

SUMMARY
Genetic tools to target microglia specifically and efficiently from the early stages of embryonic development are lacking. We generated a constitutive Cre line controlled by the microglia signature gene Crybb1 that produced nearly complete recombination in embryonic brain macrophages (microglia and border-associated macrophages (BAMs)) by the perinatal period, with limited recombination in peripheral myeloid cells. Using this tool, in combination with Flt3-Cre lineage tracer, single-cell RNA-sequencing analysis, and confocal imaging, we resolved embryonic-derived versus monocyte-derived BAMs in the mouse cortex. Deletion of the transcription factor SMAD4 in microglia and embryonic-derived BAMs using Crybb1-Cre caused a developmental arrest of microglia, which instead acquired a BAM specification signature. By contrast, the development of genuine BAMs remained unaffected. Our results reveal that SMAD4 drives a transcriptional and epigenetic program that is indispensable for the commitment of brain macrophages to the microglia fate and highlight Crybb1-Cre as a tool for targeting embryonic brain macrophages.
In brief
Tools to target microglia specifically and efficiently from the embryonic development are lacking. Brioschi et al., generate the Crybb1-Cre line which recombines in microglia and border-associated macrophages during the embryonic stage. Combining Crybb1-Cre and other tools, they resolve embryonic-derived versus monocyte-derived BAMs in the mouse cortex. Deletion of the transcription SMAD4 using Crybb1-Cre revealed that microglia require SMAD4 for differentiation.
INTRODUCTION
The central nervous system (CNS) hosts two main populations of macrophages, namely microglia and border-associated macrophages (BAMs), each of which plays different roles in brain homeostasis and immune defense [1–3]. Microglia are the most abundant myeloid population of the CNS and reside within the parenchyma. These cells emerge from yolk sac (YS) hematopoiesis during embryogenesis and infiltrate the brain rudiment at E9.5–10.5 [4–7]. Microglia are maintained by self-renewal [8, 9] with negligible input from circulating monocytes [6, 10]. Outside the encephalon and spinal cord, microglia can be found in the retina only [11–13], while peripheral nerves contain macrophages of disparate origins and phenotypes [14, 15]. BAMs are located at the CNS interfaces (dura mater, subdural meninges or leptomeninges, perivascular Virchow-Robin spaces and choroid plexus) and like microglia derive from YS progenitors [16–18]. However, unlike microglia, BAMs are partially diluted by bone marrow (BM)-derived macrophages after birth [16]. Fate-mapping studies show that monocytic input varies depending on the brain border niche. While embryonic-derived perivascular and subdural macrophages appear stable over time, macrophages in the choroid plexus and dura mater undergo more rapid turnover mediated by circulating monocytes [16, 19]. To unveil distinct features of embryonic and BM-derived brain macrophages we mostly rely on the Cre-lox system, which allows the expression of Cre recombinase under the control of a lineage-specific promoter. The constitutive Cx3cr1Cre and inducible Cx3cr1CreErt2 lines have been extensively used and have proven high efficiency of recombination in both microglia and BAMs [10, 16, 20]. However, the broad expression of Cx3cr1 within the myeloid compartment [10, 20, 21] does not allow for a selective targeting of these cells. This shortcoming has been partially resolved with the generation of tamoxifen inducible CreErt2 lines with improved specificity for either microglia (Sall1CreErt2, HexbCreErt2, Tmem119CreErt2 and P2ry12CreErt2) or BAMs (Mrc1CreErt2) [17, 22–25]. Yet, the use of these Cre constructs requires tamoxifen (TAM) administration, which is suitable for postnatal targeting. More recently, a binary split Cre system has been developed and further improved the specificity for microglia at the expense of recombination efficiency [26]. Although several Cre lines have been generated, there are currently no genetic tools to achieve efficient and specific recombination of brain macrophages during embryonic development.
Here we describe the Crybb1-Cre line, containing a codon optimized Cre (iCre) under the control of the microglia signature gene Crybb1, which is highly expressed in embryonic microglia [27]. Crybb1-Cre mice exhibited excellent recombination efficiency in microglia, with limited recombination in peripheral myeloid cells. Onset of Cre activity was detectable in the embryonic brain as early as E13.5, reaching ~100% of microglia recombination in the perinatal window. No recombination was detected in YS macrophages or erythromyeloid progenitors (EMPs). Unexpectedly, a subset of BAMs exhibited recombination in the postnatal mouse brain. Using single-cell RNA sequencing (scRNA-seq), complementary fate-mapping systems, flow cytometry and imaging techniques, we demonstrated that this subset corresponds to CD38+MHC2− BAMs and is mostly formed by embryonic-derived macrophages. Conversely, CD38−MHC2+ BAMs were monocyte-derived and therefore were minimally targeted by Crybb1-Cre. Lastly, we used Crybb1-Cre to delete SMAD4, the downstream transcription factor of TGF-β signaling. Using multiomic analysis that integrates single-cell RNA-seq and ATAC-seq in the same cell, we showed that Smad4 deletion caused an arrest of microglia specification, evidenced by a loss of the homeostatic microglia signature, upregulation of BAM genes and broad chromatin remodeling. Importantly, BAMs were not affected by Smad4 deletion, suggesting that this transcription factor is redundant for BAM maturation. At the behavioral level, mice with SMAD4-deficient microglia exhibited memory impairment, whereas locomotor activity and coordination skills remained unaffected. In sum, the Crybb1-Cre line enabled us to resolve BAM subsets with different origins and provide a valuable resource of transcriptomic and epigenetic information pertaining to the role of TGF-β signaling in microglia development.
RESULTS
Crybb1-Cre targets microglia and a subset of BAMs
We first searched for a microglia-specific gene highly expressed during embryonic development and identified Crybb1 as an ideal candidate. Crybb1 encodes Beta-crystallin B1, a protein highly expressed in the mouse and human eye and required to maintain the transparency of the eye lens [28]. Crybb1 has been reported as a microglia signature gene [29–31], while expression in other immune cells is undetectable (https://tabula-muris.ds.czbiohub.org/) [32]. Furthermore, the peak of Crybb1 expression occurs in the embryonic brain after E13.5 and decreases in adult mice [27] (Figure S1A). First, we used CRISPR/Cas9 to produce an out-of-frame deletion within the third exon of the Crybb1 locus, thus generating Crybb1 knock-out mice (Figure S1B). Abrogation of CRYBB1 expression was confirmed by immunofluorescent staining (Figure S1C). We analyzed microglia by bulk RNA-seq (Figure S1D) and concluded that lack of Crybb1 expression does not appreciably impact the microglial phenotype. Thus, Crybb1 was a good candidate gene for generating a Cre line by knock-in/knock-out strategy. We then introduced the coding sequence for codon optimized Cre (iCre) in-frame downstream of the Crybb1 promoter (hereafter Crybb1-Cre). Crybb1-Cre mice were further crossed with Rosa26-stop-tdTomato mice (hereafter Crybb1-Cre : R26-tdTomato) to determine efficiency and specificity of recombination (Figure 1A). Flow cytometry revealed 99.9% recombination in microglia (CD11b+CX3CR1hiCD45lo); however, recombination was also observed in brain BAMs (CD11b+CX3CR1loCD45hi) (Figure 1B), and dura BAMs (CD11b+CX3CR1+MERTK+) (Figure S1E) albeit to a lesser extent. Confocal imaging confirmed nearly complete recombination in both Iba1+ microglia and CD206bright BAMs from leptomeninges, perivascular space and choroid plexus (Figure 1C and D). We then assessed the percentage of recombination in myeloid cells from multiple tissues, including heart, kidney, small intestine, liver, spleen, peritoneal cavity, lung, visceral adipose tissue (VAT), skin, and blood (Figure 1E). Apart from the CNS compartment, the highest recombination frequencies were found in heart macrophages (30.0%) and kidney macrophages (11.9%). Other analyzed populations exhibited negligible recombination frequencies (Figure 1F). As expected, no microglia recombination was found in Cre negative littermates (Figure S1F). Overall, Crybb1-Cre efficiently recombined microglia and a subset of BAMs, with limited recombination in peripheral compartments, at least within the populations assessed in this study. To assess Crybb1 expression at any given time we generated a reporter mouse with tdTomato introduced in-frame downstream of the Crybb1 promoter (hereafter Crybb1-tdTomato) (Figure 1G). Young-adult Crybb1-tdTomato mice exhibited uniform tdTomato expression in Iba1+ microglia, but not in CD206+ BAMs (Figure 1H). Lack of detectable CRYBB1 protein in BAMs from young-adult mice was further confirmed by immunofluorescent staining (Figure S1G). Thus, in the adult mouse brain Crybb1 is highly expressed in microglia only. Nevertheless, Crybb1-Cre traced cells experiencing Crybb1 expression, and efficiently recombined in both microglia and CD206+ BAMs (Figure 1I), suggesting that the recombination probably occurred at early stages during development.
Figure 1. Crybb1-Cre efficiently recombines in microglia and subsets of BAMs.
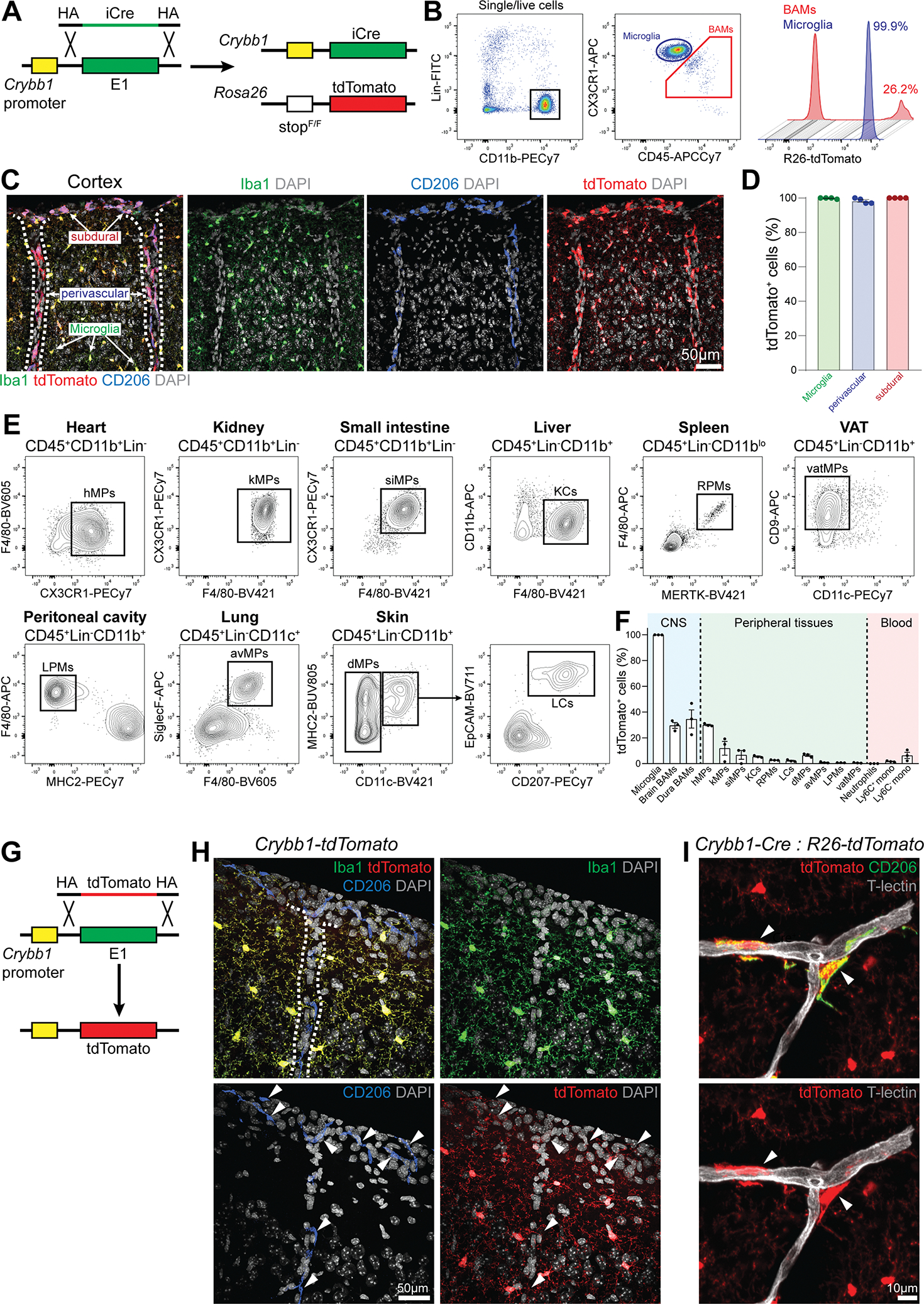
(A) Strategy used to generate the Crybb1-Cre : R26-tdTomato reporter line.
(B) Representative gating strategy for microglia and BAMs in Crybb1-Cre : R26-tdTomato mice.
(C) Representative confocal images of microglia, subdural, and perivascular BAMs expressing tdTomato in Crybb1-Cre : R26-tdTomato mice (n=4 mice, 2 months old, single experiment, dashed lines = blood vessels).
(D) Percentage of microglia (Iba1+CD206−), subdural, and perivascular BAMs (CD206bright) expressing tdTomato (n=4 mice, 2 months old, single experiment).
(E) Representative gating for all myeloid cell populations analyzed in Crybb1-Cre : R26-tdTomato mice (hMPs = heart MPs; kMPs = kidney MPs; siMPs = small intestine MPs; KCs = Kupffer cells; RPMs = red pulp MPs; vatMPs = visceral adipose tissue MPs; LPMs = large peritoneal MPs; avMPs = alveolar MPs; dMPs = dermal MPs; LCs = Langerhans cells).
(F) Percentage of tdTomato+ cells in analyzed populations (n=3 mice, 6–8 weeks old, single experiment).
(G) Strategy used to generate the Crybb1-tdTomato reporter line.
(H) Representative confocal images of microglia and BAMs in Crybb1-tdTomato mice (n=3 mice, 2 months old, single experiment, dashed lines = blood vessel).
(I) Representative confocal image of CD206+ perivascular BAMs in Crybb1-Cre : R26-tdTomato mice (white arrowheads) (n=3 mice, single experiment).
See also Figure S1
Of note, we observed sparse recombination in Iba1−CD206− cells throughout the brain sections (Figure S1H). These cells exhibited a highly ramified morphology and stained positive for OLIG2 but not for Adenomatous Polyposis Coli (APC) (Figure S1I), pointing towards oligodendrocyte progenitor cells (OPCs). Furthermore, sparse recombinant NeuN+ neurons were found in the dentate gyrus of the hippocampus (Figure S1J). We then assessed the amount of off-target recombination in different brain regions: prefrontal cortex (PFC), somatosensory cortex (SSC), striatum (STR), hippocampus (HC) and cerebellum (CER). We quantified variable numbers of recombinant OPCs in the analyzed brain regions (PFC = 14.0/mm2; STR = 11.3/mm2; SSC = 5.7/mm2; CER = 4.0/mm2; HC = 0.8/mm2), whereas a substantial number of recombinant neurons could be found in HC and CER (10.8/mm2 and 26.6/mm2 respectively) (Figure S1K). No off-target recombination was observed in the choroid plexus (Figure S1L) or dura mater (not shown). To check for recombination leakage in peripheral compartments we inspected liver, kidney, and small intestine by imaging, and found no evidence of recombination in stromal, epithelial, or vascular cells (Figure S1M–O) except for sparse recombination in bone marrow stromal cells lining on the endosteal surface (Figure S1P).
Crybb1-Cre recombines microglia and BAMs during embryonic brain development
We assessed the recombination in microglia and BAMs in Crybb1-Cre mice throughout embryonic brain development, up to the first postnatal week (Figure 2A). Embryonic brain macrophages were gated as CD45+CD11b+Gr.1−CX3CR1+F4/80+. Because embryonic BAMs highly express Mrc1 and Folr2 [18], we gated microglia as CD206−FOLR2− population, while BAMs were gated as CD206+FOLR2+ cells. We found no tdTomato+ cells in either population at E10.5, which is the time when yolk sac-derived macrophages disseminate the embryonic tissues [4, 6, 7]. However, the frequency of recombinant cells sharply increased from E13.5 in a time-dependent manner, plateauing during the first postnatal days (Figure 2B and 2C). Whereas the absolute number of tdTomato+ microglia kept increasing during development (presumably due the active proliferation of these cells), the number of tdTomato+ BAMs peaked at the late embryonic stage (Figure 2D). tdTomato expression in Iba1+ microglia and CD206+ BAMs was confirmed by confocal imaging before and after birth (Figure 2E). No recombination was found in YS EMPs, macrophage precursors (pre-Mac) or macrophages (Figure S2A–S2C) [7, 33]. Furthermore, confocal imaging of Crybb1-tdTomato reporter mice at E18.5 confirmed expression of Crybb1 in both microglia and BAMs (Figure 2F and S2D), indicating that Crybb1 is highly expressed in both populations before birth. Next, we assessed the percentage of recombination in microglia as well as CD206+ and CD206−/lo BAMs at multiple timepoints starting from weaning age (Figure 2G). In the adult brain, BAMs were gated as CD11b+Lin−CX3CR1intCD45hi population (Figure S2E). Recombination frequencies remained high and stable in both microglia and CD206+ BAMs (Figure 2H). By contrast, recombination of CD206−/lo BAMs decreased over time. Together, these data indicate that embryonic-derived brain macrophages highly express Crybb1 between E13.5 and perinatal period, thus Crybb1-Cre mediated recombination occurred during this developmental window. Microglia and CD206+ BAMs maintained stable recombination till adulthood, whereas CD206−/lo BAMs did not, suggesting that these cells were either replaced or diluted by monocyte-derived macrophages. Thus, we propose that Crybb1-Cre efficiently recombines embryonic-derived brain macrophages, but not monocyte-derived BAMs.
