

Abstract
Rationale
Eosinophils are associated with airway inflammation in respiratory disease. Eosinophil production and survival is controlled partly by interleukin-5: anti-interleukin-5 agents reduce asthma and response correlates with baseline eosinophil counts. However, whether raised eosinophils are causally related to chronic obstructive pulmonary disease (COPD) and other respiratory phenotypes is not well understood.
Objectives
We investigated causality between eosinophils and: lung function, acute exacerbations of COPD, asthma-COPD overlap (ACO), moderate-to-severe asthma and respiratory infections.
Methods
We performed Mendelian randomisation (MR) using 151 variants from genome-wide association studies of blood eosinophils in UK Biobank/INTERVAL, and respiratory traits in UK Biobank/SpiroMeta, using methods relying on different assumptions for validity. We performed multivariable analyses using eight cell types where there was possible evidence of causation by eosinophils.
Measurements and main results
Causal estimates derived from individual variants were highly heterogeneous, which may arise from pleiotropy. The average effect of raising eosinophils was to increase risk of ACO (weighted median OR per SD eosinophils, 1.44 (95%CI 1.19 to 1.74)), and moderate-severe asthma (weighted median OR 1.50 (95%CI 1.23 to 1.83)), and to reduce forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) and FEV1 (weighted median estimator, SD FEV1/FVC: −0.054 (95% CI −0.078 to −0.029), effect only prominent in individuals with asthma).
Conclusions
Broad consistency across MR methods may suggest causation by eosinophils (although of uncertain magnitude), yet heterogeneity necessitates caution: other important mechanisms may be responsible for the impairment of respiratory health by these eosinophil-raising variants. These results could suggest that anti-IL5 agents (designed to lower eosinophils) may be valuable in treating other respiratory conditions, including people with overlapping features of asthma and COPD.
Keywords: Asthma Epidemiology, Asthma Genetics, Asthma Mechanisms, COPD epidemiology, COPD exacerbations mechanisms, Eosinophil Biology, Respiratory Infection
Key messages.
What is already known on this topic?
Blood eosinophil counts are predictive of response to anti-interleukin-5 (IL5) drugs used to treat asthma. However, the causal nature of the relationship between eosinophils and a broad range of respiratory traits related to asthma and chronic obstructive pulmonary disease (COPD) is not fully understood.
What this study adds?
In this Mendelian randomisation study, while the average effect of raising eosinophils was to increase risk of asthma-COPD overlap and asthma, and worsen forced expiratory volume in 1 s (FEV1) and FEV1/forced vital capacity in individuals with asthma, heterogeneity of individual causal estimates means caution is needed when interpreting these results causally, as these results could also be consistent with eosinophil-raising genetic variants impairing respiratory health via other causal pathways.
How this study might affect research, practice or policy?
These results could suggest that anti-IL5 agents (designed to lower eosinophils) may be valuable in treating other respiratory conditions, including people overlapping features of both asthma and COPD. Future work should seek to explore other potential mechanisms besides eosinophils by which anti-IL5 agents may improve respiratory health, to inform whether the clinical indications for anti-IL5 agents or biomarkers for stratifying their use could be extended.
Introduction
Eosinophils are proinflammatory granulocytes associated with symptom severity and exacerbation frequency in asthma and chronic obstructive pulmonary disease (COPD).1–3 The degree of eosinophilia (raised eosinophils) in these obstructive lung diseases varies: while eosinophil inflammation due to allergic sensitisation has been considered characteristic of asthma, not all patients with asthma have eosinophilia.1 4 Moreover, while airway inflammation in COPD is typically mediated by neutrophils, some individuals with COPD have raised eosinophils.1 5
The production and survival of eosinophils is partly regulated by interleukin-5 (IL-5), and anti-IL5 therapies (eg, mepolizumab, reslizumab, and the anti-IL5Rα agent, benralizumab) are now licensed in many countries for the treatment of severe eosinophilic asthma.6–12 The decision to treat asthma with these drugs is currently based on blood eosinophil count, among other factors,1 since post-hoc analyses of clinical trials stratified by eosinophil levels have shown increased efficacy of mepolizumab for treating severe asthma in those with higher baseline eosinophils.2 Results from Mendelian randomisation (MR) analyses have also provided evidence for a role of eosinophils in asthma (estimated OR 1.70 (95% CI 1.53 to 1.91).13 MR analyses use genetic variants as instrumental variables (IVs) to investigate causality between exposure and outcome, and under certain assumptions may obviate problems with traditional observational epidemiology (eg, reverse causation, confounding), permitting causal inference.
In addition to asthma, blood eosinophils are associated with quantitative lung function in general populations (ie, including individuals without asthma).14 However, causality has yet to be established: an inverse relationship between eosinophils and lung health has been suggested, yet a previous MR of lung function (plus another including asthma and COPD) were of small sample size, with imprecise estimates precluding confident inference.15 16 Moreover, causality of eosinophils on other respiratory phenotypes, for example, asthma-COPD overlap (ACO), and respiratory infections are yet to be investigated. COPD is diagnosed by spirometry if the ratio of the forced expiratory volume in 1 s (FEV1) to forced vital capacity (FVC), FEV1/FVC, is <0.7, with airflow obstruction graded by predicted FEV1. Therefore, studying eosinophils as determinants of quantitative lung function is a powerful way of understanding their role in the development of fixed airflow obstruction such as in COPD.17 18 Investigating causality between eosinophils and fixed airflow obstruction is pertinent given interest in the potential use of mepolizumab in COPD9–12; evidence for causality of eosinophils in a wider range of respiratory phenotypes could suggest that anti-IL5 agents (designed to lower eosinophils) might be helpful in conditions beyond asthma.
