced widespread transcriptional changes, with significant changes (>1.5-fold; Padj<0.05) in 1386 genes compared to vehicle treated wildtype controls (Fig. 4A). One of the most significantly upregulated genes was Hspb8 (Fig. 4A,B). Hspb8 encodes the small heat shock protein B8, which is a member of the Bag3-Hspb8-Hsp70-CHIP complex, which promotes trafficking of damaged and misfolded proteins to aggresomes42,43. Aggresomes are perinuclear inclusion bodies in which misfolded and aggregated proteins are concentrated and sequestered in a cage of intermediate filaments (e.g. vimentin), and are typically substrates for a selective form of autophagy (aggrephagy)44. The Bag3 complex is opposed by the Bag1 complex, which promotes substrate delivery to proteasomes (Fig. 4C)45. Bag3 competes with Bag1 for Hsp70 bound clients, and thus the ratio of Bag3 to Bag1 can dictate the destination of misfolded proteins42,45. Similar to Hspb8, Bag3 expression was significantly increased in HSCs from Atg5−/− mice treated with bortezomib, but Bag1 expression was unchanged (Fig. S4A,B). This resulted in a significant increase in the Bag3 to Bag1 ratio (Fig. 4D). These data suggest that in the absence of proteasome and autophagy function, HSCs may increasingly direct misfolded proteins to aggresomes.
Figure 4.

HSCs preferentially traffic misfolded proteins to aggresomes for removal by aggrephagy.
A Volcano plot highlighting 20 most upregulated (red) and downregulated (blue) genes in HSCs from bortezomib-treated Atg5−/− mice relative to vehicle (DMSO)-treated wildtype (WT) HSCs.
B,D Hspb8 (B) and ratio of Bag3/Bag1 (D) expression in vehicle-treated WT, vehicle-treated Atg5−/−, bortezomib-treated WT, and bortezomib-treated Atg5−/− HSCs (n=3). Data show mean transcripts per million (TPM).
C The Bag3 complex delivers protein substrates to aggresomes by microtubule-dependent retrograde transport while the Bag1 complex shuttles unfolded/misfolded proteins to the proteasome.
D Ratio of Bag3/Bag1 expression in vehicle-treated WT, vehicle-treated Atg5−/−, bortezomib-treated WT, and bortezomib-treated Atg5−/− HSCs (n=3).
E-F Hspb8 (E) and ratio of Bag3/Bag1 (F) expression in HSCs, progenitors, and unfractionated bone marrow (BM) at steady state in vivo (n=3).
G Ratio of BAG3/BAG1 expression in human cord blood-derived HSCs, CMPs and GMPs26,46 (n=3). Data show mean counts per million.
H Representative images of young adult HSCs containing aggresomes (polarized, perinuclear co-localization of ProteoStat with vimentin).
I Frequency of young adult HSCs and myeloid progenitors (MyP; CD127−Lineage−Sca-1c-kit+) containing aggresomes (n=36–47 cells/population).
J Representative images of aggresome staining in WT and Bag3−/− HSCs.
K Frequency of WT and Bag3−/− HSCs containing aggresomes (n=17–62 HSCs).
L Aggregated proteins (relative ProteoStat MFI) in WT and Bag3−/− HSCs (n=3).
M-P Frequency of donor-derived hematopoietic (M), B (N), T (O), and myeloid (P) cell chimerism when 25 WT or Bag3−/− HSCs were transplanted with 3×105 bone marrow cells into irradiated mice (n=16 recipients/genotype from 4 experiments).
Q Lineage distribution of donor-derived cells from transplants in (M-P). Breakdown by individual recipient is shown in the right panel.
R Frequency of secondary recipients of WT and Bag3−/− HSCs that exhibit long-term multilineage reconstitution (n=13–19 recipients/genotype from 3 experiments).
S-T Frequency (S) and number (T) of HSCs in the bone marrow (1 femur + 1 tibia) of young (2–3 month) and old (22–25 month) WT and Bag3−/− mice (n=3–6).
U Representative images of aggresome staining in old (22–24 month) WT HSCs.
V Frequency of HSCs and myeloid progenitors containing aggresomes in young (3 month) and old (22–24 month) mice (n= 21–84).
Data show individual replicates/mice and/or means ± SD (B,D–G, L, Q, S–T) or SEM (M–P). Data were assessed by ordinary one-way ANOVA with Dunnett’s test relative to WT (D–G), chisquared test (I, K, R, V) or student’s t-test (L–Q, S–T). *P≤ 0.05, **P≤ 0.01, ***P≤ 0.001, ****P≤ 0.0001.
Aggresomes have been reported to form specifically under stress conditions, such as when the proteasome is overwhelmed. Given their low proteasome activity, we wondered if HSCs might form aggresomes at steady state. We mined previous RNA-sequencing data comparing mouse and human HSCs and restricted progenitors16,26,46. Strikingly, HSCs expressed significantly more Hspb8 than other hematopoietic cell populations (Fig. 4E). In fact, Hspb8 was minimally or not expressed by most other hematopoietic cells and was specifically expressed by HSCs. Furthermore, HSCs expressed a 2–3-fold higher ratio of Bag3 to Bag1 than each of the other progenitor populations (Fig. 4F). Similarly, cord blood-derived human HSCs (Lin−CD34+CD38−CD45RA−CD90+CD49f+) express significantly more BAG3 and a higher ratio of BAG3 to BAG1 than CMPs (Lin−CD34+CD38+CD10−CD7−CD45RA−CD135+) and GMPs (LinCD34+CD38+CD10−CD7−CD45RA+)26,46 (Fig. 4G, S4C, D). These data suggest that at steady state, mouse and human HSCs may preferentially traffic misfolded proteins to aggresomes.
We used confocal microscopy to quantify the frequency of aggresome-containing HSCs. Aggresomes are identified based on co-localization of protein aggregates (detected using ProteoStat, a fluorescent rotor molecule that binds protein aggregates)47 with vimentin35,48. Strikingly, ~68% of young adult HSCs contained aggresomes in vivo, which was significantly higher than restricted myeloid progenitors (Fig. 4H, I). To test if aggresome formation in HSCs is dependent on Bag3, we conditionally deleted Bag3 in young adult Mx1-Cre;Bag3fl/fl (Bag3−/−) mice. Bag3 deletion significantly reduced the frequency of HSCs with aggresomes from 68% to 17% (Fig. 4J, K). These data indicate that young adult HSCs readily and preferentially form aggresomes at steady state in vivo in a Bag3-dependent manner.
HSCs preferentially depend on autophagy to maintain proteostasis by degrading protein aggregates
Aggresome formation is thought to be cytoprotective by locally concentrating protein aggregates and preventing their dissemination throughout the cell44. While protein aggregates can be degraded either by proteasomes or chaperone-mediated autophagy (preferentially active in HSCs23), they must first be selectively degraded into soluble single peptides via macroautophagy in a process referred to as aggrephagy49. The accumulation of misfolded/aggregated proteins in aggresomes thus serves to facilitate their clearance13,49. Since HSCs preferentially form aggresomes, we hypothesized that they might depend upon aggrephagy to prevent the widespread accumulation of protein aggregates.
To test this, we assessed the impact of Bag3- and Atg5-deficiency on protein aggregate levels in HSCs by quantifying ProteoStat fluorescence. The decline in aggresome formation in Bag3−/− HSCs was associated with a significant ~1.8-fold accumulation of protein aggregates (Fig. 4L). Disabling autophagy similarly resulted in protein aggregate accumulation within Atg5−/− HSCs, but not restricted progenitors (Fig. S4H). In contrast, proteasome inhibition had no effect on protein aggregate abundance in HSCs or progenitors (Fig. S4I). While Bag3−/− HSCs did not exhibit overall changes in proteasome activity, they became increasingly reliant on the proteasome to maintain proteostasis. Bortezomib treatment significantly increased polyubiquitinated protein in Bag3−/− HSCs but not in wildtype controls or Bag3−/− progenitors that had relatively normal levels of misfolded/unfolded protein (Fig. S4E,F), which is reminiscent of effects of bortezomib on Atg5−/− HSCs (Fig. 3B,C). These data demonstrate that HSCs specifically depend on aggrephagy to eliminate protein aggregates and maintain proteostasis.
