

Abstract
Background & Aims:
Despite recent progress, non-invasive tests for the diagnostic assessment and monitoring of non-alcoholic fatty liver disease (NAFLD) remain an unmet need. Herein, we aimed to identify diagnostic signatures of the key histological features of NAFLD.
Methods:
Using modified-aptamer proteomics, we assayed 5,220 proteins in each of 2,852 single serum samples from 636 individuals with histologically confirmed NAFLD. We developed and validated dichotomized protein-phenotype models to identify clinically relevant severities of steatosis (grade 0 vs. 1–3), hepatocellular ballooning (0 vs. 1 or 2), lobular inflammation (0–1 vs. 2–3) and fibrosis (stages 0–1 vs. 2–4).
Results:
The AUCs of the four protein models, based on 37 analytes (18 not previously linked to NAFLD), for the diagnosis of their respective components (at a clinically relevant severity) in training/paired validation sets were: fibrosis (AUC 0.92/0.85); steatosis (AUC 0.95/0.79), inflammation (AUC 0.83/0.72), and ballooning (AUC 0.87/0.83). An additional outcome, at-risk NASH, defined as steatohepatitis with NAFLD activity score ≥4 (with a score of at least 1 for each of its components) and fibrosis stage ≥2, was predicted by multiplying the outputs of each individual component model (AUC 0.93/0.85). We further evaluated their ability to detect change in histology following treatment with placebo, pioglitazone, vitamin E or obeticholic acid. Component model scores significantly improved in the active therapies vs. placebo, and differential effects of vitamin E, pioglitazone, and obeticholic acid were identified.
Conclusions:
Serum protein scanning identified signatures corresponding to the key components of liver biopsy in NAFLD. The models developed were sufficiently sensitive to characterize the longitudinal change for three different drug interventions. These data support continued validation of these proteomic models to enable a “liquid biopsy”-based assessment of NAFLD.
Clinical Trial Number:
Not applicable.
Keywords: Nonalcoholic fatty liver disease (NAFLD), Nonalcoholic steatohepatitis (NASH), NAFLD activity score (NAS), fibrosis stage, cirrhosis, steatohepatitis, steatosis, hepatocellular ballooning, lobular inflammation, fibrosis, proteomics, aptamers
Graphical Abstract
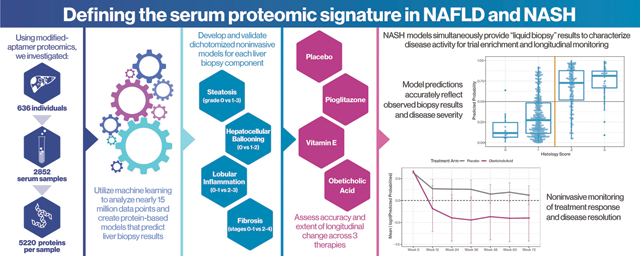
Introduction
Non-alcoholic fatty liver disease (NAFLD) affects 20–30% of the adult population.1 The majority have a relatively stable form, i.e. non-alcoholic fatty liver (NAFL), while those with non-alcoholic steatohepatitis (NASH) are more likely to progress to cirrhosis.2 The burden of end-stage liver disease due to NAFLD is expected to increase 2- to 3-fold by 2030.3,4 These data underscore the need to identify those who are likely to progress or are progressing so that they may be targeted for treatment.
Liver histology is the reference standard for identification of those with NAFLD at increased risk of liver-related outcomes.5 There are several limitations of a biopsy-based approach, including its invasive nature and attendant risks,6 sampling and observer variability, and cost, which render it suboptimal for application in routine clinical practice.2 This provides a strong rationale for the development of non-invasive tests (NITs) for the assessment of NAFLD. While many NITs exist, only the enhanced liver fibrosis test has been approved as a prognostic biomarker and the development of NITs for various purposes in NAFLD remains an unmet need.
Large-scale serum/plasma protein scanning has recently become available for identification of changes in the proteome in disease states.7 In a preliminary single-center study, this enabled identification of a circulating protein signature associated with advanced fibrosis in patients with NASH.8 This study is limited by its single-center nature and small sample size, which can increase the risk of missing relevant biomarkers with smaller effect sizes.
We therefore conducted a study to identify and validate a proteomic signature, using the SomaScan® platform for the following a priori defined contexts of use: (1) for the diagnosis of individual components of NAFLD at a clinically relevant severity, and (2) for disease monitoring to identify features reflective of disease activity and fibrosis over time with and without specific drug intervention.
Materials and methods
This study was an ancillary study of the NIDDK NASH Clinical Research Network (CRN) under a collaborative agreement with SomaLogic Operating Co. The proposal was reviewed and approved by both the Ancillary Studies and Steering Committees of the CRN and NIDDK. The data analysis was performed by SomaLogic and verified by the NASH CRN investigators. The investigators take full responsibility for the contents of this manuscript. All participants whose samples were used provided written informed consent for their biosamples to be used for research. Results are reported in alignment with TRIPOD guidance for reporting biomarker data.9
Study population
Serum samples were from adult participants in the NASH CRN NAFLD Database (DB), Adult DB2 registries, PIVENS and FLINT clinical trials (NCT#01030484, NCT#00063622, NCT# 01265498), enrolled from 2004 through 2014 at eight tertiary sites. The DB and DB2 registries are non-interventional long-term follow-up studies of patients with histologically confirmed NAFLD. The PIVENS trial tested the efficacy of pioglitazone or vitamin E vs. placebo over a 96-week treatment period whereas the FLINT trial evaluated the efficacy of obeticholic acid vs. placebo over 72 weeks.10,11 Our study required participants to have biopsy-confirmed NAFLD and corresponding serum samples. An overview of the sampling and biopsy schedule is shown in Table S1 and Fig. S1.
To address aim 1 (diagnosis of histological components of NASH at a clinically relevant severity) a training set comprising 559 unique serum samples from 315 of the trial participants and 244 participants in the Natural History Studies (234 baseline DB/DB2 and 10 who had previously participated in PIVENS but were not included in the trial sample sets noted above) was used. Half the samples from PIVENS and FLINT were baseline samples and temporally related to baseline biopsy histology, while half the samples were end-of-treatment samples and temporally related to the end of treatment biopsy. The rationale for this was both to capture a mix of populations with and without prior therapy and to maximize the likelihood that models developed would be impervious to any potential treatment effects.
Two separate and independent validation sets were used to assess model performance: a “paired validation” set included (1) the remaining baseline and end of treatment samples from each of the PIVENS and FLINT participants (n = 392) that were not used in the training model, and (2) an independent “hold-out” model validation set of samples from 77 trial participants not included in the training data or paired validation data set. These sample sets were also used for the post hoc analyses, i.e. the diagnosis of cirrhosis and at-risk NASH.
To address aim 2 (disease monitoring), a total of up to seven serum samples (see Table S1 for sampling schedule) were available per participant from PIVENS and FLINT, including those obtained at baseline and end of treatment with accompanying biopsies, and additional interval time-points defined by protocol. Approximately 91% of PIVENS and 99% of FLINT participants had six or seven samples available, resulting in 1,333 evaluable samples from PIVENS and 1,275 samples from FLINT.
