

Summary
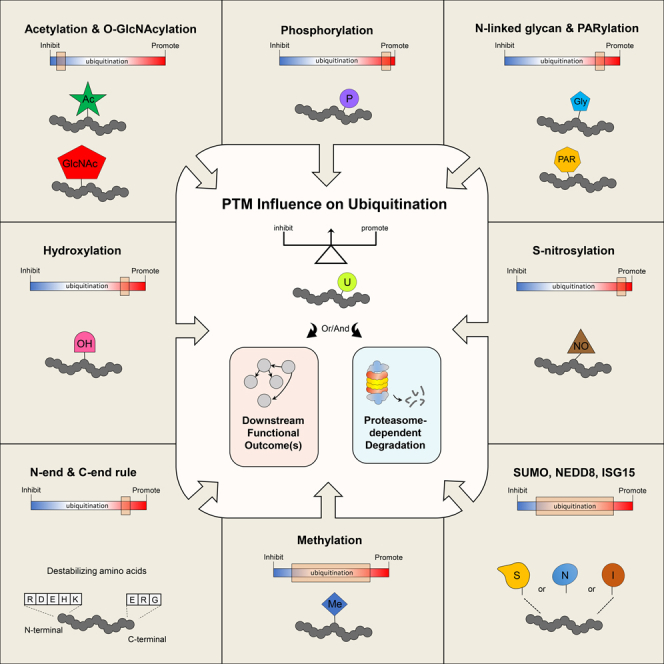
Ubiquitination is an important post-translational modification (PTM) that regulates a large spectrum of cellular processes in eukaryotes. Abnormalities in ubiquitin signaling underlie numerous human pathologies including cancer and neurodegeneration. Much progress has been made during the last three decades in understanding how ubiquitin ligases recognize their substrates and how ubiquitination is orchestrated. Several mechanisms of regulation have evolved to prevent promiscuity including the assembly of ubiquitin ligases in multi-protein complexes with dedicated subunits and specific post-translational modifications of these enzymes and their co-factors. Here, we outline another layer of complexity involving the coordinated access of E3 ligases to substrates. We provide an extensive inventory of ubiquitination crosstalk with multiple PTMs including SUMOylation, phosphorylation, methylation, acetylation, hydroxylation, prolyl isomerization, PARylation, and O-GlcNAcylation. We discuss molecular mechanisms by which PTMs orchestrate ubiquitination, thus increasing its specificity as well as its crosstalk with other signaling pathways to ensure cell homeostasis.
Subject areas: Biological sciences, Biochemistry, Molecular biology, Cell biology
Graphical abstract
Biological sciences; Biochemistry; Molecular biology; Cell biology
Introduction
Ubiquitination is a post-translational modification (PTM) that plays critical roles in regulating protein stability, activity, and localization; and constitutes a major biochemical process coordinating a vast majority of cell signaling networks.1 This modification is catalyzed by the concerted action of three distinct enzymes, E1 ubiquitin-activating, E2 ubiquitin-conjugating, and E3 ubiquitin ligase culminating in the covalent attachment of the 76 amino acids ubiquitin protein to an internal lysine or the N-terminal residue of substrates2,3 (Figure 1). Monoubiquitination corresponds to the attachment of one ubiquitin molecule to the substrate, while the attachment of individual ubiquitin molecules to several lysines of the substrate corresponds to multi-monoubiquitination. Signaling events through monoubiquitination and multi-monoubiquitination regulate diverse signaling pathways that are associated with numerous biological processes including membrane trafficking, DNA replication, and DNA repair. These monoubiquitination events have also been shown to induce proteasomal degradation. In contrast, polyubiquitination corresponds to the formation of a ubiquitin chain as ubiquitin itself contains seven lysines, in addition to the N-terminus methionine that can be used for ubiquitin attachment1 (Figure 2). Notably, polyubiquitination can involve eight different ubiquitin linkage types, i.e., M1, K6, K11, K27, K29, K33, K48, and K63. Although the mechanisms that govern the signaling outcomes of chain topology are not well understood, the various ubiquitin chains have been associated with numerous functional and biological events. For instance, polyubiquitination through lysine 48 (K48) is associated with protein degradation by the proteasome, while lysine 63 (K63) ubiquitin chain formation is often involved in protein complex assembly and activation.1 Of note, polyubiquitination can generate homotypic (e.g., K48 only), heterotypic (e.g., K48 & K63) or heterologous (e.g., Ubiquitin & SUMO) chains that endow the ubiquitin system with further possibilities for tight regulation of ubiquitin signaling pathways3 (Figure 2). Moreover, ubiquitin itself is subjected to several post-translational modifications (phosphorylation, acetylation, ADP-ribosylation, and deamidation), which can modulate the ubiquitination reaction and modulate ubiquitin chain elongation.3,4,5,6 Finally, monoubiquitin and ubiquitin chains could be recognized by a wide variety of proteins through their ubiquitin-binding domains (UBD)7 (Figure 2). These UBD could have selective affinities for specific ubiquitin chain topologies, supporting the notion that the various ubiquitination events can be read and translated into distinct signaling outcomes.
Figure 1.

Chemical mechanism or global reaction of various post-translational modifications
(A) Ubiquitination mechanism on the lysine residue of a target protein. The deprotonated amino group of the lysine engages a nucleophilic attack on the carbonyl of the thioester group linking ubiquitin to the E2 ubiquitin-conjugating enzyme or E3 ubiquitin ligase. An isopeptide bond is therefore formed after rearrangement, attaching the ubiquitin to the target protein and freeing the E2. SUMOylation, NEDDylation, and ISGylation use the same mechanism.
(B) Phosphorylation mechanism on the serine residue of a target protein. A phosphoserine is formed by nucleophilic substitution on the phosphorus of the gamma phosphate group of ATP (adenosine triphosphate) with the deprotonated alcohol as nucleophile which results in ADP release.
(C) Acetylation mechanism on the lysine residue of a target protein. Acetyl-CoA is a common source of acetyl groups. The deprotonated amino group of the lysine makes a nucleophilic attack on the carbonyl group of the acetyl-CoA, fusing it to the target protein.
(D) Mono-methylation mechanism on the lysine residue of a target protein. SAM (S-adenosyl methionine) is a common donor of methyl groups for the successive methylation of a target residue. By nucleophilic substitution, the methyl group of SAM is transferred to a target protein with a deprotonated amino group, resulting in SAH (S-adenosyl homocysteine) as a free product. The deprotonated amino group of the lysine residue acts as a nucleophile.
(E) O-GlcNAcylation mechanism on the serine residue of a target protein. By nucleophilic substitution, the N-acetylglucosamine of the UDP-GlcNAc is attached to the target protein with the deprotonated alcohol as nucleophile, resulting in the formation of a β-glycosidic bond and the release of UDP.
(F) PARylation mechanism on the serine residue of a target protein. By nucleophilic substitution, the ADP-ribose from the NAD+ (nicotinamide adenine dinucleotide) is attached to the target protein with the deprotonated alcohol as nucleophile.
(G) Hydroxylation mechanism on the proline residue of a target protein. The shown dioxygenase has an iron cofactor and complex with two histidines and aspartic acid. In the presence of O2 (dioxygen) and 2-oxoglutarate, the proline residue is targeted for hydroxylation at the end of the catalytic cycle (step 7).
(H) S-nitrosylation reaction on the cysteine residue of a target protein in the presence of NO. For each mechanism, only the reactive functional group of residues implicated is shown. The B group (for phosphorylation, PARylation, and O-GlcNAcylation) represents the enzyme base that transfer the proton to the substrate. The resulting modification is colored in blue. Chemical structures were drawn with ChemDraw.
Figure 2.

Ubiquitin chains and their functions
Ubiquitination of proteins is catalyzed by the action of E1 ubiquitin-activating, E2 ubiquitin-conjugating, and E3 ubiquitin-ligating enzymes. The concerted action of E2 and E3 enzymes dictates the nature of ubiquitin modification. Ubiquitination is reversed by deubiquitinases (DUBs). A protein target (in blue) can be monoubiquitinated, multi-monoubiquitinated, or polyubiquitinated. Monoubiquitination and multi-monoubiquitination are associated with diverse signaling pathways and processes including intracellular membrane trafficking, transcription regulation, DNA damage signaling and repair as well as the regulation of protein subcellular localization. Monoubiquitination events have also been shown to induce proteasomal degradation. Polyubiquitination involves eight ubiquitin linkage types, i.e., M1, K6, K11, K27, K29, K33, K48, and K63. The various ubiquitin chains are associated with numerous functional and biological outcomes. K63 polyubiquitin chains are associated with protein recruitment and activation of signaling cascades, K48 polyubiquitin chains generally promote the proteasomal degradation of substrates. Polyubiquitination can generate homotypic, heterotypic, or heterologous chains that add another layer of complexity to ubiquitin signaling pathways. Moreover, post-translational modifications of ubiquitin itself (phosphorylation, acetylation, ADP-ribosylation, and deamidation) can modulate E3 ligase activity and interfere with ubiquitin ligation or ubiquitin chain elongation. Following substrate ubiquitination, ubiquitin chains could be recognized by a wide variety of ubiquitin-binding proteins through their ubiquitin-binding domains (UBD). Examples of UBDs are represented by the ubiquitin interacting motifs (UIM) of RAP80, the Ubiquitin-associated domains (UBA) of Rad23, or the Npl4-like Zinc Finger (NZF) of the deubiquitinase TRABID. A wide range of signaling events and biological processes are associated with ubiquitin binding such as the DNA damage response, cell death, immune signaling, and autophagy.
Akin to other PTMs, ubiquitination is reversible, and ubiquitin removal from substrates is catalyzed by deubiquitinases (DUBs). DUBs play important roles in ubiquitin maturation, recycling and the maintenance of adequate pools of free and conjugated ubiquitin in the cell.8 DUBs have also emerged as highly selective regulators of ubiquitination events and as such, control diverse cellular processes.8,9
Due to its bulky nature, ubiquitination can have a major impact on protein function. Moreover, ubiquitination often mediates cellular events with rapid spatiotemporal dynamics.10 Thus, this reaction must be well controlled to avoid unwanted proteolysis or promiscuous change in activity. Here, we summarize and discuss the current state of knowledge on the crosstalk between ubiquitination and other PTMs in the regulation of protein stability and function. Central to this review is the impact of multiple PTMs on the ubiquitination of the substrate itself. We outline an extensive compilation of crosstalk between ubiquitination and other abundant PTMs. We discuss examples of intricate mechanisms of recognition and ubiquitination of substrates during the execution of cellular processes. Finally, we also stress some of the unaddressed questions and highlight new directions for future studies. Please refer to the glossary outlined as Data S1.
Cooperation and antagonism between SUMO, NEDD8, ISG15, and ubiquitin
Despite low sequence conservation, the 3D structures of ubiquitin and the ubiquitin-like proteins (UBLs), including SUMO (Small Ubiquitin-like MOdifier), NEDD8 (Neural Precursor Cell Expressed, Developmentally Down-Regulated 8), and ISG15 (Interferon-Stimulated Gene 15), are remarkably similar and all share a core β-grasp (β-Golgi Reassembly Stacking Protein) fold containing secondary structure elements arranged in a ββαβββ order11 (Figure 3A). Although, there are substantial mechanistic similarities between ubiquitination and UBLs-mediated reactions, specific properties of each system can selectively determine the fate of the modified protein11 (Figure 1). As ubiquitin and UBLs target lysine residues, it is expected that a certain degree of crosstalk would occur between these modifications (Tables S1, S2, and S3).
Figure 3.

Examples of crosstalk between ubiquitin-like proteins (UBLs) and ubiquitin
(A) Ubiquitin and Ubiquitin-like proteins (UBLs), SUMO2 (PDB: 1WM3), NEDD8 (PDB: 2BKR) and ISG15 (PDB: 1Z2M) share significant structural similarities with ubiquitin (PDB: 1ubq). These proteins share a core β-grasp (β-Golgi Reassembly Stacking Protein) fold containing secondary structure elements arranged in a ββαβββ order.
(B–C) Ubiquitin-like proteins compete with ubiquitination resulting in variable outcomes on protein stability and function. (B) Polyubiquitination of the NF-κB inhibitor IKBα, following its phosphorylation by the IKK kinase, leads to its proteasomal degradation and release of NF-κB to execute its transcriptional activity. SUMOylation of the same residue of IκBα blocks its degradation, while hybrid SUMO-Ubiquitin chain extension can re-engage the proteasomal degradation. (C) Attachment of SUMO, ubiquitin, NEDD8, and ISG15 moieties on PCNA is associated with different outcomes on DNA replication and repair. PCNA K164 modifications play central roles in the recruitment of UBLs binding proteins, which in turn dictate the choice or termination of DNA repair pathways. 1) At the replication fork, PCNA K164 SUMOylation allows its interaction with Srs2 which inhibits Rad51 filament formation and homologous recombination (HR). 2) During DNA damage, PCNA K164 monoubiquitination results in the recruitment of Polη for the translesion DNA synthesis (TLS) process. 3) PCNA polyubiquitinated on K164 after replication stress promotes the recruitment of ZRANB3, replication fork reversal, and protection, thus maintaining genomic integrity. 4) The execution of TLS can be inhibited by the NEDDylation of PCNA K164. This inhibits its ubiquitination and the subsequent recruitment of Polη. 5) ISGylation of PCNA is a signal for TLS termination. Following Polη recruitment, PCNA K164/K168 is ISGylated by EFP leading to the recruitment of the DUB USP10. This results in the deubiquitination of PCNA, release of Polη from the replication fork and termination of TLS.
