

SUMMARY
Small ribonucleoproteins (sRNPs) target nascent precursor RNAs to guide folding, modification, and splicing during transcription. Yet, rapid co-transcriptional folding of the RNA can mask sRNP sites, impeding target recognition and regulation. To examine how sRNPs target nascent RNAs, we monitored binding of bacterial Hfq•DsrA sRNP to rpoS transcripts using single-molecule co-localization co-transcriptional assembly (smCoCoA). We show that Hfq•DsrA recursively samples the mRNA before transcription of the target site to poise it for base pairing with DsrA. We adapted smCoCoA to precisely measure when the target site is synthesized and revealed that Hfq•DsrA often binds the mRNA during target site synthesis close to RNA polymerase (RNAP). We suggest that targeting transcripts near RNAP allows an sRNP to capture a site before the transcript folds, providing a kinetic advantage over post-transcriptional targeting. We propose that other sRNPs may also use RNAP-proximal targeting to hasten recognition and regulation.
Keywords: bacterial small RNA, co-transcriptional RNA folding, single-molecule fluorescence, RNA chaperone, Hfq
Graphical Abstract
eTOC Blurb
Rodgers et al. use single-molecule fluorescence microscopy to study how bacterial small RNA-Hfq complexes find a complementary site during transcription of a target mRNA. The results reveal that small RNA-Hfq complexes recognize RNA sequences close to the RNA polymerase elongation complex, making targeting more efficient during transcription than after transcription.
INTRODUCTION
Many small ribonucleoproteins (sRNPs) base pair with target RNAs during transcription to chaperone assembly, guide chemical modifications and processing, or regulate target expression.1 RNA-guided enzymes, such as miR-Argonaute and CRISPR-Cas, efficiently search for their target sites in mature RNAs.2 Other sRNPs, however, act on immature transcripts. For these sRNPs, the search for sequences complementary to the guide RNA must be coordinated with the elongation and folding of nascent target RNA.
It has been hypothesized that some sRNPs establish base pairing within a ‘time window of opportunity’ that is demarcated by rate of RNA elongation. For instance, the spliceosomal small nuclear RNPs (snRNPs) are proposed to find and base pair with the splice sites soon after they are transcribed to carry out splicing shortly after intron synthesis.3,4 Similarly, small nucleolar RNPs (snoRNPs) target sites in the pre-ribosomal RNA that are inaccessible in the mature rRNA, suggesting that they bind the pre-rRNA before the ribosomal subunits assemble.5,6 These examples imply that sRNPs must either rearrange the structure of the target RNA or capture the target RNA before it can fold.
The opportunity for capturing an open target is dictated by the speed of transcription and the probability of RNA folding during transcription.7 Local secondary structures form as soon as nucleotides emerge from the RNA polymerase, as evidenced by the formation of intrinsic terminator stem loops within the RNA polymerase exit channel.8 The order in which the RNA domains are transcribed can determine which structures are formed.9,10 Furthermore, studies on transcriptional riboswitches have shown that secondary and tertiary structures can rearrange as the transcript is elongated.11,12 Therefore, even if an sRNP target site is not masked by initial structures as soon as it is transcribed, the target site could become masked during transcription of downstream regions. However, recent studies have shown that variable folding of nascent RNAs during transcription can delay the acquisition of stable structure13,14, possibly enlarging the window for target recognition by sRNPs during transcription.
Here, we use the well-studied bacterial small non-coding RNAs (sRNAs) as a model for RNA-guided targeting during transcription. Bacterial sRNAs, in complex with their chaperone Hfq, base pair with mRNA targets to regulate gene expression within minutes of an inducing signal.15 The general stress response sigma factor, RpoS, is up-regulated by three different sRNAs in E. coli 16. During normal exponential growth, the 5’ untranslated region (UTR) of the rpoS mRNA folds into an inhibitory secondary structure that masks the ribosome binding site (Fig. 1A).17,18 DsrA sRNA increases rpoS translation by base pairing with the rpoS mRNA 5’ UTR, unfolding the inhibitory structure and unmasking the ribosome binding site.17-19
Fig. 1. Targeting by DsrA sRNA during transcription of the rpoS 5’ UTR.

A) Post-transcriptional up-regulation of rpoS expression by DsrA sRNA.18 Secondary structure of the 599 nt rpoS 5’ UTR masks the ribosome binding site (RBS, purple). DsrA opens the inhibitory stem by base pairing with the target site (pink). Hfq facilitates DsrA annealing by binding an upstream (AAN)4 sequence (teal).24
B) smCoCoA simultaneously monitors rpoS transcription and binding of Hfq•DsrA-Cy5 (red star). Stalled transcription elongation complexes (TECs) are immobilized through a biotinylated DNA complementary to the 5’ end of the transcript and located with a Cy3 (green star) attached near the end of the DNA template. Protein induced fluorescence enhancement (PIFE) marks the end of transcription.
C) Example of stable Hfq•DsrA-Cy5 colocalization with a single rpoS transcript before the complete rpoS sequence has been transcribed. Top scheme shows key features of the rpoS DNA template, including the T7A1 promoter and C-less cassette used to stall transcription. Dotted lines designate the position of RNAP at the start and end of the transcription window (light green shading). Transcription is restarted by injection of NTPs and 5 nM Hfq-DsrA-Cy5, indicated by an increase in the Cy5 background signal (red). The end of transcription is measured by PIFE (green) as the RNAP traverses through the Cy3 fluorophore located at +52. The arrow marks co-transcriptional binding of Hfq•DsrA-Cy5. Fluctuations in Cy5 intensity likely represent changes in its local environment within the complex. See also Fig. S1.
D) Rastergram illustrating the timing and duration of Hfq•DsrA-Cy5 binding (grey bars), to randomly selected full-length rpoS301 5’UTR transcripts. Dotted line indicates injection of NTPs and Hfq•DsrA-Cy5; green circles (PIFE) and light green shading demarcate the transcription window. 50% of analyzed TECs experienced long-lived Hfq•DsrA-Cy5 binding; the other 50% experienced only transient binding or no binding.
The chaperone Hfq accelerates base pairing between DsrA and the rpoS 5′ UTR by bringing the two RNAs together in a ternary Hfq•DsrA-rpoS mRNA complex (Fig. 1A).20 When recruited to an upstream (AAN)4 motif in rpoS, Hfq also restructures the rpoS 5′ UTR, orienting the target site for base pairing with DsrA.21-24 The relative positions of the (AAN)4 Hfq binding site and the sRNA binding site are essential for sRNA base pairing in vitro and up-regulation of rpoS expression in vivo 22,23, suggesting that orientation of these elements in the Hfq•DsrA-rpoS RNP plays a role in the mechanism of annealing.
Although sRNAs are a major form of post-transcriptional gene control in bacteria, recent studies indicate they also act during transcription.25-27 As translation can be coupled to transcription in E. coli and other bacteria28, DsrA may target rpoS mRNA during transcription to pre-empt formation of the inhibitory stem and unmask the ribosome binding site. In addition to regulating translation, it was recently proposed that sRNAs including DsrA block Rho-dependent transcription termination in the rpoS 5′ UTR, further supporting a role for sRNA target recognition during transcription.26
Here, we use single-molecule fluorescence microscopy to understand how RNPs recognize sequences as they are being transcribed. By monitoring both transcription and binding of Hfq•DsrA in real time, we find that Hfq•DsrA transiently samples the elongating transcript via the (AAN)4 motif, which primes Hfq•DsrA to capture the target site as it emerges from RNA polymerase. We propose that many RNPs utilize similar recursive sampling mechanisms to sense transcription of the target site, permitting rapid recognition of the nascent RNA near the elongation machinery.
RESULTS
Hfq•DsrA RNPs target elongating rpoS transcripts in vitro
To examine the mechanism of sRNA targeting on nascent mRNAs, we adapted the single-molecule colocalization co-transcriptional assembly (smCoCoA) platform to correlate association of Hfq•DsrA RNPs with transcription of specific sequences in the rpoS mRNA, in real time (Fig. 1B) 13. To monitor transcription of the rpoS 5’ UTR sequence, we utilized a Cy3-labeled DNA template comprising the entire 599 bp 5’ UTR of the rpoS mRNA that was shown to be sufficient for sRNA regulation of a rpoS reporter; (Fig. 1C, top) 23. Stalled E. coli RNAP Transcription Elongation Complexes (TECs) containing the rpoS 5’ UTR Cy3-labeled DNA were immobilized on the slide surface and transcription elongation was restarted during imaging by addition of nucleotide triphosphates (NTPs) to the slide chamber (Fig. 1B). When E. coli RNAP approaches a Cy3 fluorophore on the DNA template, a large increase in Cy3 fluorescence is observed due to protein induced fluorescence enhancement (PIFE) as described previously 13,29. PIFE is observed when RNAP is within ~1 – 3 nm of the Cy3 fluorophore, which only occurs as the nucleotide conjugated to Cy3 traverses through the RNAP active site. The position of the Cy3 fluorophore and the timing of the initial rise in Cy3 signal during PIFE therefore reports on the position of RNAP on the DNA and the length of RNA transcribed.
To visualize binding of the Hfq•DsrA RNP, full-length DsrA sRNA was labeled at the 5’ end with a Cy5 fluorophore and preloaded with Hfq in solution prior to use. Although rpoS is regulated by two other sRNAs, ArcZ and RprA30-32, we chose DsrA sRNA for these experiments because it is known to rapidly bind the rpoS 5’ UTR at room temperature.24 In smCoCoA experiments, 5 nM Hfq•DsrA-Cy5 RNPs were delivered to the slide chamber together with the NTPs to watch binding of single RNPs during transcription of the rpoS 5’ UTR. We often observed colocalization of the Cy5 signal with immobilized Cy3-labeled TECs before the PIFE signal, indicating Hfq•DsrA binding during transcription (Fig. 1C, bottom). We compared Cy5-labeled Hfq•DsrA RNPs binding to the nascent rpoS transcripts before and after the PIFE signal, to delineate binding of DsrA during and after transcription. Most (67%) of the rpoS 5’ UTR transcripts interacted with at least one Hfq•DsrA-Cy5 RNP molecule for ≥ 1 s before the rpoS 5’ UTR was completely transcribed (Fig. 2A).
Fig. 2. Stable binding correlates with transcription of rpoS target sites.

A) DNA template and B) Secondary structure of minimal rpoS 5’UTR (rpoS301) lacking nt 9–305 upstream of the functional sites. See Table S3 for DNA sequence.
C) Timing and duration of Hfq•DsrA-Cy5 binding to rpoS301 for 61 randomly selected transcripts. Time axis is synchronized to injection of NTPs (t = 0).
D) Example trajectory for a single DsrA binding event illustrating the intervals between the moment of stable binding and restart of transcription (ton stable) or the end of transcription (ton stable – tPIFE). See Fig. S2 for further data.
E) and F) Association kinetics. Cumulative probability density of the start of stable Hfq•DsrA binding (t > 100 s) with respect to transcription restart (E; ton stable) or transcription end (F; Δtstable). Nevents (rpoS) = 28; Nevents (rpoS301) = 124.
The 5’ end of the rpoS 5’ UTR can be truncated by ~300 nucleotides (rpoS301) while maintaining Hfq-mediated sRNA regulation.24,33 When the template was shortened, the majority of rpoS301 transcripts (77%) still interacted with at least one Hfq•DsrA complex during the transcription, demonstrating that sequences upstream of the (AAN)4 Hfq binding site did not contribute to DsrA targeting during transcription (Fig. 2B). Because the rpoS301 5′ UTR was shown to fold homogenously into the native inhibitory structure in vitro22, this shortened form of the rpoS mRNA was used for subsequent experiments.
To validate the results of the smCoCoA assay, we carried out native gel electrophoresis mobility shift assays under similar conditions, to examine the formation of ternary complexes between Hfq•DsrA and newly made rpoS301 mRNA (Fig. S1A). The results showed that ternary Hfq•DsrA-rpoS301 complexes formed within 2 minutes of the restart of transcription (Fig. S1A). This timing correlated with the appearance of full-length rpoS301 RNA in parallel single round transcription reactions (Fig. S1B). Together, these data supported the conclusion that Hfq and DsrA form stable complexes with newly made rpoS301 mRNAs under the conditions of our smCoCoA experiments.
Long-lived interactions represent successful targeting
Using maximum likelihood analysis, we measured three characteristic lifetimes for Hfq•DsrA-Cy5 RNPs colocalizing with rpoS301 transcripts (Table S1; t1 ~ 0.5 s, t2 ~ 5 s, and t3 > 100 s), indicating that at least three different types of Hfq•DsrA•rpoS ternary complexes are formed. Previous work showed that sRNA target recognition can abort at intermediate steps, whereas complete base pairing between a model sRNA and mRNA pair results in stable binding.34 Therefore, the long-lived complexes (t3 > 100 s) likely represent successful base pairing between DsrA and rpoS mRNA. In our smCoCoA experiments, about half of the complete rpoS 5′ UTR transcripts (48%) and truncated rpoS301 transcripts (50%) formed a long-lived complex with Hfq•DsrA-Cy5 sometime during the 10 min movie (Fig. 2A, B), indicating the two RNAs are targeted with similar efficiency. Further experiments confirmed that stable binding depended on Hfq, DsrA-rpoS complementarity, and transcription of the rpoS301 5′ UTR (Fig. S2; Soper et al., 2011). Overall, these data showed that stable binding of Hfq•DsrA reports on successful targeting of Hfq•DsrA to a nascent rpoS transcript.
