

Significance
We present a method to recapitulate endochondral bone formation, the process through which long bones develop and grow, in cell culture. Our method begins with induced pluripotent stem cells (iPSCs) and produces uniform cartilage that progressively matures, mirroring development. Hypertrophic chondrocytes can transition to osteoblasts in our culture system, and we provide detailed analysis of the gene expression profile and changes during maturation to support this claim. Our method can be used to interrogate cartilage differentiation and development, including chondrocyte transition to osteoblasts, model genetic cartilage and bone disorders, and search for therapies.
Keywords: cartilage, bone, iPSC, genetic skeletal disorder
Abstract
Chondrocytes and osteoblasts differentiated from induced pluripotent stem cells (iPSCs) will provide insights into skeletal development and genetic skeletal disorders and will generate cells for regenerative medicine applications. Here, we describe a method that directs iPSC-derived sclerotome to chondroprogenitors in 3D pellet culture then to articular chondrocytes or, alternatively, along the growth plate cartilage pathway to become hypertrophic chondrocytes that can transition to osteoblasts. Osteogenic organoids deposit and mineralize a collagen I extracellular matrix (ECM), mirroring in vivo endochondral bone formation. We have identified gene expression signatures at key developmental stages including chondrocyte maturation, hypertrophy, and transition to osteoblasts and show that this system can be used to model genetic cartilage and bone disorders.
Genetic skeletal disorders are a significant disease burden, but there are few effective treatments. An in vitro disease model system in which all the steps in endochondral cartilage maturation and bone formation are faithfully recapitulated would contribute to our quest for a better understanding of the molecular pathology and the search for therapeutic options. A model system that can do this does not currently exist.
Endochondral bone development begins when mesenchymal precursors condense and form a cartilage template (anlagen) which then undergoes an ordered transition to bone. Complex temporal and spatial regulatory circuits direct chondrocytes, the cartilage cells, to differentiate, proliferate, and mature (1–4). From the cartilage anlagen, chondrocytes are directed down two general pathways. Chondrocytes at the developing joint surfaces form a distinct population of articular chondrocytes which produce the permanent articular cartilages. Articular chondrocytes synthesize the characteristic extracellular components of cartilage, collagen II, and aggrecan, and a specific repertoire and stoichiometry of cartilage extracellular matrix (ECM) components that endow articular cartilage with the specialized properties that provide resilience to repetitive mechanical loading.
Embryonic chondrocytes can also follow a developmental pathway to form growth plate cartilage that drives longitudinal bone growth. Chondrocytes entering this pathway sequentially mature producing the morphologically distinct growth plate zones: resting, proliferative and hypertrophic. Resting and proliferative zone chondrocytes produce an abundant cartilage ECM containing collagen II, aggrecan, and other cartilage proteins, subtly, but functionally distinct to articular cartilage. Following the proliferative phase, chondrocytes enter hypertrophy where they begin to express the hypertrophic chondrocyte-specific network-forming collagen X. Hypertrophic chondrocytes regulate growth plate remodeling and mineralization by secreting matrix metalloproteinases and proangiogenic factors to promote vascular invasion. While vascular invasion was thought to be key in bringing in osteoprogenitors and signaling perichondrium-derived osteoprogenitors to migrate, mature, and replace the hypertrophic cartilage with mineralized bone, it is now clear that terminal hypertrophic chondrocytes can transdifferentiate to osteoblasts and, indeed, can also generate the stromal and adipocyte elements of the forming bone marrow (4–9). Thus, chondrocytes in the cartilage growth plate have, when provided with the correct temporal and spatial regulatory circuitry, the plasticity to undergo all the key steps in cartilage formation and transition to functional bone.
Differentiation of human pluripotent stem cells (hPSCs) provides a unique platform to study human development. However, to model the genesis of complex tissues and diseases, the developmental circuitry underpinning lineage-specific differentiation must be understood. In the human embryo, cartilage and bone develop from mesenchymal condensations derived from lateral plate mesoderm (the source of the appendicular skeleton), paraxial mesoderm (axial skeleton), and cranial neural crest ectomesoderm (craniofacial skeleton), each of which is uniquely specified (10–12). A number of methodologies have been used to differentiate pluripotent stem cells in vitro to chondrocytes (reviewed in ref. 13). These initially differentiated hPSCs to PDGFRa+ primitive streak mesoderm, which was then directed to form SOX9+ cartilage precursors, passing via either lateral plate or paraxial mesoderm intermediates. These protocols utilized growth factors involved in embryonic axial patterning including WNT, BMPs, NODAL/activin A, and FGFs. At the paraxial mesoderm stage, articular chondrogenesis was induced with a combination of BMP and TGF-β (14–18). More recent iterations (19–21) used combinations of small-molecule growth factor agonists and inhibitors to specify paraxial mesoderm which was then directed to the presomitic mesoderm and sclerotome of the ventral somite, the precursor to axial skeleton cartilage and bone (reviewed in ref. 12). Blocking BMP at the paraxial mesoderm stage followed by augmenting HEDGEHOG signaling resulted in a rapid and robust method that generated sclerotome in vitro (21). Recent studies of chondrocyte pellets differentiated from sclerotome precursors reveal a core of chondrocytes surrounded by a layer of noncartilage tissue expressing COL1A1 (22, 23). These protocols aimed to derive articular chondrocytes for regenerative medicine rather than recapitulating bone formation from a hypertrophic cartilage precursor.
Here, we describe an in vitro method that reproduces the cell differentiation and maturation events that occur during endochondral ossification, the process responsible for long bone formation. We show our method produces pure cartilage organoids that can be directed to either an articular phenotype or to growth plate cartilage that matures to hypertrophy. Hypertrophic chondrocytes can transition in vitro to osteoblasts that synthesize bone-specific ECM molecules and mineralize this ECM. Our protocol provides a way to explore human skeletal development in vitro and to model genetic skeletal disorders to identify pathogenic mechanisms and test drug therapies.
Results
A Reproducible iPSC Differentiation Method to Produce Growth Plate or Articular Cartilage Organoids.
During development, vertebral and rib cartilage and bone are generated from the ventral somite (sclerotome). To recapitulate this process in vitro, we based the first 6 d of our iPSC differentiation protocol (Fig. 1A) on a previously described method that generates sclerotome from iPSCs with high efficiency (21). We monitored the expression of stage-specific markers and saw the pluripotency marker OCT4 gradually down-regulated, and the primitive streak, paraxial mesoderm, somitic mesoderm, and sclerotome markers, MIXL1, MSGN1, MEOX1, and PAX1, respectively, up- and down-regulated at the appropriate developmental stages (SI Appendix, Fig. S1A). To promote sclerotome to chondrocyte differentiation, day 4 cells were formed into high-density pellets, a configuration known to favor maintenance of the chondrocyte phenotype ex-vivo. We compared supplementing postsclerotome pellets with 20 ng/mL BMP4 as used previously (21, 22), or 20 ng/mL FGF2 for 14 d followed by extended culture without additional growth factors. FGF2-treated pellets showed strong toluidine blue staining throughout at day 48, reflecting the presence of ECM proteoglycans typical for cartilage, and uniform cartilage collagen II and X immunostaining, while BMP4-treated pellets were much smaller and only isolated areas were toluidine blue and collagen II and X positive (SI Appendix, Fig. S1B). Differentiation was more uniform in iPSC lines adapted to feeder-free conditions (SI Appendix, Fig. S1C). Subsequent differentiation experiments all used feeder-free iPSCs, included pellet culture from the end of day 4, and supplemented with 20 ng/mL FGF2 for 14 to 28 d post sclerotome.
Fig. 1.
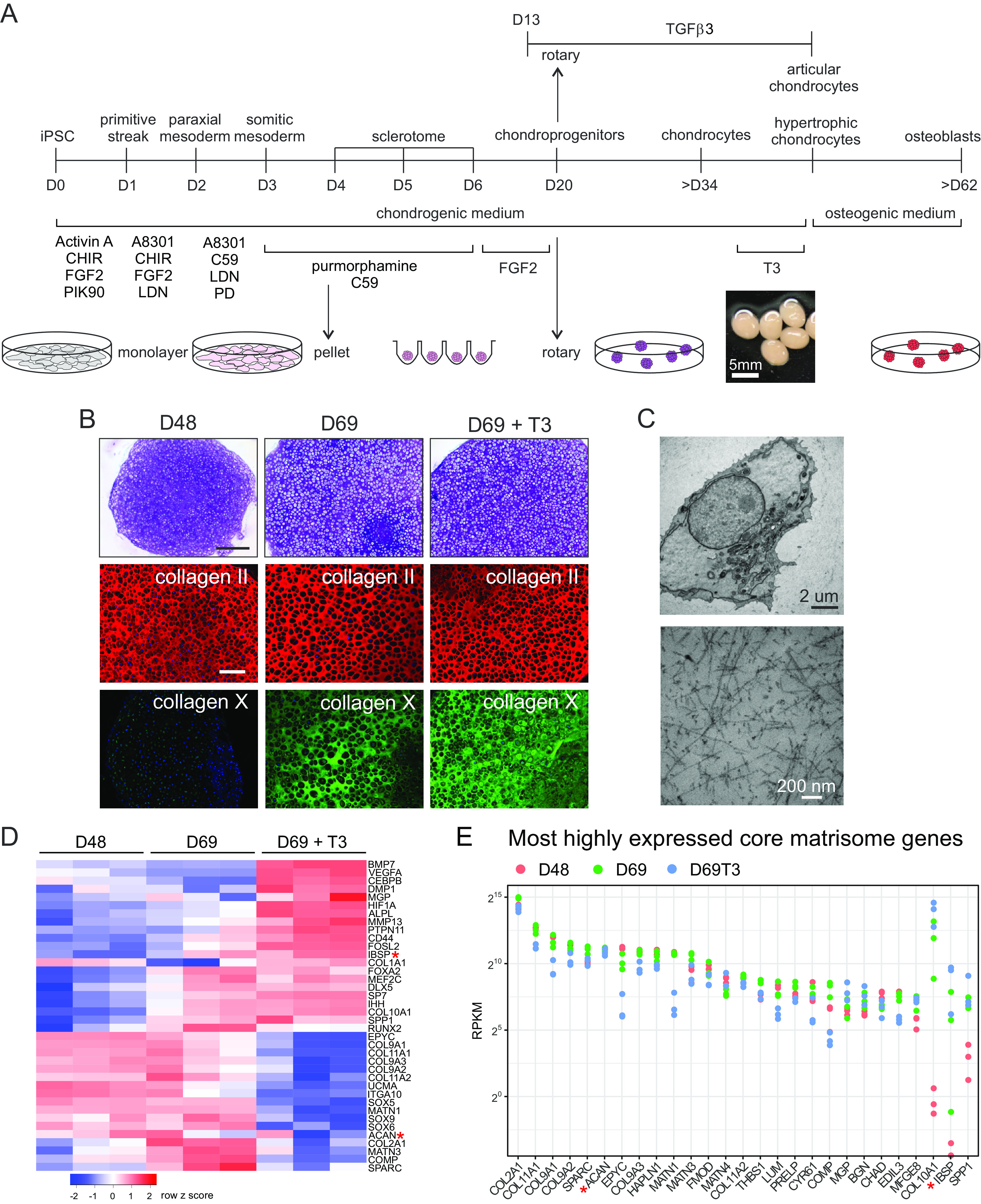
Directed iPSC differentiation to skeletal cells. (A) Schematic showing the differentiation stages from iPSCs to articular and hypertrophic chondrocytes and then toward osteoblasts. Differentiation to sclerotome in the first 6 d includes aggregating monolayer cells into pellets at day 4. Culture medium and conditions are shown below and above the timeline and culture formats are illustrated at the bottom. (B) Chondronoids spontaneously mature toward hypertrophy, and hypertrophy can be enhanced with T3. Toluidine blue-stained sections (MCRIi019-A) at days 48 and 69. Some chondronoids were treated with T3 for 3 wk before harvesting at day 69. Scale bar is 500 µm. Immunostaining shows that the chondronoids contain an extensive collagen II-rich ECM throughout the time course. Collagen X is not apparent at day 48 but has been deposited into the ECM by day 69. (Scale bar is 200 µm.) (C) TEM at day 52 showing a typical chondrocyte and an extensive network of collagen II fibrils in the ECM. (D) Changes in mRNA abundance of selected cartilage, hypertrophic cartilage, and bone proteins. N = 3 independent differentiation experiments. (E) Most highly expressed core matrisome genes. Those not reaching the statistical threshold for differential expression (adj.P.value < 0.05) in at least one of the comparisons are indicated with a red asterisk in D and E.