We undertook two-sample MR analyses using summary-level genome-wide association study (GWAS) data to assess causality between eosinophils and conditions encompassing fixed and reversible airflow obstruction, using genetic variants associated with blood eosinophils as IVs.13 We investigated causality of eosinophils on three quantitative lung function spirometry traits, and four clinical phenotypes (moderate-to-severe asthma, acute exacerbations of COPD (AECOPD), ACO and respiratory infections). We used MR approaches relying on different assumptions for validity, and followed up traits showing evidence of possible causality to assess evidence that the IVs affected lung function via eosinophil counts and not via other blood cell types. Overall, our aim was to provide a comprehensive assessment of the causal role of blood eosinophil counts in relation to respiratory health and disease.
Methods
We assessed causality between eosinophils and other blood cell counts in relation to respiratory outcomes using MR.19 20 MR involves using genetic variants (here single-nucleotide polymorphisms, SNPs), as IVs for an exposure of interest, in this case eosinophil counts, by comparing the magnitude of the effect of the SNPs on the outcome to the effect of the SNPs on the exposure.19 20 All analyses reported are two-sample MR analyses, since SNP–exposure and SNP–outcome associations were extracted from different (yet overlapping21) samples. Core MR assumptions for inferring causality between are that: (1) the genetic variants are associated with the exposure of interest; (2) there are no unmeasured confounders of the associations between genetic variants and outcome; and (3) the genetic variants affect the outcome only via the exposure of interest (figure 1).19 Additional assumptions for accurate point estimation of effect sizes are discussed in online supplemental file 1, and elsewhere.22
Figure 1.

Mendelian randomisation (MR): core assumptions Mendelian randomisation may be used to test for causality between an exposure (eg, eosinophils) and outcome (eg, a respiratory outcome such as FEV1/FVC), if the following core assumptions hold (see 1–3 on the figure): (1) the genetic variation (single nucleotide polymorphisms in this work) used as instrumental variables are associated with the exposure of interest; the genetic variants are not associated with unobserved confounders of the exposure-outcome association (straight dashed arrow). Genetic variants are allocated randomly at conception (Mendel’s law of independent assortment) and so typically should not be associated with these confounding variables; association between the genetic variants and the outcome is via the exposure, and not via an alternate pathway (ie, there is no ‘horizontal pleiotropy’, see curved dashed arrow). While difficult to verify, reassurance that this assumption holds can be provided using biological knowledge of how the SNP functions, and by checking whether multiple MR methods, each relying on different assumptions for validity, give consistent results (known as triangulation).20 FEV1, forced expiratory volume in 1 s, FVC, forced vital capacity; SNP, single-nucleotide polymorphisms.
thoraxjnl-2021-217993supp001.pdf (2.2MB, pdf)
All GWAS datasets analysed included UK Biobank, a prospective cohort study including spirometry, biological assays, questionnaire data, and linked healthcare records, and 450 000 participants with genotype data.23 Other studies were incorporated where available, and all GWAS data were from individuals of European ancestry. Datasets are summarised below, and descriptions of covariate adjustments, and exposure-outcome GWAS overlap are given in the extended methods (online supplemental file 1).
Exposure GWAS data sets (blood cell parameters)
We used summary-level data from eight published GWASs of blood cell counts13 in the initial release of UK Biobank genetic data (N up to 132,959, that is, around 30% of participants with genotype data), plus the INTERVAL study (N up to 40 521)).13 GWASs were of blood eosinophils, basophils, neutrophils, monocytes, lymphocytes, platelets, red blood cells and reticulocytes, with adjustments for technical and seasonal covariates, plus age, menopausal status, height, weight, smoking and alcohol (online supplemental file 1).
Outcome GWAS data sets (respiratory outcomes)
See also online supplemental file 1.
Quantitative lung function GWASs
We used published summary-level data from three GWAS of FEV1, FVC and FEV1/FVC, in UK Biobank (n=3 21 047) and the SpiroMeta consortium (n=79 055).18 Prior to GWAS, traits were preadjusted for age, age2, sex, height, smoking status and other covariates as appropriate, for example, ancestry principal components. Residuals were inverse-normal rank transformed.
Clinical outcome GWAS
Moderate-to-severe asthma
We used a published GWAS of moderate-to-severe asthma within the Genetics of Asthma Severity and Phenotypes initiative, the U-BIOPRED asthma cohort, and UK Biobank.24 Cases (n=5135) were taking asthma medication, and met criteria for moderate-to-severe asthma (British Thoracic Society 2014 guidelines). Controls (n=25 675) excluded those with a doctor diagnosis of asthma, rhinitis, eczema, allergy, emphysema, or chronic bronchitis, or missing medication data. Analyses were adjusted for 10 ancestry principal components.
Acute exacerbations of COPD
We defined AECOPD in UK Biobank; the eligible sample was restricted to individuals with FEV1/FVC<0.7. Exacerbation cases (n=2771) had an ICD-10 code for AECOPD or a lower respiratory tract infection in Hospital Episode Statistics data (online supplemental table 1). Controls (n=42 052) had FEV1/FVC<0.7, without an AECOPD code. Associations were adjusted for age (at recruitment), age2, sex, smoking status (ever/never), genotyping array and 10 principal components.