Liver histology assessment
The liver biopsies were read centrally by the NASH CRN Pathology Committee, who were masked to all clinical and proteomic data. The protocol and methodology used by the Committee as well as the case definitions have been described previously.12,13 It is also known that those with NASH and fibrosis stage ≥2 have higher all-cause and liver-related mortality.14 Further, disease activity drives fibrosis.2,15 We therefore defined a sub-phenotype of NAFLD with steatohepatitis, NAFLD activity score (NAS) ≥4 (with a score of at least 1 for each of its components) and fibrosis stage ≥2 as “at-risk” NASH. Fibrosis stage ≥2 was referred to as clinically significant fibrosis.16
Proteomic analyses
The modified aptamer binding reagents,17 SomaScan assay,18,19 and its performance characteristics20 have been described previously. The median intra- and inter-assay coefficients of variation are ~5%20 and median lower limit of detection is in the femtomolar range. The proteins were assayed blinded. Following normalization, calibration, and data quality control processes, the proteomic data were provided to the NASH CRN before the clinical and biopsy data were made available for machine learning, as described in the next section.
Model building and statistical analyses
Aim 1: cross-sectional protein-based model development of liver histology
For aim 1, the histological readout was dichotomized based on Pathology Committee consensus as follows: steatosis (grade 0 vs. 1, 2 or 3, training n = 72 vs. 486), lobular inflammation (0 or 1 vs. 2 or 3, training n = 361 vs. 198), hepatocellular ballooning (0 vs. 1 or 2, training n = 244 vs. 315) and fibrosis (stage 0 or 1 vs. 2, 3 or 4, training n = 330 vs. 228). The dichotomization of fibrosis stages was based on literature indicating increased risk of liver-related outcomes in such individuals.14 Proteome-based models were developed to predict these dichotomized histological readouts. These dichotomized models were developed individually for each histological component because they could not only be used to infer the presence of active disease and clinically significant fibrosis (stage ≥2) but also NASH resolution, which requires ballooning resolution and lobular inflammation to be absent or minimal (grades 0–1).21 All analyses were done on a complete-case basis.
Using the training set, univariate t-tests were used to assess associations of analytes with each histological parameter. Multiple testing correction was completed using the Benjamini-Hochberg procedure for the false discovery rate (FDR).22 As an initial feature selection step, analytes were filtered based on the minimal FDR-corrected p values using an alpha of 0.1. Remaining analytes were then centered and scaled to enable standardized coefficient values in methods that utilized regularization. After univariate filtering, a multi-variable feature selection method, stability selection23 with an L1-logistic regression kernel, was used to identify candidate lists of analytes for each histological component. Final binary prediction models consisted of an elastic net logistic regression classifier that utilized a mixture of L1 and L2 regularization24 to do a final feature reduction and to account for correlated features. Repeated k-fold cross-validation with 5 repeats of 10 folds was performed on the training set to assess initial performance, potential overfitting, and to select a final model for each of the four biopsy components individually (steatosis, inflammation, ballooning and fibrosis). Specifically, the training data was split into 10 “folds”, then a model is fit to 9 of the folds and performance assessed on the 10th. This analysis was repeated 10 times, where each fold becomes the assessment set. The entire process was repeated 5 times.
The performance of the model was further evaluated in those where samples were taken from baseline visits in clinical trials and end of treatment visits for both placebo and active treatment arms to determine if they were affected by prior treatment. The DeLong’s test for differences in AUC with a two-sided alternative was used to identify any statistically significant differences in model performance at baseline vs. end of treatment.
Aim 2: proteomic models for monitoring the course of disease over time
The characterization and monitoring of the impact of active therapy vs. placebo over time was performed using the output of models developed in aim 1 for each longitudinal sample. Though the models were trained on dichotomized scores, the output was a continuous score reflecting the probability of being in the positive category. These values were used to assess the ability to monitor change. For determination of significant treatment effects over time, linear mixed effects models that specifically explored the interaction effect of treatment by time were used with the logit-transformed predicted probability as the outcome measure.
Post hoc analyses that were not part of the original plan of analysis for this study
Diagnosing at-risk NASH
A specific model to mimic the pathologists’ diagnosis of NASH was not planned because of the diverse permutations of individual histological findings that could be associated with a NASH diagnosis. Instead, a combination of the four component models (multiplication of each of the predicted probability model outputs) was used to assess the presence of at-risk NASH, where a positive result was defined as NAS ≥4 with a score of at least 1 each for steatosis, ballooning and inflammation and fibrosis stage ≥2. The analysis was performed with the same training (n = 558), holdout (n = 77) and paired validation (n = 391) data sets as the histology component models and the prevalence of at-risk NASH was 31%, 38%, and 39%, respectively.
Diagnosis of cirrhosis (F4)
For the exploratory diagnosis of cirrhosis, we evaluated whether using a different threshold for the probability output of the dichotomous fibrosis model developed in aim 1, where the negative class represents individuals predicted to have a fibrosis stage of 0–3 and the positive class represents stages 4, could be used to identify participants with F4 results. Results are presented for a post hoc threshold predicted probability greater than 0.95. All analyses were completed using R (v3.5.2) and various R packages including the tidyverse, caret, tidy-models, and glmnet.
Results
A total of 332 participants from the DB and DB2 studies and 209 and 197 participants from PIVENS and FLINT trials were screened for this analysis. After removing duplicate and non-evaluable samples, there were 636 unique participants, including 234 from DB/DB2, 215 from PIVENS and 187 from FLINT (Fig. 1 and Table S2). The mean age and sex distribution across these modeling subsets were not significantly different compared to the Natural History cohort (Table S2). At baseline, the adult participants’ mean age was 48.6 (12.1) years, 37% were male, 13% were of Hispanic ethnicity and predominantly Caucasian race, reflecting the patient populations seen at the participating centers.
Fig. 1.