While ubiquitination generally does not target a canonical peptide sequence, SUMOylation targets a consensus motif, ΨKXE/D (where Ψ represents a large hydrophobic residue and X any amino acid), in which the lysine serves as the SUMO acceptor site. SUMOylation of target proteins is catalyzed by a unique E2 enzyme called UBC9 (ubiquitin-conjugating enzyme 9), which relies on a dozen of E3 SUMO ligases to ensure substrate specificity.12 Substrate SUMOylation is reversed by SUMO-specific proteases termed Sentrin-specific proteases (SENPs).12 SUMOylation can antagonize ubiquitination by competing for the same lysine in substrates (Table S1). This is the case of IκBα, an inhibitor of the transcription factor NF-κB, which is targeted for proteasomal degradation upon TNF receptor stimulation by the phosphorylation-mediated ubiquitination of K21 and K22 residues.13,14,15 Instead, SUMOylated IκBα on K21 is resistant to signal-induced degradation, resulting in the sequestration of inactive NF-κB in the cytoplasm and hence, limiting its transcriptional functions in the nucleus16 (Figure 3B). In addition, while the phosphorylation of IκBα on S32 and S36 is required for ubiquitination, these PTMs appear to inhibit SUMOylation indicating a hierarchy in signaling events orchestrating IκBα stability and NF-κB activation.16 In addition, deSUMOylation could also potentially orchestrate IκBα stability and function, as NF-κB promotes feedback mechanisms that involve transcriptional regulation of SENPs.17
SUMOylation and ubiquitination can also synergize to ensure the orchestration of signal transduction. For instance, IκBα could be also simultaneously targeted by SUMO and ubiquitin, resulting in the formation of SUMO-ubiquitin hybrid chains that promote IκBα degradation.18 Recent evidence indicated that SUMO and ubiquitin often act sequentially to induce substrate degradation. This is exemplified by promyelocytic leukemia (PML) whose polySUMOylation on K160 triggers its interaction with the SUMO-targeted Ubiquitin ligase (STUbL) RNF4. This E3 ligase recognizes SUMOylated PML through SUMO-interacting motifs (SIM) and catalyzes the polyubiquitination of SUMO and subsequent PML proteasomal degradation.19,20 Mechanistically, RNF4 activity is regulated by dimerization through binding to SUMO chains.21 A local increase of SUMO modification, during stress conditions, results in SIM-mediated recruitment of RNF4, inducing RING finger dimerization and ubiquitin ligase activation. Structural studies indicated that the RING dimer of RNF4 binds the E2-ubiquitin complex and facilitates catalysis.22
Analogous to ubiquitination and SUMOylation, NEDDylation also uses E1 and E2 enzymes as well as multiple E3 ligases to ensure substrate modification.23 It is well recognized that the multi-components Cullin-RING ubiquitin Ligases (CRLs) are major targets of NEDD8 which stimulates their E3 ligase activity.24,25,26 NEDDylation of non-Cullin targets has also been described and this modification can protect proteins from ubiquitin-mediated degradation (Table S2). For instance, the proto-oncogene and E3 ubiquitin ligase C-CBL, a downstream effector of TGFβ antiproliferative signaling, NEDDylates TGFβ type II receptor (TβRII), ensuring its stabilization.27 Interestingly, TβRII is internalized through two endocytic pathways, clathrin-mediated or caveolin-mediated, leading to two opposite outcomes. While clathrin-mediated TβRII internalization results in the maintenance of signal transduction, caveolin-mediated TβRII compartmentalization triggers receptor degradation and signal termination.28 C-CBL-mediated NEDDylation triggers TβRII internalization through clathrin-dependent endocytosis, thus sustaining TGFβ signaling.27 In contrast, the degradation of the proto-oncogene c-SRC involves the cooperation between NEDDylation and ubiquitination, resulting in the inhibition of the PI3K-AKT signaling and reduction of cell migration and metastasis.29 Here, C-CBL NEDDylates c-SRC, which results in its polyubiquitination and proteasomal degradation. Whether a conformational change or the recruitment of additional proteins mediates this NEDDylation-ubiquitination crosstalk remains to be determined.
The crosstalk between ubiquitin and multiple UBLs within the same substrate is best illustrated by PCNA, which is subjected to several PTMs ensuring coordination between DNA replication and repair machineries. Although established primarily in yeast, the regulation of PCNA by UBLs is highly conserved in eukaryotes. PCNA is a homotrimeric ring-shaped DNA clamp complex acting as a processivity factor for DNA polymerase δ/ϵ and is required for replication. PCNA could be modified by ubiquitin or other UBLs on the same lysine residue, but with different functional consequences30,31 (Figure 3C). In normally growing yeast, SUMOylation of PCNA K164 induces the recruitment of Srs2 protein to the replication fork, blocking unwanted homologous recombination. Srs2 interacts with PCNA through a PCNA-interacting protein (PIP) box-like motif within the carboxy-terminal domain and a SIM domain that binds SUMOylated PCNA. This consolidated Srs2-PCNA interaction prevents Rad51 filament formation and subsequent homologous recombination.32,33,34,35 In contrast, during DNA damage, the E2-E3 complex Rad6-Rad18 mediates the monoubiquitination of PCNA at K164 triggering the recruitment of Y-family damage-tolerant DNA polymerases (e.g., polymerase η), in order to bypass the lesion in a process called translesion DNA synthesis (TLS).31,36 The binding of monoubiquitinated PCNA by the TLS DNA polymerase requires the combined involvement of their PIP motifs and ubiquitin-binding domains, respectively.36,37 Interestingly, PCNA can also be polyubiquitinated on K164, through K63-chains, by Ubc13-Mms2 (E2) and Rad5 (E3), and this event has been involved in error-free DNA repair.38,39 Recent studies in mammalian cells indicated that the polyubiquitination of PCNA facilitates the recruitment of ZRANB3 translocase, which stimulates replication fork reversal and inhibits recombination events that could take place during template switching, thus maintaining genomic integrity during periods of replication stress.40,41,42,43
While the exact molecular mechanisms of PCNA modifications by ubiquitin and SUMO remain incompletely defined, this factor was recently shown to be targeted by other UBLs, notably NEDD8 and ISG15.44,45 During oxidative stress, PCNA K164 is NEDDylated, inhibiting its ubiquitination and the subsequent recruitment of TLS polymerase η.44 It was proposed that PCNA NEDDylation might counteract TLS to prevent an excessive engagement of an error-prone DNA repair mechanism. Interestingly, NEDDylation of PCNA is reversed by the deNEDDylase NEDP1, suggesting a dynamic role of this modification in negatively regulating TLS.44 Finally, another layer of complexity was recently provided by PCNA ISGylation, a protein PTM that also involves a dedicated E1/E2/E3 enzymatic cascade46,47 (Table S3). PCNA ISGylation on K164 and/or K168 residues is catalyzed by EFP ISG15 E3 ligase and takes place after the recruitment of TLS polymerase to promote TLS termination. Mechanistically, PCNA K164 monoubiquitination stimulates the recruitment of EFP and this event leads to PCNA ISGylation. In turn, PCNA ISGylation promotes the recruitment of USP10 for PCNA deubiquitination, leading to polymerase η release from DNA. Finally, DeISGylation of PCNA is mediated by UBP43 ensuring TLS termination and reestablishment of normal DNA replication.45 Nonetheless, how these ISGylation/DeISGylation events are exactly orchestrated with ubiquitin and other UBLs during the repair process remain unknown.
In summary, although much progress has been made during the last decades, the exact mechanisms of action coordinating the crosstalk between ubiquitin and UBLs remain incompletely understood. In particular, UBL crosstalk often involve coordination by small PTMs whose characterization, through functional and structural studies, should help in understanding the interplay between signaling pathways in the regulation of cellular processes.
Extensive crosstalk between protein phosphorylation and ubiquitination
Phosphorylation is a widespread PTM that consists of the attachment of a phosphoryl group on serine, threonine, or tyrosine residues of target proteins (Figure 1). Being probably the most studied protein PTM, much work has been done to determine how phosphorylation regulates cellular processes. The link between phosphorylation and ubiquitination has been studied in diverse cellular processes (Table S4). Generally, the interplay between phosphorylation and ubiquitination is often associated with protein degradation. Protein phosphorylation on a conserved short motif of amino acids, called phosphodegron, is recognized by E3 ligase complexes leading to substrate polyubiquitination and proteasomal degradation. In particular, the multi-protein E3 ligase family SKP1-Cullin-F-box (SCF) has been shown to be one of the main players in mediating phosphorylation-inducing degradation (Figure 4A). The F box protein family are responsible for the recognition and binding of the phosphorylated degron motif with one of the first F box proteins identified being the Saccharomyces cerevisiae CDC4.48 Of note, substrate phosphorylation-dependent ubiquitination is usually preceded by priming phosphorylation events that occur on adjacent sites, involving other kinases than those phosphorylating the degron, indicating the tight control of the phospho-ubiquitin signaling cascades.
Figure 4.

Model of the interplay between protein phosphorylation and ubiquitination
(A) Phosphorylation of a target protein creates a phosphodegron, recognized by E3 ligase complexes (in this case the SCF or Skip-Cullin-F-box complex family). The F box factor positions the targeted protein in the vicinity of the Cullin ligase (RBX1/2) and the E2 enzyme, leading to its ubiquitination and proteasomal degradation.
(B) Crystal structure of the quaternary complex: SKP1-SKP2-CKS1-Phospho p27Kip1 (PDB: 2AST). Left panel, overall representation of the complex showing the specific positioning of the p27 peptide within CKS1 and SKP2 binding pockets. Right panel, close up view of the phosphorylated p27Kip1 interaction with CKS1 and SKP2. Phosphorylated p27Kip1 intercalates into a CKS1/SKP2 pocket formed by SKP2 leucine-rich repeat (LRR) and CKS1 phospho binding site. pT187 is recognized by CKS1 phospho binding site residues whereas E185 binds to both CKS1 and SKP2. The hydrogen bounds between amino acids are shown at the bottom by the dashed lines.
(C) DLK stability is regulated through the action of the PHR1 E3 ligase and the DUB USP9X. DLK phosphorylation by JNK blocks its ubiquitination and degradation to reinforce the JNK signaling pathway and promote neuronal apoptosis.
(D) Phosphorylation-mediated monoubiquitination of RECQL4 regulates pathway choice of DSB repair. Following DNA damage, RECQL4 associates with the DSB binding protein Ku70 to allow non-homologous end-joining (NHEJ) repair. CDK1/2 phosphorylates RECQL4 which leads to its ubiquitination by CUL4ADDB1 and induces its interaction with MRE11 to promote homologous recombination (HR).
SCF-mediated ubiquitination covers a wide range of phosphorylated target proteins including many cell cycle effectors. One example is the Cyclin-dependent kinase inhibitor 1B (p27Kip1), which binds and prevents the activation of Cyclin E-CDK2 or Cyclin A-CDK2 complexes.49 Previous studies established that the phosphorylation of p27Kip1 on T187 is essential for its proteasomal degradation by the E3 ligase SKP2,50,51 and that this concerted reaction requires the CDK subunit 1 (CKS1).52 The crystal structure of the quaternary complex: SKP1-SKP2-CKS1-Phospho p27Kip1 revealed that CKS1 binds to both the leucine-rich repeat domain and the C-terminal tail of SKP2 (Figure 4B). p27Kip1 establishes contacts with both CKS1 and SKP2, a configuration that positions CKS1 phospho-binding motif for interaction with the phosphorylated T187 side chain of p27Kip1.53 Interestingly, p27Kip1 binding to SKP1-SKP2-CKS1 is mainly coordinated by intramolecular interactions between the p27Kip1 phospho T187 and CKS1 and p27Kip1 E185 with both CKS1 and SKP2. This conformation allows p27Kip1 phospho T187 recognition and its ubiquitination. Additionally, p27Kip1 is also phosphorylated by the oncogenic tyrosine kinase Src on Y88, promoting a conformational change disrupting p27Kip1 interaction with the CDK2 catalytic cleft. Released cyclin A-CDK2 becomes active, promoting p27Kip1 phosphorylation on T187, which then stimulates its ubiquitination and proteasomal degradation.54,55
On the other hand, phosphorylation is also known to prevent substrate ubiquitination. This is exemplified by the dual leucine zipper-bearing kinase (DLK/MAP3K12), an evolutionarily conserved member of the mixed lineage kinase (MLK) family that plays an important role in c-Jun N-terminal kinases (JNK) signaling and apoptosis. DLK protein stability is regulated through the action of the E3 ubiquitin ligase PHR1 and the DUB USP9X.56 During retrograde signaling, neurons respond to axonal damage by inducing site-specific phosphorylation of DLK by JNK, an event that prevents DLK ubiquitination (Figure 4C). Stabilized DLK acts in turn to promote downstream JNK signaling and apoptosis.56 This signaling cascade illustrates how ubiquitination and phosphorylation crosstalk can be exploited to amplify a stress response, thereby quickly responding to extracellular cues.