Figure 2. Crybb1-Cre recombination activity peaks during the embryonic brain development.
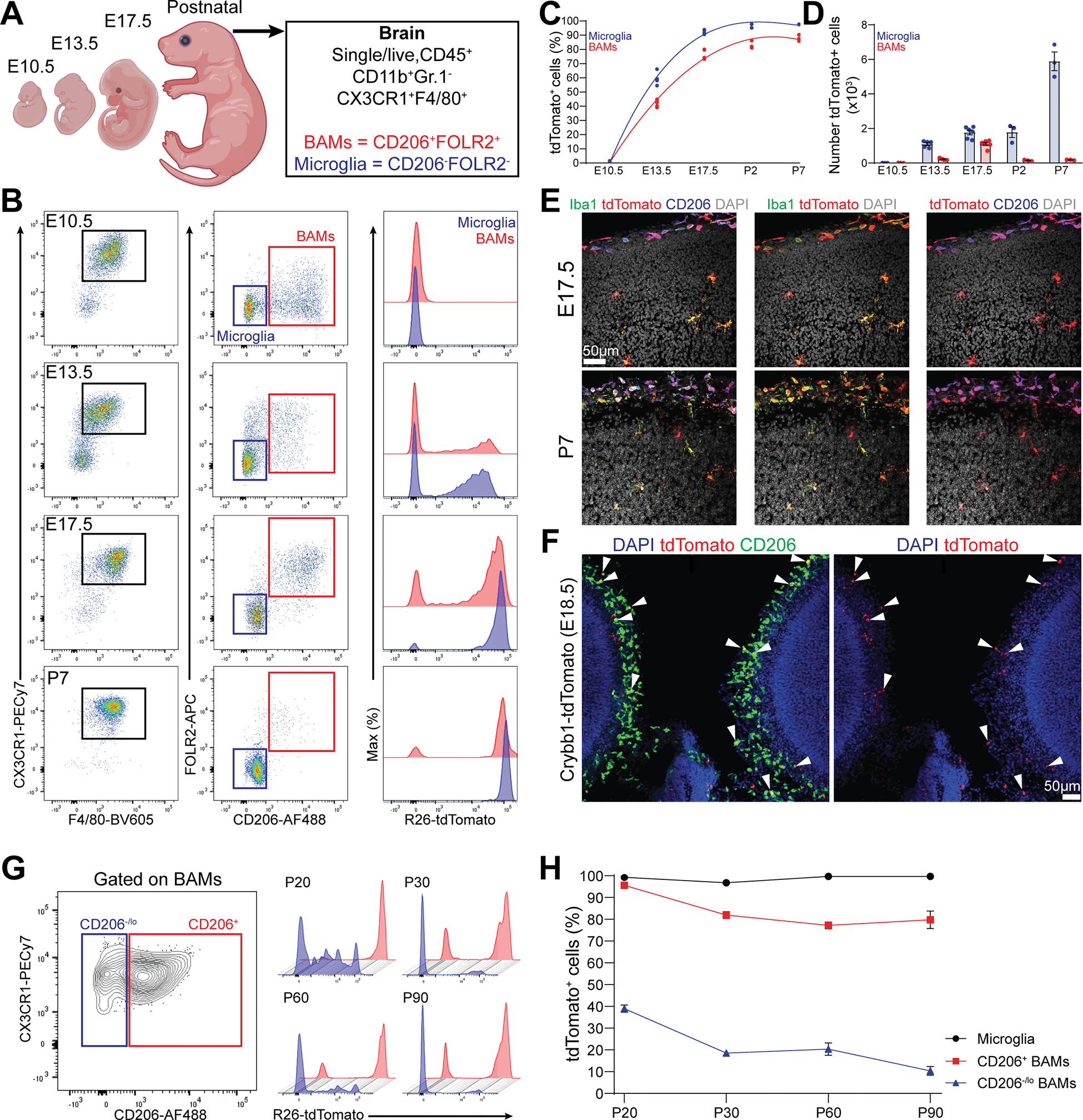
(A) Cartoon describing the experimental design and gating strategy.
(B) Representative gating strategy for microglia and BAMs in Crybb1-Cre : R26-tdTomato mice at different timepoints.
(C) Percentage of tdTomato+ microglia and BAMs from E10.5 to P7 (n=3–6 mice/timepoint, single experiment).
(D) Absolute numbers of microglia and BAMs from E10.5 to P7 (n=3–6 mice/timepoint, single experiment).
(E) Representative confocal images of microglia and BAMs expressing tdTomato in the forebrain cortex of E17.5 embryos and P7 Crybb1-Cre : R26-tdTomato mice (n=3 embryos and n=2 pups, single experiment).
(F) Representative confocal images of microglia and BAMs expressing tdTomato in the forebrain cortex of E18.5 Crybb1-tdTomato embryos (white arrowheads) (n=5 embryos, single experiment).
(G) Representative gating strategy for CD206+ and CD206− BAMs in Crybb1-Cre : R26-tdTomato mice at different timepoints.
(H) Percentage of tdTomato+ microglia, CD206+ and CD206− BAMs in Crybb1-Cre : R26-tdTomato mice at different timepoints (n=3 mice/timepoint, single experiment).
See also Figure S2
The adult brain at steady state contains two distinct subsets of BAMs expressing either CD38 or MHC2
To gain deeper insights into the phenotypic diversity of brain BAMs we performed scRNA-seq of brain macrophages (sorted as CD11b+Lin− cells) using the 10x Genomics platform (Figure 3A). For all the following experiments, we excluded the dura mater and focus on BAMs in the brain proper. Brain macrophages clustered into three distinct populations, namely microglia and two subsets of BAMs, annotated as BAM-1 and BAM-2, which we visualized using Uniform Manifold Approximation and Projection (UMAP) (Figure 3B). These BAM subsets exhibited a partially overlapping expression profile, yet several genes were differentially expressed (Figure 3C and S3A). Specifically, BAM-1 were highly enriched for MHC class-II (MHC2) genes (H2-Aa, H2-Ab1, Cd74), whereas BAM-2 were enriched for prototypical BAM signature genes (Mrc1, Pf4, Stab1, Cd209f, Ms4a7, Cd163, Lyve1, Folr2) (Figure 3D and 3E). We next wanted to leverage this transcriptomic information to identify surface markers to accurately distinguish BAM-1 and -2 by flow cytometry. MHC2 and CD38 staining allowed the resolution of four distinct BAM subsets, pre-gated as CD11b+Lin−CX3CR1intCD45hi from the whole brain (Figure 3F). Furthermore, we dissected choroid plexus (containing Cp BAMs) and cortex (enriched for subdural and perivascular BAMs) and analyzed these two compartments separately. Given the technical challenge of distinguishing perivascular and subdural BAMs by flow cytometry, we collectively refer to these populations as “cortical BAMs”. Choroid plexus mostly contained CD38+MHC2+ and CD38+MHC2− BAMs. By contrast, cortex appeared enriched for CD38−MHC2+ and CD38+MHC2− BAMs. The CD38−MHC2− population in the cortex could be further stratified into MERTK−CD45hi and MERTK+CD45lo populations (Figure 3G and 3H). We believe that CD38−MHC2−MERTK+CD45lo population may represent some microglia-like cells that fell into the BAM gate (hereafter Mg-like). Indeed, these cells expressed no CD206 and FOLR2 (not shown), but abundant TMEM119 and P2RY12 (Figure S3B). Furthermore, this population appeared greatly reduced in Fire (fms-intronic regulatory element) knock-out mice (Figure S3C), featuring a complete lack of microglia and reduced choroid plexus macrophages [34–36]. We then analyzed the CD38−MHC2−MERTK−CD45hi population more in detail. These cells did not express CD206 and FOLR2 (Figure 3G) but were CD64−CD44hi (Figure S3D) and highly expressed Nr4a1 (Nur77) (Figure S3E). Thus, we conclude that these cells are Ly6C− monocytes [37–40] which were incompletely excluded with the lineage staining. Notably, MHC2+ cortical BAMs exhibited low expression of CD206 and no expression of FOLR2 (Figure 3G). Conversely, CD38+ cortical BAMs highly expressed of both CD206 and FOLR2. Therefore, MHC2+ and CD38+ BAMs respectively recapitulated the phenotype of BAM-1 and BAM-2 subsets observed in scRNA-seq. Furthermore, the BAM-1 cluster was enriched for Ccr2, suggesting a monocytic origin. Using Ccr2GFP mice, we confirmed Ccr2 expression in MHC2+ BAMs and monocytes (Figure 3I and Figure S3F). Consistently, MHC2+ BAMs expanded significantly from 4 to 12 weeks of age, indicating that this population is established postnatally (Figure 3J and 3K). Of note, cDC2s in meninges express CD11b and CX3CR1 [19, 41, 42], therefore CD38−MHC2+ BAMs could be partially contaminated by these cells. To test this, we analyzed Zbtb46GFP reporter mice [43] and concluded that ~10% of CD38−MHC2+ BAMs are indeed cDC2s (Figure S3G). Thus, the homeostatic brain contains two major subsets of BAMs exhibiting distinct molecular signatures. This observation prompted two questions: a) is the BAM phenotype affected during neurodegeneration? b) Do BAM subsets have different hematopoietic origins?
Figure 3. Two major BAM subsets populate the homeostatic brain.
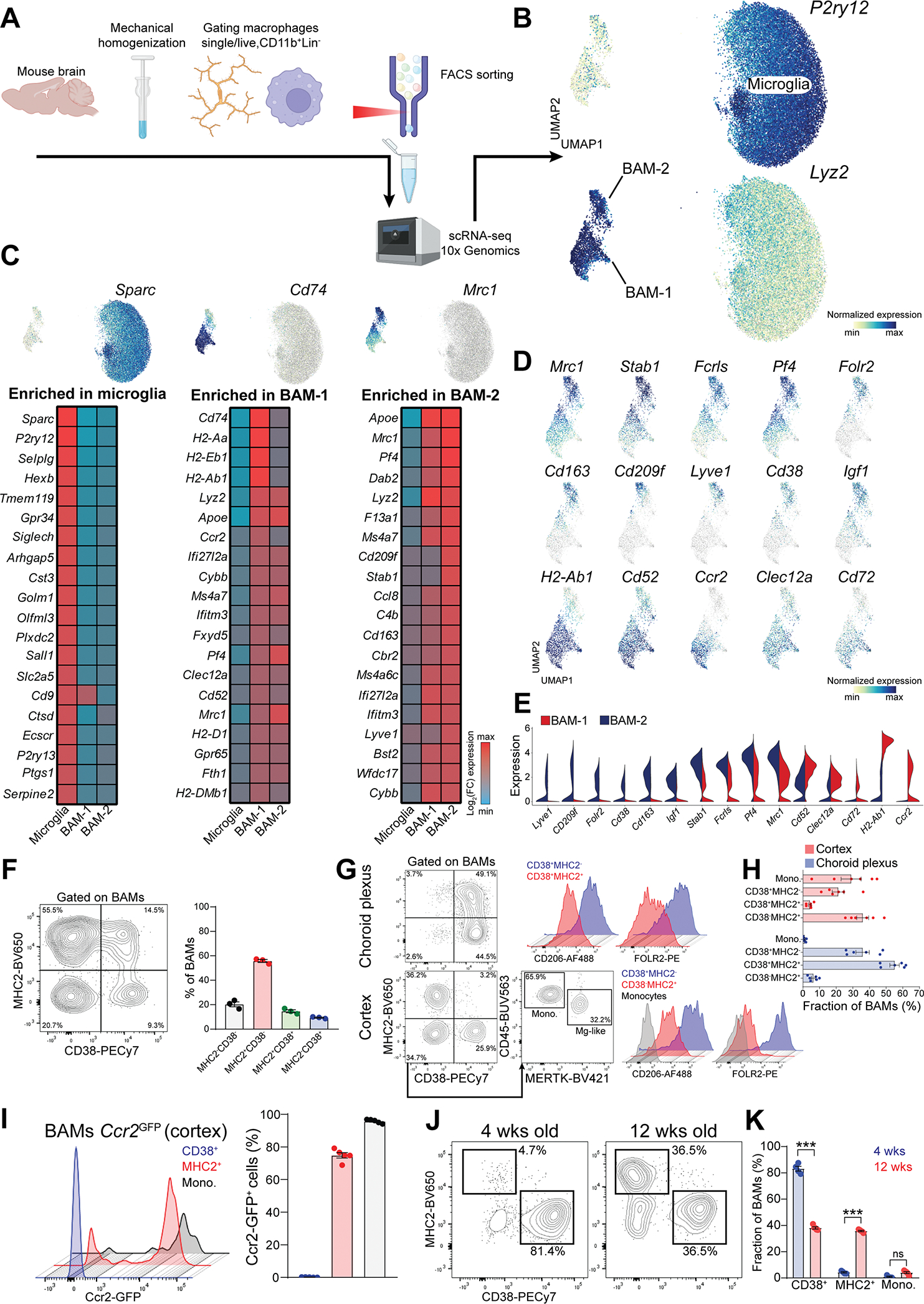
(A) Cartoon describing the experimental design.
(B) UMAP plot of microglia and BAMs (n=5 wild-type and n=4 5xFAD littermate mice, 8month-old).
(C) Heatmap of the top 20 signature genes for microglia, BAM-1 and BAM-2 populations (n=5 wild-type mice, 8-month-old).
(D) Enrichment of selected signature genes for BAM-1 and BAM-2 populations.
(E) Expression of selected signature genes for BAM-1 and BAM-2 populations.
(F) Representative FACS plot and percentage of different BAM subsets defined based on MHC2 and CD38 surface expression (n=3 wild-type mice, 3-month-old, single experiment).
(G) Representative gating for different BAM subsets in choroid plexus and cortex. Surface expression of CD206 and FOLR2 in each population is displayed.
(H) Frequency of different BAM subsets in choroid plexus and cortex, within the total BAM population (n=6 mice, 2-month-old, pool of two independent experiments).
(I) Percentage of GFP+ BAMs in the cortex of Ccr2GFP mice (n=5 mice, 3-month-old, single experiment).
(J) Representative FACS plot of MHC2+ and CD38+ BAM subsets in the cortex of 4- and 12-week-old mice.
(K) Frequency of different BAM subset within the total BAM population in the cortex of 4-and 12-week-old mice (n=4–5 mice/group, two-way ANOVA with Bonferroni post-hoc test, ***P<0.001, single experiment).
See also Figure S3
Unlike microglia, BAM phenotype is unaffected in 5xFAD mice
Having characterized the BAM phenotype in homeostatic conditions, we wanted to better explore composition and phenotypic changes of these cells in a disease model. In the 5xFAD mouse model of amyloid pathology [44], plaque associated microglia develop a specific transcriptional signature known as disease-associated microglia (DAM) [45–52]. However, whether BAMs acquire a disease-specific signature during amyloid pathology is unclear. To answer this, we analyzed microglia and BAMs from 8-month-old 5xFAD and wild-type littermates by scRNA-seq. Microglia from both mice formed multiple clusters which could be grouped into two main subsets, namely homeostatic microglia (HM) and DAM. As expected, DAM upregulated immune-related and lipid metabolism genes, like Apoe, Cst7, and Lpl (Figure S3H), and were mostly contributed by 5xFAD mice (Figure S3I). BAM-1 and BAM-2 clusters also received greater input from 5xFAD mice (Figure S3J); however differential gene expression analysis (Log2(FC)>0.5, p<0.01) revealed no differentially expressed genes in either population between 5xFAD and wild-type littermates (Figure S3K). Of note, BAM-1 cluster exhibited a mild, but significant increase of type-I IFN signature genes (Ifitm3, Bst2, Isg15) in 5xFAD mice (not shown), as recently reported [53]. However, none of these transcripts passed the chosen cutoff. Taken together, this analysis suggested that, although microglia exhibit a DAM signature under amyloid pathology, minimal transcriptional changes can be found in BAMs, at least in this model and at the assessed timepoint.