Since chondrocytes respond to biomechanical stimuli, and fluid flow-induced shear stress can promote chondrogenesis (24, 25), we explored whether introducing shear stress would influence chondrocyte differentiation. We compared pellets in static culture with those transferred to rotary orbital culture at days 6, 13, 20, and 27. At day 48, pellets that were transferred to rotary culture at day 6 retained a layer of noncartilage cells on the outside, while pellets from all the other conditions contained only cartilage tissue (SI Appendix, Fig. S1D). Pellets that were in rotary culture from day 13, day 20, and day 27 had enlarged chondrocytes that expressed collagen X, key characteristics of hypertrophic chondrocytes (SI Appendix, Fig. S1D). This finding was replicated in a second iPSC line (SI Appendix, Fig. S1E). The need for early extended static pellet culture for efficient sclerotome-to-chondrocyte transition is consistent with previous observations that mesenchymal stem cells differentiate to chondrocytes more efficiently in hypoxic conditions (26–28), and there is a low oxygen environment in pellet cultures (29). However, hypoxia inhibits chondrocyte hypertrophy and collagen X expression (30), and this is consistent with faster progression to hypertrophy in pellets that experience initial static/hypoxic culture but are then transferred to rotary culture in a larger medium volume with constant movement and aeration. These data indicate that transferring pellets to rotary culture in the day 13 to 20 window accelerates chondrocyte maturation and the timing of this transfer can be used to control how quickly the chondrocytes become hypertrophic. To differentiate this chondrogenic rotary culture format from traditional static pellet culture, we have named these chondrogenic organoids “chondronoids.”
Cell lineage tracing experiments show that articular and growth plate chondrocytes are derived from common mesenchymal precursors and that a specific fate decision to form articular chondrocytes occurs in early chondrocytes (31–33). To investigate the capacity of our system to generate articular chondrocytes, we treated iPSC-derived chondroprogenitors with TGF-β3 (17, 22, 23) for 5 wk starting at day 13. At day 48, we saw profound differences in cell morphology and gene expression between TGFβ3-treated and -untreated chondronoids. Untreated D48 chondronoids contained large chondrocytes strongly expressing collagens II and X but not PRG4 (lubricin), an articular chondrocyte marker (SI Appendix, Fig. S1F). In contrast, TGF-β3-treated chondrocytes were smaller; they did not express collagen X but were positive for collagen II and PRG4, consistent with an articular chondrocyte identity (SI Appendix, Fig. S1F).
The primary roles of permanent articular cartilage are weight-bearing and facilitating joint articulation, whereas transient growth plate cartilage drives longitudinal bone growth through endochondral ossification. These different functions are reflected in their ECM composition. Thus, we compared the gene expression profile of day 48 chondronoids that had been treated with TGF-β3 to promote articular cartilage development with untreated chondronoids which had progressed toward chondrocyte hypertrophy, a prelude to bone formation. Canonical cartilage components COL2A1, COMP, COL9A1, COL9A2, COL9A3, and COL11A1 were highly expressed in both TGFβ3-treated and -untreated chondronoids (SI Appendix, Fig. S1 G and H). In contrast, many core matrisome (34) genes were strongly up-regulated in TGF-β3-treated compared to untreated chondronoids. These included the sentinel articular cartilage marker, PRG4, articular cartilage proteins POSTN, ASPN, COL1A1, COL1A2, COL3A1, CILP, and CILP2, and MFAP4, a protein associated with elastic fibers (SI Appendix, Fig. S1 G and H). Other elastic fiber component genes such as ELN, LTBP2, TGFBI, and MFAP5 were highly up-regulated (SI Appendix, Fig. S1H), consistent with an articular cartilage phenotype (35). Importantly, ECM proteins related to cartilage hypertrophy and endochondral ossification, such as COL10A1, IBSP, SPP1, and MATN3, were among the most down-regulated in chondronoids differentiated toward articular cartilage with TGF-β3 (SI Appendix, Fig. S1H). Other changes consistent with an articular cartilage ECM included downregulation of ACAN and the associated link protein HAPLN (SI Appendix, Fig. S1H).
Articular cartilage-associated transcription factors (TFs) were up-regulated in response to TGF-β3 treatment. Specifically, ERG, a TF enriched in articular cartilage (36) and critical for articular cartilage maintenance (37), was highly expressed and up-regulated in TGF-β3-treated chondronoids (SI Appendix, Fig. S1 I and J). Transcription factor genes ELK3, ERF, TRPS1, PLAGL1, MEOX1, and MEOX2 were highly expressed and/or highly up-regulated in our TGFβ3-treated dataset (SI Appendix, Fig. S1 I and J). These genes are all expressed in somites (38) or skeletal tissues (39–42) and ERF (40), TRPS1 (41), and PLAGL1 (43) play important roles in growth plate and skeletal development, but their roles in articular cartilage development have not been elucidated.
Triiodothyronine (T3) is crucial for terminal chondrocyte hypertrophy and transdifferentiation into osteoblasts in the developing mouse secondary ossification center (44). Given spontaneous hypertrophy had been observed in previous experiments, we examined whether treating chondronoids with T3 from day 48 to 69 was able to promote uniform and synchronous hypertrophy. At day 48, untreated chondronoids stained homogeneously with toluidine blue and contained an extensive proteoglycan and collagen II-rich ECM (Fig. 1B and SI Appendix, Fig. S2A). Electron microscopy analysis of day 52 chondronoids (Fig. 1C) revealed long, thin, randomly orientated cartilage collagen fibrils in the ECM, characteristic of cartilage. By day 69, untreated chondronoids comprised enlarged chondrocytes and an ECM containing abundant collagen X indicative of hypertrophy (Fig. 1B and SI Appendix, Fig. S2A). Although T3-treated chondronoids appeared morphologically similar to their untreated counterparts at day 69, transcriptional profiling revealed substantial T3-stimulated gene expression changes. While early hypertrophic cartilage markers such as COL10A1, IHH, MEF2C, and SPP1 were not changed by T3 (Fig. 1D and SI Appendix, Fig. S2B), T3 increased the expression of late hypertrophic cartilage markers including SP7, VEGFA, DMP1, HIF1A, MMP13, and ALPL and decreased the expression of canonical cartilage genes such as COL2A1, COL9A1, COL11A1, SOX9, SOX6, and SOX5.
To further explore the developmental accuracy of iPSC-derived chondrocyte maturation in vitro, we examined our RNA-Seq data at days 48, 69, and day 69+T3 for expression of core matrisome genes (34). Cartilage collagens (COL2A1, COL11A1, COL9A1, COL9A2, and COL9A3) were highly expressed over this differentiation time span (Fig. 1E and SI Appendix, Fig. S2C). Likewise, other key cartilage ECM molecules including aggrecan (ACAN), HAPLN1, and matrilins (MATN1, MATN3) were strongly expressed throughout this developmental sequence. The hypertrophy marker, COL10A1, was up-regulated in the untreated day 69 samples, whereas sentinel cartilage genes down-regulated by T3 at day 69 included COL11A1, COL9A1, COL9A2, COL9A3, EPYC, MATN1, LUM, COMP, and EDIL3 (Fig. 1E and SI Appendix, Fig. S2C), potentially anticipating their transition to osteoblasts.