We next asked whether the onset of stable Hfq•DsrA binding correlates with the time needed to transcribe the DsrA target site. To address this question, we compared the timing of stable Hfq•DsrA binding to full-length rpoS 5’ UTR and to truncated rpoS301 transcripts (ton,stable; Fig. 2C), which differ by the number of nucleotides upstream of the Hfq and DsrA binding sites. Stable binding generally occurred sooner after injection on the shorter rpoS301 transcripts compared to rpoS 5’UTR transcripts, as evidenced by a shift in the cumulative probability for binding to each RNA (Fig. 2D). By contrast, when we accounted for the different transcription times of each transcript by comparing the moment of binding to the end of transcription signified by PIFE (Δtstable; Fig. 2C), the cumulative probability densities for rpoS301 and rpoS 5’UTR transcripts were alike within error, suggesting that the Hfq•DsrA binding kinetics is similar once the sRNA target site is synthesized (Fig. 2E and Fig. S2F-H). Thus, these results demonstrated that stable binding of Hfq•DsrA depends on the presence of DsrA target site, and more importantly, that targeting likely occurs soon after the target site is synthesized by E. coli RNAP.
Targeting during transcription circumvents restructuring of rpoS 5’ UTR
Hfq restructures the rpoS 5′ UTR to facilitate annealing of DsrA to its target site in the inhibitory stem21 that otherwise sequesters the ribosome binding site (Fig. 1A).18 Restructuring could pose a kinetic barrier to sRNA targeting and subsequent regulation of rpoS expression. Therefore, we wondered if base pairing with the nascent rpoS mRNA during transcription circumvents this remodeling step and provides an advantage over post-transcriptional targeting. To test this model, we measured binding of Hfq•DsrA RNPs to refolded full-length rpoS301 mRNA to evaluate the likelihood of targeting post-transcription (Fig. 3A). When Hfq•DsrA was added to the immobilized refolded rpoS301 under the same conditions used for co-transcriptional binding experiments, we observed transient and stable complexes with lifetimes similar to complexes with rpoS301 transcripts (Table S1). However, fewer stable Hfq•DsrA complexes formed on refolded rpoS301 mRNAs compared to nascent transcripts (46% and 29% respectively; Fig. 3B), suggesting that targeting is more efficient during transcription. Binding was also less efficient if the transcripts were allowed to fold for 45 min before Hfq•DsrA was added (Fig. S3).
Fig. 3. Downstream rpoS inhibitory stem does not influence co-transcriptional targeting.

A) rpoS variants with a very stable inhibitory stem, rpoS301GC, or no inhibitory stem, rpoS301Δ3’IS.
B) Fraction of immobilized rpoS variants that are stably bound by DsrA at some point during the 10 min movie. Bars compare targeting during transcription (coT) and refolded RNAs (R). Targeting during transcription is insensitive to the stability of the downstream inhibitory stem-loop (dark colors), whereas targeting is impeded by the refolded inhibitory stem (light colors). Symbols, values from independent replicates; bar, mean. See Table S2 for details.
C). Cumulative probability of stable Hfq•DsrA binding to rpoS301 variants during transcription reports the binding kinetics. The association time (ton stable) was measured from NTP injection. Therefore, binding lags ~100-200 s until the target site is transcribed. Dotted lines indicate the average time for complete transcription of each rpoS301 variant. The distributions were weighted by the fraction of transcripts that encountered stable Hfq•DsrA binding. Co-transcriptional targeting of rpoS301 and rpoS301Δ3’IS and rpoS301 and rpoS301GC are similar (p = 0.23 and p = 0.20, respectively; K-S test).
D). Cumulative probability of stable Hfq•DsrA binding to refolded rpoS301 variants, as in C. ton stable was measured relative to DsrA injection. Targeting is statistically different; (rpoS301 and rpoS301Δ3’IS: p = 2.0 x 10−4; rpoS301 and rpoS301GC: p = 0.03; K-S test). Data from 2 or 3 independent experiments were combined in C and D. See also Fig. S3.
We hypothesized that targeting was more prevalent during transcription because Hfq•DsrA captures the target site before the inhibitory stem forms. To test this, we altered the stability of the inhibitory stem (Figure 3A), and compared Hfq•DsrA targeting during and after transcription. When the inhibitory stem was stabilized by a “GC-clamp” mutation (rpoS301GC), post-transcriptional targeting of the refolded RNA was significantly diminished (Figure 3B), consistent with previous experiments.21 During transcription, however, targeting of rpoS301GC was similar to WT rpoS301 (Figure 3B). Conversely, when rpoS mRNA was truncated (rpoS301Δ3’IS) so that the inhibitory stem cannot form, there was little difference between post-transcriptional and co-transcriptional targeting (Figure 3B, green bars). Combined, these data show that the folded inhibitory stem hinders sRNA binding as expected, yet the potential to form downstream structure has little impact on sRNA binding during transcription.
To determine if there is a kinetic advantage to targeting rpoS mRNAs during transcription, we next compared the cumulative fraction of stable (> 100 s) Hfq•DsrA association with each rpoS301 variant during transcription (co-T) and when refolded (R; Fig. 3C). Association times (t on stable) were measured relative to the time of DsrA injection (Fig. S3B). We found that the cumulative probability densities for stable association of DsrA with rpoS301, rpoS301Δ3’IS, and rpoS301GC transcripts were statistically similar suggesting that the kinetics of targeting during transcription is not influenced by the stability of the inhibitory stem. In contrast, targeting refolded RNA was significantly faster < 200 s after injection when the inhibitory stem was unable to form (rpoS301Δ3’IS) and much slower when the inhibitory stem was stabilized (rpoS301GC; Figure 3D) than on refolded WT rpoS301. Altogether, our data indicate that targeting likely occurs prior to proper folding of the rpoS 5’ UTR thereby circumventing the need for remodeling the rpoS mRNA structure.
Faster transcription reduces Hfq•DsrA targeting
Since targeting during transcription is advantageous, we next wondered if the likelihood of DsrA binding is influenced by transcription speed and pausing, which determine the time window between synthesis of the target site and folding of the inhibitory stem-loop. In our smCoCoA assay, phage T7 RNAP transcribed the rpoS301 mRNA ~ 4 – 7-times faster than E. coli RNAP at the same NTP concentration (Fig. 4A). The fraction of rpoS301 transcripts successfully targeted by Hfq•DsrA decreased about two-fold when transcribed by T7 RNAP (Fig. 4B, C), compared to E. coli RNAP (Fig. 1E). This result was mirrored in the maximum likelihood analysis of the distribution of binding events, which showed that the characteristic lifetimes of the complexes remained the same in each transcription condition, yet the likelihood of forming a stable complex (a3) decreased when the transcription rate was increased (Fig. S4). Interestingly, although we observed fewer stable Hfq•DsrA binding events, nearly half of these (40 ± 5%) still occurred before transcription ended, despite the four times shorter T7 transcription window (Fig. 4B, striped bars). When the rate of T7 RNAP transcription was reduced by lowering the NTP concentration (2 μM), the fraction of rpoS301 mRNAs targeted by DsrA increased to the level during E. coli transcription (Fig. 4C, D). This rescue suggested that successful targeting depended primarily on the overall transcription window.
Fig. 4. Transcription rate influences Hfq•DsrA targeting in vitro.

A) Distribution of rpoS301 transcription times determined by the start of PIFE, comparing the rate of synthesis by E. coli RNAP and T7 RNAP. (E. coli RNAP, Nmol = 246; T7 RNAP + 20 μM NTP, Nmol = 312; T7 RNAP + 2 μM NTP, Nmol = 240). Fitted gaussians (smooth lines) yield mean transcription rates, 95% CI (LL, UL): E. coli RNAP k1,obs = 1.84 nt/s (1.7, 2.0) and k2,obs = 1.09 nt/s (0.8, 1.9); T7 RNAP + 20 μM NTP k,obs = 4.43 nt/s (4.2, 4.7); T7 RNAP + 2 μM NTP k1,obs = 1.42 nt/s (1.4, 1.4) and k2,obs = 0.96 nt/s (0.9, 1.0).
B) and C) Rastergrams of Hfq•DsrA binding to single rpoS301 T7 transcripts as in (A). Green shading indicates the transcription window. Compare to E. coli transcripts in Fig. 2C.
D) Fraction of rpoS301 transcripts that experience stable Hfq•DsrA binding during the 10 min movie (solid bars) or during the transcription window (striped bars); mean and s.d. from three independent trials. Student’s t test: not significant (n.s.): p > 0.05, * : 0.05 > p > 0.01, and ** : p < 0.01. See Fig. S4 for further data.
E) Radiolabeled single round transcription of rpoS301 mRNA using E. coli RNAP and 20 μM NTP analyzed by denaturing 6% PAGE. RNA ladder was generated using 3’-O-methyl chain terminator NTPs to map the locations of functional motifs and intrinsic pause sites (asterisks). See Fig. S4B for a diagram of pause sites.
We noted that E. coli RNAP pauses in vitro on the rpoS301 DNA downstream of the sRNA target site but upstream of the ribosome binding site (Fig. 4E). These pause sites are located near sequences that match the consensus for transcription pause sites in E. coli (Fig. S4B).35 Therefore, pausing by RNAP could further delay transcription of the inhibitory stem, widening the interval in which Hfq•DsrA can target the nascent mRNA.
Upstream Hfq recruitment is needed for efficient sRNA targeting
We next sought to understand the mechanism of the Hfq•DsrA target search during transcription. Previous work established that Hfq recognizes a (AAN)4 sequence in the rpoS mRNA 5′ UTR that is required for Hfq to facilitate sRNA regulation of rpoS.(Soper and Woodson, 2008; Soper et al., 2011) The AAN motif must be positioned upstream of the target site for Hfq to act on rpoS mRNA both in vitro and in vivo.23 To test if the AAN Hfq binding motif is required for sRNA annealing during transcription, we repeated the co-transcriptional targeting experiment using rpoS301 variants in which the AAN motif was deleted or repositioned.23
During transcription, we found that rpoS transcripts lacking the AAN motif (rpoS301ΔAAN) formed five-fold fewer stable DsrA complexes than WT transcripts, indicating that the AAN motif is still required for efficient targeting (9% and 46% respectively; Fig. 5A, D). These few events likely occurred independently of Hfq, since a similar fraction of WT rpoS transcripts bound DsrA in the absence of Hfq (Fig. S2A, D). Inserting a strong (AAA)4 motif 20 nucleotides downstream of the target site (rpoS301A12-484) also decreased stable targeting to a level equivalent to no AAN motif (Fig. 5B, D). Supporting this, maximum likelihood analysis revealed that while the all rpoS301 AAN variants exhibited characteristic lifetimes comparable to those for WT rpoS301 transcripts, the amplitude of the longest lifetime (a3) decreased about three-fold (Fig. S5A and Table S1). Therefore, in agreement with earlier experiments on refolded mRNAs23, these results showed that the (AAN)4 motif is needed to recruit Hfq during transcription and that it must be upstream of the sRNA target site to be effective.
Fig. 5. Upstream Hfq recruitment is required for co-transcriptional Hfq•DsrA targeting.

(A-C) Hfq•DsrA binding to variants of rpoS301 mRNA as cartooned (top). (A) deletion of upstream (AAN)4 Hfq binding site; (B) insertion of (AAA)4 Hfq binding site in downstream loop23; (C) truncation at nt 441 before the DsrA target. The stable interactions with rpoSΔtarget may represent residual base pairing between DsrA and a downstream extension (see Table S3).
D) Fraction of rpoS transcripts in (A-C) with stable (t > 100 s) Hfq•DsrA targeting. Symbols represent values from two experiments.
E) Number of unstable (t < 50 s) Hfq•DsrA binding events per mRNA on rpoS301 variants. Bars represent mean and s.d.; Nmol (WT) = 182, Nmol (ΔAAN) = 207, Nmol (A12-484) = 120, Nmol (ΔTarget) = 156.
F). Cumulative probability of association times, illustrating the onset of Hfq•DsrA transient binding relative to rpoS301 transcription as indicated by PIFE (Δttransient). Data shown for rpoS301 variants in A-C targeted by WT DsrA, and WT rpoS301 with DsrA-RBM that is not complementary to rpoS301.21 See also Fig. S5.
Hfq•DsrA transiently binds the AAN motif during transcription
We noticed that ~50% of rpoS301 transcripts that ultimately formed stable complexes with Hfq•DsrA had previously encountered Hfq•DsrA for a brief period. We postulated that these short-lived complexes represented interactions between Hfq and the AAN motif that did not result in DsrA annealing. If true, this would suggest that upstream recruitment of Hfq creates opportunities for sRNA-mRNA base pairing downstream (whether these attempts succeed or not).
To examine binding of Hfq•DsrA to the AAN motif alone, we truncated the rpoS301 mRNA (rpoS301Δtarget) after the AAN motif and just before the target site (Fig. 5C). rpoS301Δtarget formed primarily short-lived complexes with Hfq•DsrA, and substantially fewer stable complexes than full-length rpoS301 mRNA (23% and 46% respectively; Fig. 5D). The few stable complexes observed may represent partial base-pairing between DsrA and a downstream sequence appended to rpoS301Δtarget.