Crybb1-Cre efficiently targets embryonic-derived, but not monocyte-derived BAMs
We sought to determine whether these BAM subsets have different hematopoietic origins. Previous studies demonstrated that choroid plexus and dura BAMs undergo monocyte-mediated turnover after birth [16, 19]. Furthermore, our analysis revealed a considerable phenotypic heterogeneity of cortical BAMs, of which origin and distribution are currently unexplored. Therefore, we specifically focused on cortical BAMs. Flt3-Cre : R26-Yfp fate-mapper traces BM-derived immune cells [54, 55]; indeed we found approximately 95% of YFP+ blood monocytes in these mice (Figure S4A–C). Therefore, YFP expression in BAMs reflects monocytic origin. We assessed the percentage of recombination in cortical BAM subsets using Crybb1-Cre : R26-tdTomato or Flt3-Cre : R26-Yfp mice (Figure 4A). CD38+ BAMs exhibited the highest recombination frequency with Crybb1-Cre and poor recombination with Flt3-Cre (96.3% and 36.2% respectively). Conversely, MHC2+ BAMs and monocytes were poorly targeted with Crybb1-Cre (11.8% and 11.7% respectively), but efficiently targeted with Flt3-Cre (93.2% and 95.4% respectively) (Figure 4B). Recombination of CD38+ BAMs in Crybb1-Cre mice was further validated by imaging (Figure 4C). To provide additional validation, we performed confocal imaging on both Crybb1-Cre : R26-tdTomato and Flt3-Cre : R26-Yfp mice (Figure 4D and 4E), and quantified the percentage of recombination in MHC2+ BAMs for either fate-mapping system. This analysis confirmed that MHC2+ BAMs were efficiently traced with Flt3-Cre, but not with Crybb1-Cre (Figure 4F).
Figure 4. CD38+ and MHC2+ BAM subsets have different origins.
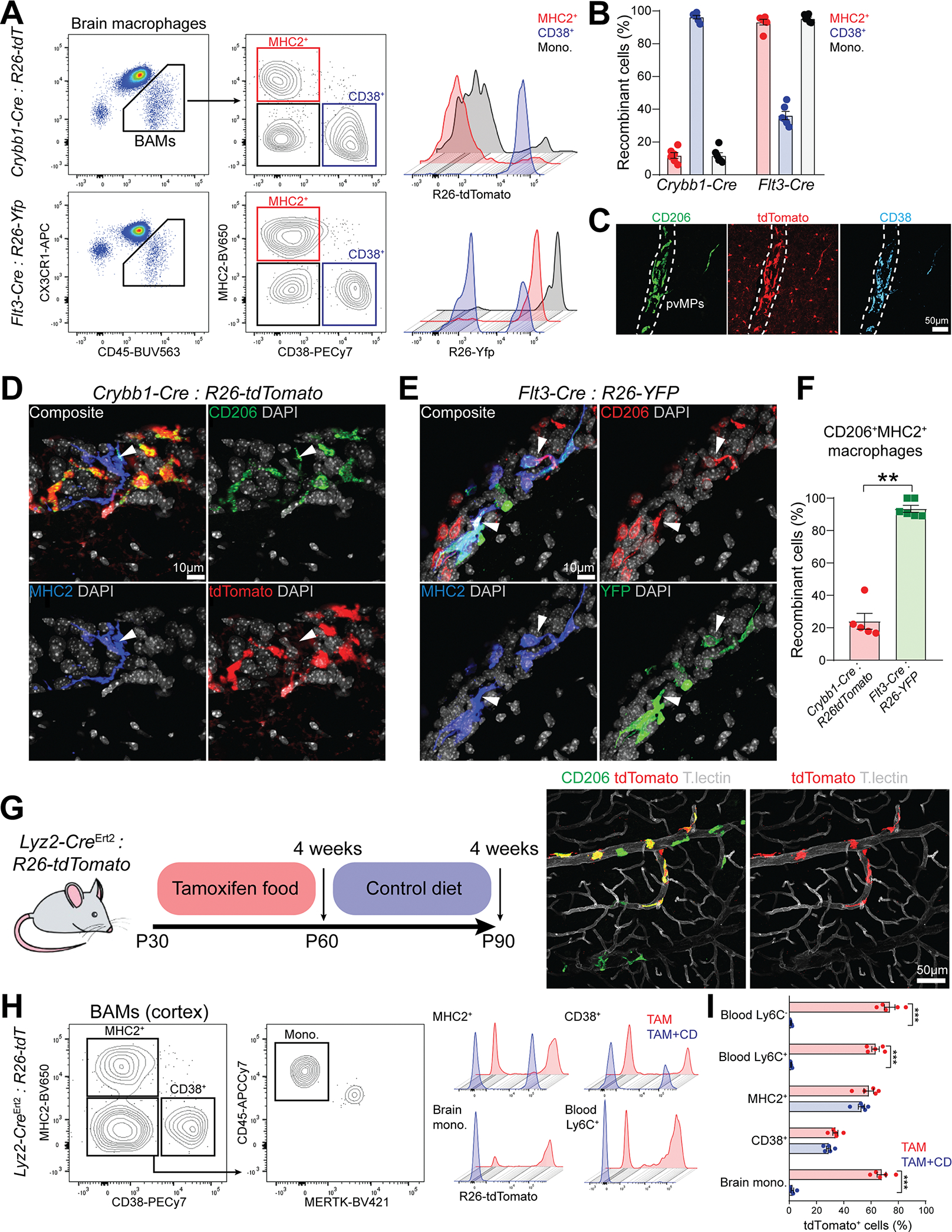
(A) Representative FACS plot of different BAM subsets defined based on MHC2 and CD38 surface expression from Crybb1-Cre : R26-tdTomato and Flt3-Cre : R26-Yfp mice.
(B) Percentage of recombination in all BAM subsets from either Crybb1-Cre : R26-tdTomato or Flt3-Cre : R26-Yfp mice (n=6 mice/group, 2–3 months old, pool of two independent experiments).
(C) Representative confocal images of CD206 and CD38 staining on perivascular BAMs in Crybb1-Cre : R26-tdTomato mice (n=3 mice, 2–3 months old, single experiment).
(D) Representative confocal images of CD206+MHC2+ BAMs in Crybb1-Cre : R26-tdTomato mice (white arrowheads, n=5 mice/group, 2–3 months old, two independent experiments).
(E) Representative confocal images of CD206+MHC2+ BAMs in Flt3-Cre : R26-Yfp mice (white arrowheads, n=6 mice/group, 2–3 months old, two independent experiments).
(F) Percentage of recombination in CD206+MHC2+ cortical BAMs from either Crybb1-Cre : R26-tdTomato and Flt3-Cre : R26-Yfp mice (n=5–6 mice/group, 2–3 months old, pool of two independent experiments).
(G) Cartoon describing the experimental design. A representative confocal image of recombinant CD206+ perivascular BAMs from Lyz2CreErt2 : R26-tdTomato after four weeks of TAM treatment is displayed.
(H) Representative FACS plot of different BAM subsets defined based on MHC2 and CD38 surface expression from Lyz2CreErt2 : R26-tdTomato mice that underwent TAM treatment regimen.
(I) Percentage of recombination in all BAM subsets and blood Ly6C+ and Ly6C− monocytes from Lyz2CreErt2 : R26-tdTomato mice upon four weeks of TAM treatment and after additional four weeks of control diet (n=5 mice/group, 2–3 months old, two-way ANOVA with Bonferroni post-hoc test, ***P<0.001, single experiment).
See also Figure S4
Given that virtually all CD38+ BAMs in the cortex were labeled with the Crybb1-Cre, and about one third of this population was also labeled with the Flt3-Cre fate mapper, it is possible that some monocyte-derived BAMs transiently expressed Crybb1 and underwent recombination. To accurately assess Crybb1 expression in brain macrophages from adult mice, we performed flow cytometry analysis of microglia and BAMs in the Crybb1-tdTomato reporter (Figure S4D). 90% of microglia was tdTomato positive, whereas ~17% of CD38+ BAMs expressed detectable tdTomato, although at much lower intensity compared to microglia (Figure S4E). Expression in MHC2+ BAMs was negligible. To further test whether BM-derived macrophages may transiently upregulate Crybb1 upon infiltration in the brain, we transplanted Crybb1-Cre : R26-tdTomato BM cells into Bl6 Cd45.1 recipient mice (Figure S4F). Eight weeks post-transplant, we obtained a nearly complete chimerism of blood myeloid cells (Figure S4G). Nevertheless, the percentage of tdTomato+ BAMs remained low and comparable to that of circulating monocytes (Figure S4H and S4I), indicating that Crybb1-Cre minimally recombined BM-derived BAMs. Given that Crybb1-Cre labels BAMs during embryonic development, while Flt3-Cre labels hematopoietic stem cell (HSC)-derived leukocytes, we conclude that CD38+ BAMs from the brain cortex are mostly of embryonic origin, whereas MHC2+ BAMs are monocyte-derived. However, it should be noted that Flt3-Cre recombines in both fetal liver and BM HSCs [6, 56], therefore the specific contribution of each hematopoietic wave to this population remains uncertain.
Next, we wanted to determine the turnover rate for each BAM subset. For this analysis we relied on the TAM inducible Lyz2-CreErt2 line [57], since Lyz2 is highly expressed in BAMs and monocytes, but not in microglia. We then fed Lyz2-CreErt2 : R26-tdTomato mice with TAM-containing food for four weeks. One cohort of mice was analyzed immediately at the end of the TAM treatment. A second cohort was returned to normal food for an additional four weeks (Figure 4G). We assessed the percentage of tdTomato+ cells in blood monocytes and BAMs at the two timepoints (Figure 4H). Blood Ly6C− and Ly6C+ monocytes were respectively 63.3% and 73.7% tdTomato+ after TAM diet. This percentage dropped to ~1% four weeks after TAM withdrawal, indicating a nearly complete turnover of circulating monocytes after this period. Consistently, brain monocytes almost completely lost tdTomato labeling during this period. By contrast, MHC2+ and CD38+ cortical BAMs remained equally labeled at the two timepoints, indicating minimal turnover (Figure 4I). Importantly, negligible recombination was found in BAMs from TAM free mice (Figure S4J). To conclude, Crybb1-Cre efficiently recombined microglia and a subset of CD38+MHC2− subdural/perivascular BAMs (Figure S4K). These cells were mostly of embryonic origin and were not continuously replaced by monocytes in adult mice, at least under steady state conditions.
SMAD4 critically drives microglia but not BAM transcriptional signatures
Starting from their entry in the embryonic brain up to the second postnatal week, microglia undergo a stepwise maturation program which eventually gives rise to the adult microglial phenotype [27, 58–61]. Several studies demonstrated that TGF-β signaling critically shapes microglial signature along this process [18, 62–66], although the exact mechanism is yet unclear. SMAD4 acts as a central hub of canonical TGF-β signaling. Activation of TGF-β-receptor induces phosphorylation of SMAD proteins (i.e. SMAD2 and SMAD3), which translocate into the nucleus upon dimerization with SMAD4. Thus, the SMAD complex regulates epigenetic modifications and transcription of TGF-β responsive genes. In parallel, a non-canonical TGF-β signaling induces a SMAD4-independent activation of a signaling cascade including MAPKs, Pi3K, ROCK and TAK1 [67]. However, whether phenotypic maturation of microglia occurs via a SMAD4-dependent or -independent pathway is unknown. To investigate this, we crossed Crybb1-Cre mice with Smad4F/F mice, thus generating Smad4 cKO mice harboring Smad4 deletion in microglia and embryonic BAMs (Figure 5A). To assess transcriptomic and epigenetic changes in these cells we performed the multiomic (scRNA-seq and ATAC-seq) protocol from 10x Genomics on brain CD45+Ly6G− cells sorted from Smad4 cKO and Smad4F/F littermates (Figure 5B). After quality filtering we obtained 108,947 single cells. Unsupervised clustering of scRNA-seq data identified microglia/macrophages, monocytes, T/NK cells and B cells (Figure S5A). To increase resolution on the microglia/macrophages population we re-clustered these cells separately and identified three clusters of Smad4F/F microglia (MgF/F 1–3), four clusters of Smad4 cKO microglia (MgcKO 1–4), two clusters of BAMs (BAM-1 and BAM-2) and one cluster of mitotic microglia (Figure 5C). MgF/F clusters were almost entirely derived from Smad4F/F mice (96.5–99.5%), while MgcKO clusters stemmed from Smad4 cKO mice (98.1–99.8%). Mitotic microglia were more contributed from Smad4 cKO compared to Smad4F/F mice (67.6% and 32.4% respectively), suggesting increased microglia proliferation in absence of SMAD4 (Figure 5D). Monocytes, T/NK cells and B cells appeared equally contributed from each genotype (Figure S5B). Differential gene expression analysis (Log2(FC)>0.5, p<0.01, expressed in >10% of cells/cluster) revealed broad transcriptional differences in microglia from Smad4 cKO mice compared to Smad4F/F microglia, while BAMs and monocytes were only marginally affected (Figure 5E). Inspecting differentially expressed genes, we found that microglia exhibited a loss of their homeostatic signature, with downregulation of virtually all microglia marker genes (Sparc, Selplg, Hexb, P2ry12, Tmem119). In parallel, BAM signature genes (Apoe, Mrc1, Lyz2, Pf4, Ms4a7) were upregulated (Figure 5F and 5G). Confocal imaging confirmed CD206 upregulation in Smad4 cKO microglia (Figure 5H, S5C and SD). Downregulation of P2RY12 and TMEM119 was validated by flow cytometry (Figure S5E and S5F). Therefore, SMAD4 deletion in embryonic brain macrophages disrupted the physiological maturation of microglia, which instead acquired a BAM-2 signature. Comparing Smad4 cKO microglia to BAM-2, only a few transcripts appeared enriched in the latter population (not shown). Among these, Cd163 and Lyve1 retained their strict specificity for BAMs (Figure 5I), indicating that the expression of these genes is not regulated by SMAD4. Importantly, we found no differences in the expression of macrophage lineage genes (C1qa, Spi1, Fcgr3, Fcgr1) between the two genotypes (Figure S5G). This indicates that SMAD4 is redundant for the lineage commitment of embryonic brain macrophages, but it is required to activate the microglial transcriptional program. It should however be noted that deletion of SMAD4 may amplify non-canonical TGF-β signaling, as previously described in NK cells [68]. Whether a similar mechanism occurs in SMAD4-deficient microglia should be tested in future studies.
Figure 5. Maturation failure of SMAD4-deficient microglia.
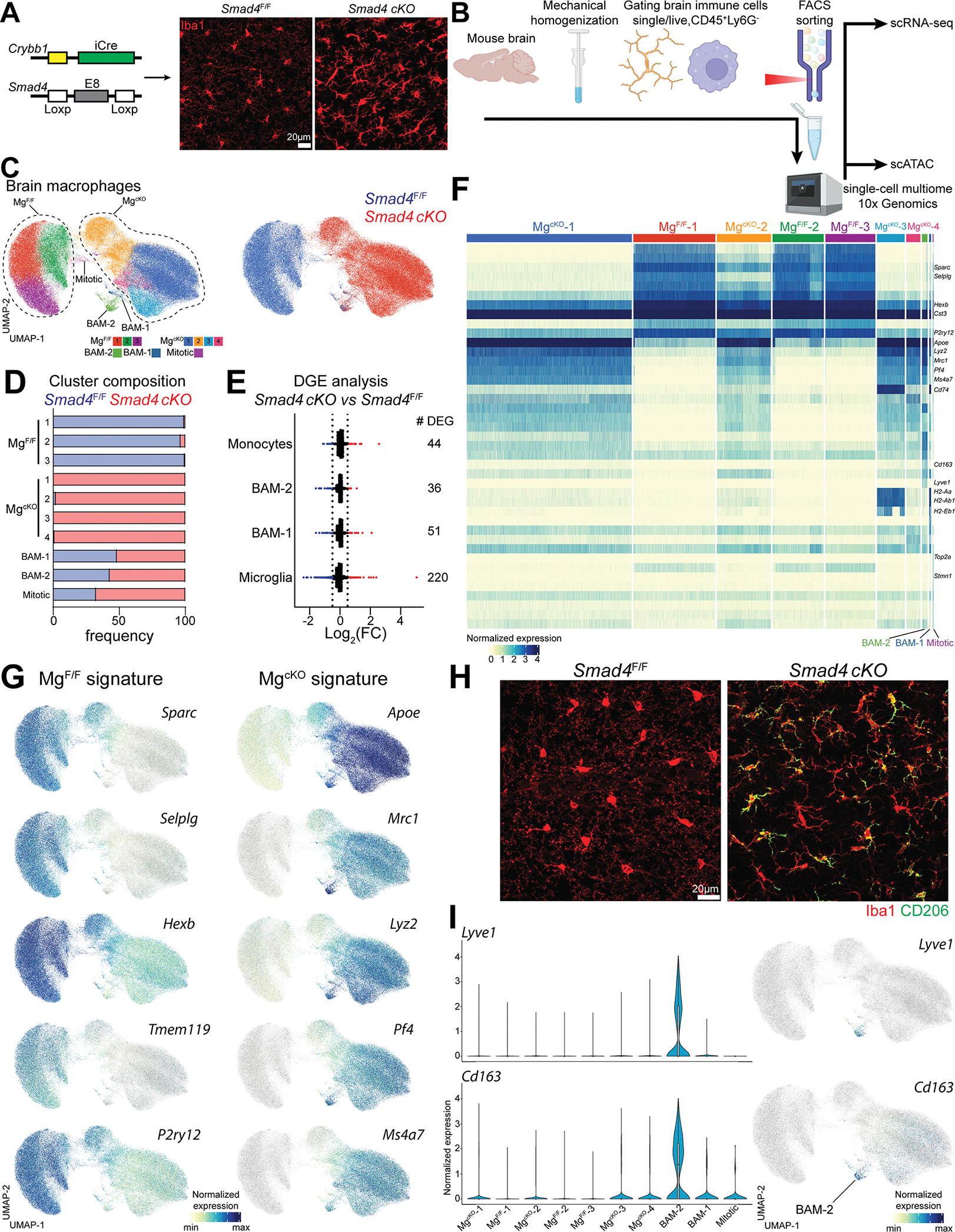
(A) Strategy used to generate the Smad4 cKO line. Representative confocal image of microglia from either Smad4F/F and Smad4 cKO (n=5 mice/genotype, 4-week-old, two independent experiments).
