

Abstract
Presented is a light-promoted C–C bond forming reaction yielding sulfone and phosphate derivatives at room temperature in the absence of metals or photoredox catalyst. This transformation proceeds in neat conditions through an auto-oxidation mechanism which is maintained through the leaching of trace amounts of O2 as sole green oxidant.
Introduction
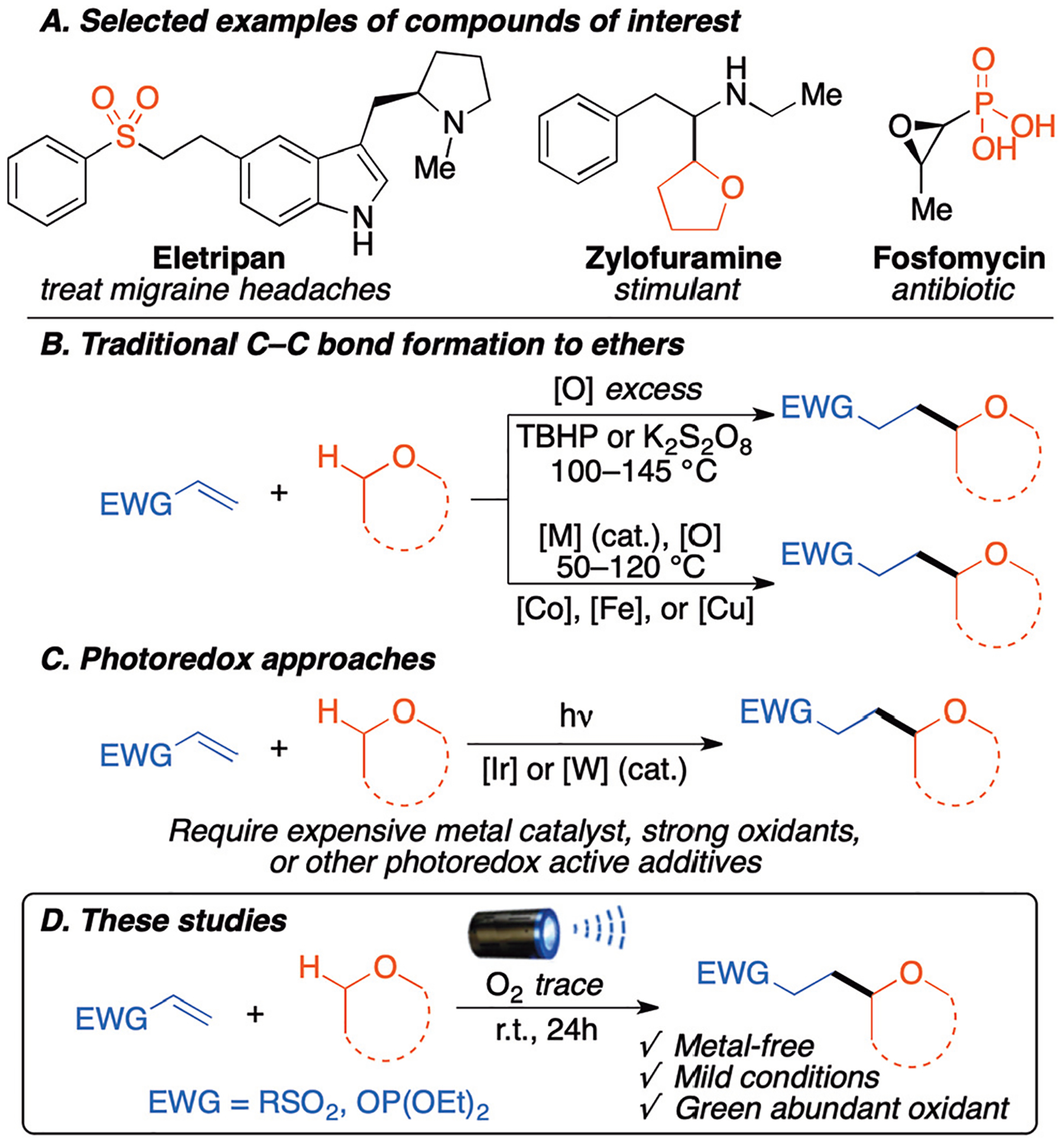
Sulfone and phosphate moieties are of great interest in medicinal and biological chemistry.1 Sulfone derivatives have attracted widespread attention since many exhibit biological activity and are found in pharmaceutical compounds.1a–i Similarly, many alkyl phosphate derivatives such as fosfomycin have been used as antibacterial compounds (Scheme 1A).1j–o
Scheme 1.
A. Examples of FDA approved drugs containing groups of interest. B. Traditional C–H activations of ethers. C. Photoredox methods to C–C bond formation of ethers. D. Presented work.
Cyclic ethers are another molecular moiety of interest in medicinal chemistry. In particular, the tetrahydrofuran (THF) motif is a valuable building blocks ubiquitous in natural products, bioactive molecules, and pharmaceuticals.2 Thus selective C–H functionalization of ethers at their α-position has attracted considerable interest.3 Recently, many selective C–C bond forming transformations have been developed.4,5 However, these methodologies always require high temperature, transition metal catalysts, or a stoichiometric amount of peroxides (Scheme 1B).4 Photoredox-catalyzed C–H functionalization of ethers has also been developed to enable selective C–C bond formations, but these photocatalysts can be costly and also contain transition-metals (Scheme 1C).5
Despite all these available technologies, the need for greener and more environmentally friendly approaches to C–H functionalization has been identified as one of the top three research areas that still require advancement by the ACS.6 Therefore, there is a dearth of simple and metal-free methodologies that use green and abundant reagents for C–H activation.
Atmospheric oxygen (O2) is an abundant and sustainable oxidant that has the potential to selectively activate C–H bonds. It has been recognized as an ideal oxidant for many oxidative transformations and has been used for the industrial production of commodity chemicals.7 Auto-oxidation has attracted considerable attention in recent years, since it does not require any additional reagents or catalysts. Molecular oxygen can directly interact with certain C–H bonds to generate carbon radicals without any additional initiator species or additives, which allows for C–H functionalization through reaction of transient radical species.8
Our interest in radical-base C–H functionalization of ethers3m and photo-induced processes,9 recently led us to the development of a regioselective amination of ethers using N-haloimides and LiOtBu.9a Herein, we report the first metal-free selective photochemical C–C bond forming reaction between 5-membered cyclic ethers and vinyl sulfones and vinyl phosphates in which trace amounts of oxygen (O2) act as sole oxidant under blue light (Scheme 1D). This method proceeds using neat conditions in the absence of any additives, making it the mildest and most environmentally benign C–C bond forming reaction for these substrates.