Phosphorylation can also regulate substrate ubiquitination, independently of proteolysis. The pathways of DNA double-strand break (DSB) repair are subjected to an intricate level of regulation during initiation, repair pathway choice, and termination. The extent of DNA-end resection highly influences the pathway choice between non-homologous end-joining (NHEJ) and homologous recombination (HR).57,58,59 While NHEJ can function in all phases of the cell cycle, HR is highly active in S and G2 phases, as sister chromatids must be used for recombination.60,61 The increase of Cyclin-dependent kinases (CDKs) activity at G1/S transition results in the coordinated phosphorylation of key chromatin-associated and DNA repair factors, thus promoting DNA resection and HR pathway. For instance, RECQL4, a RecQ-type helicase required for genomic integrity, regulates both NHEJ and HR depending on cell cycle phases.61 Mechanistically, RECQL4 forms a complex with the DSB binding protein Ku70 and facilitates NHEJ. Increased CDK1 and CDK2 activities in S and G2 result in the phosphorylation of RECQL4 on S89 and S251, promoting its interaction with the MRN complex, which is known to promote HR. Phosphorylated RECQL4 becomes a substrate of CUL4ADDB1 E3 ubiquitin ligase, which catalyses its ubiquitination and retention at DSB sites to mediate DNA end resection and HR (Figure 4D).61 These results indicate how phosphorylation and ubiquitination can orchestrate the intervention of key factors to ensure the timely engagement of the proper DNA repair pathway.
Opposing outcomes of protein acetylation and ubiquitination: A rule with exceptions
Mostly known for its function on histone and chromatin structure, protein lysine acetylation is a widespread PTM catalyzed by lysine acetyltransferases (KATs/HATs) and reversed by lysine deacetylases (KDACs/HDACs).62,63 Acetylation plays critical roles in the regulation of protein stability, function and localization.62 Crosstalk between acetylation and ubiquitination is a critical regulatory mechanism controlling numerous vital cellular processes including chromatin-dependent processes (Table S5). Protein acetylation can promote protein stability by blocking ubiquitination-mediated proteasomal degradation. One of the first observations validating this notion is the regulation of p53 degradation, as its ubiquitination by the E3 ligase MDM2 can be inhibited through the acetylation of the same lysines of p53 C-terminal domain by p300.64 Interestingly, acetylation may also attenuate the ubiquitination of p53 by inducing potential conformational changes or possibly by preventing substrate recognition by E3 ligases.64 To counteract this mechanism, the ubiquitin ligase MDM2 functions in part by recruiting the histone deacetylase 1 (KDAC1/HDAC1) to deacetylate p53, thus allowing its ubiquitination and subsequent degradation.65 The model of negative regulation of protein ubiquitination by acetylation has been subsequently expanded to many other substrates (Table S5).
On the other hand, acetylation can also create an acetyl-degron targeting a substrate for proteasomal degradation. Indeed, in response to increased concentrations of glutamine, acetylation of glutamine synthetase induces its recognition by the CRL4CRBN E3 complex, resulting in its subsequent proteasomal degradation, thereby impacting glutamine production (Figure 5A). However, how acetylation/deacetylation events of glutamine synthetase could sense glutamine concentration remains to be determined. It is possible that the extent of glutamine synthetase acetylation might serve as a metabolic rheostat that modulates enzyme abundance as a function of glutamine availability.
Figure 5.

Crosstalk between methylation or acetylation and ubiquitination
(A) High level of cellular glutamine (Gln) leads to the acetylation of Glutamine synthetase (GS) by P300/CBP. This event creates an acetyl-degron recognized by the CRL4CRBN E3 complex leading to GS polyubiquitination and subsequent degradation by the proteasome.
(B) Bivalent recognition of unmethylated H3R2 and methylated H3K9 by the multidomain E3 ligase, UHRF1, leading to the ubiquitination of H3K23.
(C) Surface representation of the crystal structure of the E3 ubiquitin ligase UHRF1 in complex with histone H3 peptide (PDB: 3ASK). The Tudor domains and the PHD domain (TTD-PHD) used for the crystallization are shown in the left panel. Right panel, zoom in view of the interaction between histone H3 peptide with the PHD domain (top) and the Tudor1/2 domains (bottom). The H3 peptide is composed of two cassettes: cassette 1 encompassing H3R2, is positioned within the PHD acidic pocket; and cassette 2 containing H3K9me3 is recognized by an “aromatic cage” surface within Tudor 1. The hydrogen bounds between amino acids are shown by the dashed lines. The structure shows that the unmodified H3R2 intercalates into an acidic pocket within the PHD finger domain. The Tudor domain 1 accommodated the H3 peptide C-terminus residues into an “aromatic cage” involving H3K9me3 and S10.
(D) Regulation of the RORα nuclear receptor by a methylation/ubiquitination crosstalk. RORα is subjected to monomethylation by the PRC2 methyl-transferase EZH2. This monomethylated RORα is bound by the chromo-domain of DCAF1 recruiting it to the DCAF1/DDB1/CUL4B ubiquitin ligase complex, resulting in the proteasomal degradation of RORα and repression of its target genes.
The link between protein methylation and ubiquitination
Protein methylation consists of the addition of one to three methyl groups on lysines or one to two methyl groups on arginines by enzymes termed methyltransferases66,67 (Figure 1). Several examples describe intricate crosstalk between methylation and ubiquitination that result in diverse functional outcomes (Table S6). An interesting mechanism of histone methylation-induced histone ubiquitination is provided by UHRF1 E3 ligase, which ensures the propagation of DNA methylation during DNA replication, thus maintaining epigenetic gene silencing. UHRF1 contains a ubiquitin-like domain (UBL), a tandem Tudor domain (TTD), a plant homeodomain (PHD), a SET- and RING-associated (SRA) domain, and a RING finger domain. The TTD and PHD are readers of di- or tri-methylated histone H3K9 and unmodified H3R2, respectively, while SRA binds hemi-methylated DNA. Following chromatin binding at the replication fork, UHRF1 catalyzes histone H3K23 ubiquitination, an event stimulated by its UBL domain. The proper positioning of UHRF1 on nucleosomal marks and subsequent ubiquitination is a prerequisite for the recruitment of DNMT1, which, in turn, catalyzes DNA methylation (Figure 5B).68,69,70,71,72,73 The crystal structure of UHRF1 provided a molecular explanation for the bivalent recognition of histone H3 (Figure 5C).69 Notably, the PHD and the TTD are linked with 17 residues linker that plays an important role in maintaining a proper structural conformation of the PHD-Tudor module for the bivalent recognition of the H3 tail and the positioning of the RING finger in close proximity of H3K23. This conformation is inhibited by the phosphorylation of S298 within the linker of UHRF1.69 Overall, recognition of histone methylation and DNA methylation are cooperatively involved in ensuring histone H3 ubiquitination, which in turn promotes DNA methylation and mitotic propagation of gene silencing.
Methylation/ubiquitination interplay is often linked to protein stability as shown for the orphan nuclear receptor RORα whose methylation is driven by the Polycomb group protein and methyltransferase EZH274 (Figure 5D). EZH2 specifically monomethylates RORα on K38, a methyl-degron motif resembling the histone H3K27 LxxxxxRKS methyl acceptor motif, the classical target of EZH2. Once methylated, RORα(me1) is recognized and bound by the DDB1 and CUL4 Associated Factor 1 (DCAF1) which is known to act as an adapter for CUL4DDB1 E3 ubiquitin ligases.75 RORα is then polyubiquitinated and degraded by the proteasome, thus inducing transcriptional repression of RORα target genes and inhibition of its tumor suppressor properties. Interestingly, DCAF1 specifically recognizes the monomethylated RORα through its C-terminal chromodomain, but not other methylated proteins such as H3K27, which restricts the spectrum of target proteins to be ubiquitinated by CUL4DDB1.74
Finally, increasing evidence indicates that arginine methylation could also interfere with ubiquitination events76 (Table S6). For instance, PRMT5-driven arginine methylation of the transcription factor KLF4 inhibits its ubiquitination and proteasomal degradation by the CRL2pVHL E3 ubiquitin ligase complex.77 This stabilizing effect contributes to the oncogenic function of KLF4 in breast cancer initiation and invasion.
Impact of O-GLCNACYLATION on protein ubiquitination
O-GlcNAcylation consists of the O-linked attachment of β-N-acetylglucosamine (O-GlcNAc) group on S/T residues of target proteins and is catalyzed by the O-GlcNAc transferase (OGT)78 (Figure 1). The synthesis of UDP-GlcNAc, the donor of the O-GlcNAc group, involves the hexosamine biosynthesis pathway, which acts as a metabolic sensor.79 The O-GlcNAcylation of proteins generally results in a negative regulatory action on ubiquitination, thus increasing protein stability (Table S7). An interesting example of the interplay between the O-GlcNAcylation and ubiquitination pertains to the two circadian clock proteins BMAL1 and CLOCK. The circadian clock system is a machinery that living cells use to synchronize their biological processes with the external environment, exhibiting 24 h-long cycles.80 Notably, the molecular circadian clock machinery involves transcriptional and post-transcriptional control mechanisms as well as feedback loops that ensure proper synchrony of the rhythm.81 Through dimerization, BMAL1 and CLOCK form a transcription activator complex inducing the expression of the Period genes (Per1, Per2) along with two Cryptochrome genes (Cry1 and Cry2).81 Both BMAL1 and CLOCK proteins are stabilized by O-GlcNAcylation which prevents their proteasomal degradation82 (Figure 6A). The crystal structure of the BMAL1/CLOCK complex suggests that the O-GlcNAcylation site on BMAL1 (S418) is less likely to be involved in the interaction with CLOCK since it is located far from the interaction interface between the two proteins.83 It is possible that the S418 O-GlcNAcylation site, which is near K404 and K415 ubiquitination sites, prevents E3 binding and substrate ubiquitination.84 Interestingly, OGT, along with the DUB BAP1, stabilize the BMAL1/CLOCK complex in response to nutrient abundance.82 This suggests that the O-GlcNAcylation of BMAL1 promotes its deubiquitination. Overall, these findings indicate that O-GlcNAcylation and ubiquitination could orchestrate an intricate signaling cascade that fine-tunes the functions of target proteins in response to alterations in cell metabolism and nutrient abundance.
Figure 6.

Crosstalk between protein modification by mono- or oligosaccharides and ubiquitination
(A) Example of crosstalk between O-GlcNAcylation and ubiquitination regulating the circadian cycle. The CLOCK/BMAL1 complex ensures the expression of the circadian rhythm genes in a cyclic manner. Depending on the availability the UDP-GlcNAc, BMAL1 could be O-GlcNAcylated which could prevent its polyubiquitination by UBE3A ligase and enhance the activity/recruitment of deubiquitinases, BAP1/USP2, thus stabilizing the CLOCK/BMAL1 complex. Low levels of UDP-GlcNAc during the slow metabolism phase of the circadian cycle leads to BMAL1 polyubiquitination and proteasomal degradation. This results in the repression of the CLOCK/BMAL1 target genes: Per1, Per2, Cry1, and Cry2.
(B) The ERAD-L pathway regulation by oligosaccharide/ubiquitination interplay in S. cerevisiae. 1) The glycan groups on misfolded proteins in the lumen of the ER are recognized by the Yos9 protein which triggers their recruitment to the Hrd3/Hrd1 complex within the ER membrane. 2) The Hrd1 ubiquitin ligase dimerizes at the membrane and then polyubiquitinates the misfolded protein via the cytoplasmic RING domain. 3) The cytoplasmic Cdc48/p97 ATPase complex drags the polyubiquitinated polypeptide to the proteasome for degradation.
(C) N-linked glycoproteins recognition by the F box E3 ligase complex. Structural overview of the Fbs1 SBD in complex with modified RNase B (top left panel) (PDB: 2E33). Close up view of the RNase B Man3GlcNAc2 moiety binding with the SBD domain (bottom left panel). Structural model of the SCFFbs1 ubiquitin ligase complex bound to modified RNase and the E2, UBCH7 (right panel). The model was generated by superimposing the current crystal structure of the SBD/RNase B with SKP1/Cul1/RBX1 (PDB: 1LDK) and c-Cbl-UBCH7 (PDB: 1FBV) structures. The hydrogen bounds between amino acids are shown by the dashed lines.
O-GlcNAcylation may also influence ubiquitination by competing over the same site of phosphorylation as shown for the zinc-finger protein SNAIL1. SNAIL1 is an epithelial to mesenchymal transition-promoting transcription factor repressing the cell adhesion junction factor E-cadherin.85,86 SNAIL1 is phosphorylated by glycogen synthase kinase-3β (GSK-3β) on two consensus motifs, creating a docking site for SCFβ−TRCP E3 ubiquitin ligase and leading to the degradation of SNAIL1.87 SNAIL1 O-GlcNAcylation can also occur on S112 within the GSK-3β consensus motif.88 This prevents SNAIL1 polyubiquitination and degradation under hyperglycemic conditions, inducing the down-regulation of E-cadherin and promoting epithelial to mesenchymal transition.88 These findings reveal a molecular switch from phosphorylation to O-GlcNAcylation, which inhibits the recruitment of an E3 ligase responsible for targeting SNAIL1 for polyubiquitination and subsequent downregulation.
Finally, O-GlcNAcylation could also promote protein polyubiquitination and degradation, in opposition to the established view of antagonism between these two PTMs.89 Following DNA damage, the TLS DNA polymerase η (Polη) is O-GlcNAcylated on T457, which induces its polyubiquitination by CRL4CDT2 and subsequent extraction from chromatin by the p97 segregase.89 This contributes to Polη dissociation from replication forks after the completion of TLS. However, it remains unclear whether OGT specifically triggers the O-GlcNAcylation of Polη at DNA lesions. It will also be interesting to determine how O-GlcNAcylated Polη is recognized by the CRL4CDT2.
Overall, both O-GlcNAcylation and ubiquitination are important PTMs intimately involved in coordinating cellular processes. As these PTMs dynamically target a wide spectrum of substrates, it is anticipated that a significant interplay between both pathways takes place in response to alterations in nutrient availability and the microenvironment states of the cells.
N-linked glycan signals for ubiquitination-mediated degradation
Several protein quality control mechanisms have evolved to ensure that proteins are correctly folded before reaching their destination or otherwise degraded by the proteasome, thus avoiding undesirable protein interactions that can lead to protein aggregation and human diseases. For instance, in the ER improperly, folded proteins are recognized and eliminated by the Endoplasmic Reticulum-Associated Degradation (ERAD) system.90,91,92
The ERAD pathways have been extensively studied in yeast. When entering the ER, proteins are enzymatically modified en bloc on an asparagine residue by a branched oligosaccharide composed of three glucose, nine mannose, and two N-acetylglucosamine residues (GlcNAc2Man9Glc3).93 These glycan groups are trimmed during the protein journey inside the ER until their export as a fully folded protein. However, if the protein is delayed inside the ER due to a folding problem, a late glycan-processing enzyme such as the mannosidase Htm1 intervenes to generate an oligosaccharide that directs the improperly folded substrate for ubiquitination and proteasomal degradation.94 The association of the yeast protein Htm1p into an Htm1p-Pdi1p complex selectively guides Htm1p activity toward misfolded N-glycoprotein targets due to the Pdip1 adapter function.95,96 Since the Htm1 enzymatic activity is relatively slower than that of other glycan processing enzymes and that its output product (Man7GlcNAc2 with an exposed α1,6-linked mannosyl residue) is essential for ERAD pathway, it has been proposed that the ERAD machinery would involve a “reader” for this modification.94,97 For instance, Yos9 has been identified as a lectin capable of binding glycan products generated by Htm1.98,99 However, Yos9 binds a glycan attached to the unstructured region of the target protein, which should also be bound by Hrd3 (component of the Hrd1 complex)100,101 (Figure 6B). This dual action of Htm1p-Pdi1p on the one hand and Yos9 on the other hand, adds another layer of control to the ERAD pathway to carefully select the misfolded targets. After the recognition and binding steps by Yos9, the target protein is polyubiquitinated by Hrd1 and extracted through the ER membrane to be retrotranslocated to the cytoplasm where it is recognized by the Cdc48/p97 ATPase complex which pulls the substrate off, so it could be processed by the proteasome102,103,104 (Figure 6B). This example of target protein selection for polyubiquitination and proteasomal degradation shows the tight control for protein fate decisions by adopting a multi-step mechanism, which is highly conserved through evolution.
Signaling protein degradation by N-linked glycan ligation also occurs in the cytoplasm.105 An F box E3 ubiquitin ligase subfamily has been identified for its ability to recognize N-glycan.106,107 For instance, SCFFbs1 ubiquitin ligase complex that contains Fbs1/Fbx2/NFB42 was identified as an E3 ligase that recognizes N-linked glycoproteins through a sugar-binding domain (SBD).105,107 To reveal the mechanism of ubiquitination of N-glycoproteins by the SCFFbs1 complex, the crystal structures of SKP1 in complex with Fbs1 as well as SBD with Ribonuclease B (RNase B) have been solved.108,109 RNase B was used as a model glycoprotein for the co-structure as this enzyme is modified by a single oligosaccharide (Man6-8GlcNAc2). The SBD-sugar binding surface is composed of several residues that interact with Man3GlcNAc2. The overall SCFFbs1-RNase B-E2 complex model (Figure 6C), indicates that RNase B is specifically positioned at a distance that makes it accessible for ubiquitination by the E2 (UBCH7). In addition, a linker loop between the F box and SBD domains of Fbs1 would endow SCFFbs1 with a certain rotational flexibility to accommodate diverse substrates.
Protein PARYLATION and crosstalk with ubiquitination
ADP-ribosylation is catalyzed by enzymes of the 18 family members called PARPs (Poly-ADP-Ribose Polymerases). Having NAD-dependent catalytic activity, PARPs add one or multiple negatively charged ADP-ribose molecules (PAR) on target proteins, allowing the regulation of their functions.110 The majority of studies investigating the biological function(s) of poly-ADP-ribosylation (PARylation) were conducted in the context of DNA damage response (DDR) since PARylation by PARP1 is known to be among the first signals of DDR.111 As most PTMs, PARylation has multiple specific binding motifs called readers of PAR. At least three major groups of PAR-binding domains were described including the PAR-binding macrodomain, PAR-binding zinc finger (PBZ), and most recently the WWE domain linking PARylation to ubiquitin signaling.112,113,114 Many of these reader proteins (or associated complexes) are E3 ligases such as RNF146, BAL1/BBAP complex, BARD1/BRCA1 complex and Checkpoint with forkhead-associated and RING domains, CHFR,114,115,116,117 suggesting an extensive interplay between PARylation and ubiquitination (Table S8).
The mechanism by which PARylation promotes ubiquitination has been revealed by recent studies reporting the interaction of RNF146 with PARylated target proteins of the WNT signaling pathway.118,119 In the absence of the WNT ligand, β-catenin is driven to degradation through its assembly in a specific proteolysis-inducing complex.120 However, in the presence of the WNT ligand, β-catenin is released from its degradation complex and translocated into the nucleus to transduce the WNT signaling. The β-catenin destruction complex is formed by multiple subunits including AXIN, which is a concentration limiting factor essential for complex assembly.120,121 To avoid sustained activation of β-catenin, stabilization of AXIN protein is ensured through the inhibition of the Tankyrases PARPs (TNKS1 and TNKS2) responsible for AXIN PARylation and subsequent degradation.118 RNF146 is the E3 ubiquitin ligase responsible for AXIN polyubiquitination and degradation upon PARylation by TNKS1/TNKS2.122 Interestingly, RNF146 interacts with both the Tankyrase and the PARylated target protein through its WWE domain. The crystal structures of RNF146/UbcH5a (E2 conjugating enzyme) and RNF146/TNKS in the presence of PAR suggest an interesting multi-step activation mechanism by which RNF146 ubiquitinates PARylated substrates in the presence of TNKS and UbcH5a119 (Figure 7). The first step is the PARylation of the target substrate followed by RNF146 binding the TNKS’s five Ankyrin Repeat Clusters (ARCs) domain by its C-terminal domain exposing the PAR chain to the RING/WWE domains of the E3 ligase. Next, the PAR chain is immobilized by the WWE domain and the proximal RING domain. This new conformation induces an allosteric change in the E3 catalytic domain resulting in enhanced ubiquitin ligase activity.119 Therefore, this mode of regulation implies that PAR recognition ensures high specificity, thus preventing promiscuous degradation of PARylated proteins.
Figure 7.

The mechanism of PARylation-triggered ubiquitination
(Top) The crystal structure of RNF146 WWE/RING domains associated with the UBCH5A E2 conjugating enzyme shows a binding pocket for iso-ADPr within the WWE domain and with additional contact with the RING domain (PDB: 4QPL). (Bottom) PARylation of a target protein by the Tankyrase interacting with RNF146. This binding triggers an allosteric conformational change within the RING domain of RNF146 switching its E3 ligase activity from an “OFF” to an “ON” state leading to efficient recruitment of E2 enzyme and ubiquitination of the PARylated target proteins.
Hydroxylation-associated protein ubiquitination
Several studies linked protein ubiquitination and hydroxylation states (Table S9). An eminent example is provided by the hydroxylation-mediated degradation of the hypoxia-inducible factor (HIF1α) transcription factor. HIF1α is normally activated under hypoxia, which leads to the activation of hypoxia-inducible genes. However, under physiological conditions, HIF1α is targeted to ubiquitination by the pVHL, a multi-subunit E3 ligase complex containing CUL2, Elongin B, Elongin C, and Rbx1.123,124 The factors responsible for HIF1α hydroxylation correspond to the Egg Laying defective Nine family proteins (EGLN), also known as prolyl-hydroxylase-domain proteins (PHDs).125,126,127 The hydroxylation of HIF1α on its conserved proline residues (P402 or P564) creates a binding site for pVHL (Figure 8A).126 The molecular basis of pVHL-HIF1α interaction was rapidly established, and it was revealed that pVHL forms a conserved hydroxyproline binding pocket involving well-arranged hydrogen bonds for selective binding of hydroxylated prolyl residues of HIF1α123 (Figure 8B). The binding of pVHL is highly specific to hydroxylated proline on HIF1α as a non-hydroxylated peptide couldn’t form a stable complex with pVHL indicating the tight regulation of HIF-1α degradation by hydroxylation. pVHL cancer mutations occurring in the binding interface with the hydroxylated proline peptide of HIF1α have been reported, further supporting the involvement of pVHL in regulating HIF1α stability upon hydroxylation.124
Figure 8.

Oxygen-sensing dependent ubiquitination regulates the function of the hypoxia-induced factor-1 (HIF1α)
(A) During hypoxia, the HIF1α factor is translocated to the nucleus where it dimerizes with HIF1β and activates hypoxia-induced response genes in the presence of CBP/P300. During normal conditions (normoxia) and in the presence of αKG, HIF1α is rapidly targeted by different proline hydroxylating enzymes, prolyl-hydroxylase-domain (PHD) proteins and the factor inhibitor HIF (FIH). Hydroxylation by FIH on the C-terminal end of HIF1α block its interaction with CBP/P300 and inhibits transcriptional repression. Hydroxylation by the PHD enzymes generates a binding site for the von-Hippel-Lindau (pVHL) ubiquitin ligase complex. This induces its polyubiquitination and proteasomal degradation.
(B) Structural description of the recognition of hydroxylated HIF1α (HYP 564) by pVHL E3 ligase complex (PDB: 1LM8). Left panel, surface representation of the pVHL/Elongin B/Elongin C/HIF1α co-structure showing the binding of HIF1α peptide with pVHL subunit. Right panel, zoom in the HIF1α/pVHL β domain. Hydroxylated proline 564 (HYP 564) inserts into a β domain hydrophobic core region (top view). Additional contacts are made between HIF1α backbone residues and pVHL side chain group (bottom view). The hydrogen bounds between amino acids are shown by the dashed lines.
The regulation of HIF1α stability by hydroxylation appears to be more complex in vivo. For instance, HIF1α prolyl-hydroxylation by the PHDs could be assisted by third party proteins such as the Osteosarcoma Amplified 9 (OS-9) protein which binds HIF1α and PHD2 or PHD3 and is required for HIF1α hydroxylation and degradation.128 Regulation of HIF1α stability also involves SUMOylation, which promotes its degradation.129 During hypoxia, active deSUMOylation of HIF1α by SENP1 leads to HIF1α stabilization, suggesting cooperation between SUMOylation and hydroxylation in promoting ubiquitination and degradation of HIF1α by pVHL.129 Another layer of complexity is provided by SIRT2, a NAD-dependent protein deacetylase, which removes the acetyl group from the K709 residue of HIF1α, inducing its destabilization. The deacetylation of HIF1α promotes the binding of PHD2 and prolyl-hydroxylation with the subsequent ubiquitination of HIF-1α.130 Finally, protein hydroxylation can regulate protein stability by interfering with the deubiquitinating reaction. For instance, hydroxylation of the transcription factor FOXO3a induces its degradation by inhibiting its interaction with the DUB USP9X,131 highlighting the complexity of crosstalk between these two PTMs.
In conclusion, the target list of proteins harboring proline hydroxylation is likely larger than anticipated, and recent proteomics studies suggest that oxygen sensing could potentially be a major determinant in the regulation of many cellular proteins.132 Thus, further work is needed to determine the extent to which the hydroxylation/ubiquitination interplay regulates cellular processes.
Protein-prolyl isomerization and ubiquitination
Peptidyl-prolyl isomerases (PPIases) are a family of enzymes that catalyze conformation changes between cis and trans configurations of proline peptide bonds (X-P whereby X is any amino acid except P).133,134 These enzymes exert important functions in protein quality control and coordination of cellular processes (Table S10). For instance, the PPlase PIN1 contains, in addition to the C-terminal catalytic domain, an N-terminal WW domain (harboring two conserved tryptophan residues) that recognizes S-P or T-P motifs, when the S or T residues are phosphorylated. An interesting example of Peptidyl-prolyl isomerization is provided by the C-terminal binding protein (CtBP)-interacting protein (CtIP), a DNA repair factor that plays an important role in DNA end-resection and homologous recombination repair of DSBs. PIN1-mediated interaction with phosphorylated CtIP induces its ubiquitination and proteasomal degradation, thus limiting DNA end-resection and hence regulating the balance between homologous recombination and non-homologous end-joining DSB repair pathways.135 PIN1 action on CtIP involves its prolyl isomerase activity indicating that a conformational change is required for CtIP ubiquitination and degradation. On the other hand, PPlase activity can also protect from degradation, as PIN1-mediated isomerization of phosphorylated FAAP20 on S48 promotes its association with the phosphatase PP2A, resulting in FAAP20 dephosphorylation by PP2A on S113 phosphodegron.136 This event prevents FAAP20 interaction with SCFFBW7 and results in the inhibition of FAAP20 proteasomal degradation, thus promoting FANCD2 activation and DNA interstrand cross-link repair. Clearly, PPIase-mediated protein conformation changes and its impact on substrate ubiquitination can involve an intricate interplay of additional PTMs, thus dynamically orchestrating cellular processes.
N-end rule and C-end rule pathways of protein degradation
The conjugation of specific amino acids, such as arginine, at the N-terminal end of proteins (Nt-arginylation) was initially found to have a destabilizing effect on modified proteins.137 It was later found that protein Nt-arginylation defines a distinct class of protein modification-inducing degradation, which is part of a general mechanism of protein degradation termed the N-end rule pathway.138,139,140,141,142,143,144,145,146 The N-end rule pathway relies on the recognition of an N-terminal destabilizing amino acid by specific E3 ubiquitin ligases, called N-recognins, which ubiquitinate the target protein (or nascent peptide in the case of co-translational modification) leading to proteasomal degradation in most cases.140,141 The N-end rule pathway can engage substrate ubiquitination following demasking of destabilizing amino acids or protein cleavage, with or without further protein modification. In eukaryotes, two major N-end rule pathway branches involving Nt-arginylation and Nt-acetylation have been described (discussed later in discussion), in addition to three recently discovered N-end rule pathways of protein degradation associated with Nt-G, Nt-P, and formylation of Nt-M138,139,140,141,142,143,144,145 (Table S11).
Nt-arginylation occurs on destabilizing N-terminal D or E residues and oxidized C residues which fall into a type 1 class of Nt-degron along with unmodified R, H and K residues.141 The second class includes the hydrophobic amino acids I and L and the aromatic amino acids such as F, W and Y.141 Type 1 and type 2 Nt-degrons are known to be bound by the UBR-box domain of the N-recognins E3 ligase family members, which are responsible for substrate ubiquitination and subsequent proteolysis.146,147 The Nt-arginylation on D, E and oxidized C residues is promoted by a unique enzyme called the arginyltransferase ATE1, which catalyzes the transfer of the R residue from arginyl-tRNA to acceptor amino acid residue on target proteins in an ATP-independent manner.148,149 Interestingly, Nt-arginylated peptides could be either degraded by the UPS system or autophagy.150 Indeed, Nt-arginylated substrates could be bound by the ZZ-type zinc finger (ZZ domain) of the p62 autophagy adapter, and this event can induce autophagy of target proteins.151,152,153,154 The potential signaling determinants that can shift the degradation of Nt-arginylated proteins, depending on the cellular contexts, toward the UPS or the autophagy degradation system remains to be defined.
The other major and frequent branch of the N-end rule pathway is Nt-acetylation (either on Nt-Met or second residue after Nt-Met removal).141,142 Nt-acetylation is carried by several Nt-acetyltransferases including NatA, NatB, NatC, NatD, NatE and NatF, classified according to their target specificity even though some of them could overlap in function and targets.155,156 In yeast, Nt-acetylation has been linked to proteasomal degradation of several target proteins such as the APC/C complex component Hcn1 and the Conserved oligomeric golgi subunit Cog1.139,157 Interestingly, Nt-acetylation-mediated substrate degradation could be blocked by its interacting proteins or folding states, thus preventing the recognition of Nt-acetylated residue by Nt-recognins.157 Indeed, in mammals, the same mechanism of “conditional degron” has been described for the protein G regulator, RGS2 which is targeted for Nt-acetylation and ubiquitination by TEB4 ER-associated ubiquitin ligase for subsequent degradation. This results in increased Gαq protein activation leading, in turn, to an increase of PLCβ and ERK1/2 signaling. In contrast, RGS2 is stabilized through interaction with Gαq, establishing a regulatory loop for protein G signaling.158 Whether additional factors could shift RGS2 between the free and assembled forms, thus regulating the extent of Gαq signaling, remains an interesting line of inquiry. On the other hand, recent proteomics studies in yeast indicated that Nt-acetylation is rarely recognized as a degron, and might even induce the opposite effect, i.e., protects substrates from proteasomal degradation.159 Moreover, this analysis also revealed that the overall hydrophobicity of the N-terminus constitutes a critical determinant in signaling degradation, notably through the action of Doa10 E3 ubiquitin ligase.159
Underlining the importance of protein termini in protein quality control, an additional mechanism of proteolysis involving C-terminal degrons was recently discovered.160,161 Similar to N-terminal degron of the N-end rule pathway, the C-end rule pathway (also termed DesCEND) is based on the recognition of specific amino acids, notably G residues at the C-terminal end of target proteins.160,161 While part of the N-end rule pathway relies on PTMs to promote the ubiquitination of the target protein, the C-end rule pathway seems to only require the recognition of specific “codes” of C-terminal amino acids within the last few residues of the target protein.160,161 For instance, the degrons might be generated following limited proteolysis of substrates by caspases or deubiquitinases. The Cullin-RING E3 ubiquitin ligase complexes seem to be the main drivers of the C-end rule pathway with specifically CRL2 and CRL4 directed-ubiquitination using interchangeable substrate recognition modules.160,161 Protein stability assay screens using a peptide library representing the human proteome revealed the degron specificity for each of Cullin E3-adapter proteins.160,161 For example, CRL2 substrate recognition adapters KLHDC2, KLHDC3, and KLHDC10 require a G residue at the end of the protein, however, FEM1A, FEM1B, and FEM1C target C-terminal R residue.160,161 Specifically, APPBP2 adapter protein targets proteins ending with an RxxG motif for ubiquitination by CRL2, while DCAF12 and TRPC4AP adapters recognize EE-endings and R residue at −3 position respectively for CRL4 ubiquitination.160,161 There are also additional degron signals as part of the C-end rule pathway targeting alanine containing C-termini probably involving non-CRL E3 ligases161 (Table S11). Moreover, the authors made an interesting observation following an analysis of the C-termini composition of proteomes from multiple eukaryotic species showing that glycine is less likely to be coded at this position.161 This led to the hypothesis of avoidance of G residue at the C-terminal end to globally protect the gene products from ubiquitination.161 Overall, the C-end rule pathway provides a novel paradigm of regulation of protein homeostasis and further studies are required to shed light on the signaling mechanisms that orchestrate this pathway.
Interplay between S-nitrosylation and ubiquitination
S-nitrosylation is another abundant PTM consisting of covalently linking a nitric oxide group (a.k.a. nitroso group or NO) to cysteine residues on target proteins resulting in the formation of S-nitrosothiols (SNOs) (Figure 1). Recent studies indicated that a significant proportion of the human proteome (over 2,000 sites) is modified by S-nitrosylation under normal conditions.162 The enzymatic catalysis of S-nitrosylation remains largely elusive, although recent studies provided insights into the enzymatic dependency of NO transfer to substrates.163
Since most UPS enzymes involve a cysteine in their catalytic sites, a crosstalk between ubiquitination and S-nitrosylation seemed highly plausible. Indeed, multiple components of the UPS are regulated by S-nitrosylation.164,165,166,167 Several studies revealed that S-nitrosylation triggers substrate ubiquitination and degradation168,169,170,171 (Table S12). For instance, initial observations have indicated that the S-nitrosylation of iron regulatory protein 2 (IRP2), which binds iron-responsive elements found in mRNAs, induces its ubiquitination and proteasomal degradation.168 Loss or depletion of the tumor suppressor PARK2 induces mitochondria-associated metabolic defects leading to S-nitrosylation and subsequent ubiquitination of PTEN, leading in turn to the activation of the AKT signaling pathway and cellular proliferation.169 In this setting, S-nitrosylation-mediated ubiquitination plays an important role in promoting tumorigenesis. On the other hand, S-nitrosylation could also block ubiquitination. This is the case of the cell survival-promoting factor BCL-2, whose S-nitrosylation protects it from ubiquitination and proteasomal degradation,170,171 and this deregulation might partly mediate cancer cell resistance to chemotherapy.
Overall, S-nitrosylation is an important PTM with elaborate connections with ubiquitination and other PTMs. It will be interesting to identify how S-nitrosylation promotes or inhibits substrate ubiquitination and whether specific co-factors could act as “readers” of S-nitrosylated residues.
Methods to study the crosstalk between post-translational modifications and ubiquitination
To study the crosstalk between ubiquitin and other PTMs on the same protein, a variety of biochemical and biophysical techniques can be applied to in vitro or in vivo cellular systems. Depending on the questions that need to be addressed, PTMs can be probed at the level of individual proteins or, in a high-throughput manner, through Mass Spectrometry (MS)-based proteomics analysis. Importantly, while certain PTMs require specific methodologies, the identification and characterization of a wide spectrum of PTMs often involve protein purification and detection with specific antibodies, chemical probes, or MS.172,173,174
Immunoprecipitation in conjunction with immunoblotting with specific antibodies recognizing individual PTMs has been widely used. These pan-specific antibodies have been generated for a wide spectrum of modifications including anti-methyl, anti-phosphoserine, anti-phosphothreonine, anti-acetyl, anti-O-GlcNAc, anti-PAR, anti-ubiquitin, and anti-SUMO. Conversely, global immunoprecipitation of PTM-modified proteins followed by the detection of the protein of interest, with specific antibodies, can also be conducted. Moreover, a large variety of highly specific antibodies against PTMs in the context of specific proteins of interest, notably phospho-specific antibodies, can also be used for direct immunoblotting or immunofluorescence detection. Of note, a decreased signal of a given protein PTM, under specific conditions, could be due to the modification’s loss or decreased protein expression. Thus, when investigating PTM dynamics, the detection of the protein of interest with antibodies targeting the protein, independently of the modification state, is required. Anti-PTM antibodies that are highly specific to protein targets can also be used for the immunoprecipitation of the modified protein followed by the detection of other modifications including ubiquitination. This approach provides indications about the co-existence or the mutual exclusivity of two different PTMs on the same protein, thus providing insights into their crosstalk. We caution that anti-PTM-directed antibodies should be rigorously validated, as a significant proportion of commercially available antibodies are not as specific, as claimed by vendors.175,176 Mutation of specific modification sites, purification, or enrichment in conjunction with immunodetection as well as protein depletion or inactivation should be performed to ensure that a given antibody recognizes its target in a highly specific manner.
On the other hand, MS has become the technique of choice to detect PTMs and crosstalk among various types of PTMs.177 In general, a “bottom-up” strategy that involves the tryptic digestion of the purified protein or a protein mixture can be employed. This generates small peptides that are first separated through liquid chromatography and fragmented for tandem mass spectrometry analysis (LC-MS/MS). MS spectra analysis can indicate the presence of PTMs on specific peptide sequences and at precise amino acid positions. For example, residual Gly-Gly di-peptides characteristic of ubiquitin-modified lysine could be readily detected on proteins of interest. Quantitative analysis of PTMs can also be performed, for instance, through stable isotope labeling using amino acids in cell culture (SILAC).177 This technique involves the metabolic incorporation, during cell culture, of heavy amino acids (e.g., lysine or arginine labeled with Carbon 13 and Nitrogen 15 isotopes) into the cellular pool of proteins. To enable the quantification of changes between two experimental conditions, one cell culture population is labeled with heavy amino acid “heavy condition” whereas the other sample is left in normal media “light condition.” Following specific treatments, the “heavy” and “light” samples are mixed in equal quantities, and the proteins are extracted or purified for analysis by LC-MS/MS. The differential changes in specific modifications or protein levels between two experimental conditions could be distinguished by comparing the peak intensity of the “light” versus the “heavy” peptides, which can be readily distinguished due to increased molecular mass.
It is worth noting that an enrichment step could be beneficial to increase the coverage and probability of detecting PTMs. Antibodies or chemically modified matrices that have an affinity for specific PTMs could then be applied.177 Once a specific PTM is detected, the crosstalk with ubiquitination can be further investigated through biochemical and functional assays. Notably, site-directed mutagenesis of amino acid targeted by a given PTM could be conducted to determine the impact on protein stability and ubiquitination. Moreover, mutagenesis to amino acids that can act as potential PTM mimetics could also be performed, with the resulting mutants tested for functional assays. On the other hand, pharmacological inhibitors or activators of signaling pathways that modulate a given PTM can also be used to determine the impact on protein ubiquitination. Finally, depleting the proteins or co-factors as well as expressing catalytic dead mutants of the enzymes responsible for catalyzing the PTM on substrates can also be used to investigate the impact on ubiquitination states.
Concluding remarks
Decades of research efforts have led to the uncovering of many complex mechanisms underlying the interplay between ubiquitination and other PTMs. Diverse PTMs were found to promote or inhibit substrate ubiquitination in response to signaling pathways induced by cell-intrinsic or environmental changes. At the functional level, the general understanding of these PTM crosstalks and relationships has revealed unexpected connections between signaling pathways and cellular processes, and has also improved our understanding of disease conditions. As indicated throughout this review, a lot remains to be discovered regarding PTM crosstalk. Nonetheless, the knowledge obtained so far has contributed to elaborating novel diagnostic tools and therapeutic strategies targeting aberrant signaling pathways and cellular processes.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research.
Author contributions
H. B., N. S. N., B. E., C. M., S. D., and E. B. A. conceptualization; H. B., N. S. N., B. E., C. M., M. G. L., R. V., M. U., K. B., S. H., B. F., F. A. M., E. M., S. D., and E. B. A. data curation; H. B., N. S. N., S. D., and E. B. A. writing-original draft; H. B., N. S. N., B. E., C. M., M. G. L., R. V., M. U., K. B., S. H., B. F., F. A. M., E. M., S. D., and E. B. A. writing-review and editing.
Declaration of interests
The authors declare no competing interests with the content of this article.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106276.
Contributor Information
Eric Milot, Email: e.milot.1@umontreal.ca.
Frédérick A. Mallette, Email: fa.mallette@umontreal.ca.
Salima Daou, Email: sdaou@lunenfeld.ca.
El Bachir Affar, Email: el.bachir.affar@umontreal.ca.
Supplemental information
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
References
- 1.Oh E., Akopian D., Rape M. Principles of ubiquitin-dependent signaling. Annu. Rev. Cell Dev. Biol. 2018;34:137–162. doi: 10.1146/annurev-cellbio-100617-062802. [DOI] [PubMed] [Google Scholar]
- 2.Zheng N., Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu. Rev. Biochem. 2017;86:129–157. doi: 10.1146/annurev-biochem-060815-014922. [DOI] [PubMed] [Google Scholar]
- 3.French M.E., Koehler C.F., Hunter T. Emerging functions of branched ubiquitin chains. Cell Discov. 2021;7:6. doi: 10.1038/s41421-020-00237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan F., Huang C., Wang X., Tan J., Cheng S., Wan M., Wang Z., Wang S., Luo S., Li A., et al. Threonine ADP-ribosylation of ubiquitin by a bacterial effector family blocks host ubiquitination. Mol. Cell. 2020;78:641–652.e9. doi: 10.1016/j.molcel.2020.03.016. [DOI] [PubMed] [Google Scholar]
- 5.Chatrin C., Gabrielsen M., Buetow L., Nakasone M.A., Ahmed S.F., Sumpton D., Sibbet G.J., Smith B.O., Huang D.T. Structural insights into ADP-ribosylation of ubiquitin by Deltex family E3 ubiquitin ligases. Sci. Adv. 2020;6:eabc0418. doi: 10.1126/sciadv.abc0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui J., Yao Q., Li S., Ding X., Lu Q., Mao H., Liu L., Zheng N., Chen S., Shao F. Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science. 2010;329:1215–1218. doi: 10.1126/science.1193844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Husnjak K., Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 2012;81:291–322. doi: 10.1146/annurev-biochem-051810-094654. [DOI] [PubMed] [Google Scholar]
- 8.Clague M.J., Urbé S., Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019;20:338–352. doi: 10.1038/s41580-019-0099-1. [DOI] [PubMed] [Google Scholar]
- 9.Estavoyer B., Messmer C., Echbicheb M., Rudd C.E., Milot E., Affar E.B. Mechanisms orchestrating the enzymatic activity and cellular functions of deubiquitinases. J. Biol. Chem. 2022;298:102198. doi: 10.1016/j.jbc.2022.102198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grabbe C., Husnjak K., Dikic I. The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 2011;12:295–307. doi: 10.1038/nrm3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welchman R.L., Gordon C., Mayer R.J. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 2005;6:599–609. doi: 10.1038/nrm1700. [DOI] [PubMed] [Google Scholar]
- 12.Flotho A., Melchior F. Sumoylation: a regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013;82:357–385. doi: 10.1146/annurev-biochem-061909-093311. [DOI] [PubMed] [Google Scholar]
- 13.Chen Z., Hagler J., Palombella V.J., Melandri F., Scherer D., Ballard D., Maniatis T. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 14.Scherer D.C., Brockman J.A., Chen Z., Maniatis T., Ballard D.W. Signal-induced degradation of I kappa B alpha requires site-specific ubiquitination. Proc. Natl. Acad. Sci. USA. 1995;92:11259–11263. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alkalay I., Yaron A., Hatzubai A., Orian A., Ciechanover A., Ben-Neriah Y. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA. 1995;92:10599–10603. doi: 10.1073/pnas.92.23.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desterro J.M., Rodriguez M.S., Hay R.T. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell. 1998;2:233–239. doi: 10.1016/s1097-2765(00)80133-1. [DOI] [PubMed] [Google Scholar]
- 17.Lee M.H., Mabb A.M., Gill G.B., Yeh E.T.H., Miyamoto S. NF-kappaB induction of the SUMO protease SENP2: a negative feedback loop to attenuate cell survival response to genotoxic stress. Mol. Cell. 2011;43:180–191. doi: 10.1016/j.molcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aillet F., Lopitz-Otsoa F., Egaña I., Hjerpe R., Fraser P., Hay R.T., Rodriguez M.S., Lang V. Heterologous SUMO-2/3-ubiquitin chains optimize IkappaBalpha degradation and NF-kappaB activity. PLoS One. 2012;7:e51672. doi: 10.1371/journal.pone.0051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatham M.H., Geoffroy M.C., Shen L., Plechanovova A., Hattersley N., Jaffray E.G., Palvimo J.J., Hay R.T. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 2008;10:538–546. doi: 10.1038/ncb1716. [DOI] [PubMed] [Google Scholar]
- 20.Lallemand-Breitenbach V., Jeanne M., Benhenda S., Nasr R., Lei M., Peres L., Zhou J., Zhu J., Raught B., de Thé H. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008;10:547–555. doi: 10.1038/ncb1717. [DOI] [PubMed] [Google Scholar]
- 21.Rojas-Fernandez A., Plechanovová A., Hattersley N., Jaffray E., Tatham M.H., Hay R.T. SUMO chain-induced dimerization activates RNF4. Mol. Cell. 2014;53:880–892. doi: 10.1016/j.molcel.2014.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plechanovová A., Jaffray E.G., McMahon S.A., Johnson K.A., Navrátilová I., Naismith J.H., Hay R.T. Mechanism of ubiquitylation by dimeric RING ligase RNF4. Nat. Struct. Mol. Biol. 2011;18:1052–1059. doi: 10.1038/nsmb.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamitani T., Kito K., Nguyen H.P., Yeh E.T. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J. Biol. Chem. 1997;272:28557–28562. doi: 10.1074/jbc.272.45.28557. [DOI] [PubMed] [Google Scholar]
- 24.Ohh M., Kim W.Y., Moslehi J.J., Chen Y., Chau V., Read M.A., Kaelin W.G., Jr. An intact NEDD8 pathway is required for Cullin-dependent ubiquitylation in mammalian cells. EMBO Rep. 2002;3:177–182. doi: 10.1093/embo-reports/kvf028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakata E., Yamaguchi Y., Miyauchi Y., Iwai K., Chiba T., Saeki Y., Matsuda N., Tanaka K., Kato K. Direct interactions between NEDD8 and ubiquitin E2 conjugating enzymes upregulate cullin-based E3 ligase activity. Nat. Struct. Mol. Biol. 2007;14:167–168. doi: 10.1038/nsmb1191. [DOI] [PubMed] [Google Scholar]
- 26.Liu J., Furukawa M., Matsumoto T., Xiong Y. NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol. Cell. 2002;10:1511–1518. doi: 10.1016/s1097-2765(02)00783-9. [DOI] [PubMed] [Google Scholar]
- 27.Zuo W., Huang F., Chiang Y.J., Li M., Du J., Ding Y., Zhang T., Lee H.W., Jeong L.S., Chen Y., et al. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-beta type II receptor. Mol. Cell. 2013;49:499–510. doi: 10.1016/j.molcel.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Di Guglielmo G.M., Le Roy C., Goodfellow A.F., Wrana J.L. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat. Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 29.Lee G.W., Park J.B., Park S.Y., Seo J., Shin S.H., Park J.W., Kim S.J., Watanabe M., Chun Y.S. The E3 ligase C-CBL inhibits cancer cell migration by neddylating the proto-oncogene c-Src. Oncogene. 2018;37:5552–5568. doi: 10.1038/s41388-018-0354-5. [DOI] [PubMed] [Google Scholar]
- 30.Hoege C., Pfander B., Moldovan G.L., Pyrowolakis G., Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 31.Stelter P., Ulrich H.D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425:188–191. doi: 10.1038/nature01965. [DOI] [PubMed] [Google Scholar]
- 32.Haracska L., Torres-Ramos C.A., Johnson R.E., Prakash S., Prakash L. Opposing effects of ubiquitin conjugation and SUMO modification of PCNA on replicational bypass of DNA lesions in Saccharomyces cerevisiae. Mol. Cell Biol. 2004;24:4267–4274. doi: 10.1128/MCB.24.10.4267-4274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfander B., Moldovan G.L., Sacher M., Hoege C., Jentsch S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature. 2005;436:428–433. doi: 10.1038/nature03665. [DOI] [PubMed] [Google Scholar]
- 34.Papouli E., Chen S., Davies A.A., Huttner D., Krejci L., Sung P., Ulrich H.D. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell. 2005;19:123–133. doi: 10.1016/j.molcel.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 35.Armstrong A.A., Mohideen F., Lima C.D. Recognition of SUMO-modified PCNA requires tandem receptor motifs in Srs2. Nature. 2012;483:59–63. doi: 10.1038/nature10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kannouche P.L., Wing J., Lehmann A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- 37.Acharya N., Yoon J.H., Gali H., Unk I., Haracska L., Johnson R.E., Hurwitz J., Prakash L., Prakash S. Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase eta in translesion DNA synthesis. Proc. Natl. Acad. Sci. USA. 2008;105:17724–17729. doi: 10.1073/pnas.0809844105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiu R.K., Brun J., Ramaekers C., Theys J., Weng L., Lambin P., Gray D.A., Wouters B.G. Lysine 63-polyubiquitination guards against translesion synthesis-induced mutations. PLoS Genet. 2006;2:e116. doi: 10.1371/journal.pgen.0020116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Branzei D., Vanoli F., Foiani M. SUMOylation regulates Rad18-mediated template switch. Nature. 2008;456:915–920. doi: 10.1038/nature07587. [DOI] [PubMed] [Google Scholar]
- 40.Weston R., Peeters H., Ahel D. ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev. 2012;26:1558–1572. doi: 10.1101/gad.193516.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ciccia A., Nimonkar A.V., Hu Y., Hajdu I., Achar Y.J., Izhar L., Petit S.A., Adamson B., Yoon J.C., Kowalczykowski S.C., et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell. 2012;47:396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vujanovic M., Krietsch J., Raso M.C., Terraneo N., Zellweger R., Schmid J.A., Taglialatela A., Huang J.W., Holland C.L., Zwicky K., et al. Replication fork slowing and reversal upon DNA damage require PCNA polyubiquitination and ZRANB3 DNA translocase activity. Mol. Cell. 2017;67:882–890.e5. doi: 10.1016/j.molcel.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masuda Y., Mitsuyuki S., Kanao R., Hishiki A., Hashimoto H., Masutani C. Regulation of HLTF-mediated PCNA polyubiquitination by RFC and PCNA monoubiquitination levels determines choice of damage tolerance pathway. Nucleic Acids Res. 2018;46:11340–11356. doi: 10.1093/nar/gky943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guan J., Yu S., Zheng X. NEDDylation antagonizes ubiquitination of proliferating cell nuclear antigen and regulates the recruitment of polymerase eta in response to oxidative DNA damage. Protein Cell. 2018;9:365–379. doi: 10.1007/s13238-017-0455-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park J.M., Yang S.W., Yu K.R., Ka S.H., Lee S.W., Seol J.H., Jeon Y.J., Chung C.H. Modification of PCNA by ISG15 plays a crucial role in termination of error-prone translesion DNA synthesis. Mol. Cell. 2014;54:626–638. doi: 10.1016/j.molcel.2014.03.031. [DOI] [PubMed] [Google Scholar]
- 46.Hermann M., Bogunovic D. ISG15: in sickness and in health. Trends Immunol. 2017;38:79–93. doi: 10.1016/j.it.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 47.Perng Y.C., Lenschow D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018;16:423–439. doi: 10.1038/s41579-018-0020-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bai C., Sen P., Hofmann K., Ma L., Goebl M., Harper J.W., Elledge S.J. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 49.Sheaff R.J., Groudine M., Gordon M., Roberts J.M., Clurman B.E. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- 50.Tsvetkov L.M., Yeh K.H., Lee S.J., Sun H., Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 1999;9:661–664. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 51.Carrano A.C., Eytan E., Hershko A., Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 52.Ganoth D., Bornstein G., Ko T.K., Larsen B., Tyers M., Pagano M., Hershko A. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat. Cell Biol. 2001;3:321–324. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- 53.Hao B., Zheng N., Schulman B.A., Wu G., Miller J.J., Pagano M., Pavletich N.P. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol. Cell. 2005;20:9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 54.Chu I., Sun J., Arnaout A., Kahn H., Hanna W., Narod S., Sun P., Tan C.K., Hengst L., Slingerland J. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell. 2007;128:281–294. doi: 10.1016/j.cell.2006.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rath S.L., Senapati S. Mechanism of p27 unfolding for CDK2 reactivation. Sci. Rep. 2016;6:26450. doi: 10.1038/srep26450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huntwork-Rodriguez S., Wang B., Watkins T., Ghosh A.S., Pozniak C.D., Bustos D., Newton K., Kirkpatrick D.S., Lewcock J.W. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J. Cell Biol. 2013;202:747–763. doi: 10.1083/jcb.201303066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Symington L.S., Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 58.Zimmermann M., de Lange T. 53BP1: pro choice in DNA repair. Trends Cell Biol. 2014;24:108–117. doi: 10.1016/j.tcb.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ceccaldi R., Rondinelli B., D'Andrea A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Orthwein A., Noordermeer S.M., Wilson M.D., Landry S., Enchev R.I., Sherker A., Munro M., Pinder J., Salsman J., Dellaire G., et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature. 2015;528:422–426. doi: 10.1038/nature16142. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Lu H., Shamanna R.A., de Freitas J.K., Okur M., Khadka P., Kulikowicz T., Holland P.P., Tian J., Croteau D.L., Davis A.J., Bohr V.A. Cell cycle-dependent phosphorylation regulates RECQL4 pathway choice and ubiquitination in DNA double-strand break repair. Nat. Commun. 2017;8:2039. doi: 10.1038/s41467-017-02146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Verdin E., Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015;16:258–264. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- 63.Narita T., Weinert B.T., Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019;20:156–174. doi: 10.1038/s41580-018-0081-3. [DOI] [PubMed] [Google Scholar]
- 64.Li M., Luo J., Brooks C.L., Gu W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem. 2002;277:50607–50611. doi: 10.1074/jbc.C200578200. [DOI] [PubMed] [Google Scholar]
- 65.Ito A., Kawaguchi Y., Lai C.H., Kovacs J.J., Higashimoto Y., Appella E., Yao T.P. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–6245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boriack-Sjodin P.A., Swinger K.K. Protein methyltransferases: a distinct, diverse, and dynamic family of enzymes. Biochemistry. 2016;55:1557–1569. doi: 10.1021/acs.biochem.5b01129. [DOI] [PubMed] [Google Scholar]
- 67.Cheng X., Collins R.E., Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu. Rev. Biophys. Biomol. Struct. 2005;34:267–294. doi: 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nishiyama A., Yamaguchi L., Sharif J., Johmura Y., Kawamura T., Nakanishi K., Shimamura S., Arita K., Kodama T., Ishikawa F., et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature. 2013;502:249–253. doi: 10.1038/nature12488. [DOI] [PubMed] [Google Scholar]
- 69.Arita K., Isogai S., Oda T., Unoki M., Sugita K., Sekiyama N., Kuwata K., Hamamoto R., Tochio H., Sato M., et al. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc. Natl. Acad. Sci. USA. 2012;109:12950–12955. doi: 10.1073/pnas.1203701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rothbart S.B., Krajewski K., Nady N., Tempel W., Xue S., Badeaux A.I., Barsyte-Lovejoy D., Martinez J.Y., Bedford M.T., Fuchs S.M., et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 2012;19:1155–1160. doi: 10.1038/nsmb.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rothbart S.B., Dickson B.M., Ong M.S., Krajewski K., Houliston S., Kireev D.B., Arrowsmith C.H., Strahl B.D. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 2013;27:1288–1298. doi: 10.1101/gad.220467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harrison J.S., Cornett E.M., Goldfarb D., DaRosa P.A., Li Z.M., Yan F., Dickson B.M., Guo A.H., Cantu D.V., Kaustov L., et al. Hemi-methylated DNA regulates DNA methylation inheritance through allosteric activation of H3 ubiquitylation by UHRF1. Elife. 2016;5:e17101. doi: 10.7554/eLife.17101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DaRosa P.A., Harrison J.S., Zelter A., Davis T.N., Brzovic P., Kuhlman B., Klevit R.E. A bifunctional role for the UHRF1 UBL domain in the control of hemi-methylated DNA-dependent histone ubiquitylation. Mol. Cell. 2018;72:753–765.e6. doi: 10.1016/j.molcel.2018.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee J.M., Lee J.S., Kim H., Kim K., Park H., Kim J.Y., Lee S.H., Kim I.S., Kim J., Lee M., et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell. 2012;48:572–586. doi: 10.1016/j.molcel.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 75.He Y.J., McCall C.M., Hu J., Zeng Y., Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 2006;20:2949–2954. doi: 10.1101/gad.1483206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bedford M.T., Clarke S.G. Protein arginine methylation in mammals: who, what, and why. Mol. Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu D., Gur M., Zhou Z., Gamper A., Hung M.C., Fujita N., Lan L., Bahar I., Wan Y. Interplay between arginine methylation and ubiquitylation regulates KLF4-mediated genome stability and carcinogenesis. Nat. Commun. 2015;6:8419. doi: 10.1038/ncomms9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang X., Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017;18:452–465. doi: 10.1038/nrm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hardivillé S., Hart G.W. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metabol. 2014;20:208–213. doi: 10.1016/j.cmet.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mohawk J.A., Green C.B., Takahashi J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012;35:445–462. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lowrey P.L., Takahashi J.S. Genetics of circadian rhythms in Mammalian model organisms. Adv. Genet. 2011;74:175–230. doi: 10.1016/B978-0-12-387690-4.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li M.D., Ruan H.B., Hughes M.E., Lee J.S., Singh J.P., Jones S.P., Nitabach M.N., Yang X. O-GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/CLOCK ubiquitination. Cell Metabol. 2013;17:303–310. doi: 10.1016/j.cmet.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang N., Chelliah Y., Shan Y., Taylor C.A., Yoo S.H., Partch C., Green C.B., Zhang H., Takahashi J.S. Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science. 2012;337:189–194. doi: 10.1126/science.1222804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Udeshi N.D., Svinkina T., Mertins P., Kuhn E., Mani D.R., Qiao J.W., Carr S.A. Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteomics. 2013;12:825–831. doi: 10.1074/mcp.O112.027094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee J.M., Dedhar S., Kalluri R., Thompson E.W. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J. Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu Y., Lee S.H., Kim H.S., Kim N.H., Piao S., Park S.H., Jung Y.S., Yook J.I., Park B.J., Ha N.C. Role of CK1 in GSK3beta-mediated phosphorylation and degradation of snail. Oncogene. 2010;29:3124–3133. doi: 10.1038/onc.2010.77. [DOI] [PubMed] [Google Scholar]
- 87.Zhou B.P., Deng J., Xia W., Xu J., Li Y.M., Gunduz M., Hung M.C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 88.Park S.Y., Kim H.S., Kim N.H., Ji S., Cha S.Y., Kang J.G., Ota I., Shimada K., Konishi N., Nam H.W., et al. Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J. 2010;29:3787–3796. doi: 10.1038/emboj.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma X., Liu H., Li J., Wang Y., Ding Y.H., Shen H., Yang Y., Sun C., Huang M., Tu Y., et al. Poleta O-GlcNAcylation governs genome integrity during translesion DNA synthesis. Nat. Commun. 2017;8:1941. doi: 10.1038/s41467-017-02164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu C., Ng D.T.W. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol. 2015;16:742–752. doi: 10.1038/nrm4073. [DOI] [PubMed] [Google Scholar]
- 91.Ruggiano A., Foresti O., Carvalho P. Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol. 2014;204:869–879. doi: 10.1083/jcb.201312042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Olzmann J.A., Kopito R.R., Christianson J.C. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harbor Perspect. Biol. 2013;5:a013185. doi: 10.1101/cshperspect.a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Braakman I., Hebert D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harbor Perspect. Biol. 2013;5:a013201. doi: 10.1101/cshperspect.a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Clerc S., Hirsch C., Oggier D.M., Deprez P., Jakob C., Sommer T., Aebi M. Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J. Cell Biol. 2009;184:159–172. doi: 10.1083/jcb.200809198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gauss R., Kanehara K., Carvalho P., Ng D.T.W., Aebi M. A complex of Pdi1p and the mannosidase Htm1p initiates clearance of unfolded glycoproteins from the endoplasmic reticulum. Mol. Cell. 2011;42:782–793. doi: 10.1016/j.molcel.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 96.Liu Y.C., Fujimori D.G., Weissman J.S. Htm1p-Pdi1p is a folding-sensitive mannosidase that marks N-glycoproteins for ER-associated protein degradation. Proc. Natl. Acad. Sci. USA. 2016;113:E4015–E4024. doi: 10.1073/pnas.1608795113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jakob C.A., Burda P., Roth J., Aebi M. Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. J. Cell Biol. 1998;142:1223–1233. doi: 10.1083/jcb.142.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Szathmary R., Bielmann R., Nita-Lazar M., Burda P., Jakob C.A. Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol. Cell. 2005;19:765–775. doi: 10.1016/j.molcel.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 99.Kim W., Spear E.D., Ng D.T.W. Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol. Cell. 2005;19:753–764. doi: 10.1016/j.molcel.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 100.Xie W., Kanehara K., Sayeed A., Ng D.T.W. Intrinsic conformational determinants signal protein misfolding to the Hrd1/Htm1 endoplasmic reticulum-associated degradation system. Mol. Biol. Cell. 2009;20:3317–3329. doi: 10.1091/mbc.E09-03-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schoebel S., Mi W., Stein A., Ovchinnikov S., Pavlovicz R., DiMaio F., Baker D., Chambers M.G., Su H., Li D., et al. Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature. 2017;548:352–355. doi: 10.1038/nature23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ye Y., Meyer H.H., Rapoport T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 103.Ye Y., Meyer H.H., Rapoport T.A. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ye Y., Shibata Y., Yun C., Ron D., Rapoport T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 105.Yoshida Y., Adachi E., Fukiya K., Iwai K., Tanaka K. Glycoprotein-specific ubiquitin ligases recognize N-glycans in unfolded substrates. EMBO Rep. 2005;6:239–244. doi: 10.1038/sj.embor.7400351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yoshida Y., Chiba T., Tokunaga F., Kawasaki H., Iwai K., Suzuki T., Ito Y., Matsuoka K., Yoshida M., Tanaka K., Tai T. E3 ubiquitin ligase that recognizes sugar chains. Nature. 2002;418:438–442. doi: 10.1038/nature00890. [DOI] [PubMed] [Google Scholar]
- 107.Yoshida Y., Tokunaga F., Chiba T., Iwai K., Tanaka K., Tai T. Fbs2 is a new member of the E3 ubiquitin ligase family that recognizes sugar chains. J. Biol. Chem. 2003;278:43877–43884. doi: 10.1074/jbc.M304157200. [DOI] [PubMed] [Google Scholar]
- 108.Mizushima T., Hirao T., Yoshida Y., Lee S.J., Chiba T., Iwai K., Yamaguchi Y., Kato K., Tsukihara T., Tanaka K. Structural basis of sugar-recognizing ubiquitin ligase. Nat. Struct. Mol. Biol. 2004;11:365–370. doi: 10.1038/nsmb732. [DOI] [PubMed] [Google Scholar]
- 109.Mizushima T., Yoshida Y., Kumanomidou T., Hasegawa Y., Suzuki A., Yamane T., Tanaka K. Structural basis for the selection of glycosylated substrates by SCF(Fbs1) ubiquitin ligase. Proc. Natl. Acad. Sci. USA. 2007;104:5777–5781. doi: 10.1073/pnas.0610312104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gibson B.A., Kraus W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 111.Liu C., Vyas A., Kassab M.A., Singh A.K., Yu X. The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 2017;45:8129–8141. doi: 10.1093/nar/gkx565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Aravind L. The WWE domain: a common interaction module in protein ubiquitination and ADP ribosylation. Trends Biochem. Sci. 2001;26:273–275. doi: 10.1016/s0968-0004(01)01787-x. [DOI] [PubMed] [Google Scholar]
- 113.Karras G.I., Kustatscher G., Buhecha H.R., Allen M.D., Pugieux C., Sait F., Bycroft M., Ladurner A.G. The macro domain is an ADP-ribose binding module. EMBO J. 2005;24:1911–1920. doi: 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang Z., Michaud G.A., Cheng Z., Zhang Y., Hinds T.R., Fan E., Cong F., Xu W. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 2012;26:235–240. doi: 10.1101/gad.182618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kang H.C., Lee Y.I., Shin J.H., Andrabi S.A., Chi Z., Gagné J.P., Lee Y., Ko H.S., Lee B.D., Poirier G.G., et al. Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proc. Natl. Acad. Sci. USA. 2011;108:14103–14108. doi: 10.1073/pnas.1108799108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yan Q., Xu R., Zhu L., Cheng X., Wang Z., Manis J., Shipp M.A. BAL1 and its partner E3 ligase, BBAP, link Poly(ADP-ribose) activation, ubiquitylation, and double-strand DNA repair independent of ATM, MDC1, and RNF8. Mol. Cell Biol. 2013;33:845–857. doi: 10.1128/MCB.00990-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li M., Yu X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell. 2013;23:693–704. doi: 10.1016/j.ccr.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huang S.M.A., Mishina Y.M., Liu S., Cheung A., Stegmeier F., Michaud G.A., Charlat O., Wiellette E., Zhang Y., Wiessner S., et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 119.DaRosa P.A., Wang Z., Jiang X., Pruneda J.N., Cong F., Klevit R.E., Xu W. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature. 2015;517:223–226. doi: 10.1038/nature13826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Logan C.Y., Nusse R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 121.Lee E., Salic A., Krüger R., Heinrich R., Kirschner M.W. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003;1:E10. doi: 10.1371/journal.pbio.0000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang Y., Liu S., Mickanin C., Feng Y., Charlat O., Michaud G.A., Schirle M., Shi X., Hild M., Bauer A., et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol. 2011;13:623–629. doi: 10.1038/ncb2222. [DOI] [PubMed] [Google Scholar]
- 123.Hon W.C., Wilson M.I., Harlos K., Claridge T.D.W., Schofield C.J., Pugh C.W., Maxwell P.H., Ratcliffe P.J., Stuart D.I., Jones E.Y. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature. 2002;417:975–978. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- 124.Min J.H., Yang H., Ivan M., Gertler F., Kaelin W.G., Jr., Pavletich N.P. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 125.Jaakkola P., Mole D.R., Tian Y.M., Wilson M.I., Gielbert J., Gaskell S.J., von Kriegsheim A., Hebestreit H.F., Mukherji M., Schofield C.J., et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 126.Epstein A.C., Gleadle J.M., McNeill L.A., Hewitson K.S., O'Rourke J., Mole D.R., Mukherji M., Metzen E., Wilson M.I., Dhanda A., et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 127.Bruick R.K., McKnight S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 128.Baek J.H., Mahon P.C., Oh J., Kelly B., Krishnamachary B., Pearson M., Chan D.A., Giaccia A.J., Semenza G.L. OS-9 interacts with hypoxia-inducible factor 1alpha and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1alpha. Mol. Cell. 2005;17:503–512. doi: 10.1016/j.molcel.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 129.Cheng J., Kang X., Zhang S., Yeh E.T.H. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584–595. doi: 10.1016/j.cell.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Seo K.S., Park J.H., Heo J.Y., Jing K., Han J., Min K.N., Kim C., Koh G.Y., Lim K., Kang G.Y., et al. SIRT2 regulates tumour hypoxia response by promoting HIF-1alpha hydroxylation. Oncogene. 2015;34:1354–1362. doi: 10.1038/onc.2014.76. [DOI] [PubMed] [Google Scholar]
- 131.Zheng X., Zhai B., Koivunen P., Shin S.J., Lu G., Liu J., Geisen C., Chakraborty A.A., Moslehi J.J., Smalley D.M., et al. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev. 2014;28:1429–1444. doi: 10.1101/gad.242131.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhou T., Erber L., Liu B., Gao Y., Ruan H.B., Chen Y. Proteomic analysis reveals diverse proline hydroxylation-mediated oxygen-sensing cellular pathways in cancer cells. Oncotarget. 2016;7:79154–79169. doi: 10.18632/oncotarget.12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liou Y.C., Zhou X.Z., Lu K.P. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 2011;36:501–514. doi: 10.1016/j.tibs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhou X.Z., Lu K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer. 2016;16:463–478. doi: 10.1038/nrc.2016.49. [DOI] [PubMed] [Google Scholar]
- 135.Steger M., Murina O., Hühn D., Ferretti L.P., Walser R., Hänggi K., Lafranchi L., Neugebauer C., Paliwal S., Janscak P., et al. Prolyl isomerase PIN1 regulates DNA double-strand break repair by counteracting DNA end resection. Mol. Cell. 2013;50:333–343. doi: 10.1016/j.molcel.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 136.Wang J., Chan B., Tong M., Paung Y., Jo U., Martin D., Seeliger M., Haley J., Kim H. Prolyl isomerization of FAAP20 catalyzed by PIN1 regulates the Fanconi anemia pathway. PLoS Genet. 2019;15:e1007983. doi: 10.1371/journal.pgen.1007983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Soffer R.L. Enzymatic modification of proteins. 4. Arginylation of bovine thyroglobulin. J. Biol. Chem. 1971;246:1481–1484. [PubMed] [Google Scholar]
- 138.Mayer A., Siegel N.R., Schwartz A.L., Ciechanover A. Degradation of proteins with acetylated amino termini by the ubiquitin system. Science. 1989;244:1480–1483. doi: 10.1126/science.2544030. [DOI] [PubMed] [Google Scholar]
- 139.Hwang C.S., Shemorry A., Varshavsky A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science. 2010;327:973–977. doi: 10.1126/science.1183147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sriram S.M., Kim B.Y., Kwon Y.T. The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat. Rev. Mol. Cell Biol. 2011;12:735–747. doi: 10.1038/nrm3217. [DOI] [PubMed] [Google Scholar]
- 141.Tasaki T., Sriram S.M., Park K.S., Kwon Y.T. The N-end rule pathway. Annu. Rev. Biochem. 2012;81:261–289. doi: 10.1146/annurev-biochem-051710-093308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nguyen K.T., Mun S.H., Lee C.S., Hwang C.S. Control of protein degradation by N-terminal acetylation and the N-end rule pathway. Exp. Mol. Med. 2018;50:1–8. doi: 10.1038/s12276-018-0097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen S.J., Wu X., Wadas B., Oh J.H., Varshavsky A. An N-end rule pathway that recognizes proline and destroys gluconeogenic enzymes. Science. 2017;355:eaal3655. doi: 10.1126/science.aal3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kim J.M., Seok O.H., Ju S., Heo J.E., Yeom J., Kim D.S., Yoo J.Y., Varshavsky A., Lee C., Hwang C.S. Formyl-methionine as an N-degron of a eukaryotic N-end rule pathway. Science. 2018;362:eaat0174. doi: 10.1126/science.aat0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Timms R.T., Zhang Z., Rhee D.Y., Harper J.W., Koren I., Elledge S.J. A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation. Science. 2019;365:eaaw4912. doi: 10.1126/science.aaw4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tasaki T., Zakrzewska A., Dudgeon D.D., Jiang Y., Lazo J.S., Kwon Y.T. The substrate recognition domains of the N-end rule pathway. J. Biol. Chem. 2009;284:1884–1895. doi: 10.1074/jbc.M803641200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Román-Hernández G., Grant R.A., Sauer R.T., Baker T.A. Molecular basis of substrate selection by the N-end rule adaptor protein ClpS. Proc. Natl. Acad. Sci. USA. 2009;106:8888–8893. doi: 10.1073/pnas.0903614106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hu R.G., Brower C.S., Wang H., Davydov I.V., Sheng J., Zhou J., Kwon Y.T., Varshavsky A. Arginyltransferase, its specificity, putative substrates, bidirectional promoter, and splicing-derived isoforms. J. Biol. Chem. 2006;281:32559–32573. doi: 10.1074/jbc.M604355200. [DOI] [PubMed] [Google Scholar]
- 149.Wang J., Han X., Saha S., Xu T., Rai R., Zhang F., Wolf Y.I., Wolfson A., Yates J.R., 3rd, Kashina A. Arginyltransferase is an ATP-independent self-regulating enzyme that forms distinct functional complexes in vivo. Chem. Biol. 2011;18:121–130. doi: 10.1016/j.chembiol.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yoo Y.D., Mun S.R., Ji C.H., Sung K.W., Kang K.Y., Heo A.J., Lee S.H., An J.Y., Hwang J., Xie X.Q., et al. N-terminal arginylation generates a bimodal degron that modulates autophagic proteolysis. Proc. Natl. Acad. Sci. USA. 2018;115:E2716–E2724. doi: 10.1073/pnas.1719110115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cha-Molstad H., Yu J.E., Feng Z., Lee S.H., Kim J.G., Yang P., Han B., Sung K.W., Yoo Y.D., Hwang J., et al. p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat. Commun. 2017;8:102. doi: 10.1038/s41467-017-00085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cha-Molstad H., Sung K.S., Hwang J., Kim K.A., Yu J.E., Yoo Y.D., Jang J.M., Han D.H., Molstad M., Kim J.G., et al. Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat. Cell Biol. 2015;17:917–929. doi: 10.1038/ncb3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kwon D.H., Park O.H., Kim L., Jung Y.O., Park Y., Jeong H., Hyun J., Kim Y.K., Song H.K. Insights into degradation mechanism of N-end rule substrates by p62/SQSTM1 autophagy adapter. Nat. Commun. 2018;9:3291. doi: 10.1038/s41467-018-05825-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhang Y., Mun S.R., Linares J.F., Ahn J., Towers C.G., Ji C.H., Fitzwalter B.E., Holden M.R., Mi W., Shi X., et al. ZZ-dependent regulation of p62/SQSTM1 in autophagy. Nat. Commun. 2018;9:4373. doi: 10.1038/s41467-018-06878-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Aksnes H., Hole K., Arnesen T. Molecular, cellular, and physiological significance of N-terminal acetylation. Int. Rev. Cell Mol. Biol. 2015;316:267–305. doi: 10.1016/bs.ircmb.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 156.Aksnes H., Drazic A., Marie M., Arnesen T. First things first: vital protein marks by N-terminal acetyltransferases. Trends Biochem. Sci. 2016;41:746–760. doi: 10.1016/j.tibs.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 157.Shemorry A., Hwang C.S., Varshavsky A. Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol. Cell. 2013;50:540–551. doi: 10.1016/j.molcel.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Park S.E., Kim J.M., Seok O.H., Cho H., Wadas B., Kim S.Y., Varshavsky A., Hwang C.S. Control of mammalian G protein signaling by N-terminal acetylation and the N-end rule pathway. Science. 2015;347:1249–1252. doi: 10.1126/science.aaa3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kats I., Khmelinskii A., Kschonsak M., Huber F., Knieß R.A., Bartosik A., Knop M. Mapping degradation signals and pathways in a eukaryotic N-terminome. Mol. Cell. 2018;70:488–501.e5. doi: 10.1016/j.molcel.2018.03.033. [DOI] [PubMed] [Google Scholar]
- 160.Lin H.C., Yeh C.W., Chen Y.F., Lee T.T., Hsieh P.Y., Rusnac D.V., Lin S.Y., Elledge S.J., Zheng N., Yen H.C.S. C-terminal end-directed protein elimination by CRL2 ubiquitin ligases. Mol. Cell. 2018;70:602–613.e3. doi: 10.1016/j.molcel.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Koren I., Timms R.T., Kula T., Xu Q., Li M.Z., Elledge S.J. The eukaryotic proteome is shaped by E3 ubiquitin ligases targeting C-terminal degrons. Cell. 2018;173:1622–1635.e14. doi: 10.1016/j.cell.2018.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mnatsakanyan R., Markoutsa S., Walbrunn K., Roos A., Verhelst S.H.L., Zahedi R.P. Proteome-wide detection of S-nitrosylation targets and motifs using bioorthogonal cleavable-linker-based enrichment and switch technique. Nat. Commun. 2019;10:2195. doi: 10.1038/s41467-019-10182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Seth D., Hess D.T., Hausladen A., Wang L., Wang Y.J., Stamler J.S. A multiplex enzymatic machinery for cellular protein S-nitrosylation. Mol. Cell. 2018;69:451–464.e6. doi: 10.1016/j.molcel.2017.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chung K.K.K., Thomas B., Li X., Pletnikova O., Troncoso J.C., Marsh L., Dawson V.L., Dawson T.M. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 165.Iglesias M.J., Terrile M.C., Correa-Aragunde N., Colman S.L., Izquierdo-Álvarez A., Fiol D.F., París R., Sánchez-López N., Marina A., Calderón Villalobos L.I.A., et al. Regulation of SCF(TIR1/AFBs) E3 ligase assembly by S-nitrosylation of Arabidopsis SKP1-like1 impacts on auxin signaling. Redox Biol. 2018;18:200–210. doi: 10.1016/j.redox.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kumar R., Jangir D.K., Verma G., Shekhar S., Hanpude P., Kumar S., Kumari R., Singh N., Sarovar Bhavesh N., Ranjan Jana N., Kanti Maiti T. S-nitrosylation of UCHL1 induces its structural instability and promotes alpha-synuclein aggregation. Sci. Rep. 2017;7:44558. doi: 10.1038/srep44558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Amal H., Gong G., Gjoneska E., Lewis S.M., Wishnok J.S., Tsai L.H., Tannenbaum S.R. S-nitrosylation of E3 ubiquitin-protein ligase RNF213 alters non-canonical Wnt/Ca+2 signaling in the P301S mouse model of tauopathy. Transl. Psychiatry. 2019;9:44. doi: 10.1038/s41398-019-0388-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kim S., Wing S.S., Ponka P. S-nitrosylation of IRP2 regulates its stability via the ubiquitin-proteasome pathway. Mol. Cell Biol. 2004;24:330–337. doi: 10.1128/MCB.24.1.330-337.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gupta A., Anjomani-Virmouni S., Koundouros N., Dimitriadi M., Choo-Wing R., Valle A., Zheng Y., Chiu Y.H., Agnihotri S., Zadeh G., et al. PARK2 depletion connects energy and oxidative stress to PI3K/Akt activation via PTEN S-nitrosylation. Mol. Cell. 2017;65:999–1013.e7. doi: 10.1016/j.molcel.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Azad N., Vallyathan V., Wang L., Tantishaiyakul V., Stehlik C., Leonard S.S., Rojanasakul Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 2006;281:34124–34134. doi: 10.1074/jbc.M602551200. [DOI] [PubMed] [Google Scholar]
- 171.Chanvorachote P., Nimmannit U., Stehlik C., Wang L., Jiang B.H., Ongpipatanakul B., Rojanasakul Y. Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through S-nitrosylation and inhibition of Bcl-2 ubiquitination. Cancer Res. 2006;66:6353–6360. doi: 10.1158/0008-5472.CAN-05-4533. [DOI] [PubMed] [Google Scholar]
- 172.Zhao Y., Jensen O.N. Modification-specific proteomics: strategies for characterization of post-translational modifications using enrichment techniques. Proteomics. 2009;9:4632–4641. doi: 10.1002/pmic.200900398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hattori T., Koide S. Next-generation antibodies for post-translational modifications. Curr. Opin. Struct. Biol. 2018;51:141–148. doi: 10.1016/j.sbi.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dunphy K., Dowling P., Bazou D., O'Gorman P. Current methods of post-translational modification analysis and their applications in blood cancers. Cancers. 2021;13:1930. doi: 10.3390/cancers13081930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Egelhofer T.A., Minoda A., Klugman S., Lee K., Kolasinska-Zwierz P., Alekseyenko A.A., Cheung M.S., Day D.S., Gadel S., Gorchakov A.A., et al. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 2011;18:91–93. doi: 10.1038/nsmb.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rothbart S.B., Dickson B.M., Raab J.R., Grzybowski A.T., Krajewski K., Guo A.H., Shanle E.K., Josefowicz S.Z., Fuchs S.M., Allis C.D., et al. An interactive database for the assessment of histone antibody specificity. Mol. Cell. 2015;59:502–511. doi: 10.1016/j.molcel.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Witze E.S., Old W.M., Resing K.A., Ahn N.G. Mapping protein post-translational modifications with mass spectrometry. Nat. Methods. 2007;4:798–806. doi: 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.
The table contains the substrate name, the modification site(s), the effector enzymes, and the functional/biological outcome.