


Abstract
Hepatitis B viruses, or hepadnaviruses, are small DNA-containing viruses that replicate through reverse transcription. Their prototype, HBV, causes severe liver disease in humans. The hepadnaviral P protein is an unusual reverse transcriptase (RT) that initiates DNA synthesis by host-factor-dependent protein priming on a specific RNA stem–loop template, ɛ, yielding a short DNA oligonucleotide covalently attached to the RT. This priming reaction can be reconstituted with in vitro-translated duck hepatitis B virus (DHBV) P protein. No direct structural data are available for any P protein. However, P proteins share a number of conserved motifs with other polymerases. Box A contains an invariant bulky residue recently shown to be crucial for dNTP versus NTP discrimination in RTs and some DNA polymerases; its equivalent in DHBV P protein would be phenylalanine 451 (F451). Four mutants, containing glycine (F451G), alanine (F451A), valine (F451V) and aspartate (F451D), were therefore analyzed for their ability to utilize dNTPs and NTPs in in vitro priming. Priming efficiencies with dNTPs decreased with decreasing side chain size but GTP utilization increased; the wild-type enzyme was inactive with GTP. In the context of complete DHBV genomes, all mutant proteins were competent for RNA encapsidation, indicating the absence of global structural alterations. Because the function of the discriminatory residue depends on its specific spatial disposition this strongly suggests a similar architecture for the P protein dNTP-binding pocket as in other RTs.
INTRODUCTION
Hepatitis B viruses comprise a family of small enveloped DNA viruses that infect humans (1,2), some mammals like woodchucks and ground squirrels, and birds, e.g. Pekin ducks and grey herons. They all replicate their tiny 3.2 kb genome by reverse transcription (3,4) with the essential replication functions being provided by a virus encoded reverse transcriptase (RT) called P protein. Though hepadnaviral reverse transcription is fundamentally similar to that of retroviruses, it has several unique features (5,6). Initiation proceeds by protein priming rather than from a nucleic acid primer such as tRNA, and it strictly depends on cellular proteins, apparently including the heat shock proteins Hsp90 and p23 (7,8). This multi-protein complex binds to a stem–loop structure close to the 5′ end of the pregenomic RNA (pgRNA), ɛ, mediating pgRNA encapsidation (9,10) on the one hand, and initiation of DNA synthesis on the other (11–13). In the priming reaction, the active site of P protein is positioned over a bulge within the ɛ stem–loop such that the first dNTP becomes covalently attached, as dNMP, to a Tyr residue in the unique terminal protein (TP) domain of the P protein (14–16). These events are accompanied by major structural alterations in the upper part of the ɛ stem–loop (17,18) and the P protein (19). After two or three more nucleotides have been added, templated by the ɛ RNA (Fig. 1A), synthesis is arrested (20) and the resulting DNA oligonucleotide–P protein complex is translocated to a 3′-proximal RNA element for synthesis of a complete (–)-DNA strand.
Figure 1.

Replication initiation in hepatitis B viruses and sequence motifs conserved between hepadnaviral and retroviral RTs. (A) Schematic representation of the DHBV replication initiation complex. The secondary structure of the Dɛ RNA bound to P protein with its TP and polymerase/RNase H (pol/RH) domains is shown. The bulge within Dɛ contains the region that serves as template for DNA initiation. The 5′-terminal nucleotide of the DNA primer is covalently attached to a Tyr residue in TP. The cellular factors required for this priming reaction are not shown. (B) Alignment of the polymerase domains of DHBV and HBV P protein with that of HIV-1 and MoMLV RT. The linear representations (top) indicate the borders of the individual domains in DHBV and HBV P protein, and the relative positions of conserved motives 1 and 2, and A to E in the polymerase domain. Numbers are amino acid positions that, for HBV, correspond to subtype ayw. Individual sequences within the boxes are shown below their consensus motifs (23); h, hydrophobic residues; c, charged residues. Residues lining the dNTP pocket and the Mg2+ coordinating aspartates (gray background) in HIV-1 RT are boxed. F451 is highlighted by an arrowhead.
The mechanistic differences in retroviral replication are reflected in the primary structure of the P proteins (Fig. 1B). At ∼90 kDa they are substantially larger than retroviral RTs, mainly due to the presence of the additional, N-terminal TP domain, and a spacer region that connects TP to the polymerase and RNase H domains typical of all RTs (21). Although the P protein sequences are quite divergent even in these common domains, they contain several short signature boxes (22,23) that occur in the corresponding domains of other polymerases (Fig. 1B). Direct proof that these conserved primary sequence elements reflect a common structure is as yet not available because, in contrast to retroviral RTs, the P polypeptides, let alone enzymatically active complexes, cannot be produced in sufficient amounts for structural analyses.
Very recently, a molecular modeling study of the polymerase domain of HBV P protein has been reported that predicts a high extent of structural similarity to the corresponding domain of HIV-1 RT (24). Currently, however, the experimental evidence supporting this model is limited. Most mutational studies have concentrated on the box C motif containing the sequence Tyr-Met-Asp-Asp (YMDD) and confirmed the importance of the two Asp residues (25,26) which in other RTs are known to coordinate, together with a third acidic residue in box A, two essential metal ions (27). The participation of box C residues in active site formation is further underlined by the natural occurrence in this motif of mutations during chemotherapy of chronic hepatitis B in humans with nucleoside analogs such as 3′-thiacytidine (28). In contrast, few attempts have been made to establish the functional relevance of residues in the other boxes by mutagenesis. One reason is the limited interpretability of functional knock-outs induced by mutation: because P protein depends on various partners to be active, a comparably large proportion of amino acid replacements might act indirectly by affecting the interactions with the other essential components of the replication complex. Another concern is global misfolding.