Results and discussion
Our initial studies aimed at the selective C–H α-functionalization of THF with the commercially available phenyl vinyl sulfone 1a as model substrates to optimize the reaction (Table 1). Firstly, we investigated the influence of Eosin Y, a known metal-free photocatalyst, in presence of potassium carbonate as base in DCM under blue LED light irradiation (entry 1). To our delight, the desired product 3a was obtained in 76%.
Table 1.
Optimization of the reaction and its conditionsa
| ||||
|---|---|---|---|---|
| Entrya | Additives (mol%) | Base (equiv) | Solvent | Yieldb (%) |
| 1 | Eosin Y (1%) | K2CO3 (2.0) | CH2Cl2 | 76 |
| 2 | — | K2CO3 (2.0) | CH2Cl2 | 40 |
| 3 | Eosin Y (1%) | — | CH2Cl2 | 50 |
| 4 | — | — | CH2Cl2 | 72 |
| 5 | — | — | CH2Cl2 | Tracec |
| 6 | — | — | CH2Cl2 | 40d |
| 7 | — | — | PhCH3 | Trace |
| 8 | — | — | MeCN | Trace |
| 9 | — | — | Acetone | Trace |
| 10 | — | — | Neat | 95(92)e |
| 11 | — | — | Neat | n.d.f |
| 12 | — | — | Neat | 80%g |
| 13 | — | — | Neat | 84%h |
| 14 | — | — | Neat | 86%i |
| 15 | — | — | Neat | 35%j |
Reaction conditions: 1a (0.2 mmol), THF 10 equivalent, solvent 1 mL, base 2.0 equivalent, room temperature, under argon atmosphere were irradiated with 40 W LED lamp (440 nm) for 24 h.
Yields are based on 1a, determined by 1H-NMR using dibromomethane as the internal standard.
Dark.
Open to air.
Isolated yields, 1 mL THF.
Reaction performed under 100% oxygen atmosphere.
Open to air conditions.
Under nitrogen atmosphere containing 1% oxygen.
Positive pressure of argon.
Freshly distilled THF, 24 hours, 440 nm blue LED.
However, control experiments quickly revealed that a photocatalyst and a base were not required for this transformation to proceed. Indeed, in the absence of either the photocatalyst (entry 2), the base (entry 3), or both (entry 4) the desired product was formed in 40%, 50%, and 72% yields, respectively. Blue light irradiation remained essential for the generation of the desired product even in the absence of a photocatalyst and base (entry 5). It is important to note that an open-to-air reaction had a detrimental effect in yield, generating the desired product 3a in only 40% (entry 6). We presume that high concentrations of atmospheric oxygen (O2) leads to the quenching of the generated C-centered THF radicals and favours the formation of oxidized products,8e,10 in lieu of promoting a radical chain reaction with the vinyl sulfone. Finally, the solvent has a significant effect on this transformation; while dichloromethane (CH2Cl2) greatly promoted this process, other solvents, such as toluene, acetonitrile, and acetone, were not effective, and all the starting material 1a remained unreacted (entries 7–9). Performing the reaction in neat THF (1 mL) without the use of any additives provided the best overall conditions with 95% NMR yield of the desired product, further reducing the environmental footprint of the reaction (entry 10). To assess the effects of O2 in the reaction (entry 11), we attempted the reaction using an oxygen balloon; no product was detected under those conditions, which confirms that excess oxygen leads to undesired side oxidations of the ether moiety and leaves the starting vinyl sulfone unreacted. Under an open atmosphere (18% oxygen) the reaction afforded the desired product in 80% yield (entry 12). Using a nitrogen atmosphere containing 1% O2 (entry 13) gave product 3a in 84% yield, while using a positive pressure of argon in the system (entry 14) generated the desire product in 86% yield. These results indicate that trace amounts of oxygen (<1%) and peroxides (2–5 ppm) in the system are sufficient to initiate and maintain the reaction. Finally, using freshly distilled THF (containing <0.5 ppm peroxide, see ESI S21 and S22†) under identical conditions only gave product 3a in 35% yield (entry 15).
Having established the optimized reaction conditions, we started exploring the substrate scope with different vinyl sulfones, while using THF as solvent and ether source (Scheme 2). para-Substituted phenyl vinyl sulfones with electron-donating (Me, OMe, tBu) afforded the desired products in good to excellent yields (3a–d, 67–95%). Electron-withdrawing groups in the para-position (F, Br, Cl and CF3) were also well tolerated with yields ranging from 68–80%. However, 4-nitrophenyl vinyl sulfone was the exception and did not afford desired product 3h. Furthermore, ortho-substitution afforded products 3k and 3l in 56% and 75% yield, respectively. We were delighted to see that high-sterically hindered 2,4,6-trimethylphenyl vinyl sulfone reacted smoothly to afford the desired product 3m with 56% yield. The N-(4-(vinylsulfonyl) phenyl)acetamide was well tolerated in this reaction affording product 3n in good yield (70%). Furthermore, the shift to alkyl vinyl sulfones gave products 3o and 3p in excellent yields (84% and 89%). Similarly, divinyl sulfones react to generate monofunctionalized product 3q in 68% yield; di-functionalized product was not observed under these reaction conditions. Of particular interest to medicinal chemists, sulfonamides and sulfonate esters were well tolerated and afforded products 3r–x in moderate to good yields (45–82%). Gratifyingly, this reaction is compatible with tertiary sulfonamides such as N,N-dimethyl, N,N-diisopropyl and 4-methylpiperidine (3r, 3s, and 3t). Various substituted vinyl sulfonate esters afforded the corresponding products in good yields (3u, 3v, 3w, and 3x). It is important to highlight that the reaction conditions appear to be mild enough to tolerate good leaving groups such as phenol (3u, 78%) that could be easily hydrolysed, and highly oxygenated sugar substrates (3x, 71%) that could participate in the radical process. The vinyl sulfonamides and sulfonates can be conveniently obtained from commercially available 2-chloroethanesulfonyl chloride and the corresponding amine or alcohol in the presence of Et3N (see ESI†).
Scheme 2.