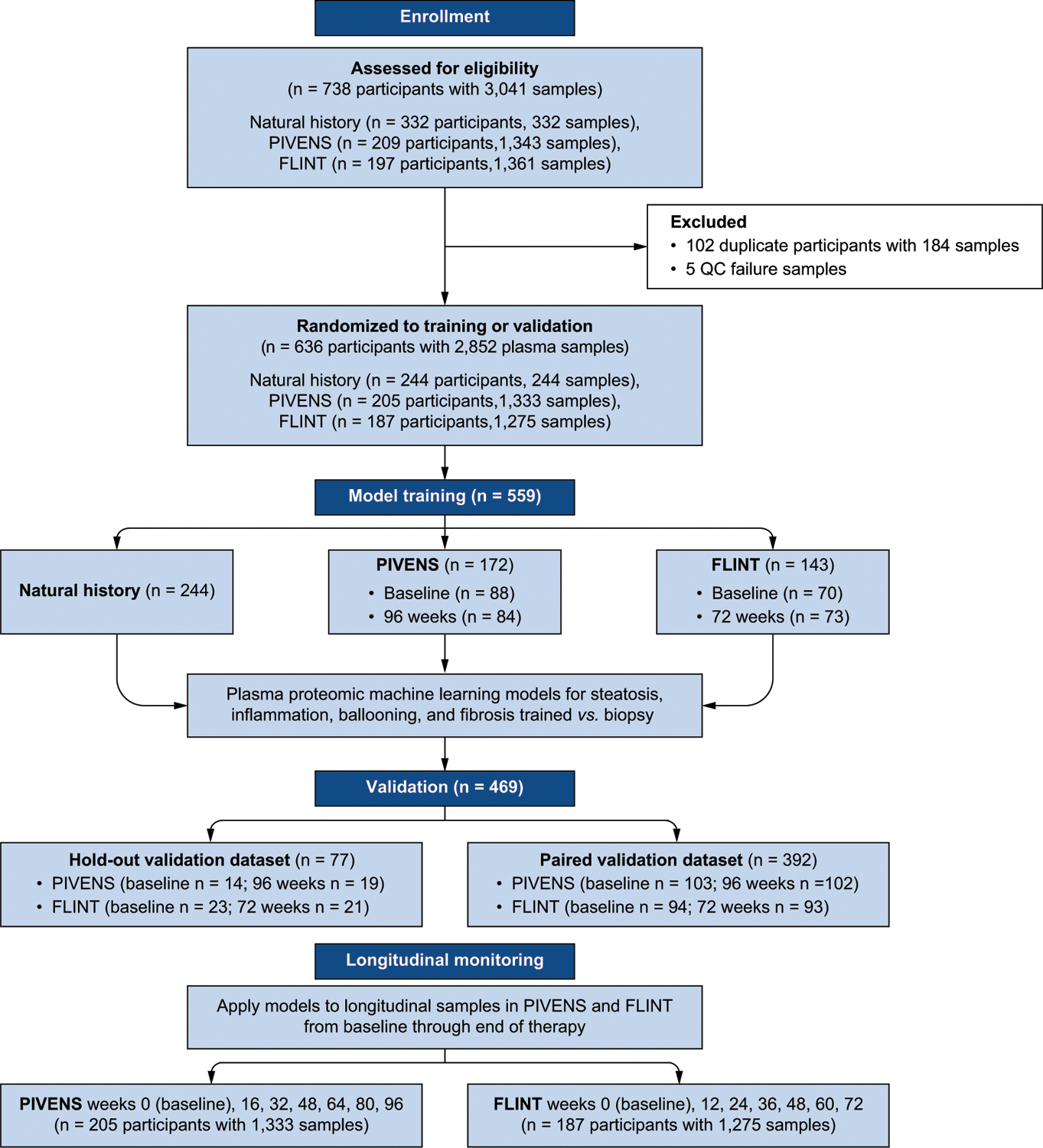
Cohort derivation, validation and longitudinal assessment.
The mean interval between baseline serum samples and biopsy was 37 days, with a maximum interval of 6 months. The mean prevalence of type 2 diabetes (27%), dyslipidemia (tri-glycerides >140 mg/dl and/or HDL-cholesterol <40 mg/dl; 59%) or hypertension (51%) was similar to that in other reported NAFLD populations,25 in addition to obesity (BMI: 34 kg/m2, weight: 97 kg). The mean [SD] baseline liver-related enzyme values (alanine aminotransferase 76 U/L [48], aspartate aminotransferase 53 U/L [32], alkaline phosphatase 84 U/L [52] and gamma-glutamyltransferase 72 U/L [128]) were also similar to other reported NAFLD populations and included large proportions of definite NASH (60%), bridging fibrosis (19%), and high activity (NAS >4 in 49%). Only 3% of participants had cirrhosis.
Cross-sectional protein-based model development of liver histology (aim 1)
Initial univariate analysis using a FDR with alpha = 0.1 yielded a large number of potential targets, including 532, 1,408, 809, and 2,201 proteins for steatosis, ballooning, lobular inflammation, and fibrosis, respectively (Table S3). Table 1 shows the analytes selected for inclusion first by stability selection and further by elastic net regularization in the four final models in rank order of their statistical association with the endpoint. There were 12 for steatosis, 14 for inflammation, 5 for ballooning, and 8 for fibrosis. Thirty-seven unique analytes were featured in the final four models. There was little overlap between analytes across models, with only two analytes shared between two models (PTGR1 in steatosis and ballooning models and ADAMTSL2 in ballooning and fibrosis models) and none in three or more. Fourteen of 37 proteins have previously been associated with various aspects of NAFLD whereas 18 proteins were previously unrecognized in relation to NAFLD (Table S4). The relationship of the predicted probabilities for each proteomic model to the ordinal biopsy result for model training and validation are shown in Figs 2 and 3 and its diagnostic performance metrics are shown in Table 2.
Table 1.
Proteins included in each model.
| Entrez Gene Symbol | Target full name | UniProt | t-statistic | p value |
|---|---|---|---|---|
| Steatosis | ||||
| PTGR1 | Prostaglandin reductase 1 | Q14914 | 11.20 | 2.2e-16 |
| GUSB | Beta-glucuronidase | P08236 | 9.29 | 5.8e-15 |
| INHBC | Inhibin beta C chain | P55103 | 6.39 | 7.1e-09 |
| HEXB | Beta-hexosaminidase subunit beta | P07686 | 6.30 | 1.0e-08 |
| RECQL | ATP-dependent DNA helicase Q1 | P46063 | 5.51 | 2.7e-07 |
| BPIFB1 | BPI fold-containing family B member 1 | Q8TDL5 | −5.07 | 2.2e-06 |
| GH2 | Growth hormone variant | P01242 | −4.40 | 3.0e-05 |
| INSL5 | Insulin-like peptide INSL5 | Q9Y5Q6 | 4.35 | 2.5e-05 |
| FABP12 | Fatty acid-binding protein 12 | A6NFH5 | 4.34 | 2.2e-05 |
| ERN1 | Serine/threonine-protein kinase/endoribonuclease IRE1 | O75460 | 4.10 | 6.0e-05 |
| GRID2 | Glutamate receptor ionotropic, delta-2 | O43424 | −3.86 | 2.2e-04 |
| CNDP1 | Beta-Ala-His dipeptidase | Q96KN2 | 3.62 | 5.0e-04 |
| Inflammation | ||||
| ACY1 | Aminoacylase-1 | Q03154 | 12.12 | 2.2e-16 |
| TXNRD1 | Thioredoxin reductase 1, cytoplasmic | Q16881 | 11.36 | 2.2e-16 |
| FCGR3B | Low affinity immunoglobulin gamma Fc region receptor III-B | O75015 | 8.29 | 1.5e-15 |
| PCOLCE2 | Procollagen C-endopeptidase enhancer 2 | Q9UKZ9 | −6.03 | 3.5e-09 |
| ADIPOQ | Adiponectin | Q15848 | −4.59 | 5.8e-06 |
| RPN1 | Dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit 1 | P04843 | 4.46 | 1.1e-05 |
| GSTZ1 | Maleylacetoacetate isomerase | O43708 | 4.39 | 1.5e-05 |
| PYY | Peptide YY | P10082 | 4.30 | 2.3e-05 |
| CCL23 | C-C motif chemokine 23 | P55773 | −4.27 | 2.5e-05 |
| ACP1 | Low molecular weight phosphotyrosine protein phosphatase | P24666 | 4.02 | 6.9e-05 |
| CTCF | Transcriptional repressor CTCF | P49711 | −3.85 | 1.3e-04 |
| SAA2 | Serum amyloid A-2 protein | P0DJI9 | 3.78 | 1.9e-04 |
| C1orf198 | Uncharacterized protein C1orf198 | Q9H425 | −3.67 | 2.7e-04 |
| TACSTD2 | Tumor-associated calcium signal transducer 2 | P09758 | −3.46 | 5.9e-04 |
| Hepatocyte ballooning | ||||
| AKR1B10 | Aldo-keto reductase family 1 member B10 | O60218 | 15.94 | 2.2e-16 |
| PTGR1 | Prostaglandin reductase 1 | Q14914 | 15.75 | 2.2e-16 |
| ADAMTSL2 | ADAMTS-like protein 2 | Q86TH1 | 15.10 | 2.2e-16 |
| CNN2 | Calponin-2 | Q99439 | 7.38 | 6.0e-13 |
| CTLA4 | Cytotoxic T-lymphocyte protein 4 | P16410 | −3.20 | 1.5e-03 |
| Fibrosis | ||||
| ADAMTSL2 | ADAMTS-like protein 2 | Q86TH1 | 18.48 | 2.2e-16 |
| COLEC11 | Collectin-11 | Q9BWP8 | 15.72 | 2.2e-16 |
| NFASC | Neurofascin | O94856 | 14.78 | 2.2e-16 |
| C7 | Complement component C7 | P10643 | 11.67 | 2.2e-16 |
| FCRL3 | Fc receptor-like protein 3 | Q96P31 | 7.15 | 6.2e-12 |
| KDR | Vascular endothelial growth factor receptor 2 | P35968 | −7.02 | 8.7e-12 |
| PLOD3 | Procollagen-lysine,2-oxoglutarate 5-dioxygenase 3 | O60568 | 6.32 | 6.2e-10 |
| WNT5A | Protein Wnt-5a | P41221 | 4.42 | 1.2e-05 |
Proteins are listed in rank order of the strength of their univariate signal with the endpoint (smallest to largest p value). p values are calculated from a two-sided t-test where a positive t-statistic indicates the mean analyte value for the case group was greater than the mean analyte value for the control group*.
Histology dichotomy for control vs. case groups are steatosis (grade 0 vs. 1, 2 or 3), lobular inflammation (0 or 1 vs. 2 or 3), hepatocellular ballooning (0 vs. 1 or 2) and fibrosis (stage 0 or 1 vs. 2, 3 or 4).
Fig. 2. Model predictions vs. observed biopsy results in the training, hold-out validation, and paired validation data sets.
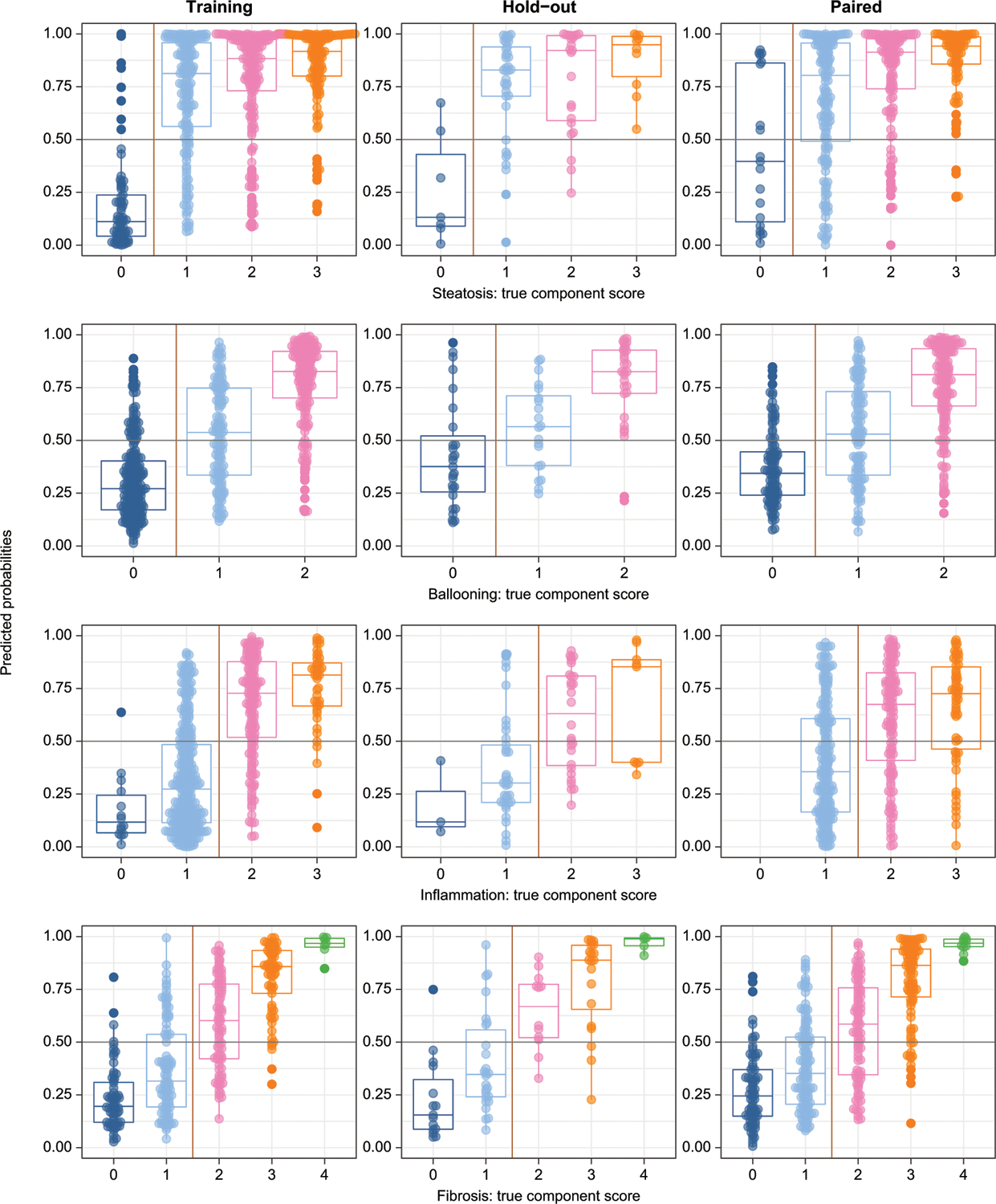
Models were trained on dichotomized variables (left and right of vertical yellow lines). Probability outputs of the models (probability of any given sample being in the positive class) are displayed by the original biopsy grade. The decision threshold for all models was greater than or equal to 0.5 (horizontal gray lines). Boxes show medians, 25th and 75th centiles. By random chance there were no zero inflammation scores in the paired validation set. Training: left panels; Hold-out validation: center panels; Paired validation: Right panels.
Fig. 3. Model predictions for at-risk NASH vs. biopsy-based composite outcome of NAS ≥4 and fibrosis ≥2 in training, hold-out validation, and paired validation data sets.
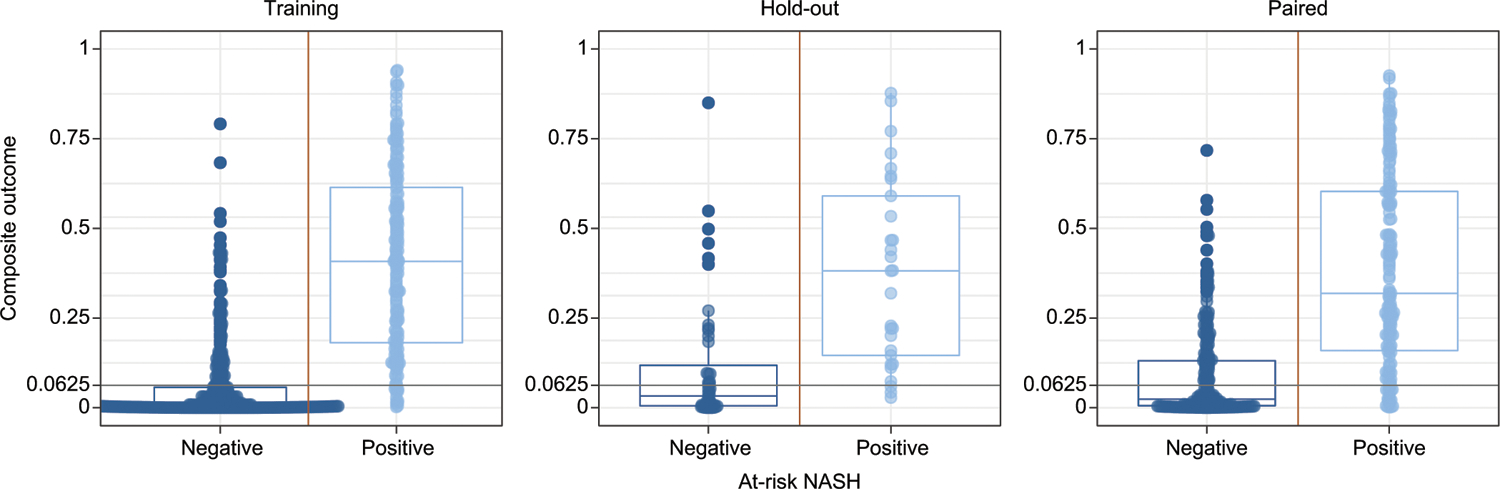
At-risk NASH predictions are calculated by multiplying the predicted probability of the models. The decision threshold was set at 0.0625 (0.54, the equivalent of multiplying the decision thresholds for each model). Yellow vertical lines indicate the binary class threshold and gray horizontal lines indicate the model decision threshold. Boxes show medians, 25th and 75th centiles. Prevalence of at-risk NASH was 31%, 38% and 39% for training/holdout/validation data sets respectively. Training: left panels; Hold-out validation: center panels; Paired validation: Right panels.
Table 2.
Performance metrics in training and the two different validation approaches.
| Model | Data set | Sample size | Cases* | AUC | Sensitivity | Specificity | Accuracy |
|---|---|---|---|---|---|---|---|
| Steatosis | |||||||
| Training | 558 | 486 | 0.95 | 0.86 | 0.88 | 0.86 | |
| Hold-out validation* | 78 | 70 | 0.67 | 0.80 | 0.50 | 0.77 | |
| Paired validation | 392 | 373 | 0.79 | 0.84 | 0.58 | 0.67 | |
| Lobular inflammation | |||||||
| Training | 559 | 198 | 0.83 | 0.78 | 0.76 | 0.76 | |
| Hold-out validation | 77 | 37 | 0.79 | 0.59 | 0.78 | 0.69 | |
| Paired validation | 392 | 179 | 0.72 | 0.69 | 0.65 | 0.67 | |
| Ballooning | |||||||
| Training | 559 | 315 | 0.87 | 0.77 | 0.82 | 0.78 | |
| Hold-out validation | 77 | 56 | 0.71 | 0.63 | 0.71 | 0.65 | |
| Paired validation | 392 | 285 | 0.83 | 0.75 | 0.79 | 0.76 | |
| Fibrosis | |||||||
| Training | 558 | 228 | 0.92 | 0.81 | 0.85 | 0.83 | |
| Hold-out validation | 77 | 38 | 0.83 | 0.79 | 0.72 | 0.75 | |
| Paired validation | 391 | 193 | 0.85 | 0.76 | 0.77 | 0.76 | |
| At-risk NASH (4 model outputs multiplied) | |||||||
| Training | 558 | 171 | 0.93 | 0.92 | 0.77 | 0.82 | |
| Hold-out validation | 77 | 29 | 0.84 | 0.90 | 0.60 | 0.71 | |
| Paired validation | 391 | 154 | 0.85 | 0.87 | 0.63 | 0.73 | |
Performance of each model was evaluated using AUC, sensitivity, specificity, and accuracy with the decision threshold at 0.5.
Histology dichotomy for control vs. case groups are steatosis (grade 0 vs. 1, 2 or 3), lobular inflammation (0 or 1 vs. 2 or 3), hepatocellular ballooning (0 vs. 1 or 2) and fibrosis (stage 0 or 1 versus 2, 3 or 4). Number of cases are shown for each data set
The AUC of the models for steatosis (0.67–0.95), lobular inflammation (0.72–0.83), hepatocellular ballooning (0.71–0.87) and fibrosis (0.83–0.92) all had higher AUCs in the training sets but expected lower values in validation sets (a typical pattern in machine learning). Further, the AUCs of the models for inflammation, hepatocellular ballooning, and fibrosis were similar when using baseline and end of study samples from the placebo and the active treatment arms, indicating that the models were not affected by the treatments provided in the trials (Table S5). The relatively large drop for the steatosis model may relate to the poor representation in the hold-out set; specifically, there were only eight individuals with no steatosis resulting in the observed drop in AUC (Table 2).
The construct specificity of the individual models was further evaluated by measuring the performance of the model for its intended purpose vs. its performance for the diagnosis of other features of NAFLD. For example, a model for steatosis would be expected to perform well for steatosis but not as well to evaluate another feature, e.g. hepatocellular ballooning.
As expected, this resulted in a decrease in average accuracy of the models (between 10–30% decrease) and odds ratio (Fisher’s exact test) obtained from applying the proteomic models to their intended vs. non-intended histological features of interest (Table S6). For example, when the fibrosis model, with an accuracy of 78%, is used to assess inflammation, ballooning or steatosis, the predicted accuracy drops to 52–66%. Conversely, when inflammation, ballooning or steatosis models are applied to fibrosis scores, the accuracy drops to 54–73%. Similarly, comparing fibrosis biopsy results from histology to ballooning, inflammation, or steatosis biopsy results, the accuracy drops to 51–65%.
Proteomic models for monitoring the course of disease over time (aim 2)
Predicted probabilities were computed using each of the four proteomic models for the individual histologic characteristics of NASH for all seven samples available (baseline, end of treatment, interim visits) for each participant in PIVENS and the FLINT trial. Linear mixed effects models to test changes in logit-transformed predicted probabilities of each of the proteomic models by group over time (i.e., the treatment group × time interaction effect) were developed for each histological component. The patterns for the predicted probabilities (mean and 95% CIs) are shown in Fig. 4.
Fig. 4. Predictions of protein models in longitudinal serum samples.
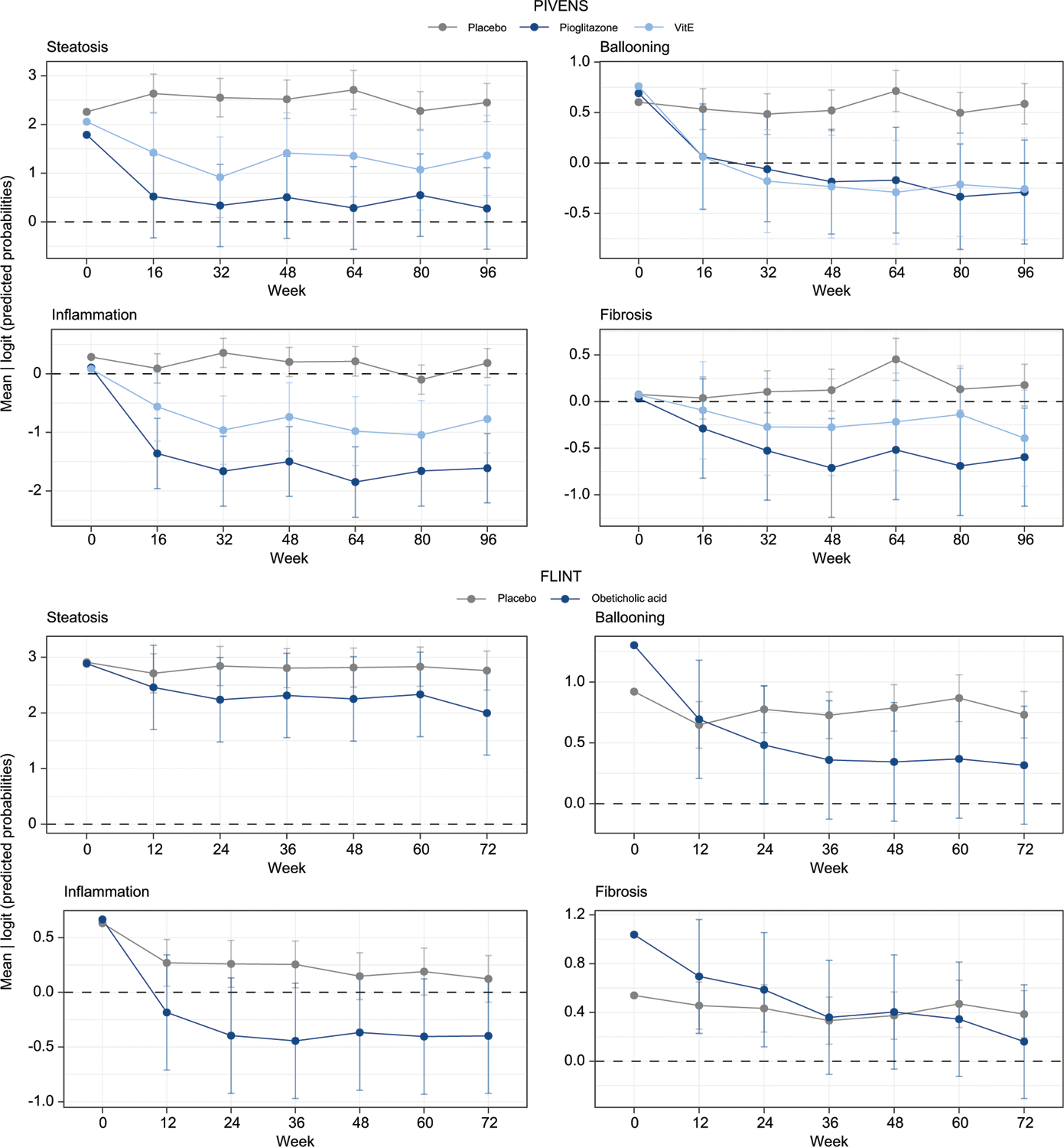
Results are for each component for the mixed effects models using continuous predicted probability (logit-transformed) for each study. Higher scores reflect greater probability of being in the positive class. The black dashed line corresponds to the decision cut-off at 0.5. The placebo groups are shown in gray. The active groups are shown in blue and teal. The 95% CIs of the mean predicted probabilities across all samples is shown for each group at each single time point. Confidence intervals were calculated using the standard error estimated from the mixed effects models with week and treatment group fixed effects and a random subject effect.
Within the PIVENS cohort, there was a significant interaction between time and treatment across all four models (fibrosis: χ211,23 = 59.27, p value = 3.06e-08; steatosis: χ211,23 = 44.48, p value = 1.27e-05; ballooning: χ211,23 = 73.84, p value = 6.1e-11; inflammation: χ211,23 = 109.13, p value = 8.89e-18). For fibrosis, the average difference in change over time between placebo and pioglitazone or vitamin E was significant starting at week 32 (p = 1.28e-03 and p = 1.26e-02, respectively). Both therapies had earlier significant impacts on steatosis, ballooning, and inflammation, starting at week 16 (steatosis: pPioglitazone = 1.27e-05, pVitamin E = 9.01e-03; ballooning: pPioglitazone = 2.68e-03, pVitamin E = 6.85e-04; inflammation: pPioglitazone = 4.94e-10, pVitamin E = 2.37e-02).
In the FLINT cohort, there was not a significant difference in the changes in steatosis probability score in the treatment group compared to placebo over time, in contrast to histological assessments which demonstrated a decrease in steatosis.11 There was however a significant decrease in the probability score of fibrosis in the treatment group over time (χ210,16 = 44.16, p = 6.87e-08) starting at week 24 (p = 1.00e-02), concordant with histological improvement at an individual participant level. Similar changes in the probability scores for lobular inflammation and ballooning were noted starting at week 12 (p = 3.41e-03, p = 2.68e-02).
Post hoc analyses
Identification of at-risk NASH
Using the multiplied outputs of each of the four component models, the AUC for identification of at-risk NASH was 0.93 in the training cohort and 0.84–0.85 in the validation cohorts (Table 2 and Fig. 3). In all cohorts, using a threshold of 0.0625 (0.54, the equivalent of multiplying the thresholds for each proteomic model), the sensitivity was high (0.87–0.92) with a specificity ranging from 0.6–0.77 at a prevalence of biopsy-confirmed at-risk NASH of 31–39%.
Identification of cirrhosis (F4)
Using a 95% probability cut-off in the fibrosis model, the current data set enabled identification of 17 of 25 individuals with cirrhosis in the training set and 2 of 3 and 7 of 10 in the two validation sets, respectively. The overall specificity for diagnosis of cirrhosis was 93–95% while 14% of those with stage 2–3 would be misclassified as having cirrhosis.
Discussion
The current study demonstrates that serum proteomic profiles are associated with various phenotypes of NAFLD and can also detect histological changes induced by multiple therapeutic interventions. These data must be considered in the context of the robustness of the assays, the biological relationship between the proteins identified and disease biology, and their overall diagnostic performance.
The robustness of the aptamer-based proteomic assays has been previously established.26 Notably, ALT – a well-known marker of liver injury – was not identified as a key analyte in the models. The classical tests for ALT do not distinguish between the ALT1 and ALT2 isoforms and report only on its functional activity, whereas the aptamer methods quantify ALT1 abundance only. The mean ALT values were low, and the limited range of ALT may have further decreased the ability of the models to relate ALT to histological severity in this study. Thus, the lack of correlation is not surprising.
The biological plausibility of the models is supported by the known connection of multiple proteins with metabolic stress and metabo-inflammation, supporting a linkage with NASH biology.27 Of note, AKR1B10 was also identified in transcriptomic analyses of NASH in other studies.28 Eighteen proteins were identified that have not been previously linked to NAFLD biology and could be further studied for their potential role in disease development and progression.
A key element in biomarker evaluation is its context of use, which defines the conditions in which it will be used. For the diagnostic context of use, the goal was to enrich the probability of having a high level of activity and/or stage 2 or higher fibrosis in a population with NAFLD. The study population was therefore appropriate. Yet, there is potential for some ascertainment bias given that the study was performed in a tertiary care setting. Spectrum bias is another important issue. While the proportions of individuals with and without inflammation, ballooning and fibrosis stages 0–3 were relatively balanced, this study is limited by a small number of individuals with cirrhosis. Also, since these studies were performed in a cohort with NAFLD, there were very few individuals with grade 1 or 0 steatosis. This potentially explains the drop in accuracy of the models for steatosis in the validation cohorts, in which only eight individuals had grade 0–1 steatosis. This limitation notwithstanding, the current data are foundational for further testing in an intended use population with risk factors for NAFLD.
Another potential source of error relates to the known inter- and intra-observer variability in histological assessment.2 In this study, the NASH CRN pathology committee followed a rigorous validated scoring of the biopsies with no knowledge of the proteomic findings. The histological reads thus meet the highest standards available for scientific rigor.
The AUC and overall accuracy of the proteomic signatures for features of NAFLD and clinically significant fibrosis are comparable to several leading biomarkers and elastography-based methods.29 It is however cautioned that the generation of predictive values for clinical decision making will also depend on the prevalence of the phenotypes in the populations where it will be tested. Identifying those with at-risk NASH is key in clinical practice and for clinical trial enrollment. This was evaluated in a post hoc manner because this entity was not formally identified at the time the project was conceptualized. The dichotomous assessment of NAS ≥4 and stage ≥2 fibrosis was leveraged to generate a probability score for the presence of these phenotypes. This would extend the dynamic range of the results and potentially allow for more refined assessment along a continuous scale in future studies. This, however, awaits independent validation in future studies such as those performed and reported in abstract form by the LIT-MUS consortium.
The current study also supports the ability of the proteomic models to detect histological changes over time. The changes predicted by the model were largely concordant in placebo and active arms of the treatment trials. It is important to note that the overall clinical endpoint in the trials of a decrease in NAS by ≥2 points was not evaluated because the size of the study population was insufficient to model the multiple combinations of changes that could lead to such a decrease. The observed disconnect in the lack of model changes in steatosis for OCA vs. the histological findings could either be a failure of the model to be sensitive to change or inaccurate histological assessment. It is noteworthy that OCA did not improve steatosis in the REGENERATE trial.30
The current study mainly included Caucasians, reflecting the study populations at the participating centers and the data cannot yet be generalized to other populations. Also, there were not enough individuals with each grade and stage of disease to model each one individually and this remains an important area for future study in a large and diverse cohort.
In summary, the current study demonstrates that a proteomics-based signature of individual features of NAFLD can detect the key histological phenotypes of NASH, including at-risk NASH. The proteomic models are sensitive to change and may enable patient selection and monitoring in clinical trials, and serve as an aid to clinical management. These results represent critical initial steps to support their further validation as biomarkers for NAFLD.
Supplementary Material
Highlights.
Aptamer proteomics and machine learning generated blood-based NASH models.
Serum models of liver steatosis, in flammation, ballooning and fibrosis were validated.
Models accurately reflect liver biopsy results and NASH severity.
Models allow for non-invasive longitudinal monitoring of treatment response.
Impact and implications.
An aptamer-based protein scan of serum proteins was performed to identify diagnostic signatures of the key histological features of non-alcoholic fatty liver disease (NAFLD), for which no approved non-invasive diagnostic tools are currently available. We also identified specific protein signatures related to the presence and severity of NAFLD and its histological components that were also sensitive to change over time. These are fundamental initial steps in establishing a serum proteome-based diagnostic signature of NASH and provide the rationale for using these signatures to test treatment response and to identify several novel targets for evaluation in the pathogenesis of NAFLD.
Acknowledgements
We acknowledge the members of the publication committee of the NIDDK NASH CRN who provided critical review of the manuscript.
Financial support
The Nonalcoholic Steatohepatitis Clinical Research Network (NASH CRN) is supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (grants U01DK061713, U01DK061718, U01DK061728, U01DK061731, U01DK061732, U01DK061734, U01DK061737, U01DK061738, U01DK061730, U24DK061730). Additional support is received from the National Center for Advancing Translational Sciences (NCATS) (grants UL1TR000439, UL1TR000436, UL1TR000006, UL1TR000448, UL1TR000100, UL1TR000004, UL1TR000423, UL1TR000058). RL receives funding support from NCATS (5UL1TR001442), NIDDK (U01DK061734, U01DK130190, R01DK106419, R01DK121378, R01DK124318, P30DK120515), and NHLBI (P01HL147835). This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute. SomaLogic funded the cost of the proteomic assays. The authors thank the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) for its support of the NASH CRN and this research. Note that the content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The biospecimens from the NASH CRN reported on here were supplied by the NIDDK Central Repository. This manuscript was not prepared in collaboration with the NIDDK Central Repository and does not necessarily reflect the opinions or views of the NIDDK Central Repository or the NIDDK. The PIVENS trial was conducted by the NASH CRN and supported in part by Takeda Pharmaceuticals North America through a Cooperative Research and Development Agreement with the NIDDK. The vitamin E and matching placebo for the PIVENS trial were provided by Pharmavite through a Clinical Trial Agreement with the NIH. The FLINT trial was conducted by the NASH CRN and supported in part by a Collaborative Research and Development Agreement (CRADA) between NIDDK and Intercept Pharmaceuticals.
Abbreviations
- ALT
alanine aminotransferase
- FDR
false discovery rate
- NAS
NAFLD activity score
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NIT
non-invasive test
- OCA
obeticholic acid
Footnotes
Conflict of interest
The authors declare no conflicts of interest that pertain to this work. Please refer to the accompanying ICMJE disclosure forms for further details.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhep.2022.11.029.
Data availability statement
Pre-existing data access policies for each of the parent cohort studies specify that research data requests can be submitted to each steering committee; these will be reviewed promptly for confidentiality or intellectual property restrictions and will not unreasonably be refused. Individual level patient or protein data may further be restricted by consent, confidentiality or privacy laws/considerations. These policies apply to both the non-publicly available clinical and the proteomic data. The NAFLD DB, PIVENS and FLINT clinical data are available at the NIDDK Central Repository: https://repository.niddk.nih.gov/home/
References
- [1].Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, et al. Global Perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology (Baltimore, Md) 2019;69:2672–2682. [DOI] [PubMed] [Google Scholar]
- [2].Kleiner DE, Brunt EM, Wilson LA, Behling C, Guy C, Contos M, et al. Association of histologic disease activity with progression of nonalcoholic fatty liver disease. JAMA Netw open 2019;2:e1912565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology (Baltimore, Md) 2018;67:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J Hepatol 2018;69:896–904. [DOI] [PubMed] [Google Scholar]
- [5].Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328–357. [DOI] [PubMed] [Google Scholar]
- [6].Rockey DC, Caldwell SH, Goodman ZD, Nelson RC, Smith AD. American association for the study of liver D. Liver biopsy. Hepatology (Baltimore, Md) 2009;49:1017–1044. [DOI] [PubMed] [Google Scholar]
- [7].Williams SA, Kivimaki M, Langenberg C, Hingorani AD, Casas JP, Bouchard C, et al. Plasma protein patterns as comprehensive indicators of health. Nat Med 2019;25:1851–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Luo Y, Wadhawan S, Greenfield A, Decato BE, Oseini AM, Collen R, et al. SOMAscan proteomics Identifies serum biomarkers associated with liver fibrosis in patients with NASH. Hepatol Commun 2021;5:760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Collins GS, Reitsma JB, Altman DG, Moons KG. Transparent Reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD): the TRIPOD statement. Ann Intern Med 2015;162:55–63. [DOI] [PubMed] [Google Scholar]
- [10].Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. The New Engl J Med 2010;362:1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 2015;385:956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology (Baltimore, Md) 2005;41:1313–1321. [DOI] [PubMed] [Google Scholar]
- [13].Kleiner DE, Brunt EM. Nonalcoholic fatty liver disease: pathologic patterns and biopsy evaluation in clinical research. Semin Liver Dis 2012;32:3–13. [DOI] [PubMed] [Google Scholar]
- [14].Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology (Baltimore, Md) 2017;65: 1557–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Brunt EM, Kleiner DE, Wilson LA, Sanyal AJ, Neuschwander-Tetri BA, Nonalcoholic Steatohepatitis Clinical Research N. Improvements in histologic features and diagnosis associated with improvement in fibrosis in nonalcoholic steatohepatitis: results from the nonalcoholic steatohepatitis clinical research network treatment trials. Hepatology (Baltimore, Md) 2019;70:522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sanyal AJ, Shankar SS, Calle RA, Samir AE, Sirlin CB, Sherlock SP, et al. Non-invasive biomarkers of nonalcoholic steatohepatitis: the FNIH NIMBLE project. Nat Med 01 Mar 2022, 28(3): 430–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Davies DR, Gelinas AD, Zhang C, Rohloff JC, Carter JD, O’Connell D, et al. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc Natl Acad Sci USA 2012;109: 19971–19976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mehan MR, Ostroff R, Wilcox SK, Steele F, Schneider D, Jarvis TC, et al. Highly multiplexed proteomic platform for biomarker discovery, diagnostics, and therapeutics. Adv Exp Med Biol 2013;735:283–300. [DOI] [PubMed] [Google Scholar]
- [19].Brody E, Gold L, Mehan M, Ostroff R, Rohloff J, Walker J, et al. Life’s simple measures: unlocking the proteome. J Mol Biol 2012;422:595–606. [DOI] [PubMed] [Google Scholar]
- [20].Kim CH, Tworoger SS, Stampfer MJ, Dillon ST, Gu X, Sawyer SJ, et al. Stability and reproducibility of proteomic profiles measured with an aptamer-based platform. Sci Rep 2018;8:8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cheung A, Neuschwander-Tetri BA, Kleiner DE, Schabel E, Rinella M, Harrison S, et al. Defining improvement in nonalcoholic steatohepatitis for treatment trial endpoints: Recommendations from the liver Forum. Hepatology (Baltimore, Md) 2019;70:1841–1855. [DOI] [PubMed] [Google Scholar]
- [22].Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stats Soc 1995;57:289–300. [Google Scholar]
- [23].Klasen JR, Barbez E, Meier L, Meinshausen N, Buhlmann P, Koornneef M, et al. A multi-marker association method for genome-wide association studies without the need for population structure correction. Nat Commun 2016;7:13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Friedman J, Hastie T, Tibshirani R. Sparse inverse covariance estimation with the graphical lasso. Biostatistics 2008;9:432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic steatohepatitis: a review. JAMA 2020;323:1175–1183. [DOI] [PubMed] [Google Scholar]
- [26].Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Genomic atlas of the human plasma proteome. Nature 2018;558:73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hoang SA, Oseini A, Feaver RE, Cole BK, Asgharpour A, Vincent R, et al. Gene Expression predicts histological severity and Reveals Distinct Molecular profiles of nonalcoholic fatty liver disease. Scientific Rep 2019;9:12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Younossi ZM, Loomba R, Anstee QM, Rinella ME, Bugianesi E, Marchesini G, et al. Diagnostic modalities for nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, and associated fibrosis. Hepatology (Baltimore, Md) 2018;68:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019;394:2184–2196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Pre-existing data access policies for each of the parent cohort studies specify that research data requests can be submitted to each steering committee; these will be reviewed promptly for confidentiality or intellectual property restrictions and will not unreasonably be refused. Individual level patient or protein data may further be restricted by consent, confidentiality or privacy laws/considerations. These policies apply to both the non-publicly available clinical and the proteomic data. The NAFLD DB, PIVENS and FLINT clinical data are available at the NIDDK Central Repository: https://repository.niddk.nih.gov/home/