Therefore, we sought a mutational target within P that would allow a gain rather than a loss of function. In addition, it should allow us to monitor, in vitro, the effects on genuine P protein activity. While no such assay is established for human HBV, the P protein of the related duck HBV (DHBV), in vitro translated in rabbit reticulocyte lysate (RRL), is able to initiate DNA synthesis from the authentic DHBV ɛ (Dɛ) RNA template (29–31). Based on these considerations we selected residue F451 in DHBV P protein as a target. F451 is part of box A, like box C common to all polymerases. DNA polymerases, including RTs, have a bulky residue at this position, either an aromatic amino acid or a glutamate (reviewed in 32). Mutational studies have indicated that this single residue is crucial for the selective use of dNTPs rather than NTPs. Recent X-ray crystallographic analyses of several polymerase complexes showed that it is part of the dNTP-binding pocket, and confirmed that its side chain acts as a steric gate preventing the larger ribose moieties of NTPs with their 2′-OH from correct positioning in the pocket (33–35). RNA polymerases, in contrast, usually have a small amino acid at this position. The large F451 side chain in DHBV P protein fits this model, suggesting its replacement by smaller residues might allow the protein to utilize NTPs instead, similarly as reported for F155 mutants of MoMLV RT (36) and Y115 variants of HIV-1 RT (37). If so, this would result in the desired positive readout and thus provide strong evidence for a similar functional and structural role of F451.
Accordingly, F451 of DHBV P protein was replaced by smaller residues, namely aspartate, valine, alanine and glycine. Using the in vitro-priming reaction in RRL the corresponding P protein mutants F415D, F451V, F451A and F451G were compared with wild-type P protein for their Dɛ-dependent priming activities in the presence of dNTPs and NTPs. In addition, the mutant P proteins were analyzed in the context of complete DHBV genomes, in transfected LMH cells, for their ability to support pgRNA packaging into nucleocapsids and synthesis of viral DNA. The results reported below show that F451 is indeed critical for dNTP versus NTP discrimination. Because this function is highly specific and closely related to the precise position of the amino acid side chain in the dNTP-binding pocket, our results strongly suggest that in P proteins this pocket has a very similar architecture to in conventional RTs.
MATERIALS AND METHODS
Bacterial strains and eukaryotic cells
All plasmids were propagated in Escherichia coli Top 10 cells (Invitrogen). For cell culture studies, the chicken hepatoma cell line LMH was used. Cells were maintained as described (38).
Plasmid constructs
Constructs for in vitro translation of DHBV P protein and its variants were based on the previously described plasmid pT7AMVpol16H6 (39). In brief, they contain, behind the T7 promoter, an unstructured leader sequence from alfalfa mosaic virus for enhanced translation, the complete P open reading frame from DHBV 16 (nucleotide positions 170–2530) plus an insertion of six His codons between codon 2 and 3. These constructs lack the Dɛ sequence. Constructs for expression of complete DHBV genomes were based on plasmid pCD16 (40) which contains a slightly overlength DHBV16 genome (nucleotide positions 2520–3021/1–2816) under control of the cytomegalovirus IE enhancer/promoter. P protein mutants were generated by standard procedures using appropriate mutagenic PCR primers introducing the following exchanges of codon 451 (TTT): GGT (F451G), GCT (F451A), GTT (F451V) and GAT (F451D). All mutations were confirmed by sequencing. The encapsidation-deficient control construct pCD4 (40) is identical to pCD16 except that DHBV nucleotides 2520–2579, encompassing part of Dɛ, are deleted. Dɛ RNA was in vitro transcribed from plasmid pBDɛwt that contains DHBV16 sequence from position 2520 to 2652 (20).
In vitro priming reactions
Assays were performed as previously described (17). In brief, wild-type and mutant P proteins were in vitro translated, in the presence of 1 µM Dɛ RNA, in nuclease-treated RRL (Promega) programmed with Dɛ deficient transcripts from the appropriate plasmids linearized with AflII (DHBV position 2526), and in a volume sufficient to allow splitting the reaction into several aliquots for parallel priming assays. Translation efficiencies were controlled by performing parallel translations in the presence of [35S]-Met (Amersham-Pharmacia-Biotech). After 2 h at 30°C, 10 µl aliquots were mixed with an equal volume of 2× priming buffer (15), and supplemented with the desired dNTPs and NTPs. For labeling, between 2.5 and 20 µCi of [α-32P]-labeled dNTPs and NTPs (3000 Ci/mmol; Amersham-Pharmacia-Biotech) per 20 µl reaction were used, corresponding to concentrations from ∼40 to 300 nM. Reactions were kept at 30°C for 60 min, and stopped by the addition of SDS containing sample buffer. Aliquots were separated by SDS–PAGE (7.5% polyacrylamide, 0.1% SDS) using the Laemmli system. Gels were dried and exposed on a Fuji BAS 1500 phosphorimager; semi-quantitations were performed using Imagequant software (both Raytest).