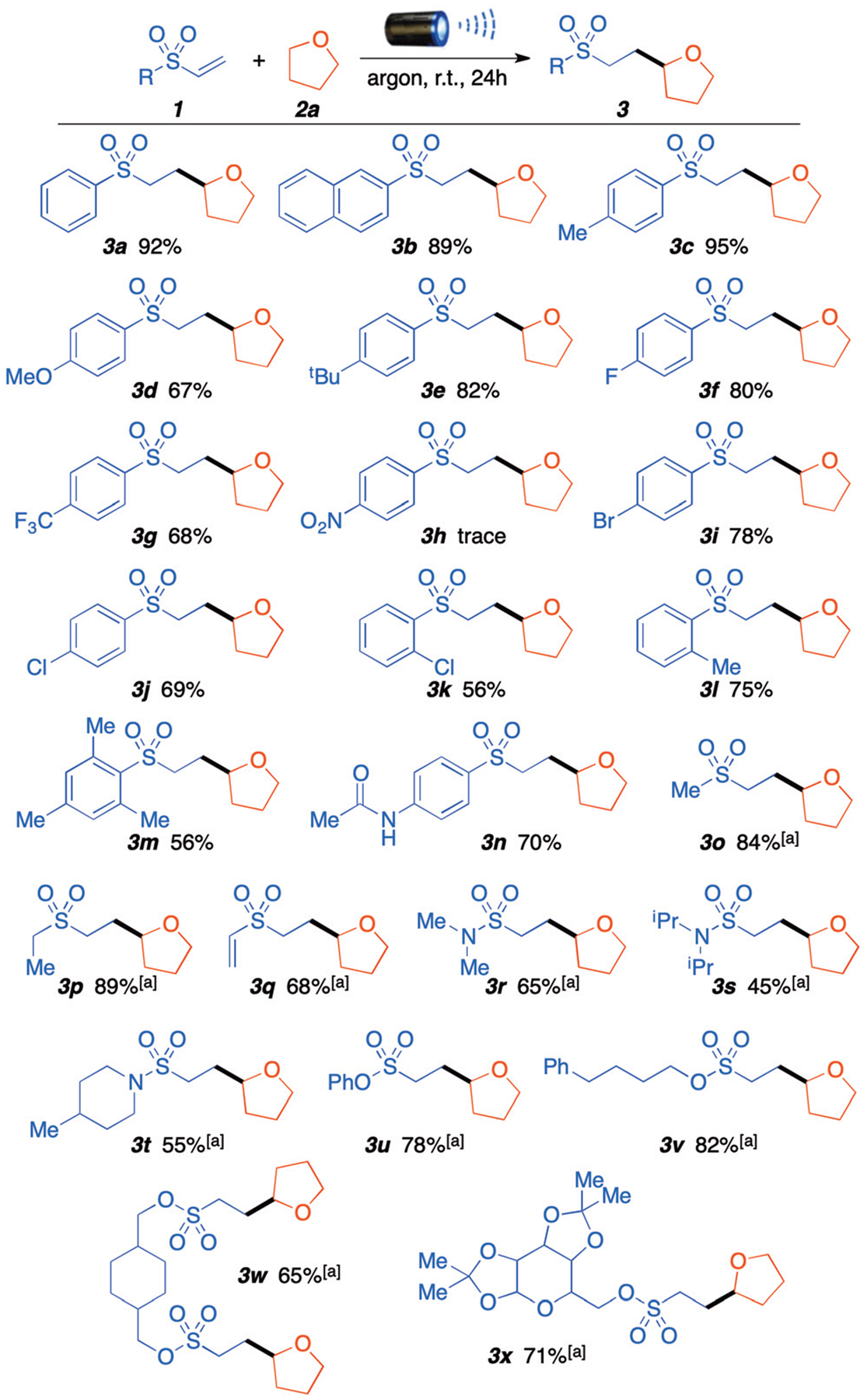
Substrate scope with differently substituted vinyl sulfones. Standard reaction conditions: THF 1 mL, vinylsulfone 0.2 mmol, 440 nm blue light, 24 hours; yields refer to isolated products. a THF 1 mL, vinylsulfone 0.2 mmol, 390 nm purple light, 24 hours.
We continued to explore the reaction performance using different ethers (Scheme 3). We were pleased to find that, along with THF, other 5-member-ring ether derivatives such as 2 methyl–THF, 1,3-dioxolane, 2-methyl-1,3-dioxolane, and tetrahydrothiophene are all suitable for this protocol, obtaining the corresponding product 4a–f in good to excellent yields. 2-Methyl–THF generated a single regioisomer of product 4a and 4e at the least sterically hindered α-position, which mimics previous observations for radical C–H activation of ethers. On the other hand, 1,3-dioxolane generated an inseparable mixture of regioisomer for products 4b and 4f in a (5 : 1) and (10 : 1) distributions, respectively. Interestingly, 2-methyl-1,3-dioxolane, which has a tertiary acetylic position, afforded desired product 4c (44%) and hydrolyzed product 4c′ (22%). Unfortunately, six membered cyclic ethers, such as tetrahydropyran and 1,4-dioxane, as well as acyclic ethers, such as 1,2-diethyl ether and methyl tert-butyl ether do not fit in this reaction system. Possible explanations are a decrease in the hydrogen atom abstraction rate due to ring size and stereoelectronic factors influence, and ring size bond dissociation energy.8a,11
Scheme 3.