Bag3−/− HSCs are myeloid-biased and have diminished self-renewal activity
To determine whether Bag3-mediated aggrephagy supports HSC function, we competitively transplanted 25 wildtype or Bag3−/− HSCs into irradiated recipients. Although donor B and T cell reconstitution were similar between wildtype and Bag3−/− HSCs (Fig. 4M-O), myeloid chimerism was significantly elevated in recipients of Bag3−/− HSCs (Fig. 4P). Analysis of lineage distribution of donor-derived cells in the bone marrow of transplant recipients confirmed that Bag3−/− HSCs have a significant myeloid bias (Fig. 4Q). We observed a >50% reduction in relative B cell chimerism and >60% increase in relative myeloid cell chimerism from Bag3−/− compared to wildtype HSCs. Moreover, Bag3−/− HSCs exhibited reduced long-term multilineage reconstituting potential upon serial transplantation. A substantially smaller fraction of secondary recipients of Bag3−/− HSCs exhibited long-term multilineage reconstitution compared to controls (Fig. 4R, S4J-M). Consistent with these data, old adult (22–25 month-old) Bag3−/− mice exhibited a ~4-fold reduction in HSC frequency and number (Fig. 4S, T), as well as significant myeloerythroid expansion in the spleen at the expense of B lineage cells (Fig. S4N). Overall, these data reveal that Bag3-mediated aggresome formation is critical to sustaining balanced differentiation and normal self-renewal capacity of HSCs.
Aging is associated with a loss of aggresomes in HSCs
The myeloid-biased differentiation and diminished self-renewal of Bag3−/− HSCs is reminiscent of changes that occur in HSCs during aging50. Furthermore, loss of proteostasis is a hallmark of aging51, and recent studies suggest that aging HSCs experience proteotoxic stress4. Consequently, we examined aggresomes in old adult HSCs. While most young adult HSCs contained aggresomes, old adult HSCs almost completely lacked aggresomes (Fig. 4U, V). The age-related loss of aggresomes was restricted to the HSC compartment, as aggresomes were present in young and old adult myeloid progenitors at similar frequencies (Fig. 4V). Loss of aggresomes in HSCs did not appear to be caused by changes in expression of Bag1 or Bag3 complex constituents (Fig. S4O), suggesting that other yet unidentified changes disrupt aggresomes in aging HSCs. Overall, these results reveal that HSCs exhibit fundamental changes in proteostasis regulation during aging, and suggest that aggresome loss and aggrephagy dysfunction contribute to age-related declines in HSC fitness.
DISCUSSION
Though the ubiquitin proteasome system is considered the canonical degradation pathway for misfolded proteins36, we found that HSCs preferentially depend on aggrephagy to maintain proteostasis in vivo. We identified Bag3 as a regulator of HSC proteostasis responsible for trafficking misfolded proteins to aggresomes and promoting aggrephagy. Autophagy is highly active within HSCs but not restricted progenitors at steady state in vivo (Fig. 1). Prior studies reported a role for autophagy in promoting HSC function, but this was largely attributed to its bulk degradative activity22,25,33,52. Recent studies have begun to uncover critical selective forms of autophagy that are preferentially important for HSCs23,24. Here, we found that HSCs preferentially depend on aggrephagy to selectively eliminate aggregated proteins and maintain proteostasis, and that disruptions in proteostasis partly contribute to HSC defects associated with impaired autophagy.
Mouse and human HSCs preferentially express components of the Bag3 complex (Fig. 3), which promotes trafficking of misfolded/unfolded proteins to aggresomes43 and chaperone assisted selective autophagy53. Why do HSCs preferentially traffic misfolded proteins to aggresomes and utilize aggrephagy to maintain proteostasis? One possibility is that it enables finer control over resource allocation. Degrading misfolded proteins via the proteasome results in immediate recycling of amino acids into the cytoplasmic pool54 that can be used for de novo protein synthesis. This may be preferred by rapidly dividing progenitors in constant need of amino acids to support high protein biogenesis demands, but may be suboptimal for HSCs that depend on lower protein synthesis rates3,4,10,16,55,56. In contrast, storing misfolded proteins in aggresomes or autophagosomes in quiescent cells preserves raw materials for when they are most needed, such as in response to proliferative or regenerative cues. It also provides a cell intrinsic cache of resources that could make HSCs less reliant on importing amino acids and thus less sensitive to fluctuations in nutrient availability. In support of this model, lysosomal biogenesis is associated with HSC quiescence and HSC cycling is associated with lysosomal activation57,58, which is required for degradation of protein aggregates. A similar phenomenon of aggregate degradation supports neural stem cell activation15,59. In addition, HSCs that asymmetrically inherit more autophagosomes are more likely to preserve their stemness60.
Disrupting aggresome assembly in Bag3−/− HSCs leads to myeloid-biased differentiation and diminished self-renewal (Fig. 4), which are two phenotypes associated with HSC aging. Declines in self-renewal were evident as Bag3−/− HSCs exhibited diminished serial long-term multilineage reconstituting activity. Interestingly, there was a bimodal effect in secondary transplants, as a substantial fraction of recipients of Bag3−/− HSCs failed to exhibit long-term multilineage reconstitution while others exhibited robust reconstitution (this effect was spread across multiple donors and experiments; Fig. S4J-M). One possible explanation is that Bag3-deficiency exerts a selective pressure that supports the emergence of an HSC subset that tolerates aggresome loss, perhaps through activation of proteotoxic stress response pathways. Consistent with this idea, aggresomes are mostly lost in old adult HSCs (Fig. 4), and aging HSCs activate the heat shock response4,61, a key proteotoxic stress response pathway that promotes HSC fitness. Together, these data raise the possibility that aggresome loss and proteostasis dysfunction apply critical selective pressures that contribute to age-related changes in HSC fitness and function.
Aging is associated with proteasome dysfunction in multiple tissues, including the brain, and compensatory activation of autophagy is critical for preventing accumulation of protein aggregates associated with age-related neurodegenerative disorders62. Interestingly, this phenomenon is reversed within HSCs, which exhibit age-related declines in autophagic flux and compensate by increasing proteasome activity during aging. Future studies uncovering the mechanism of age-related proteasome activation in HSCs could offer important insights for ameliorating proteostasis dysfunction in other tissues and cell types.
Bidirectional crosstalk between the ubiquitin proteasome system and autophagy in HSCs may also have important therapeutic implications in cancer. Proteasome inhibitors are widely used to treat multiple myeloma, and their effectiveness partly depends on proteostasis disruption and UPR activation63,64. However, proteasome inhibitors are largely ineffective in treating other cancers65. Similarly, drugs that inhibit autophagy have been largely ineffective as cancer therapeutics66. Compensatory increases in autophagy in response to proteasome inhibition and increased proteasome activity in response to autophagy inhibition may contribute to the ineffectiveness of these therapies. Dual inhibition of these pathways or targeting potential mechanisms that contribute to this crosstalk may yield improved outcomes for cancer patients.
LIMITATIONS OF THE STUDY
Our study uncovers key differences in HSC proteostasis regulation by the proteasome and autophagy. One limitation of this comparison is that autophagy disruption was examined via genetic intervention (Atg5−/−), while proteasome disruption was examined acutely in response to pharmacological inhibition.
Our data indicate that the HSC autophagic state is plastic, and that HSCs can transition between high, low, and no autophagic flux states after transplantation (Fig. 2). However, to what extent this occurs at steady state remains unknown. Furthermore, transplantation reduces autophagic activity in HSCs. At steady state, ~75% of HSCs exhibit ongoing autophagy (Fig. 1), but after transplantation that number is reduced to ~50% (Fig. 2). Similarly, autophagic flux is reduced in HSCs during aging (Fig. 3). It remains unclear how these prolonged and chronic stressors dysregulate autophagy. Elucidating how and why autophagic activity changes in HSCs could reveal strategies to preserve or enhance autophagy to improve HSC proteostasis, fitness, and longevity.
While aggresome formation depends upon Bag3 in young adult HSCs, it does not appear that changes in expression of Bag3 complex members underly declines in aggresomes in old adult HSCs. The mechanism underlying the age-related loss of aggresomes in HSCs is a limitation that should be addressed in future studies. One possibility is that since aggresome formation is microtubule-dependent 44, old adult HSCs may be unable to support aggresome assembly as a consequence of their loss of tubulin polarization67. Uncovering mechanisms of aggresome dysfunction could have important implications for rejuvenating aged HSCs.
STAR METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Robert Signer (rsigner@ucsd.edu).
MATERIALS AVAILABILITY
This study did not generate new unique reagents.