Formation of viral particles and DNA synthesis in LMH cells
LMH cells were transfected with the appropriate plasmids using FuGene6 (Roche Diagnostics) according to the manufacturer’s recommendations. Cells were harvested 4 days post transfection. For monitoring formation of DNA containing nucleocapsids, the cells were lysed, and aliquots corresponding to 20 µl of 1 ml cytoplasmic lysate from 6 × 106 cells were subjected to electrophoresis in 1% agarose gels in 1× TAE (40 mM Tris–acetate, 1 mM EDTA, pH 7.5) buffer as described (41). For protein detection, the gels were blotted onto a polyvinyliden fluoride membrane, and DHBV core protein was identified using a polyclonal rabbit antiserum raised against recombinant DHBV capsids, followed by a goat-anti-rabbit peroxidase conjugate and a chemiluminescent substrate (ECL+, Amersham-Pharmacia-Biotech). For detection of core-borne DNA, gels run in parallel were blotted to a nylon membrane (Roche Diagnostics) and hybridized with a digoxygenin (DIG)-labeled DNA probe obtained by random priming on a cloned DHBV genome. Specific products were detected using the chemiluminescent substrate CDP-Star (Roche Diagnostics).
Competence of mutant P proteins to package pgRNA
Packaged RNA was isolated from DHBV core particles obtained by immunoprecipitation with the polyclonal anti-DHBV capsid antiserum from cytoplasmic lysates of the transfected LMH cells as previously described (42). In brief, non-encapsidated nucleic acids were digested by incubation with micrococcal nuclease in the presence of 10 mM CaCl2. After washing the immobilized core particles twice with lysis buffer, encapsidated RNA was released by treatment with proteinase K in the presence of 5 mM EDTA and 1% SDS, and recovered by phenol extraction and ethanol precipitation. Total cytoplasmic RNA was obtained using the RNeasy Mini kit (Qiagen) according to the manufacturer’s instructions. DHBV-specific RNA was detected by northern blotting using the same DIG-labeled DNA probe as described above.
RESULTS
Smaller side chains at P protein position 451 decrease primer initiation with dGTP
As a first functional test the four P protein variants F451G, F451A, F451V and F451D were subjected to in vitro priming assays with dNTPs, using conditions previously established to lead to maximal labeling of wild-type P protein (43). After in vitro translation of P protein in RRL, in the presence of 1 µM in vitro-transcribed Dɛ RNA, the reactions were supplemented with priming buffer and the desired dNTPs. [α-32P]-labeled dNTPs were used at ∼80 nM concentration (see Materials and Methods). The reaction products were analyzed, after incubation for an additional hour, by SDS–PAGE and subsequent autoradiography. The oligonucleotide primer produced should have the sequence 5′-GTA(A), complementary to the 5′-(U)UAC template sequence within Dɛ (whether the second U residue is copied has not firmly been established), with dGMP becoming covalently fixed to P protein (Fig. 1). Therefore, provision of [α-32P]dGTP should lead to [32P]-labeling of P protein independent of other dNTPs. [35S]-Met-labeling showed that all proteins were translated with similar efficiency (Fig. 2). The priming reactions generated in all cases [32P]-labeled protein bands at the expected position of ∼90 kDa (Fig. 2) but their intensities decreased strongly with decreasing size of the amino acid side chain. Semi-quantitative estimates of the signal intensities by phosphorimaging, compared with the wild-type set at 100%, showed 20% activity for the F451D, and 5, 2 and 1% for the V, A and G variants. Similar values were obtained in repeat experiments. Hence the mutations reduced, either directly or indirectly, the efficiency of dGTP utilization in the variant proteins.
Figure 2.

In vitro priming with dGTP. Wild-type DHBV P protein (wt) and the indicated F451 variants were in vitro translated and subjected to priming assays with [α-32P]dGTP as detailed in Materials and Methods. Reaction products were analyzed by SDS–PAGE and subsequent autoradiography. Molecular masses of protein size markers in kDa are given on the right. Numbers below each lane indicate the relative priming efficiencies as determined by phosphorimager analysis of the band intensities. Control translations in the presence of [35S]-Met (bottom) demonstrated equal translation efficiencies for all variants.
Next [α-32P]dATP, corresponding to the third residue of the DNA primer, was used instead of labeled dGTP. dAMP incorporation requires that synthesis of the oligonucleotide primer has already advanced to the second nucleotide (18,29). As previously shown, the concentration of the endogenous dNTP pool is sufficiently high to allow this reaction to proceed with the wild-type enzyme where the specific requirement for dGTP as the starting nucleotide becomes apparent only after isolation of the initiation complex from the RRL (18). To explicitly test this notion, [α-32P]dATP-priming reactions were supplemented by additional, unlabeled dNTPs. Variant F451D was only weakly stimulated by 500 µM dTTP (data not shown), but a signal of similar intensity as with the wild-type enzyme was obtained with 500 µM dGTP plus 500 µM dTTP (∼75% of the wild-type intensity in the experiment shown in Figure 3A, and ∼105% in an independent repeat experiment). This was not further increased when dCTP was also present (data not shown), consistent with the synthesis of an authentic ɛ-templated DNA primer. No signals were detected with the other variants. To more quantitatively investigate the dNTP concentration dependence of F451D, the priming assays were repeated in the presence of varying concentrations of dGTP plus dTTP. The signals increased in a dose-dependent manner and saturated at ∼5 µM dNTP concentration (Fig. 3B); the signal strength was comparable with that obtained with the wild-type enzyme, suggesting that the lower activity of the F451D variant under standard dGTP-priming conditions was due to a decreased dNTP affinity rather than a compromised catalytic activity or a folding problem. The absence of signals with the V, A and G variants suggests either an even further decreased affinity for at least one of the dNTPs or a lack of processivity, or both.