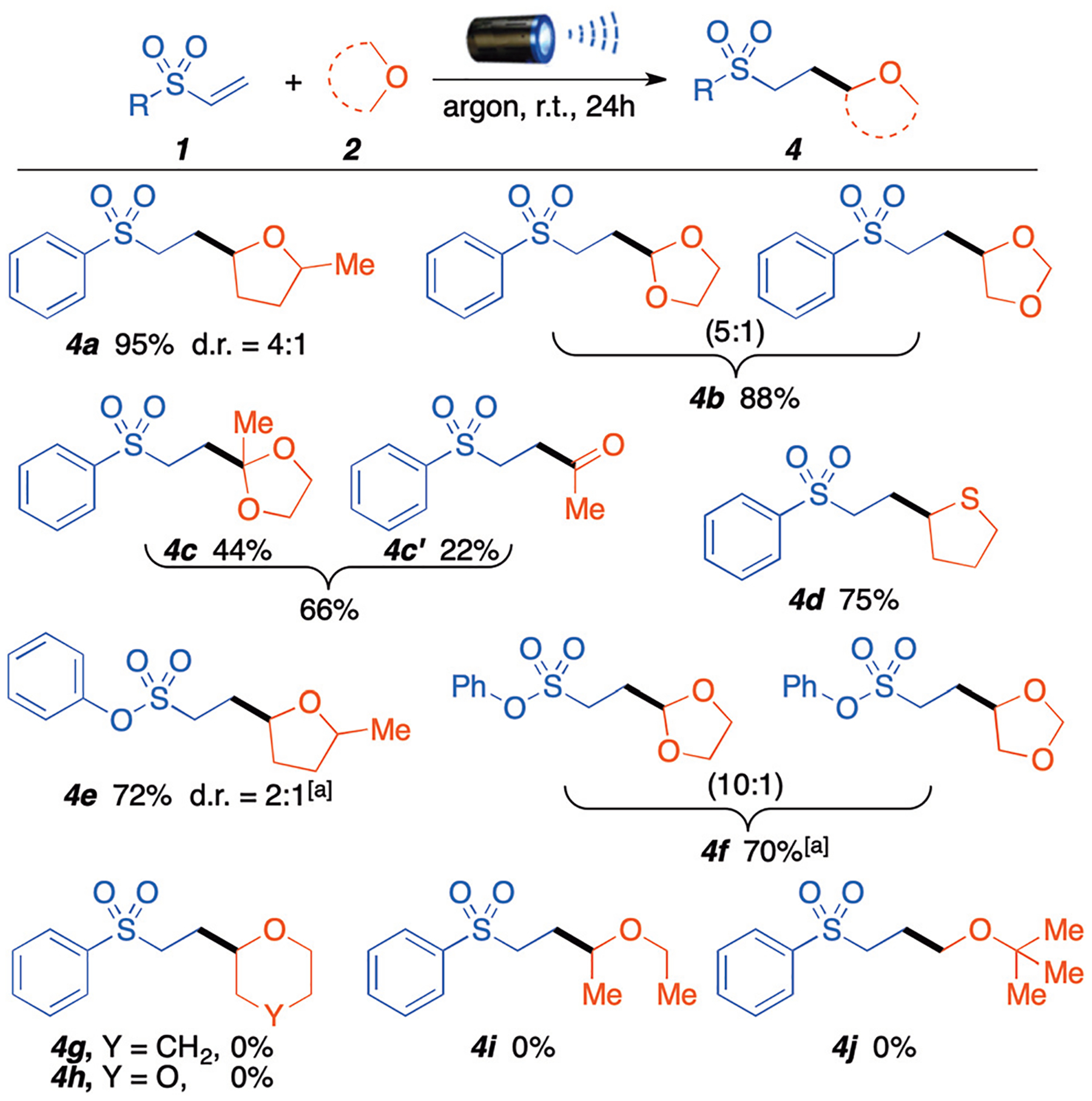
Substrate scope for different ether derivatives. Standard reaction conditions: ethers 1 mL, vinylsulfone 0.2 mmol, 440 nm blue light, 24 hours. a Ethers 1 mL, vinylsulfone 0.2 mmol, 390 nm purple light, 24 hours. All yields are from isolated products.
Further exploration of the substrate scope for this C–H activation coupling revealed that this methodology could also be extended to other Michael acceptors and electron-deficient heteroarenes (Scheme 4). Under standard reaction conditions, quinoxaline was coupled to THF to give product 5a in low yield (35%). Diethyl azodicarboxylate (DEAD) was also successfully coupled with in good yield (5b, 58%). Then, we switched our attention to vinyl phosphonate esters. We were pleased to observe that THF and 2-methyl-1,3-dioxolane afforded products 5c and 5d in 62% yield. Interestingly, product 5d was isolated as a single regioisomer distinct from when using vinyl sulfone. Similarly, 2-methyl–THF was coupled to give an inseparable mixture of regioisomer (5e) in a (1 : 2) ratio between both α-positions in good yield (61%). Finally, 1,3-dioxolane also reacted with vinyl phosphate ester to give mixture 5f in good yield (68%).
Scheme 4.
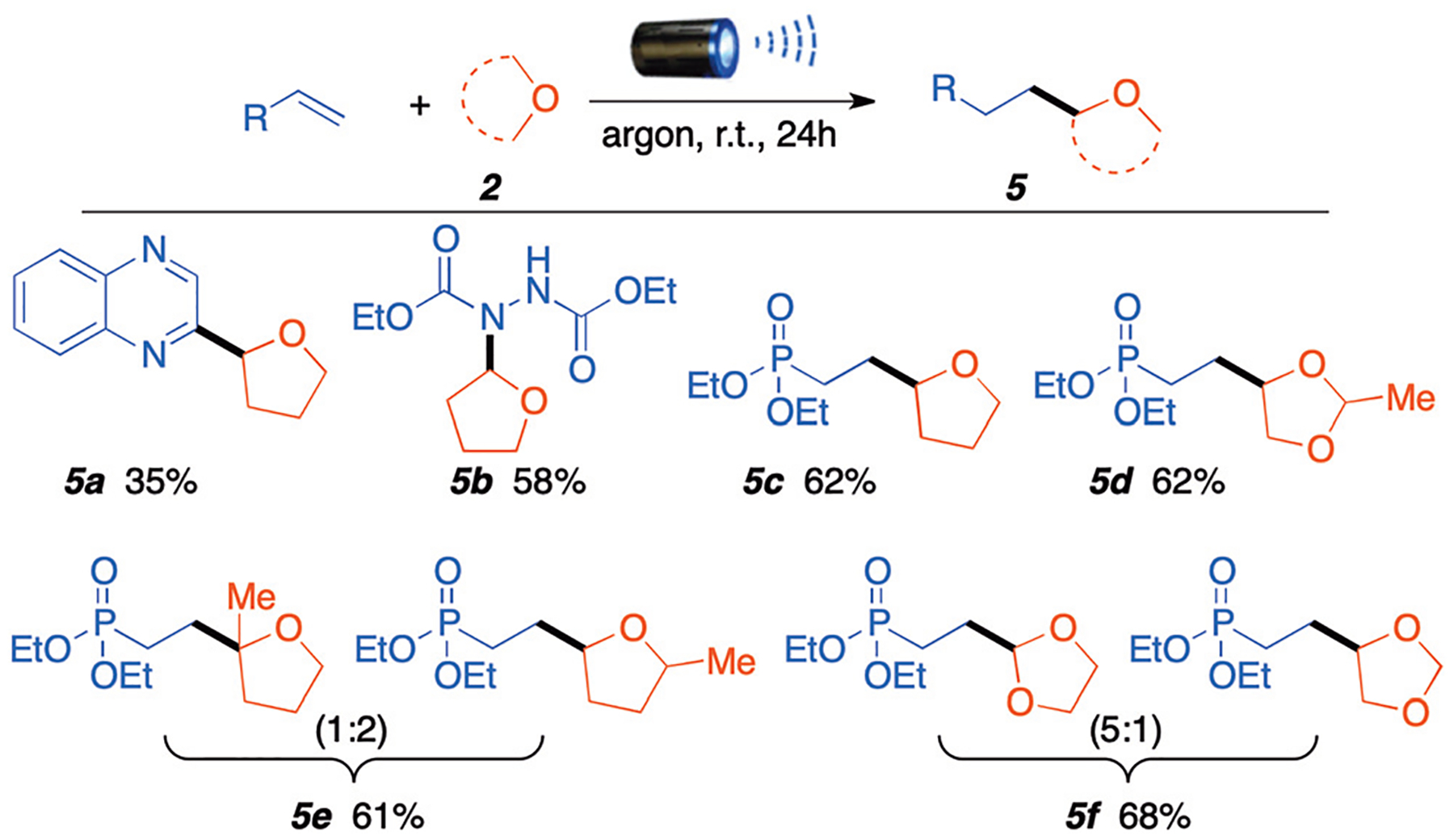
Substrates scope for different Michael acceptors. Reaction conditions: ethers 1 mL, Michael acceptor 0.2 mmol, under 390 nm purple light, 24 hours. All yields are from isolated products.