DATA AND CODE AVAILABILITY
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC Anti-mouse CD3ε (17A2) | BioLegend | Cat #100236; RRID:AB_2561456 |
| FITC Anti-mouse CD3ε (17A2) | BioLegend | Cat #100204; RRID:AB_312661 |
| PE Anti-mouse CD3ε (17A2) | BioLegend | Cat #100206; RRID:AB_312663 |
| Brilliant Violet 650 Anti-mouse CD3ε (17A2) | BioLegend | Cat #100229 RRID: AB_11204249 |
| APC Anti-mouse CD4 (GK1.5) | BioLegend | Cat #100412; RRID:AB_312697 |
| FITC Anti-mouse CD4 (GK1.5) | BioLegend | Cat #100406; RRID:AB_312691 |
| PE Anti-mouse CD4 (GK1.5) | BioLegend | Cat #100408; RRID:AB_312693 |
| APC Anti-mouse CD5 (53–7.3) | BioLegend | Cat #100626; RRID:AB_2563929 |
| FITC Anti-mouse CD5 (53–7.3) | BioLegend | Cat #100606; RRID:AB_312735 |
| PE Anti-mouse CD5 (53–7.3) | BioLegend | Cat #100608; RRID:AB_312737 |
| APC Anti-mouse CD8α (53–6.7) | eBioscience | Cat #17–0081-82; RRID:AB_469335 |
| FITC Anti-mouse CD8α (53–6.7) | eBioscience | Cat #11–0081-85; RRID:AB_464916 |
| PE Anti-mouse CD8α (53–6.7) | eBioscience | Cat #12–0081-83; RRID:AB_465531 |
| APC Anti-mouse CD11b (M1/70) | eBioscience | Cat #17–0112-82; RRID:AB_469343 |
| APC efluor780 Anti-mouse CD11b (M1/70) | eBioscience | Cat #47–0112-82; RRID:AB_1603093 |
| FITC Anti-mouse CD11b (M1/70) | eBioscience | Cat #12–0112-83; RRID:AB_2734870 |
| PE Anti-mouse CD11b (M1/70) | eBioscience | Cat #11–0112-85; RRID:AB_464936 |
| PE/Cy7 Anti-mouse CD16/32 (FcγRII/III; 93) | BioLegend | Cat #101318; RRID:AB_2104156 |
| PerCP/Cy5.5 Anti-mouse CD16/32 (FcγRII/III; 93) | BioLegend | Cat #101324; RRID:AB_1877267 |
| Biotin Anti-mouse CD34 (RAM34) | eBioscience | Cat #13–0341-85; RRID:AB_466426 |
| Alexa Fluor 700 Anti-mouse CD34 (RAM34) | eBioscience | Cat #56–0341-82; RRID:AB_493998 |
| FITC Anti-mouse CD34 (RAM34) | eBioscience | Cat #11–0341-85; RRID:AB_465022 |
| APC Anti-mouse CD41 (MWReg30) | Biolegend | Cat #133913; RRID:AB_1112675 1 |
| FITC Anti-mouse CD43 (R2/60) | eBioscience | Cat #11–0431-85; RRID:AB_465041 |
| PE Anti-mouse CD43 (R2/60) | eBioscience | Cat #12–0431-83; RRID:AB_465660 |
| APC eFluor 780 Anti-mouse CD45.1 (A20) | eBioscience | Cat #47–0453-82; RRID:AB_1582228 |
| Alexa Fluor 700 Anti-mouse CD45.2 (104) | BioLegend | Cat #109822; RRID:AB_493731 |
| FITC Anti-mouse CD45.2 (104) | BioLegend | Cat #109806; RRID:AB_313443 |
| APC Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #17–0452-83; RRID:AB_469396 |
| FITC Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #11–0452-85; RRID:AB_465055 |
| PE Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #12–0452-85; RRID:AB_465673 |
| PerCP-Cyanine5 Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #45–0452-82; RRID:AB_1107006 |
| APC Anti-mouse CD48 (HM48–1) | BioLegend | Cat #103412; RRID:AB_571997 |
| FITC Anti-mouse CD48 (HM48–1) | BioLegend | Cat #103404; RRID:AB_313019 |
| PE Anti-mouse CD48 (HM48–1) | BioLegend | Cat #103406; RRID:AB_313021 |
| PE/Cy7 Anti-mouse CD48 (HM48 | BioLegend | Cat #103424; RRID:AB_2075049 |
| FITC Anti-mouse CD71 (R17217) | eBioscience | Cat #11–0711-85; RRID:AB_465125 |
| APC Anti-mouse CD117 (cKit) (2B8) | eBioscience | Cat #17–1171-83; RRID:AB_469431 |
| APC eFluor 780 Anti-mouse CD117 (cKit) (2B8) | eBioscience | Cat #47–1171-82; RRID:AB_1272177 |
| PE-Cyanine7 Anti-mouse CD117 (cKit) (2B8) | eBioscience | Cat #25–1171-82; RRID:AB_469644 |
| PE Anti-mouse CD127 (IL7Rα; A7R34) | BioLegend | Cat #135010; RRID:AB_1937251 |
| APC Anti-mouse CD150 (TC15–12F12.2) | BioLegend | Cat #115910; RRID:AB_493460 |
| PE Anti-mouse CD150 (TC15–12F12.2) | BioLegend | Cat #115904; RRID:AB_313683 |
| PE-Cyanine7 Anti-mouse CD150 (TC15–12F12.2) | BioLegend | Cat #115914; RRID:AB_439797 |
| Brilliant Violet 650 Anti-mouse CD150 (TC15–12F12.2) | BioLegend | Cat #115931 RRID: AB_2562402 |
| APC Anti-mouse Ter119 (TER-119) | Biolegend | Cat #116212; RRID:AB_313713 |
| FITC Anti-mouse Ter119 (TER-119) | Biolegend | Cat #116206; RRID:AB_313709 |
| PE Ter119 (TER-119) | Biolegend | Cat #116208; RRID:AB_313709 |
| APC Anti-mouse Sca-1 (D7, E13–161.7)\ | eBioscience | Cat #17–5981-82; RRID:AB_469487 |
| Alexa Fluor 700 Anti-mouse Sca-1 (D7, E13–161.7) | eBioscience | Cat #56–5981-82; RRID:AB_657836 |
| FITC Anti-mouse Sca-1 (D7, E13–161.7) | eBioscience | Cat #11–5981-85; RRID:AB_465334 |
| PerCp-Cyanine5.5 Anti-mouse Sca-1 (D7, E13–161.7) | eBioscience | Cat #45–5981-82; RRID:AB_914372 |
| APC Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108412; RRID:AB_313377 |
| FITC Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108406; RRID:AB_313371 |
| PE Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108408; RRID:AB_313373 |
| PE/Cy7 Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108416; RRID:AB_312663 |
| APC Anti-mouse IgM (II/41) | eBioscience | Cat #17–5790-82; RRID:AB_469458 |
| PE Anti-mouse IgM (II/41) | eBioscience | Cat #12–5790-83; RRID:AB_465941 |
| Recombinant Alexa Fluor 647 Anti-SQSTM1/p62 antibody (EPR4844) | Abcam | Cat # ab194721 |
| Anti-vimentin antibody | Millipore Sigma | Cat #AB5733 |
| Goat anti-chicken IgY (H&L) Alexa Fluor 488 | Invitrogen | Cat #A-11039 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PIPC | GE Healthcare Biosciences Corp | Cat #27473201 |
| Anti-mouse CD117 microbeads | Miltenyi | Cat #130–091-224 |
| 2-mercaptoethanol | Sigma | Cat #M3148 |
| Heat-inactivated bovine serum | Gibco | Cat #26170043 |
| DMSO | Sigma | Cat #D2650 |
| Baytril | Covetrus | Cat #37713 |
| Ammonium chloride potassium buffer | Covetrus | Cat #37713 |
| Paraformaldehyde | Affymetrix | Cat #19943 |
| 4% paraformaldehyde aqueous solution | Electron Microscopy Sciences | Cat #157–4 |
| PBS | Corning | Cat #MT21040CV |
| Tween 20 | Sigma | Cat #P9416 |
| 4’,6-diamidino-2-phenylindole (DAPI ) | Life Technologies | Cat #62247 |
| Saponin | Sigma | Cat #47036 |
| Fetal Bovine Serum | Life Technologies | Cat #16000044 |
| Bortezomib | Cell Signaling | Cat #2204S |
| Critical Commercial Assays | ||
| Proteasome-Glo chymotrypsin-like cell based assay | Promega | Cat #G8660 |
| RNeasy Plus Micro Kit | Qiagen | Cat #74034 |
| PROTEOSTAT Aggresome detection kit | Enzo | Cat #ENZ-51035-K100 |
| ProLong Diamond Antifade Mountant | Thermo | Cat #P36965 |
| Deposited Data | ||
| RNA-sequencing data | This manuscript | GEO GSE179415 |
| RNA-sequencing data for human HSCs and progenitors | Xie et al, 2019 | GEO: GSE125345 |
| RNA-sequencing data for human HSCs and progenitors | Laurenti et al, 2013 | GEO: GSE42414 |
| RNA-sequencing data for mouse HSCs and progenitors | Signer et al, 2016 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6: C57BL/6J | The Jackson Laboratory |
Cat #000664; RRID:IMSR_JAX:0 00664 |
| Mouse: B6SJL: B6.SJL | The Jackson Laboratory | Cat #002014; RRID:IMSR_JAX:0 02014 |
| Mouse: C57BL/6-Tg(CAG- RFP/EGFP/Map1lc3b)1Hill/J |
The Jackson Laboratory | Cat #027139; RRID:IMSR_JAX:0 27139 |
| Mouse: Bag3fl/fl | Fang et al, 2017 | N/A |
| Mouse: Atg5fl/fl | Hara et al, 2006 | N/A |
| Mouse: Mx1-Cre: B6.Cg-Tg(Mx1-cre)1Cgn/J | The Jackson Laboratory | Cat #003556 RRID:IMSR_JAX:0 03556 |
| Software and Algorithms | ||
| FlowJo | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo/downloads |
| FACSDiva | BD Bioscience | https://www.bdbiosciences.com/enus/products/software/instrumentsoftware/bdfacsdiva-software |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Image Lab 6.0.1 | Bio-Rad | https://www.biorad.com/enus/product/image-lab-software?ID=KRE6P5E8Z |
| TopHat 1.4.1 | JHU CCB | https://ccb.jhu.edu/software/tophat/downloads/ |
| STAR 2.6.1 | N/A | https://github.com/alexdobin/STAR |
| PRINSEQ Lite 0.20.3 | N/A | https://sourceforge.net/projects/prinseq/files/standalone/ |
| Htseq-count 0.7.1 | N/A | https://htseq.readthedocs.io/en/master/install.html |
| featureCounts 1.6.5 | N/A | https://sourceforge.net/projects/subre ad/files/subread1.6.5/ |
| Bioconductor package DESeq2 1.24.0 | Bioconductor | https://git.bioconductor.org/packages/DESeq2 |
| Matplotlib | J.D Hunter, 2007 | https://matplotlib.org/stable/index.html |
| ELDASoftware | Hu and Smyth, 2009 | https://bioinf.wehi.edu.au/software/elda/ |
| ZEN (black edition) | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
| ZEN (blue edition) | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice.