Figure 3.

In vitro priming with dATP. (A) dATP priming in the presence of dGTP + dTTP. Because dA is the third nucleotide in the DNA primer the [α-32P]dATP-priming reactions were supplemented with 500 µM each of dGTP and dTTP. Under these conditions, but not without supplementation, variant F451D produced a signal of similar intensity as the wild-type protein. (B) Dependence of F451D dATP-priming activity on the concentration of added dGTP + dTTP. Equal amounts of in vitro-translated F451D protein were subjected to [α-32P]dATP priming in the presence of the indicated concentrations of non-labeled dGTP + dTTP. Reaction products were analyzed as described in the legend of Figure 2. Molecular masses (in kDa) of protein size markers loaded in lane ma are indicated on the left.
Smaller side chains at position 451 allow for primer initiation with GTP
If indeed F451 was involved in dNTP versus NTP discrimination, the F451 mutants, especially those with the smallest side chains, were predicted to be able to use NTPs instead of dNTPs. Priming reactions were set up as before, except that ∼300 nM [α-32P]GTP was used instead of dGTP. As shown in Figure 4A, labeled bands of the expected size were observed for all variants. Labeling was generally low but most efficient for F451G, followed by the A, V and D mutants. As determined by phosphorimager analysis the relative signal intensities, with F451G set at 100%, were 90, 70 and 10%, respectively, for the A, V and D mutants. Importantly, no signal was observed for the wild-type enzyme. Hence, the order of labeling efficiencies with GTP was reversed compared with that of dGTP, consistent with a crucial role of the side chain at position 451 in discrimination against NTPs. The relative efficiencies of GTP priming were also semi-quantitatively related, by phosphorimaging, to a dGTP priming sample with the wild- type protein run on the same gel and found to be 1.0, 0.8, 0.7 and 0.1% for the variants G, A, V and D, respectively. To corroborate that these weak signals were specific, parallel GTP-priming assays with the F451G and the wild-type enzyme were performed in the presence, or in the absence, of Dɛ RNA. Labeling occurred only with the mutant, and only when Dɛ RNA was present (Fig. 4B).
Figure 4.

In vitro priming with NTPs. (A) Relative GTP-priming activities. Priming reactions were performed as before except that [α-32P]GTP was used as labeled nucleoside triphosphate. Band intensities were semi-quantitatively compared by phosphorimaging; relative intensities within the GTP series, and versus a standard [α-32P]dGTP-priming reaction with wild-type protein from the same gel are given below each lane. Molecular masses (in kDa) of protein size markers loaded in lane ma are indicated on the left. nd, not determined. (B) Dɛ dependence of GTP priming with variant F451G. To confirm specificity of the GTP-priming signal with the F451G variant, the reactions with wild-type P protein and the variant were repeated in the presence and absence of Dɛ RNA and analyzed as in (A). (C) UTP incorporation. Priming assays were performed as before except that [α-32P]UTP and 500 µM dGTP were used.
In a second set of experiments, [α-32P]UTP was used, corresponding to the dT at the second oligonucleotide primer position. With UTP only, no signals were obtained. However, when the reactions were supplemented with 0.5 mM dGTP, weak but clearly detectable signals were observed for the F451G and, still weaker, for the F451A variant (Fig. 4C). These data show that in particular the F451G mutation allowed the protein to incorporate a ribonucleoside triphosphate in the second position of the primer.
All F451 P protein mutants support pgRNA encapsidation in transfected cells
To independently confirm that the differences in in vitro priming efficiency between the variants reflected local perturbations rather than the proteins’ global structure and/or interactions with the cofactors, we took advantage of a second function of P protein, i.e. packaging of the viral pgRNA, which also depends on binding of the P protein–host factor complex to ɛ (42). Hence, a wild-type-like packaging efficiency of the F451 mutants would indicate that these interactions are not disturbed.
Because RNA encapsidation cannot be analyzed in vitro we generated expression constructs for complete DHBV genomes that encode the mutant P proteins F451D, F451V and F451G. Upon transfection into the chicken hepatoma cell line LMH the wild-type construct produces, under the control of the CMV promoter, relatively large amounts of pgRNA which serves as template for the translation of core and P protein that co-assemble into authentic DHBV nucleocapsids. As a control, a mutant with a deletion in the ɛ signal (ɛ–) that renders the pgRNA encapsidation defective (40) was analyzed in parallel.
First, nucleocapsid formation was monitored by native gel electrophoresis of aliquots from cytoplasmic lysates. In this assay both genome-containing and empty capsids migrate as distinct bands of essentially identical mobility through the gel (44). Capsid proteins are detected by western blotting, and the membrane can also be probed for co-migrating, i.e. encapsidated, virus-specific DNA by molecular hybridization (41). In contrast, encapsidated RNA is unstable during the procedure. As shown in Figure 5A, all constructs led to similarly intense core protein signals, demonstrating similar core protein expression and assembly efficiencies. When probed for DHBV nucleic acids, a strong signal was observed for the wild-type construct. In contrast, none of the mutants gave signals above the background produced by the ɛ– control, indicating that these capsids did not contain significant amounts of DHBV DNA. To distinguish between an RNA-packaging and a DNA-synthesis defect causing this phenotype, cores from equal aliquots of the same cell lysates were enriched by immunoprecipitation with anti-core antiserum, and potentially contained RNA was extracted and analyzed by northern blotting; total RNA from the same transfected cells served as a control. All samples gave similar signals for core-borne pgRNA except, as expected, the one from transfection with the packaging-defective ɛ– construct (Fig. 5B). Hence, the F451 mutations did not induce detectable packaging defects, suggesting the mutations neither impeded the interactions of P protein with ɛ or the cellular factors in the complex nor induce global misfolding.