To gain insight into the reaction mechanism, a series of control experiments were investigated. As expected, when the radical scavengers were added to the reaction mixture, the transformation was halted (Scheme 5A). The use of 2,2,6,6-tetramethyl-piperidinyloxyl (TEMPO) afforded only trace amounts of the desired product and THF radical was trapped and observed via GC-MS as a TEMPO adduct (see ESI, S24†). The use of 2,6-di-tert-butyl-4-methylphenol (BHT) also completely quenched the reaction. Finally, 1,1-diphenylethylene also afforded the THF radical adduct and the desired product 3a was not detected. These experiments indicate that a radical process is most likely involved in this transformation. Given the possibility of radical chain processes, we performed light on/off experiments (see ESI, S23†) to evaluate whether product was being formed in the absence of light once the radical process was initiated. Indeed we observed 4–5% product formation in the dark, suggesting a short-lived radical chain process.
Scheme 5.
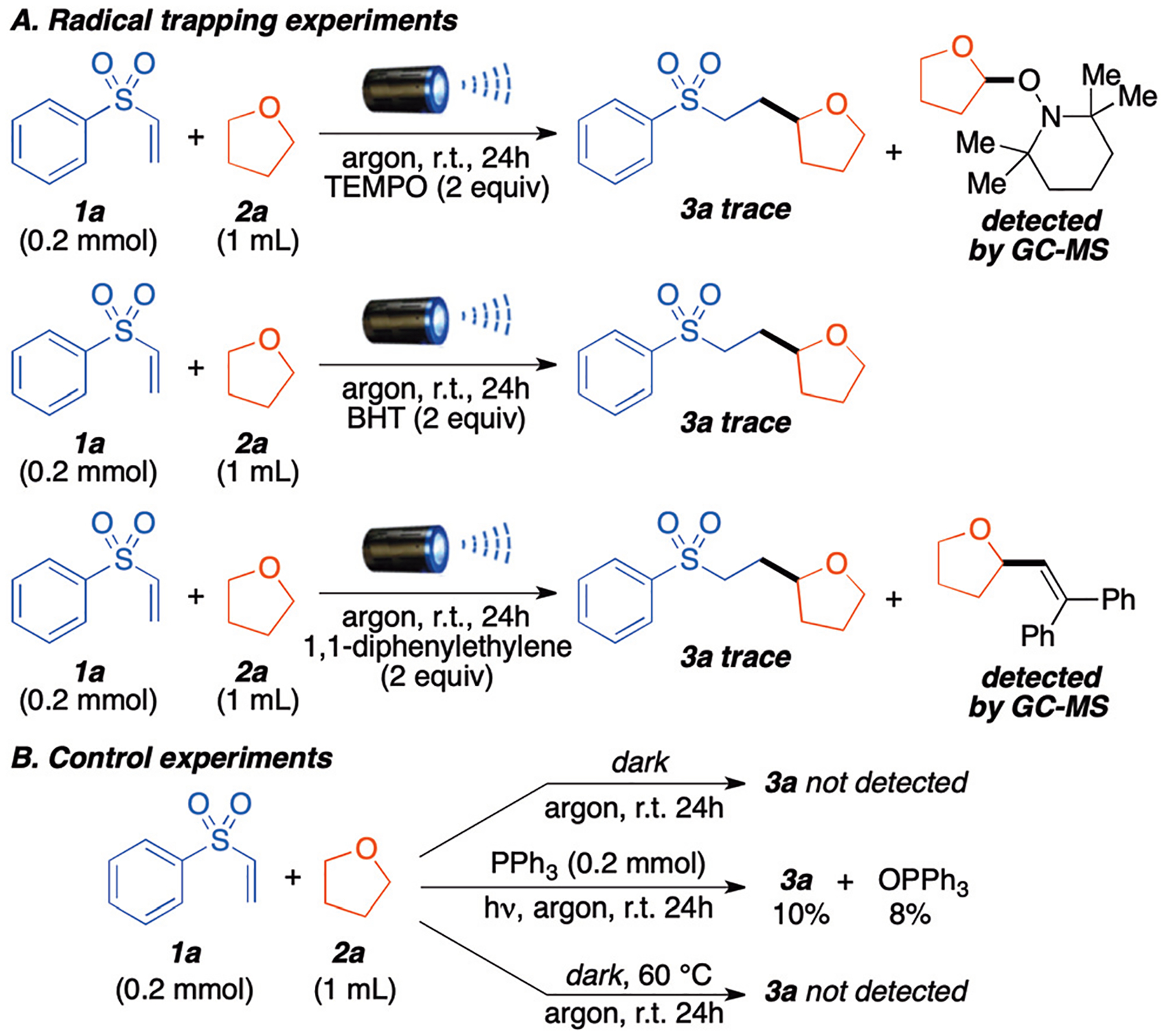
Mechanistic study: A. Reaction in the presence of radical scavenger. B. Control experiments.