Skeletal ontogeny is orchestrated by complex TF circuitry that is regulated temporally and spatially to coordinate cell fate decisions with tissue morphogenesis. Examination of global TF expression (45) during chondrocyte differentiation in vitro indicated that many displayed an expression profile that mirrored their regulation during skeletal development and maturation (SI Appendix, Fig. S2D). For example, the master transcriptional mediator of the adaptive response to hypoxia, HIF1A (HIF1α), and EPAS1 (HIF2α), a regulator of VEGF expression, were up-regulated with hypertrophy (SI Appendix, Fig. S2D). Similarly, paralleling the regulation of their counterparts in vivo, the transition to hypertrophy in vitro is marked by upregulation of SP7 (osterix), RARG (retinoic acid receptor) (46), and MEF2C, the last of which is also important for endochondral differentiation of mesenchymal progenitor cells (47). Likewise, upregulation of CEBPB (C/EBPβ) during hypertrophy in vitro is consistent with its suggested role in suppressing early chondrocyte differentiation and stimulating hypertrophic markers (48). Conversely, SOX5 and SOX6, part of the SOX9/SOX5/SOX6 master chondrogenic trio, were coordinately down-regulated (SI Appendix, Fig. S2D) along with SOX9 (Fig. 1D and SI Appendix, Fig. S2B) in response to T3 treatment.
We also identified TFs with less well-recognized roles in cartilage development that were strongly up- or down-regulated with hypertrophy (SI Appendix, Fig. S2 D–F). Up-regulated TFs included ETS1, STAT3, DDIT3 and members of the KLF family (KLF2, KLF5, and KLF10). Also up-regulated during maturation were TFs associated with the circadian rhythm pathway, NFIL3, NR1D1, BHLHE41, ARNTL, CLOCK, NPAS2, NR1D2, DBP, and RORA. TFs with poorly described roles in chondrogenesis and chondrocyte maturation that are potentially important during skeletal development, DLX3 and CXXC5, as well as TFs with recently described roles in skeletal development, IRX3 and IRX5 (49, 50), were also up-regulated in our growth plate chondronoids.
Our iPSC-chondrocyte differentiation protocol reliably produced ~2.5 to 3 mm diameter organoids (SI Appendix, Fig. S3A) comprising uniform cartilage without evidence for contaminating noncartilage tissue (SI Appendix, Fig. S3B). To further demonstrate the reproducibility of our differentiation protocol, we compared the transcriptome in three independent differentiations of two iPSC lines. At D48, there were no differentially expressed genes, at D69, 156 genes were differentially expressed (1% of the transcriptome, Adj.P.val ≤ 0.05), and in T3-treated D69 samples, there were 669 differentially expressed genes (4% of the transcriptome) (SI Appendix, Fig. S4A). Principal component analysis revealed that the largest contributors to variance were the differentiation time and treatment, PC1 and PC2, respectively, accounting for 69% of the variance (SI Appendix, Fig. S4 B–D). PC3, contributing 7.3% of the variance, reflects differences between the iPSC lines (SI Appendix, Fig. S4 B and D). Thus, while the data show that the trajectory of chondrogenic differentiation and maturation is reproducible, at later development stages (D69) and hypertrophy (D69 +T3), there could be small transcriptome differences that would need to be accounted for in experimental development or disease models.
iPSC-Derived Hypertrophic Chondrocytes Can Transition to Osteoblasts and Osteocytes In Vivo and In Vitro.
Endochondral ossification was long thought to involve hypertrophic chondrocyte cell death, followed by cartilage remodeling and vascular invasion. However, it has now become clear that an alternative pathway exists whereby hypertrophic chondrocytes transdifferentiate to osteoblasts (5, 8). To test whether our iPSC-derived chondrocytes could transition to osteoblasts in vivo, we transplanted hypertrophic chondronoids (D42) subcutaneously into immunodeficient mice. The resultant grafts were harvested after 13 wk and analyzed by immunohistochemistry. This revealed that implants contained some residual cartilage with characteristic hypertrophic chondrocytes embedded in the cartilage ECM. Significantly, however, much of the implant formed histologically recognizable bone (Fig. 2A) that was mineralized (SI Appendix, Fig. S5A) and, like in vivo mouse bone, expressed the osteoblast/osteocyte proteins BGLAP (osteocalcin) and SOST (sclerostin) (SI Appendix, Fig. S5 B–E). Staining with a human-specific Ku80 antibody showed that both cartilage and bone in the implant were derived from human cells (Fig. 2A and SI Appendix, Fig. S6), suggesting that the in vitro differentiated hypertrophic chondrocytes transitioned into osteoblasts when provided with signaling cues in vivo.
Fig. 2.
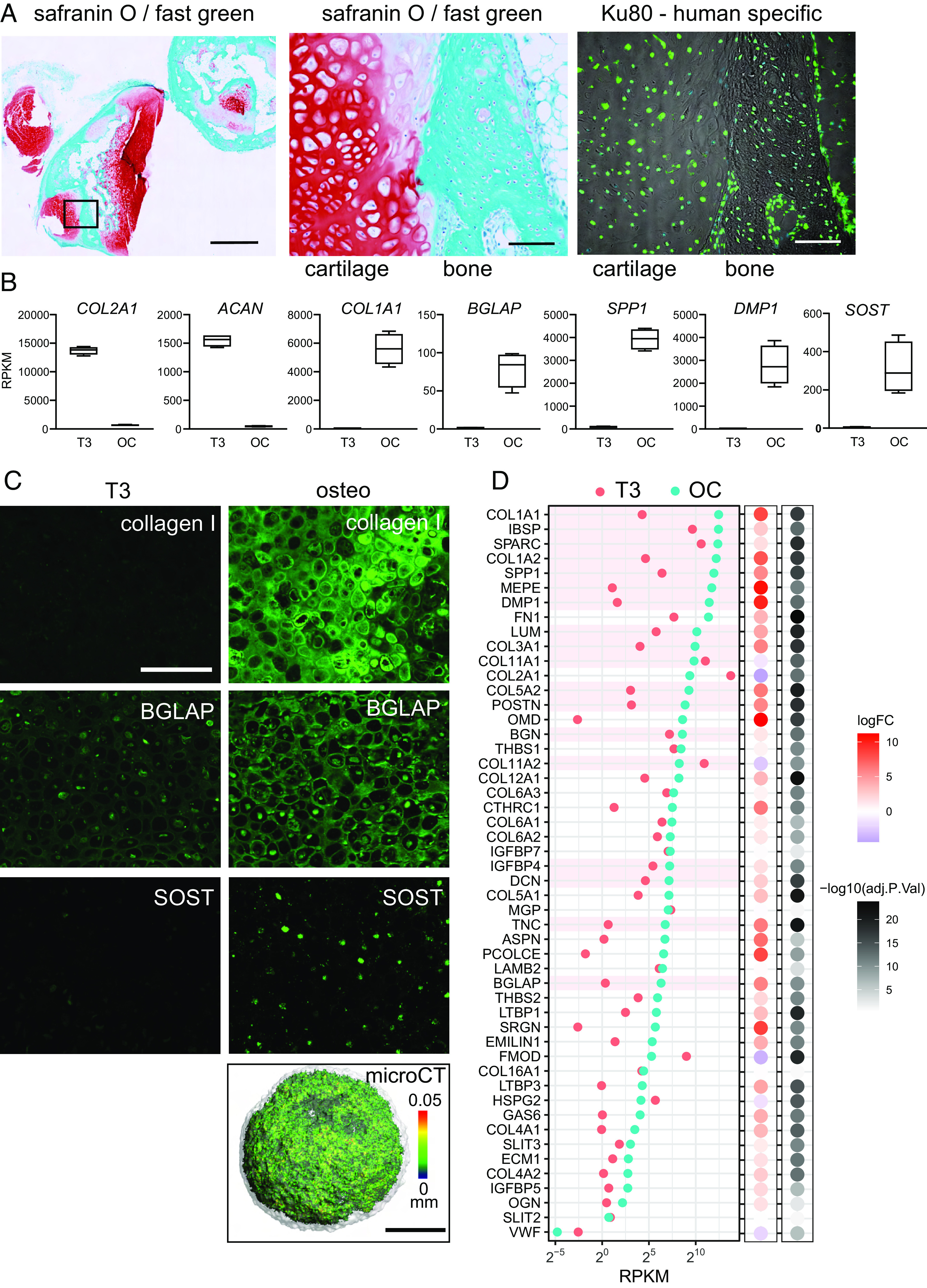
Hypertrophic chondrocytes can transition to osteoblasts. (A) Transdifferentiation in vivo. The iPSC line MCRIi001-A-BFP was differentiated to cartilage and then treated with 10 nM T3 for 7 d from day 35. At day 42, hypertrophic chondronoids were implanted subcutaneously into immunocompromised mice and harvested after 13 wk. Implants were decalcified and then sectioned and stained with safranin O for cartilage proteoglycans and fast green to highlight bone. Scale bar on left-hand image is 1000 µm. The box shows the region enlarged in the middle image. The image on the right shows the same region from an adjacent section immunostained with a human-specific Ku80 antibody. Scale bars on middle and right images are 100 µm. (B) Hypertrophic chondrocyte-to-osteoblast transition in vitro. From day 38, MCRIi018-B chondronoids were treated with 10 nM T3 for 14 d (T3) and then transferred to osteogenic medium for a further 3 wk (OC). Abundance (RPKM) of mRNA for cartilage genes COL2A1 and ACAN declines, and mRNA for osteoblast/osteocyte genes COL1A1BGLAPSPP1 DMP1, and SOST is significantly more abundant in osteogenic organoids. (C) Immunostaining shows collagen I and BGLAP deposited in the ECM in osteogenic organoids. Cells also express the mature osteocyte marker SOST (Scale bar is 200 µm.). The osteogenic organoid ECM is mineralized [microCT, (Scale bar is 500 µm.), color scale indicates mineral structure size]. (D) The 50 most highly expressed core matrisome genes in osteocytes isolated from mouse tibia (51) and their relative average expression in iPSC-derived hypertrophic chondronoids (T3) and osteogenic organoids (OC). N = 4 parallel differentiations. The 18 most highly expressed genes in vivo are highlighted in pink for comparison.