Next, we tallied the frequency of unstable interactions (DsrA lifetimes < 50 s) to examine how often the AAN motif is recognized by Hfq•DsrA during transcription. Deletion of the AAN motif (rpoS301ΔAAN) resulted in fewer short-lived Hfq•DsrA interactions per mRNA relative to WT rpoS301 transcripts, and 60% of rpoS301ΔAAN transcripts experienced no observable binding during the 10 min movie (Fig. 5A, E). Thus, many unstable events on WT rpoS301 transcripts likely represent Hfq binding to the AAN motif without sRNA-mRNA annealing. In agreement with this interpretation, the number of unstable binding events per mRNA increased about three times when the target was removed (AAN only), or when an (AAA)4 binding site was re-introduced downstream of the target in rpoS301A12-484 (Fig. 5E). The downstream Hfq binding site is sampled by Hfq•DsrA but cannot support stable annealing (Fig. 5D).
The first appearance of unstable complexes correlated with transcription of the AAN motif, suggesting that the synthesis of the Hfq binding site dictates when sRNAs can begin attempting to target the mRNA. Firstly, we found that transient complexes first appeared at similar points during transcription (Δttransient) for WT DsrA and for DsrARBM that cannot base pair with the rpoS target (cyan and black, Fig. 5F). Secondly, deletion of the AAN motif shifted the cumulative probability density curve to later times during transcription consistent with fewer binding events overall (blue, Fig. 5F). Thirdly, relocating the AAN motif downstream (rpoSA12-484) reduced transient binding early in transcription (−400 to −100 s), but increased binding near the end of transcription (~ −100 s), coinciding with the expected synthesis of the repositioned AAN motif in rpoS301A12-484 (cherry, Fig. 5F). This change in timing provided evidence that Hfq•DsrA is recruited to the rpoS mRNA once the AAN motif is transcribed.
Monitoring waypoints of RNA synthesis in real time using PIFE
Next, we wanted to ask how closely successful targeting correlates with transcription of the sRNA target site. Because our pausing assays (Fig. 4E) indicated that the rate of elongation is not uniform across the rpoS gene, we developed a double-PIFE method to pinpoint the position of the TEC more precisely (Fig. 6A). For this, we generated two doubly labeled DNA templates for transcription that incorporate Cy3 fluorophores at different sites (Fig. 6B and S6A). The first Cy3 fluorophore was attached to the downstream end of the template to mark the end of transcription as before. The second Cy3 fluorophore was incorporated in the middle of the template, either just after the AAN motif and before the target site (2x-Cy3-rpoSafterAAN) or just after the target site (2x-Cy3-rpoSafterTarget). In 2x-Cy3-rpoSafterTarget DNA, the second Cy3 is located adjacent to an intrinsic E. coli RNAP pause site at A473 (Fig. 4E).
Fig. 6. Hfq•DsrA targeting coincides with transcription of the target site.

A) Double-PIFE smCoCoA monitors specific points of RNA synthesis in real time.
B) Schematic of the 2x-Cy3-rpoSafterTarget DNA template with two Cy3 fluorophores: one located internally to mark transcription of the target site and the other located near the end of the template to mark the end of transcription.
C) Representative single-molecule time trace showing two distinct PIFE peaks (green, top). Dotted lines indicate the start of PIFE 1, the end of PIFE 1, the onset of stable Hfq•DsrA targeting, and the start of PIFE 2. Grey arrows indicate transient binding before the onset of stable Hfq•DsrA targeting. Different levels of Cy5 intensity may reflect differences in the fluorophore microenvironment.
D-F) Distribution of the onset of stable Hfq•DsrA binding relative to (D) the start and (E) the end of the first PIFE peak denoting the end of complete target synthesis; (F) the onset of transient Hfq•DsrA binding relative to the start of the first PIFE peak.
G) Model of single molecule experiments showing that stable Hfq•DsrA targeting occurs near RNA polymerase during the first PIFE signal. This model is based on the single nanometer distance of PIFE36,54,55 and the structure of the E. coli RNAP elongation complex.56 See Fig. S7 for sequence details.
H) Cumulative probability density plot comparing the onset of stable Hfq•DsrA targeting during transcription (relative to transcription of the target site; cyan) and after transcription (relative to injection of Hfq•DsrA; red). Successful targeting by Hfq•DsrA occurs more rapidly during transcripton. t1/2, time required for 50% saturation of immobilized rpoS RNAs. Co-transcriptional targeting: t1/2 = 46 s, 95% CI (34.6 s, 55.2 s) and refolded rpoS mRNA: t1/2 = 191 s, 95% CI (177 s, 234 s).
In smCoCoA experiments on doubly labeled DNA templates, we observed two well-separated PIFE signals representing the passage of RNAP over each fluorophore (Fig. 6C and S6). We confirmed that the PIFE signals correlated with the Cy3 locations by comparing the cumulative probability for onset of the first PIFE signal relative to NTP injection, which occurred earlier on 2x-Cy3-rpoSafterAAN DNA than 2x-Cy3-rpoSafterTarget DNA, as expected (Fig. S6C). Because PIFE is most prominent when the protein is within ~ 1 nm of the fluorophore36, we interpret the start of highest intensity plateau in the PIFE signal as the moment when the leading edge of RNAP encounters the Cy3 fluorophore on the DNA template strand. The cumulative probabilities for the onset of the second PIFE signal in the two doubly labeled templates overlapped significantly, further evidence that PIFE accurately reports on transcription progression (Fig. S6C).
Stable targeting of Hfq•DsrA occurs just after the target site is transcribed
To pinpoint when successful targeting occurred during transcription, we measured the difference between the onset of stable Hfq•DsrA targeting and synthesis of the sRNA binding site, which is reported by the first PIFE signal on 2x-Cy3-rpoSafterTarget DNA (Fig. 6C, blue shaded region). Upon inspection of the rpoS transcripts that were ultimately targeted by Hfq•DsrA, we found that more than 80% formed a stable Hfq•DsrA complex after the start of the first PIFE signal (Fig. 6D). This further supports that stable targeting depends on synthesis of the target site. Moreover, > 60% rpoS transcripts formed a stable Hfq•DsrA complex before the end of the first PIFE signal (Fig. 6E), when the target site is within or near the polymerase RNA exit channel (Fig. S7). Combined, these data demonstrated that Hfq•DsrA frequently recognizes the target site concomitant with its exit from the TEC.
Because Hfq•DsrA binds AAN motifs frequently and independently of the sRNA target (Fig. 5), we hypothesized that recruitment of the sRNP positions DsrA to recursively sample the elongating mRNA until a target site complementary to DsrA is transcribed. To evaluate this model, we analyzed the frequency of transient Hfq•DsrA binding prior to successful targeting. We found that ~60% of the rpoS transcripts that were ultimately stably targeted by Hfq•DsrA had experienced earlier transient encounters. On this subset of rpoS transcripts, the first transient encounter nearly always occurred before the start of the first PIFE signal (Fig. 6F), whereas stable binding typically occurred after the start of the first PIFE signal. Combined with the results in Fig. 5, these data were consistent with a model in which recruitment of Hfq•DsrA to the upstream AAN motif poises the sRNA to immediately base pair with the target site as soon as it is transcribed.
Targeting in proximity to polymerase enables fast recognition
We hypothesized that the accessibility of the target site as it emerges from RNA polymerase (Fig. 6G) underlies the kinetic advantage of co-transcriptional targeting. We compared the binding kinetics during transcription with the binding kinetics on refolded rpoS mRNA from Fig. 2. The cumulative probability of stable targeting immediately following transcription of the target site rose substantially faster than the stable targeting on refolded rpoS mRNA (Fig. 6H). During transcription, a 50% probability of successful DsrA-Hfq targeting was reached at t1/2 = 46 s following transcription of the target site (Fig. 6H). Whereas on refolded rpoS mRNA, t1/2 = 190.8 s (Fig. 6H). In total, these data suggested that capturing the target site soon after it emerges from RNA polymerase (Fig. 6G) provides a kinetic advantage over targeting after the rpoS 5′ UTR has folded.
DISCUSSION
Variable folding of RNA in the wake of an elongating polymerase can affect recognition by RNA binding proteins, processing enzymes or small RNAs during transcription. For example, recent studies showed that incomplete folding of newly made RNA delays binding of ribosomal proteins, which recognize the native rRNA structure.7,13,14 Here, we show that a sRNA binds faster during transcription by accessing the RNA close to RNAP, during the interval between synthesis and stable folding.
Although previous reports showed that sRNAs can act on certain targets during transcription26,27, how this could be achieved was not known. Our results show that this kinetic advantage depends on the action of Hfq, which chaperones targeting by bacterial sRNAs (Fig. 7). Hfq is first recruited to an upstream site in the transcript, positioning the sRNA to anneal with complementary sequences. Then, Hfq•sRNA captures the target site concurrent with its transcription, forming a stably annealed complex. As discussed below, this efficient search is enabled by features of the Hfq chaperone that it shares with other RNA-guided enzymes, perhaps explaining why many sRNPs are capable of acting on nascent RNAs.
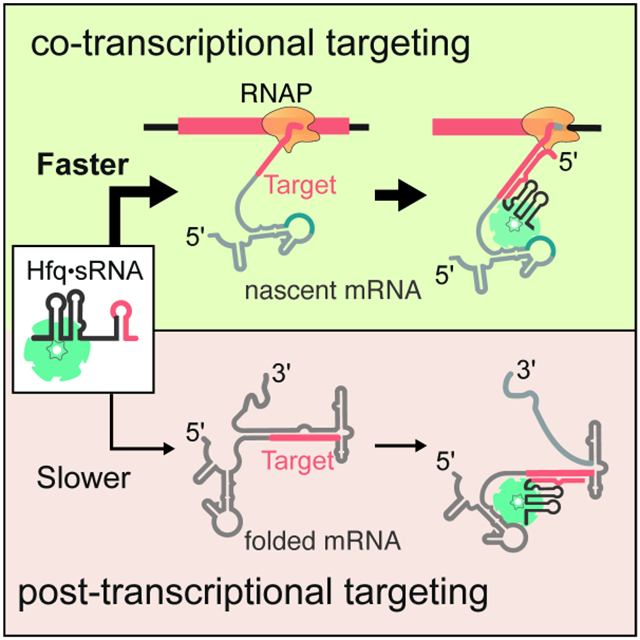
Fig. 7. Recursive sampling of nascent RNA leads to efficient sRNA targeting.

Model for co-transcriptional binding of Hfq•DsrA sRNP with the rpoS 5′ UTR. After the AAN Hfq motif is transcribed, transient binding enables recursive base pairing between DsrA sRNA and the rpoS transcript. As soon as the complementary target site is accessible, it is captured by Hfq•DsrA, forming a stable ternary complex that persists long after the remaining mRNA transcription. Co-transcriptional targeting circumvents the need for rearranging the rpoS mRNA structure by allowing Hfq•DsrA to capture the target site before the inhibitory stem structure has a chance to fold.
Targeting an RNA before it folds
Studies with model Hfq-sRNA complexes demonstrated that the sRNA is less likely to anneal with structured targets than unstructured ones.34 Our results suggest that targeting of the rpoS mRNA 5′ UTR is likewise more efficient during transcription because the 5′ UTR has not yet folded into a stable structure that masks the sRNA target site. First, we show that the Hfq•DsrA sRNPs can stably bind residues close to RNAP (Fig. 6 and S7), capturing the target when it is still relatively unstructured. Based on the footprint of E. coli RNAP 37, we estimate that stable Hfq•DsrA binding can be first observed when RNAP has synthesized 15-20 nt of the target site (Fig. 6 and Fig. S7). RNA within the exit channel as close as 11-15 nt from the insertion site is accessible for base-pairing, as evidenced by the structure of the putL anti-termination complex and an intrinsic terminator hairpin.38,39 Thus, at this stage of elongation, 3-8 nt of the target site would be available to base pair with DsrA. Short duplexes of 8 bp are sufficient for sRNA regulation in vivo.40 Second, we observe that co-transcriptional targeting is insensitive to downstream structure (Fig. 3). Although Hfq can remodel the folded rpoS 5′ UTR post-transcription, this imposes an additional barrier that reduces the chance of successful sRNA annealing (Fig. 3).21,41 In this way, the time window for sRNP targeting is analogous to that of transcriptional riboswitches, in which folding of the nascent RNA dictates the window of opportunity for ligand binding and regulation.11
Elongation sets the timer for RNP targeting
The time window for sRNA binding and co-transcriptional folding is dependent on the elongation rate, which we find has a large impact on the likelihood of sRNA targeting during transcription (Fig. 4). In this work, we used slow transcription speeds (0.5-2 nt/s) to observe RNP binding at the low concentrations suitable for single molecule studies. Modeling of gene specific transcription rates determined that the rpoS mRNA is transcribed at a rate of ~ 8 nt/s during exponential growth in rich media 42, which is in the range reported for other bacterial mRNAs (10 - 20 nt/s).42 However, significant RNAP pausing near translation start sites35 may widen the time window for co-transcriptional recognition of target sites. Importantly, during stationary phase and under stress conditions when sRNAs upregulate rpoS expression43, an increase in the concentration of the stringent response alarmone, ppGpp, may increase RNAP pausing37, thereby increasing the likelihood that sRNAs anneal with the rpoS 5′ UTR during transcription.