Mx1-Cre 68, Atg5fl 31, Bag3fl 69, and CAG-RFP-EGFP-LC327 mice have been previously described. Mice were all backcrossed for at least ten generations onto a C57BL background. To generate Mx1-Cre+;Atg5fl/fl mice we intercrossed Mx1-Cre and Atg5fl/fl mice. Once generated, Mx1-Cre+;Atg5fl/fl mice were bred with Atg5fl/fl mice to maintain the colony. To generate Mx1-Cre+;Bag3fl/fl mice we intercrossed Mx1-Cre and Bag3fl/fl mice. Once generated, Mx1-Cre+;Bag3fl/fl mice were bred with Bag3fl/fl mice to maintain the colony. CAG-RFP-EGFP-LC3 mice were maintained by breeding CAG-RFP-EGFP-LC3Tg/+ mice with C57BL6/J mice. C57BL6/J were used throughout the study. C57BL6.SJL (CD45.1) mice were used in transplantation experiments. Both male and female mice between 6 and 12 weeks (young adult) or 22–25 months (old adult) of age were used in these studies. All mice were housed in the vivarium at the UC San Diego Moores Cancer Center in specific pathogen-free conditions. All protocols were approved by the UC San Diego Institutional Animal Care and Use Committee.
METHOD DETAILS
PIPC treatment.
Expression of Mx1-Cre was induced by three injections of 10 mg PIPC administered intraperitoneally every other day to 6-week-old mice. Subsequent analyses were performed at least 7 days after the final PIPC injection.
Cyclophosphamide and GCSF treatment.
Mice were injected with 4 mg of cyclophosphamide intraperitoneally followed by two daily subcutaneous injections of 5 μg of GCSF. Mice were analyzed one day after the final GCSF injection.
Flow cytometry and cell sorting.
For flow cytometric analysis and isolation of specific hematopoietic progenitors, cells were incubated with combinations of antibodies to the following cell-surface markers, conjugated to FITC, PE, PerCP-Cy5.5, APC, PE-Cy7, Alexa Fluor 700, APC-eFluor 780, or biotin (antibody clones are given in brackets in the following list): CD3ε (17A2), CD4 (GK1.5), CD5 (53–7.3), CD8a (53–6.7), CD11b (M1/70), CD16/32 (FcgRII/III; 93), CD34 (RAM34), CD43 (R2/60), CD45.1 (A20), CD45.2 (104), CD45R (B220; RA3–6B2), CD48 (HM48–1), CD71 (R17217), CD117 (cKit; 2B8), CD127 (IL7Ra; A7R34), CD150 (TC15–12F12.2), Ter119 (TER-119), Sca-1 (D7, E13–161.7), Gr-1 (RB6–8C5), IgM (II/41), and CD135 (A2F10). For isolation of HSCs and MPPs, Lineage markers included CD3, CD5, CD8, B220, Gr-1 and Ter119. For isolation of CMPs, GMPs and MEPs, these Lineage markers were supplemented with additional antibodies against CD4 and CD11b. Biotinylated antibodies were visualized by incubation with PE-Cy7 conjugated streptavidin. All reagents were acquired from eBiosciences, or BioLegend. All incubations were for 30–90 min on ice.
HSCs, MPPs, CD34+CD16/32lowCD127−Lineage−Sca-1−c-kit+ CMPs70, CD34+CD16/32highCD127−Lineage−Sca- 1−c-kit+ GMPs19, CD34−CD16/32−/lowCD127−Lineage−Sca-1−c-kit+ MEPs19 were pre-enriched by selecting c-kit+ cells using paramagnetic microbeads and an autoMACS magnetic separator (Miltenyi Biotec) before sorting. CD127+Flt3+Lin−Sca1lowcKitlow CLPs71,72 were sorted directly. Non-viable cells were excluded from sorts and analyses using 4’,6-diamidino-2-phenylindole (DAPI).
Cell sorting was performed on BD FACSAria I and BD FACSAria II, or BD FACSAria Fusion. Flow cytometry samples were acquired using a BD LSR I, BD LSR II, or BD FACSymphony A1. Data were analyzed using FlowJo (TreeStar) software.
Proteasome inhibition.
Mice were administered 10mg/kg of bortezomib (Cell Signaling) diluted in 10% (v/v) DMSO retro-orbitally 48 hours and 24 hours before analysis.
Transplantation assays.
For bone marrow transplants, 5×105 donor (CD45.2+) bone marrow cells were competitively transplanted with 5×105 (CD45.1+) whole bone marrow cells into lethally irradiated congenic recipients (CD45.1+). For HSC transplants, 25 donor HSCs (CD45.2+) were competitively transplanted with 3×105 whole bone marrow cells (CD45.1+) into lethally irradiated congenic recipients (CD45.1+). A Mark I Cesium source irradiator (J.L. Shepherd) was used to deliver two doses of 550 rad 4 hours apart for a total lethal dose of 1,100 rad.
Peripheral blood from tail veins of recipient mice was collected every 4 weeks for a total of 16 weeks. Red blood cells were lysed with ammonium chloride potassium buffer. Cells were stained with antibodies against CD45.2, CD45.1, CD45R (B220), CD11b, CD3, and Gr-1 to evaluate donor cell engraftment. Sixteen weeks post-transplant, bone marrow was analyzed for HSC engraftment and multilineage chimerism.
For secondary transplants, 3×106 bone marrow cells collected from primary recipients were transplanted non-competitively into irradiated recipient mice. Primary recipients used for secondary transplantation had long-term multilineage reconstitution by donor cells and median levels of donor-cell reconstitution for the treatments from which they originated. Transplanted mice were administered drinking water with Baytril (250 mg/L) for the first 4 weeks posttransplant. Mice were considered long-term multilineage reconstituted if they exhibited >0.5% donor derived peripheral blood B, T and myeloid cells 16 weeks post-transplant.
Proteasome activity.
5,000–10,000 cells were isolated and transferred to solid opaque 96-well plates. Proteasome activity was measured using Proteasome-Glo Chymotrypsin-Like Cell-Based Assay (Promega). Luminescence was measured using Tecan Spark plate reader.
Confocal microscopy.