We then tested whether culture conditions supporting osteogenesis (52) would permit chondrocyte-to-osteoblast transition in vitro. After 3 wk in osteogenic medium, transcriptional profiling revealed that cartilage collagen II (COL2A1) and aggrecan (ACAN) were strongly down-regulated and hallmark osteoblast genes COL1A1, BGLAP, SPP1, and DMP1, and the osteocyte marker gene SOST, were strongly up-regulated (Fig. 2B). Collagen I, the major collagen type in bone, and BGLAP (osteocalcin), a mature osteoblast protein, were deposited into the ECM (Fig. 2C). Cells also expressed the mature osteocyte protein SOST consistent with osteoblast to osteocyte maturation (Fig. 2C). Further, microcomputed tomography (microCT) analysis confirmed extensive mineral deposition in the organoids, suggesting transition to a bone-like state (Fig. 2C). Importantly, our results show that prior T3 treatment to drive chondrocyte terminal hypertrophy is essential for optimal transition to bone-like organoids (SI Appendix, Fig. S7A).
Osteoblasts and osteocytes express a characteristic set of genes encoding ECM proteins that includes collagens COL1A1, COL1A2, COL3A1, COL5A1, and COL5A2; osteocalcin (BGLAP); proteoglycans, BGN, DCN, KERA, and ASPN; glycoproteins, SPARC and THBS1; and SIBLING proteins, IBSP, SPP1, DMP1, and MEPE (53). Many of these are among the most highly expressed and most highly up-regulated core matrisome genes in osteogenic conditions (Fig. 3 A and B). We identified the 50 most highly expressed core matrisome genes in mouse osteocytes in vivo (51) and found that 40 were more highly expressed in osteogenic organoids than in hypertrophic chondrocytes and those most highly expressed in vivo were also highly expressed in vitro (Figs. 2D and 3A). This pattern of gene expression changes was similar in experiments using chondronoids derived from a second iPSC line which had been induced to transition to osteoblasts (SI Appendix, Fig. S7 B–D).
Fig. 3.

Transcriptome changes during chondrocyte-to-osteoblast transition in vitro are consistent with in vivo skeletal development. (A) The most highly expressed core matrisome genes after 3 wk in osteogenic conditions (OC) (MCRIi018-B) and their relative expression in hypertrophic cartilage (T3). (B) The 20 most up-regulated and 20 most down-regulated core matrisome genes in osteogenic conditions vs hypertrophic cartilage. (C) Expression of the top transcripts that correspond to osteoblast precursor, osteoblast, and mature osteoblast clusters in scRNAseq of cells isolated from mouse calvaria (54) during in vitro transdifferentiation. Bubble plots show logFC and adj.P.value during in vitro chondrocyte-to-osteoblast transition. (D) Expression of genes that mark skeletal cell subpopulations in scRNAseq of in vivo mouse transdifferentiation (5) during in vitro chondrocyte-to-osteoblast transition. (E) Expression of TFs known to regulate osteoblast differentiation (55) during in vitro chondrocyte–to-osteoblast transition. N = 4 parallel differentiations. (F) The most highly expressed transcription factor genes after 3 wk in osteogenic conditions (OC) and their relative expression in hypertrophic cartilage (T3). (G) The 20 most up-regulated and 20 most down-regulated transcription factor genes in osteogenic conditions vs hypertrophic cartilage.
To provide further support for an in vitro hypertrophic chondrocyte-to-osteoblast/osteocyte transition, we examined the gene expression profile of cells generated during this stage of our differentiation protocol for markers that define osteoblast precursor, osteoblast, and more mature osteoblast cell clusters in single-cell RNA-Seq analysis of cells isolated from mouse bone (54). Osteoblast precursor markers did not differ greatly between T3-treated hypertrophic chondrocytes and osteogenic organoids (Fig. 3C). In contrast, many osteoblast and mature osteoblast markers were highly up-regulated under osteogenic conditions. For example, POSTN, TNC, CCDC3, FAP, and SFRP4 were up-regulated 3-21 fold in osteogenic organoids when compared to hypertrophic chondrocytes. The mature osteoblast markers COL1A1, COL1A2, BGLAP, IBSP, DMP1, and IFITM5 were similarly highly up-regulated under osteogenic conditions and were some of the most highly expressed marker genes in our dataset (Fig. 3C). These gene expression data are consistent with T3-treated hypertrophic chondrocytes transitioning to osteoblasts under osteogenic conditions in vitro.
We next examined the expression of 20 genes that mark hypertrophic chondrocyte, preosteoblast, and osteoblast cell clusters during mouse in vivo transdifferentiation and skeletal development (5). Under osteogenic conditions in vitro, gene expression patterns closely followed those identified during transdifferentiation of chondrocytes during mouse development (Fig. 3D), with concordant upregulation of osteogenic genes and downregulation of hypertrophic chondrocyte markers. Twelve of 13 well-characterized osteogenic TFs (55) were more highly expressed in osteogenic conditions than in hypertrophic chondrocytes (Fig. 3E). The reproducibility of this result was confirmed in osteogenic experiments using a second independent iPSC line (SI Appendix, Fig. S7 E–G).
These gene expression data indicate that our in vitro chondrocyte-to-osteoblast transition protocol recapitulates known key aspects of osteoblast development in vivo and thus presents an opportunity to further understand how hypertrophic chondrocyte-to-osteoblast transition is regulated. The 20 most highly expressed TFs under osteogenic conditions include 11 with known roles in osteoblast differentiation or bone development, SP7, MEF2A, ATF4, NFE2L1, VDR, HIF1A, XBP1, EGR1, CREB3L1, DLX3, and SKIL (Fig. 3E) (55–62). Other highly expressed TFs such as YBX1, HMGN3, HMGA1, MAZ, CREB3, SON, TRAFD1, ELK3, and DRAP1 have yet to be implicated in osteoblast differentiation. Similarly, the 20 most highly up-regulated TFs (Fig. 3G) include many with known roles in osteoblasts and bone, PRRX1, SATB2, RARB, TBX2, MAFB, PRRX2, TWIST1, FOS, JUNB, ZHX3 (55, 63–68), but others such as EGR3, CENPX, SNAI2, and ELK3 have as yet unknown osteogenic roles. Similar TF gene expression patterns were seen in experiments using a second independent iPSC line (SI Appendix, Fig. S7 H and I).
Modeling Human Skeletal Disorders.
We then examined the feasibility of using our iPSC differentiation protocol to model genetic skeletal disorders. Compared to an isogenic control, cartilage derived from iPSCs with a heterozygous COL2A1 p.G1113C mutation that causes hypochondrogenesis (69) had less collagen II in the ECM and was comprised of chondrocytes with strong intracellular collagen II staining, reflecting collagen misfolding and retention (Fig. 4A). The collagen fibrils in the ECM were disorganized and sparse (Fig. 4B), appearing similar to collagen II fibrils in human patient cartilage (70) and mice with Col2a1 mutations (71, 72). When iPSCs with a perinatal lethal osteogenesis imperfecta mutation, COL1A1 p.W1312C (73, 74), were differentiated to chondrocytes and then osteoblasts, collagen I staining in the ECM was less intense than that in the isogenic control (Fig. 4C), mirroring the reduced collagen I deposited by fibroblasts from this patient (74). The patient’s long bones displayed an osteogenesis maturation defect, with persistent cartilage in the cortical bone and only a thin layer of overlying osteoid more typical of early embryonic bone (75). Tellingly, osteogenic organoids derived from iPSCs carrying this mutation were unable to deposit mineral that could be detected with microCT (Fig. 4 D–F), consistent with the in vivo osteogenesis maturation defect. This ability to reproduce key features of these COL1A1 and COL2A1 skeletal disease phenotypes in vitro promises to facilitate detailed studies describing disease mechanisms, identifying therapeutic targets, and screening for novel drug treatments for patients.
Fig. 4.
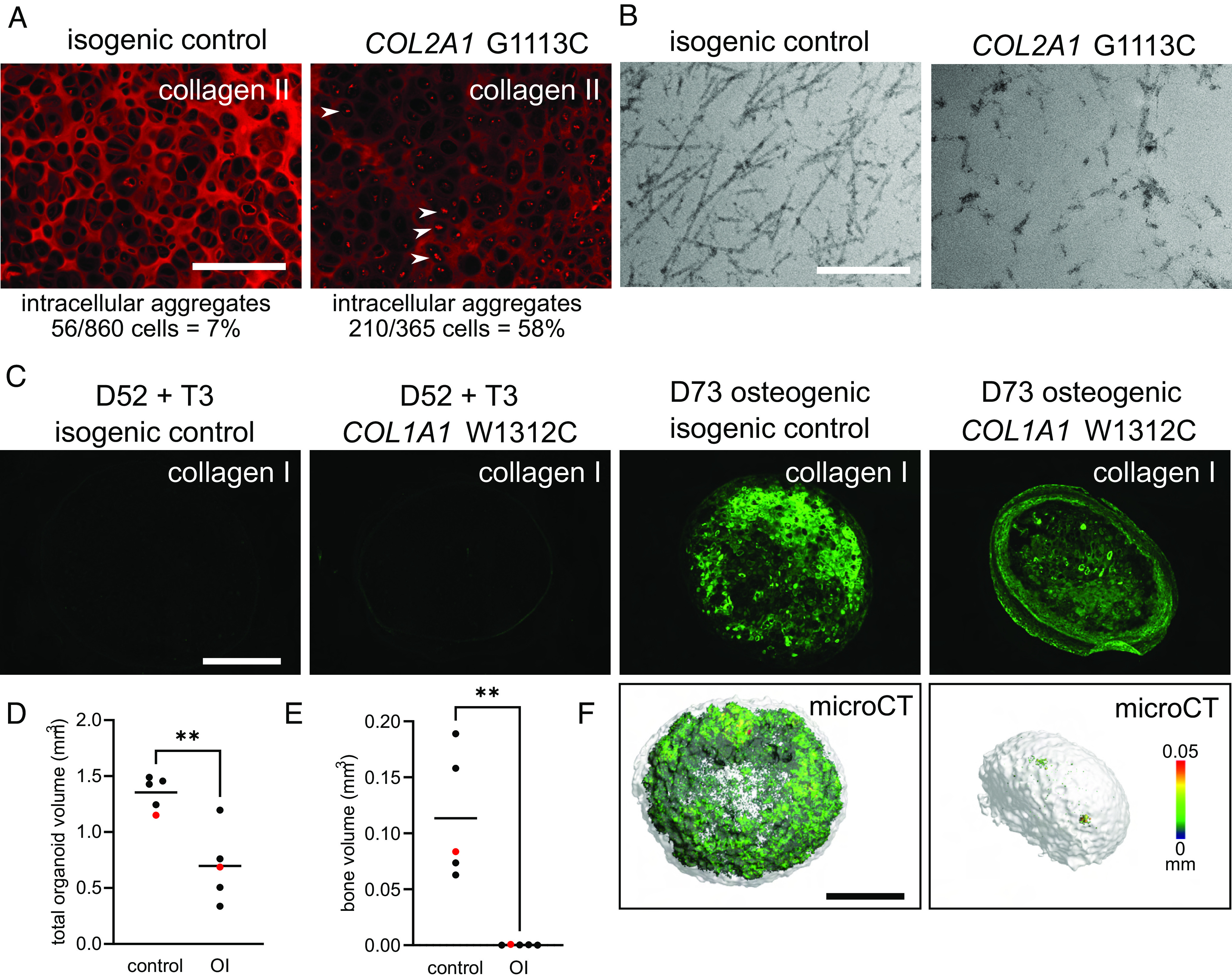
Modeling human genetic skeletal disorders in vitro. (A) Modeling the human cartilage disorder, hypochondrogenesis. COL2A1 G1113C mutant (MCRIi019-A-7) iPSC-derived chondronoids at D48 have reduced collagen II ECM immunostaining compared to the isogenic control (MCRIi019-A) and intracellular collagen II aggregates (white arrows). (Scale bar is 200 µm.) (B) TEM shows reduced and disorganized collagen II fibrils in COL2A1 G1113C mutant cartilage. (Scale bar is 500 nm.) (C) Modeling the human brittle bone disorder, osteogenesis imperfecta. COL1A1 W1312C mutant (MCRIi018-A) iPSC-derived osteogenic organoids at D73 have reduced collagen I ECM immunostaining compared to the isogenic control (MCRIi018-B). Organoids were fixed and then scanned with microCT. Representative images (F) are the samples shown in red in D and E. (Scale bars are 500 µm.), color scale indicates mineral structure size. (D) Total organoid volume. (E) Mineral volume. Data in D and E were compared using a Student’s t test, N = 5, **P < 0.005.