Repeated sampling of elongating transcripts increases the efficiency of targeting
To capture a target site soon after it emerges from RNA polymerase, an sRNA must rapidly search the transcript for complementary sites without being trapped by near-complementary sites. Our results show how Hfq enhances this search during transcription. First, prior to transcription of the target site, Hfq recognizes an upstream AAN motif (Fig. 5), limiting the sRNA search to sequences near an Hfq binding site. Second, Hfq facilitates reversible base pairing between the sRNA and the emerging transcript. In our experiments, Hfq-DsrA binding is unstable until the target sequence is transcribed (Fig. 5), which likely reflects attempted but incomplete base pairing with DsrA.34 As a result, the ternary complex dissociates if DsrA cannot form an extended duplex with the rpoS transcript, leaving the Hfq binding motif available for the next Hfq•DsrA RNP (Fig. 7). This dynamic sampling would allow different sRNPs to scan an elongating mRNA until the target site becomes accessible. In cells, active cycling of sRNAs on Hfq is likely to accelerate the exchange of sRNA-Hfq-rpoS mRNA complexes.44,45
The actual speed of Hfq•sRNA recruitment will depend on the sRNA copy number and the availability of Hfq in the cell. Nevertheless, Hfq binds its RNA substrates near the rate of diffusion (~108 M−1s−1; 46), about 1,000 times faster than the association of two unchaperoned RNA strands. Thus, recruitment of Hfq to an upstream AAN motif is expected to speed up co-transcriptional recognition of downstream targets. In some targets of sRNA regulation, the AAN motif lies downstream of the sRNA binding site. It will be interesting to know whether these targets are only recognized after the AAN motif has been transcribed, or whether Hfq is temporarily recruited to the plentiful suboptimal AAN motifs upstream.
Implications for sRNA regulation
Several sRNAs have been proposed to associate with mRNAs during transcription in vivo27 suggesting that regulation of mRNAs during transcription may provide an additional layer of sRNA-mediated regulation. Previous studies found that biochemical measures of sRNA binding correlate well with up-regulation of rpoS expression in E. coli.47 In our experiments, stable Hfq•DsrA complexes (≥ 1 min) persist long after the ribosome binding site in rpoS is transcribed. As a result, early targeting of rpoS transcripts before the inhibitory stem forms could facilitate transcription-coupled translation 28. In the same manner, binding of Hfq-sRNAs close to RNAP could facilitate negative regulation by blocking 30S subunits from initiating on nascent transcripts. sRNAs may also act by preventing or enhancing Rho-dependent transcription termination.26,48 Click or tap here to enter text. We hypothesize that both post-transcriptional and co-transcriptional regulation by sRNAs operate simultaneously to optimize the response to stress. Based on our observation that co-transcriptional targeting is sensitive to transcription rate (Fig. 4), the balance between these pathways could be altered by elongation factors that regulate the accessibility and timing of RNP targeting during transcription. The three sRNAs that upregulate rpoS mRNA under different forms of stress may each have a different propensity to bind the target site during transcription or after transcription, allowing for further optimization of the regulatory interactions.
Co-transcriptional target search in other RNPs
The main features of sRNP targeting, including recruitment of protein partners upstream of the target site and dynamic base pairing, may be used by other RNA-guided complexes to capitalize on the accessibility of nascent transcripts. For example, during spliceosome assembly, protein-protein interactions between the U1 snRNP and the cap binding complex (CBC) help accelerate recognition of the 5’ splice site (5’ SS) in single molecule assays.49 Furthermore, a recent structure of a U1 snRNP-TEC complex shows that U1 snRNP proteins interact with core components of RNAP and the U1-snRNA forms a duplex with the 5’ SS close to the exit channel of RNAP.50 Therefore, during transcription, transient association of U1 with RNAP and CBC may have an analogous recruitment function to the AAN motif by increasing the chance that a U1 snRNP successfully forms a complex as soon as a 5’ SS is transcribed. Type III-A CRISPR Cas systems may use similar strategies for targeting and cleaving nascent RNAs close to a TEC.51,52 Finally, the immediate capture of nascent sequences suggests how snoRNAs may act to redirect folding of pre-ribosomal RNA during transcription.5,6,53 Future work examining the principles governing competition between upstream interactions and RNP association during transcription will be crucial to our understanding how RNPs can alter the co-transcriptional folding landscape.
LIMITATIONS OF THE STUDY
The efficiency of targeting in this assay is measured by amount of stable Hfq-DsrA complexes formed on the slide surface, which may not perfectly reflect up-regulation of rpoS translation in cells. In the future, it will be interesting to examine the contributions of co-transcriptional and post-transcriptional targeting to regulation by sRNAs. This study provides limited information on how pausing may influence targeting because only transcription elongation complexes that reached the end of the template within the single-molecule movie were analyzed. Longer imaging and direct detection of paused TECs will be required to answer these questions. Finally, we studied only one isolated sRNA:mRNA pair. The kinetics of co-transcriptional targeting may be different for other sRNAs, especially if different sRNAs compete for the same mRNA target.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sarah Woodson (swoodson@jhu.edu).
Materials Availability:
All unique and stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability:
All microscopy data generated in this study have been deposited and are publicly available as of the date of publication. DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli DH5 | NEB | Cat# C2987H |
| Escherichia coli BL21(DE3) | NEB | Cat# C2527H |
| Chemicals, peptides, and recombinant proteins | ||
| T7 RNA polymerase | This study; purified as in (Butler and Chamberlin, 1982; Davanloo et al., 1984) | N/A |
| E. coli RNA polymerase holoenzyme | NEB | Cat# M0551 |
| NTP set | Thermo Fisher | Cat# R0481 |
| Dichlorodimethylsilane (DDS) | Sigma | Cat# 440272 |
| Biotin-conjugated bovine serum albumin | Sigma | Cat# A8549 |
| Tween-20 | Fisher BioReagents | Cat# BP337 |
| Neutravidin | Thermo Fisher | Cat# 31000 |
| Trolox | ACROS Organics | Cat# AC218940010 |
| Glucose oxidase | Sigma | Cat# G2133 |
| Catalase | Sigma | Cat# C9322 |
| Glucose | Sigma | Cat#G8270 |
| RNasin plus | Promega | Cat# N2611 |
| ATP, [gamma-32P]-6000Ci/mmol | Perkin Elmer | Cat# BLU502Z500UC |
| UTP, [alpha-32P]-6000Ci/mmol | Perkin Elmer | Cat# BLU007H250UC |
| Sulfo-Cyanine3 NHS ester | Lumiprobe | Cat# 21320 |
| Sulfo-Cyanine5 NHS ester | Lumiprobe | Cat# 23320 |
| Q5 DNA polymerase | NEB | Cat# M0491 |
| Q5U DNA polymerase | NEB | Cat# M0515 |
| HiFi Taq Ligase | NEB | Cat# M0647 |
| USER enzyme | NEB | Cat# M5505 |
| Rifampicin | Sigma | Cat# R3501 |
| Heparin Sodium Salt | Sigma | Cat#H3393 |
| 3'-O-Methyluridine-5'-Triphosphate | TriLink | Cat# N-1059-1 |
| 3'-O-Methylcytidine-5'-Triphosphate | TriLink | Cat# N-1057-1 |
| 3'-O-Methylguanosine-5'-Triphosphate | TriLink | Cat# N-1058-1 |
| 3'-O-Methyladenosine-5'-Triphosphate | TriLink | Cat# N-1056-1 |
| Critical commercial assays | ||
| NucleoSpin Gel and PCR Clean-up | Takara | Cat# 740609 |
| Deposited data | ||
| Raw microscopy movies | This paper | https://doi.org/10.7281/T1/AVEV7M |
| Figure source data | This paper | https://doi.org/10.7281/T1/AVEV7M |
| Oligonucleotides | ||
| Tether_T3_33nts_3’ BIO: 5’ – CTAACTCTCTACCCA TCCATCTCTCACTCACCC /3BIO/ – 3’ | (Rodgers and Woodson, 2019) | N/A |
| SA5_aadU35: 5’ –CCTGTGTCCTGTGTGTCCTGTCCAAAGTGTGTCG/iAmMC6T/CC – 3’ | (Rodgers and Woodson, 2019) | N/A |
| T7Pro_Tether_FOR: 5’ –GATCCTAATACGACTCACTATAGGGTGAGTGAGAGATGGATGGGTAGAGAGTTAGTAGTA – 3’ | (Rodgers and Woodson, 2019) | N/A |
| T7A1_FOR: 5’ – TATCAAAAAGAGTATTGACTTAAAGTCTAACCTATAGGATACTTACAGCC – 3’ | This study | N/A |
| Term_REV_29_aadT25: 5’ – CAAAAAACCCCTCAAGACCCGTT/iAmMC6T/AGAGG – 3’ | This study | N/A |
| rpoSREV2_dT180_dUx2: 5’ – AAA/ideoxyU/AACGCAGCGGG/ideoxyU/T/iAmMC6T/ACGGATTTCC – 3’ | This study | N/A |
| rpoSFOR2_dUx2_197: 5’ – ACCCGC/ideoxyU/GCGTTATT/ideoxyU/GCCGCAGCGATAAATCG – 3’ | This study | N/A |
| RpoS301_REV_aadT121_dU130: 5’ – ATGCAAGCGTGTTGAACTGG/ideoxyU/TCCGGTGC/iAmMC6T/ACCC – 3’ | This study | N/A |
| RpoS301_FOR_dU150: 5’ – ACCAGTTCAACACGCTTGCA/ideoxyU/UTTTGAAATTCG – 3’ | This study | N/A |
| RpoS_REV_SA5_nt432: 5’ – GTGTCCTGTCCAAAGTGTGTCGTCCTGCAAGCGTGTTGAACTGGTTCCGGTGCTACCC – 3’ | This study | N/A |
| RpoS_REV2_SA5_NIS_nt562: 5’ – GTGTCCTGTCCAAAGTGTGTCGTCCGACGGAACATTCAAGCAAAAGCCTGGTTCC – 3’ | This study | N/A |
| Recombinant DNA | ||
| pET21b-EcHfq | (Zhang et al., 2002) | N/A |
| pAR1219-T7RNAP | (Davanloo et al., 1984) | N/A |
| Sequences for linear DNA templates listed in Table S2 | This study | N/A |
| Software and algorithms | ||
| Single-molecule fluorescence acquisition software (smCamera) | Ha Lab, Custom made | https://github.com/Ha-SingleMoleculeLab/Data-Aquisition |
| MATLAB | Mathworks | R2022a |
| GLIMPSE/IMSCROLL | (Friedman and Gelles, 2015) | N/A |
| FIJI | (Schindelin et al., 2012) | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial Strains
Hfq protein, DsrA small RNA, and rpoS mRNA sequences were derived from the K-12 strain of Escherichia coli. Hfq protein was overexpressed on a plasmid and purified from E. coli BL21(DE3) cells.
METHOD DETAILS
RNA preparation and fluorescent labeling
Fluorescently labeled DsrA sRNA and unlabeled rpoS mRNAs for refolding experiments were prepared by in vitro transcription with T7 RNA polymerase and purified on denaturing polyacrylamide gels. The DNA template for DsrA was made by PCR amplification with Q5 DNA polymerase (NEB) using overlapping primers as previously described45; Key Resources Table). DsrA sRNAs were transcribed in the presence of GMP and labeled at the 5’ end with a Cy5-NHS ester fluorophore using previously established protocols.45,57
Protein purification
Hexameric wild type Hfq protein was overexpressed in E. coli BL21(DE3)Δhfq::cat-sacB cells and purified as previously described.34
Single-round transcription and pause site mapping
Radiolabeled TECs stalled in absence of CTP were assembled in 20 μL (total volume) containing 50 nM DNA template, 40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 50 nM E. coli RNAP (NEB), 2 μM GTP, 2 μM ATP, 0.5 μM UTP, 1 U/μL RNasin Plus (Promega), 20 μCi 32P-α-ATP. Transcription reactions were incubated at room temperature for 10 min and then rifampicin was added to a final concentration of 25 μg/μL. Transcription was restarted by diluting reactions to 40 μL with 2X restart mixture: 40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 40 μM NTPs (GTP, ATP, UTP, CTP), 2 U RNasin Plus. Aliquots (3-5 μL) were taken from the reaction mixture at various times, quenched in 95% formamide, 25 mM EDTA, and placed on ice. For pause site mapping, stalled TEC samples were prepared in the same manner and split into four 5 μL reactions. Transcription was restarted by diluting each sample to 10 μL with 2X restart buffer plus 50 μM of one of the 3’-O-methyl NTPs (TriLink). Chain terminator reactions were incubated for 20 min at RT before being quenched with 95% formamide, 25 mM EDTA and placed on ice. All samples were heated to 95 °C and loaded onto a sequencing 6% polyacrylamide gel. After loading, the gel was run at 55 W for 1 – 2 hours. Sequencing gels were transferred to filter paper, dried, exposed to a phosphor storage screen overnight, and imaged (GE Typhoon). Gels were quantified using FIJI.58
DsrA and Hfq binding by native gel mobility shift
DsrA sRNA was 5' end labeled with γ-[32P]-ATP using T4 polynucleotide kinase (NEB) and purified using a Chroma TE-30 spin column (Takara). Radiolabeled DsrA (2 nM) was incubated with Hfq protein (70 nM) in a 40 μL reaction containing: 50 mM Tris-HCl pH 7.5, 20 mM MgCl2, 100 mM NaCl, 100 mM KCl, 40 μM NTPs (GTP, ATP, UTP, CTP), 2 U RNasin Plus, 20% glycerol (v/v). Unlabeled rpoS301 TECs stalled in absence of CTP were assembled in 40 μL (total volume) containing 25 nM DNA template, 40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 25 nM T7 RNAP, 2 μM GTP, 2 μM ATP, 0.5 μM UTP, 1 U/μL RNasin Plus (Promega). Stalled TECs were incubated at RT for 5 minutes and heparin was added to a final concentration of 1 mg/mL to inhibit reinitiation. Hfq-DsrA binding was monitored during transcription by adding the Hfq-DsrA-NTP restart mixture to the stalled TECs in a 1:1 ratio. In the control lanes, 2 nM radiolabeled DsrA was incubated in annealing buffer 50 mM Tris-HCl pH 7.5, 20 mM MgCl2, 100 mM NaCl, 100 mM KCl, 1 U/μL RNasin Plus (Promega) with 25 nM rpoS RNA and/or 35 nM Hfq as indicated. Aliquots were taken out and immediately loaded onto a pre-run 8% native polyacrylamide gel at 15W for 1.5 hr at 4°C in 1X TBE. Gels were dried, exposed overnight to a phosphorescence storage screen, and imaged (GE Typhoon).