Unfractionated, RFP+EGFP−, RFP+EGFP+, and RFP−EGFP−CD150+CD48−LSK HSCs were FACS isolated from the bone marrow of CAG-RFP-EGFP-LC3 mice. Cells were fixed in 4% PFA (Electron Microscopy Sciences) for 10 min at room temperature before incubation with DAPI for 5 min. Cells were washed with PBS and then affixed onto coverslips coated with 0.1% poly-L-lysine (Sigma) diluted 1:5 in molecular biology grade water. Coverslips were mounted onto slides using ProLong Diamond Antifade Mountant (Fisher). Images were acquired on Zeiss LSM880 microscope equipped with Airyscan using a 63X objective. Images were processed with Zeiss ZEN blue/black software and analyzed using FIJI (ImageJ).
For aggresome staining, sorted cells were spun onto coated slides at 450 RPM for 5 min (Cytospin). Cells were then fixed in 4% paraformaldehyde in PBS for 15 min at room temperature. Cells were washed with PBS then permeabilized with PBS supplemented with 0.1% Triton X-100, 0.03% Tween 20 and 1% BSA for 20 min at room temperature. Cells were washed with PBS supplemented with 0.03% Tween 20. Cells were blocked with PBS supplemented with 1% BSA and 0.03% Tween 20 for 1 hour at room temperature. Samples were incubated with anti-vimentin antibody (Millipore AB5733, 1:1000) overnight at 4°C. Cells were washed and incubated with goat anti-chicken IgY Alexa Fluor 488 (Thermo A-11039, 1:500) and ProteoStat (Enzo, 1:5000) for 1 hour at room temperature. Cells were washed with PBS. Before the last wash, nuclei were stained with DAPI for 5 min at room temperature. Cells were mounted with ProLong Diamond Antifade Mountant (Fisher). Raw images were processed using ZEN Blue and maximum intensity projection of z-stacks was generated using FIJI. Blue (DAPI), green (vimentin), and red (ProteoStat) channels were merged and aggresome presence was determined based on co-localization of vimentin and ProteoStat structures. Cells were considered aggresome-positive if they exhibited co-localization of a vimentin cage with ProteoStat-positive puncta. Cells were considered aggresome-free if they did not exhibit co-localization.
Misfolded (polyubiquitinated) protein.
6×106 bone marrow cells were stained with cell surface markers to identify HSCs and progenitor populations. Cells were fixed in 1% PFA and permeabilized in saponin before incubation with 1:500 primary pan-ubiquitin antibody, clone FK2 (Sigma) followed by 1:500 goat anti-mouse IgG AF-488 secondary antibody (Thermo). Cells were analyzed by flow cytometry.
Unfolded protein.
6×106 bone marrow cells were stained with cell surface markers to identify HSCs and progenitor populations. Samples were washed twice in Ca2+- and Mg2+-free PBS. Tetraphenylethene maleimide37 (TMI; stock 2 mM in DMSO) was diluted in PBS (50 mM final concentration) and added to each sample. Samples were incubated at 37°C for 45 min. Samples were washed twice in PBS and analyzed by flow cytometry. DAPI was omitted from these samples because of spectral overlap.
Aggregated protein.
2,000–5,000 HSCs or progenitors were isolated by FACS then fixed in 1% PFA followed by permeabilization with saponin. Samples were stained with ProteoStat (Enzo) (1:2500) for 30 min then resuspended in PBS with DAPI for flow cytometric analysis.
p62 staining.
6×106 bone marrow cells were stained with cell surface markers to identify HSCs and progenitor populations. Cells were fixed and permeabilized using BD Cytofix/Cytoperm solutions and washed with BD Perm/Wash buffer. Samples were incubated with anti-p62 antibody (ab194721) for 30 min at room temperature73.
Annexin V staining.
6×106 bone marrow cells from CAG-RFP-EGFP-LC3 mice were stained with cell surface markers to identify HSCs. Annexin V Apoptosis Detection Kit APC (eBioscience) was used to detect Annexin V.
Cell cycle status.
For Ki67 analysis of autophagy subsets, approximately 2,000 RFP−EGFP− or RFP+ CD48−LSK cells were FACS isolated from the bone marrow of CAG-RFP-EGFP-LC3 mice.
Cells were fixed and permeabilized as described above and stained with AlexaFluor 647 mouse anti-Ki67 (BD; Clone B56).
RNA-sequencing.
For RNA-sequencing analysis, total RNA was extracted from up to 10,000 HSCs using the RNeasy Plus Micro Kit (Qiagen). Illumina mRNA libraries were prepared using the SMARTseq2 protocol74. 2.6μl of total RNA was used in the SMARTSeq2 protocol (70pg to 1.62ng). 18 cycles of PCR were performed for the cDNA preamplification step and 12 cycles were performed for the tagmentation library preparation. The resulting libraries were pooled and deep sequenced on the Illumina NovaSeq using paired-end reads with both forward and reverse read lengths of 50 nucleotides (60–100M reads per condition). The reads that passed Illumina filters were filtered for reads aligning to tRNA, rRNA, adapter sequences, and spike-in controls. The reads were then aligned to the GRCm38 reference genome using STAR (v2.6.1)75. DUST scores were calculated with PRINSEQ Lite (v 0.20.3)76 and low-complexity reads (DUST > 4) were removed from the BAM files. The alignment results were parsed via the SAMtools77 to generate SAM files. Read counts to each genomic feature were obtained with the featureCounts program (v 1.6.5)78 using the default options along with a minimum quality cut off (Phred > 10). After removing absent features (zero counts in all samples), the raw counts were then imported to Bioconductor package DESeq2 (v 1.24.0)79 to identify differentially expressed genes among samples. P-values for differential expression are calculated using the Wald test for differences between the base means of two conditions. These P-values are then adjusted for multiple test correction using Benjamini Hochberg algorithm. We considered genes differentially expressed between two groups of samples when the DESeq2 analysis resulted in an adjusted P-value of <0.05 and the difference in gene expression was at least 1.5-fold.
Gene set enrichment analysis was done using the “GseaPreranked” method with “classic” scoring scheme. MSigDB Gene sets were downloaded in GMT format for mouse using R package msigdbr v7.4.1. Rank files for each DE comparison of interest were generated by assigning a rank of negative log10(pValue) to genes with log2FoldChange greater than zero and a rank of positive log10(pValue) to genes with log2FoldChange less than zero
QUANTIFICATION AND STATISTICAL ANALYSIS
Group data are represented by mean ± standard deviation, except for transplantation data which are represented as mean ± standard error of the mean. The specific type of statistical test used for each figure panel is described in the figure legends. For t-tests, a confidence interval of 95% was used. For normalized data, means were calculated and statistical tests were performed using log10-transformed data and then means were back-transformed to prevent data skewing. No randomization or blinding was used in any experiments. The only mice excluded from any experiment were those that died after transplantation. We performed multiple independent experiments with multiple biological replicates to ensure the reproducibility of our findings.
Supplementary Material
Table S1. Expression of UPR related genes in HSCs with disrupted protein degradation. Related to Figure 3. Expression of genes in a UPR gene set (Fig. 3D-F) in vehicle- (DMSO) or bortezomib-treated wildtype and Atg5−/− HSCs (n=3). Data show mean transcripts per million (TPM).
Highlights.
Ex vivo cultured HSCs rapidly and massively increase protein synthesis
Hsf1 is activated in culture to promote ex vivo HSC maintenance and proteostasis
Small molecule activators of Hsf1 enhance HSC maintenance and fitness in culture
Hsf1 is activated in aging HSCs in vivo to promote proteostasis and fitness
ACKNOWLEDGMENTS
Work in the Signer Laboratory is supported by the NIH/NIDDK (R01DK116951; R01DK124775), NIH/NCI (U01CA267031), the Blood Cancer Discoveries Grant program (8025-20) through The Leukemia & Lymphoma Society, The Mark Foundation for Cancer Research and The Paul G. Allen Frontiers Group, the American Cancer Society (CSCC-RSG-23-994830-01-CSCC), the Sanford Stem Cell Institute, and the UC San Diego Moores Cancer Center which is supported by the NCI Cancer Center Support Grant (P30CA023100). The LJI and Moores Cancer Center Flow Cytometry and Microscopy core facilities are supported by the NIH Shared Instrumentation Grant Program (S10 RR027366, S10OD032316, S10OD021831). Some figures were generated using Biorender. We thank N. Mizushima, J. Chen, Y. Hong, J. Magee, K. Dorshkind, S. Morrison, and C. Jamieson for reagents and/or advice.