Figure 5.
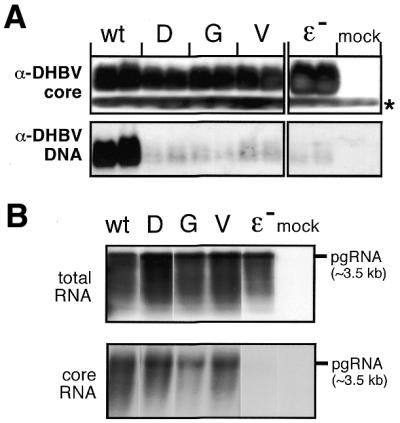
P protein variants in the context of complete DHBV genomes. (A) DHBV nucleocapsid formation. LMH cells were transfected with expression plasmids for DHBV genomes encoding wild-type P protein, the indicated variants, an encapsidation-deficient construct (ɛ–) or with empty plasmid vector (mock). Equal aliquots from cytoplasmic lysates were analyzed in duplicate by native agarose gel electrophoresis. After blotting DHBV core protein was detected using an anti-DHBV capsid antiserum (top) and a secondary antibody-peroxidase conjugate, followed by a chemiluminescent substrate. The faster migrating band (asterisk) corresponds to a non-specific cross-reaction product. DHBV DNA was detected by Southern hybridization using a DIG-labeled DNA probe (bottom) and a chemiluminescent substrate. (B) Encapsidation competence of variant DHBV P proteins. Capsids were enriched from cytoplasmic lysates of LMH cells transfected with the indicated constructs by immunopreciptiation with anti-DHBV capsid antiserum, and core-associated RNA was isolated. For comparison, total RNA was prepared from the same transfections. RNAs were separated by agarose gel electrophoresis, and hybridized with a DIG-labeled DNA probe specific for DHBV pgRNA. The position of a 3.5 kb marker RNA is indicated on the right.
Given the weak in vitro priming activities of the mutant P proteins, the lack of detectable core-borne viral DNA was not surprising. Nonetheless, we also employed a sensitive alternative assay to detect DNA synthesis activity, the endogenous polymerase reaction. There, dNTPs, one of them [α-32P]-labeled, are allowed to diffuse into isolated core particles and are used by the packaged P protein to further extend initiated DNA strands (45). Because many radioactively labeled nucleotides are incorporated the products are of high specific activity. However, only the wild-type virus but none of the mutants produced the expected 3 kb DNA bands (data not shown). Hence, whether the P protein mutants, under in vivo conditions, are not at all able to synthesize DNA, or whether some low-level DNA synthesis leading to short strands does occur, remains to be established.
DISCUSSION
Structural information on the hepadnaviral P proteins would help to better understand the peculiarities of hepatitis B virus replication, and would also be of considerable medical significance because, as the only virus-encoded enzyme, P protein is a prime target for chemotherapy of chronic hepatitis B. In particular, the host-factor dependence makes it unlikely that direct approaches will yield this information in the near future. Therefore, we used in vitro translation of DHBV P protein (17,18,29,30) to show, biochemically, that residue F451 is involved in discriminating ribo- from deoxyribonucleotides. The in vitro data were corroborated by cell culture experiments indicating that the mutant P proteins are properly folded. These data experimentally support the relevance of the conservation of the box A residues in DHBV P protein and, as discussed below, they strongly suggest that the dNTP-binding pocket in DHBV as well as in HBV P protein has a common architecture with that of other RTs.
Ribo- versus deoxyribonucleoside triphosphate discrimination by P protein
The fundamental similarities between all polynucleotide polymerases are reflected in some invariant sequence motives (22,23,46), especially the residues in box A and C coordinating the two metal ions which are essential for catalysis (27,47). The functional importance of the YMDD motif in box C of hepadnaviral P proteins (25,26) indicates a similar role for these two Asp residues; the third aspartate would be located in box A. However, there must also be a means as to how the different polymerases distinguish their specific templates and monomeric building blocks (32). Mutational studies with various DNA polymerases and RTs (48–51) showed, surprisingly at the time, that a single bulky residue in the conserved box A motif is critical for maintaining selectivity for 2′-deoxyribose rather than ribose in the nucleoside triphosphate. For instance, replacement of Y115 in HIV-1 RT (51), and of F155 in MoMLV RT (36), by smaller amino acids strongly decreased discrimination against NTPs, allowing the mutant enzymes to extend preformed primer-templates by one to a few NTPs. The concept of the bulky residue acting as a steric gate has been fully confirmed by recent crystallographic studies of several polymerase complexes (see below). Our results demonstrate that F451 plays this role in DHBV P protein.