Additional control experiments (Scheme 5B) were performed in the dark, using heat, and by adding triphenylphosphine (PPh3) in the reaction mixtures. When the reaction was carried in the dark or at 60 °C, the desired product 3a was not observed. Initially, we believed that the vinyl sulfone may be absorbing visible light and serving as a sensitizer to excite triplet oxygen (3O2) into reactive singlet oxygen (1O2). However, our UV-Vis spectroscopic measurements did not show absorption by the vinyl sulfone within the light spectrum used in the reaction, thereby ruling out the triplet state hypothesis (ESI S21†). Then, we turned our attention to the possibility of peroxy-radical formation using the peroxide test strips (see ESI, S21†). The results demonstrate that blue light irradiation significantly increases the generation of ethereal radicals over the reaction time. To verify this, PPh3 was added to the reaction mixture, as it is known to react with peroxides to form triphenylphosphine oxide (OPPh3). Under normal reaction conditions 8% OPPh3 formation was observed after 24 h, which suggests that small amounts of peroxides are generated throughout the reaction through the leaching of O2 in the reaction. Indeed, while the reactions are set up under argon, they are not maintained under a positive pressure of argon. We performed a series of experiments to measure oxygen leaching over the course of the reaction (see ESI, S22†) and we determined that 0.004 mmol of O2 leach every 12 h. Finally, we measured peroxides in the THF used for the reaction prior to light irradiation. Bottled THF used contains 2–5 ppm peroxides, while freshly distilled THF contained <0.5 ppm (see ESI, S22†). These trace amounts of ethereal peroxides are responsible for the initiation of the reaction.
Based on literature reports8d,12 and our experimental results, we proposed a possible reaction mechanism as shown in Scheme 6. First, trace amounts of ether peroxides A homolytically cleave in presence of blue light to generate reactive oxygen species in the initiation step. These hydroxyl radicals then react with THF to generate carbon-centered radical B. Radical intermediate B then reacts with vinyl sulfone 1a to form species C. Through a radical chain process, species C can further abstract a H-atom from THF to form the desired product 3a and regenerate ethereal radical B.
Scheme 6.
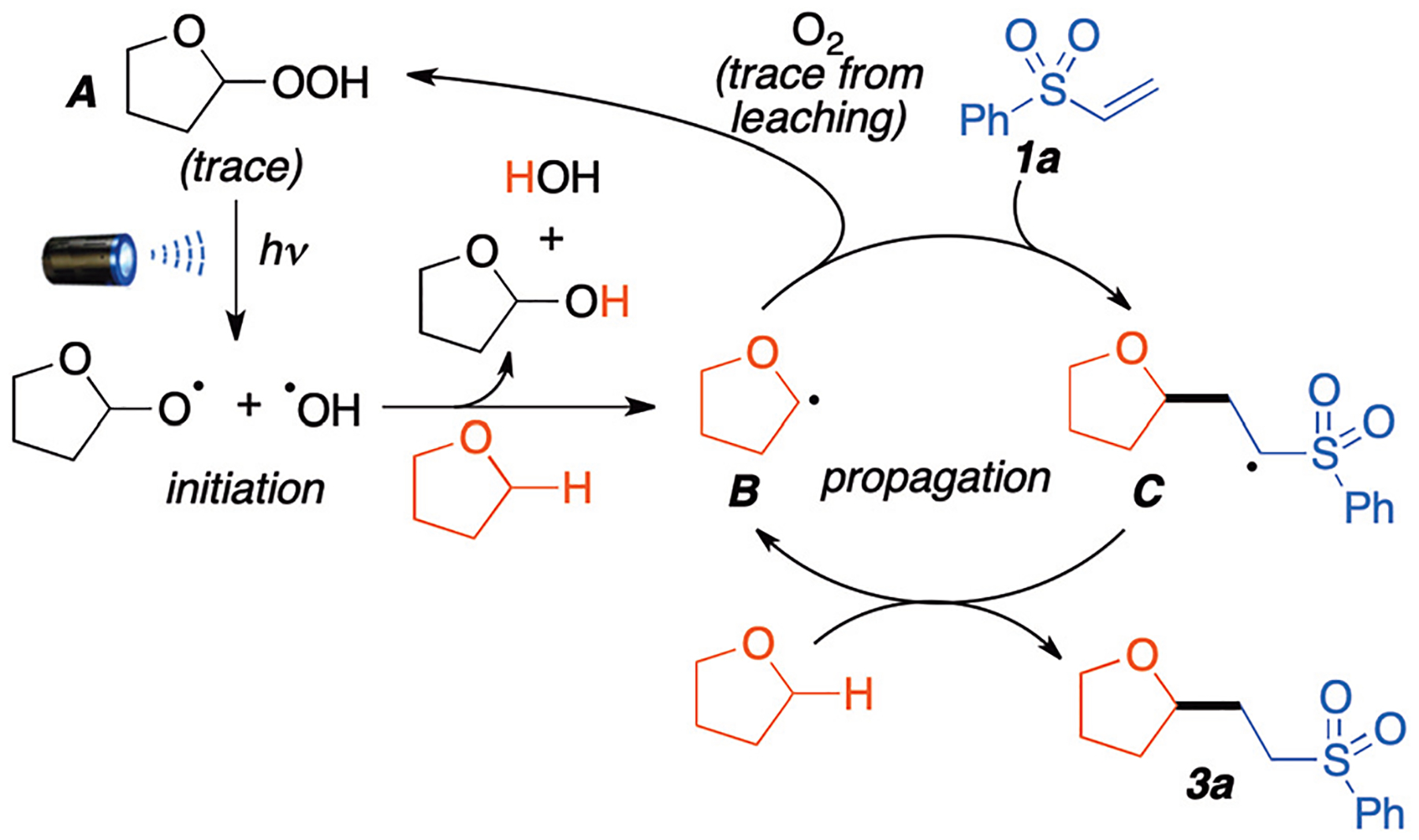
Proposed mechanism.
As shown in the table of optimization, excess oxygen negatively impacted the yield of this reaction (Table 1, entries 6 and 11). This is most likely due to intermediate B reacting with O2 to generate peroxyethers and oxidized species8e,10 instead of reacting with Michael acceptors. However, trace O2 from leaching at a rate of 0.004 mmol every 12 h is probably responsible for continued generation of trace peroxide that maintains the process going. Finally, reaction initiation due to peroxides was demonstrated through a series of experiments (see Table 1, entry 15 and ESI, S6†), which show that amounts as low as 2–5 ppm of peroxides are able to initiate the reaction.