thoraxjnl-2021-217993supp003.xlsx (86.2KB, xlsx)
Asthma-COPD overlap
We defined ACO in UK Biobank (N=8068) as individuals self-reporting a doctor diagnosis of asthma, with FEV1/FVC<0.7 and FEV1 <80% predicted at any study visit. Controls (N=40 360) were selected in approximately a 5:1 ratio, from participants reporting no asthma or COPD, (FEV1 >80% predicted, FEV1/FVC>0.7). Associations were adjusted for age (at recruitment), sex, smoking status and 10 principal components.25
Respiratory infections
We defined respiratory tract infections requiring hospital admission in UK Biobank, using the ICD-10 codes in online supplemental table 2. Cases had ≥1 admission for respiratory infections (N=19 459). Controls had no admissions for respiratory infections and were selected in approximately a 5:1 ratio (N=101 438). Associations were adjusted for age (at recruitment), age2, sex, smoking status, genotyping array, and 10 principal components.26
Statistical methods
Univariable MR of eosinophils and respiratory traits and diseases
We performed separate MR analyses of eosinophils on three quantitative lung function traits (FEV1, FVC, FEV1/FVC); and four clinical phenotypes (asthma, AECOPD, ACO, respiratory infections) using genetic IVs from the work of Astle and colleagues.13 Selection of 151 eosinophil IVs and harmonisation of SNP-exposure and SNP-outcome datasets is detailed in the online supplemental file 1. The primary MR analysis used the inverse-variance weighted (IVW) method and a random-effects model, which will return a valid causal estimate provided that the average pleiotropic effect is zero. We investigated the ‘no pleiotropy’ assumption using MR-Egger regression,27 the weighted median estimator28 and MR-PRESSO29 (see online supplemental file 1 for details on assumptions relied on for validity by each method). Further sensitivity analyses: (1) investigated robustness of findings to heterogeneity using MR-PRESSO (for traits with some evidence of causation by eosinophils), (2) restricted to non-UKB FEV1/FVC GWAS data, to assess sensitivity to sample overlap and (3) restricted to FEV1/FVC GWAS data in UKB, stratifying by asthma status.
Multivariable MR analyses of multiple blood cell types and respiratory outcomes
Since SNPs affecting eosinophils also affect other blood cell types,13 we used multivariable MR to estimate the influence of multiple cell types on respiratory outcomes, after conditioning on the effects of the SNPs on other cell types. Multivariable MR analyses were performed for respiratory outcomes with evidence of eosinophil causation in the IVW MR analyses above, and with broadly consistent effect estimates in the weighted median and MR-Egger analyses. We also performed an analysis of FEV1/FVC in UKB (stratifying by asthma status).
There were 1166 SNPs associated with at least one of eight blood traits reported by Astle and colleagues13 at a genome-wide threshold. These SNPs were LD clumped, and effect sizes extracted from each blood cell GWAS, and each outcome GWAS. Effects for 318 clumped SNPs were harmonised, that is, so effect sizes for SNP-exposure and SNP-outcome effects corresponded to the same allele (online supplemental table 3, online supplemental file 1). Conditional F-statistics were estimated using the strength_mvmr() function of the ‘MVMR’ R package.30
For IVW multivariable MR analyses, we used the mv_multiple() function of the ‘TwoSampleMR’ R package.31–33 This analysis aimed to further investigate the possibility of horizontal pleiotropy affecting the results of the univariable eosinophil MR; and to establish whether other blood cell types besides eosinophils could affect the respiratory outcomes studied.
Sensitivity MVMR methods (online supplemental file 1) included: (1) use of an MVMR method more robust to pleiotropy in the presence of weak instruments (using the qhet_mvmr() function of the ‘MVMR’ R package,30—standard errors calculated by a jack-knife approach) and (2) recalculation of IVW MVMR estimates after removal of SNPs contributing most to heterogeneity (SNPs identified using the pleiotropy_mvmr() function).
Results
Univariable MR analyses of eosinophils and respiratory outcomes
There were 151 SNPs available for the univariable MR analyses of three quantitative traits (FEV1, FVC and FEV1/FVC), and four respiratory disease phenotypes (moderate-to-severe asthma, AECOPD, ACO and respiratory infections). Details of SNP selection are described in figure 2.
Figure 2.

Selection of SNPs for univariable MR analyses of eosinophils and respiratory outcomes flow chart describing the analysis workflow for initial MR analyses of eosinophils. Of 209 SNPs associated with eosinophil count, 167 were available in lung function GWASs (missingness is due to some SpiroMeta studies not being imputed to the HRC panel).18 LD proxies at R2 >0.8 were retrieved for 24/42 missing variants. Of the resulting 191 SNPs, 188 were successfully harmonised between the SNP-eosinophil and SNP-lung function data sets, and 151* remained after LD clumping at an R2 threshold of 0.01. These 151 SNPs were used in analyses. *One SNP, rs9974367, was missing in the moderate-severe asthma GWAS. AECOPD, acute exacerbation of COPD; ACO, asthma COPD overlap; COPD, chronic obstructive pulmonary disease; FEV1, forced expiratory volume in 1 s, FVC, forced vital capacity; GWAS, genome-wide association study; MR, Mendelian randomisation; SNPs, single-nucleotide polymorphisms.
Results are presented in figure 2. Among the quantitative traits, there was evidence for an effect of eosinophils on FEV1/FVC (SD change in FEV1/FVC per SD eosinophils, IVW estimate=−0.049 (95% CI −0.079 to–0.020)), with a smaller effect on FEV1 (IVW estimate=−0.028 (95% CI −0.054 to –0.003)). However, there was substantial heterogeneity of SNP-specific causal estimates, as evidenced by the large values of Cochran’s Q statistic, suggesting that core MR assumptions were violated for at least some SNPs. Scatterplots of SNP-outcome against SNP-exposure effects are given in online supplemental figure 1).
thoraxjnl-2021-217993supp002.pdf (813.9KB, pdf)
Among the respiratory disease phenotypes (figure 3), there was evidence for an effect of eosinophils on asthma (OR per SD eosinophil count, IVW method=2.46 (95% CI 1.98 to 3.06)), and ACO (IVW OR=1.86 (95% CI 1.52 to 2.27)). There was substantial heterogeneity of SNP-specific causal estimates for these two traits, and weighted median estimates were of smaller magnitude than IVW estimates (weighted median OR: 1.50 (95% CI 1.23 to 1.83) for asthma, and 1.44 (95% CI 1.19 to 1.74) for ACO). While confidence intervals for the MR Egger estimates were still broad, estimates were generally similar to weighted median estimates. The asthma estimates in particular may have been inflated by overlap between the SNP-exposure and SNP-outcome datasets (see online supplemental file 1). Scatterplots of SNP-outcome against SNP-exposure effects for these outcomes are given in online supplemental figure 2.