In the initial standard dGTP-priming assays, all variants exerted a reduced priming efficiency, with smaller side chains leading to a stronger decrease. This suggested, but did not prove, that position 451 is involved in dNTP binding. A similar decrease in dATP affinities with decreasing side chain size has been reported for Y115 mutants of HIV-1 RT. There, the Km values for dATP incorporation into a preformed primer-template increased from ∼8 nM for the wild type over 0.4 µM for the Y115V variant to ∼10 µM for the Y115A and Y115G mutants (37). With [α-32P]dATP as the only priming dNTP, only the wild-type P protein produced a specific signal. Virtually wild-type levels of priming could, however, be restored with mutant F451D when the reactions were supplemented with at least 5 µM dGTP and dTTP. Hence, although kinetic contributions cannot be excluded, its reduced priming efficiency in the standard dGTP reaction is most likely caused by a lowered dNTP affinity. The seemingly paradoxical dATP labeling of the wild-type enzyme without addition of dGTP and dTTP has previously been shown to be due to the dNTPs endogenously present in the RRL (18). Unfortunately, their concentrations are not known, hence quantitations are difficult. However, the full activity of the wild-type enzyme without supplementation, and the requirement of the F451D variant for 5 µM dGTP + dTTP for maximal priming while 50 nM did not lead to a detectable increase over background, indicates that its dNTP affinity is at least 100-fold lower than that of the wild-type protein. For the other mutants no definitive conclusions as to the cause of their strongly reduced activities could be made from these experiments.
Most revealing regarding the potential involvement of F451 in dNTP versus NTP discrimination were the GTP-priming assays. All mutants, but not the wild-type protein, yielded clearly detectable signals which for F451G were explicitly shown to be Dɛ dependent. Contrary to dNTP priming, their intensities increased with decreasing side chain size. F451G displayed the strongest labeling which was ∼1% as intense as dGTP priming with the wild-type enzyme. This mutant and, albeit at very low levels, F451A but not the others, were also able to incorporate UTP at the second primer position when the reactions were supplemented with dGTP. Although exact quantitations are difficult, at least two informative conclusions can be drawn: first, the relative labeling efficiencies with the F451G variant were similar for both dGTP and GTP priming, demonstrating a lack of discrimination between dNTPs and NTPs. This strongly resembles the nearly identical Km values for dNTPs versus NTPs reported for the Y115G and F155G variants of HIV-1 RT (37) and MoMLV RT (52). Compared with the F451G variant, the A, V and D mutants had an increased ability to incorporate dNTP whereas, in the same order, they were less efficient in GTP utilization. These data are fully consistent with the discriminatory importance of the side chain size at position 451.
Secondly, absolutely no signal was observed for the wild-type enzyme upon GTP priming; in contrast, the GTP signal with the F451D mutant, at ∼0.1% of the intensity of the wild-type dGTP signal, was clearly above background. Therefore, wild-type P protein discriminates at least 1000-fold against NTPs. Given that this is a lower limit, it compares well with the 104–105-fold selectivities for dNTPs over NTPs reported for the wild-type HIV-1 (37) and MoMLV RTs (36). Together, these data further corroborate the involvement of residue F451 in dNTP versus NTP discrimination.
Indirect effects of the mutations on the folding of the mutant P proteins or their interactions with the other complex components could largely be excluded by the cell culture experiments. Even those P protein mutants with a greatly diminished priming activity were pgRNA-packaging proficient, consistent with the ability of active site variants to encapsidate RNA (42,53). This strongly suggests that none of the mutations had a major effect on complex formation, and that the observed differences in the priming and dNTP versus NTP utilization abilities of the mutants were caused by local alterations in the dNTP-binding pocket of P protein.
A side aspect was whether the mutant P proteins were able to synthesize viral DNA under physiological conditions. However, we did not obtain positive evidence for specific DNAs of sufficient length to be detectable by Southern blotting or by the endogenous polymerase reaction. Notably, of several variants of Y115 in HIV-1 RT, and of F155 in MoMLV RT, only those maintaining an aromatic side chain were able to produce viable virus even if the other mutants had detectable in vitro polymerase activity (54). In view of these results, our failure to detect in vivo DNA synthesis by the P protein variants is not surprising.
Structural implications
The above described results demonstrated that F451 of DHBV P protein is functionally equivalent to Y115 in the HIV-1 and F155 in the MoMLV RT. Recent crystallographic analyses revealed how the specific disposition of a single bulky residue involved in forming the dNTP-binding pocket, i.e. an aromatic residue in DNA polymerases α and RTs, e.g. Y115 in the HIV-1 enzyme (35), and a glutamate in DNA polymerases I (33,34) can control specificity for dNTPs over NTPs. Hence, the discriminatory function of F451 in the hepadnaviral protein strongly suggests that its spatial arrangement with respect to the architecture of the dNTP-binding pocket is similar to that in the retroviral RTs although, regarding the active site as a whole, the structural differences between the TP domain containing the priming Tyr residue on the one hand and a tRNA primer on the other must be accounted for. Especially revealing is a comparison with the crystal structure of the HIV-1 enzyme in a covalently trapped catalytic complex including a DNA template primer and dTTP (35). A schematic representation of the essential amino acids lining the dNTP pocket, and the catalytic aspartate triade (D110, D185, D186) coordinating the two Mg2+ ions at the active site is shown in Figure 6. The aromatic ring of Y115 is located below the ribose ring of the dTTP such that the 2′-OH group of an NTP would clash with its side chain. Inspection of the other motifs in the DHBV as well as the HBV P protein shows that not only F451, and its HBV equivalent F423, but many of the residues crucial for dNTP binding are conserved. D185 and 186 in the HIV-1 enzyme are part of the YMDD motif which is identically present in the hepadnaviral proteins (amino acids 511–514 in DHBV, amino acids 538–541 in HBV); the third active site aspartate D110 has its counterparts in D446 (DHBV) and D418 (HBV) in box A. The triphosphate moiety of the incoming dNTP is held in close proximity to the active site by coordination with the two Mg2+ ions and by the side chains of K65 and R72, both of which have identical counterparts in K395 and R404 of DHBV, and K367 and R376 of HBV. Further contacts exist to the main chains of A114, conserved in both hepadnaviral proteins (A450 and A422), and that of D113; this residue differs in DHBV (Q449) and HBV (A421) but also in other RTs. The side chains of R72 and Q151 pack against the opposite part of the dNTP; the equivalents to Q151 are M480 (DHBV) and M506 (HBV), hence glutamine is replaced by methionine with its similar steric requirements. The ribose pocket is defined by the side chains of D113, Y115, F116 and Q151. Apart from the already discussed residues, the F116 position has its counterparts in Y452 in DHBV, and Y424 in HBV. A caveat is that these structural data relate to dNTP/NTP discrimination on a DNA rather than on an RNA template as in our experiments. The congruency of the data argues that the discriminatory mechanism is not fundamentally different, although the structural differences between an A form RNA–RNA duplex and an RNA–DNA hybrid duplex could contribute to discrimination.