Conclusions
In conclusion, we have developed an environmentally benign and efficient selective photochemical C–C bond forming reaction between cyclic ethers and vinyl sulfones utilizing trace amounts of aerobic oxygen as sole green oxidant under blue light at room temperature. Broad scopes of vinyl sulfone are compatible with cyclic ethers and provided desired products in good to excellent yields. This method is also applicable to vinyl phosphonate esters and other Michael acceptors. Visible blue light was utilized to promote the reaction without transition-metals or photocatalysts. Further studies on the detailed reaction mechanism and applications are currently ongoing in our laboratories.
Supplementary Material
Acknowledgements
This publication was made possible with support from the National Institute of Dental & Craniofacial Research grant number 1R21DE029156-02.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03482k
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.(a) Feng M, Tang B, Liang H and Jiang X, Curr. Top. Med. Chem, 2016, 16, 1200; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang N, Saidhareddy P and Jiang X, Nat. Prod. Rep, 2020, 37, 246; [DOI] [PubMed] [Google Scholar]; (c) Aziz J, Messaoudi S, Alami M and Hamze A, Org. Biomol. Chem, 2014, 12, 9743; [DOI] [PubMed] [Google Scholar]; (d) Meadows D and Gervay-Hague J, Med. Res. Rev, 2007, 26, 793; [DOI] [PubMed] [Google Scholar]; (e) Doherty W, Adler N, Knox A, Nolan D, McGouran J, Nikalje A, Lokwani D, Sarkate A and Evans P, Eur. J. Org. Chem, 2017, 175; [Google Scholar]; (f) Carmine A, Brogden RN and Heel RC, Drugs, 1982, 24, 85; [DOI] [PubMed] [Google Scholar]; (g) He F, Mai LH, Longeon A, Copp B, Loaëc N, Bescond A, Meijer L and Bourguet-Kondracki M, Mar. Drugs, 2015, 13, 2617; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) IIardi E, Vitaku E and Njardarson J, J. Med. Chem, 2014, 57(7), 2832; [DOI] [PubMed] [Google Scholar]; (i) Scott K and Njardarson J, Top. Med. Chem, 2018, 376, 5; [DOI] [PubMed] [Google Scholar]; (j) Palte M, Davis A, McGrath N, Spiegel C and Raines R, ChemMedChem, 2012, 7, 1361; [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) McGrath J, Chin J and Quinn J, Nat. Rev. Microbiol, 2013, 11, 412; [DOI] [PubMed] [Google Scholar]; (l) Horsman G and Zechel D, Chem. Rev, 2017, 117, 5704; [DOI] [PubMed] [Google Scholar]; (m) Chin J, McGrath J and Quinn J, Curr. Opin. Chem. Biol, 2016, 31, 50; [DOI] [PubMed] [Google Scholar]; (n) Yu H, Yang H, Shi E and Tang W, Med. Drug Discovery, 2020, 8, 100063; [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Yeganeh-Sefidan F, Ghotaslou R, Akhi M, Sadeghi M, Mohammadzadeh-Asl Y and Baghi H, Iran. J. Microbiol, 2016, 8, 125. [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Miles S, Marsden S, Leatherbarrow RJ and Coates W, J. Org. Chem, 2004, 69, 6874; [DOI] [PubMed] [Google Scholar]; (b) Lu QL, Harmalkar D, Choi Y and Lee K, Molecules, 2019, 24, 3778; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nakata T, Chem. Rev, 2005, 105(12), 4314; [DOI] [PubMed] [Google Scholar]; (d) Yeung K and Paterson L, Chem. Rev, 2005, 105, 4237; [DOI] [PubMed] [Google Scholar]; (e) Wu JL, Li N, Hasegawa T, Sakai J, Kakuta S, Tang W, Oka S, Kiuchi M, Ogura H, Kataoka T, Tomida A, Tsuruo T and Ando M, J. Nat. Prod, 2005, 68, 1656; [DOI] [PubMed] [Google Scholar]; (f) Piper B, Templin T, Kirsner R and Birk T, Wound Repair Regener, 2009, 17, 485; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Lorente A, Lamariano-Merketegi J, Albericio F and Álvarez M, Chem. Rev, 2013, 113, 4567; [DOI] [PubMed] [Google Scholar]; (h) Tikad A, Delbrouck J and Vincent S, Chem. – Eur. J, 2016, 22, 9456; [DOI] [PubMed] [Google Scholar]; (i) Mihovilovic M, Bianchi D and Rudroff F, Chem. Commun, 2006, 3214; [DOI] [PubMed] [Google Scholar]; (j) Bai Y, Davis D and Dai M, Angew. Chem., Int. Ed, 2014, 53, 6519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Zhang S, Zhang F and Tu Y, Chem. Soc. Rev, 2011, 40, 1937; [DOI] [PubMed] [Google Scholar]; (b) Cheng K, Huang L and Zhang Y, Org. Lett, 2009, 11, 2908; [DOI] [PubMed] [Google Scholar]; (c) Du P, Li H, Wang Y, Cheng J and Wan X, Org. Lett, 2014, 16, 6350; [DOI] [PubMed] [Google Scholar]; (d) Liu D, Liu C, Li H and Lei A, Chem. Commun, 2014, 50, 3623; [DOI] [PubMed] [Google Scholar]; (e) Jin L, Wan L, Feng J and Cai C, Org. Lett, 2015, 17, 4726; [DOI] [PubMed] [Google Scholar]; (f) Kawade R, Huple D, Lin R and Liu R, Chem. Commun, 2015, 51, 6625; [DOI] [PubMed] [Google Scholar]; (g) Sølvhøj A, Ahlburg A and Madsen R, Chem. – Eur. J, 2015, 21, 16272; [DOI] [PubMed] [Google Scholar]; (h) Yamada K, Fujihara H, Yamamoto Y, Miwa Y, Taga T and Tomioka K, Org. Lett, 2002, 4, 3509; [DOI] [PubMed] [Google Scholar]; (i) Yamada K, Yamamoto Y and Tomioka K, Org. Lett, 2003, 5, 