Figure 3.

MR analyses of eosinophils (exposure) on three quantitative lung function traits (top) and four respiratory disease phenotypes (bottom), using 151 eosinophil-associated SNPs top: results of MR analyses of eosinophil counts (exposure) on three quantitative lung function traits (outcome), FEV1, FVC and FEV1/FVC. A forest plot of three estimates for each traits is shown (IVW, MR Egger, weighted median), along with the maximum sample size in the outcome GWAS (N), the effect size in SD change in outcome trait per SD increase eosinophil count, and 95% CI, values for Cochran’s Q statistic (Q) and the associated df (Q_df), and the p value for the MR Egger intercept (Intercept_P). Boxes of the forest plot represent effect sizes, whiskers are 95% CIs. Bottom: results of MR analyses of eosinophil counts (exposure) on four respiratory disease phenotypes (outcome), moderate-to-severe asthma, acute exacerbations of COPD (AECOPD), asthma-COPD overlap (ACO), and respiratory infection (Resp. IX). A forest plot of three estimates for each traits is shown (IVW, MR Egger, weighted median), along with sample size in the outcome GWAS for cases and controls, respectively (N), the effect size as OR per SD eosinophil count, and 95% CI, values for Cochran’s Q statistic (Q) and the associated df (Q_df), and the p value for the Mr Egger intercept (Intercept_P). Boxes of the forest plot represent ORs, whiskers are 95% CIs. Nb only 150/151 of the eosinophil SNPs were available in the moderate-to-severe asthma GWAS. COPD, chronic obstructive pulmonary disease; FEV1, forced expiratory volume in 1 s, FVC, forced vital capacity; GWAS, genome-wide association study; IVW, inverse-variance weighted; MR, Mendelian randomisation; SNPs, single-nucleotide polymorphisms.
There was no evidence of association of eosinophils with AECOPD or respiratory infections. CIs for all three MR methods included the null, and point estimates approached the null. See online supplemental table 4 for results for all models and all traits.
Sensitivity analysis to assess further the robustness of findings to heterogeneity, using MR-PRESSO
For FEV1, FEV1/FVC, ACO and asthma (traits showing strongest evidence of causation), we used MR-PRESSO to identify possible pleiotropic outliers (online supplemental table 5). Results were qualitatively similar to IVW estimates (higher eosinophils consistent with respiratory morbidity), but ACO and asthma effect estimates attenuated after MR-PRESSO outlier correction; MR-PRESSO estimates were most similar to weighted median causal estimates.
Sensitivity analysis to assess the effects of sample overlap for quantitative lung function traits
UK Biobank featured in all GWAS datasets used, although the blood cell count GWAS and asthma GWAS included only approximately one third of the UK Biobank genotype data.13 We conducted sensitivity analyses to assess for the effect of sample overlap, since we had access to quantitative lung function GWAS data without UK Biobank participants (see online supplemental file 1). Results were generally consistent (SD change in FEV1/FVC per SD eosinophil count, IVW estimate=−0.041 (95% CI −0.072 to –0.009); SD change FEV1 per SD eosinophil count=−0.043 (95% CI −0.077 to –0.010)) (online supplemental table 6).
Sensitivity analysis to assess the effect on FEV1/FVC in individuals with and without asthma
The causal effect of eosinophils on FEV1/FVC was recalculated using data from UK Biobank, stratifying by asthma status (37 868 cases, 283 179 controls). The effect size was larger in individuals with asthma (IVW −0.083 (95% CI −0.139 to –0.028)) than in those without asthma, in whom there was no effect (IVW −0.013 (95% CI −0.041 to 0.015)). However, confidence intervals for both subgroups overlapped one another (see online supplemental table 7).
Multivariable MR analyses of blood cell counts and respiratory outcomes
To further explore causality between blood cell parameters and FEV1, FEV1/FVC, moderate-to-severe asthma and ACO, and to see if other exposures could have accounted for the heterogeneity observed in the previous analyses, we carried out multivariable MR analyses, using eight cell type exposures (eosinophils, basophils, neutrophils, monocytes, lymphocytes, platelets, red blood cells and reticulocytes).
Selection of 318 SNP IVs for multivariable MR is described in online supplemental file 1, online supplemental table 3. SNPs used in the univariable and multivariable MR are listed in online supplemental tables 8 and 9. Briefly, 1166 unique SNPs were associated with at least one of the eight cell types at a genome-wide level in the cell type GWAS, and were available in outcome GWAS. After LD-clumping, 329 SNPs remained, and after harmonising SNP-exposure and SNP-outcome effects, 318 remained (see online supplemental table 3) for conditional F statistics, which were all F>10, except for basophils (Fconditional=8).
Multivariable MR results for FEV1 and FEV1/FVC are presented in figure 4. Even after conditioning on the effects of the SNPs on other cell types, the average effect of the eosinophil-lowering IVs was to reduce lung function as measured by FEV1/FVC (multivariable estimate, SD change in FEV1/FVC per SD eosinophils adjusted for other cell types: −0.065 (95% CI −0.104 to –0.026)). The eosinophil point estimate for FEV1 (−0.032 (95% CI −0.068 to 0.005)) was consistent with the univariable estimate (figure 3), but CIs for all cell types were consistent with the null. When asthma cases were excluded from SNP-FEV1/FVC results, the eosinophil estimate attenuated, and confidence intervals overlapped the null (−0.028 (95% CI −0.069 to 0.013)), consistent with the causal effect of eosinophils on lung function being of greater magnitude in people with a history of asthma (online supplemental figure 3).
Figure 4.

Multivariable MR analyses of eight cell types and forced expiratory volume in 1 s (FEV1) and FEV1/forced vital capacity (FVC) forest plot showing multivariable MR estimating the causal effect of multiple cell types on quantitative lung function outcomes, after conditioning on the effects of the SNPs on other cell types. Models were run for each of FEV1 and the ratio of FEV1 to FVC separately, but effect sizes are shown next to one another for comparison. Effect sizes (beta, 95% CI) are in SD change in lung function outcome per SD cell count (adjusted for the effects of other cell types). Points of the forest plot represent effect size estimate; whiskers are 95% CIs. MR, Mendelian randomisation.
Results of the multivariable MR analysis for ACO and asthma are presented in figure 5. There was an association of eosinophil count with both ACO (OR 1.95 (95% CI 1.57 to 2.42)) and asthma (OR 2.90 (95% CI 2.31 to 3.65)), after adjusting for the effects of the SNPs on other cell types. Confidence intervals for other cell type estimates were consistent with the null, with the exception of neutrophils for ACO. None of the additional seven cell types showed strong evidence of causality.
Figure 5.
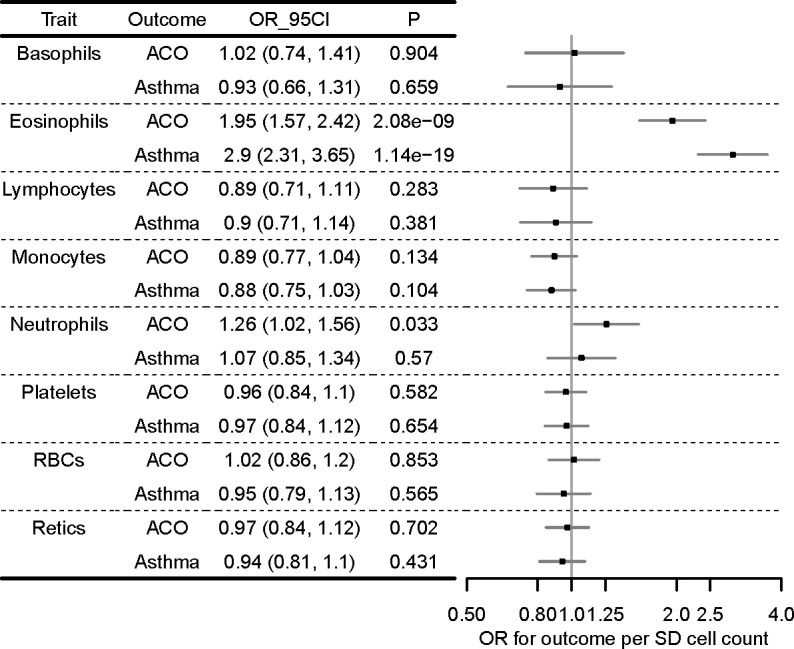
Multivariable MR analyses of eight cell types and two respiratory disease outcomes, ACO and asthma forest plot showing multivariable MR estimating the causal effect of multiple cell types on respiratory disease outcomes, after conditioning on the effects of the SNPs on other cell types. Models were run for each of ACO and asthma separately, but effect sizes are shown next to one another for comparison. ORs (95% CI) are per SD cell count (adjusted for the effects of other cell types). Points of the forest plot represent ORs; whiskers are 95% CIs. ACO, asthma-COPD overlap; MR, Mendelian randomisation; SNP, single-nucleotide polymorphisms.
Sensitivity multivariable MR analyses
Sensitivity MVMR analyses (1) used an estimation technique more robust to balanced pleiotropy and (2) repeated IVW MVMR, omitting SNP IVs with the most evidence of heterogeneity. Effect directions of sensitivity analyses and the main MVMR analyses were concordant for FEV1, FEV1/FVC, ACO, and asthma. However, CIs for FEV1 and FEV1/FVC were broad, and overlapped the null. For ACO and asthma estimates, there was still evidence of an effect, although attenuated in both analyses (estimates from analysis more robust to pleiotropy; ACO OR 1.57 (95% CI 1.07 to 2.30); asthma OR 2.66 (95% CI 1.65 to 4.33); estimates after omitting the most heterogeneous SNPs: ACO OR 1.51 (95% CI 1.23 to 1.85); asthma OR 2.29 (95% CI 1.84 to 2.86)).
Discussion
In MR analyses, we found that the average effect of raising eosinophils was to decrease FEV1/FVC and FEV1, and to increase ACO and asthma risk, and there was broad consistency across MR methods. However, causal estimates of individual variants were highly heterogeneous, suggesting that caution is needed in concluding causal inference: some IVs may have violated MR assumptions, and other important genetically correlated mechanisms could be responsible for the effect on lung health and disease by the eosinophil-raising variants studied.
To our knowledge, this is the largest MR of eosinophils and lung function, and the first to investigate eosinophils and AECOPD, ACO and respiratory infections. Terminology of ACO has changed over time, yet recognition that asthma and COPD coexist in some patients has not changed,34 and this is what our analysis aimed to capture.
A previous two-sample MR of eosinophils and asthma was undertaken by the authors of the GWAS that discovered the eosinophil IVs used; this MR analysis used asthma GWAS data from the GABRIEL study.13 We are aware of one other small MR of eosinophils and asthma, COPD, FEV1 and FEV1/FVC, conducted in the LifeLines cohort (N=13 301, 5 SNPs IVs).15 In that study, CIs for causal estimates of eosinophils overlapped the null, although point estimates were consistent with a harmful effect for FEV1/FVC, asthma and COPD. We used a larger eosinophil GWAS (N=172 275)13 to derive IVs, and found that the average effect of eosinophil-raising IVs was to reduce FEV1/FVC, the trait used in COPD diagnosis and FEV1, used to grade COPD airflow limitation. However, sensitivity analyses highlighted a larger causal estimate of eosinophils on FEV1/FVC among those with asthma, with effect estimates attenuating when excluding this group. These findings may highlight the importance of eosinophils as a marker of impaired lung function and airflow obstruction in people with a history of asthma.
We highlight a need for caution in inferring simple causation between eosinophils and these phenotypes, since high degrees of heterogeneity in our results may arise from pleiotropy. To investigate, we compared MR methods relying on differing assumptions for validity (Methods section). Attenuation of some results when using the MR-Egger, weighted median, and MR-PRESSO approaches suggests that some SNP IVs are associated with asthma and ACO via pathways other than eosinophils, which is a known challenge in MR studies (see also Methods section).
Since many of the eosinophil SNP IVs are also associated with other cell counts,13 we performed multivariable MR to estimate the influence of multiple cell types simultaneously, after conditioning on the effects of the SNPs on other cell types. While we did not find substantial evidence for a harmful effect of neutrophils on asthma, nor a protective effect of monocytes and lymphocytes, as reported previously,13 effect directions in our IVW multivariable MR were consistent with the previous study for neutrophils, monocytes and lymphocytes. We observed a larger effect of eosinophils on asthma than reported previously: this could be because our SNP-outcome dataset was of moderate-to-severe asthma (which has a higher point estimate of genetic correlation with eosinophils), but also, around half of the cases and the majority of controls were also included in the exposure GWAS, which may make this analysis closer to a one-sample MR, and inflate causal effect estimates. Notably, effect sizes partly attenuated in sensitivity analyses which may be more robust to heterogeneity. The MR estimates from multivariable analyses, and the MR-Egger regression and weighted median univariable analyses were consistent with the previous estimate reported for asthma in multivariable analysis by Astle et al.13 Nevertheless, these limitations may preclude precise estimation of effect sizes, and our results may be more useful in terms of assessing whether there is causality between eosinophils and the phenotypes studied, as opposed to providing estimates of the magnitude of any causal effect between phenotypes.
While we did not find strong evidence for causality of eosinophils on AECOPD and respiratory infections, point estimates were consistent with a harmful effect on AECOPD, and may have been limited by power. The effects of anti-IL5 drugs that have been attributed to the reduction of eosinophils have been noted to be smaller in AECOPD compared with asthma.2 35
Key strengths are that we used MR methods with differing sensitivities to underlying assumptions. We a large GWAS of eosinophil counts, to provide a comprehensive assessment of the role of blood eosinophils in relation to multiple respiratory health and disease outcomes. Another strength is that we undertook multivariable MR to investigate causality between multiple cell types and the outcomes studied, while controlling for the effects of IVs that may have had pleiotropic effects via other cell types.
We acknowledge several limitations. We did not have post-bronchodilator measures of spirometry. We used GOLD Stage 2–4 COPD (prebronchodilator FEV1 <80% predicted) when defining ACO; using the same prebronchodilator spirometry definition of COPD, a positive predictive value of 98% for diagnosis of postbronchodilation-defined COPD has been shown.36 Sample overlap between the SNP-eosinophil and SNP-outcome datasets (all included participants from UK Biobank) could bias estimates towards the observational eosinophil-outcome association21; we repeated the univariable MR analysis of eosinophils using SNP-lung function results excluding UK Biobank participants, and observed a consistent IVW estimate. Nevertheless, our other analyses (particularly the asthma analysis) could be vulnerable to some non-conservative bias.19 21 GWAS analyses of cell counts have, since analysis, been extended to a larger sample across UKB, and future work deriving IVs from this study would be valuable.37 UK BiLEVE participants (a subset of UK Biobank selected for extremes of respiratory traits), were overrepresented in Astle et al, which used the interim release of UKB data. While correlation between effect sizes from the two GWAS for the 151 IVs used in this analyses were high, the possibility of selection effects remains. Our MR analyses also use genome-wide results adjusted for covariates, and therefore may be susceptible to collider bias.19 38 There is also potential bias in the causal estimates for binary outcomes due to non-collapsibility of the OR,22 and we did not consider the possibility of non-linear effects. The multivariable analyses may still be vulnerable to pleiotropy via pathways other than the eight cell types studied, so while we cannot strongly assert causality of eosinophils on lung function, neither do we rule it out, as our results are consistent with a causal effect.
At present, treatment with anti-IL5/anti-IL5Rα agents in asthma is initiated according to eosinophil counts and other factors,8 yet it is possible that a more proximal factor may be an even better predictor of drug response. Future work could seek therefore to identify whether particular pathways upstream of eosinophil counts might help design better methods for deciding on treatment initiation. In addition, use of suitable IVs for IL5 levels would permit two-step MR analyses, assessing for a mediating effect of eosinophils on the action of anti-IL5 agents in reducing respiratory morbidity.
To conclude, using MR, we found that the average effect of raising eosinophils was to increase risk of ACO and asthma, and to reduce FEV1/FVC (the latter association was only prominent in individuals with asthma). Broad consistency across MR methods is suggestive of a causal effect of eosinophils on asthma overall, and in individuals with features of both asthma and fixed airflow obstruction, although of uncertain magnitude. However, given heterogeneity in results derived from individual IVs, which may indicate violation of MR assumptions, we highlight a need for caution, since alternative mechanisms may be responsible for the impairment of respiratory health by these eosinophil-raising variants. These results could suggest that anti-IL5 agents (designed to lower eosinophils) may be of value in a wider range of respiratory traits, including people with features of both asthma and COPD. Future work should seek to explore other potential mechanisms besides eosinophils by which anti-IL5 agents may improve respiratory health.
Acknowledgments
We are grateful to Dr William J Astle for advice. This research used ALICE/SPECTRE High Performance Computing Facilities at the University of Leicester, and summary data available via the BREATHE Digital Innovation Hub at SAIL databank (https://saildatabank.com/).
Footnotes
Collaborators: SpiroMeta Consortium: James P Cook, Ian J Deary, Stefan Enroth, Ralf Ewert, Christian Gieger, Ulf Gyllensten, Sarah E Harris, Caroline Hayward, Georg Homuth, Jennie Hui, Medea Imboden, Alan L James, Marjo-Riitta Jarvelin, Peter K Joshi, Mika Kahonen, Stefan Karrasch, Katherine A Kentistou, Shona M Kerr, Meena Kumari, Claudia Langenberg, Terho Lehtimaki, Lars Lind, Jian’an Luan, Anubha Mahajan, Jonathan Marten, Andrew P Morris, Ozren Polasek, David J Porteous, Nicole M Probst-Hensch, Olli T Raitakari, Rajesh Rawal, Igor Rudan, Holger Schulz, Blair H Smith, David P Strachan, Beate Stubbe, Ida Surakka, Paul RJH Timmers, Véronique Vitart, Nick Wareham, Stefan Weiss, Matthias Wielscher, James F Wilson, Eleftheria Zeggini, Jing Hua Zhao, Understanding Society Scientific Group.
Contributors: AG, NuS, FD and MDT contributed to the conception and design of the study. AG undertook data analysis and produced the first draft of the manuscript, and revised it with CJ, IH, LVW, NuS, FD and MDT. AG, CJ, ATW, NiS, NFR, SpiroMeta consortium, IS, IH, LVW and MDT contributed to data acquisition. AG, CJ, IS, IH, LVW, NuS, FD and MDT contributed to data interpretation. All authors critically reviewed the manuscript before submission. AG and MDT act as guarantors for the content of the manuscript.
Funding: AG: Wellcome Trust Institutional Strategic Support Fund (204801/Z/16/Z), BHF Accelerator Award (AA/18/3/34220). CJ: Medical Research Council Clinical Research Training Fellowship (MR/P00167X/1). ATW: BBSRC CASE studentship with GSK. LVW: GSK/British Lung Foundation Chair in Respiratory Research. MDT: Wellcome Trust Investigator Award (WT202849/Z/16/Z). MDT and LVW: MRC (MR/N011317/1). MDT and IH hold NIHR Senior Investigator Awards. The research was supported by BREATHE — The Health Data Research Hub for Respiratory Health (MC_PC_19004). The research was partially supported by the NIHR Leicester Biomedical Research Centre and the NIHR Nottingham Biomedical Research Centre; views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. This research was funded in whole, or in part, by the Wellcome Trust. For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Disclaimer: The funders had no role in the design of the Mendelian randomisation analyses.
Competing interests: MDT and LVW receive funding from GSK for collaborative research projects outside of the submitted work. IH has funded research collaborations with GSK, Boehringer Ingelheim and Orion.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Contributor Information
Collaborators: SpiroMeta consortium, James P Cook, Ian J Deary, Stefan Enroth, Ralf Ewert, Christian Gieger, Ulf Gyllensten, Sarah E Harris, Caroline Hayward, Georg Homuth, Jennie Hui, Medea Imboden, Alan L James, Marjo-Riitta Jarvelin, Peter K Joshi, Mika Kahonen, Stefan Karrasch, Katherine A Kentistou, Shona M Kerr, Meena Kumari, Claudia Langenberg, Terho Lehtimaki, Lars Lind, Jian’an Luan, Anubha Mahajan, Jonathan Marten, Andrew P Morris, Ozren Polasek, David J Porteous, Nicole M Probst-Hensch, Olli T Raitakari, Rajesh Rawal, Igor Rudan, Holger Schulz, Blair H Smith, David P Strachan, Beate Stubbe, Ida Surakka, Paul RJH Timmers, Véronique Vitart, Nick Wareham, Stefan Weiss, Matthias Wielscher, James F Wilson, Eleftheria Zeggini, and Jing Hua Zhao
Data availability statement
Code used in the analysis for this paper is available on request. Summary-level statistics for the IVs used in this paper are available in the relevant published papers, or are available on request where not already published. Summary-level lung function GWAS and blood cell GWAS data are available from https://www.ebi.ac.uk/gwas/.
Ethics statements
Patient consent for publication
Not applicable.
References
- 1. George L, Brightling CE. Eosinophilic airway inflammation: role in asthma and chronic obstructive pulmonary disease. Ther Adv Chronic Dis 2016;7:34–51. 10.1177/2040622315609251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ortega HG, Yancey SW, Mayer B, et al. Severe eosinophilic asthma treated with mepolizumab stratified by baseline eosinophil thresholds: a secondary analysis of the DREAM and MENSA studies. Lancet Respir Med 2016;4:549–56. 10.1016/S2213-2600(16)30031-5 [DOI] [PubMed] [Google Scholar]
- 3. Pavord ID, Chanez P, Criner GJ, et al. Mepolizumab for eosinophilic chronic obstructive pulmonary disease. N Engl J Med 2017;377:1613–29. 10.1056/NEJMoa1708208 [DOI] [PubMed] [Google Scholar]
- 4. Eltboli O, Brightling CE. Eosinophils as diagnostic tools in chronic lung disease. Expert Rev Respir Med 2013;7:33–42. 10.1586/ers.12.81 [DOI] [PubMed] [Google Scholar]
- 5. Saetta M, Di Stefano A, Maestrelli P, et al. Airway eosinophilia in chronic bronchitis during exacerbations. Am J Respir Crit Care Med 1994;150:1646–52. 10.1164/ajrccm.150.6.7952628 [DOI] [PubMed] [Google Scholar]
- 6. Pavord ID, Korn S, Howarth P, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet 2012;380:651–9. 10.1016/S0140-6736(12)60988-X [DOI] [PubMed] [Google Scholar]
- 7. Haldar P, Brightling CE, Hargadon B, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med 2009;360:973–84. 10.1056/NEJMoa0808991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. National Institute for Health and Care Excellence . Mepolizumab for treating severe refractory eosinophilic asthma. Technology appraisal guidance [TA431], 2017. Available: https://www.nice.org.uk/guidance/ta431
- 9. U.S. Food and Drug Administration (FDA) . Drug trials snapshots: NUCALA, 2016. Available: https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trials-snapshots-nucala [Accessed 27 Jun 2019].
- 10. U.S. Food and Drug Administration (FDA) . Drug trials snapshots: FASENRA, 2017. Available: https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trials-snapshots-fasenra [Accessed 27 Jun 2019].
- 11. European medicines Agency . Nucala (mepolizumab), 2019. Available: https://www.ema.europa.eu/en/documents/overview/nucala-epar-medicine-overview_en.pdf [Accessed 06 Jan 2020].
- 12. European medicines Agency . Fasenra (benralizumab), 2019. Available: https://www.ema.europa.eu/en/documents/overview/fasenra-epar-medicine-overview_en.pdf [Accessed 06 Jan 2020].
- 13. Astle WJ, Elding H, Jiang T, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 2016;167:e19:1415–29. 10.1016/j.cell.2016.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hancox RJ, Pavord ID, Sears MR. Associations between blood eosinophils and decline in lung function among adults with and without asthma. Eur Respir J 2018;51:1702536. 10.1183/13993003.02536-2017 [DOI] [PubMed] [Google Scholar]
- 15. Amini M, Vonk JM, Abbasi A, et al. Blood eosinophil count and metabolic, cardiac and pulmonary outcomes: a Mendelian randomization study. Twin Res Hum Genet 2018;21:89–100. 10.1017/thg.2018.6 [DOI] [PubMed] [Google Scholar]
- 16. Wu X, Wang C, Li H, et al. Circulating white blood cells and lung function impairment: the observational studies and Mendelian randomization analysis. Ann Med 2021;53:1119–29. 10.1080/07853890.2021.1948603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sakornsakolpat P, Prokopenko D, Lamontagne M, et al. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat Genet 2019;51:494–505. 10.1038/s41588-018-0342-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shrine N, Guyatt AL, Erzurumluoglu AM, et al. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet 2019;51:481–93. 10.1038/s41588-018-0321-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 2018;362:k601. 10.1136/bmj.k601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 2014;23:R89–98. 10.1093/hmg/ddu328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol 2016;40:597–608. 10.1002/gepi.21998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sheehan NA, Didelez V, Epidemiology DV. Epidemiology, genetic epidemiology and Mendelian randomisation: more need than ever to attend to detail. Hum Genet 2020;139:121–36. 10.1007/s00439-019-02027-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–9. 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shrine N, Portelli MA, John C, et al. Moderate-to-severe asthma in individuals of European ancestry: a genome-wide association study. Lancet Respir Med 2019;7:20–34. 10.1016/S2213-2600(18)30389-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. John C, Guyatt AL, Shrine N. Genetic associations and architecture of asthma-chronic obstructive pulmonary disease overlap. medRxiv 2020;2. 10.1101/2020.11.26.20236760v1 [DOI] [Google Scholar]
- 26. Williams A, Shrine N, Naghra-van Gijzel H. Genome-wide association study of susceptibility to hospitalised respiratory infections [version 1; peer review: awaiting peer review]. Wellcome Open Research 2021;6 https://wellcomeopenresearch.org/articles/6-290 10.12688/wellcomeopenres.17230.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–25. 10.1093/ije/dyv080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bowden J, Davey Smith G, Haycock PC, et al. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–14. 10.1002/gepi.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Verbanck M, Chen C-Y, Neale B, et al. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018;50:693–8. 10.1038/s41588-018-0099-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanderson E, Spiller W, Bowden J. Testing and correcting for weak and pleiotropic instruments in two-sample multivariable Mendelian randomization. Stat Med 2021;40:5434–52. 10.1002/sim.9133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. 10.7554/eLife.34408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Burgess S, Dudbridge F, Thompson SG. Re: "Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects". Am J Epidemiol 2015;181:290–1. 10.1093/aje/kwv017 [DOI] [PubMed] [Google Scholar]
- 33. Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol 2015;181:251–60. 10.1093/aje/kwu283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Global Initiative for Chronic Obstructive Lung Disease I . Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease (2020 report), 2020. Available: https://goldcopd.org/wp-content/uploads/2019/12/GOLD-2020-FINAL-ver1.2-03Dec19_WMV.pdf
- 35. Brightling C, Greening N. Airway inflammation in COPD: progress to precision medicine. Eur Respir J 2019;54. 10.1183/13993003.00651-2019. [Epub ahead of print: 01 08 2019]. [DOI] [PubMed] [Google Scholar]
- 36. Soler Artigas M, Loth DW, Wain LV, et al. Genome-Wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat Genet 2011;43:1082–90. 10.1038/ng.941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vuckovic D, Bao EL, Akbari P, et al. The polygenic and monogenic basis of blood traits and diseases. Cell 2020;182:1214–31. 10.1016/j.cell.2020.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Munafò MR, Tilling K, Taylor AE, et al. Collider scope: when selection bias can substantially influence observed associations. Int J Epidemiol 2018;47:226–35. 10.1093/ije/dyx206 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
thoraxjnl-2021-217993supp001.pdf (2.2MB, pdf)
thoraxjnl-2021-217993supp003.xlsx (86.2KB, xlsx)
thoraxjnl-2021-217993supp002.pdf (813.9KB, pdf)
Data Availability Statement
Code used in the analysis for this paper is available on request. Summary-level statistics for the IVs used in this paper are available in the relevant published papers, or are available on request where not already published. Summary-level lung function GWAS and blood cell GWAS data are available from https://www.ebi.ac.uk/gwas/.