Construction and fluorescent labeling of DNA templates
Primers were purchased from Integrated DNA technologies, Inc. (IDT). Reverse primers (see Key Resources Table) containing an internal amino-alkyl modified nucleotide (IDT; see Table S1) were fluorescently labeled with Cy3-NHS dye (Lumiprobe) as follows: 0.5 mg of Cy3 dye was dissolved in 33 μL of DMSO and added to a reaction mixture containing 5 nmol modified primer adjusted to 100 μL final volume with 100 mM sodium bicarbonate pH 8.5. Reactions were incubated overnight at room temperature and then free dye was removed using a Chroma TE-10 spin column (Takara Bio) followed by clean-up on a Monarch DNA column using the oligonucleotide clean-up protocol.
Fluorescent DNA templates for transcription were generated by PCR using Q5 high fidelity polymerase (NEB). Synthetic G-blocks containing the rpoS mRNA sequence were purchased from IDT. Following PCR, fluorescently labeled DNA templates were separated on 1% agarose, gel purified using the Nucleospin gel purification kit (Takara) and eluted in 20 μL of water.
For site-specific double labeling of the DNA templates with attachment of a second fluorophore to an internal site, two overlapping DNAs were designed for ligation which each contained a Cy3 fluorophore in the template strand. Internal sites for fluorophore placement were chosen based on the proximity to sites of interest and sequence context for ligation junctions by HiFi Taq Ligase59; NEB). Fluorophores were incorporated in the reverse primers for each fragment as above. To generate long ssDNA overhangs for ligation, dU residues were incorporated in the reverse primer downstream of the fluorophore (Fragment 1) or in the forward primer (Fragment 2). DNA fragments containing the desired fluorophore and dU substitutions were amplified by PCR using Q5U high fidelity polymerase master mix (NEB) and gel purified using the Nucleospin gel purification kit (Takara). ssDNA overhangs were generated by treating the DNA fragments with USER enzyme (NEB) in a 20 μL reaction containing 400 nM DNA at 37 °C for 30 min. Overhang fragments were then combined in 1:1 molar ratio and annealed slowly by incubating at 95 °C and cooling to 42 °C over 30 min. HiFi Ligase buffer and 5 μL of Hifi Taq Ligase (NEB) were added to a final reaction volume of 50 μL and ligation was carried out at 42 °C for 2 h. Ligation reactions were cleaned up using Nucleospin gel purification kit (Takara), and the DNA concentration was determined as above. The doubly labeled fluorescent DNA was stored at −20 C in the dark.
Co-transcriptional colocalization single-molecule experiments
smCoCoA experiments were performed as previously described13 with the following modifications: stalled TECs were prepared in 20 μL containing 50-100 nM Cy3-labeled DNA template, 40 mM Tris-HCl pH 7.9, 20 mM MgCl2, 50 nM E. coli RNAP, 200 μM GTP, 200 μM ATP, 50 μM UTP, 2 U RNasin Plus, 100 nM biotinylated tether oligomer (Tether_T3_33nts_3’BIO). Stalled TECs were incubated at RT for 10 min and then immobilized on the slide surface to generate sufficient and well-separated spot density. Hfq•DsrA-Cy5 complexes were prepared at room temperature (20 °C) by mixing Hfq and DsrA-Cy5 1:1 at 200 nM final concentration and diluting the complexes to 5 nM in the restart imaging buffer immediately before injection as previously described.34 Under these conditions, nearly all the sRNA is complexed with Hfq.34
All single-molecule imaging was performed on a homebuilt prism total internal reflection fluorescence (TIRF) microscope using an alternating excitation scheme between 532 nm (green laser) and 630 nm (red laser) excitation at the acquisition frame rate of 100 ms. After the start of imaging, transcription elongation was restarted by injection of restart imaging buffer containing 20 μM NTPs, 5 nM Hfq•DsrA-Cy5, 50 mM Tris HCl pH 7.9, 20 mM MgCl2, 50 mM NaCl, 50 mM KCl, 2 U RNasin Plus, 4 mM Trolox, 1 w/v % glucose, 165 U/mL glucose oxidase. Frames were taken every 100 ms for ~7000 frames (~12 minutes).
QUANTIFICATION AND STATISTICAL ANALYSIS
Single-molecule colocalization analysis
Single-molecule colocalization was analyzed as previously described13 using the Imscroll software developed by the Gelles lab and implemented in MATLAB.60 Briefly, Cy3-labeled rpoS TECs were selected as areas of interest (AOIs) shortly after injection to account for any drift during injection. The intensity from each AOI was integrated over all frames of the movie and plotted as a single-molecule time trajectory. Using a mapping function that relates the position of a single molecule in both green and red channels, the Cy3 AOIs were translated to the red channel, intensity was integrated over all frames of the movie, and a single-molecule trace was generated to show colocalization of Cy5-labeled Hfq•DsrA.
Marking transcription time using one or multiple PIFE signals
The timing for transcription of a particular sequence was approximated using the timing of the start of a protein induced fluorescence enhancement (PIFE) signal similarly to previous work.13 Transcription by E. coli RNAP in vitro at 20 μM NTP concentration is slow compared to the frame rate of the camera with a range of ~ 2.5 – 8 frames/nucleotide (Fig. 3A). Only E. coli RNAP TECs that exhibited a PIFE signal indicating complete transcription of the template (typically 31 ± 5%) were analyzed for DsrA colocalization. TECs that do not exhibit a PIFE signal may have failed to restart, or failed to complete transcription during our observation window (~10 min) due to pausing, premature termination or slow elongation at low NTP concentrations.
At the beginning of a PIFE signal, there was often a gradual increase in fluorescence consistent with RNAP approaching the Cy3 fluorophore (Fig. 1C, Fig. S6, and Fig. S7). This was followed by a plateau consistent with the Cy3 fluorophore being translocated through the RNA/DNA hybrid in the active site of RNAP. Because of the nanometer dependence of PIFE, the beginning of the PIFE signal was taken to be the first frame of the plateau (Fig. S7). Near the end of the PIFE signal, the fluorescence signal decreased gradually to an intensity similar to the intensity prior to the start of PIFE, indicating that the elongating RNAP had moved past the Cy3 fluorophore (Fig. 1C, Fig. S6, and Fig. S7). In some cases, loss in Cy3 fluorescence was observed in a single frame resulting in an intensity at a lower baseline fluorescence compared to the intensity before the start of the PIFE signal. This single step loss in fluorescence was likely due to Cy3 photobleaching during PIFE. Due to the possibility of photobleaching during PIFE, the duration of the PIFE event is likely underestimated and provides a lower limit for the moment of transcription past the Cy3 fluorophore. The appearance of the second PIFE signal was delayed by 50 – 100 s on DNA templates containing two Cy3 fluorophores compared to DNA containing a single Cy3 located at the end of the template (Fig. S6). This may be due to slow bypass of the bulky fluorophore attached to the template strand, which must traverse through the active site of RNAP. The magnitude of this slowdown was the same for both doubly labeled DNA templates, indicating no other effects of sequence context. This slowdown does not affect analysis of DsrA binding relative to the position of RNAP on the template.
Analysis of DsrA dwell times and arrival times
Binding of Hfq•DsrA-Cy5 was analyzed for all AOIs that exhibited a PIFE signal, indicating restart of the stalled TEC. For DNAs that contained two Cy3 fluorophores, only AOIs that exhibited two distinct PIFE signals and a stable (> 100 s) Hfq•DsrA-Cy5 colocalization event were analyzed for arrival times of the stable Hfq•DsrA-Cy5 complex. Dwell times for Hfq•DsrA were generated as previously described.13,60 Association times (ton) were determined from the interval between injection of Hfq•DsrA-Cy5 and the starting frame of a Hfq•DsrA-Cy5 binding event as indicated by an increase in fluorescence intensity of Cy5 above background. The moment of injection was reported by a small rise in the Cy5 background intensity due to the presence of unbound DsrA-Cy5 in the solution. Cumulative density plots and K-S tests were implemented in MATLAB.
Maximum likelihood analysis of the unbinned data for triple exponential kinetic binding behavior was used to determine characteristic lifetimes of the Hfq•DsrA•rpoS mRNA complexes. Only traces which exhibited a transcription signature indicated by a PIFE signal were analyzed for colocalization of Hfq•DsrA-Cy5. All Hfq•DsrA-Cy5 binding events detected with these valid transcripts were included in the analysis. Timing of Hfq•DsrA-Cy5 and Hfq•DsrA-Cy5 binding lifetimes were measured as previously described.60 Equation 1 describes triple exponential kinetic binding behavior for maximum likelihood analysis, where is the total time duration of the movie; is the minimum resolvable time interval in the experiment; is the maximum time interval; , , , , represent characteristic lifetimes; and and are the amplitudes associated with the fitted lifetimes.
| (eq. 1) |
Errors were determined by bootstrapping to obtain 95% confidence bounds as previously described.60 Histograms were generated in MATLAB (the Mathworks) by unequal binning of the data to minimize empty bins and visualize the data with the maximum likelihood fits. Error bars in the histogram represent the standard deviation in a binomial distribution, , where N is the number of observations and P is the event probability.
Supplementary Material
Supplemental Figure S1. Hfq•DsrA form stable ternary complexes with nascent rpoS mRNA during transcription. Related to Figure 1.
A) Unlabeled T7 RNA polymerase TECs stalled on a template encoding rpoS301 RNA were restarted in the presence of 20 μM NTPs and trace 32P-labeled DsrA•Hfq to monitor association of Hfq•DsrA with rpoS mRNAs during and after transcription. Samples were loaded onto a native 8% PAGE at 0 – 300 s after transcription restart. The gel was running continuously during the experiment, so lanes on the right were loaded later than those on the left. Left, a super-shift corresponding to the DsrA•rpoS•Hfq ternary complex is observed after 90 s. We only resolve ternary complexes with full-length rpoS301 because nascent transcripts tethered to T7 RNA polymerase remain in the wells of the gel. Right, binding to refolded, full-length rpoS301 RNA (60 min, RT). DsrA•Hfq can associate to form a complex with full-length wild type rpoS301 mRNA but forms less complex with rpoS301GC mRNAs containing a GC-clamp mutation as previously reported.21 DsrA binding is only observed in the presence of Hfq. Excess unlabeled rpoS301GC mRNA competes Hfq away from DsrA, increasing the amount of free DsrA in the second to last lane.
B) Single round transcription kinetics for rpoS301 mRNA under the same transcription conditions as in panel A. Transcripts were resolved by denaturing 6% PAGE. Ladder, radiolabeled Riboruler Low Range (Thermo). Full-length rpoS301 mRNA is first observed at 90 s, corresponding to the time when DsrA-Hfq first associates with rpoS mRNA during transcription.
Supplemental Figure S2. Stable Hfq•DsrA targeting depends on Hfq, base pairing, and rpoS synthesis. Related to Figure 1, 2.
A) Rastergram of DsrA-Cy5 association with rpoS301 transcripts in the absence of Hfq, showing little stable binding. Colored as in Fig. 1.
B) Rastergram of non-complementary Hfq•DsrARBM-Cy5 binding to rpoS301 transcripts. There were fewer stable complexes compared to WT DsrA, in agreement with earlier results (Soper et al., 2011). The presence of a few stable complexes (> 100 s) may indicate residual base-pairing between DsrARBM and other regions of the rpoS301 mRNA or may indicate a low probability of long-lived interactions between Hfq•DsrARBM and the rpoS301AAN motif in absence of sRNA-mRNA base pairing.
C) Rastergram of DsrA-Cy5 binding to rpoS301 stalled TECs in the absence of NTPs. The lack of stable binding indicates that these events likely represent interactions between Hfq•DsrA and nascent rpoS301 transcripts as opposed to interactions with RNAP or the DNA template. Colored as in Fig. 1.