(B) Cartoon describing the experimental design.
(C) UMAP plot of microglia and BAMs from Smad4F/F and Smad4 cKO littermate mice, split either by cluster or genotype (n=4 mice/genotype, 6–8 weeks old).
(D) Percentage of genotypes in each cluster.
(E) Number and fold-change of differentially expressed genes in brain myeloid populations from Smad4F/F and Smad4 cKO mice.
(F) Heatmap of the top 10 signature genes per cluster.
(G) UMAP plot showing the differential expression of signature genes in Smad4F/F and Smad4 cKO mice.
(H) Representative confocal images of CD206 staining on microglia from Smad4F/F and Smad4 cKO mice (n=5 mice/genotype, 4-week-old, two independent experiments).
(I) UMAP and violin plots showing the expression of Lyve1 and Cd163 in each cluster.
See also Figure S5
SMAD4 deletion reshapes chromatin accessibility in microglia but not in BAMs
The 10x multiomic protocol allowed us to perform scATAC-seq (Figure 6A) on the same cells analyzed by scRNA-seq. After quality filtering we obtained 118,507 single cells with high quality ATAC-seq signal in all clusters and samples (Figure S6A and SB). Based on the ATAC accessibility for lineage specific genes, we identified clusters of macrophages, BAMs, monocytes, T cells and B cells (Figure S6C). Since RNA and ATAC data originating from the same cell have a unique barcode, we integrated the scRNA-seq cluster annotations into our ATAC-seq dataset (Figure S6D). Then, we performed re-clustering of the macrophage population only (containing microglia and BAMs) and analyzed these cells in more detail (Figure 6B). Again, we obtained clusters of MgF/F (1–3), MgcKO (1–4), BAM-1, BAM-2, and mitotic microglia. Microglia from Smad4 cKO and Smad4F/F littermates clustered on non-overlapping UMAP territories (Figure 6C). Compared to MgF/F, MgcKO exhibited a vast number of differentially accessible genes and acquired an epigenetic signature akin to BAM-2 (Figure 6D). Analysis of the open chromatin region (OCRs) between genotypes (FDR < 0.05) revealed 14,774 different OCRs in microglia, only two in BAM-2 and zero in BAM-1 populations (Figure 6E). For example, MgcKO exhibited minimal accessibility in the locus coding for the microglia signature gene Tmem119, while open chromatin was found in the loci coding for the BAM signature genes Apoe and Mrc1. No differences were found in the BAM-2 population between Smad4F/F and Smad4 cKO genotypes (BAM-2F/F and BAM-2cKO respectively) (Figure 6F and 6G). Next, we performed analysis of cis-regulatory elements to identify transcription factors (TF) binding motifs differentially enriched in MgF/F and MgcKO. Investigated populations were clustered based on the 16,270 OCRs detected in our dataset. Then, we searched for known TF motifs enriched in these OCRs and found several TF binding sites with different enrichment for each population (Figure 6H). Specifically, the MgcKO population exhibited an abundance of binding motifs for Spi1 (PU.1), IRF1, and several members of the MAF and C/EBP families (Figure 6I). Conversely, the MgcKO population was poorly enriched for SMAD2/3 binding motifs (Figure S6E). This data indicates that SMAD4 deletion reshaped the chromatin landscape in microglia, but not in BAMs. Furthermore, lack of SMAD4 increased accessibility to genomic loci containing binding sites for several MAF-family TFs. Whether MAF-family TFs drive the BAM-like signature in SMAD4-deficient microglia should be assessed in future studies.
Figure 6. Broad epigenetic changes in SMAD4-deficient microglia.
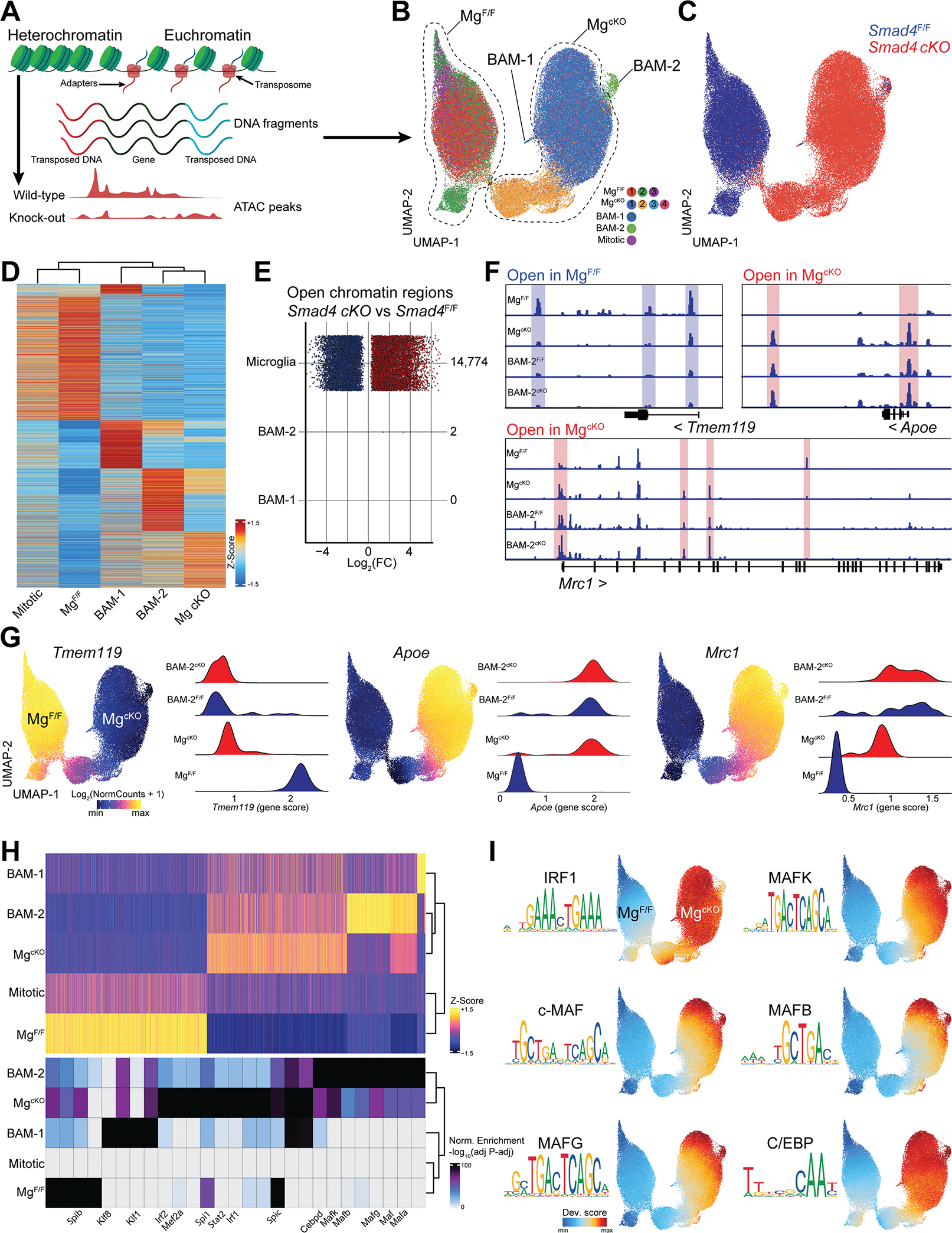
(A) Cartoon describing scATAC-seq protocol and analysis.
(B) UMAP of the scATAC-seq profiles of microglia and BAMs from Smad4F/F and Smad4 cKO littermate mice split by cluster (n=4 mice/genotype, 6–8 weeks old).
(C) UMAP of the scATAC-seq profiles of microglia and BAMs split by genotype.
(D) Heatmap displaying accessibility of 6,335 marker genes for the indicated cell populations.
(E) Number and fold-change of differential OCRs in Smad4 cKO vs Smad4F/F for the indicated cell types.
(F) Pseudo-bulk ATAC-seq coverage of selected gene loci.
(G) UMAP of the scATAC-seq profiles colored by accessibility of the indicated gene, and quantification of locus accessibility (gene score) by cell population and genotype.
(H) Heatmap of 16,270 OCRs in the indicated cell populations and top enriched motifs for each population. Up to 8 motifs are shown per population.
(I) UMAP of the scATAC-seq profiles colored by enrichment of the indicated TF motifs.
See also Figure S6
SMAD4 deletion with Crybb1-Cre causes memory impairment in mice
Apart from broad phenotypic changes in microglia, we observed widespread astrogliosis in Smad4 cKO mice, evidenced by GFAP upregulation throughout the cortical areas (Figure 7A and B). This suggested that SMAD4 deletion in microglia may perturb the CNS microenvironment in a cell-extrinsic manner. Thus, we performed a battery of behavioral tests to assess basal locomotor and exploration activity, motor learning and memory skills in these mice. Smad4 cKO mice exhibited slightly increased locomotor activity at the Open Field test (OFT) compared to Smad4F/F littermates (Figure 7C). Exploratory behavior at the Elevated Plus Maze (EPS) was also increased in Smad4 cKO mice (Figure 7D). Furthermore, no difference was observed between genotypes at the accelerated Rotarod test, assessing motor learning and coordination (Figure 7E). These data indicate that deletion of SMAD4 in microglia did not impair mouse locomotor ability. At the Morris Water Maze, however, Smad4 cKO mice exhibited a delayed learning curve, although it did not reach statistical significance (Figure 7F), and a significant impairment of memory recall (Figure 7G). Latency to locate the visible platform and swimming speed were unchanged between genotypes (Figure S6F), indicating intact visual and swimming ability. Together, these data indicate that SMAD4 deletion in microglia impaired learning and memory skills. Further studies are needed to determine whether SMAD4-deficient microglia alter physiological brain wiring or synaptic activity.
Figure 7. Memory impairment in mice with SMAD4-deficient microglia.
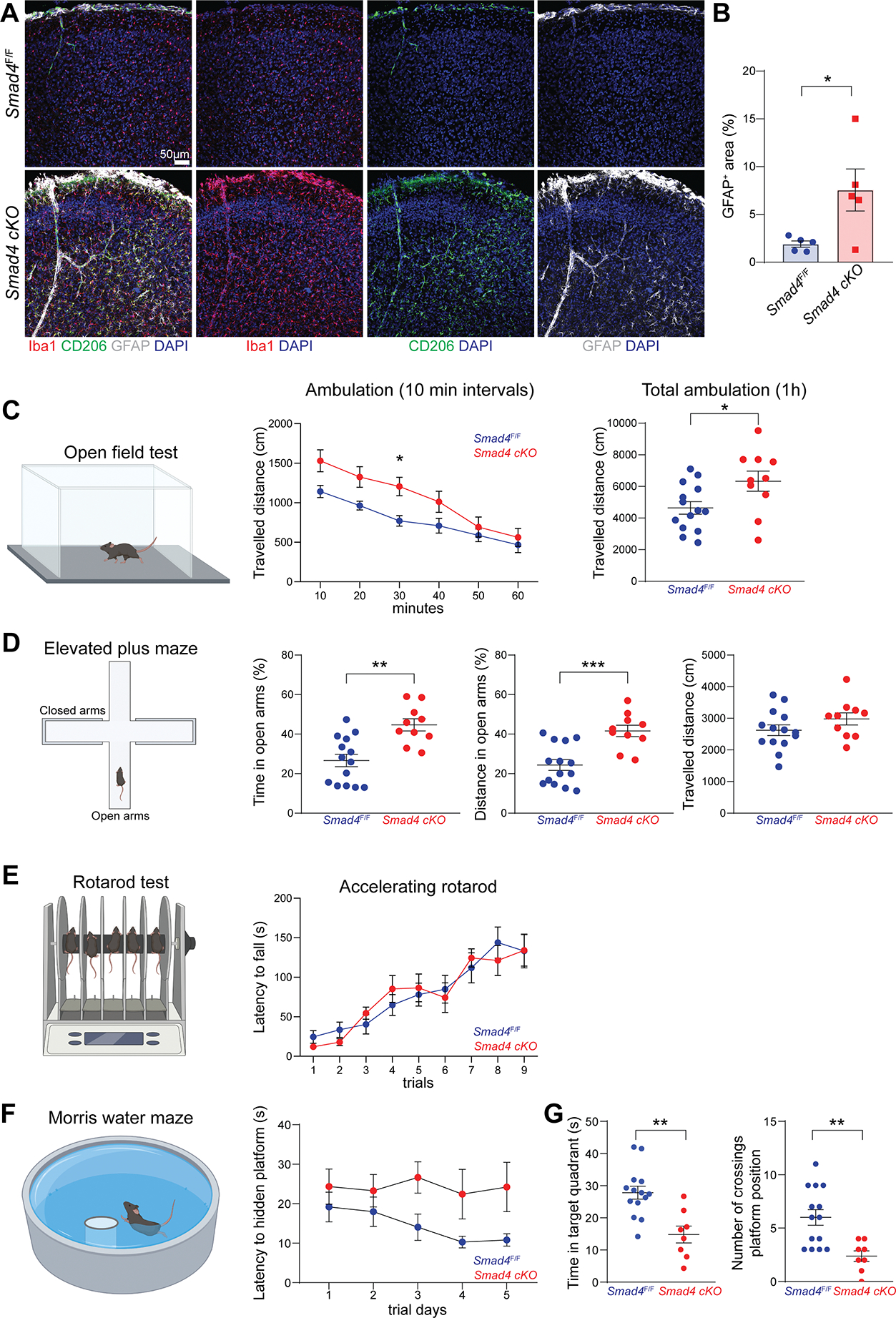
(A) Representative confocal images of Iba1, CD206 and GFAP staining on brains from Smad4F/F and Smad4 cKO mice (n=5 mice/genotype, 4-week-old, two independent experiments).
(B) Percentage of GFAP covered area in the brain of Smad4F/F and Smad4 cKO mice (n=5 mice/genotype, 4-week-old, displayed mean values of three sections per mouse, Mann-Whitney U test, *P<0.05, pool of two independent experiments).
(C) Open Field Test on Smad4F/F and Smad4 cKO littermate mice assessing travelled distance every 10 min intervals, and total travelled distance during 1h test (n=14 Smad4F/F and n=10 Smad4 cKO, 2–3-month-old, two-way ANOVA with Bonferroni post-hoc test and Mann-Whitney U test, *P<0.05, pool of three independent experiments).
(D) Elevated Plus Maze test on Smad4F/F and Smad4 cKO littermate mice assessing percentage of time and percentage of travelled distance in open arms, and total travelled during 5 min test (n=14 Smad4F/F and n=10 Smad4 cKO, 2–3-month-old, Mann-Whitney U test, **P<0.01, ***P<0.001, pool of three independent experiments).
(E) Rotarod test on Smad4F/F and Smad4 cKO littermate mice assessing latency time to fall from accelerated rod during 9 trials (n=14 Smad4F/F and n=10 Smad4 cKO, 2–3-month-old, Mann-Whitney U test, pool of three independent experiments).
(F) Morris Water Maze test on Smad4F/F and Smad4 cKO littermate mice assessing latency time to locate hidden platform during 5 days of training (n=14 Smad4F/F and n=8 Smad4 cKO, 3–4-month-old, two-way ANOVA with Bonferroni post-hoc test, pool of three independent experiments).
(G) Morris Water Maze test on Smad4F/F and Smad4 cKO littermate mice assessing swimming time in target quadrant and number of crossings over platform position during probe trial (n=14 Smad4F/F and n=8 Smad4 cKO, 3–4-month-old, Mann-Whitney U test, **P<0.01, pool of three independent experiments).