Footnotes
AUTHOR INFORMATION
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Chua BA, Van Der Werf I, Jamieson C, and Signer RAJ (2020). Post-Transcriptional Regulation of Homeostatic, Stressed, and Malignant Stem Cells. Cell Stem Cell 26, 138–159. 10.1016/j.stem.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, and Serrano AL (2016). Autophagy maintains stemness by preventing senescence. Nature 529, 37–42. [DOI] [PubMed] [Google Scholar]
- 3.Hidalgo San Jose L, Sunshine MJ, Dillingham CH, Chua BA, Kruta M, Hong Y, Hatters DM, and Signer RAJ (2020). Modest Declines in Proteome Quality Impair Hematopoietic Stem Cell Self-Renewal. Cell Rep 30, 69–80.e66. 10.1016/j.celrep.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kruta M, Sunshine MJ, Chua BA, Fu Y, Chawla A, Dillingham CH, Hidalgo San Jose L, De Jong B, Zhou FJ, and Signer RAJ (2021). Hsf1 promotes hematopoietic stem cell fitness and proteostasis in response to ex vivo culture stress and aging. Cell Stem Cell 28, 1950–1965.e1956. 10.1016/j.stem.2021.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vilchez D, Simic MS, and Dillin A (2014). Proteostasis and aging of stem cells. Trends Cell Biol 24, 161–170. 10.1016/j.tcb.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Balch WE, Morimoto RI, Dillin A, and Kelly JW (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 7.Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J, Sajini A, Tanna H, Cortés-Garrido R, Gkatza N, et al. (2016). Stem cell function and stress response are controlled by protein synthesis. Nature 534, 335–340. 10.1038/nature18282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Llorens-Bobadilla E, Zhao S, Baser A, Saiz-Castro G, Zwadlo K, and Martin-Villalba A (2015). Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell 17, 329–340. 10.1016/j.stem.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez Carlos G., Teixeira Felipe K., Czech B, Preall, Jonathan B, Zamparini, Andrea L, Seifert, Jessica RK, Malone, Colin D, Hannon, Gregory J, and Lehmann R (2016). Regulation of Ribosome Biogenesis and Protein Synthesis Controls Germline Stem Cell Differentiation. Cell Stem Cell 18, 276–290. 10.1016/j.stem.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Signer RA, Magee JA, Salic A, and Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. 10.1038/nature13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zismanov V, Chichkov V, Colangelo V, Jamet S, Wang S, Syme A, Koromilas, Antonis E, and Crist C (2016). Phosphorylation of eIF2α Is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell 18, 79–90. 10.1016/j.stem.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 12.van Galen P., Kreso A., Mbong N., Kent DG., Fitzmaurice T., Chambers JE., Xie S., Laurenti E., Hermans K., Eppert K., et al. (2014). The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 510, 268272. 10.1038/nature13228. [DOI] [PubMed] [Google Scholar]
- 13.Dikic I (2017). Proteasomal and Autophagic Degradation Systems. Annu Rev Biochem 86, 193–224. 10.1146/annurev-biochem-061516-044908. [DOI] [PubMed] [Google Scholar]
- 14.Vilchez D, Boyer L, Morantte I, Lutz M, Merkwirth C, Joyce D, Spencer B, Page L, Masliah E, Berggren WT, et al. (2012). Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 489, 304–308. 10.1038/nature11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leeman DS, Hebestreit K, Ruetz T, Webb AE, McKay A, Pollina EA, Dulken BW, Zhao X, Yeo RW, Ho TT, et al. (2018). Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 359, 1277–1283. 10.1126/science.aag3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Signer RA, Qi L, Zhao Z, Thompson D, Sigova AA, Fan ZP, DeMartino GN, Young RA, Sonenberg N, and Morrison SJ (2016). The rate of protein synthesis in hematopoietic stem cells is limited partly by 4E-BPs. Genes Dev 30, 1698–1703. 10.1101/gad.282756.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, and Morrison SJ (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121. 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 18.Challen GA, Pietras EM, Wallscheid NC, and Signer RAJ (2021). Simplified murine multipotent progenitor isolation scheme: Establishing a consensus approach for multipotent progenitor identification. Exp Hematol 104, 55–63. 10.1016/j.exphem.2021.09.007. [DOI] [PubMed] [Google Scholar]
- 19.Oguro H, Ding L, and Morrison SJ (2013). SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 13, 102–116. 10.1016/j.stem.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Z, and Klionsky DJ (2010). Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22, 124–131. 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murrow L, and Debnath J (2013). Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol 8, 105–137. 10.1146/annurev-pathol-020712-163918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, and Passegué E (2013). FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494, 323–327. 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong S, Wang Q, Kao YR, Diaz A, Tasset I, Kaushik S, Thiruthuvanathan V, Zintiridou A, Nieves E, Dzieciatkowska M, et al. (2021). Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 591, 117–123. 10.1038/s41586-020-03129-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, and Passegué E (2017). Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210. 10.1038/nature21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortensen M., Soilleux EJ., Djordjevic G., Tripp R., Lutteropp M., Sadighi-Aksh E., Stranks AJ., Glanville J., Knight S., Jacobsen SE., et al. (2011). The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med 208, 455467. 10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie SZ, Garcia-Prat L, Voisin V, Ferrari R, Gan OI, Wagenblast E, Kaufmann KB, Zeng AGX, Takayanagi SI, Patel I, et al. (2019). Sphingolipid Modulation Activates Proteostasis Programs to Govern Human Hematopoietic Stem Cell Self-Renewal. Cell Stem Cell 25, 639–653.e637. 10.1016/j.stem.2019.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Wang ZV, Hill JA, and Lin F (2014). New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol 25, 305–315. 10.1681/asn.2013040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, and Yoshimori T (2004). LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 117, 2805–2812. 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 29.Yoshii SR, and Mizushima N (2017). Monitoring and Measuring Autophagy. Int J Mol Sci 18. 10.3390/ijms18091865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, and Johansen T (2009). Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 452, 181–197. 10.1016/s0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 31.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, and Mizushima N (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 32.Morrison SJ, Wright DE, and Weissman IL (1997). Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proc Natl Acad Sci U S A 94, 1908–1913. 10.1073/pnas.94.5.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu F, Lee JY, Wei H, Tanabe O, Engel JD, Morrison SJ, and Guan JL (2010). FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 116, 4806–4814. 10.1182/blood-2010-06-288589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nomura N, Ito C, Ooshio T, Tadokoro Y, Kohno S, Ueno M, Kobayashi M, Kasahara A, Takase Y, Kurayoshi K, et al. (2021). Essential role of autophagy in protecting neonatal haematopoietic stem cells from oxidative stress in a p62independent manner. Scientific Reports 11, 1666. 10.1038/s41598-021-81076-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnston JA, Ward CL, and Kopito RR (1998). Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143, 1883–1898. 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, and Goldberg AL (1994). Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78, 761–771. 10.1016/S0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 37.Chen MZ, Moily NS, Bridgford JL, Wood RJ, Radwan M, Smith TA, Song Z, Tang BZ, Tilley L, and Xu X (2017). A thiol probe for measuring unfolded protein load and proteostasis in cells. Nature communications 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen D, Frezza M, Schmitt S, Kanwar J, and Dou QP (2011). Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets 11, 239–253. 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia R, and Bonifacino JS (2020). Regulation of LC3B levels by ubiquitination and proteasomal degradation. Autophagy 16, 382–384. 10.1080/15548627.2019.1709766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, Miller FJ Jr., Rothermel BA, and Hill JA (2008). Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation 117, 3070–3078. 10.1161/circulationaha.107.763870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang XJ., Yu J., Wong SH., Cheng AS., Chan FK., Ng SS., Cho CH., Sung JJ., and Wu WK. (2013). A novel crosstalk between two major protein degradation systems: regulation of proteasomal activity by autophagy. Autophagy 9, 1500–1508. 10.4161/auto.25573. [DOI] [PubMed] [Google Scholar]