D) Fraction of immobilized rpoS301 transcripts that are stably bound by DsrA at some point during the 10 min movie for the components shown. The fractions from two replicate experiments are shown as scatter points and the bar indicates the mean. Details of the number of molecules for each experiment and replicates can be found in Table S2.
E) Probability density histogram with overlayed maximum likelihood triple exponential fit illustrating the distribution of DsrA lifetimes for different conditions. The amplitude of the longest characteristic lifetime among all binding events was diminished in the absence of Hfq or by mutations in DsrA that disrupt base pairing with rpoS mRNA. Equations for fitting can be found in Star Methods. Fit parameters and errors are outlined in Table S1.
F and G) Cumulative probability density plots as in Figure 2B and C including 95% confidence bounds (dotted lines) to illustrate the error in the probability.
H) Cumulative probability density plots for the measurement of ton stable for three independent replicates of the rpoS301 co-transcriptional experiments indicating 95% confidence intervals for each (dotted lines). The co-transcriptional binding replicate experiments have statistically similar kinetics demonstrating repeatability (WT 1 vs WT 2: p = 0.656; WT 1 vs WT 3: p = 0.241; WT 2 vs WT 3: p = 0.699; K-S test).
Supplemental Figure S3. Stability of the inhibitory stem affects Hfq-DsrA binding to renatured rpoS RNAs and nascent rpoS RNAs during transcription. Related to Figure 3.
A) Single-molecule colocalization experiment for monitoring post-transcriptional association of Hfq•DsrA-Cy5 to refolded, full length rpoS301 mRNAs. Refolded mRNA was hybridized with a Cy3-labeled anti-sense oligomer (green star) prior to immobilization on the slide.
B) Example time trace for a single refolded rpoS301 mRNA molecule showing the Hfq•DsrA-Cy5 binding interval, Δt, relative to injection.
C) Rastergrams of Hfq•DsrA-Cy5 binding during transcription to rpoS301Δ3’IS transcripts (left) and rpoS301GC transcripts (right). Colored as in Fig. 1. Rastergrams were assembled from 52-55 randomly selected transcripts from a total combined dataset of > 100 total transcripts from at least two independent replicate experiments. The time axis was synchronized with the start of injection (t = 0) for each transcript.
D) Rastergram of Hfq•DsrA-Cy5 binding to WT rpoS301 mRNA 45 minutes post-transcription.
E) Rastergrams of Hfq•DsrA-Cy5 binding to refolded mRNAs: rpoS301 WT (right), rpoS301Δ3’IS (middle), and rpoS301GC (left).
F) A cumulative probability density plot illustrating the relative timing of the onset of stable Hfq•DsrA binding to refolded WT rpoS mRNA and co-transcriptionally folded rpoS mRNAs 45 minutes after addition of NTPs to the slide surface (WT post-txn). Above 100 s, the distributions lie within overlapping 95% confidence intervals (right). Before 100 s, excess binding of Hfq•DsrA to the co-transcriptionally folded RNA compared to refolded RNA indicates that a fraction of transcripts remain poorly folded 45 min after transcription and are readily targeted by DsrA.
Supplemental Figure S4. Transcription speed influences the likelihood of stable Hfq•DsrA binding. Related to Figure 4. A) Probability density histogram with overlayed maximum likelihood triple exponential fit illustrating the distribution of DsrA lifetimes for each transcription condition. Faster transcription by T7 RNAP in 20 μM NTPs (purple) reduces the likelihood of forming a stable complex. Equations for fitting can be found in Star Methods. Fit parameters for rpoS301 mRNA transcribed by E. coli RNAP at 20 uM can be found in Table S1. Fit parameters for T7 RNAP are as follows:
20 μM NTPs: , , , , , ;
2 μM: , , , , ,
B) Secondary structure of rpoS301 with sequences around observed in vitro pause sites. A consensus pause sequence35 can be found near these mapped sites suggesting these may be used in vivo.
Supplemental Figure S5. Hfq binds transiently to AAN motif during transcription. Related to Figure 5.
A) Probability density histogram with overlayed maximum likelihood triple exponential fit illustrating the distribution of DsrA lifetimes for rpoS301 variants lacking an Hfq binding site (ΔAAN), a downstream Hfq binding site (A12-484), or the DsrA binding site (ΔTarget). Inset shows that the probability of forming short-lived complexes (t2) is greatest when Hfq can bind an AAN motif but DsrA cannot base pair with the target. Equations for fitting can be found in Star Methods. Fit parameters for each rpoS301 variant can be found in Table S1.
B) Cumulative probability density plot as in Figure 5F showing 95% confidence bounds for each variant. DsrARBM was omitted for clarity.
Supplemental Figure S6. PIFE monitors the progress of active transcription. Related to Figure 6.
A) Schematic of doubly labeled DNA templates indicating the positions of Cy3 fluorophores relative to the Hfq binding motif (AAN) and target site.
B) Example single molecule trajectory illustrating the detection of double PIFE events based on fluorescence intensity. (top) Single frame images of the Cy3 fluorescence indicating increased fluorescence observed for one or two Cy3 fluorophores during PIFE 1 and PIFE 2. Notches at the right of the trace indicate the standard intensities of 0, 1, 2 active Cy3 fluorophores.
C) A cumulative probability density plot illustrating the relative timing for the start of PIFE 1 and PIFE 2 for the 2X-Cy3 labeled rpoS DNAs (after AAN and after Target shown in part A) compared to PIFE from a singly labeled DNA template with Cy3 located at 599+52 (green, Figure 1C). A significant delay in the onset of PIFE 1 is observed when the Cy3 is incorporated at position 473 (after target, orange) relative to position 413 (after AAN, red), demonstrating that the appearance of PIFE reflects the distance traveled by RNAP before reaching the fluorophore. The cumulative probability density becomes similar for both 2X-Cy3 labeled rpoS DNAs for PIFE 2 (blue and purple) where the Cy3 fluorophore is located on the same nucleotide (599+52) in each template. This occurs later than PIFE on a single-labeled DNA template (green) containing Cy3 at the same position (599+52), suggesting the upstream fluorophore impedes elongation to some degree. This effect is the same for both doubly labeled templates.
D) Example single molecule traces of Hfq•DsrA binding relative to elongation of 2x-Cy3-rpoSafterTarget. Fluorescence intensity from Cy3 fluorophores located on the 2x-Cy3-rpoSafterTarget DNA template is shown in the top trace (green) and intensity from a Cy5 fluorophore on the 5’ end of DsrA sRNA is shown in the bottom trace (red). Numbers indicate first and second PIFE signals from Cy31 and Cy32 respectively.
Supplemental Figure S7. Interpretation of the start and end of PIFE signals. Related to Figure 6.
A) Single molecule trace for transcription on 2x-Cy3-rpoSafterTarget DNA (green, top) and DsrA-Cy5 (red, bottom).
B) Expansion of first PIFE signal shown in A, illustrating the identification of the start time and end time from the duration of the PIFE signal plateau (dotted lines). A gradual increase in Cy3 intensity can be seen until the signal reaches the plateau before the start of PIFE 1 and after the end of PIFE 1. Based on the single nanometer distance dependence of PIFE 36,54,55, we interpret the gradual increase as RNAP approaching the Cy3 fluorophore and the plateau in the PIFE signal as confinement of the Cy3 fluorophore within the active site of E. coli RNAP during translocation of the template strand (see also Figure 6G).
C) Map of DsrA target site in rpoS mRNA (green letters), relative to the TEC at various stages of rpoS transcription. In the structure of the E. coli RNAP elongation complex 56, nucleobases −15 to +14 of the template relative to the insertion site are highly ordered and within 1 – 2 nm of protein residues. Therefore, a Cy3 fluorophore attached to a nucleobase within this 29 bp window would experience an environment conducive to PIFE. We estimate that the PIFE plateau begins when the Cy31 fluorophore is located on the +14 position in the RNAP active site (top). During the plateau signal, the Cy31 fluorophore traverses through nucleotide −15 in the template strand before the signal starts to gradually decrease as polymerase moves away from the fluorophore (bottom). Potential base pairing with the DsrA-Cy5 sRNA is shown on the nascent rpoS301 mRNA for each scenario, assuming DsrA can extend into the exit channel up to the position of the DNA-RNA hybrid (magenta). This extent of base pairing is consistent with recent structures of E. coli termination complexes.39,61,62
Highlights.
Hfq and small RNAs target mRNAs during transcription before the mRNA folds.
Co-transcriptional targeting by Hfq and small RNA is faster and more efficient.
Transient Hfq binding poises the small RNA to base pair with complementary sites.
Successful targeting often occurs close to the RNA polymerase exit channel.
ACKNOWLEDGEMENTS
The authors thank Jorjethe Roca for help preparing fluorescent sRNAs, Debapratim Dutta for preparing Hfq protein, and all members of the Woodson lab for helpful discussions. This work was supported by grants from the National Institute of General Medicine [K99GM140204 to M.L.R.; R35GM136351 to S.A.W.; and T32GM080189 for support of B.M.O.]
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Gerovac M, Vogel J, and Smirnov A (2021). The World of Stable Ribonucleoproteins and Its Mapping With Grad-Seq and Related Approaches. Frontiers Mol Biosci 8, 661448. 10.3389/fmolb.2021.661448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorski SA, Vogel J, and Doudna JA (2017). RNA-based recognition and targeting: sowing the seeds of specificity. Nat Rev Mol Cell Bio 18, 215–228. 10.1038/nrm.2016.174. [DOI] [PubMed] [Google Scholar]
- 3.Oesterreich FC, Herzel L, Straube K, Hujer K, Howard J, and Neugebauer KM (2016). Splicing of Nascent RNA Coincides with Intron Exit from RNA Polymerase II. Cell 165, 1 11. 10.1016/j.cell.2016.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts GC, Gooding C, Mak HY, Smith CWJ, and Proudfoot NJ (1998). Co-transcriptional commitment to alternative splice site selection. Nucleic Acids Res 26, 5568–5572. 10.1093/nar/26.24.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steitz JA, and Tycowski KT (1995). ENHANCED PERSPECTIVE: Small RNA Chaperones for Ribosome Biogenesis. Science 270, 1626–1626. 10.1126/science.270.5242.1626. [DOI] [PubMed] [Google Scholar]
- 6.Kos M, and Tollervey D (2010). Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol Cell 37, 809–820. 10.1016/j.molcel.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodgers ML, and Woodson SA (2021). A roadmap for rRNA folding and assembly during transcription. Trends Biochem Sci. 10.1016/j.tibs.2021.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, and Landick R (2016). A Two-Way Street: Regulatory Interplay between RNA Polymerase and Nascent RNA Structure. Trends Biochem Sci 41, 293–310. 10.1016/j.tibs.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heilman-Miller SL, and Woodson SA (2003). Effect of transcription on folding of the Tetrahymena ribozyme. RNA 9, 722–733. 10.1261/rna.5200903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan T, Artsimovitch I, Fang XW, Landick R, and Sosnick TR (1999). Folding of a large ribozyme during transcription and the effect of the elongation factor NusA. Proceedings of the National Academy of Sciences of the United States of America 96, 9545 9550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scull CE, Dandpat SS, Romero RA, and Walter NG (2021). Transcriptional Riboswitches Integrate Timescales for Bacterial Gene Expression Control. Front Mol Biosci 7, 607158. 10.3389/fmolb.2020.607158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bushhouse DZ, Choi EK, Hertz LM, and Lucks JB (2022). How does RNA fold dynamically? J Mol Biol, 167665. 10.1016/j.jmb.2022.167665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodgers ML, and Woodson SA (2019). Transcription Increases the Cooperativity of Ribonucleoprotein Assembly. Cell 179, 1370–1381.e12. 10.1016/j.cell.2019.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duss O, Stepanyuk GA, Puglisi JD, and Williamson JR (2019). Transient Protein-RNA Interactions Guide Nascent Ribosomal RNA Folding. Cell 179, 1357–1369.e16. 10.1016/j.cell.2019.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner EGH, and Romby P (2015). Chapter Three Small RNAs in Bacteria and Archaea Who They Are, What They Do, and How They Do It. Adv Genet 90, 133–208. 10.1016/bs.adgen.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Gottesman S (2019). Trouble is coming: Signaling pathways that regulate general stress responses in bacteria. J Biol Chem 294, 11685–11700. 10.1074/jbc.rev119.005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lease RA, Cusick ME, and Belfort M (1998). Riboregulation in Escherichia coli: DsrA RNA acts by RNA:RNA interactions at multiple loci. Proc National Acad Sci 95, 12456–12461. 10.1073/pnas.95.21.12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majdalani N, Cunning C, Sledjeski D, Elliott T, and Gottesman S (1998). DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc National Acad Sci 95, 12462–12467. 10.1073/pnas.95.21.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sledjeski DD, Gupta A, and Gottesman S (1996). The small RNA, DsrA, is essential for the low temperature expression of RpoS during exponential growth in Escherichia coli. Embo J 15, 3993–4000. 10.1002/j.1460-2075.1996.tb00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang A, Wassarman KM, Ortega J, Steven AC, and Storz G (2002). The Sm-like Hfq Protein Increases OxyS RNA Interaction with Target mRNAs. Mol Cell 9, 11–22. 10.1016/s1097-2765(01)00437-3. [DOI] [PubMed] [Google Scholar]