Figure 6.

Similarity of the dNTP-binding pocket in hepadnaviral P proteins to that in HIV-1 RT. The structural disposition of essential residues in the active site and the dNTP-binding pocket in HIV-1 RT is shown, as a stereo pair, according to Huang et al. (35) (PDB accession code 1RTD); the incoming dTTP is depicted as a wireframe model. The aspartates from the catalytic triade coordinating the two essential Mg2+ ions (spheres) are located in the upper left quadrant. The discriminatory Y115, to which F451 of the DHBV P protein is functionally equivalent, is at the bottom below the sugar ring of the dTTP; a 2′-OH would sterically clash with the side chain. The identity of each residue in the HIV-1 enzyme is indicated, followed in parentheses by the corresponding conserved residues in the DHBV and HBV P protein. The graphic representation was generated using Rasmac v.2.6.
Together, the conservation of essential residues and our experiments imply a common structure of the dNTP-binding pockets in retroviral and hepadnaviral RTs. This adds substantially to the validity of attempts to molecularly model the structure of the entire P protein polymerase domain using HIV-1 RT as a template (24). However, lacking a retroviral equivalent, the unique features of hepadnaviral replication, i.e. the priming role of TP, the template specificity for ɛ, and the dependence on cellular factors, will not yield to this approach but rather require experiments directly addressing the underlying interactions.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported in part by grants from the Deutsche Forschungsgemeinschaft and the Zentrum für Klinische Forschung I (ZKFI-B7).
REFERENCES
- 1.Blumberg B.S. (1997) Hepatitis B virus, the vaccine and the control of primary cancer of the liver. Proc. Natl Acad. Sci. USA, 94, 7121–7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hilleman M.R. (2001) Overview of the pathogenesis, prophylaxis and therapeusis of viral hepatitis B, with focus on reduction to practical applications. Vaccine, 19, 1837–1848. [DOI] [PubMed] [Google Scholar]
- 3.Nassal M. (2000) Macromolecular interactions in hepatitis B virus replication and particle assembly. In Cann,A.J. (ed.), Frontiers in Molecular Biology: DNA Virus Replication. Oxford University Press, Oxford, UK, Vol. 26, pp. 1–40.
- 4.Seeger C. and Mason,W.S. (2000) Hepatitis B virus biology. Microbiol. Mol. Biol. Rev., 64, 51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nassal M. and Schaller,H. (1996) Hepatitis B virus replication—an update. J. Viral Hepatol., 3, 217–226. [DOI] [PubMed] [Google Scholar]
- 6.Nassal M. (1999) Hepatitis B virus replication: novel roles for virus–host interactions. Intervirology, 42, 100–116. [DOI] [PubMed] [Google Scholar]
- 7.Hu J. and Seeger,C. (1996) Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl Acad. Sci. USA, 93, 1060–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu J., Toft,D.O. and Seeger,C. (1997) Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J., 16, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knaus T. and Nassal,M. (1993) The encapsidation signal on the hepatitis B virus RNA pregenome forms a stem–loop structure that is critical for its function. Nucleic Acids Res., 21, 3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pollack J.R. and Ganem,D. (1993) An RNA stem–loop structure directs hepatitis B virus genomic RNA encapsidation. J. Virol., 67, 3254–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G.H. and Seeger,C. (1993) Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol., 67, 6507–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pollack J.R. and Ganem,D. (1994) Site-specific RNA binding by a hepatitis B virus reverse transcriptase initiates two distinct reactions: RNA packaging and DNA synthesis. J. Virol., 68, 5579–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nassal M. and Rieger,A. (1996) A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J. Virol., 70, 2764–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lanford R.E., Notvall,L., Lee,H. and Beames,B. (1997) Transcomplementation of nucleotide priming and reverse transcription between independently expressed TP and RT domains of the hepatitis B virus reverse transcriptase. J. Virol., 71, 2996–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weber M., Bronsema,V., Bartos,H., Bosserhoff,A., Bartenschlager,R. and Schaller,H. (1994) Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J. Virol., 68, 2994–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zoulim F. and Seeger,C. (1994) Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J. Virol., 68, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beck J. and Nassal,M. (1997) Sequence- and structure-specific determinants in the interaction between the RNA encapsidation signal and reverse transcriptase of avian hepatitis B viruses. J. Virol., 71, 4971–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beck J. and Nassal,M. (1998) Formation of a functional hepatitis B virus replication initiation complex involves a major structural alteration in the RNA template. Mol. Cell. Biol., 18, 6265–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tavis J.E., Massey,B. and Gong,Y. (1998) The duck hepatitis B virus polymerase is activated by its RNA packaging signal, epsilon. J. Virol., 72, 5789–5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaaf S.G., Beck,J. and Nassal,M. (1999) A small 2′-OH- and base-dependent recognition element downstream of the initiation site in the RNA encapsidation signal is essential for hepatitis B virus replication initiation. J. Biol. Chem., 274, 37787–37794. [DOI] [PubMed] [Google Scholar]
- 21.Xiong Y. and Eickbush,T.H. (1990) Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J., 9, 3353–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sousa R. (1996) Structural and mechanistic relationships between nucleic acid polymerases. Trends Biochem. Sci., 21, 186–190. [PubMed] [Google Scholar]
- 23.Eickbush T.H. (1994) Origin and evolutionary relationships of retroelements. In Morse,S.S. (ed.), The Evolutionary Biology of Viruses. Raven Press, New York, NY, pp. 121–160.