- 42.Carra S, Seguin SJ, and Landry J (2008). HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 4, 237–239. 10.4161/auto.5407. [DOI] [PubMed] [Google Scholar]
- 43.Gamerdinger M, Kaya AM, Wolfrum U, Clement AM, and Behl C (2011). BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep 12, 149–156. 10.1038/embor.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kopito RR (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10, 524–530. 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 45.Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, and Behl C (2009). Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. Embo j 28, 889–901. 10.1038/emboj.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laurenti E, Doulatov S, Zandi S, Plumb I, Chen J, April C, Fan JB, and Dick JE (2013). The transcriptional architecture of early human hematopoiesis identifies multilevel control of lymphoid commitment. Nat Immunol 14, 756–763. 10.1038/ni.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Navarro S, and Ventura S (2014). Fluorescent dye ProteoStat to detect and discriminate intracellular amyloid-like aggregates in Escherichia coli. Biotechnol J 9, 1259–1266. 10.1002/biot.201400291. [DOI] [PubMed] [Google Scholar]
- 48.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, and Goldberg AL (1994). Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78, 761–771. 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 49.Lamark T, and Johansen T (2012). Aggrephagy: Selective Disposal of Protein Aggregates by Macroautophagy. International Journal of Cell Biology 2012, 736905. 10.1155/2012/736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Signer RA, and Morrison SJ (2013). Mechanisms that regulate stem cell aging and life span. Cell Stem Cell 12, 152–165. 10.1016/j.stem.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nguyen-McCarty M, and Klein PS (2017). Autophagy is a signature of a signaling network that maintains hematopoietic stem cells. PLoS One 12, e0177054. 10.1371/journal.pone.0177054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Fürst DO, Saftig P, Saint R, Fleischmann BK, et al. (2010). Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol 20, 143–148. 10.1016/j.cub.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 54.Suraweera A, Münch C, Hanssum A, and Bertolotti A (2012). Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell 48, 242–253. 10.1016/j.molcel.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keyvani Chahi A, Belew MS, Xu J, Chen HTT, Rentas S, Voisin V, Krivdova G, Lechman E, Marhon SA, De Carvalho DD, et al. (2022). PLAG1 dampens protein synthesis to promote human hematopoietic stem cell self-renewal. Blood 140, 992–1008. 10.1182/blood.2021014698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Magee JA, and Signer RAJ (2021). Developmental Stage-Specific Changes in Protein Synthesis Differentially Sensitize Hematopoietic Stem Cells and Erythroid Progenitors to Impaired Ribosome Biogenesis. Stem Cell Reports 16, 20–28. 10.1016/j.stemcr.2020.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.García-Prat L., Kaufmann KB., Schneiter F., Voisin V., Murison A., Chen J., ChanSeng-Yue M., Gan OI., McLeod JL., Smith SA., et al. (2021). TFEB-mediated endolysosomal activity controls human hematopoietic stem cell fate. Cell Stem Cell 28, 1838–1850.e1810. 10.1016/j.stem.2021.07.003. [DOI] [PubMed] [Google Scholar]
- 58.Liang R, Arif T, Kalmykova S, Kasianov A, Lin M, Menon V, Qiu J, Bernitz JM, Moore K, Lin F, et al. (2020). Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 26, 359–376.e357. 10.1016/j.stem.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morrow CS, Porter TJ, Xu N, Arndt ZP, Ako-Asare K, Heo HJ, Thompson EAN, and Moore DL (2020). Vimentin Coordinates Protein Turnover at the Aggresome during Neural Stem Cell Quiescence Exit. Cell Stem Cell 26, 558–568.e559. 10.1016/j.stem.2020.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loeffler D, Wehling A, Schneiter F, Zhang Y, Müller-Bötticher N, Hoppe PS, Hilsenbeck O, Kokkaliaris KD, Endele M, and Schroeder T (2019). Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 573, 426–429. 10.1038/s41586-019-1531-6. [DOI] [PubMed] [Google Scholar]
- 61.Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, and Goodell MA (2007). Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol 5, e201. 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seidel K, Vinet J, Dunnen WF, Brunt ER, Meister M, Boncoraglio A, Zijlstra MP, Boddeke HW, Rüb U, Kampinga HH, and Carra S (2012). The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathol Appl Neurobiol 38, 39–53. 10.1111/j.1365-2990.2011.01198.x. [DOI] [PubMed] [Google Scholar]
- 63.Lee A-H, Iwakoshi NN, Anderson KC, and Glimcher LH (2003). Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proceedings of the National Academy of Sciences 100, 9946–9951. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr., Lee KP, and Boise LH (2006). Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 107, 4907–4916. 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Manasanch EE, and Orlowski RZ (2017). Proteasome inhibitors in cancer therapy. Nature Reviews Clinical Oncology 14, 417–433. 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manic G, Obrist F, Kroemer G, Vitale I, and Galluzzi L (2014). Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol 1, e29911. 10.4161/mco.29911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Florian MC, Dörr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, Filippi MD, Hasenberg A, Gunzer M, Scharffetter-Kochanek K, et al. (2012). Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 10, 520–530. 10.1016/j.stem.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kühn R, Schwenk F, Aguet M, and Rajewsky K (1995). Inducible gene targeting in mice. Science 269, 1427–1429. 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 69.Fang X, Bogomolovas J, Wu T, Zhang W, Liu C, Veevers J, Stroud MJ, Zhang Z, Ma X, Mu Y, et al. (2017). Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest 127, 3189–3200. 10.1172/jci94310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akashi K, Traver D, Miyamoto T, and Weissman IL (2000). A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404, 193–197. 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 71.Kikuchi K, Kasai H, Watanabe A, Lai AY, and Kondo M (2008). IL-7 specifies B cell fate at the common lymphoid progenitor to pre-proB transition stage by maintaining early B cell factor expression. J Immunol 181, 383–392. 10.4049/jimmunol.181.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kondo M, Weissman IL, and Akashi K (1997). Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 91, 661–672. 10.1016/s0092-8674(00)80453-5. [DOI] [PubMed] [Google Scholar]
- 73.Ames K, Kaur I, Shi Y, Tong M, Sinclair T, Hemmati S, Glushakow-Smith SG, Tein E, Gurska L, Dubin R, et al. (2020). Deletion of PI3-Kinase Promotes Myelodysplasia Through Dysregulation of Autophagy in Hematopoietic Stem Cells. bioRxiv, 2020.2012.2004.412593. 10.1101/2020.12.04.412593. [DOI] [Google Scholar]
- 74.Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9, 171–181. 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 75.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schmieder R., and Edwards R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 79.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Expression of UPR related genes in HSCs with disrupted protein degradation. Related to Figure 3. Expression of genes in a UPR gene set (Fig. 3D-F) in vehicle- (DMSO) or bortezomib-treated wildtype and Atg5−/− HSCs (n=3). Data show mean transcripts per million (TPM).