Discussion
To model human skeletal development and facilitate identifying the pathogenic mechanisms that result from disease-causing mutations, we have developed an iPSC differentiation protocol that follows normal mesoderm developmental pathways to sequentially produce sclerotome, then chondrocyte progenitors before branching to either articular chondrocytes, or chondrocytes that mature to hypertrophic chondrocytes and transition to osteoblasts producing a mineralized ECM. Our protocol thus recapitulates the key steps in growth plate development and endochondral ossification in vitro. We have documented global gene expression and identified unique gene expression patterns for ECM genes and TFs at different developmental stages in vitro. These gene expression patterns mirror what is known about in vivo gene expression and can provide insights into how cartilage development and endochondral bone growth are regulated.
Our iPSC-chondrocyte differentiation method has several advantages over current methods (see ref. 13 for a detailed review). It is relatively simple, uses defined media, and produces uniform cartilage organoids without requiring an intermediate flow sorting step. Our protocol includes cartilage pellet formation at the end of day 4, during the transition of somitic mesoderm to sclerotome, providing cells with the 3D structure essential to maintain their phenotype (76). Introducing fluid shear stress with rotary orbital culture accelerates chondrogenic differentiation and maturation and simplifies medium changes. We saw no evidence from histology or transcriptomics of off-target cell types such as melanocytes and neural cells that occur when BMP4 is used to stimulate chondroprogenitors postsclerotome (23). While these off-target cell types can be suppressed by inhibiting WNT and melanocyte-inducing transcription factor signaling, inhibiting WNT signaling prevents chondrocyte hypertrophy (23), a major disadvantage for modeling endochondral ossification.
After the sclerotome stage, an FGF2 pulse and prolonged culture supplemented with TGF-β3 produce articular chondrocytes that express the sentinel articular cartilage marker PRG4 as well as a range of other articular cartilage ECM genes such as CILP, ASPN, COL1A1, COL3A1, and elastic fiber components.
The alternative postsclerotome cell fate, growth plate hypertrophic chondrocytes, can be induced with an FGF2 pulse followed by culture in our chondrogenic basal medium. Some hypertrophic markers such as COL10A1, RUNX2, SPP1, SP7, MMP13, IHH, and MEF2C are up-regulated with spontaneous hypertrophy and are therefore likely to be early hypertrophy markers, while others including ALPL, FOSL2, VEGFA, BMP7, and DMP1 are only up-regulated after stimulation with T3 (triiodothyronine). T3 down-regulates the chondrogenic TFs SOX9/SOX5/SOX6 and cartilage matrisome genes COL9A1, COMP, and MATN1. In mouse skeletal development, T3 is required to promote hypertrophy around the tibial secondary ossification centers and allows hypertrophic chondrocyte-to-osteoblast transition (44). Others have used T3 to promote hypertrophy in iPSC chondrocyte differentiation (77); however, that protocol did not follow normal postsclerotome developmental pathways and included sequential and overlapping dexamethasone, PDGF, TGF-β3, BMP4, T3, and β-glycerophosphate addition. Gene expression data show that our differentiation program from chondroprogenitors to chondrocytes and chondrocyte hypertrophy follows the expected matrisome and transcription factor expression patterns during growth plate maturation. This gives us confidence that our system can be used to identify important players in these processes and explore the consequences of perturbing normal gene expression.
We identified dynamic gene expression changes in circadian rhythm genes and IRX3/IRX5, consistent with their roles in chondrocyte differentiation and skeletal development, highlighting the developmental relevance of our differentiation protocol. The chondrocyte-intrinsic circadian clock is essential to maintain articular cartilage structure and integrity (78), and many studies show that the chondrocyte circadian clock is dysregulated in osteoarthritis (79). The circadian clock is also important in early chondrogenesis, and synchronizing the clock with daily cyclic mechanical load (80) or serum shock (81) enhances cartilage ECM production. Our transcriptome data showing that circadian rhythm TFs are up-regulated as chondrocytes mature to hypertrophy are consistent with an emerging role for the circadian clock in the growth plate illustrated using a cartilage-specific Arntl (BMAL1) knockout in mouse (82). Deleting this core circadian rhythm gene led to a smaller proliferative zone, an enlarged hypertrophic zone, and reduced postnatal long bone growth (82). Similarly, Iroquois homeobox genes IRX3 and IRX5 were up-regulated in our hypertrophic chondrocytes. Human IRX5 mutations cause Hamamy syndrome, a recessive disorder with severe skeletal defects (83), and in the mouse, IRX3 and IRX5 are up-regulated in hypertrophic chondrocytes and are critical in the hypertrophic chondrocyte-to-osteoblast transition (50).
Stimulating in vitro hypertrophic chondrocyte transition to osteoblasts is an important and significant advance that will enhance our understanding of this fundamental pathway and allow in vitro bone disease modeling. The gene expression changes during this in vitro transition mirror the documented changes during in vivo hypertrophic chondrocyte to osteoblast transdifferentiation in mice (5), and expression in osteoblast lineage cells in mouse bone (54). For example, the osteocyte transcriptome genes most enriched in mouse bone osteocytes are Dmp1 and Mepe (51). These, along with the highly osteocyte-enriched Sost (51), are similarly highly enriched and highly expressed in our human osteoblast/osteocyte organoids.
Our iPSC–chondrocyte–osteocyte differentiation protocol could be further improved. Chondrocyte differentiation could be enhanced and/or the time to hypertrophy shortened if compressive mechanical forces were incorporated into the protocol. Cyclic compression of chondroprogenitors for 1 h a day entrains the circadian clock, increases cartilage marker gene expression, and enhances cartilage ECM production (80). Osteocytes also sense and respond to mechanical forces and conversely, bone is rapidly lost when not under load (84). This suggests that applying mechanical forces during the osteogenic induction phase could also be beneficial. Extending the osteogenic differentiation beyond 3 wk would allow further time to remodel the cartilage ECM and enhance deposition and organization of the bone-like ECM and thus likely improve the model. Blood vessel invasion into the hypertrophic cartilage in vivo brings in signals to promote osteogenesis, and so we used serum in the osteogenic medium. Defining the growth factors and signaling molecules critical for the hypertrophic chondrocyte-to-osteoblast transition would be a significant advance and allow the entire differentiation protocol to be done in defined medium suitable for producing cells/tissues for regenerative medicine.
Our protocol provides a way to explore human skeletal development in vitro and to model genetic skeletal disorders to identify pathogenic mechanisms and test drug therapies. Key pathogenic features of the collagen II disorder, hypochondrogenesis, and the collagen I disorder, osteogenesis imperfecta, were reproduced in our cartilage and bone organoids, suggesting that our system will be able to provide insights into genetic skeletal disorders, specifically those caused by mutations in structural ECM genes and TFs and signaling molecules important for chondrocyte differentiation and maturation. There are few effective therapies for genetic skeletal disorders, and our protocol recapitulating endochondral ossification in vitro promises to be an important tool in the search for treatments. The in vivo growth plate contains chondrocytes at different maturation stages, proliferative, prehypertrophic, and hypertrophic, as well as hypertrophic chondrocytes in the process of transitioning to osteoblasts. This zonal architecture is not present in our chondronoids, rather, the chondrocytes mature in a relatively synchronous fashion, providing a “snapshot” of each stage, and potentially making it easier to define the developmental stage first or most affected in disease and how this is moderated by drug therapies. The alternative differentiation pathway from chondroprogenitors to articular cartilage will facilitate arthritis-related studies, and we anticipate that it will be used to produce cartilage biomaterials for tissue regeneration.
Materials and Methods
Human-Induced Pluripotent Stem Cell Lines and Maintenance.