See also Figure S6
DISCUSSION
In this study we introduced a Cre deleter (Crybb1-Cre) which recombines embryonic brain macrophages (microglia and BAMs) with high efficiency and specificity. BAMs are highly heterogeneous macrophages [19, 41, 69] and originate from both embryonic and adult hematopoiesis [16–18]. Previous studies broke down BAM diversity in homeostasis and neuroinflammation using high-dimensional cytometry [41, 42, 70] and scRNA-seq [19, 69, 71, 72]. For example, Mrdjen et al. identified four different BAM subsets with diverse expression of MHC2 and CD38 [41]. These populations resemble those described by Van Hove et al. using MHC2 and FOLR2 as discrimination markers [19]. More recently, three distinct BAM subsets were reported based on TIM4, LYVE1, FOLR2, MHC2 and CCR2 expression [71], or CD206, MHC2 and CD11a expression [73]. Although different staining panels have been used across these studies, we believe that these authors have consistently identified the same populations. Delving into our scRNA-seq data, we chose MHC2 and CD38 as ideal surface markers to resolve heterogeneity as proposed by Mrdjen et al. We showed that CD38+MHC2+ and CD38+MHC2− BAMs are mostly found in the choroid plexus, whereas CD38−MHC2+ and CD38+MHC2− BAMs are enriched in the cortex (leptomeninges and perivascular spaces). Focusing on the cortex (excluding dura mater and choroid plexus BAMs), CD38+MHC2− BAMs were maximally labeled with our Crybb1-Cre lineage tracer, whereas CD38−MHC2+ BAMs were efficiently targeted with the Flt3-Cre fate-mapping system. This indicates that CD38+MHC2− BAMs in the brain proper have mostly embryonic origin, whereas CD38−MHC2+ BAMs are monocyte-derived. The phenotype of tissue resident macrophages heavily relies on the environmental cues within the tissue niche [65]. Indeed, monocyte-derived macrophages have shown a high phenotypic plasticity and capacity to acquire a niche specific signature in different compartments [74–80]. Nevertheless, our data suggest that ontogeny strictly dictates the phenotype of BAMs, at least in steady state.
We used Crybb1-Cre to delete SMAD4 from microglia and embryonic BAMs. Microglia underwent a developmental arrest, with nearly complete loss of their homeostatic signature (Sparc, Selplg, Hexb, P2ry12, Tmem119), and upregulation of BAMs signature genes (Apoe, Mrc1, Lyz2, Pf4, Ms4a7). Alongside, microglia exhibited broad chromatin remodeling, which exposed genomic loci containing BAM signature genes and MAF family binding motifs. By contrast, embryonic-derived BAMs (alias BAM-2) were unaffected by SMAD4 deletion. This phenotype is similar, but not identical, to that described after Tgfbr2 deletion in embryonic hematopoietic cells using Vav1iCre mice [18].
Unlike this group, we did not observe reduced microglia numbers, reduced proliferation, or upregulation of inflammatory genes (not shown). These differences may stem from the simultaneous disruption of SMAD4-dependent and independent pathways under Tgfbr2 deficiency. Lastly, mice with SMAD4-deficient microglia exhibited impaired memory skills and widespread astrogliosis compared to littermate controls. This data contributes to the growing body of evidence that phenotypic maturation of microglia is tightly linked to the physiological brain development and function, and vice versa [81–90].
Microglia and embryonic BAMs share the same origin [17]. Therefore, there must be some environmental factors driving either the BAMs or microglia fates locally in the CNS. We showed that in the absence of SMAD4, the microglial transcriptome and epigenome become almost indistinguishable from that of BAMs, thus SMAD4 crucially controls the microglia specification program. By contrast, SMAD4 appears redundant for BAM maturation. To conclude, the Crybb1-Cre line described here is an ideal tool to assess the role of genes (including TFs) potentially involved in microglia and BAM development.
LIMITATIONS OF THE STUDY
Crybb1-Cre : R26-tdTomato mice exhibited recombination in OPCs and neurons. Off-target recombination of non-microglia brain cells may represent a bias for functional studies. Furthermore, we did not determine if substantial recombination occurred in tissues other than those assessed here. About 30% of CD38+ BAMs were labeled with the Flt3-Cre, whereas ~95% of the same population was labeled with the Crybb1-Cre. At present, we cannot completely exclude that some monocyte-derived BAMs may transiently express Crybb1 during differentiation and therefore undergo recombination. Although our bone marrow chimera experiment seems to exclude this scenario, future studies are warranted to determine if fetal liver HSCs can supply monocyte-derived macrophages to the CD38+ BAM subset. Postnatal deletion of SMAD4 in microglia should be performed to completely clarify the role of SMAD4 after microglia differentiation has occurred. Lastly, our study is limited to subdural and perivascular (cortical) BAMs. A detailed analysis of BAMs from choroid plexus and dura mater should be addressed in future studies.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Marco Colonna (mcolonna@wustl.edu).
Materials availability statement
The following mouse lines generated in this study have been deposited at the Washington University Pathology Transgenic and Knockout Mouse Core Cryo facility: Crybb1 knock-out (Colonna F2–12-3–13), Crybb1-tdTomato (Colonna F2–13-2–7), Crybb1-Cre (Colonna F2–13-2–6).
Data and code availability
Single-cell RNA-seq and ATAC-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the Key Resources Table.
Original codes have been deposited at GitHub and are publicly available as of the date of publication. DOIs are listed in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Primary antibodies and dyes flow cytometry | ||
|
| ||
| CD45-APCCy7 | Biolegend | RRID: AB_312981 |
| CD45-PE | Biolegend | RRID: AB_2563598 |
| CD45-BUV563 | BD Bioscience | RRID: AB_2870209 |
| CD45-AF700 | Biolegend | RRID: AB_493715 |
| CD45.2-APC | Biolegend | RRID: AB_389211 |
| CD45.1-Biotin | Biolegend | RRID: AB_313493 |
| CD11b-PECy7 | Biolegend | RRID: AB_312799 |
| CD11b-BV421 | Biolegend | RRID: AB_10897942 |
| CD11b-APCCy7 | Biolegend | RRID: AB_830642 |
| CD11b-BB515 | BD Biosciences | RRID: AB_2665392 |
| CD11b-APC | Biolegend | RRID: AB_312795 |
| CD11b-PerCPCy5.5 | Biolegend | RRID: AB_2129374 |
| CD11c-APCCy7 | Biolegend | RRID: AB_830649 |
| CD11c-BV421 | Biolegend | RRID: AB_10897814 |
| CD11c-PECy7 | Biolegend | RRID: AB_493568 |
| Ly6C-FITC | Biolegend | RRID: AB_1186135 |
| Ly6C-PE | Biolegend | RRID: AB_1186132 |
| Ly6C-APCCy7 | Biolegend | RRID: AB_10640120 |
| Ly6G-FITC | Biolegend | RRID: AB_1236494 |
| Ly6G-APC | Biolegend | RRID: AB_2227348 |
| Gr-1-PerCPCy5.5 | Biolegend | RRID: AB_893557 |
| NK1.1-FITC | Biolegend | RRID: AB_313393 |
| NK1.1-PerCPCy5.5 | Biolegend | RRID: AB_2132707 |
| CD43-FITC | Biolegend | RRID: AB_10960745 |
| CD43- PerCPCy5.5 | Biolegend | RRID: AB_2286556 |
| CD44-FITC | Biolegend | RRID: AB_312957 |
| CD44-PE | Biolegend | RRID: AB_312959 |
| F4/80-APC | Biolegend | RRID: AB_2832549 |
| F4/80-Biotin | Biolegend | RRID: AB_893501 |
| F4/80-BV605 | Biolegend | RRID: AB_2562305 |
| CX3CR1-PECy7 | Biolegend | RRID: AB_2565700 |
| CX3CR1-APC | Biolegend | RRID: AB_2564492 |
| MERTK-BV421 | Biolegend | RRID: AB_2832533 |
| I-A/I-E(MHC-M)-PECy7 | Biolegend | RRID: AB_313327 |
| I-A/I-E(MHC-II)-BV650 | Biolegend | RRID: AB_2565975 |
| I-A/I-E(MHC-II)-BUV805 | BD Biosciences | RRID: AB_2873247 |
| Siglec-F-APC | Biolegend | RRID: AB_2750237 |
| CD115-BV421 | Biolegend | RRID: AB_2562667 |
| CD206-AF488 | Biolegend | RRID: AB_10900445 |
| CD207-PECy7 | Biolegend | RRID: AB_2876490 |
| FOLR2-APC | Biolegend | RRID: AB_2721313 |
| FOLR2-PE | Biolegend | RRID: AB_2721344 |
| CD38-PECy7 | Biolegend | RRID: AB_2275531 |
| CD64-PE | Biolegend | RRID: AB_10612740 |
| TMEM119-PE | Invitrogen | RRID: AB_2848262 |
| P2RY12-Biotin | Biolegend | RRID: AB_2749906 |
| cKit-BB515 | BD Biosciences | RRID: AB_2738826 |
| CD93-APC | Biolegend | RRID: AB_2275868 |
| EpCAM-BV711 | Biolegend | RRID: AB_2632775 |
|
| ||
| Primary antibodies immunofluorescence | ||
|
| ||
| Rabbit Iba1 | Cell signaling | RRID: AB_2820254 |
| Rabbit CD206 | Cell signaling | RRID: AB_2892682 |
| Rat CD206-AF488 | Biolegend | RRID: AB_10900445 |
| Rat MHC2-AF647 | Biolegend | RRID: AB_493526 |
| Rat EpCAM-AF647 | Biolegend | RRID: AB_1134104 |
| Rat CD38-AF647 | Biolegend | RRID: AB_2073334 |
| Mouse GFAP-AF594 | Cell signaling | RRID: AB_10998775 |
| Mouse GFAP-AF488 | Invitrogen | RRID: AB_10598515 |
| Rabbit CRYBB1 | Cell signaling | Cat# 95666 |
| Rabbit Olig2 | MilliporeSigma | RRID: AB_570666 |
| Mouse APC(CC-1) | MilliporeSigma | RRID: AB_2057371 |
| Mouse NeuN-AF488 | MilliporeSigma | RRID: AB_2149209 |
| Rat CD45-AF488 | Biolegend | RRID: AB_493531 |
| Rat CD45-AF647 | Biolegend | RRID: AB_2876569 |
| Rat CD34-eFluor 660 | Invitrogen | RRID: AB_10596826 |
| Rat GFP-AF488 | Biolegend | RRID: AB_2563288 |
|
| ||
| Secondary antibodies immunofluorescence | ||
|
| ||
| Donkey anti-rat IgG AF647 | Life technologies | RRID: AB_2896338 |
| Chicken anti-rat IgG AF488 | Life technologies | RRID: AB_2535873 |
| Donkey anti-rabbit IgG AF647 | Life technologies | RRID: AB_2536183 |
| Donkey anti-rabbit IgG AF488 | Life technologies | RRID: AB_2535792 |
| Donkey anti-rabbit IgG AF555 | Life technologies | RRID: AB_162543 |
| Goat anti-mouse IgG AF555 | Life technologies | RRID: AB_2535844 |
| Goat anti-mouse IgG1 AF488 | Life technologies | RRID: AB_2535764 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| EDTA | Corning | Cat# 46-034-Cl |
| Triton X-100 | Sigma | Cat# T8787 |
| Tween 20 | Fisher Bioreagents | Cat# BP337 |
| Paraformaldehyde 32% | Electron Microscopy Science | Cat# 15714-S |
| NP-40 Substitute | Sigma | Cat# 74385 |
| 5% Digitonin | Thermo Fisher | Cat# BN2006 |
| DL-Dithiothreitol solution (DTT) | Sigma | Cat# 646563 |
| RNase inhibitor | Promega | Cat# N2515 |
| 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) | Sigma | Cat# D9542 |
| Tris-HCl (pH 7.4) | Sigma | Cat# T2194 |
| NaCl | Sigma | Cat# 59222C |
| MgCl2 | Sigma | Cat# M1028 |
| Tamoxifen diet (500 mg tamoxifen/Kg chow) | Envigo | Cat# TD.130857 |
| Streptavidin BV421 | Biolegend | Cat# 405225 |
| Streptavidin AF488 | Biolegend | Cat# 405235 |
| Zombie Aqua Fixable Viability Kit | Biolegend | Cat# 423102 |
| 2.4G2 CD16/32 Fc block from 197 hybridomas | ATCC | Cat# HB-197 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Chromium Next GEM Single Cell 3’ Kit v3.1 | 10× Genomics | PN-1000268 |
| Chromium Next GEM Chip G Single Cell Kit | 10× Genomics | PN-1000120 |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle | 10× Genomics | PN-1000283 |
| Chromium Next GEM Chip J Single Cell Kit | 10× Genomics | PN-1000234 |
| Single Index Kit N Set A | 10× Genomics | PN-1000212 |
| Dual Index Kit TT Set A | 10× Genomics | PN-1000215 |
| NovaSeq6000 | Illumina | S4 Flow Cell |
|
| ||
| Deposited data | ||
|
| ||
| Single cell transcriptomic and ATAC sequencing data | This paper | GEO: GSE213020 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mice | Jackson laboratory | JAX:000664 |
| Flt3-Cre mice | Benz et al.54 | MGI: 4462354 |
| Rosa26-STOP-EYFP mice (Ai2) | Madisen et al.91 | JAX:007920 |
| Rosa26-STOP-tdTomato mice (Ai14) | Madisen et al.91 | JAX: 007908 |
| Smad4-flox mice | Yang et al.92 | JAX: 017462 |
| Lyz2-CreEr2 mice | Canli et al.57 | JAX: 032291 |
| Nur77GFP mice | Zikherman et al.93 | MMRRC: 012015 |
| Zbtb46GFP mice | Satpathy et al.43 | JAX: 027618 |
| Crybb1 knock-out mice | This paper | Colonna F2-12-3-13 |
| Crybb1-tdTomato mice | This paper | Colonna F2-13-2-7 |
| Crybb1-Cre mice | This paper | Colonna F2-13-2-6 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Crybb1 gRNA exon1: AGCACCAGGAACCATGTCCCNGG | This paper | N/A |
| Crybb1 gRNAs exon3: GTGACCGGCTCATGTCCTTCNGG; GTGGGTACTCGCCCTTCTCCNGG | This paper | N/A |
| Crybb1-Cre Fwd. primer: AGACAATAGCAGGCATGCTGG | This paper | N/A |
| Crybb1-Cre Rev. primer: GGATCAGTACAGCCCAGCTC | This paper | N/A |
| Crybb1 knock-out Fwd. primer: GGGTGGCCTTTGAGCAATCT | This paper | N/A |
| Crybb1 knock-out Rev. primer: ACGTCACATCTTCCCCCAAA. | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Cell Ranger v2.0.0 and v6.0.0 | 10× Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| R project 4.1.3 | http://www.r-project.org/ | RRID: SCR_001905 |
| Rstudio | https://posit.co | RRID: SCR_000432 |
| Seurat v4.1.1 | Hao et al.94 | RRID: SCR_007322 |
| ArchR v1.0.1 | Granja et al.95 | http://www.archrproject.com/ |
| Deposited algorithms | This paper | https://doi.org/10.5281/zenodo.7558104 |
| ImageJ/Fiji | Schneider et al.96 | RRID: SCR_002285 |
| Imaris V8.3 | Bitplane | RRID: SCR_007370 |
| ANY-maze Video Tracking Software | Stoelting | RRID: SCR_014289 |
|
| ||
| Other | ||
|
| ||
| PBS | Corning | Cat# 21-040-CM |
| DMEM | Gibco | Cat# 11965-084 |
| RPMI | Sigma | Cat# R8758 |
| HBSS | Gibco | Cat# 14185-052 |
| HEPES | Corning | Cat# 25-060-Cl |
| Bovine calf serum (BCS) | Cytiva | Cat# SH30072.04 |
| BSA | Rockland | Cat# BSA-1000 |
| 10X red blood cells (RBC) lysis buffer | Biolegend | Cat# 420302 |
| Percoll | GE Healthcare | Cat# 17089101 |
| Collagenase-D | Sigma | Cat# 11088882001 |
| Collagenase-II | Gibco | Cat# 17101015 |
| Collagenase-IV | Sigma | Cat# C4-22-1G |
| Liberase TM | Sigma | Cat# 5401127001 |
| Hyaluronidase | Sigma | Cat# H1115000 |
| Dispase-II | Gibco | Cat# 17105-041 |
| DNase-I | Sigma | Cat# 10104159001 |
| Tomato-lectin Dylight 649 | Vector Laboratories | Cat# DL-1178 |
| Tomato-lectin Dylight 488 | Vector Laboratories | Cat# DL-1174-1 |
| Superfrost glass slides | Fisher Scientific | Cat# 12-550-15 |
| Prolong Glass anti-fade mounting media | Thermo Fisher | Cat# P36980 |
| Fluoromount-G mounting media | SouthernBiotech | Cat# 0100-01 |
| Nuclei Buffer | 10× Genomics | PN-2000207 |
| Ultra-Pure BSA | Thermo Fisher | Cat# AM2616 |
| Nuclease-free water | Invitrogen | Cat# AM9937 |
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS
Animals
All mice used in this study were C57BL/6J background housed under specific pathogen free conditions at Washington University School of Medicine animal facility. Both male and female mice were used between P20 and P90. Embryos were analyzed between E10.5 and E18.5. Crybb1 knock-out, Crybb1-tdTomato and Crybb1-Cre mice were generated by the Genome Engineering and iPSC Center (GEiC), and the Transgenic, Knockout and Micro-Injection Core at Pathology and Immunology department, Washington University in St. Louis. Crybb1-tdTomato and Crybb1-Cre mice were generated by knock-in/knock-out strategy using CRISPR/Cas9 technology. One gRNA complementary to the 5’ end of exon 1 with predicted high on-target activity score was used (AGCACCAGGAACCATGTCCCNGG). B6/J zygotes at 0.5 days post oocytes fertilization were transduced with an AAV6 to deliver either tdTomato or codon-optimized iCre sequences with 3’ poly(A) signal and flanked by homology arms. CRISPR/Cas9 proteins and gRNA (RPMs) were introduced into the zygote by electroporation. Embryos were subsequently transferred into a D0.5 pseudo-pregnant B6/J recipient female. Resulting founders were screened by next-generation sequencing (NGS) and male mice with targeted integration only were bred to B6/J female mice from Jackson Laboratory (JAX stock # 000664). Crybb1-Cre mice were further intercrossed with homozygous Rosa26-STOP-tdTomato mice (Ai14; JAX stock # 007908) [91] or Smad4-flox mice (JAX stock # 017462) [92], both bred in house. For genotyping of locus specific iCre the following primers were used; Fwd. AGACAATAGCAGGCATGCTGG; Rev. GGATCAGTACAGCCCAGCTC. Crybb1-tdTomato mice did not require PCR for genotyping. Crybb1 is highly expressed in the eye lens and Crybb1-tdTomato carriers exhibited bright red pupils.
Crybb1 knock-out mice were generated by CRISPR/Cas9 technology. Two gRNAs complementary to exon 3 with predicted high on-target activity score were used (GTGACCGGCTCATGTCCTTCNGG; GTGGGTACTCGCCCTTCTCCNGG). CRISPR/Cas9 proteins and gRNAs (RPMs) were introduced into B6/J zygotes at 0.5 days post oocytes fertilization by electroporation. Embryos were subsequently transferred into a D0.5 pseudo-pregnant B6J recipient female. Resulting founders were screened by NGS. A male founder with germline out-of-frame deletion of 177bp in exon 3 was bred to C57BL/6J female mice from Jackson Laboratory. For genotyping of Crybb1 knock-out the following primers were used; Fwd. GGGTGGCCTTTGAGCAATCT; Rev. ACGTCACATCTTCCCCCAAA.
Flt3-Cre mice [54] crossed with Rosa26-STOP-EYFP mice (Ai2; JAX stock # 007920) [91] bred in house were kindly provided by Dr. David DeNardo.
Lyz2-CreErt2 males purchased from Jackson Laboratory (JAX stock # 032291) [57] were crossed with Rosa26-STOP-tdTomato mice (Ai14). At the age of P30, CreErt2 carriers were fed with tamoxifen-containing chow (500 mg tamoxifen/Kg, Envigo) for four weeks. This formula is expected to provide ~80 mg tamoxifen per Kg body weight per day in 20–25g mice.
Nur77GFP mice [93] were bred in house, and Zbtb46GFP mice [43] were kindly provided by Dr. Kenneth Murphy.
All experiments involving laboratory animals were performed under the approval of the Institutional Animal Care and Use Committee at Washington University in St. Louis (protocol #19–0981).
METHODS DETAILS
Sample preparation for flow cytometry or FACS sorting
Except for dura, kidney, heart, small intestine and lung, all sample preparation was carried out on ice and no enzymatic digestion was used. Mice were deeply anesthetized with a lethal dose of Ketamine/Xylazine cocktail injected ip. Peritoneal lavage was collected by flushing 5ml of ice-cold phosphate buffer saline (PBS, Corning) in the peritoneal cavity. Blood was sampled from the right ventricle prior perfusion and underwent red blood cells (RBC) lysis (Biolegend) for 5 min at room temperature. All other samples were collected after transcardial perfusion with 20ml ice-cold PBS. Brain was harvested and collected in ice-cold DMEM (Gibco). When specified, cortices and choroid plexus from lateral and IV ventricles were separated. Cortices or whole brains were mechanically dissociated with dounce homogenizer, filtered through a 70 μm strainer into a 50 ml tube and spun down for 10 min/300g at 4°C. Brain cell pellet was further resuspended in 30% isotonic percoll (GE Healthcare), and myelin fraction was depleted by centrifugation for 20 min/800g at 4°C (acceleration 1, break 1). Choroid plexus was collected in ice-cold DMEM (Gibco) and mechanically homogenized with a 3ml syringe, using in sequence 19- and 23-gauge needles. Dura was dissected from the inner surface of the skull and collected in complete RPMI (Sigma) supplemented with 10% bovine calf serum (BCS) and Collagenase-D (0.25 U/ml, Sigma) and incubated at 37°C for 20 min. After digestion, dura was mechanically dissociated with dounce homogenizer, filtered through a 70 μm strainer into a 50 ml tube and spun down for 10 min/300g at 4°C. Spleen and liver were mashed on a 70 μm strainer and collected into a 50 ml tube, followed by RBC lysis for 2 min on ice. Hepatocytes were depleted by sedimentation under low gravity centrifugation (5 min/30g at 4°C). Supernatant containing liver immune cells was collected. Kidneys were collected in complete RPMI supplemented with 10% BCS and Collagenase-D (0.25 U/ml, Sigma), finely chopped with scissors and incubated at 37°C for 30 min. Digested kidneys were mashed on a 70 μm strainer and collected into a 50 ml tube, followed by RBC lysis for 2 min on ice. Heart was collected in DMEM supplemented with Liberase TM (0.26 U/ml, Sigma), Hyaluronidase (100 U/ml, Sigma) and DNase-I (50 U/ml, Sigma) and incubated at 37°C for 45 min, as previously described [94]. Digested heart tissue was filtered through a 40 μm strainer and collected into a 50 ml tube, followed by RBC lysis for 5 min on ice. Small intestine was collected in HBSS, flushed to remove luminal contents, and opened lengthwise. Peyer’s patches were removed. To remove epithelial cells, intestine tissue was shacked for 20 min at room temperature in HBSS (Gibco) supplemented with 10 % BCS, 15mM HEPES (Corning), and 5 mM EDTA (Corning). The tissue was rinsed in HBSS and underwent additional 20 min of shacking as above. Intestinal tissue depleted of epithelial cells was rinsed with HBSS and digested in complete RPMI supplemented with 10% BCS and Collagenase IV (100 U/ml, Sigma) for 40 min at 37°C under mild shacking, as previously described [95]. Digested tissue was filtered through a 100 μm strainer and collected into a 50 ml tube. Lungs were collected in PBS and finely chopped with scissors. The tissue was transferred into a 50 ml tube and digested in 3 ml of complete RPMI supplemented with 10% BCS, Liberase TM (0.26 U/ml, Sigma), Hyaluronidase (10 U/ml, Sigma), DNase-I (50 U/ml, Sigma), at 37°C for 40 minutes. Tubes were inverted few times every 10 minutes. Digested tissue was mashed on a 70 μm strainer and collected into a 50 ml tube. Skin was peeled off from the mouse ears and collected in PBS supplemented with 5% BCS, Dispase-II (0.6 U/ml, Gibco) and Collagenase D (0.25 U/ml, Sigma) and minced into pieces of 2–4 mm with scissors. Samples were then incubated at 37°C for 2 hours under mild stirring. Digested skin tissue was filtered through a 40 μm strainer and collected into a 50 ml tube. Visceral adipose tissue (VAT) was excised above the epididymis, collected in PBS, and minced with scissors. The tissue was digested in complete RPMI supplemented with 10% BCS and Collagenase-II (100U/ml, Gibco) at 37°C for 30 minutes, as previously described [96]. The digested tissue was filtered through a 100μm strainer and collected into a 50ml tube. Prior staining, all samples were washed once in FACS buffer with 1% bovine serum albumin (BSA, Rockland) and 5mM EDTA.
Flow cytometry analysis and sorting
For flow cytometry analyses, single cell suspensions underwent live/dead staining (Zombie Aqua, Biolegend) at 1:1000 dilution in PBS for 20 min at 4°C. Fc-receptor blockade was performed using CD16/32 blocking antibody (clone 2.4G2, made in house from HB-197 hybridoma cells) incubated 10 min on ice. Surface staining was performed for 30 min to 1 hour on ice. Flow cytometry analysis was performed on LSR Fortessa or Symphony A3 (BD Bioscience). Raw data were analyzed with FlowJo v10. For sorting experiments, cells underwent Fc-receptor blockade as above, followed by surface staining for 20 min on ice. To sort brain macrophages for scRNA-seq experiment, anti-CD11b and linage antibody cocktail (anti-Ly6C, anti-Ly6G, anti-CD43, anti-CD44 and anti-NK1.1) were used. To sort brain immune cells for multiomics protocol, anti-CD45 and anti-Ly6G antibodies were used. 1mg/ml DAPI (4′,6-diamidino-2-phenylindole, Sigma) at 1:5000 dilution was used for dead cells exclusion. Sorting was performed on FACS Aria-II (BD Bioscience).
Bone marrow chimeras
Eight-week-old C57BL/6J Cd45.1 mice (JAX stock # 002014) received 11 Gy of gamma irradiation, split into two doses 4 hours apart. After the second dose, mice were injected iv. with 2.5×106 Crybb1-Cre : R26-tdTomato bone marrow cells (Cd45.2). Mice were returned to their home cage for eight weeks before analysis. Percentage of chimerism was determined in blood myeloid cells by CD45.2 and CD45.1 staining.
Immunofluorescence staining
Upon perfusion, tissues were harvested and fixed in 4% paraformaldehyde (Electron Microscopy Science) at 4°C overnight. Fixed specimens were dehydrated in 30% sucrose solution for at least 48hours and then cut into 60 μm-thick sections at the cryostat (Leica). Staining was performed on free- floating sections. Cryosections were blocked for 4 hours in PBS + 5% BSA and 0.5% Triton X-100. Primary antibody staining was performed in PBS + 1% BSA and 0.5% Triton X-100, for 48 hours at 4°C. Secondary staining with fluorochrome-conjugated antibodies was performed at room temperature for 2 hours. Tomato-lectin (TL) staining was performed by retroorbital injection of 100 μl of 1mg/ml fluorochrome-conjugated TL (Vector Laboratories) in deeply anesthetized mice 5 min prior perfusion. Stained sections were mounted on Superfrost glass slides (Fisher Scientific) and embedded in Prolong Glass anti-fade mounting media (Thermo Fisher). After fixation, femurs, tibias, and skull bones underwent decalcification in 0.5M EDTA (Corning) for additional 72h. Staining was performed as described above. Dura maters were embedded in Fluoromount-G mounting media (SouthernBiotech) as whole mount preparation.
Confocal imaging
Confocal imaging of brain cryosections was performed using a Zeiss LSM880 airyscan inverted confocal microscope equipped with a 34-channel GaAsp (gallium arsenide phosphide) detector. 10–20 μm-thick z-stack images were acquired with a 20X/NA0.8 air objective or 40X/NA1.4 oil-immersion objective at 2048×2048-pixel resolution, z-step=1μm, line averaging=2, using ZEN Black (ZEISS Efficient Navigation) software (Zeiss). Maximal projections were rendered in Imaris V8.3 (Bitplane, Zurich, Switzerland). Number of cells and percentages of covered area were measured with ImageJ/Fiji [97]. Default automatic threshold was used, and manual adjustments were applied if necessary.
10x Genomics single-cell RNAseq library preparation
Sorted brain macrophages were resuspended in PBS + 0.04% BSA at a final concentration of ~1000 cells/μl. Single cells gene expression libraries and sequencing were generated by the McDonnell Genome Institute (MGI) at Washington University using the Next GEM Single Cell 3’ Reagents Kit from 10x Genomics. For sample preparation on the 10x Genomics platform, the Chromium Next GEM Single Cell 3’ Kit v3.1, 16 rxns (PN-1000268), Chromium Next GEM Chip G Single Cell Kit, 48 rxns (PN-1000120), and Dual Index Kit TT Set A, 96 rxns (PN-1000215) were used. Briefly, up to 16,500 cells were partitioned into nanoliter Gel-bead-in-Emulsions (GEMs) droplets containing retro-transcriptase reaction mix. Single-cell cDNA received a unique 12 nucleotide barcode and unique molecular identifier (UMI). GEM cDNA was amplified for 11 cycles before being purified using SPRIselect beads. GEX libraries were prepared as recommended by the 10x Genomics Chromium Single Cell 3’ Reagent Kits User Guide (v3.1 Chemistry Dual Index) with appropriate modifications to the PCR cycles based on the calculated cDNA concentration. The concentration of each library was accurately determined through qPCR utilizing the KAPA library Quantification Kit according to the manufacturer’s protocol (KAPA Biosystems/Roche). Normalized cDNA libraries were sequenced on a NovaSeq6000 S4 Flow Cell using the XP workflow and a 28×10×10×150 sequencing recipe according to manufacturer protocol (Illumina). A median sequencing depth of 50,000 reads/cell was targeted for each gene expression library.
10x Genomics multiomics library preparation
CD45+Ly6G− brain immune cells (>100,000) were sorted into collection tubes with 1% BSA, 1U/μl RNase inhibitor (Promega). Nuclei extraction has been performed as described in the 10x Genomics demonstrated protocol (CG000365). Briefly, cells were spun down 5 min at 500g at +4°C, followed by 3 min cell lysis on ice to extract nuclei. Cell lysis buffer contained 10mM Tris-HCl (pH 7.4), 10mM NaCl, 3mM MgCl2, 0.1% Tween-20 (Fisher Bioreagents), 0.1% NP-40 Substitute (Sigma), 0.01% Digitonin (Thermo Fisher), 1% UltraPure BSA (Thermo Fisher), 1mM DTT (Sigma), 1U/μl RNase inhibitor (Promega), diluted in nuclease-free water (Invitrogen). Nuclei were then washed three times in wash buffer (10mM Tris-HCl, 10mM NaCl, 3mM MgCl2, 0.1% Tween-20, 1% UltraPure BSA, 1mM DTT, 1U/μl RNase inhibitor). Nuclei were resuspended in 5 μl of 1x Nuclei Buffer (10x Genomics) with 1mM DTT and 1U/μl RNase inhibitor.
For sample preparation on the 10x Genomics platform, the Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle, 16 rxns (PN-1000283), Chromium Next GEM Chip J Single Cell Kit, 48 rxns (PN-1000234), Single Index Kit N Set A, 96 rxns (ATAC) (PN-1000212), Dual Index Kit TT Set A, 96 rxns (PN-1000215) (3v3.1 GEX), were used. Barcoded cDNA was amplified for 6 cycles before being purified using SPRIselect beads. The concentration of each library was accurately determined through qPCR utilizing the KAPA library Quantification Kit according to the manufacturer’s protocol (KAPA Biosystems/Roche) to produce cluster counts appropriate for the Illumina NovaSeq6000 instrument. Normalized GEX libraries were pooled and run over 0.05 of a NovaSeq6000 S4 flow cell using the XP workflow and running a 28×10×10×150 sequencing recipe in accordance with manufacturer’s protocol (Illumina). Target coverage was 500M reads per sample. Barcoded transposed DNA was amplified for 7 cycles before being purified using SPRIselect beads. ATAC libraries were pooled and run over 0.167 of a NovaSeq6000 S1 flow cell using the XP workflow and running a 51×8×16×51 sequencing recipe in accordance with manufacturer’s protocol (Illumina). Target coverage was 250M reads per sample.
Single-cell RNAseq analysis
Cell Ranger Software Suite (v6.0.0) from 10x Genomics was used for sample demultiplexing, barcode processing, and single cell counting. Cell Ranger count was used to align samples to the mm39 v104 reference genome, quantify reads, and filter reads and barcodes. An average of 100,817 GEX reads per cells was obtained. The Seurat (v4.1.1) package in R was used for downstream analysis. For quality control, cells with mitochondrial content <7.5% were removed. Cells with low UMI and gene number per cell were filtered out. Cutoffs of >1000 UMI and >800 genes were applied. Genes expressed in fewer than 10 cells were removed from the dataset. Data were normalized using the SCTransform method regressed on mitochondrial gene percentage using the glmGamPoi method. After QC, a median of 5159 UMIs and 2250 genes per cell was obtained. The top 30 principle components and a resolution of 0.4 were used for dimensionality reduction using the RunUMAP, FindNeighbors, and FindClusters functions. For data visualization, differential expression analysis and display of scRNA-seq data BBrowser version 3 was used. Differential gene expression analysis was performed using Venice from Bioturing (https://github.com/bioturing/signac).
Single-cell Multiomics analysis
Cell Ranger pipeline (v2.0.0) from 10x Genomics was used for sample demultiplexing, aligning scRNA and scATAC reads to the mm10 2020-A reference genome, and initial quality control. An average of 32,242 GEX and 23,218 ATAC reads per cell was obtained. For scRNA-seq analysis, counts matrices were imported into Seurat (v4.1.1) package in R for downstream processing [98]. After QC, a median of 2007 UMIs and 1224 genes per cell was obtained. For scATAC-seq analysis, fragments files were imported into ArchR (v1.0.1) for downstream processing [99]. For snRNA-seq data, barcodes were called as cells based on > 250 features and < 5% mitochondrial reads. For scATAC-seq data, barcodes were called as cells based on > 2000 but less than 106 fragments, and a minimum TSS enrichment of 4. After QC, a median of 26.383 TSS enrichment and 10,147 fragments per cell was obtained. After initial clustering of the scRNA-seq data using the top 10 principle components, a resolution of 0.15 and the “FindNeighbors”, “FindClusters”, and “RunUMAP” functions, microglia and BAMs were re-clustered to exclude non-macrophage populations as well as artificial clusters containing either low quality cells or cells enriched for immediate early genes (IEGs) induced by cell isolation procedure. For data visualization, differential expression analysis and display of scRNA-seq data BBrowser version 3 was used. scATAC-seq data was then subsetted based on barcodes present in the subsetted snRNA-seq dataset, and scRNA cluster labels were added onto the scATAC object by matching 10x barcodes. For scATAC-seq dimensionality reduction, ArchR functions “addIterativeLSI” and “addUMAP” were used. Default ArchR parameters, including imputation, were used for visualizing motif and gene accessibility in single cells. Normalized pseudo-bulk genome coverage tracks were exported using ArchR command “getGroupBW” and then visualized using the Integrative Genomics Viewer (IGV, available at https://software.broadinstitute.org/software/igv/).
Behavioral testing
All behavioral testing was conducted in the Animal Behavior Core facility at Washington University in St. Louis. Tests were performed during the light cycle by an experimenter blinded to experimental groups. Equipment was cleaned with 2% chlorhexidine diacetate between animals. Mouse video tracking was performed using a digital video camera and the computer program ANY-maze (Stoelting Co.). For behavioral studies, only littermates have been used. The order of tests conducted for each cohort were as follows: Open Field Test > Elevated Plus Maze > accelerated rotarod > Morris Water Maze.
Open field test (OFT)
To assess general locomotor activity mice were allowed to freely explore a transparent polystyrene arena (47.6L, 25.4W, 20.6H cm) a were monitored over a 1h period. Ambulation over time, total travelled distance, and time spent in the center zone of the arena were measured.
Elevated plus maze (EPM)
Each mouse was allowed to freely move for 300s on a cross maze (Kinder Scientific, LLC), elevated 63 cm above the floor and equipped with four arms (35L, 5W cm) extended from the center of the maze. Two arms were completely open and two were enclosed by 15cm high walls. Number of entries and traveled distance in closed and open arms were measured.
Accelerating Rotarod
Motor function and motor learning were evaluated using an accelerating rotarod test (AccuRotor EZrod, Omnitech Electronics, Inc). Mice were placed on a 30mm diameter rod that was set to accelerate from 4–40rpm over 300s. Each mouse was given 3 trials/day for 3 consecutive days, and latency to fall from the rod was recorded for each trial.
Morris water maze
Morris water maze was performed as previously described [100]. Trials were conducted in a galvanized steel pool, measuring 120 cm in diameter, and filled with opaque water (diluted nontoxic white tempera paint). The escape platform measured 11.5 cm in diameter. The swimming pathway of the mouse was tracked and path length, latency to platform, and swimming speeds were measured.
On two consecutive days, animals received four cued trials to habituate mice to the swimming task procedure and control for any differences in swimming, visual, or motivational performance in the test. A red tennis ball atop a rod was attached to the escape platform and served as a visual cue for the platform. Three days following visible platform testing, the cue was removed, and the platform was submerged 1 cm under the water for the hidden platform tests to evaluate spatial learning. Animals received four trials per day, on five consecutive days. Visual cues were placed on the walls of the testing room to facilitate spatial learning. Each mouse was allowed 60s swimming time to locate the escape platform and 10s resting time on the platform before being returned to its home cage. One hour following completion of the last trials on the 5th day of training, the escape platform was removed, and one 60s probe trial was conducted to assess memory retention for the platform location. Mice exhibiting swimming difficulties were excluded from the test.
QUANTIFICATION AND STATISTICAL ANALYSIS
For flow cytometry and imaging data, graphs and statistics were produced using the GraphPad Prism 8 software package. Statistical difference between two groups was determined by two-tailed Mann Whitney U test. When the effects of two independent variables were considered, the two-way analysis of variance (ANOVA) with Bonferroni post-hoc test was used. When more than two groups were compared, one-way analysis of variance (ANOVA) with Bonferroni post-hoc test was used. All statistical analyses display mean value ± standard error of the mean (SEM). Statistical significance was set at p-value <0.05.
Supplementary Material
Highlights.
Generated Crybb1-Cre line that recombines in microglia and BAMs in embryonic development
Embryonic BAMs are CD38+MHC2−, while monocyte-derived BAMs are CD38−MHC2+
Used Crybb1-Cre to delete SMAD4 in microglia and embryonic BAMs
SMAD4 deletion arrests microglia development and impairs mouse memory skills
ACKNOWLEDGMENTS
We thank Dr. E. Lantelme for the excellent management of the flow cytometry core facility at the Pathology and Immunology Department (Washington University in St. Louis). We thank Dr. W. Beatty from the Molecular Microbiology Department (Washington University in St. Louis) for the excellent management of the imaging core. We thank Drs. M. Blurton-Jones (University of California Irvine) and D. Hume (University of Queensland) for providing the Fire knock-out mice, Dr. K. Murphy (Washington University in St. Louis) for providing the Zbtb46GFP mice and B. Bhattarai for sharing the Smad4F/F mice. We also thank Dr. Y. Zhou for helping set up the nuclei extraction protocol, J. Sun for helping collect preliminary data, A. Swain (10x Genomics) for helpful suggestions regarding the multiomics protocol, and Richard Cho (Cell Signaling) for the generous supply of antibodies. Lastly, we thank the Genome Engineering and iPSC Center (GEiC) (Washington University in St. Louis) for gRNA validation services, as well as Mike White and the Transgenic, Knockout and Micro-Injection Core at the Pathology and Immunology Department (Washington University in St. Louis) for generating the genetically modified mice used in this study. M. Colonna is supported by the Rheumatic Diseases Research Resource-Based Center at Washington University (NIH/NIAMS P30AR073752). The Animal Behavior Core is supported in part by funding from the McDonnell Centers for Cellular and Molecular, and Systems Neuroscience. This work was supported by NIH RF1AG05148501, RF1AG059082, R21AG059176, R01AI158579, and Cure Alzheimer’s Fund (to Dr. M. Colonna). A. Satpathy is supported by a Career Award for Medical Scientists from the Burroughs Wellcome Fund and a Pew-Stewart Scholars Award. J. Belk is supported by a Stanford Graduate Fellowship and the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1656518. Cartoons and graphical abstract were created with BioRender.com.
Footnotes
DECLARATION OF INTERESTS
A.T.S. is a founder of Immunai and Cartography Biosciences and receives research funding from Allogene Therapeutics and Merck Research Laboratories. All other authors declare no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Brioschi S, Zhou Y, Colonna M (2020) Brain Parenchymal and Extraparenchymal Macrophages in Development, Homeostasis, and Disease. J Immunol 204:294–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kierdorf K, Masuda T, Jordão MJC, Prinz M (2019) Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci 20:547–562 [DOI] [PubMed] [Google Scholar]
- 3.Prinz M, Masuda T, Wheeler MA, Quintana FJ (2021) Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu Rev Immunol 39:251–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ginhoux F, Greter M, Leboeuf M, et al. (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulz C, Gomez Perdiguero E, Chorro L, et al. (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336:86–90 [DOI] [PubMed] [Google Scholar]
- 6.Gomez Perdiguero E, Klapproth K, Schulz C, et al. (2015) Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518:547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kierdorf K, Erny D, Goldmann T, et al. (2013) Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci 16:273–280 [DOI] [PubMed] [Google Scholar]
- 8.Tay TL, Mai D, Dautzenberg J, et al. (2017) A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci 20:793–803 [DOI] [PubMed] [Google Scholar]
- 9.Askew K, Li K, Olmos-Alonso A, et al. (2017) Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep 18:391–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldmann T, Wieghofer P, Müller PF, et al. (2013) A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci 16:1618–1626 [DOI] [PubMed] [Google Scholar]
- 11.Wieghofer P, Hagemeyer N, Sankowski R, et al. (2021) Mapping the origin and fate of myeloid cells in distinct compartments of the eye by single-cell profiling. EMBO J 40:e105123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Koren EG, Yu C, Klingeborn M, et al. (2019) Microglial Function Is Distinct in Different Anatomical Locations during Retinal Homeostasis and Degeneration. Immunity 50:723–737.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Margeta MA, Yin Z, Madore C, et al. (2022) Apolipoprotein E4 impairs the response of neurodegenerative retinal microglia and prevents neuronal loss in glaucoma. Immunity 55:1627–1644.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ydens E, Amann L, Asselbergh B, et al. (2020) Profiling peripheral nerve macrophages reveals two macrophage subsets with distinct localization, transcriptome and response to injury. Nat Neurosci 23:676–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang PL, Yim AKY, Kim K-W, Avey D, Czepielewski RS, Colonna M, Milbrandt J, Randolph GJ (2020) Peripheral nerve resident macrophages share tissue-specific programming and features of activated microglia. Nat Commun 11:2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldmann T, Wieghofer P, Jordão MJC, et al. (2016) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17:797–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masuda T, Amann L, Monaco G, et al. (2022) Specification of CNS macrophage subsets occurs postnatally in defined niches. Nature 604:740–748 [DOI] [PubMed] [Google Scholar]
- 18.Utz SG, See P, Mildenberger W, et al. (2020) Early Fate Defines Microglia and Non-parenchymal Brain Macrophage Development. Cell 181:557–573.e18 [DOI] [PubMed] [Google Scholar]
- 19.Van Hove H, Martens L, Scheyltjens I, et al. (2019) A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22:1021–1035 [DOI] [PubMed] [Google Scholar]
- 20.Yona S, Kim KW, Wolf Y, et al. (2013) Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 38:79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20:4106–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, Nishinakamura R, Becher B, Greter M (2016) Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol 17:1397–1406 [DOI] [PubMed] [Google Scholar]
- 23.Masuda T, Amann L, Sankowski R, et al. (2020) Novel Hexb-based tools for studying microglia in the CNS. Nat Immunol 21:802–815 [DOI] [PubMed] [Google Scholar]
- 24.Kaiser T, Feng G (2019) Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia. eNeuro 6:0448–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKinsey GL, Lizama CO, Keown-Lang AE, Niu A, Santander N, Larpthaveesarp A, Chee E, Gonzalez FF, Arnold TD (2020) A new genetic strategy for targeting microglia in development and disease. Elife. 10.7554/eLife.54590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J-S, Kolesnikov M, Peled-Hajaj S, et al. (2021) A Binary Cre Transgenic Approach Dissects Microglia and CNS Border-Associated Macrophages. Immunity 54:176–190.e7 [DOI] [PubMed] [Google Scholar]
- 27.Matcovitch-Natan O, Winter DR, Giladi A, et al. (2016) Microglia development follows a stepwise program to regulate brain homeostasis. Science 353:aad8670. [DOI] [PubMed] [Google Scholar]
- 28.Graw J (2009) Genetics of crystallins: cataract and beyond. Exp Eye Res 88:173–189 [DOI] [PubMed] [Google Scholar]
- 29.Gautier EL, Shay T, Miller J, et al. (2012) Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13:1118–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang L, Means TK, El Khoury J (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16:1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolf SA, Boddeke HWGM, Kettenmann H (2017) Microglia in Physiology and Disease. Annu Rev Physiol 79:619–643 [DOI] [PubMed] [Google Scholar]
- 32.The Tabula Muris Consortium (2018) Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562:367–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mass E, Ballesteros I, Farlik M, et al. (2016) Specification of tissue-resident macrophages during organogenesis. Science. 10.1126/science.aaf4238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rojo R, Raper A, Ozdemir DD, et al. (2019) Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun 10:3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiani Shabestari S, Morabito S, Danhash EP, et al. (2022) Absence of microglia promotes diverse pathologies and early lethality in Alzheimer’s disease mice. Cell Rep 39:110961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munro DAD, Bradford BM, Mariani SA, Hampton DW, Vink CS, Chandran S, Hume DA, Pridans C, Priller J (2020) CNS macrophages differentially rely on an intronic Csf1r enhancer for their development. Development. 10.1242/dev.194449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, Koren T, Rolls A (2017) High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci 20:1300–1309 [DOI] [PubMed] [Google Scholar]
- 38.Mildner A, Schönheit J, Giladi A, et al. (2017) Genomic Characterization of Murine Monocytes Reveals C/EBPβ Transcription Factor Dependence of Ly6C(−) Cells. Immunity 46:849–862.e7 [DOI] [PubMed] [Google Scholar]
- 39.Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, Geissmann F, Hedrick CC (2011) The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol 12:778–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas GD, Hanna RN, Vasudevan NT, et al. (2016) Deleting an Nr4a1 Super-Enhancer Subdomain Ablates Ly6C(low) Monocytes while Preserving Macrophage Gene Function. Immunity 45:975–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mrdjen D, Pavlovic A, Hartmann FJ, et al. (2018) High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 48:380–395.e6 [DOI] [PubMed] [Google Scholar]
- 42.Mundt S, Mrdjen D, Utz SG, Greter M, Schreiner B, Becher B (2019) Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci Immunol 4:eaau8380. [DOI] [PubMed] [Google Scholar]
- 43.Satpathy AT, KC W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, Murphy KM (2012) Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med 209:1135–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oakley H, Cole SL, Logan S, et al. (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci Off J Soc Neurosci 26:10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keren-Shaul H, Spinrad A, Weiner A, et al. (2017) A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169:1276–1290.e17 [DOI] [PubMed] [Google Scholar]
- 46.Krasemann S, Madore C, Cialic R, et al. (2017) The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47:566–581.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ellwanger DC, Wang S, Brioschi S, et al. (2021) Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 118:e2017742118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S, Sudan R, Peng V, et al. (2022) TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell 185:4153–4169.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, De Jager PL, Ransohoff RM, Regev A, Tsai LH (2017) Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep 21:366–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Friedman BA, Srinivasan K, Ayalon G, et al. (2018) Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep 22:832–847 [DOI] [PubMed] [Google Scholar]
- 51.Sala Frigerio C, Wolfs L, Fattorelli N, et al. (2019) The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep 27:1293–1306.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Y, Song WM, Andhey PS, et al. (2020) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26:131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Drieu A, Du S, Storck SE, et al. (2022) Parenchymal border macrophages regulate the flow dynamics of the cerebrospinal fluid. Nature 611:585–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benz C, Martins VC, Radtke F, Bleul CC (2008) The stream of precursors that colonizes the thymus proceeds selectively through the early T lineage precursor stage of T cell development. J Exp Med 205:1187–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boyer SW, Schroeder AV, Smith-Berdan S, Forsberg EC (2011) All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell 9:64–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoeffel G, Chen J, Lavin Y, et al. (2015) C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42:665–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Canli Ö, Nicolas AM, Gupta J, et al. (2017) Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 32:869–883.e5 [DOI] [PubMed] [Google Scholar]
- 58.Hammond TR, Dufort C, Dissing-Olesen L, et al. (2019) Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50:253–271.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Masuda T, Sankowski R, Staszewski O, et al. (2019) Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566:388–392 [DOI] [PubMed] [Google Scholar]
- 60.Bennett ML, Bennett FC, Liddelow SA, et al. (2016) New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 113:E1738–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Q, Cheng Z, Zhou L, et al. (2019) Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 101:207–223.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Butovsky O, Jedrychowski MP, Moore CS, et al. (2014) Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci 17:131–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lund H, Pieber M, Parsa R, et al. (2018) Fatal demyelinating disease is induced by monocyte-derived macrophages in the absence of TGF-β signaling. Nat Immunol 19:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zöller T, Schneider A, Kleimeyer C, Masuda T, Potru PS, Pfeifer D, Blank T, Prinz M, Spittau B (2018) Silencing of TGFβ signalling in microglia results in impaired homeostasis. Nat Commun 9:4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gosselin D, Link VM, Romanoski CE, et al. (2014) Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159:1327–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA (2017) Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron 94:759–773.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Massagué J (2012) TGFβ signalling in context. Nat Rev Mol Cell Biol 13:616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, White AJ, Gilfillan S, Cella M, Colonna M (2017) SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat Immunol 18:995–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jordão MJC, Sankowski R, Brendecke SM, et al. (2019) Neuroimmunology: Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science (80- ) 363:eaat7554. [DOI] [PubMed] [Google Scholar]
- 70.Ajami B, Samusik N, Wieghofer P, Ho PP, Crotti A, Bjornson Z, Prinz M, Fantl WJ, Nolan GP, Steinman L (2018) Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat Neurosci 21:541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dick SA, Wong A, Hamidzada H, et al. (2022) Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci Immunol 7:eabf7777. [DOI] [PubMed] [Google Scholar]
- 72.De Vlaminck K, Van Hove H, Kancheva D, et al. (2022) Differential plasticity and fate of brain-resident and recruited macrophages during the onset and resolution of neuroinflammation. Immunity 55:2085–2102.e9 [DOI] [PubMed] [Google Scholar]
- 73.Silvin A, Uderhardt S, Piot C, et al. (2022) Dual ontogeny of disease-associated microglia and disease inflammatory macrophages in aging and neurodegeneration. Immunity 55:1448–1465.e6 [DOI] [PubMed] [Google Scholar]
- 74.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I (2014) Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159:1312–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van de Laar L, Saelens W, De Prijck S, et al. (2016) Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity 44:755–768 [DOI] [PubMed] [Google Scholar]
- 76.Scott CL, Zheng F, De Baetselier P, et al. (2016) Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun 7:10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, Plowey ED, Barres BA (2018) A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 98:1170–1183.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guilliams M, Scott CL (2017) Does niche competition determine the origin of tissue-resident macrophages? Nat Rev Immunol 17:451–460 [DOI] [PubMed] [Google Scholar]
- 79.Gundra UM, Girgis NM, Gonzalez MA, et al. (2017) Vitamin A mediates conversion of monocyte-derived macrophages into tissue-resident macrophages during alternative activation. Nat Immunol 18:642–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shemer A, Grozovski J, Tay TL, et al. (2018) Engrafted parenchymal brain macrophages differ from microglia in transcriptome, chromatin landscape and response to challenge. Nat Commun 9:5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Badimon A, Strasburger HJ, Ayata P, et al. (2020) Negative feedback control of neuronal activity by microglia. Nature 586:417–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, Low D, Bessis A, Ginhoux F, Garel S (2014) Microglia modulate wiring of the embryonic forebrain. Cell Rep 8:1271–1279 [DOI] [PubMed] [Google Scholar]
- 83.Paolicelli RC, Bolasco G, Pagani F, et al. (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–1458 [DOI] [PubMed] [Google Scholar]
- 84.Zhan Y, Paolicelli RC, Sforazzini F, et al. (2014) Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17:400–406 [DOI] [PubMed] [Google Scholar]
- 85.Stogsdill JA, Kim K, Binan L, Farhi SL, Levin JZ, Arlotta P (2022) Pyramidal neuron subtype diversity governs microglia states in the neocortex. Nature 608:750–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Filipello F, Morini R, Corradini I, et al. (2018) The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 48:979–991.e8 [DOI] [PubMed] [Google Scholar]
- 87.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74:691–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ayata P, Badimon A, Strasburger HJ, et al. (2018) Epigenetic regulation of brain region-specific microglia clearance activity. Nat Neurosci 21:1049–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gunner G, Cheadle L, Johnson KM, et al. (2019) Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat Neurosci 22:1075–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW (2016) Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci 19:504–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Madisen L, Zwingman TA, Sunkin SM, et al. (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang X, Li C, Herrera P-L, Deng C-X (2002) Generation of Smad4/Dpc4 conditional knockout mice. Genesis 32:80–81 [DOI] [PubMed] [Google Scholar]
- 93.Zikherman J, Parameswaran R, Weiss A (2012) Endogenous antigen tunes the responsiveness of naive B cells but not T cells. Nature 489:160–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zaman R, Hamidzada H, Kantores C, et al. (2021) Selective loss of resident macrophage-derived insulin-like growth factor-1 abolishes adaptive cardiac growth to stress. Immunity 54:2057–2071.e6 [DOI] [PubMed] [Google Scholar]
- 95.Bando JK, Gilfillan S, Song C, et al. (2018) The Tumor Necrosis Factor Superfamily Member RANKL Suppresses Effector Cytokine Production in Group 3 Innate Lymphoid Cells. Immunity 48:1208–1219.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jaitin DA, Adlung L, Thaiss CA, et al. (2019) Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178:686–698.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hao Y, Hao S, Andersen-Nissen E, et al. (2021) Integrated analysis of multimodal single-cell data. Cell 184:3573–3587.e29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, Greenleaf WJ (2021) ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet 53:403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yuede CM, Wallace CE, Davis TA, et al. (2021) Pimavanserin, a 5HT(2A) receptor inverse agonist, rapidly suppresses Aβ production and related pathology in a mouse model of Alzheimer’s disease. J Neurochem 156:658–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq and ATAC-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the Key Resources Table.