Data Availability Statement
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC Anti-mouse CD3ε (17A2) | BioLegend | Cat #100236; RRID:AB_2561456 |
| FITC Anti-mouse CD3ε (17A2) | BioLegend | Cat #100204; RRID:AB_312661 |
| PE Anti-mouse CD3ε (17A2) | BioLegend | Cat #100206; RRID:AB_312663 |
| Brilliant Violet 650 Anti-mouse CD3ε (17A2) | BioLegend | Cat #100229 RRID: AB_11204249 |
| APC Anti-mouse CD4 (GK1.5) | BioLegend | Cat #100412; RRID:AB_312697 |
| FITC Anti-mouse CD4 (GK1.5) | BioLegend | Cat #100406; RRID:AB_312691 |
| PE Anti-mouse CD4 (GK1.5) | BioLegend | Cat #100408; RRID:AB_312693 |
| APC Anti-mouse CD5 (53–7.3) | BioLegend | Cat #100626; RRID:AB_2563929 |
| FITC Anti-mouse CD5 (53–7.3) | BioLegend | Cat #100606; RRID:AB_312735 |
| PE Anti-mouse CD5 (53–7.3) | BioLegend | Cat #100608; RRID:AB_312737 |
| APC Anti-mouse CD8α (53–6.7) | eBioscience | Cat #17–0081-82; RRID:AB_469335 |
| FITC Anti-mouse CD8α (53–6.7) | eBioscience | Cat #11–0081-85; RRID:AB_464916 |
| PE Anti-mouse CD8α (53–6.7) | eBioscience | Cat #12–0081-83; RRID:AB_465531 |
| APC Anti-mouse CD11b (M1/70) | eBioscience | Cat #17–0112-82; RRID:AB_469343 |
| APC efluor780 Anti-mouse CD11b (M1/70) | eBioscience | Cat #47–0112-82; RRID:AB_1603093 |
| FITC Anti-mouse CD11b (M1/70) | eBioscience | Cat #12–0112-83; RRID:AB_2734870 |
| PE Anti-mouse CD11b (M1/70) | eBioscience | Cat #11–0112-85; RRID:AB_464936 |
| PE/Cy7 Anti-mouse CD16/32 (FcγRII/III; 93) | BioLegend | Cat #101318; RRID:AB_2104156 |
| PerCP/Cy5.5 Anti-mouse CD16/32 (FcγRII/III; 93) | BioLegend | Cat #101324; RRID:AB_1877267 |
| Biotin Anti-mouse CD34 (RAM34) | eBioscience | Cat #13–0341-85; RRID:AB_466426 |
| Alexa Fluor 700 Anti-mouse CD34 (RAM34) | eBioscience | Cat #56–0341-82; RRID:AB_493998 |
| FITC Anti-mouse CD34 (RAM34) | eBioscience | Cat #11–0341-85; RRID:AB_465022 |
| APC Anti-mouse CD41 (MWReg30) | Biolegend | Cat #133913; RRID:AB_1112675 1 |
| FITC Anti-mouse CD43 (R2/60) | eBioscience | Cat #11–0431-85; RRID:AB_465041 |
| PE Anti-mouse CD43 (R2/60) | eBioscience | Cat #12–0431-83; RRID:AB_465660 |
| APC eFluor 780 Anti-mouse CD45.1 (A20) | eBioscience | Cat #47–0453-82; RRID:AB_1582228 |
| Alexa Fluor 700 Anti-mouse CD45.2 (104) | BioLegend | Cat #109822; RRID:AB_493731 |
| FITC Anti-mouse CD45.2 (104) | BioLegend | Cat #109806; RRID:AB_313443 |
| APC Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #17–0452-83; RRID:AB_469396 |
| FITC Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #11–0452-85; RRID:AB_465055 |
| PE Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #12–0452-85; RRID:AB_465673 |
| PerCP-Cyanine5 Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #45–0452-82; RRID:AB_1107006 |
| APC Anti-mouse CD48 (HM48–1) | BioLegend | Cat #103412; RRID:AB_571997 |
| FITC Anti-mouse CD48 (HM48–1) | BioLegend | Cat #103404; RRID:AB_313019 |
| PE Anti-mouse CD48 (HM48–1) | BioLegend | Cat #103406; RRID:AB_313021 |
| PE/Cy7 Anti-mouse CD48 (HM48 | BioLegend | Cat #103424; RRID:AB_2075049 |
| FITC Anti-mouse CD71 (R17217) | eBioscience | Cat #11–0711-85; RRID:AB_465125 |
| APC Anti-mouse CD117 (cKit) (2B8) | eBioscience | Cat #17–1171-83; RRID:AB_469431 |
| APC eFluor 780 Anti-mouse CD117 (cKit) (2B8) | eBioscience | Cat #47–1171-82; RRID:AB_1272177 |
| PE-Cyanine7 Anti-mouse CD117 (cKit) (2B8) | eBioscience | Cat #25–1171-82; RRID:AB_469644 |
| PE Anti-mouse CD127 (IL7Rα; A7R34) | BioLegend | Cat #135010; RRID:AB_1937251 |
| APC Anti-mouse CD150 (TC15–12F12.2) | BioLegend | Cat #115910; RRID:AB_493460 |
| PE Anti-mouse CD150 (TC15–12F12.2) | BioLegend | Cat #115904; RRID:AB_313683 |
| PE-Cyanine7 Anti-mouse CD150 (TC15–12F12.2) | BioLegend | Cat #115914; RRID:AB_439797 |
| Brilliant Violet 650 Anti-mouse CD150 (TC15–12F12.2) | BioLegend | Cat #115931 RRID: AB_2562402 |
| APC Anti-mouse Ter119 (TER-119) | Biolegend | Cat #116212; RRID:AB_313713 |
| FITC Anti-mouse Ter119 (TER-119) | Biolegend | Cat #116206; RRID:AB_313709 |
| PE Ter119 (TER-119) | Biolegend | Cat #116208; RRID:AB_313709 |
| APC Anti-mouse Sca-1 (D7, E13–161.7)\ | eBioscience | Cat #17–5981-82; RRID:AB_469487 |
| Alexa Fluor 700 Anti-mouse Sca-1 (D7, E13–161.7) | eBioscience | Cat #56–5981-82; RRID:AB_657836 |
| FITC Anti-mouse Sca-1 (D7, E13–161.7) | eBioscience | Cat #11–5981-85; RRID:AB_465334 |
| PerCp-Cyanine5.5 Anti-mouse Sca-1 (D7, E13–161.7) | eBioscience | Cat #45–5981-82; RRID:AB_914372 |
| APC Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108412; RRID:AB_313377 |
| FITC Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108406; RRID:AB_313371 |
| PE Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108408; RRID:AB_313373 |
| PE/Cy7 Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108416; RRID:AB_312663 |
| APC Anti-mouse IgM (II/41) | eBioscience | Cat #17–5790-82; RRID:AB_469458 |
| PE Anti-mouse IgM (II/41) | eBioscience | Cat #12–5790-83; RRID:AB_465941 |
| Recombinant Alexa Fluor 647 Anti-SQSTM1/p62 antibody (EPR4844) | Abcam | Cat # ab194721 |
| Anti-vimentin antibody | Millipore Sigma | Cat #AB5733 |
| Goat anti-chicken IgY (H&L) Alexa Fluor 488 | Invitrogen | Cat #A-11039 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PIPC | GE Healthcare Biosciences Corp | Cat #27473201 |
| Anti-mouse CD117 microbeads | Miltenyi | Cat #130–091-224 |
| 2-mercaptoethanol | Sigma | Cat #M3148 |
| Heat-inactivated bovine serum | Gibco | Cat #26170043 |
| DMSO | Sigma | Cat #D2650 |
| Baytril | Covetrus | Cat #37713 |
| Ammonium chloride potassium buffer | Covetrus | Cat #37713 |
| Paraformaldehyde | Affymetrix | Cat #19943 |
| 4% paraformaldehyde aqueous solution | Electron Microscopy Sciences | Cat #157–4 |
| PBS | Corning | Cat #MT21040CV |
| Tween 20 | Sigma | Cat #P9416 |
| 4’,6-diamidino-2-phenylindole (DAPI ) | Life Technologies | Cat #62247 |
| Saponin | Sigma | Cat #47036 |
| Fetal Bovine Serum | Life Technologies | Cat #16000044 |
| Bortezomib | Cell Signaling | Cat #2204S |
| Critical Commercial Assays | ||
| Proteasome-Glo chymotrypsin-like cell based assay | Promega | Cat #G8660 |
| RNeasy Plus Micro Kit | Qiagen | Cat #74034 |
| PROTEOSTAT Aggresome detection kit | Enzo | Cat #ENZ-51035-K100 |
| ProLong Diamond Antifade Mountant | Thermo | Cat #P36965 |
| Deposited Data | ||
| RNA-sequencing data | This manuscript | GEO GSE179415 |
| RNA-sequencing data for human HSCs and progenitors | Xie et al, 2019 | GEO: GSE125345 |
| RNA-sequencing data for human HSCs and progenitors | Laurenti et al, 2013 | GEO: GSE42414 |
| RNA-sequencing data for mouse HSCs and progenitors | Signer et al, 2016 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6: C57BL/6J | The Jackson Laboratory |
Cat #000664; RRID:IMSR_JAX:0 00664 |
| Mouse: B6SJL: B6.SJL | The Jackson Laboratory | Cat #002014; RRID:IMSR_JAX:0 02014 |
| Mouse: C57BL/6-Tg(CAG- RFP/EGFP/Map1lc3b)1Hill/J |
The Jackson Laboratory | Cat #027139; RRID:IMSR_JAX:0 27139 |
| Mouse: Bag3fl/fl | Fang et al, 2017 | N/A |
| Mouse: Atg5fl/fl | Hara et al, 2006 | N/A |
| Mouse: Mx1-Cre: B6.Cg-Tg(Mx1-cre)1Cgn/J | The Jackson Laboratory | Cat #003556 RRID:IMSR_JAX:0 03556 |
| Software and Algorithms | ||
| FlowJo | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo/downloads |
| FACSDiva | BD Bioscience | https://www.bdbiosciences.com/enus/products/software/instrumentsoftware/bdfacsdiva-software |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Image Lab 6.0.1 | Bio-Rad | https://www.biorad.com/enus/product/image-lab-software?ID=KRE6P5E8Z |
| TopHat 1.4.1 | JHU CCB | https://ccb.jhu.edu/software/tophat/downloads/ |
| STAR 2.6.1 | N/A | https://github.com/alexdobin/STAR |
| PRINSEQ Lite 0.20.3 | N/A | https://sourceforge.net/projects/prinseq/files/standalone/ |
| Htseq-count 0.7.1 | N/A | https://htseq.readthedocs.io/en/master/install.html |
| featureCounts 1.6.5 | N/A | https://sourceforge.net/projects/subre ad/files/subread1.6.5/ |
| Bioconductor package DESeq2 1.24.0 | Bioconductor | https://git.bioconductor.org/packages/DESeq2 |
| Matplotlib | J.D Hunter, 2007 | https://matplotlib.org/stable/index.html |
| ELDASoftware | Hu and Smyth, 2009 | https://bioinf.wehi.edu.au/software/elda/ |
| ZEN (black edition) | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
| ZEN (blue edition) | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |