

Summary
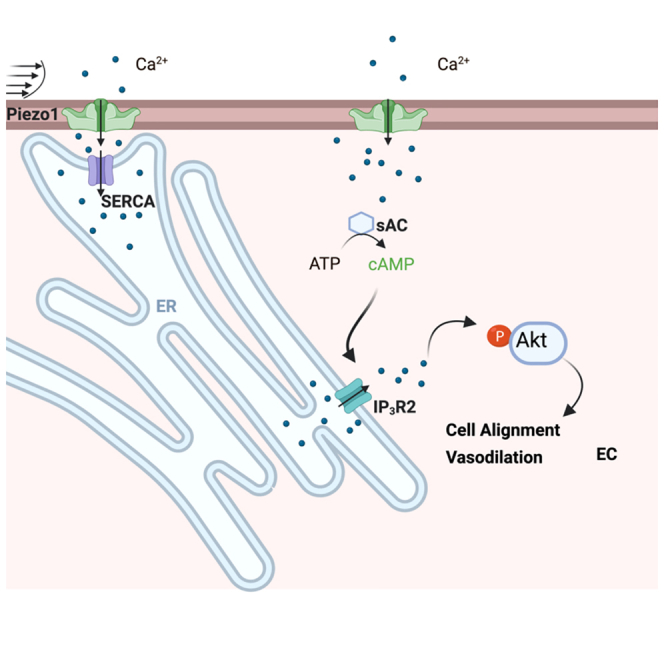
Endothelial cells (ECs) continuously sense and adapt to changes in shear stress generated by blood flow. Here, we show that the activation of the mechanosensitive channel Piezo1 by defined shear forces induces Ca2+ entry into the endoplasmic reticulum (ER) via the ER Ca2+ ATPase pump. This entry is followed by inositol trisphosphate receptor 2 (IP3R2)-elicited ER Ca2+ release into the cytosol. The mechanism of ER Ca2+ release involves the generation of cAMP by soluble adenylyl cyclase (sAC), leading to IP3R2-evoked Ca2+ gating. Depleting sAC or IP3R2 prevents ER Ca2+ release and blocks EC alignment in the direction of flow. Overexpression of constitutively active Akt1 restores the shear-induced alignment of ECs lacking Piezo1 or IP3R2, as well as the flow-induced vasodilation in endothelial restricted Piezo1 knockout mice. These studies describe an unknown Piezo1-cAMP-IP3R2 circuit as an essential mechanism activating Akt signaling and inducing adaptive changes in ECs to laminar flow.
Subject areas: cellular physiology, molecular biology, cell biology
Graphical abstract
Highlights
-
•
Piezo1 activation leads to the rapid sequestration of Ca2+ into the ER lumen through SERCA
-
•
Piezo1 activation leads to Ca2+ release from cAMP sensitive IP3R2 stores
-
•
ER Ca2+ release induces Akt activity and EC alignment in response to shear stresses
-
•
Piezo1-mediated Akt activity induces vasodilation in response to increased flow
Cellular physiology; Molecular biology; Cell biology
Introduction
Endothelial cells (ECs) lining the vessel intima are continuously exposed to recurring alterations in local fluid shear stress,1,2 variations in hydrostatic pressure in the capillaries,3,4 and changes in vessel wall tensile strain during increased vascular pulsations.5 These localized changes in mechanical forces, such as fluid shear stress, are integral to normal perfusion. Fluid shear stress, the frictional force generated by distinct spatial and temporal blood flow patterns, is the most relevant factor in developmental and physiological vascular processes.1,6,7 These processes include angiogenesis,6 as well as the maintenance of blood pressure and flow patterns1,2,7 needed to ensure normal blood perfusion in vital organs such as the brain and heart.8,9,10
It is now apparent that ECs sense forces such as shear stress by engaging multiple mechanosensitive proteins, including G-protein-coupled receptors,11,12 adherens junction proteins,13,14 and flow-sensitive ion channels including the recently discovered Piezo-type mechanosensitive ion channel component 1 (Piezo1).15,16,17 Piezo1 is a well-established mechanosensitive channel, which non-selectively transports Ca2+ from the extracellular milieu to the cytosol of ECs in response to mechanical forces.18,19,20 Deleting the Piezo1 gene in mice reduces the level of shear-evoked increases in intracellular Ca2+18 as well as prevents the morphological and cytoprotective responses of ECs to shear force.18,21 Impairment or loss of Piezo1 activity severely compromises vascular development and angiogenesis18,19 and induces hypertension in adult mice.22 However, signaling mechanisms that control EC adaptation to shear stress downstream of Piezo1 remain poorly understood.
Shear stress induces both Ca2+ entry from the extracellular milieu through mechanosensitive channels, and Ca2+ release from inositol 1,4,5-trisphosphate receptors (IP3Rs) localized in the ER membrane.23 A fundamental question is whether these two mechanotransduction pathways are intrinsically linked for regulating EC adaptation to sheer stress. Piezo1 channel, which interacts with and modulates the activity of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) pump,24 may activate both Ca2+ entry from the extracellular milieu and Ca2+ release from the ER stores. Here, the data show that the activation of Piezo1 by shear stress leads to the rapid mobilization of intracellular Ca2+ into the ER lumen followed by ER Ca2+ release via a sAC-cAMP sensitive mechanism. In contrast to previous studies showing phospholipase C (PLC)-mediated Ca2+ release from the ER stores via an IP3-IP3R cascade,25,26 the data, described here, link Piezo1 mechanosensing to a cAMP-dependent ER Ca2+ release through IP3R2. This Piezo1-sAC-IP3R2 mechanotransduction circuit plays a critical role in the activation of Akt signaling as a part of the adaptive morphological response of ECs to laminar shear stress causing vessel vasodilation.
Results
Shear stress activation of Piezo1 in endothelial cells induces rapid mobilization of intracellular Ca2+ into the endoplasmic reticulum lumen
To determine the role of Piezo1 in shear stress-induced ER Ca2+ signaling, we exposed endothelial monolayers to laminar fluid shear forces of 5 and 10 dyn/cm2, stresses known to stimulate adaptive morphological response in ECs.1 We also exposed endothelial cell monolayers to lower level laminar flow of 2 dyn/cm2, as it does not induce EC adaptive morphological changes.1 The temporal changes in [Ca2+]ER were measured using the ER-targeted Ca2+ sensor G-CEPIA1er, which emits fluorescence when bound to ER Ca2+.27 Exposure of human pulmonary arterial endothelial (HPAE) monolayers to 5 and 10 dyn/cm2 shear forces for 60s increased fluorescence in the ER, indicative of increased [Ca2+]ER (Figures 1A and 1B). These changes were dependent on the shear forces applied and were far less apparent in cells exposed to 2 dyn/cm2 shear (Figures 1A and 1B). Furthermore, the amplitude of [Ca2+]ER increased proportionally to the shear force applied (Figures S1A and S1B), whereas the rate constants calculated for ER Ca2+ rise and decay were lower at higher shear force (Figure 1C). Similar kinetics were also observed in human aortic endothelial (HAE) monolayers subjected to 10 dyn/cm2 laminar shear stress (Figures S1C–S1F), indicating the generalizability of our finding to arterial ECs of different vascular beds.
Figure 1.

Shear stress activation of Piezo1 induces rapid mobilization of Ca2+ into the ER lumen
(A) Live-cell images of G-CEPIA1er, used to measure changes in [Ca2+]ER, upon the application of 10 dyn/cm2 laminar shear for 60 s in untransfected (UT) or siRNA-treated endothelial monolayers as indicated; color-coding scale is shown below; times in seconds; scale bar, 20 μm. Arrow indicates the application of shear stress.
(B) Time-dependent changes of [Ca2+]ER in endothelial monolayers before and after stimulation with 2, 5, and 10 dyn/cm2 shear stress for 60 s (highlighted area) as in (A); n = 5-7 cells per group from 3 independent experiments; mean ± SEM.
(C) Rate constants of ER Ca2+ rise and decay in (B), mean ± SD; ∗, p < 0.05; ∗∗∗, p < 0.001, ANOVA with Tukey’s post hoc test.
(D) Time-dependent changes of [Ca2+]ER in control and Piezo1-depleted endothelial monolayers upon the application of 10 dyn/cm2 shear stress as in (A); n = 6 cells per group from 3 independent experiments; mean ± SEM.
(E) Rate constants of ER Ca2+ rise and decay in D, mean ± SD; left, ∗, p < 0.05; ∗∗∗, p < 0.001, Student’s t test.
Depletion of Piezo1 abrogated the transient increase in [Ca2+]ER, leading to the reduction of both rise and decay constants in ECs exposed to the stresses applied (Figures 1A, 1D, 1E, S1G, and S1H). Consistent with previous reports,18 depletion of Piezo1 also markedly diminished the cytosolic Ca2+ rise in response to shear stress (Figures S1I and S1J). Thus, these results revealed the central role of Piezo1 in regulating Ca2+ homeostasis via a rapid mobilization of Ca2+ into the ER in response to shear stresses applied.
Piezo1 cooperates with sarco/endoplasmic reticulum Ca2+ ATPase to induce the rapid mobilization of Ca2+ into the endoplasmic reticulum
We next addressed whether the activation of Piezo1 can induce changes in [Ca2+]ER independent of mechanical stress. Here using Yoda1, a specific Piezo1 activator,28 we also observed transient increases in [Ca2+]ER (Figures 2A–2C). Similar to cells exposed to laminar flow, activation of Piezo1 with Yoda1 led to a rapid transient increase in ER Ca2+ (Figures 2A–2C). This response was prevented by Piezo1 knockdown, indicative of the specificity of Yoda1 in activating Piezo1 (Figures 2A–2C). Depletion of extracellular Ca2+ also abolished the rapid mobilization of Ca2+ into the ER (Figures S2A and S2B). Furthermore, the rate constants of ER Ca2+ rise and decay were significantly greater in Yoda1-stimulated ECs as compared to ECs treated with diacylglycerol (DAG) analogue 1-oleoyl-2-acetyl-sn-glycerol (OAG) (Figures S2C and S2D), the activator of receptor-operated Ca2+ entry,29 suggesting a different mechanism of Ca2+ mobilization into the ER induced by Piezo1 as compared to receptor-operated Ca2+ entry.
Figure 2.

Activation of Piezo1 with Yoda induces SERCA-dependent uptake of Ca2+ in the ER lumen
(A) Live-cell images of G-CEPIA1er in endothelial monolayers pre-treated with control or Piezo1 siRNA and stimulated with the Piezo1 activator Yoda1 at time 0; color-coding as in Figure 1A; time in seconds; scale bar, 10 μm.
(B) Time-dependent changes of [Ca2+]ER upon Piezo1 activation using Yoda1 in (A); n = 6 cells per group from 3 independent experiments; mean ± SEM.
(C) Rate constants of ER Ca2+ rise and decay calculated from data in (B); mean ± SD; ∗, p < 0.05, Student’s t test; ∗∗∗, p < 0.001, Student’s t test.
(D) Time course of [Ca2+]ER in endothelial monolayers pre-treated with control or STIM1 siRNA and stimulated with Yoda1 (arrow); n = 6 cells per group from 2 independent experiments; mean ± SEM.
(E and F) Rate constants of the first (E) and the second (F) rise of ER Ca2+ calculated from data in (D), mean ± SD; ns, not significant, Student’s t test; ∗∗, p < 0.01, Student’s t test.
(G) Time-dependent changes of cytosolic [Ca2+]i on the activation of Piezo1 with Yoda1 (arrow) in endothelial monolayers pre-treated with control or STIM1 siRNA; n = 10-17 cells per group from 2 independent experiments; mean ± SEM.
(H) Changes of [Ca2+]i within the first 350s (area under the curve) after Yoda1 stimulation calculated from data in (F); mean ± SD; ns, not significant, Student’s t test; ∗∗∗, p < 0.001, Student’s t test.
(I) Live-cell images of G-CEPIA1er before and after the activation of Piezo1 with Yoda1 in endothelial monolayers pre-treated with vehicle (DMSO) or thapsigargin (Th) to inhibit SERCA; color-coding as in Figure 1A; note, different scale ranges are used to demonstrate ER Ca2+ kinetics in cells inhibited of SERCA; time in seconds; scale bar, 10μm.
(J) Time-dependent changes of [Ca2+]ER in (H); n = 6-7 cells per group from 3 independent experiments; mean ± SEM.
(K and L) Changes in ER Ca2+ (area under the curve) following Yoda1 treatment (K) and rate constants of ER Ca2+decay (L) calculated from data in (J); mean ± SD; ∗∗∗, p < 0.001, Student’s t test.
To investigate the role of STIM1, which translocates to the plasma membrane upon ER Ca2+ depletion to activate store-operated Ca2+ entry30,31 in rapid mobilization of Ca2+ into the ER lumen upon the activation of Piezo1, we repeated these studies in STIM1 depleted cells (Figures 2D and 2E). Interestingly, STIM1 depletion (Figures S2E and S2F) did not affect the kinetics of rapid rise in the ER Ca2+ but decreased the rate constant of the second much slower rise in ER Ca2+ (Figures 2D–2F). Consistent with the role of STIM1 in activating store-operated Ca2+ entry, the depletion of STIM1 also altered the kinetics of Yoda1-induced increases in intracellular Ca2+ (Figures 2G and 2H). Whereas control ECs showed a sustained increase in intracellular Ca2+ upon challenge with Yoda1, cells depleted of STIM1 showed a transient increase in intracellular Ca2+ (Figures 2G and 2H). We concluded that STIM1 did not contribute to the rapid rise in ER Ca2+ following Piezo1 activation but was necessary to replenish the ER Ca2+ upon depletion.
We also addressed the role of the SERCA pump in regulating Piezo1-mediated ER Ca2+ entry. As Piezo1 and SERCA interact both in vitro and in cells,24 Piezo1 function may be coupled to the activation of the SERCA pump. Thus, we inhibited SERCA using the specific non-competitive inhibitor thapsigargin,32 and determined the role of SERCA in the Piezo1-induced mobilization of Ca2+ into the ER. We demonstrated that thapsigargin abrogated Yoda1-induced rise in the ER Ca2+ (Figures 2I–2K), but it had no significant effect on ER Ca2+ decay (Figures 2J and 2L), which occurred at the same rate as in DMSO-treated cells. ECs treated with thapsigargin showed a trend toward higher decay constants as the result of passive leakage of ER Ca2⁺, which became more apparent when SERCA pump activity was inhibited. Thus, SERCA activity was required for Piezo1-mediated rise in ER Ca2⁺ but not ER Ca2⁺ decay.
Activation of Piezo1 induces Ca2+ release through IP3R2
To address the mechanism of ER Ca2+ decay downstream of Piezo1, we depleted IP3R2 and IP3R3, two inositol trisphosphate receptors highly expressed on the ER membrane of ECs33,34. Depletion of IP3R3 had no effect on Piezo1-induced ER Ca2+ decay since the rate constants were similar to control siRNA-treated cells (Figures 3A–3C and S3A–S3C). In contrast, the depletion of IP3R2 significantly reduced the rate constant of ER Ca2+ decay without affecting the rate constant of ER Ca2+ rise (Figure 3A–3C, S3C, and S3D). Similarly, depletion of IP3R2 significantly diminished the rate constant of ER Ca2+ decay upon the application of laminar shear stress (Figures 3D–3F).
Figure 3.

Activation of Piezo1 induces ER Ca2+ store release via IP3R2 channel
(A) Live-cell images of G-CEPIA1er upon the activation of Piezo1 with Yoda1 of endothelial monolayers pre-treated with control, IP3R2, or IP3R3 siRNA; color-coding as in Figure 1A; time in seconds; scale bar, 10 μm.
(B) Time course of [Ca2+]ER in (A); n = 8-12 cells per group from 3 independent experiments; mean ± SEM.
(C) Rate constants of ER Ca2+ decay calculated form data in (B); mean ± SD; ∗∗, p < 0.01; ANOVA with Tukey’s post hoc test.
(D) Live-cell images of G-CEPIAer in control and IP3R2 deficient endothelial monolayers upon the application of 10 dyn/cm2 laminar shear for 60 s; time in seconds; scale bar, 10 μm.
(E) Time course of [Ca2+]ER in (D); n = 7-9 cells per group from 3 independent experiments; mean ± SEM.
(F) Rate constants of ER Ca2+ decay calculated form data in (E); mean ± SD; ∗∗, p < 0.01, Student’s t test.
Depletion of either receptor did not result in the compensatory upregulation of the other receptor (Figures S3A–S3D). Furthermore, in contrast to IP3R3 depletion, which had no effect on Piezo1-mediated changes in intracellular [Ca2+]i (Figures S3E and S3F), depletion of IP3R2 significantly reduced increases in [Ca2+]i following the activation of Piezo1 with Yoda1 or by applying laminar shear stresses (Figures S3G–S3J). These data suggest that the IP3R2 pathway was essential for the release of ER Ca2+ into the cytosol following mechanical or pharmacological activation of Piezo1.
Piezo1 activation of soluble adenyl cyclase induces cAMP-dependent endoplasmic reticulum Ca2+ release
To investigate the signaling mechanism of Ca2+ release from the ER downstream of Piezo1, we studied the role of cAMP in sensitizing ER calcium release through IP3R2 stores. The second messengers IP3 and free intracellular Ca2+ activate all IP3Rs,33,35 whereas cAMP exclusively sensitizes IP3R2.36,37,38 Since Ca2+ directly stimulates soluble-adenylate cyclase (sAC),39 we determined whether the influx of Ca2+ through Piezo1 activated sAC and induced the production of cAMP. We measured relative changes in cAMP concentrations using the cAMP sensor EPAC1, a guanine nucleotide exchange factor directly activated by cAMP, tagged to a circularly permuted GFP protein.40 Yoda1 increased GFP intensity of the cAMP sensor in endothelial monolayers (Figure 4A), indicative of the increased generation of cAMP.
Figure 4.

Activation of Piezo1 induces sAC-dependent generation of cAMP and cAMP-evoked ER Ca2+ release
(A) Time-dependent changes of cAMP concentrations upon the activation of Piezo1 with Yoda1 (arrow) in control and sAC-depleted endothelial monolayers; n = 18 cells per group from 3 independent experiments; mean ± SEM.
(B) Relative changes of cAMP concentrations in (A); mean ± SD; ∗∗∗∗, p < 0.0001, Student’s t test.
(C) Live-cell images of G-CEPIA1er upon the activation of Piezo1 with Yoda1 (arrow) in control and sAC-depleted endothelial monolayers; color-coding as in Figure 1A; times in seconds; scale bar, 10 μm.
(D) Time-dependent changes of [Ca2+]ER in (C); n = 7-10 cells per group from 3 independent experiments; mean ± SEM.
(E) The rate constants of ER Ca2+ decay calculated from data in (C); mean ± SD; ∗, p < 0.05, Student’s t test.
Furthermore, depletion of sAC significantly reduced Yoda1-induced production of cAMP (Figures 4A, 4B, S4A, and S4B), suggesting that sAC was crucial in mediating cAMP generation downstream of Piezo1 activation. Furthermore, depletion or pharmacological inhibition of sAC with bithionol markedly reduced the rate constant of ER Ca2+ decay (Figures 4C–4E and S4C–S4E) as well as delayed the increase in cytosolic [Ca2+]i following pharmacological or shear-mediated activation of Piezo1 (Figures S4F–S4I). Thus, sAC activation was essential for Piezo1-induced generation of cAMP and Ca2+ release through cAMP-sensitized IP3R2 stores.
Endoplasmic reticulum Ca2+ release through IP3R2 channel is required for the Akt-dependent response of endothelial cells to shear stress
ECs lacking Piezo1 failed to induce Akt1 phosphorylation and became aligned in the direction of laminar flow.18,19,22 Thus, we determined whether ER Ca2+ release as well as Akt1 activity were required for the adaptive reorientation of ECs as induced by flow. As compared to endothelial monolayers grown under static conditions, both HPAE and HAE monolayers subjected to 10 dyn/cm2 shear stress for 24h had greater incidence of F-actin filaments with a smaller angle (0°-30°) relative to the direction of flow, indicative of cell alignment shift (Figures 5A, 5B and S5A–S5F). The alignment, however, was lost in cells depleted of either Piezo1 or IP3R2, as apparent from a significant decrease in the incidence of filaments aligned to flow (0°-30°) and an increase in randomly distributed filaments (forming larger angles to flow) (Figures 5A, 5B and S5A–S5F). Similar to Piezo1 depletion,22 IP3R2 deficiency prevented shear-induced phosphorylation of Akt at S473 (Figures 5C and 5D), the site required for the maximal enzymatic activation of Akt.41
Figure 5.

IP3R2-evoked ER Ca2+ release is required for shear-induced EC alignment and activation of Akt
(A) Representative images of F-actin in endothelial monolayers depleted of indicated proteins and grown under static or 10 dyn/cm2 laminar shear flow conditions for 24 h; red arrow indicates the direction of flow; scale bar 20 μm.
(B) Frequency of F-actin distribution within 0-30° relative to the direction of flow, a measure of cell alignment; n = 6-15 field per condition, from 2 to 4 independent experiment on static group and 17-24 fields per condition from 4 independent experiments on shear group; mean ± SD; ∗, p < 0.05; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001, ANOVA with Tukey’s post hoc test.
(C) Western blot analysis of Akt (Ser 473) phosphorylation in endothelial monolayers depleted of indicated proteins and subjected to static or 10 dyn/cm2 shear stress conditions for 30 min.
(D) Fold increase in Akt phosphorylation in (C); data are presented as 3 biological replicates from 3 independent experiments; mean ± SD; ∗, p < 0.05; ∗∗, p < 0.01; ANOVA with Tukey’s post hoc test.
Since the depletion of Piezo1 or IP3R2 prevented both shear-induced activation of Akt and alignment of ECs in the direction of flow, we next addressed the role of Akt kinase in mediating the adaptive morphological response to shear stresses. Expression of an activation-deficient Akt1 mutant42 prevented the alignment of ECs in the direction of flow (Figures 6A, 6B, S5G, and S5H). We additionally determined whether ectopic expression of constitutively active (myr)-Akt142 would restore the response of ECs lacking Piezo1 or IP3R2 to shear stress. We observed that myr-Akt1 restored the alignment of ECs depleted of either Piezo1 or IP3R2 (Figures 6C, 6D, S5I, and S5J). However, this response was not seen in Piezo1-or IP3R2-depleted cells expressing the empty vector (Figures 6C, 6D, S5I, and S5J). Thus, the loss of either Piezo1 or IP3R2 each prevented the adaptive response of ECs to shear stress by limiting Akt signaling.
Figure 6.

Activation of Akt downstream of Piezo1-sAC-IP3R2 circuit is required for the adaptation of endothelial cells to laminar shear stress and vasodilation
(A) Representative images of F-actin, GFP, and Akt in endothelial monolayers ectopically expressing GFP or activity deficient AD-Akt. Cells were grown at static or 10 dyn/cm2 laminar shear stress conditions for 24 h; red arrow indicates the direction of flow; scale bar 20 μm.
(B) Frequency of F-actin distribution within 0-30° angle relative to the direction of flow in (A); n = 5-10 fields per condition from 2 independent experiments; mean ± SD; ∗∗, p < 0.01, ANOVA with Tukey’s post hoc test.
(C) Representative images of F-actin and phospho-Akt (p-Akt) in endothelial monolayers depleted of Piezo1 or IP3R2 and ectopically expressing empty vector or myr-Akt1. Cells were grown at static or 10 dyn/cm2 laminar shear stress conditions for 24 h; ref arrows indicate the direction of flow; scale bar 20 μm.
(D) Frequency of F-actin distribution within 0-30° angle relative to the direction of flow; n = 14-18 fields per condition across 3 independent experiments; mean ± SD; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001, ANOVA with Tukey’s post hoc test.
(E) Flow-induced vasodilation of mesenteric arteries isolated from wild type (WT) or Piezo1ΔEC mice infected with adenoviral particles encoding empty vector or myr-Akt1 as indicated. n = 4 and 7 for Piezo1ΔEC and WT mice, respectively; ∗, p < 0.05 using two-way ANOVA, pap: papaverine.
(F) Quantification of data in (E) at Δ100 cm H2O; ∗∗∗∗, p < 0.0001, ANOVA with Tukey’s post hoc test.
Furthermore, to address the physiological implication of Piezo1 in regulating the Akt-mediated response of ECs to laminar shear stress, we transduced either empty control vector or myr-Akt1 in ECs of mesenteric vessels isolated from endothelial-restricted knockout of Piezo1 mice (Piezo1ΔEC), which showed markedly reduced vasodilation response to increased fluid shear as compared to control wild-type (WT) mice (Figures 6E and 6F). We observed that the expression of myr-Akt1, but not the control vector, restored the flow-induced vasodilation response of mesenteric vessels ex vivo to the level of control vessels (Figures 6E and 6F). These data cumulatively suggest that the Piezo1-sAC-IP3R2 circuit is responsible for the activation of Akt1 signaling in ECs and thereby, the spatial and temporal response of blood vessels to changes in blood flow.
Discussion
Here, we describe a unique role of Piezo1 in activating ER Ca2+ release that plays a fundamental role in the adaptation of ECs to shear stresses (as described in the graphic abstract). The data presented here show that the activation of Piezo1 by the application of transient or continuous laminar shear stresses (data not shown) not only induces Ca2+ entry from the extracellular milieu into the cytosol but also activates rapid mobilization of Ca2+ into the ER lumen in a force-dependent manner. The latter event is followed by a rapid ER Ca2+ release from cAMP-sensitive IP3R2 stores, allowing the amplification and propagation of calcium signaling throughout the cytosol to induce the adaptive response of ECs to shear applied. Thus, Piezo1 regulates flow-induced increases in cytosolic Ca2+ by two independent mechanisms: through the initial Ca2+ mobilization from the extracellular milieu and the release of Ca2+ through cAMP-sensitive stores.
The initial Ca2+ ions entering through the Piezo1 channel are promptly mobilized into the ER lumen by SERCA, the Ca2+ pump that recycles cytosolic Ca2+ into the ER lumen.43 ECs express all mammalian SERCA isoforms, including SERCA2a and SERCA3,44 which exhibit high Ca2+ turnover rates45,46 and might be involved in regulating the rapid mobilization of Ca2+ into the ER lumen downstream of Piezo1 mechanosensing. We speculate that the activation of Piezo1, which forms a complex with all SERCA isoforms to inhibit their activity,24 may modulate SERCA function by relieving the inhibitory effects on SERCA activity.24 The rapid kinetics of Ca2+ mobilization into the ER also suggests that Piezo1 might be recruited to specific plasma membrane microdomains to form a transient interaction with the ER membrane. Therefore, we posit that Piezo1-mediated mobilization of Ca2+ into the ER resembles the STIM1-Orai pathway, which refills the ER stores.31 In the latter case, SERCA is recruited to STIM1 and Orai clusters47,48 to pump Ca2+ into the ER lumen upon the influx of extracellular Ca2+ through Orai.49 However, Piezo1 induces prompt mobilization of extracellular Ca2+ into the ER through the activation of SERCA. The present findings also show functional differences by which Orai and Piezo1 operate to mediate extracellular Ca2+ influx. While Orai is activated by STIM1 upon ER store depletion,30,31,50 rapid mobilization of Ca2+ in the ER downstream of Piezo1 is activated by mechanical stimuli independent of ER store depletion. We show that STIM1 is dispensable for the initial rapid mobilization of extracellular Ca2+ into the ER but is involved in ER Ca2+ replenishment secondary to IP3R2-evoked Ca2+ release downstream of Piezo1 mechanosensing.
ER Ca2+ storage accounts for 75% of intracellular Ca2+ in ECs.51 The build-up of ER Ca2+ can influence the sensitivity of IP3R and RyR channels to their ligands35,52 and concomitantly activate ER Ca2+ release. The ER Ca2+ release downstream of Piezo1 described here was independent of the initial increase in luminal ER Ca2+. Treatment of cells with thapsigargin, a potent inhibitor of SERCA, blocked Ca2+ uptake into the ER lumen but did not alter the kinetics of ER Ca2+ decay downstream of Piezo1 activation. Further analysis of the pathways involved in ER Ca2+ release revealed that Piezo1-mediated influx of extracellular Ca2+ subsequently activated ER Ca2+ release into the cytosol through the IP3R2 channel. This rapid ER Ca2+ release was an essential part of the mechanotransduction response required for rapid increase in cytosolic Ca2+.
In contrast to IP3R3, which is activated by IP3 and free intracellular Ca2+, IP3R2 activity can be potentiated by cAMP.53,54 Production of both cAMP and ATP are commonly induced by mechanical forces, including shear stresses.55,56 We show that the activation of Piezo1 with Yoda1 induced a transient increase in cAMP production. Depletion of sAC suppressed the Piezo1-mediated increase in cAMP, consistent with the crucial role of sAC, which is activated by free intracellular Ca2+,39 in the generation of cAMP and cAMP-dependent ER Ca2+ release downstream of Piezo1 mechanosensing. cAMP potentiates IP3-evoked Ca2+ release by unmasking hypersensitive IP3R2 channels and potentiates IP3-evoked Ca2+ release from a discrete ER Ca2+ stores.54 We speculate that Piezo1 downstream signaling is spatially controlled and restricted to a subset of IP3R2, which has a distinct intracellular distribution compared to IP3R3 precluding the activation of calcium-induced calcium release from IP3R3 stores. cAMP may also induce Ca2+ release by phosphorylating all IP3Rs including IP3R3 in a PKA-dependent manner57; however, this mechanism does not explain our results because the depletion of IP3R3 had no effect on ER Ca2+ release. Thus, our data support the role of the cAMP-IP3R2 pathway, consistent with the specific effect of cAMP on IP3R2 channels.53,54 sAC tethered to the mitochondria and microtubules58 can generate cAMP in close proximity to IP3R2, which localizes at ER-mitochondrial interaction sites.59 Therefore, the subcellular organization of cAMP signals60,61 distinguishes the functional role of sAC from the transmembrane AC1, AC3, and AC8 isoforms that are also activated by Ca2+ signaling through binding to calmodulin.62
Transient receptor potential cation channel V4 (TRPV4) also contributes to shear-evoked Ca2+ mobilization.18,63 Deletion of the Trpv4 gene significantly reduces shear-induced Ca2+ influx in ECs and vasodilation of small mesenteric arteries in mice.63 Recent work showed that TRPV4 activation might be coupled to Piezo1 signaling.64,65 Therefore, our results describing the mechanism of IP3R2 activation downstream of Piezo1 may also inform how Piezo1 induced Ca2+ mobilization contributes to consequent activation of TRPV4 by shear stresses.63 We posit here that release of ER Ca2+ through sAC-sensitive IP3R2 stores can play an essential role in the amplification of calcium signaling downstream of Piezo1, a fast-inactivating channel,16 which cannot produce, as we show here, a sufficient increase in cytosolic calcium to activate EC response to shear stress. It is possible that IP3R2-evoked release is required for the activation of phospholipase A (PLA2), which is responsible for the generation of 5′,6′-epoxyeicosatrienoic acid from arachidonic acid66,67 and the sequential activation of TRPV4 by this natural ligand.64
ECs exposed to laminar shear stress undergo adaptive morphological changes through the reorientation of cells parallel to the direction of flow.68 Our findings show that these changes rely on a Piezo1-mediated increase in cytosolic Ca2+ since ECs lacking Piezo1 failed to adapt to shear stress18 and induce flow-mediated vasodilation.69 We demonstrate here that the genetic ablation of IP3R2 or sAC not only mitigate the increase in cytosolic Ca2+ but also prevents the alignment of ECs in the direction of flow, emphasizing the importance of the sAC-IP3R2 pathway in signaling the adaptive changes of ECs. Strikingly, ectopic expression of myr-Akt1 restores the morphological response of Piezo1- and IP3R2-deficient cells as well as flow-induced vasodilation of mesenteric vessels of Piezo1ΔEC mice, showing the critical role of Akt signaling in the shear-induced regulation of vascular tone.
The serine-threonine protein kinase Akt plays a crucial role in integrin-mediated signaling, endothelial cell survival, and endothelial response to laminar shear.70,71,72 Akt translocates from the perinuclear region to the upstream edge of cell-cell junctions relative to flow,73 where it phosphorylated immunoglobulin and proline-rich receptor-1 (IGPR-1) at S220, causing a lateral redistribution of IGPR-1 along the cell-cell junctions parallel to the direction of flow.74 In our studies, loss of either Piezo1 or IP3R2 prevented phosphorylation of Akt at S473 in response to shear stresses. Thus, ER Ca2+ release was required to activate Akt signaling mediated by sequential phosphorylation at Thr308 and S473 by phosphoinositide-dependent kinases PDK1 and PDK2, and integrin-linked kinase (ILK), respectively.75,76 Whereas the mechanism of ER Ca2+ release in activating Akt signaling remains unclear, shear-induced activation of Akt was essential for the formation of a mechanosensory complex between platelet-endothelial cell adhesion molecule 1 (PECAM1), vascular endothelial (VE)-cadherin, and vascular endothelial growth factor receptors VEGFR2/VEGFR3.77 In addition, Akt might promote Piezo1 localization at the plasma membrane microdomains, thus providing a positive feedback regulation of Piezo1 mechanosensing.78 However, the data presented here show that expression of constitutively active Akt1 suffices to restore EC response to shear stress even in absence of Piezo1 or IP3R2.
The results described here suggest a unique sAC-IP3R2 signaling pathway activated downstream of Piezo1 mechanosensing in ECs. We propose Piezo1 acts as an essential adaptive sensor for ECs that reorient ECs through regulating ER Ca2+ dynamics and Akt signaling.
Limitations of the study
The current study addresses a key signaling mechanism downstream of Piezo1 mechanosensing that promotes EC adaptation to physiological shear stresses using cell culture and ex vivo vessel models. A potential limitation to the findings described here is a lack of analyses of the sAC-IP3R2 pathway in regulating blood pressure, a physiologically relevant response of blood vessels to local and temporal changes in shear stresses in vivo. From this perspective, it will be important to investigate the role of the sAC-IP3R2 pathway using transgenic mouse models allowing a deletion of sAC or IP3R2 in ECs. On the basis of our findings, it is anticipated that similar to Piezo1 EC-KO mice,22 animals deficient of sAC or IP3R2 in endothelial cells would be prone to hypertension.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-rabbit Piezo1 | Biorbyt | orb41297 |
| anti-rabbit IP3R2 | In house against CPDYRDAQNEGKTVRDGELP | NA |
| Anti-mouse IP3R3 | BD Biosciences | 610312; RRID: AB_397704 |
| anti-rabbit sAC | Thermo Fisher | PA5-25756; RRID: AB_2543256 |
| anti-rabbit Stim1 | Cell Signaling | 4916; RRID: AB_2271287 |
| anti-goat VE-cadherin | Santa Cruz | sc-6458; RRID: AB_2077955 |
| anti-rabbit Total AKT | Cell Signaling | 9272S; RRID: AB_329827 |
| anti-rabbit pAKT (p-Ser 473) | Cell Signaling | 9271T; RRID: AB_329825 |
| anti-mouse β actin | Santa Cruz | sc47778; RRID: AB_626632 |
| anti-mouse α tubulin | Sigma-Aldrich | T6074; RRID: AB_477582 |
| anti-mouse GAPDH | Proteintech | 60004-1-Ig; RRID: AB_2107436 |
| Alexa Fluor 488 Phalloidin | Invitrogen | A12379 |
| anti-mouse GFP | Fisher Scientific | NC9777966 |
| Experimental models: Cell lines | ||
| Human Pulmonary Arterial Endothelial Cells | Lonza | CC-2530, Lot NO:0000662151 CC-2530, Lot NO: 20TL209928 |
| Human Aortic Endothelial Cells | Lonza | CC-2535, Lot NO: 20TL122171 |
| Experimental models: Organisms/strain | ||
| Piezo1 flox/flox | Li et al.18 | NA |
| Cdh5-Cre Piezo1 flox/flox | Friedrich et al.20 | NA |
| Recombinant DNA | ||
| G-CEPIA1er | Suzuki et al.27 | Addgene 58215 |
| mCherry-er | Suzuki et al.27 | NA |
| myr-Akt | Fulton et al.42 | NA |
| AA-Akt | Fulton et al.42 | NA |
| pmaxGFP® Vector | Lonza | VPI-1001 |
| Bacterial and virus strains | ||
| Ad-hVECadp-myr-AKT1 | Vector Biolabs | N/A |
| Ad-GFP | Vector Biolabs | #1060 |
| Chemicals, peptides, and recombinant proteins | ||
| EGM2 | Lonza | CC-4176 |
| Fibronectin | Sigma-Aldrich | F1141 |
| Dextran from Leuconostoc mesenteroides | Millipore-Sigma | D1662 |
| Gene Silencer siRNA Transfection Reagent | Genlantis | T500750 |
| Basic Endothelial Cells Nucleofactor Kit | Lonza | VPI-1001 |
| Fura 2 AM | Life Technologies | F1221 |
| Ionomycin | Life Technologies | I657 |
| MnCl2 | Life Technologies | M1787 |
| Calcium Calibration Buffer Kit | Life Technologies | C3008MP |
| EC media w/o Phenol Red | Cell Applications | 211PR-500 |
| Yoda1 | Sigma-Aldrich | SML1558 |
| Bithionol | Selleck Chemicals | S4552 |
| 1-Oleoyl-2-acetyl-sn-glycerol (OAG) | Sigma-Aldrich | O6754 |
| Thapsigargin | Thermo Fisher | T7458 |
| MINIPULS 3 Peristaltic Pumps | Gilson | |
| 5 m Silicone Tubing 0.8 mm | ibidi | 10841 |
| 1. 6 mm serial connector for μ-Slides | ibidi | 10830 |
| Elbow Luer connector male | ibidi | 10802 |
| μ-Slide I 0.2 | ibidi | 80161 |
| μ-Slide I 0.4 | ibidi | 80171 |
| μ-Slide VI 0.4 | ibidi | 80601 |
| Ripa Buffer | Thermo Fisher | R027 |
| Laemmli Sample Buffer | Bio Rad | 1610747 |
| Critical commercial assays | ||
| Green Up cADDis cAMP Assay Kit | Montana Molecular | U0200G |
| BCA Protein Assay Kit | Thermo Fisher | 23225 |
| Oligonucleotides | ||
| ON-Targe Plus SMART pool Piezo1 siRNA: GCAGCAUGACAGACGACAU UGGAGUAUGCCAACGAGAA UGGCUGAUGUUGUCGACUU GCGCAUCAGUCUACGUUUU |
Horizon Discovery | L-020870-03-0005 |
| ON-Targe Plus SMART pool IP3R2 siRNA: CGAAAUGGCCGCUCUAUUA GCAAUGGGUUGGAGACUAU GGAAUGAAAGGGCAAUUAA GAGCAAUAACUACCGGAUU |
Horizon Discovery | L-006208-02-0005 |
| ITPR3 GCAUGGAGCAGAUCGUGU | Thermo Fischer | 4392420 |
| ADCY10 CCAAAGAACUGGACUCGUA | Horizon Discovery | J-006353-09-0005 |
| Silencer Negative Control siRNA #1 | Thermo Fischer | AM4635 |
| Software and algorithms | ||
| ImageJ (FIJI) | National Institutes of Health | https://fiji.sc/ |
| Prism 7 | Graph Pad | https://www.graphpad.com/scientific-software/prism/ |
Resource availability
Lead contact
Any additional information and requests for resources and materials should be addressed and will be fulfill by the lead contact, Yulia A. Komarova (ykomarov@uic.edu).
Materials availability
Any request for resources and materials utilized in this paper should be addressed and fulfill by the lead contact, Yulia A. Komarova (ykomarov@uic.edu).
Experimental model and subject details
Cell culture
Human Pulmonary Artery Endothelial cells (cat# CC-2530; Lots: 0000662151 and 20TL209928) and Human Aortic Endothelial cells (Catalog #: CC-2535) purchased from Lonza were grown in EGM-2 media (cat# CC-4176; Lonza) supplemented with EGM2 Bulletkit and 10% fetal bovine serum (FBS). Cells were cultured at 37 with 5% CO2 and used from passages 4–7.
Method details
Transfections
Cells were treated with IP3R2, IP3R3, Piezo1 and sAC siRNA duplexes using Gene Silencer transfection reagent (cat# T500750; Genlantis) and used for experiments at 72–96 h post-treatment. Cells were transfected with G-CEPIA1er sensor and ER marker mCherry-er27 by electroporating DNA vectors using Basic Endothelial Cells Nucleofactor Kit (cat# VPI-1001; Lonza) and a Nucleofactor Device (Lonza) according to the manufacturer’s instructions. Following DNA electroporation, cells were seeded at 70–75% confluency and confluent monolayers were imaged 24–48 hours post transfection. For cell alignment experiments cells were transfected with myr-Akt and activation deficient AD-Akt.42 Myr-AKT and AD-AKT constructs were a gift from Dr. Monica Lee’s laboratory.
Measurements of intracellular calcium
Intracellular calcium concentrations were measured as previously described by us.34 HPAE cells grown on glass-bottom dishes (Becton Dickinson) were loaded with 3 μM Fura 2 AM (Life technologies) for 20 minutes at 37°C in EGM-2 media without supplements. Cells were washed with Hank’s balanced salt solution (HBSS) containing Ca2+ and Mg2+ and images were acquired at room temperature in phenol-free media (Cell Applications, cat# 211PR-500). Fura-2 AM fluorescence was excited at 340 and 380 nm and collected at 510 ± 80 nm using an Axiovert 100 inverted microscope (Carl Zeiss) equipped with Plan-Apo 60× with the numerical aperture (NA) 1.4 oil immersion objective, Lambda DG-4 switcher illumination system (Sutter Instruments), AxioCom Hsm camera (Zeiss), fura-2 filter set (Chroma), and AxioVision Physiology Acquisition module. Images were collected at 2-s intervals. Fluorescence ratios (F340/F380) were calculated within a circular region of interest (radius 3 μm) for each cell after subtraction of intracellular background, which was determined after quenching Fura-2 AM fluorescence by adding a mixture of 3 μM ionomycin and 5 mM MnCl2 in HBSS. [Ca2+]c was calculated from F340/F380 ratios using references to Ca2+ standard solutions (Calcium Calibration Buffer Kits; Life technologies). Stored Operated Calcium Entry (SOCE) was determined in HPAE cells loaded with Fura 2AM, incubated in Ca2+ free HBSS media, and stimulated with 2 μM thapsigargin. Ca2+ entry was determined by addition of 2 mM Ca2+.
Measurement of ER calcium
Live-cell images of G-CEPIA1er and mCherry-er27 were simultaneously acquired at 37°C before and after treatment of HPAECs with 5 μM Yoda1 using a confocal microscope (Zeiss LSM 710, Axio Observer Z1) equipped with a 63× 1.4 NA Plan-Apochromat oil immersion as well as a 10× 0.5 NA Plan-Apochromat dry objectives and a diode-pumped solid-state laser. The 63× objective was used to obtain high resolution images of the ER structure whereas the images in shear stress experiments were acquired with 10× objective to correct for rapid changes in focal plane. Dual-color images were captured simultaneously using λ = 561nm and 488nm lasers and two BiG (binary gallium arsenide phosphide photomultiplier tube (GaAsP-PMT) detectors. For comparisons of relative change in [Ca2+]ER, a cross-sectional view intensity values of G-CEPIA1er normalized to mCherry intensity values was generated at each time point, and the relative changes in [Ca2+]ERwere calculated.
In the experiments with SERCA inhibitor, cells were pretreated with 2 μM thapsigargin prior to starting live-cell imaging experiments to ensure that SERCA is inhibited at the time of Yoda1 treatment without causing a significant decrease in [Ca2+]ER.
Measurement of cAMP production
cAMP production was measured using a genetically encoded Green Up cADDis cAMP sensor40 packed in a BackMam viral vector (Montana Molecular; cat# U0200G), according to the manufacturer’s protocol. The changes in cAMP fluorescence upon 5 μM Yoda1 treatment were assessed using a Nikon Eclipse Ti microscope equipped with a 20X/0.40 objective and NIS-Elements image software at 48 hours post-transduction.
Shear stress
For live-cell imaging and immunofluorescent staining experiments, pulsatile laminar shear stress was applied on cells grown on μ-Slide I 0.2, μ-Slide I 0.4 or μ-Slide VI 0.4 to achieve different ranges of shear stress levels (IBIDI, cat# 80161, 80171, and 80601) using a MINIPULS 3 Peristaltic Pump (Gilson) connected with a 1.6 mm serial connector for μ-Slides and elbow luer connector male (IBIDI; cat# 10830 and 10802). Shear stress application was made accordingly to IBIDI shear rate calculation for μ-Slides. For measurements of relative [Ca2+]ER, cells were imaged before and after application of 2, 5 or 10 dyn/cm2 shear stresses for 60 seconds or duration of the experiment (data not shown). For cell alignment assays, cells were subjected to flow or static condition for 24 hours.
For biochemical assessment of Akt signaling, cells were incubated in a 6 well plate and subjected to 10 dyn/cm2 shear stress for 30 minutes. The laminar shear flow was replicated using a flow device consisting of a cone and plate apparatus describe elsewhere.79,80 The flow device consisted of a computerized stepper motor UMD-17 (Arcus Technology) and a 1 deg tapered stainless steel cone.81
Immunofluorescent staining and image analysis
Immunofluorescent staining was made as published by us.82 In brief, HPAE cells were fixed with 4% formaldehyde in phosphate buffered saline (PBS) for 10 min at room temperature, washed once with PBS, and permeabilized for 15 min with 0.01% Triton X-100 in PBS. Nonspecific sites were blocked with 3% Bovine Serum Albumin (BSA) solution in PBS for 2 hours at room temperature. Cells were incubated with primary antibody for 1 hour at room temp (or overnight at 4°C). After several washes with PBS, samples were incubated with secondary antibodies for 2 hours at room temperature. Cells were mounted Flouromount-G with DAPI. Images were collected with a confocal microscope (Zeiss LSM880 Confocal Microscope) equipped with a 63× 1.4 NA Plan-Apochromat oil immersion objective.
To analyze the orientation of cells, a z-projected image of samples stained with phalloidin was rotated to the direction of flow. Automated quantification by the ImageJ plugin OrientationJ83,84 evaluated the orientation of actin filaments using a Gaussian function of standard deviation with a Cubic Spline gradient to produce an event distribution of all local angles relative to flow. The distribution of orientation as a frequency of events at 30 degree intervals was determined. The OrientationJ plugin was also used to generate a visual of the local directional analysis using the same Gaussian window σ and Cubic Spline gradient.
Western blotting analysis
Western blotting was made as described by us.82 For knock down experiments, cell lysates were collected using Radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher, Cat # R027). For determination of AKT and ERK phosphorylation, cell lysates were washed 2 times with cold PBS and collected using lysis buffer (20 mM HEPES-KOH, pH 7.8, 50 mM KCl, 1 mM EGTA, 1% NP40, 1 mM NaF, 1 mM Na3VO4). Total protein concentrations were measured using a Bicinchoninic acid (BCA) protein assay kit (Thermo Scientific; cat# 23225). Samples were boiled in Laemmli sample buffer (Bio-Rad; cat# 1610747) for 10 min and separated using a SDS-PAGE system. Proteins were transferred to nitrocellulose membranes overnight at 4°C. Membranes were blocked in 5% BSA for 1–2 hours. Proteins of interest were then detected by probing with the indicated primary and horseradish peroxidase–conjugated secondary antibodies. In the experiments where greater protein separation was needed, for example, for Piezo1 detection, vinculin and α-tubulin were used as loading controls because of their higher molecular weight. In the experiments detacting AKT and ERK phosphorylation, GAPDH was used as loading control due to variability in β-actin protein expression under flow conditions. Following incubation with secondary antibodies, proteins were detected using chemiluminescence and Premium Autoradiography film (lab Force, cat# 1141J52). Band intensities were quantified using densitometry analysis using ImageJ software (National Institute of Health).
Curve fit and quantification of rate constants
GraphPad Prism9 software was used to fit a linear regression to the ER Ca2+ rise and one phase exponential decay regression to fit ER Ca2+ decay. To calculate the rate constant for ER Ca2+ rise, a linear regression defined as y = yIntercept + Slope∗x was used. To calculate the rate constant of ER Ca2+ decay, a one phase exponential decay regression defined as y= (y0 - Plateau)∗exp(-K∗x) + Plateau, where K is the rate constant for ER Ca2+ decay was used.
Vasodilation measurement in mouse mesenteric arteries
Mesenteric arteries from EC-restricted Piezo1 knockout mice and wild type C57BL/6 mice were used to measure flow-induced vasodilation as described previously.85 Briefly, mice were euthanized by CO2 inhalation followed by cervical dislocation. Mesenteries were extracted and removed of all fatty tissues to isolate arterioles. Arterioles were divided into two pieces and incubated with adenoviral particles carrying the empty vector or encoding myr-Akt1 for 48 hours at 37 °C, 5% CO2. First order arteries (120–180 μm in diameter) were cannulated into a flow-chamber, and rested for 30 minutes under 60 cm H2O internal pressure (resting diameter). Endothelin-1 (120 pM) was applied to pre-constrict the vessel to respond to flow (pre-constrict diameter). Internal flow was generated by a pressure gradient between two reservoirs connected to each end of the cannulated vessel at Δ10, Δ20, Δ40, Δ60, and Δ100 cm H2O, while the internal pressure was maintained at 60 cm H2O. Flow-induced vasodilation was measured after 3 minutes of each pressure gradient applied (flow-induced diameter). Papaverine (100 μM), an endothelial independent vasodilator, was applied after vasodilation measurement at Δ100 cm H2O to confirm smooth muscle functionality in vasodilation. Data were calculated using following equation:
Vasodilation (%) = (flow-induced diameter – pre-constrict diameter)/(resting diameter – pre-constrict diameter)∗100.
Quantification and statistical analysis
Statistical significance was calculated using GraphPad Prism9 software (GraphPad). Student’s t-test was used for experiments with two experimental groups. One-way ANOVA with Tukey’s test was used for experiments with two or more experimental groups. For flow-induced vasodilation experiments statistical analysis was performed using two-way ANOVA, and the data were presented as mean ± standard error.
Acknowledgments
This work made use of Confocal Microscopy Facility (Research Resources Center, UIC). The authors would like to thank Jae-Won Shin for suggestions related to the analysis of calcium kinetics. The work is supported by NHLBI grants T32HL007829, and R01HL045638 and R01HL045638 supplement.
Author contributions
YAK and DS designed research studies, DS, SJA, and ACM conducted experiments and acquired data; DL assisted with flow experiments, YAK, QL, SJA, and DS analyzed and interpreted data; YAK and DS wrote the article. DM, IL, and ABM provided feedback and discussion of the data and edited the article.
Declaration of interests
The authors have declared that no conflict of interest exists.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: April 11, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106661.
Supplemental information
Data and code availability
-
•
All raw data or information related to western blot quantification as well as image analysis presented in this paper is available from the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Dewey C.F., Jr., Bussolari S.R., Gimbrone M.A., Jr., Davies P.F. The dynamic response of vascular endothelial cells to fluid shear stress. J. Biomech. Eng. 1981;103:177–185. doi: 10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- 2.Davies P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009;6:16–26. doi: 10.1038/ncpcardio1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartz E.A., Bizios R., Medow M.S., Gerritsen M.E. Exposure of human vascular endothelial cells to sustained hydrostatic pressure stimulates proliferation. Circ. Res. 1999;84:315–322. doi: 10.1161/01.RES.84.3.315. [DOI] [PubMed] [Google Scholar]
- 4.Acevedo A.D., Bowser S.S., Gerritsen M.E., Bizios R. Morphological and proliferative responses of endothelial cells to hydrostatic pressure: role of fibroblast growth factor. J. Cell. Physiol. 1993;157:603–614. doi: 10.1002/jcp.1041570321. [DOI] [PubMed] [Google Scholar]
- 5.Awolesi M.A., Sessa W.C., Sumpio B.E. Cyclic strain upregulates nitric oxide synthase in cultured bovine aortic endothelial cells. J. Clin. Invest. 1995;96:1449–1454. doi: 10.1172/JCI118181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galie P.A., Nguyen D.H.T., Choi C.K., Cohen D.M., Janmey P.A., Chen C.S. Fluid shear stress threshold regulates angiogenic sprouting. Proc. Natl. Acad. Sci. USA. 2014;111:7968–7973. doi: 10.1073/pnas.1310842111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunningham K.S., Gotlieb A.I. The role of shear stress in the pathogenesis of atherosclerosis. Lab. Invest. 2005;85:9–23. doi: 10.1038/labinvest.3700215. [DOI] [PubMed] [Google Scholar]
- 8.Huo Y., Linares C.O., Kassab G.S. Capillary perfusion and wall shear stress are restored in the coronary circulation of hypertrophic right ventricle. Circ. Res. 2007;100:273–283. doi: 10.1161/01.Res.0000257777.83431.13. [DOI] [PubMed] [Google Scholar]
- 9.Mairey E., Genovesio A., Donnadieu E., Bernard C., Jaubert F., Pinard E., Seylaz J., Olivo-Marin J.C., Nassif X., Duménil G. Cerebral microcirculation shear stress levels determine Neisseria meningitidis attachment sites along the blood-brain barrier. J. Exp. Med. 2006;203:1939–1950. doi: 10.1084/jem.20060482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Polite F., Martorell J., Del Rey-Puech P., Melgar-Lesmes P., O'Brien C.C., Roquer J., Ois A., Principe A., Edelman E.R., Balcells M. Pulsatility and high shear stress deteriorate barrier phenotype in brain microvascular endothelium. J. Cerebr. Blood Flow Metabol. 2017;37:2614–2625. doi: 10.1177/0271678x16672482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chachisvilis M., Zhang Y.-L., Frangos J.A. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc. Natl. Acad. Sci. USA. 2006;103:15463–15468. doi: 10.1073/pnas.0607224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J., Mathur J., Vessières E., Hammack S., Nonomura K., Favre J., Grimaud L., Petrus M., Francisco A., Li J., et al. GPR68 senses flow and is essential for vascular Physiology. Cell. 2018;173:762–775.e16. doi: 10.1016/j.cell.2018.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noria S., Cowan D.B., Gotlieb A.I., Langille B.L. Transient and steady-state effects of shear stress on endothelial cell adherens junctions. Circ. Res. 1999;85:504–514. doi: 10.1161/01.RES.85.6.504. [DOI] [PubMed] [Google Scholar]
- 14.Langille B.L. Morphologic responses of endothelium to shear stress: reorganization of the adherens junction. Microcirculation. 2001;8:195–206. doi: 10.1038/sj/mn/7800085. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe H., Vriens J., Janssens A., Wondergem R., Droogmans G., Nilius B. Modulation of TRPV4 gating by intra- and extracellular Ca2+ Cell Calcium. 2003;33:489–495. doi: 10.1016/s0143-4160(03)00064-2. [DOI] [PubMed] [Google Scholar]
- 16.Coste B., Mathur J., Schmidt M., Earley T.J., Ranade S., Petrus M.J., Dubin A.E., Patapoutian A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerhold K.A., Schwartz M.A. Ion channels in endothelial responses to fluid shear stress. Physiology. 2016;31:359–369. doi: 10.1152/physiol.00007.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J., Hou B., Tumova S., Muraki K., Bruns A., Ludlow M.J., Sedo A., Hyman A.J., McKeown L., Young R.S., et al. Piezo1 integration of vascular architecture with physiological force. Nature. 2014;515:279–282. doi: 10.1038/nature13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ranade S.S., Qiu Z., Woo S.H., Hur S.S., Murthy S.E., Cahalan S.M., Xu J., Mathur J., Bandell M., Coste B., et al. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc. Natl. Acad. Sci. USA. 2014;111:10347–10352. doi: 10.1073/pnas.1409233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedrich E.E., Hong Z., Xiong S., Zhong M., Di A., Rehman J., Komarova Y.A., Malik A.B. Endothelial cell Piezo1 mediates pressure-induced lung vascular hyperpermeability via disruption of adherens junctions. Proc. Natl. Acad. Sci. USA. 2019;116:12980–12985. doi: 10.1073/pnas.1902165116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duchemin A.-L., Vignes H., Vermot J. Mechanically activated piezo channels modulate outflow tract valve development through the Yap1 and Klf2-Notch signaling axis. Elife. 2019;8:e44706. doi: 10.7554/eLife.44706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S., Chennupati R., Kaur H., Iring A., Wettschureck N., Offermanns S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Invest. 2016;126:4527–4536. doi: 10.1172/JCI87343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nollert M.U., Eskin S.G., McIntire L.V. Shear stress increases inositol trisphosphate levels in human endothelial cells. Biochem. Biophys. Res. Commun. 1990;170:281–287. doi: 10.1016/0006-291x(90)91271-s. [DOI] [PubMed] [Google Scholar]
- 24.Zhang T., Chi S., Jiang F., Zhao Q., Xiao B. A protein interaction mechanism for suppressing the mechanosensitive Piezo channels. Nat. Commun. 2017;8:1797. doi: 10.1038/s41467-017-01712-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majerus P.W., Connolly T.M., Deckmyn H., Ross T.S., Bross T.E., Ishii H., Bansal V.S., Wilson D.B. The metabolism of phosphoinositide-derived messenger molecules. Science. 1986;234:1519–1526. doi: 10.1126/science.3024320. [DOI] [PubMed] [Google Scholar]
- 26.Rebecchi M.J., Pentyala S.N. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol. Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki J., Kanemaru K., Ishii K., Ohkura M., Okubo Y., Iino M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014;5:4153. doi: 10.1038/ncomms5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Syeda R., Xu J., Dubin A.E., Coste B., Mathur J., Huynh T., Matzen J., Lao J., Tully D.C., Engels I.H., et al. Chemical activation of the mechanotransduction channel Piezo1. Elife. 2015;4:e07369. doi: 10.7554/eLife.07369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hofmann T., Obukhov A.G., Schaefer M., Harteneck C., Gudermann T., Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 30.Xu P., Lu J., Li Z., Yu X., Chen L., Xu T. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem. Biophys. Res. Commun. 2006;350:969–976. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- 31.Calloway N., Vig M., Kinet J.P., Holowka D., Baird B. Molecular clustering of STIM1 with Orai1/CRACM1 at the plasma membrane depends dynamically on depletion of Ca2+ stores and on electrostatic interactions. Mol. Biol. Cell. 2009;20:389–399. doi: 10.1091/mbc.e07-11-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thastrup O., Cullen P.J., Drøbak B.K., Hanley M.R., Dawson A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc. Natl. Acad. Sci. USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foskett J.K., White C., Cheung K.H., Mak D.O.D. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geyer M., Huang F., Sun Y., Vogel S.M., Malik A.B., Taylor C.W., Komarova Y.A. Microtubule-associated protein EB3 regulates IP3 receptor clustering and Ca(2+) signaling in endothelial cells. Cell Rep. 2015;12:79–89. doi: 10.1016/j.celrep.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finch E.A., Turner T.J., Goldin S.M. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- 36.Tovey S.C., Dedos S.G., Rahman T., Taylor E.J.A., Pantazaka E., Taylor C.W. Regulation of inositol 1,4,5-trisphosphate receptors by cAMP independent of cAMP-dependent protein kinase. J. Biol. Chem. 2010;285:12979–12989. doi: 10.1074/jbc.M109.096016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor C.W. Regulation of IP(3) receptors by cyclic AMP. Cell Calcium. 2017;63:48–52. doi: 10.1016/j.ceca.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdullaev I.F., Bisaillon J.M., Potier M., Gonzalez J.C., Motiani R.K., Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008;103:1289–1299. doi: 10.1161/01.Res.0000338496.95579.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Litvin T.N., Kamenetsky M., Zarifyan A., Buck J., Levin L.R. Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J. Biol. Chem. 2003;278:15922–15926. doi: 10.1074/jbc.M212475200. [DOI] [PubMed] [Google Scholar]
- 40.Tewson P.H., Martinka S., Shaner N.C., Hughes T.E., Quinn A.M. New DAG and cAMP sensors optimized for live-cell assays in automated laboratories. J. Biomol. Screen. 2016;21:298–305. doi: 10.1177/1087057115618608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alessi D.R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., Hemmings B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 42.Fulton D., Gratton J.P., McCabe T.J., Fontana J., Fujio Y., Walsh K., Franke T.F., Papapetropoulos A., Sessa W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brini M., Carafoli E. Calcium pumps in health and disease. Physiol. Rev. 2009;89:1341–1378. doi: 10.1152/physrev.00032.2008. [DOI] [PubMed] [Google Scholar]
- 44.Mountian I., Manolopoulos V.G., De Smedt H., Parys J.B., Missiaen L., Wuytack F. Expression patterns of sarco/endoplasmic reticulum Ca(2+)-ATPase and inositol 1,4,5-trisphosphate receptor isoforms in vascular endothelial cells. Cell Calcium. 1999;25:371–380. doi: 10.1054/ceca.1999.0034. [DOI] [PubMed] [Google Scholar]
- 45.Verboomen H., Wuytack F., De Smedt H., Himpens B., Casteels R. Functional difference between SERCA2a and SERCA2b Ca2+ pumps and their modulation by phospholamban. Biochem. J. 1992;286:591–595. doi: 10.1042/bj2860591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dode L., Andersen J.P., Leslie N., Dhitavat J., Vilsen B., Hovnanian A. Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) 1 and 2 isoforms and characterization of Darier disease (SERCA2) mutants by steady-state and transient kinetic analyses. J. Biol. Chem. 2003;278:47877–47889. doi: 10.1074/jbc.M306784200. [DOI] [PubMed] [Google Scholar]
- 47.Jousset H., Frieden M., Demaurex N. STIM1 knockdown reveals that store-operated Ca2+ channels located close to sarco/endoplasmic Ca2+ ATPases (SERCA) pumps silently refill the endoplasmic reticulum. J. Biol. Chem. 2007;282:11456–11464. doi: 10.1074/jbc.M609551200. [DOI] [PubMed] [Google Scholar]
- 48.Manjarrés I.M., Rodríguez-García A., Alonso M.T., García-Sancho J. The sarco/endoplasmic reticulum Ca(2+) ATPase (SERCA) is the third element in capacitative calcium entry. Cell Calcium. 2010;47:412–418. doi: 10.1016/j.ceca.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 49.Alonso M.T., Manjarrés I.M., García-Sancho J. Privileged coupling between Ca(2+) entry through plasma membrane store-operated Ca(2+) channels and the endoplasmic reticulum Ca(2+) pump. Mol. Cell. Endocrinol. 2012;353:37–44. doi: 10.1016/j.mce.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 50.Liou J., Kim M.L., Heo W.D., Jones J.T., Myers J.W., Ferrell J.E., Jr., Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tran Q.K., Ohashi K., Watanabe H. Calcium signalling in endothelial cells. Cardiovasc. Res. 2000;48:13–22. doi: 10.1016/s0008-6363(00)00172-3. [DOI] [PubMed] [Google Scholar]
- 52.Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu. Rev. Physiol. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- 53.Tovey S.C., Dedos S.G., Taylor E.J.A., Church J.E., Taylor C.W. Selective coupling of type 6 adenylyl cyclase with type 2 IP3 receptors mediates direct sensitization of IP3 receptors by cAMP. J. Cell Biol. 2008;183:297–311. doi: 10.1083/jcb.200803172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Konieczny V., Tovey S.C., Mataragka S., Prole D.L., Taylor C.W. Cyclic AMP recruits a discrete intracellular Ca(2+) store by unmasking hypersensitive IP(3) receptors. Cell Rep. 2017;18:711–722. doi: 10.1016/j.celrep.2016.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reich K.M., Gay C.V., Frangos J.A. Fluid shear stress as a mediator of osteoblast cyclic adenosine monophosphate production. J. Cell. Physiol. 1990;143:100–104. doi: 10.1002/jcp.1041430113. [DOI] [PubMed] [Google Scholar]
- 56.Yamamoto K., Furuya K., Nakamura M., Kobatake E., Sokabe M., Ando J. Visualization of flow-induced ATP release and triggering of Ca2+ waves at caveolae in vascular endothelial cells. J. Cell Sci. 2011;124:3477–3483. doi: 10.1242/jcs.087221. [DOI] [PubMed] [Google Scholar]
- 57.Wojcikiewicz R.J., Luo S.G. Phosphorylation of inositol 1,4,5-trisphosphate receptors by cAMP-dependent protein kinase. Type I, II, and III receptors are differentially susceptible to phosphorylation and are phosphorylated in intact cells. J. Biol. Chem. 1998;273:5670–5677. doi: 10.1074/jbc.273.10.5670. [DOI] [PubMed] [Google Scholar]
- 58.Zippin J.H., Chen Y., Nahirney P., Kamenetsky M., Wuttke M.S., Fischman D.A., Levin L.R., Buck J. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J. 2003;17:82–84. doi: 10.1096/fj.02-0598fje. [DOI] [PubMed] [Google Scholar]
- 59.Bartok A., Weaver D., Golenár T., Nichtova Z., Katona M., Bánsághi S., Alzayady K.J., Thomas V.K., Ando H., Mikoshiba K., et al. IP(3) receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019;10:3726. doi: 10.1038/s41467-019-11646-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valsecchi F., Konrad C., D'Aurelio M., Ramos-Espiritu L.S., Stepanova A., Burstein S.R., Galkin A., Magranè J., Starkov A., Buck J., et al. Distinct intracellular sAC-cAMP domains regulate ER Ca(2+) signaling and OXPHOS function. J. Cell Sci. 2017;130:3713–3727. doi: 10.1242/jcs.206318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mewes M., Lenders M., Stappers F., Scharnetzki D., Nedele J., Fels J., Wedlich-Söldner R., Brand S.M., Schmitz B., Brand E. Soluble adenylyl cyclase (sAC) regulates calcium signaling in the vascular endothelium. FASEB J. 2019;33:13762–13774. doi: 10.1096/fj.201900724R. [DOI] [PubMed] [Google Scholar]
- 62.Halls M.L., Cooper D.M.F. Regulation by Ca2+-signaling pathways of adenylyl cyclases. Cold Spring Harb. Perspect. Biol. 2011;3:a004143. doi: 10.1101/cshperspect.a004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mendoza S.A., Fang J., Gutterman D.D., Wilcox D.A., Bubolz A.H., Li R., Suzuki M., Zhang D.X. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am. J. Physiol. Heart Circ. Physiol. 2010;298:H466–H476. doi: 10.1152/ajpheart.00854.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Swain S.M., Liddle R.A. Piezo1 acts upstream of TRPV4 to induce pathological changes in endothelial cells due to shear stress. J. Biol. Chem. 2021;296:100171. doi: 10.1074/jbc.RA120.015059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swain S.M., Romac J.M.J., Shahid R.A., Pandol S.J., Liedtke W., Vigna S.R., Liddle R.A. TRPV4 channel opening mediates pressure-induced pancreatitis initiated by Piezo1 activation. J. Clin. Invest. 2020;130:2527–2541. doi: 10.1172/JCI134111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Loot A.E., Popp R., Fisslthaler B., Vriens J., Nilius B., Fleming I. Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc. Res. 2008;80:445–452. doi: 10.1093/cvr/cvn207. [DOI] [PubMed] [Google Scholar]
- 67.Watanabe H., Vriens J., Prenen J., Droogmans G., Voets T., Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- 68.Davies P.F. Flow-mediated endothelial mechanotransduction. Physiol. Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Albarrán-Juárez J., Iring A., Wang S., Joseph S., Grimm M., Strilic B., Wettschureck N., Althoff T.F., Offermanns S. Piezo1 and G(q)/G(11) promote endothelial inflammation depending on flow pattern and integrin activation. J. Exp. Med. 2018;215:2655–2672. doi: 10.1084/jem.20180483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen J., Somanath P.R., Razorenova O., Chen W.S., Hay N., Bornstein P., Byzova T.V. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat. Med. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kobayashi H., Butler J.M., O'Donnell R., Kobayashi M., Ding B.S., Bonner B., Chiu V.K., Nolan D.J., Shido K., Benjamin L., Rafii S. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat. Cell Biol. 2010;12:1046–1056. doi: 10.1038/ncb2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsuoka T., Yashiro M., Nishioka N., Hirakawa K., Olden K., Roberts J.D. PI3K/Akt signalling is required for the attachment and spreading, and growth in vivo of metastatic scirrhous gastric carcinoma. Br. J. Cancer. 2012;106:1535–1542. doi: 10.1038/bjc.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Melchior B., Frangos J.A. Distinctive subcellular Akt-1 responses to shear stress in endothelial cells. J. Cell. Biochem. 2014;115:121–129. doi: 10.1002/jcb.24639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ho R.X.Y., Tahboub R., Amraei R., Meyer R.D., Varongchayakul N., Grinstaff M., Rahimi N. The cell adhesion molecule IGPR-1 is activated by and regulates responses of endothelial cells to shear stress. J. Biol. Chem. 2019;294:13671–13680. doi: 10.1074/jbc.RA119.008548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chan T.O., Tsichlis P.N. PDK2: a complex tail in one Akt. Sci. STKE. 2001;2001:pe1. doi: 10.1126/stke.2001.66.pe1. [DOI] [PubMed] [Google Scholar]
- 76.Persad S., Attwell S., Gray V., Delcommenne M., Troussard A., Sanghera J., Dedhar S. Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells. Proc. Natl. Acad. Sci. USA. 2000;97:3207–3212. doi: 10.1073/pnas.97.7.3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fleming I., Fisslthaler B., Dixit M., Busse R. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J. Cell Sci. 2005;118:4103–4111. doi: 10.1242/jcs.02541. [DOI] [PubMed] [Google Scholar]
- 78.Lai A., Chen Y.C., Cox C.D., Jaworowski A., Peter K., Baratchi S. Analyzing the shear-induced sensitization of mechanosensitive ion channel Piezo-1 in human aortic endothelial cells. J. Cell. Physiol. 2021;236:2976–2987. doi: 10.1002/jcp.30056. [DOI] [PubMed] [Google Scholar]
- 79.Blackman B.R., García-Cardeña G., Gimbrone M.A., Jr. A new in vitro model to evaluate differential responses of endothelial cells to simulated arterial shear stress waveforms. J. Biomech. Eng. 2002;124:397–407. doi: 10.1115/1.1486468. [DOI] [PubMed] [Google Scholar]
- 80.Dai G., Kaazempur-Mofrad M.R., Natarajan S., Zhang Y., Vaughn S., Blackman B.R., Kamm R.D., García-Cardeña G., Gimbrone M.A., Jr. Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc. Natl. Acad. Sci. USA. 2004;101:14871–14876. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ahn S.J., Fancher I.S., Bian J.T., Zhang C.X., Schwab S., Gaffin R., Phillips S.A., Levitan I. Inwardly rectifying K(+) channels are major contributors to flow-induced vasodilatation in resistance arteries. J. Physiol. 2017;595:2339–2364. doi: 10.1113/jp273255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kruse K., Lee Q.S., Sun Y., Klomp J., Yang X., Huang F., Sun M.Y., Zhao S., Hong Z., Vogel S.M., et al. N-cadherin signaling via Trio assembles adherens junctions to restrict endothelial permeability. J. Cell Biol. 2019;218:299–316. doi: 10.1083/jcb.201802076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fonck E., Feigl G.G., Fasel J., Sage D., Unser M., Rüfenacht D.A., Stergiopulos N. Effect of aging on elastin functionality in human cerebral arteries. Stroke. 2009;40:2552–2556. doi: 10.1161/strokeaha.108.528091. [DOI] [PubMed] [Google Scholar]
- 84.Püspöki Z., Storath M., Sage D., Unser M. Transforms and operators for directional bioimage analysis: a survey. Adv. Anat. Embryol. Cell Biol. 2016;219:69–93. doi: 10.1007/978-3-319-28549-8_3. [DOI] [PubMed] [Google Scholar]
- 85.Ahn S.J., Fancher I.S., Granados S.T., Do Couto N.F., Hwang C.L., Phillips S.A., Levitan I. Cholesterol-induced suppression of endothelial kir channels is a driver of impairment of arteriolar flow-induced vasodilation in humans. Hypertension. 2022;79:126–138. doi: 10.1161/hypertensionaha.121.17672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All raw data or information related to western blot quantification as well as image analysis presented in this paper is available from the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.