Original codes have been deposited at GitHub and are publicly available as of the date of publication. DOIs are listed in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Primary antibodies and dyes flow cytometry | ||
|
| ||
| CD45-APCCy7 | Biolegend | RRID: AB_312981 |
| CD45-PE | Biolegend | RRID: AB_2563598 |
| CD45-BUV563 | BD Bioscience | RRID: AB_2870209 |
| CD45-AF700 | Biolegend | RRID: AB_493715 |
| CD45.2-APC | Biolegend | RRID: AB_389211 |
| CD45.1-Biotin | Biolegend | RRID: AB_313493 |
| CD11b-PECy7 | Biolegend | RRID: AB_312799 |
| CD11b-BV421 | Biolegend | RRID: AB_10897942 |
| CD11b-APCCy7 | Biolegend | RRID: AB_830642 |
| CD11b-BB515 | BD Biosciences | RRID: AB_2665392 |
| CD11b-APC | Biolegend | RRID: AB_312795 |
| CD11b-PerCPCy5.5 | Biolegend | RRID: AB_2129374 |
| CD11c-APCCy7 | Biolegend | RRID: AB_830649 |
| CD11c-BV421 | Biolegend | RRID: AB_10897814 |
| CD11c-PECy7 | Biolegend | RRID: AB_493568 |
| Ly6C-FITC | Biolegend | RRID: AB_1186135 |
| Ly6C-PE | Biolegend | RRID: AB_1186132 |
| Ly6C-APCCy7 | Biolegend | RRID: AB_10640120 |
| Ly6G-FITC | Biolegend | RRID: AB_1236494 |
| Ly6G-APC | Biolegend | RRID: AB_2227348 |
| Gr-1-PerCPCy5.5 | Biolegend | RRID: AB_893557 |
| NK1.1-FITC | Biolegend | RRID: AB_313393 |
| NK1.1-PerCPCy5.5 | Biolegend | RRID: AB_2132707 |
| CD43-FITC | Biolegend | RRID: AB_10960745 |
| CD43- PerCPCy5.5 | Biolegend | RRID: AB_2286556 |
| CD44-FITC | Biolegend | RRID: AB_312957 |
| CD44-PE | Biolegend | RRID: AB_312959 |
| F4/80-APC | Biolegend | RRID: AB_2832549 |
| F4/80-Biotin | Biolegend | RRID: AB_893501 |
| F4/80-BV605 | Biolegend | RRID: AB_2562305 |
| CX3CR1-PECy7 | Biolegend | RRID: AB_2565700 |
| CX3CR1-APC | Biolegend | RRID: AB_2564492 |
| MERTK-BV421 | Biolegend | RRID: AB_2832533 |
| I-A/I-E(MHC-M)-PECy7 | Biolegend | RRID: AB_313327 |
| I-A/I-E(MHC-II)-BV650 | Biolegend | RRID: AB_2565975 |
| I-A/I-E(MHC-II)-BUV805 | BD Biosciences | RRID: AB_2873247 |
| Siglec-F-APC | Biolegend | RRID: AB_2750237 |
| CD115-BV421 | Biolegend | RRID: AB_2562667 |
| CD206-AF488 | Biolegend | RRID: AB_10900445 |
| CD207-PECy7 | Biolegend | RRID: AB_2876490 |
| FOLR2-APC | Biolegend | RRID: AB_2721313 |
| FOLR2-PE | Biolegend | RRID: AB_2721344 |
| CD38-PECy7 | Biolegend | RRID: AB_2275531 |
| CD64-PE | Biolegend | RRID: AB_10612740 |
| TMEM119-PE | Invitrogen | RRID: AB_2848262 |
| P2RY12-Biotin | Biolegend | RRID: AB_2749906 |
| cKit-BB515 | BD Biosciences | RRID: AB_2738826 |
| CD93-APC | Biolegend | RRID: AB_2275868 |
| EpCAM-BV711 | Biolegend | RRID: AB_2632775 |
|
| ||
| Primary antibodies immunofluorescence | ||
|
| ||
| Rabbit Iba1 | Cell signaling | RRID: AB_2820254 |
| Rabbit CD206 | Cell signaling | RRID: AB_2892682 |
| Rat CD206-AF488 | Biolegend | RRID: AB_10900445 |
| Rat MHC2-AF647 | Biolegend | RRID: AB_493526 |
| Rat EpCAM-AF647 | Biolegend | RRID: AB_1134104 |
| Rat CD38-AF647 | Biolegend | RRID: AB_2073334 |
| Mouse GFAP-AF594 | Cell signaling | RRID: AB_10998775 |
| Mouse GFAP-AF488 | Invitrogen | RRID: AB_10598515 |
| Rabbit CRYBB1 | Cell signaling | Cat# 95666 |
| Rabbit Olig2 | MilliporeSigma | RRID: AB_570666 |
| Mouse APC(CC-1) | MilliporeSigma | RRID: AB_2057371 |
| Mouse NeuN-AF488 | MilliporeSigma | RRID: AB_2149209 |
| Rat CD45-AF488 | Biolegend | RRID: AB_493531 |
| Rat CD45-AF647 | Biolegend | RRID: AB_2876569 |
| Rat CD34-eFluor 660 | Invitrogen | RRID: AB_10596826 |
| Rat GFP-AF488 | Biolegend | RRID: AB_2563288 |
|
| ||
| Secondary antibodies immunofluorescence | ||
|
| ||
| Donkey anti-rat IgG AF647 | Life technologies | RRID: AB_2896338 |
| Chicken anti-rat IgG AF488 | Life technologies | RRID: AB_2535873 |
| Donkey anti-rabbit IgG AF647 | Life technologies | RRID: AB_2536183 |
| Donkey anti-rabbit IgG AF488 | Life technologies | RRID: AB_2535792 |
| Donkey anti-rabbit IgG AF555 | Life technologies | RRID: AB_162543 |
| Goat anti-mouse IgG AF555 | Life technologies | RRID: AB_2535844 |
| Goat anti-mouse IgG1 AF488 | Life technologies | RRID: AB_2535764 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| EDTA | Corning | Cat# 46-034-Cl |
| Triton X-100 | Sigma | Cat# T8787 |
| Tween 20 | Fisher Bioreagents | Cat# BP337 |
| Paraformaldehyde 32% | Electron Microscopy Science | Cat# 15714-S |
| NP-40 Substitute | Sigma | Cat# 74385 |
| 5% Digitonin | Thermo Fisher | Cat# BN2006 |
| DL-Dithiothreitol solution (DTT) | Sigma | Cat# 646563 |
| RNase inhibitor | Promega | Cat# N2515 |
| 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) | Sigma | Cat# D9542 |
| Tris-HCl (pH 7.4) | Sigma | Cat# T2194 |
| NaCl | Sigma | Cat# 59222C |
| MgCl2 | Sigma | Cat# M1028 |
| Tamoxifen diet (500 mg tamoxifen/Kg chow) | Envigo | Cat# TD.130857 |
| Streptavidin BV421 | Biolegend | Cat# 405225 |
| Streptavidin AF488 | Biolegend | Cat# 405235 |
| Zombie Aqua Fixable Viability Kit | Biolegend | Cat# 423102 |
| 2.4G2 CD16/32 Fc block from 197 hybridomas | ATCC | Cat# HB-197 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Chromium Next GEM Single Cell 3’ Kit v3.1 | 10× Genomics | PN-1000268 |
| Chromium Next GEM Chip G Single Cell Kit | 10× Genomics | PN-1000120 |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle | 10× Genomics | PN-1000283 |
| Chromium Next GEM Chip J Single Cell Kit | 10× Genomics | PN-1000234 |
| Single Index Kit N Set A | 10× Genomics | PN-1000212 |
| Dual Index Kit TT Set A | 10× Genomics | PN-1000215 |
| NovaSeq6000 | Illumina | S4 Flow Cell |
|
| ||
| Deposited data | ||
|
| ||
| Single cell transcriptomic and ATAC sequencing data | This paper | GEO: GSE213020 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mice | Jackson laboratory | JAX:000664 |
| Flt3-Cre mice | Benz et al.54 | MGI: 4462354 |
| Rosa26-STOP-EYFP mice (Ai2) | Madisen et al.91 | JAX:007920 |
| Rosa26-STOP-tdTomato mice (Ai14) | Madisen et al.91 | JAX: 007908 |
| Smad4-flox mice | Yang et al.92 | JAX: 017462 |
| Lyz2-CreEr2 mice | Canli et al.57 | JAX: 032291 |
| Nur77GFP mice | Zikherman et al.93 | MMRRC: 012015 |
| Zbtb46GFP mice | Satpathy et al.43 | JAX: 027618 |
| Crybb1 knock-out mice | This paper | Colonna F2-12-3-13 |
| Crybb1-tdTomato mice | This paper | Colonna F2-13-2-7 |
| Crybb1-Cre mice | This paper | Colonna F2-13-2-6 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Crybb1 gRNA exon1: AGCACCAGGAACCATGTCCCNGG | This paper | N/A |
| Crybb1 gRNAs exon3: GTGACCGGCTCATGTCCTTCNGG; GTGGGTACTCGCCCTTCTCCNGG | This paper | N/A |
| Crybb1-Cre Fwd. primer: AGACAATAGCAGGCATGCTGG | This paper | N/A |
| Crybb1-Cre Rev. primer: GGATCAGTACAGCCCAGCTC | This paper | N/A |
| Crybb1 knock-out Fwd. primer: GGGTGGCCTTTGAGCAATCT | This paper | N/A |
| Crybb1 knock-out Rev. primer: ACGTCACATCTTCCCCCAAA. | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Cell Ranger v2.0.0 and v6.0.0 | 10× Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| R project 4.1.3 | http://www.r-project.org/ | RRID: SCR_001905 |
| Rstudio | https://posit.co | RRID: SCR_000432 |
| Seurat v4.1.1 | Hao et al.94 | RRID: SCR_007322 |
| ArchR v1.0.1 | Granja et al.95 | http://www.archrproject.com/ |
| Deposited algorithms | This paper | https://doi.org/10.5281/zenodo.7558104 |
| ImageJ/Fiji | Schneider et al.96 | RRID: SCR_002285 |
| Imaris V8.3 | Bitplane | RRID: SCR_007370 |
| ANY-maze Video Tracking Software | Stoelting | RRID: SCR_014289 |
|
| ||
| Other | ||
|
| ||
| PBS | Corning | Cat# 21-040-CM |
| DMEM | Gibco | Cat# 11965-084 |
| RPMI | Sigma | Cat# R8758 |
| HBSS | Gibco | Cat# 14185-052 |
| HEPES | Corning | Cat# 25-060-Cl |
| Bovine calf serum (BCS) | Cytiva | Cat# SH30072.04 |
| BSA | Rockland | Cat# BSA-1000 |
| 10X red blood cells (RBC) lysis buffer | Biolegend | Cat# 420302 |
| Percoll | GE Healthcare | Cat# 17089101 |
| Collagenase-D | Sigma | Cat# 11088882001 |
| Collagenase-II | Gibco | Cat# 17101015 |
| Collagenase-IV | Sigma | Cat# C4-22-1G |
| Liberase TM | Sigma | Cat# 5401127001 |
| Hyaluronidase | Sigma | Cat# H1115000 |
| Dispase-II | Gibco | Cat# 17105-041 |
| DNase-I | Sigma | Cat# 10104159001 |
| Tomato-lectin Dylight 649 | Vector Laboratories | Cat# DL-1178 |
| Tomato-lectin Dylight 488 | Vector Laboratories | Cat# DL-1174-1 |
| Superfrost glass slides | Fisher Scientific | Cat# 12-550-15 |
| Prolong Glass anti-fade mounting media | Thermo Fisher | Cat# P36980 |
| Fluoromount-G mounting media | SouthernBiotech | Cat# 0100-01 |
| Nuclei Buffer | 10× Genomics | PN-2000207 |
| Ultra-Pure BSA | Thermo Fisher | Cat# AM2616 |
| Nuclease-free water | Invitrogen | Cat# AM9937 |
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.