- 21.Soper TJ, Doxzen K, and Woodson SA (2011). Major role for mRNA binding and restructuring in sRNA recruitment by Hfq. Rna 17, 1544–1550. 10.1261/rna.2767211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng Y, Curtis JE, Fang X, and Woodson SA (2014). Structural model of an mRNA in complex with the bacterial chaperone Hfq. Proc National Acad Sci 111, 17134–17139. 10.1073/pnas.1410114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng Y, Soper TJ, and Woodson SA (2014). Positional Effects of AAN Motifs in rpoS Regulation by sRNAs and Hfq. J Mol Biol 426, 275–285. 10.1016/j.jmb.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soper TJ, and Woodson SA (2008). The rpoS mRNA leader recruits Hfq to facilitate annealing with DsrA sRNA. Rna 14, 1907–1917. 10.1261/rna.1110608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoyos M, Huber M, Förstner KU, and Papenfort K (2020). Gene autoregulation by 3’ UTR-derived bacterial small RNAs. Elife 9, e58836. 10.7554/elife.58836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sedlyarova N, Shamovsky I, Bharati BK, Epshtein V, Chen J, Gottesman S, Schroeder R, and Nudler E (2016). sRNA-Mediated Control of Transcription Termination in E. coli. Cell 167, 111–121.e13. 10.1016/j.cell.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reyer MA, Chennakesavalu S, Heideman EM, Ma X, Bujnowska M, Hong L, Dinner AR, Vanderpool CK, and Fei J (2021). Kinetic modeling reveals additional regulation at co-transcriptional level by post-transcriptional sRNA regulators. Cell Reports 36, 109764. 10.1016/j.celrep.2021.109764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irastortza-Olaziregi M, and Amster-Choder O (2021). Coupled Transcription-Translation in Prokaryotes: An Old Couple With New Surprises. Front Microbiol 11, 624830. 10.3389/fmicb.2020.624830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chatterjee S, Chauvier A, Dandpat SS, Artsimovitch I, and Walter NG (2021). A translational riboswitch coordinates nascent transcription–translation coupling. Proc National Acad Sci 118, e2023426118. 10.1073/pnas.2023426118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCullen CA, Benhammou JN, Majdalani N, and Gottesman S (2010). Mechanism of Positive Regulation by DsrA and RprA Small Noncoding RNAs: Pairing Increases Translation and Protects rpoS mRNA from Degradation. J Bacteriol 192, 5559–5571. 10.1128/jb.00464-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandin P, and Gottesman S (2010). Integrating anaerobic/aerobic sensing and the general stress response through the ArcZ small RNA. Embo J 29, 3094–3107. 10.1038/emboj.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Majdalani N, Chen S, Murrow J, John KS, and Gottesman S (2001). Regulation of RpoS by a novel small RNA: the characterization of RprA. Mol Microbiol 39, 1382–1394. 10.1111/j.1365-2958.2001.02329.x. [DOI] [PubMed] [Google Scholar]
- 33.Cunning C, Brown L, and Elliott T (1998). Promoter Substitution and Deletion Analysis of Upstream Region Required for rpoS Translational Regulation. J Bacteriol 180, 4564–4570. 10.1128/jb.180.17.4564-4570.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Małecka EM, and Woodson SA (2021). Stepwise sRNA targeting of structured bacterial mRNAs leads to abortive annealing. Mol Cell. 10.1016/j.molcel.2021.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larson MH, Mooney RA, Peters JM, Windgassen T, Nayak D, Gross CA, Block SM, Greenleaf WJ, Landick R, and Weissman JS (2014). A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science 344, 1042 1047. 10.1126/science.1251871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hwang H, and Myong S (2014). Protein induced fluorescence enhancement (PIFE) for probing protein-nucleic acid interactions. Chem Soc Rev 43, 1221–1229. 10.1039/c3cs60201j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang JY, Mishanina TV, Landick R, and Darst SA (2019). Mechanisms of Transcriptional Pausing in Bacteria. J Mol Biol 431, 4007–4029. 10.1016/j.jmb.2019.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dey S, Batisse C, Shukla J, Webster MW, Takacs M, Saint-André C, and Weixlbaumer A (2022). Structural insights into RNA-mediated transcription regulation in bacteria. Mol Cell 82, 3885–3900.e10. 10.1016/j.molcel.2022.09.020. [DOI] [PubMed] [Google Scholar]
- 39.You L, Omollo EO, Yu C, Mooney RA, Shi J, Shen L, Wu X, Wen A, He D, Zeng Y, et al. (2023). Structural basis for intrinsic transcription termination. Nature 613, 783–789. 10.1038/s41586-022-05604-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poddar A, Azam MS, Kayikcioglu T, Bobrovskyy M, Zhang J, Ma X, Labhsetwar P, Fei J, Singh D, Luthey-Schulten Z, et al. (2021). Effects of individual base-pairs on in vivo target search and destruction kinetics of bacterial small RNA. Nat Commun 12, 874. 10.1038/s41467-021-21144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hwang W, Arluison V, and Hohng S (2011). Dynamic competition of DsrA and rpoS fragments for the proximal binding site of Hfq as a means for efficient annealing. Nucleic Acids Res 39, 5131–5139. 10.1093/nar/gkr075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Großmann P, Lück A, and Kaleta C (2017). Model-based genome-wide determination of RNA chain elongation rates in Escherichia coli. Sci Rep-uk 7, 17213. 10.1038/s41598-017-17408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Battesti A, Majdalani N, and Gottesman S (2011). The RpoS-Mediated General Stress Response in Escherichia coli. Microbiology+ 65, 189–213. 10.1146/annurev-micro-090110-102946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fender A, Elf J, Hampel K, Zimmermann B, and Wagner EGH (2010). RNAs actively cycle on the Sm-like protein Hfq. Gene Dev 24, 2621 2626. 10.1101/gad.591310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roca J, Santiago-Frangos A, and Woodson SA (2022). Diversity of bacterial small RNAs drives competitive strategies for a mutual chaperone. Nat Commun 13, 2449. 10.1038/s41467-022-30211-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hopkins JF, Panja S, and Woodson SA (2011). Rapid binding and release of Hfq from ternary complexes during RNA annealing. Nucleic Acids Res 39, 5193–5202. 10.1093/nar/gkr062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soper T, Mandin P, Majdalani N, Gottesman S, and Woodson SA (2010). Positive regulation by small RNAs and the role of Hfq. Proc National Acad Sci 107, 9602–9607. 10.1073/pnas.1004435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J, Morita T, and Gottesman S (2019). Regulation of Transcription Termination of Small RNAs and by Small RNAs: Molecular Mechanisms and Biological Functions. Front Cell Infect Mi 9, 201. 10.3389/fcimb.2019.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larson JD, and Hoskins AA (2017). Dynamics and consequences of spliceosome E complex formation. Elife 6. 10.7554/elife.27592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang S, Aibara S, Vos SM, Agafonov DE, Lührmann R, and Cramer P (2021). Structure of a transcribing RNA polymerase II–U1 snRNP complex. Science 371, 305–309. 10.1126/science.abf1870. [DOI] [PubMed] [Google Scholar]
- 51.Samai P, Pyenson N, Jiang W, Goldberg GW, Hatoum-Aslan A, and Marraffini LA (2015). Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell 161, 1164–1174. 10.1016/j.cell.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu TY, Liu J-J, Aditham AJ, Nogales E, and Doudna JA (2019). Target preference of Type III-A CRISPR-Cas complexes at the transcription bubble. Nat Commun 10, 3001. 10.1038/s41467-019-10780-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karbstein K (2011). Inside the 40S ribosome assembly machinery. Curr Opin Chem Biol 15, 657–663. 10.1016/j.cbpa.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lerner E, Ploetz E, Hohlbein J, Cordes T, and Weiss S (2016). A Quantitative Theoretical Framework For Protein-Induced Fluorescence Enhancement–Förster-Type Resonance Energy Transfer (PIFE-FRET). J Phys Chem B 120, 6401–6410. 10.1021/acs.jpcb.6b03692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ploetz E, Lerner E, Husada F, Roelfs M, Chung S, Hohlbein J, Weiss S, and Cordes T (2016). Förster resonance energy transfer and protein-induced fluorescence enhancement as synergetic multi-scale molecular rulers. Sci Rep-uk 6, 1 18. 10.1038/srep33257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kang JY, Olinares PDB, Chen J, Campbell EA, Mustaev A, Chait BT, Gottesman ME, and Darst SA (2017). Structural basis of transcription arrest by coliphage HK022 Nun in an Escherichia coli RNA polymerase elongation complex. Elife 6, e25478. 10.7554/elife.25478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rinaldi AJ, Suddala KC, and Walter NG (2014). Native Purification and Labeling of RNA for Single Molecule Fluorescence Studies. In Methods in Molecular Biology. (Springer; New York: ), p. 63 95. 10.1007/978-1-4939-1896-6_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676 682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lohman GJS, Bauer RJ, Nichols NM, Mazzola L, Bybee J, Rivizzigno D, Cantin E, and Evans TC (2016). A high-throughput assay for the comprehensive profiling of DNA ligase fidelity. Nucleic Acids Res 44, e14–e14. 10.1093/nar/gkv898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Friedman LJ, and Gelles J (2015). Multi-wavelength single-molecule fluorescence analysis of transcription mechanisms. Methods 86, 27 36. 10.1016/j.ymeth.2015.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Molodtsov V, Wang C, Firlar E, Kaelber JT, and Ebright RH (2023). Structural basis of Rho-dependent transcription termination. Nature, 1–8. 10.1038/s41586-022-05658-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Said N, Hilal T, Sunday ND, Khatri A, Bürger J, Mielke T, Belogurov GA, Loll B, Sen R, Artsimovitch I, et al. (2021). Steps toward translocation-independent RNA polymerase inactivation by terminator ATPase ρ. Science 371, eabd1673. 10.1126/science.abd1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Hfq•DsrA form stable ternary complexes with nascent rpoS mRNA during transcription. Related to Figure 1.
A) Unlabeled T7 RNA polymerase TECs stalled on a template encoding rpoS301 RNA were restarted in the presence of 20 μM NTPs and trace 32P-labeled DsrA•Hfq to monitor association of Hfq•DsrA with rpoS mRNAs during and after transcription. Samples were loaded onto a native 8% PAGE at 0 – 300 s after transcription restart. The gel was running continuously during the experiment, so lanes on the right were loaded later than those on the left. Left, a super-shift corresponding to the DsrA•rpoS•Hfq ternary complex is observed after 90 s. We only resolve ternary complexes with full-length rpoS301 because nascent transcripts tethered to T7 RNA polymerase remain in the wells of the gel. Right, binding to refolded, full-length rpoS301 RNA (60 min, RT). DsrA•Hfq can associate to form a complex with full-length wild type rpoS301 mRNA but forms less complex with rpoS301GC mRNAs containing a GC-clamp mutation as previously reported.21 DsrA binding is only observed in the presence of Hfq. Excess unlabeled rpoS301GC mRNA competes Hfq away from DsrA, increasing the amount of free DsrA in the second to last lane.
B) Single round transcription kinetics for rpoS301 mRNA under the same transcription conditions as in panel A. Transcripts were resolved by denaturing 6% PAGE. Ladder, radiolabeled Riboruler Low Range (Thermo). Full-length rpoS301 mRNA is first observed at 90 s, corresponding to the time when DsrA-Hfq first associates with rpoS mRNA during transcription.
Supplemental Figure S2. Stable Hfq•DsrA targeting depends on Hfq, base pairing, and rpoS synthesis. Related to Figure 1, 2.
A) Rastergram of DsrA-Cy5 association with rpoS301 transcripts in the absence of Hfq, showing little stable binding. Colored as in Fig. 1.
B) Rastergram of non-complementary Hfq•DsrARBM-Cy5 binding to rpoS301 transcripts. There were fewer stable complexes compared to WT DsrA, in agreement with earlier results (Soper et al., 2011). The presence of a few stable complexes (> 100 s) may indicate residual base-pairing between DsrARBM and other regions of the rpoS301 mRNA or may indicate a low probability of long-lived interactions between Hfq•DsrARBM and the rpoS301AAN motif in absence of sRNA-mRNA base pairing.
C) Rastergram of DsrA-Cy5 binding to rpoS301 stalled TECs in the absence of NTPs. The lack of stable binding indicates that these events likely represent interactions between Hfq•DsrA and nascent rpoS301 transcripts as opposed to interactions with RNAP or the DNA template. Colored as in Fig. 1.
D) Fraction of immobilized rpoS301 transcripts that are stably bound by DsrA at some point during the 10 min movie for the components shown. The fractions from two replicate experiments are shown as scatter points and the bar indicates the mean. Details of the number of molecules for each experiment and replicates can be found in Table S2.
E) Probability density histogram with overlayed maximum likelihood triple exponential fit illustrating the distribution of DsrA lifetimes for different conditions. The amplitude of the longest characteristic lifetime among all binding events was diminished in the absence of Hfq or by mutations in DsrA that disrupt base pairing with rpoS mRNA. Equations for fitting can be found in Star Methods. Fit parameters and errors are outlined in Table S1.