Six fully characterized and validated control human-induced pluripotent stem cell (iPSC) lines were used–feeder-dependent RM3.5 derived from deidentified fibroblasts purchased from the American Type Culture Collection (ATCC) (85); peripheral blood mononuclear cell-derived MCRIi001-A (86), and gene-edited subclones, MCRIi001-A-2, expressing a SOX9-tdTomato reporter (87), and MCRIi001-A-BFP expressing blue fluorescent protein (MCRIi001-A and derivatives produced with approval from the Royal Children’s Hospital Human Research Ethics Committee, HREC 35121A); and fibroblast-derived feeder-independent lines MCRIi018-B (HREC 33118A) (73) and MCRIi019-A derived from deidentified fibroblasts purchased from ATCC (88). For skeletal disease modeling, we used iPSC line MCRIi018-A with a heterozygous COL1A1 p.W1312C mutation and gene-corrected isogenic control MCRIi018-B (HREC 33118A) (73), and MCRIi019-A-7 (69) with a heterozygous COL2A1 p.G1113C mutation and isogenic control MCRIi019-A (88). Feeder-dependent cells were routinely expanded on mitotically inactivated mouse embryonic fibroblasts (MEFs) in DMEM/F-12 with 20% KnockOut Serum Replacement, 2 mM GlutaMAX, 1% Non-Essential Amino Acid Solution, 0.1 mM β-mercaptoethanol (all from ThermoFisher Scientific), and 50 ng/mL FGF2 (PeproTech) at 37 °C with 95% air/5% CO2. The media was changed daily, and cells passaged (1:6 split) every ~3 d with 0.5 mM EDTA in PBS. The feeder-free cells were routinely expanded on Matrigel (Corning)-coated plates in Essential 8 (E8) medium (ThermoFisher Scientific). The media was changed daily, and cells passaged (1:4 to 1:6) every 3 to 4 d with 0.5 mM EDTA in PBS.
Sclerotome Differentiation.
Undifferentiated iPSCs at 70 to 90% confluency were dissociated into fine cell clusters using 0.5 mM EDTA in PBS and typically passaged into 6-well plates (~2 × 105 cells per well for RM3.5, MCRIi0018-B and MCRIi019-A and ~0.8 × 105 cells per well for MCRIi001-A and MCRIi001-A-2), either preseeded with MEFs or precoated with Matrigel, and cultured for 24 to 48 h in the appropriate expansion media. Differentiation to sclerotome was as described (21) modified by substituting the CDM2 basal medium with APEL2 (StemCell Technologies) containing 5% Protein-Free Hybridoma Medium (PFHM II; ThermoFisher Scientific). On day 0, the medium was changed to anterior primitive streak-inducing medium containing 30 ng/mL Activin A (R&D Systems), 4 µM CHIR99021 (Tocris), 20 ng/mL FGF2 (PeproTech), and 100 nM PIK90 (Merck Millipore). After 24 h, this medium was replaced with medium containing 3 µM CHIR99021, 20 ng/mL FGF2, 1 µM A8301 (Tocris), and 0.25 µM LDN193189 (Cayman Chemical) to induce paraxial mesoderm for 24 h. Early somite development was then induced with medium containing 1 µM A8301 and 0.25 µM LDN193189, 3 µM C59 (Tocris), and 0.5 µM PD0325901 (Selleck Chemicals). After 24 h, sclerotome induction was initiated with 1 µM C59 and 2 µM purmorphamine (Sigma-Aldrich). After the first day of sclerotome induction (day 4), cells were dissociated from monolayer culture using TrypLE Select (ThermoFisher Scientific); resuspended in sclerotome differentiation media; 300 μL aliquots containing 2 × 105 cells were dispensed into 96-well low-attachment, round-bottomed plates (Corning); and pelleted by centrifugation at 400 g for 3 min using a swing-out rotor. The pellets were incubated in sclerotome differentiation media for a further 48 h to complete sclerotome differentiation in the 96-well static culture format. Importantly, cells at the sclerotome stage before pelleting could be frozen in medium containing 10% DMSO, stored under liquid nitrogen, and retrieved for subsequent studies, allowing well-characterized sclerotome to be stockpiled for differentiation consistency and larger-scale experiments.
Chondrocyte Differentiation and Maturation.
Sclerotome-to-chondrocyte differentiation was routinely done in APEL2 supplemented with 5% PFHM II, 20 ng/mL FGF2, and 1× Penicillin-Streptomycin (ThermoFisher Scientific). In some experiments, 20 ng/mL BMP4 (R & D Systems) was used for comparison. Pellets were routinely transferred to 6 cm nonadherent dishes (Greiner) at the end of either day 6 or day 20; 15 to 20 pellets per dish in 5 mL of medium with orbital rotation at 60 rpm. The medium was changed every 2 to 3 d. From day 20 (after 2 wk of FGF2 treatment), differentiation/maturation continued in APEL2 / 5% PFHM II with medium replaced every 2 to 3 d for the duration of the chondrogenic differentiation experiments as indicated. In some experiments, pellets were treated with FGF2 for 4 wk postsclerotome. Triiodothyronine (T3) is crucial for chondrocyte hypertrophy (44); thus in some experiments, differentiated chondrocytes were treated with 10 nM T3 (Sigma-Aldrich) for 10 to 21 d to induce chondrocyte hypertrophy. T3 was added when the pellets had consistent and even cartilage histology, which ranged between 4 and 6 wk postsclerotome chondrocyte differentiation. To generate articular cartilage, pellets were treated with 20 ng/mL FGF2 from the end of day 6 for 14 d, and with 10 ng/mL TGFβ3 (R&D Systems) from day 13.
Osteogenic Differentiation.
Hypertrophic chondrocytes can transdifferentiate into osteoblasts and osteocytes in vivo (7, 8). To recapitulate this later stage of endochondral bone formation in vitro, hypertrophic chondrocyte pellets in orbital culture were rinsed in PBS and then cultured in DMEM, high glucose, GlutaMAX Supplement, pyruvate (ThermoFisher Scientific), 20% FBS, 10 mM β-glycerophosphate, 50 μg/mL ascorbic acid 2-phosphate, 50 µg/mL sodium ascorbate, and 100 nM dexamethasone (all from Sigma-Aldrich) (osteogenic differentiation media) (52) for up to 3 wk. Some cultures were supplemented with 10 μM CHIR99021 for the first 7 d. The media was replaced every 2 to 3 d.
In Vivo Transplantation to Generate Human Ectopic Bone.
Cartilage organoids were treated with T3 for 1 wk from day 35, or for 2 wk from day 28 followed by 2 wk in osteogenic medium, then implanted subcutaneously into NBSGW mice at day 42 or day 56, respectively (89). The organoid grafts were harvested after 13 wk. All animal procedures were approved by the Murdoch Children’s Research Institute Animal Ethics Committee (Approval A863).
Histological Analyses.
Cultured and in vivo grafted pellet organoids were fixed overnight at 4 °C in Confix (Australian Biostain) neutral-buffered formalin, then washed in 70% ethanol, processed on a tissue processor (Excelsior AS, ThermoFisher Scientific), and paraffin-embedded (HistoStar Embedding Machine, ThermoFisher Scientific). After fixing, some in vivo grafted pellets were washed in water for 10 min and then decalcified in 14% EDTA, pH 7.4 at 4 °C for 5 d before embedding. Serial 5 μm sections cut from the pellet centers were mounted on Superfrost Plus slides (Menzel-Gläser) and heated at 60 °C for 1 h. The sections were treated with xylene to remove the paraffin wax and an ethanol series comprising 100%, 90%, 70% ethanol and then water to rehydrate the sections. The sections were stained with toluidine blue to detect a cartilage proteoglycan matrix or safranin O /fast green for cartilage and bone following standard protocols. Osteogenic pellet sections were also stained with von Kossa to detect mineralization. Images were captured using a Leica DM 2000 LED microscope with Leica Application Suite (LAS) software version 4.9.0 or Zeiss Axio Imager Z2 with Zen 3.1 Blue Edition software.
Immunostaining.
Formalin-fixed and paraffin-embedded sections were immunostained for collagen II (MAB8887; Sigma-Aldrich; 1:150 dilution), collagen I (LF68; Kerafast ENH018-FP; 1:1000), collagen X (90) (1:100), PRG4 (MABT401, Sigma-Aldrich) (1:100), BGLAP (LF-126; Kerafast ENH091-FP) (1:100), Ku80 (Cell Signaling) (1:300), and SOST (AF1589; R&D Systems; 1:100). For collagen II staining, antigen retrieval used 2 mg/mL porcine pepsin (Sigma-Aldrich) freshly prepared in 1 M Tris/HCl, pH 2.0 for 30 min at 37 °C, followed by digestion for 30 min at 37 °C with 0.2% hyaluronidase (Sigma-Aldrich) in PBS. Collagen X and PRG4 antigen retrieval involved heating to 60 °C in 10 mM Tris/1 mM EDTA/0.05% Tween-20, pH 9.0 (Tris/EDTA) for 30 min, cooling to room temperature for 30 min, followed by hyaluronidase digestion; collagen I and Ku80 staining followed antigen retrieval at 600C in Tris/EDTA as above. Sections to be stained with Ku80 were then permeabilized with 0.05% Triton X-100. Antigen retrieval for BGLAP was digestion with 20 µg/mL Proteinase K in 50 mM Tris/1 mM EDTA/5 mM CaCl2, pH 8 for 15 min at 37 °C. The sections were blocked with 3% BSA (Sigma-Aldrich) in PBS for 1 h at room temperature, and then incubated with primary antibodies in 1% BSA in PBS for 16 h at 4 °C. Appropriate species-specific Alexa Fluor 488 and 594 secondary antibodies (ThermoFisher Scientific) were diluted 1:200 in 1 % BSA in PBS with 2.5 µg/mL DAPI, and the sections were incubated for 1 h at room temperature. Slides were washed with PBS and coverslips mounted using Shandon Immu-Mount (ThermoFisher Scientific). Images were captured as described above.
Transmission Electron Microscopy.
Pellets were washed with PBS and fixed in 0.1 M sodium cacodylate containing 2.5% glutaraldehyde at 4 °C. Samples were prepared using the ROTO technique (91), dehydrated in an alcohol series, and embedded in Epon 812. Ultrathin sections (70 nm) were observed on a Tecnai F30 (FEI) with an extraction voltage of 200 kV. Micrographs were taken with a CETA camera (ThermoFisher Scientific).
MicroCT.
Pellets were formalin fixed, stored in 70% ethanol at 4 °C, then scanned using microCT (µCT50, Scanco Medical AG) with an 0.5-mm Al filter at an energy of 70 kVp, intensity of 200 μA, integration time of 300 ms, and a voxel (native) resolution of 3 μm. Bone morphometry parameters were calculated using a common threshold of 210/1,000 for bone tissue within the volume of interest.
Bulk Population RNA-Seq.
For RNA-Seq, organoids (2 to 3 pooled) were snap frozen in liquid nitrogen, pulverized using a liquid nitrogen-cooled tissue grinder, and RNA extracted with TRIzol, followed by purification using Direct-zol RNA Microprep kit spin columns (Zymo Research) according to the manufacturer’s instructions. RNA samples were quality controlled and sequenced at the Translational Genomics Unit, Murdoch Children’s Research Institute. When samples were directly compared, RNA was prepared from all samples at the same time and RNA sequencing libraries were made at the same time with the same batch of reagents. Libraries were constructed using Illumina Stranded mRNA Prep kits and sequenced using a NextSeq 500 to obtain ~20 × 106 75 bp paired-end reads per sample or a NovaSeq 6000 for 150 bp paired-end reads. Reads were aligned to hg38 using a Bpipe (92) RNA-Seq pipeline that incorporated FastQC quality control, adaptor trimming with Trimmomatic v.0.35 (93), mapping with STAR 2.7.3a (94), summarizing reads over genes with featureCounts (95), and MultiQC (96) to summarize the analyses. Downstream analyses and identification of differentially expressed genes used the EdgeR Bioconductor package (97). Genes with expression levels of at least one count per million in at least three samples were kept for further analysis. The data were TMM normalized and voom transformed. Differential expression was identified with robust paired moderated t tests using limma (97). Graphical visualizations used the gplots, tidyverse, and ggplot2 packages.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We would like to thank Zlatan Trifunovic, from the Bio21 Institute Ian Holmes Imaging Center, for technical assistance. This work was supported by the National Health and Medical Research Council of Australia through project grants awarded to S.R.L. and J.F.B. (GNT2003393); S.R.L., A.G.E., and J.F.B. (GNT1144807 and GNT1146952); E.G.S. and A.G.E. (GNT1068866, GNT1129861); S.R.L. and E.S.N. (GNT1164577); and research fellowships awarded to A.G.E. (GNT1117596) and E.G.S. (GNT1079004) and project grants awarded by the Australian Research Council Special Research Initiative in Stem Cells (Stem Cells Australia) and by the Stafford Fox Medical Research Foundation. Additional infrastructure funding to the Murdoch Children’s Research Institute was provided by the Australian Government National Health and Medical Research Council Independent Research Institute Infrastructure Support Scheme and the Victorian Government’s Operational Infrastructure Support Program. Work in the laboratories of E.S.N., E.G.S, and A.G.E. in the Novo Nordisk Foundation Center for Stem Cell Medicine is supported by Novo Nordisk Foundation grants (NNF21CC0073729).
Author contributions
S.R.L., E.S.N., T.L.C., L.H.W.K., J.L., Y.N.P., K.S.S., E.G.S., A.G.E., and J.F.B. designed research; S.R.L., E.S.N., T.L.C., L.H.W.K., L.S., L.R., J.L., Y.N.P., T.S., E.H., R.S., K.S.S., and J.F.B. performed research; S.R.L., K.M.B., and J.F.B. analyzed data; and S.R.L., E.S.N., E.G.S., A.G.E., and J.F.B. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
The RNAseq data discussed in this paper have been deposited in NCBI Gene Expression Omnibus with series accession number GSE219215 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE219215) (98).
Supporting Information
References
- 1.Karsenty G., Kronenberg H. M., Settembre C., Genetic control of bone formation. Annu. Rev. Cell Dev. Biol. 25, 629–648 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Kronenberg H. M., Developmental regulation of the growth plate. Nature 423, 332–336 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Long F., Ornitz D. M., Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 5, a008334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsang K. Y., Tsang S. W., Chan D., Cheah K. S. E., The chondrocytic journey in endochondral bone growth and skeletal dysplasia. Birth Defects Res. C Embryo Today 102, 52–73 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Haseeb A., et al. , SOX9 keeps growth plates and articular cartilage healthy by inhibiting chondrocyte dedifferentiation/osteoblastic redifferentiation. Proc. Natl. Acad. Sci. U.S.A. 118, e2019152118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park J., et al. , Dual pathways to endochondral osteoblasts: A novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol. Open 4, 608–621 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang L., Tsang K. Y., Tang H. C., Chan D., Cheah K. S. E., Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. U.S.A. 111, 12097–12102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou X., et al. , Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 10, e1004820 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long J. T., et al. , Hypertrophic chondrocytes serve as a reservoir for marrow-associated skeletal stem and progenitor cells, osteoblasts, and adipocytes during skeletal development. eLife 11, e76932 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fell Honor B., The histogenesis of cartilage and bone in the long bones of the embryonic fowl. J. Morphol. Physiol. 40, 417–459 (1925). [Google Scholar]
- 11.Hall B. K., Miyake T., All for one and one for all: Condensations and the initiation of skeletal development. BioEssays 22, 138–147 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Tani S., Chung U., Ohba S., Hojo H., Understanding paraxial mesoderm development and sclerotome specification for skeletal repair. Exp. Mol. Med. 52, 1166–1177 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Kinderen P., et al. , Differentiation of induced pluripotent stem cells into chondrocytes: Methods and applications for disease modeling and drug discovery. J. Bone Mineral Res. 37, 397–410 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Craft A. M., et al. , Specification of chondrocytes and cartilage tissues from embryonic stem cells. Development 140, 2597–2610 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Kawata M., et al. , Simple and robust differentiation of human pluripotent stem cells toward chondrocytes by two small-molecule compounds. Stem. Cell Rep. 13, 530–544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oldershaw R. A., et al. , Directed differentiation of human embryonic stem cells toward chondrocytes. Nat. Biotechnol. 28, 1187–1194 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Craft A. M., et al. , Generation of articular chondrocytes from human pluripotent stem cells. Nat. Biotechnol. 33, 638–645 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Umeda K., et al. , Human chondrogenic paraxial mesoderm, directed specification and prospective isolation from pluripotent stem cells. Sci. Rep. 2, 455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakajima T., et al. , Modeling human somite development and fibrodysplasia ossificans progressiva with induced pluripotent stem cells. Development 145, dev165431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xi H., et al. , In vivo human somitogenesis guides somite development from hPSCs. Cell Rep. 18, 1573–1585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loh K. M., et al. , Mapping the pairwise choices leading from pluripotency to human bone, heart, and other mesoderm cell types. Cell 166, 451–467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adkar S. S., et al. , Step-wise chondrogenesis of human induced pluripotent stem cells and purification via a reporter allele generated by CRISPR-Cas9 genome editing. Stem. Cells 37, 65–76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu C.-L., et al. , Single cell transcriptomic analysis of human pluripotent stem cell chondrogenesis. Nat. Commun. 12, 362 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Limraksasin P., et al. , Shaking culture enhances chondrogenic differentiation of mouse induced pluripotent stem cell constructs. Sci. Rep. 10, 14996 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salinas E. Y., et al. , Shear stress induced by fluid flow produces improvements in tissue-engineered cartilage. Biofabrication 12, 045010 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee H.-H., et al. , Hypoxia enhances chondrogenesis and prevents terminal differentiation through PI3K/Akt/FoxO dependent anti-apoptotic effect. Sci. Rep. 3, 2683 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robins J. C., et al. , Hypoxia induces chondrocyte-specific gene expression in mesenchymal cells in association with transcriptional activation of Sox9. Bone 37, 313–322 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Shang J., Liu H., Li J., Zhou Y., Roles of hypoxia during the chondrogenic differentiation of mesenchymal stem cells. Curr. Stem Cell Res. Ther. 9, 141–147 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Galeano-Garces C., et al. , Molecular validation of chondrogenic differentiation and hypoxia responsiveness of platelet-lysate expanded adipose tissue-derived human mesenchymal stromal cells. Cartilage 8, 283–299 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirao M., Tamai N., Tsumaki N., Yoshikawa H., Myoui A., Oxygen Tension regulates chondrocyte differentiation and function during endochondral ossification*. J. Biol. Chem. 281, 31079–31092 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Decker R. S., Koyama E., Pacifici M., Genesis and morphogenesis of limb synovial joints and articular cartilage. Matrix Biol. 39, 5–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soeda T., et al. , Sox9-expressing precursors are the cellular origin of the cruciate ligament of the knee joint and the limb tendons. Genesis 48, 635–644 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Q., et al. , Expression of doublecortin reveals articular chondrocyte lineage in mouse embryonic limbs. Genesis 49, 75–82 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Naba A., et al. , The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 49, 10–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyanich R., et al. , Application of confocal, SHG and atomic force microscopy for characterizing the structure of the most superficial layer of articular cartilage. J. Microsc. 275, 159–171 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Iwamoto M., et al. , Transcription factor ERG and joint and articular cartilage formation during mouse limb and spine skeletogenesis. Dev. Biol. 305, 40–51 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohta Y., et al. , Articular cartilage endurance and resistance to osteoarthritic changes require transcription factor Erg. Arthritis Rheumatol. 67, 2679–2690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reijntjes S., Stricker S., Mankoo B. S., A comparative analysis of Meox1 and Meox2 in the developing somites and limbs of the chick embryo. Int. J. Dev. Biol. 51, 753–759 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Ayadi A., Suelves M., Dollé P., Wasylyk B., Net, an Ets ternary complex transcription factor, is expressed in sites of vasculogenesis, angiogenesis, and chondrogenesis during mouse development. Mech. Dev. 102, 205–208 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Raouf A., Seth A., Ets transcription factors and targets in osteogenesis. Oncogene 19, 6455–6463 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Napierala D., et al. , Uncoupling of chondrocyte differentiation and perichondrial mineralization underlies the skeletal dysplasia in tricho-rhino-phalangeal syndrome. Hum. Mol. Genet. 17, 2244–2254 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsuda T., et al. , Zinc finger protein Zac1 is expressed in chondrogenic sites of the mouse. Dev. Dyn. 229, 340–348 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Varrault A., et al. , Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 11, 711–722 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Aghajanian P., Xing W., Cheng S., Mohan S., Epiphyseal bone formation occurs via thyroid hormone regulation of chondrocyte to osteoblast transdifferentiation. Sci. Rep. 7, 10432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lambert S. A., et al. , The human transcription factors. Cell 172, 650–665 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Shimo T., et al. , Retinoic receptor signaling regulates hypertrophic chondrocyte-specific gene expression. In Vivo 33, 85–91 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dreher S. I., Fischer J., Walker T., Diederichs S., Richter W., Significance of MEF2C and RUNX3 Regulation for endochondral differentiation of human mesenchymal progenitor cells. Front. Cell Dev. Biol. 8, 81 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okuma T., et al. , Regulation of mouse chondrocyte differentiation by CCAAT/enhancer-binding proteins. Biomed. Res. 36, 21–29 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Tan Z., et al. , Synergistic co-regulation and competition by a SOX9-GLI-FOXA phasic transcriptional network coordinate chondrocyte differentiation transitions. PLoS Genet. 14, e1007346 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan Z., et al. , IRX3 and IRX5 inhibit adipogenic differentiation of hypertrophic chondrocytes and promote osteogenesis. J. Bone Mineral Res. 35, 2444–2457 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Youlten S. E., et al. , Osteocyte transcriptome mapping identifies a molecular landscape controlling skeletal homeostasis and susceptibility to skeletal disease. Nat. Commun. 12, 2444 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langenbach F., Handschel J., Effects of dexamethasone, ascorbic acid and β-glycerophosphate on the osteogenic differentiation of stem cells in vitro. Stem. Cell Res. Ther. 4, 117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin X., Patil S., Gao Y.-G., Qian A., The bone extracellular matrix in bone formation and regeneration. Front. Pharmacol. 11, 757 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ayturk U. M., et al. , Single-cell RNA sequencing of calvarial and long-bone endocortical cells. J. Bone Miner. Res. 35, 1981–1991 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan W. C. W., Tan Z., To M. K. T., Chan D., Regulation and role of transcription factors in osteogenesis. Int. J. Mol. Sci. 22, 5445 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim J., Xing W., Wergedal J., Chan J. Y., Mohan S., Targeted disruption of nuclear factor erythroid-derived 2-like 1 in osteoblasts reduces bone size and bone formation in mice. Physiol. Genom. 40, 100–110 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Leupin O., et al. , Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J. Bone. Miner. Res. 22, 1957–1967 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pang L., et al. , Mutant dlx3b disturbs normal tooth mineralization and bone formation in zebrafish. PeerJ 8, e8515 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Price J. A., Bowden D. W., Wright J. T., Pettenati M. J., Hart T. C., Identification of a mutation in DLX3 associated with tricho-dento-osseous (TDO) syndrome. Hum. Mol. Genet. 7, 563–569 (1998). [DOI] [PubMed] [Google Scholar]
- 60.Symoens S., et al. , Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet. J. Rare. Dis. 8, 154 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tohmonda T., et al. , The IRE1α-XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix. EMBO Rep. 12, 451–457 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Q., et al. , RUNX2 co-operates with EGR1 to regulate osteogenic differentiation through Htra1 enhancers. J. Cell Physiol. 235, 8601–8612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dobreva G., et al. , SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell 125, 971–986 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Liu N., et al. , Functional variants in TBX2 are associated with a syndromic cardiovascular and skeletal developmental disorder. Hum. Mol. Genet. 27, 2454–2465 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suehiro F., et al. , Impact of zinc fingers and homeoboxes 3 on the regulation of mesenchymal stem cell osteogenic differentiation. Stem. Cells Dev. 20, 1539–1547 (2011). [DOI] [PubMed] [Google Scholar]
- 66.ten Berge D., Brouwer A., Korving J., Martin J. F., Meijlink F., Prx1 and Prx2 in skeletogenesis: Roles in the craniofacial region, inner ear and limbs. Development 125, 3831–3842 (1998). [DOI] [PubMed] [Google Scholar]
- 67.Wagner E. F., Functions of AP1 (Fos/Jun) in bone development. Anna. Rheumatic Dis. 61, ii40–ii42 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zankl A., et al. , Multicentric carpotarsal osteolysis is caused by mutations clustering in the amino-terminal transcriptional activation domain of MAFB. Am. J. Hum. Genet. 90, 494–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lilianty J., Bateman J. F., Lamandé S. R., Generation of a heterozygous COL2A1 (p.G1113C) hypochondrogenesis mutation iPSC line, MCRIi019-A-7, using CRISPR/Cas9 gene editing. Stem Cell Res. 56, 102515 (2021). [DOI] [PubMed] [Google Scholar]
- 70.Freisinger P., et al. , Mutation in the COL2A1 gene in a patient with hypochondrogenesis. Expression of mutated COL2A1 gene is accompanied by expression of genes for type I procollagen in chondrocytes. J. Biol. Chem. 269, 13663–13669 (1994). [PubMed] [Google Scholar]
- 71.Liang G., et al. , Endoplasmic reticulum stress-unfolding protein response-apoptosis cascade causes chondrodysplasia in a col2a1 p.Gly1170Ser mutated mouse model. PLoS One 9, e86894 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kimura M., et al. , Endoplasmic reticulum stress-mediated apoptosis contributes to a skeletal dysplasia resembling platyspondylic lethal skeletal dysplasia, torrance type, in a novel Col2a1 mutant mouse line. Biochem. Biophys. Res. Commun. 468, 86–91 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Howden S., et al. , The use of simultaneous reprogramming and gene correction to generate an osteogenesis imperfecta patient COL1A1 c. 3936 G>T iPSC line and an isogenic control iPSC line. Stem Cell Res. 38, 101453 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Lamandé S. R., et al. , Endoplasmic reticulum-mediated quality control of type I collagen production by cells from osteogenesis imperfecta patients with mutations in the proα1(I) chain carboxyl-terminal propeptide which impair subunit assembly *. J. Biol. Chem. 270, 8642–8649 (1995). [DOI] [PubMed] [Google Scholar]
- 75.Cole W. G., Chow C. W., Bateman J. F., Sillence D. O., The phenotypic features of osteogenesis imperfecta resulting from a mutation of the carboxyl-terminal pro alpha 1 (I) propeptide that impairs the assembly of type I procollagen and formation of the extracellular matrix. J. Med. Genet. 33, 965–967 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ecke A., et al. , Tissue specific differentiation of human chondrocytes depends on cell microenvironment and serum selection. Cells 8, 934 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pretemer Y., et al. , Differentiation of hypertrophic chondrocytes from human iPSCs for the in vitro modeling of chondrodysplasias. Stem Cell Rep. 16, 610–625 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dudek M., The chondrocyte clock gene Bmal1 controls cartilage homeostasis andintegrity (2016). 10.1172/JCI82755. Accessed 14 November 2022. [DOI] [PMC free article] [PubMed]
- 79.Snelling S. J. B., Forster A., Mukherjee S., Price A. J., Poulsen R. C., The chondrocyte-intrinsic circadian clock is disrupted in human osteoarthritis. Chronobiol. Int. 33, 574–579 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Vágó J., et al. , Cyclic uniaxial mechanical load enhances chondrogenesis through entraining the molecular circadian clock. J. Pineal Res. 73, e12827 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alagha M. A., et al. , A synchronized circadian clock enhances early chondrogenesis. Cartilage 13, 53S–67S (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma Z., et al. , Deletion of clock gene Bmal1 impaired the chondrocyte function due to disruption of the HIF1α-VEGF signaling pathway. Cell Cycle 18, 1473–1489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bonnard C., et al. , Mutations in IRX5 impair craniofacial development and germ cell migration via SDF1. Nat. Genet. 44, 709–713 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Li X., Kordsmeier J., Xiong J., New advances in osteocyte mechanotransduction. Curr. Osteoporos. Rep. 19, 101–106 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kao T., et al. , GAPTrap: A simple expression system for pluripotent stem cells and their derivatives. Stem Cell Rep. 7, 518–526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vlahos K., et al. , Generation of iPSC lines from peripheral blood mononuclear cells from 5 healthy adults. Stem Cell Res. 34, 101380 (2019). [DOI] [PubMed] [Google Scholar]
- 87.Nur Patria Y., et al. , Generation of a SOX9-tdTomato reporter human iPSC line, MCRIi001-A-2, using CRISPR/Cas9 editing. Stem Cell Res. 42, 101689 (2020). [DOI] [PubMed] [Google Scholar]
- 88.Kung L. H. W., et al. , CRISPR/Cas9 editing to generate a heterozygous COL2A1 p. G1170S human chondrodysplasia iPSC line, MCRIi019-A-2, in a control iPSC line, MCRIi019-A. Stem Cell Res. 48, 101962 (2020). [DOI] [PubMed] [Google Scholar]
- 89.McIntosh B. E., et al. , B6.SCID Il2rγ-/- Kit(W41/W41) (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Rep. 4, 171–180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cameron T. L., et al. , Cartilage-specific ablation of XBP1 signaling in mouse results in a chondrodysplasia characterized by reduced chondrocyte proliferation and delayed cartilage maturation and mineralization. Osteoarthritis Cartilage 23, 661–670 (2015). [DOI] [PubMed] [Google Scholar]
- 91.Hanssen E., et al. , Electron tomography of plasmodium falciparum merozoites reveals core cellular events that underpin erythrocyte invasion. Cell. Microbiol. 15, 1457–1472 (2013). [DOI] [PubMed] [Google Scholar]
- 92.Sadedin S. P., Pope B., Oshlack A., Bpipe: A tool for running and managing bioinformatics pipelines. Bioinformatics 28, 1525–1526 (2012). [DOI] [PubMed] [Google Scholar]
- 93.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dobin A., et al. , STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Ewels P., Magnusson M., Lundin S., Käller M., MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Law C. W., et al. , RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res 5, ISCB Comm J-1408 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lamandé S. R., Ng E. S., Stanley E. G., Elefanty A. G., Bateman J. F., Human iPSC can be differentiated in vitro to articular cartilage or to osteogenic organoids via the endochondral ossification transdifferentiation pathway. NCBI Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE219215. Deposited 2 December 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
The RNAseq data discussed in this paper have been deposited in NCBI Gene Expression Omnibus with series accession number GSE219215 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE219215) (98).