1797; [DOI] [PubMed] [Google Scholar]; (j) Nielsen M, Shields B, Liu J, Williams MJ, Zacuto M and Doyle A, Angew. Chem., Int. Ed, 2017, 56, 7191; [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Guo S, Kumar P and Yang M, Adv. Synth. Catal, 2017, 359, 2; [Google Scholar]; (l) Zhang Y and Li C, Angew. Chem, 2006, 118, 1983; [Google Scholar]; (m) Gasonoo M, Thom Z and Laulhé S, J. Org. Chem, 2019, 84, 8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Liu D, Liu C, Li H and Lei A, Angew. Chem., Int. Ed, 2013, 52, 4453; [DOI] [PubMed] [Google Scholar]; (b) Chu X, Meng H, Zi Y and Xu X, Chem. Commun, 2014, 50, 9718; [DOI] [PubMed] [Google Scholar]; (c) Ambala S, Thatikonda T, Sharma S, Munagala G, Yempalla K, Vishwakarma R and Singh P, Org. Biomol. Chem, 2015, 13, 11341; [DOI] [PubMed] [Google Scholar]; (d) Li Y, Wang M, Fan W, Qian F, Li G and Lu H, J. Org. Chem, 2016, 81(23), 11743; [DOI] [PubMed] [Google Scholar]; (e) Liu S, Liu A, Zhang Y and Wang W, Chem. Sci, 2017, 8, 4044; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhou J, Zou Y, Zhou P, Chen Z and Li J, Org. Chem. Front, 2019, 6, 1594; [Google Scholar]; (g) Niu B, Zhao W, Ding Y, Bian Z, Pittman C Jr., Zhou A and Ge H, J. Org. Chem, 2015, 80(14), 7251; [DOI] [PubMed] [Google Scholar]; (h) Xie Z, Cai Y, Hu H, Lin C, Jiang J, Chen Z, Wang L and Pan Y, Org. Lett, 2013, 15(17), 4600; [DOI] [PubMed] [Google Scholar]; (i) Correa A, Fiser B and Gómez-Bengoa E, Chem. Commun, 2015, 51, 13365; [DOI] [PubMed] [Google Scholar]; (j) Guo S-R, Yuan Y-Q and Xiang J-N, Org. Lett, 2013, 15, 4654; [DOI] [PubMed] [Google Scholar]; (k) Sun W, Xie Z, Liu J and Wang L, Org. Biomol. Chem, 2015, 13, 4596; [DOI] [PubMed] [Google Scholar]; (l) Ji P, Liu Y, Xu J, Luo W, Liu Q and Guo C, J. Org. Chem, 2017, 82, 2965; [DOI] [PubMed] [Google Scholar]; (m) Liu Y, Wang Q, Zhou C, Xiong B, Zhang P, Yang C and Tang K, J. Org. Chem, 2017, 82, 7394. [DOI] [PubMed] [Google Scholar]
- 5.(a) Jin J and MacMillan D, Angew. Chem., Int. Ed, 2015, 54, 1565; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Devari S and Shah B, Chem. Commun, 2016, 52, 1490; [DOI] [PubMed] [Google Scholar]; (c) Ravelli D, Montanaro S, Zema M, Fagnoni M and Albini A, Adv. Synth. Catal, 2011, 353, 3295; [Google Scholar]; (d) Yamada K, Fukuyama T, Fujii S, Ravelli D, Fagnoni M and Ryu I, Chem. – Eur. J, 2017, 23, 8615; [DOI] [PubMed] [Google Scholar]; (e) Raviola C and Ravelli D, Synlett, 2019, 30, 803; [Google Scholar]; (f) Deng H-P, Zhou Q and Wu J, Angew. Chem., Int. Ed, 2018, 57, 12661; [DOI] [PubMed] [Google Scholar]; (g) Paul S and Guin J, Green Chem, 2017, 19, 2530; [Google Scholar]; (h) Jiang S, Tian X, Feng S, Li J, Li Z, Lu C, Li C and Liu W, Org. Lett, 2021, 23, 692. [DOI] [PubMed] [Google Scholar]
- 6.(a) Bryan MC, Dunn P, Entwistle D, Gallou F, Koenig S, Hayler JD, Hickey M, Hughes S, Kopach M, Moine G, Richardson P, Roschangar F, Steven A and Weiberth F, Green Chem, 2018, 20, 5082; [Google Scholar]; (b) Dalton T, Faber T and Glorius F, ACS Cent. Sci, 2021, 7(2), 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Campbell AN and Stahl S, Acc. Chem. Res, 2012, 45(6), 851; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bryliakov K-P, Chem. Rev, 2017, 117, 11406; [DOI] [PubMed] [Google Scholar]; (c) Liang Y and Jiao N, Acc. Chem. Res, 2017, 50, 1640. [DOI] [PubMed] [Google Scholar]
- 8.(a) Matsubara H, Suzuki S and Hirano S, Org. Biomol. Chem, 2015, 13, 4686; [DOI] [PubMed] [Google Scholar]; (b) Shamsabadi A and Chudasama V, Org. Biomol. Chem, 2019, 17, 2865; [DOI] [PubMed] [Google Scholar]; (c) Rawat D, Kumar R and Adimurthy S, Green Chem, 2020, 22, 6170; [Google Scholar]; (d) Di Tommaso S, Rotureau P, Crescenzi O and Adamo C, Phys. Chem. Chem. Phys, 2011, 13, 14636; [DOI] [PubMed] [Google Scholar]; (e) Sagadevan A, Hwang K and Su M, Nat. Commun, 2017, 8, 1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Pan L, Elmasry J, Osccorima T, Cooke MV and Laulhé S, Org. Lett, 2021, 23, 3389; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pan L, Cooke MV, Spencer A and Laulhé S, Adv. Synth. Catal, 2021, 363, DOI: 10.1002/adsc.202101052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Decker C and Jenkins AD, Macromolecules, 1985, 18(6), 1241. [Google Scholar]
- 11.(a) Malatesta V and Scaiano J, J. Am. Chem. Soc, 1981, 103(3), 61; [Google Scholar]; (b) Hallen RT, Gleicher G, Mahiou B and Clapp G, J. Phys. Org. Chem, 1989, 2, 367. [Google Scholar]
- 12.(a) Di Tommaso S, Rotureau P, Sirjean B, Fournet R, Benaissa W, Gruez P and Adamo C, Process Saf. Prog, 2014, 33, 64; [Google Scholar]; (b) Barks JM, Gilbert B, Parsons A and Upeandran B, Tetrahedron Lett, 2000, 41, 6249; [Google Scholar]; (c) Troisi L, Granito C, Ronzini L, Rosato F and Videtta V, Tetrahedron Lett, 2010, 51, 5980. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.