F and G) Cumulative probability density plots as in Figure 2B and C including 95% confidence bounds (dotted lines) to illustrate the error in the probability.
H) Cumulative probability density plots for the measurement of ton stable for three independent replicates of the rpoS301 co-transcriptional experiments indicating 95% confidence intervals for each (dotted lines). The co-transcriptional binding replicate experiments have statistically similar kinetics demonstrating repeatability (WT 1 vs WT 2: p = 0.656; WT 1 vs WT 3: p = 0.241; WT 2 vs WT 3: p = 0.699; K-S test).
Supplemental Figure S3. Stability of the inhibitory stem affects Hfq-DsrA binding to renatured rpoS RNAs and nascent rpoS RNAs during transcription. Related to Figure 3.
A) Single-molecule colocalization experiment for monitoring post-transcriptional association of Hfq•DsrA-Cy5 to refolded, full length rpoS301 mRNAs. Refolded mRNA was hybridized with a Cy3-labeled anti-sense oligomer (green star) prior to immobilization on the slide.
B) Example time trace for a single refolded rpoS301 mRNA molecule showing the Hfq•DsrA-Cy5 binding interval, Δt, relative to injection.
C) Rastergrams of Hfq•DsrA-Cy5 binding during transcription to rpoS301Δ3’IS transcripts (left) and rpoS301GC transcripts (right). Colored as in Fig. 1. Rastergrams were assembled from 52-55 randomly selected transcripts from a total combined dataset of > 100 total transcripts from at least two independent replicate experiments. The time axis was synchronized with the start of injection (t = 0) for each transcript.
D) Rastergram of Hfq•DsrA-Cy5 binding to WT rpoS301 mRNA 45 minutes post-transcription.
E) Rastergrams of Hfq•DsrA-Cy5 binding to refolded mRNAs: rpoS301 WT (right), rpoS301Δ3’IS (middle), and rpoS301GC (left).
F) A cumulative probability density plot illustrating the relative timing of the onset of stable Hfq•DsrA binding to refolded WT rpoS mRNA and co-transcriptionally folded rpoS mRNAs 45 minutes after addition of NTPs to the slide surface (WT post-txn). Above 100 s, the distributions lie within overlapping 95% confidence intervals (right). Before 100 s, excess binding of Hfq•DsrA to the co-transcriptionally folded RNA compared to refolded RNA indicates that a fraction of transcripts remain poorly folded 45 min after transcription and are readily targeted by DsrA.
Supplemental Figure S4. Transcription speed influences the likelihood of stable Hfq•DsrA binding. Related to Figure 4. A) Probability density histogram with overlayed maximum likelihood triple exponential fit illustrating the distribution of DsrA lifetimes for each transcription condition. Faster transcription by T7 RNAP in 20 μM NTPs (purple) reduces the likelihood of forming a stable complex. Equations for fitting can be found in Star Methods. Fit parameters for rpoS301 mRNA transcribed by E. coli RNAP at 20 uM can be found in Table S1. Fit parameters for T7 RNAP are as follows:
20 μM NTPs: , , , , , ;
2 μM: , , , , ,
B) Secondary structure of rpoS301 with sequences around observed in vitro pause sites. A consensus pause sequence35 can be found near these mapped sites suggesting these may be used in vivo.
Supplemental Figure S5. Hfq binds transiently to AAN motif during transcription. Related to Figure 5.
A) Probability density histogram with overlayed maximum likelihood triple exponential fit illustrating the distribution of DsrA lifetimes for rpoS301 variants lacking an Hfq binding site (ΔAAN), a downstream Hfq binding site (A12-484), or the DsrA binding site (ΔTarget). Inset shows that the probability of forming short-lived complexes (t2) is greatest when Hfq can bind an AAN motif but DsrA cannot base pair with the target. Equations for fitting can be found in Star Methods. Fit parameters for each rpoS301 variant can be found in Table S1.
B) Cumulative probability density plot as in Figure 5F showing 95% confidence bounds for each variant. DsrARBM was omitted for clarity.
Supplemental Figure S6. PIFE monitors the progress of active transcription. Related to Figure 6.
A) Schematic of doubly labeled DNA templates indicating the positions of Cy3 fluorophores relative to the Hfq binding motif (AAN) and target site.
B) Example single molecule trajectory illustrating the detection of double PIFE events based on fluorescence intensity. (top) Single frame images of the Cy3 fluorescence indicating increased fluorescence observed for one or two Cy3 fluorophores during PIFE 1 and PIFE 2. Notches at the right of the trace indicate the standard intensities of 0, 1, 2 active Cy3 fluorophores.
C) A cumulative probability density plot illustrating the relative timing for the start of PIFE 1 and PIFE 2 for the 2X-Cy3 labeled rpoS DNAs (after AAN and after Target shown in part A) compared to PIFE from a singly labeled DNA template with Cy3 located at 599+52 (green, Figure 1C). A significant delay in the onset of PIFE 1 is observed when the Cy3 is incorporated at position 473 (after target, orange) relative to position 413 (after AAN, red), demonstrating that the appearance of PIFE reflects the distance traveled by RNAP before reaching the fluorophore. The cumulative probability density becomes similar for both 2X-Cy3 labeled rpoS DNAs for PIFE 2 (blue and purple) where the Cy3 fluorophore is located on the same nucleotide (599+52) in each template. This occurs later than PIFE on a single-labeled DNA template (green) containing Cy3 at the same position (599+52), suggesting the upstream fluorophore impedes elongation to some degree. This effect is the same for both doubly labeled templates.
D) Example single molecule traces of Hfq•DsrA binding relative to elongation of 2x-Cy3-rpoSafterTarget. Fluorescence intensity from Cy3 fluorophores located on the 2x-Cy3-rpoSafterTarget DNA template is shown in the top trace (green) and intensity from a Cy5 fluorophore on the 5’ end of DsrA sRNA is shown in the bottom trace (red). Numbers indicate first and second PIFE signals from Cy31 and Cy32 respectively.
Supplemental Figure S7. Interpretation of the start and end of PIFE signals. Related to Figure 6.
A) Single molecule trace for transcription on 2x-Cy3-rpoSafterTarget DNA (green, top) and DsrA-Cy5 (red, bottom).
B) Expansion of first PIFE signal shown in A, illustrating the identification of the start time and end time from the duration of the PIFE signal plateau (dotted lines). A gradual increase in Cy3 intensity can be seen until the signal reaches the plateau before the start of PIFE 1 and after the end of PIFE 1. Based on the single nanometer distance dependence of PIFE 36,54,55, we interpret the gradual increase as RNAP approaching the Cy3 fluorophore and the plateau in the PIFE signal as confinement of the Cy3 fluorophore within the active site of E. coli RNAP during translocation of the template strand (see also Figure 6G).
C) Map of DsrA target site in rpoS mRNA (green letters), relative to the TEC at various stages of rpoS transcription. In the structure of the E. coli RNAP elongation complex 56, nucleobases −15 to +14 of the template relative to the insertion site are highly ordered and within 1 – 2 nm of protein residues. Therefore, a Cy3 fluorophore attached to a nucleobase within this 29 bp window would experience an environment conducive to PIFE. We estimate that the PIFE plateau begins when the Cy31 fluorophore is located on the +14 position in the RNAP active site (top). During the plateau signal, the Cy31 fluorophore traverses through nucleotide −15 in the template strand before the signal starts to gradually decrease as polymerase moves away from the fluorophore (bottom). Potential base pairing with the DsrA-Cy5 sRNA is shown on the nascent rpoS301 mRNA for each scenario, assuming DsrA can extend into the exit channel up to the position of the DNA-RNA hybrid (magenta). This extent of base pairing is consistent with recent structures of E. coli termination complexes.39,61,62
Data Availability Statement
All microscopy data generated in this study have been deposited and are publicly available as of the date of publication. DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli DH5 | NEB | Cat# C2987H |
| Escherichia coli BL21(DE3) | NEB | Cat# C2527H |
| Chemicals, peptides, and recombinant proteins | ||
| T7 RNA polymerase | This study; purified as in (Butler and Chamberlin, 1982; Davanloo et al., 1984) | N/A |
| E. coli RNA polymerase holoenzyme | NEB | Cat# M0551 |
| NTP set | Thermo Fisher | Cat# R0481 |
| Dichlorodimethylsilane (DDS) | Sigma | Cat# 440272 |
| Biotin-conjugated bovine serum albumin | Sigma | Cat# A8549 |
| Tween-20 | Fisher BioReagents | Cat# BP337 |
| Neutravidin | Thermo Fisher | Cat# 31000 |
| Trolox | ACROS Organics | Cat# AC218940010 |
| Glucose oxidase | Sigma | Cat# G2133 |
| Catalase | Sigma | Cat# C9322 |
| Glucose | Sigma | Cat#G8270 |
| RNasin plus | Promega | Cat# N2611 |
| ATP, [gamma-32P]-6000Ci/mmol | Perkin Elmer | Cat# BLU502Z500UC |
| UTP, [alpha-32P]-6000Ci/mmol | Perkin Elmer | Cat# BLU007H250UC |
| Sulfo-Cyanine3 NHS ester | Lumiprobe | Cat# 21320 |
| Sulfo-Cyanine5 NHS ester | Lumiprobe | Cat# 23320 |
| Q5 DNA polymerase | NEB | Cat# M0491 |
| Q5U DNA polymerase | NEB | Cat# M0515 |
| HiFi Taq Ligase | NEB | Cat# M0647 |
| USER enzyme | NEB | Cat# M5505 |
| Rifampicin | Sigma | Cat# R3501 |
| Heparin Sodium Salt | Sigma | Cat#H3393 |
| 3'-O-Methyluridine-5'-Triphosphate | TriLink | Cat# N-1059-1 |
| 3'-O-Methylcytidine-5'-Triphosphate | TriLink | Cat# N-1057-1 |
| 3'-O-Methylguanosine-5'-Triphosphate | TriLink | Cat# N-1058-1 |
| 3'-O-Methyladenosine-5'-Triphosphate | TriLink | Cat# N-1056-1 |
| Critical commercial assays | ||
| NucleoSpin Gel and PCR Clean-up | Takara | Cat# 740609 |
| Deposited data | ||
| Raw microscopy movies | This paper | https://doi.org/10.7281/T1/AVEV7M |
| Figure source data | This paper | https://doi.org/10.7281/T1/AVEV7M |
| Oligonucleotides | ||
| Tether_T3_33nts_3’ BIO: 5’ – CTAACTCTCTACCCA TCCATCTCTCACTCACCC /3BIO/ – 3’ | (Rodgers and Woodson, 2019) | N/A |
| SA5_aadU35: 5’ –CCTGTGTCCTGTGTGTCCTGTCCAAAGTGTGTCG/iAmMC6T/CC – 3’ | (Rodgers and Woodson, 2019) | N/A |
| T7Pro_Tether_FOR: 5’ –GATCCTAATACGACTCACTATAGGGTGAGTGAGAGATGGATGGGTAGAGAGTTAGTAGTA – 3’ | (Rodgers and Woodson, 2019) | N/A |
| T7A1_FOR: 5’ – TATCAAAAAGAGTATTGACTTAAAGTCTAACCTATAGGATACTTACAGCC – 3’ | This study | N/A |
| Term_REV_29_aadT25: 5’ – CAAAAAACCCCTCAAGACCCGTT/iAmMC6T/AGAGG – 3’ | This study | N/A |
| rpoSREV2_dT180_dUx2: 5’ – AAA/ideoxyU/AACGCAGCGGG/ideoxyU/T/iAmMC6T/ACGGATTTCC – 3’ | This study | N/A |
| rpoSFOR2_dUx2_197: 5’ – ACCCGC/ideoxyU/GCGTTATT/ideoxyU/GCCGCAGCGATAAATCG – 3’ | This study | N/A |
| RpoS301_REV_aadT121_dU130: 5’ – ATGCAAGCGTGTTGAACTGG/ideoxyU/TCCGGTGC/iAmMC6T/ACCC – 3’ | This study | N/A |
| RpoS301_FOR_dU150: 5’ – ACCAGTTCAACACGCTTGCA/ideoxyU/UTTTGAAATTCG – 3’ | This study | N/A |
| RpoS_REV_SA5_nt432: 5’ – GTGTCCTGTCCAAAGTGTGTCGTCCTGCAAGCGTGTTGAACTGGTTCCGGTGCTACCC – 3’ | This study | N/A |
| RpoS_REV2_SA5_NIS_nt562: 5’ – GTGTCCTGTCCAAAGTGTGTCGTCCGACGGAACATTCAAGCAAAAGCCTGGTTCC – 3’ | This study | N/A |
| Recombinant DNA | ||
| pET21b-EcHfq | (Zhang et al., 2002) | N/A |
| pAR1219-T7RNAP | (Davanloo et al., 1984) | N/A |
| Sequences for linear DNA templates listed in Table S2 | This study | N/A |
| Software and algorithms | ||
| Single-molecule fluorescence acquisition software (smCamera) | Ha Lab, Custom made | https://github.com/Ha-SingleMoleculeLab/Data-Aquisition |
| MATLAB | Mathworks | R2022a |
| GLIMPSE/IMSCROLL | (Friedman and Gelles, 2015) | N/A |
| FIJI | (Schindelin et al., 2012) | N/A |