- 24.Das K., Xiong,X., Yang,H., Westland,C.E., Gibbs,C.S., Sarafianos,S.G. and Arnold,E. (2001) Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J. Virol., 75, 4771–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radziwill G., Tucker,W. and Schaller,H. (1990) Mutational analysis of the hepatitis B virus P gene product: domain structure and RNase H activity. J. Virol., 64, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang L.J., Hirsch,R.C., Ganem,D. and Varmus,H.E. (1990) Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase. J. Virol., 64, 5553–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steitz T.A. (1999) DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem., 274, 17395–17398. [DOI] [PubMed] [Google Scholar]
- 28.Mutimer D. (2001) Hepatitis B virus infection: resistance to antiviral agents. J. Clin. Virol., 21, 239–242. [DOI] [PubMed] [Google Scholar]
- 29.Wang G.H. and Seeger,C. (1992) The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell, 71, 663–670. [DOI] [PubMed] [Google Scholar]
- 30.Beck J. and Nassal,M. (2001) Reconstitution of a functional duck hepatitis B virus replication initiation complex from separate reverse transcriptase domains expressed in Escherichia coli. J. Virol., 75, 7410–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu J. and Anselmo,D. (2000) In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J. Virol., 74, 11447–11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joyce C.M. (1997) Choosing the right sugar: how polymerases select a nucleotide substrate. Proc. Natl Acad. Sci. USA, 94, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doublie S., Tabor,S., Long,A.M., Richardson,C.C. and Ellenberger,T. (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature, 391, 251–258. [DOI] [PubMed] [Google Scholar]
- 34.Li Y., Korolev,S. and Waksman,G. (1998) Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J., 17, 7514–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang H., Chopra,R., Verdine,G.L. and Harrison,S.C. (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science, 282, 1669–1675. [DOI] [PubMed] [Google Scholar]
- 36.Gao G., Orlova,M., Georgiadis,M.M., Hendrickson,W.A. and Goff,S.P. (1997) Conferring RNA polymerase activity to a DNA polymerase: a single residue in reverse transcriptase controls substrate selection. Proc. Natl Acad. Sci. USA, 94, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cases-Gonzalez C.E., Gutierrez-Rivas,M. and Menendez-Arias,L. (2000) Coupling ribose selection to fidelity of DNA synthesis. The role of Tyr-115 of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem., 275, 19759–19767. [DOI] [PubMed] [Google Scholar]
- 38.Protzer U., Nassal,M., Chiang,P.W., Kirschfink,M. and Schaller,H. (1999) Interferon gene transfer by a hepatitis B virus vector efficiently suppresses wild-type virus infection. Proc. Natl Acad. Sci. USA, 96, 10818–10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beck J. and Nassal,M. (1996) A sensitive procedure for mapping the boundaries of RNA elements binding in vitro translated proteins defines a minimal hepatitis B virus encapsidation signal. Nucleic Acids Res., 24, 4364–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Obert S., Zachmann-Brand,B., Deindl,E., Tucker,W., Bartenschlager,R. and Schaller,H. (1996) A splice hepadnavirus RNA that is essential for virus replication. EMBO J., 15, 2565–2574. [PMC free article] [PubMed] [Google Scholar]
- 41.König S., Beterams,G. and Nassal,M. (1998) Mapping of homologous interaction sites in the hepatitis B virus core protein. J. Virol., 72, 4997–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartenschlager R., Junker-Niepmann,M. and Schaller,H. (1990) The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J. Virol., 64, 5324–5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beck J., Bartos,H. and Nassal,M. (1997) Experimental confirmation of a hepatitis B virus (HBV) epsilon-like bulge-and-loop structure in avian HBV RNA encapsidation signals. Virology, 227, 500–504. [DOI] [PubMed] [Google Scholar]
- 44.Birnbaum F. and Nassal,M. (1990) Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J. Virol., 64, 3319–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nassal M. (1992) The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol., 66, 4107–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joyce C.M. and Steitz,T.A. (1994) Function and structure relationships in DNA polymerases. Annu. Rev. Biochem., 63, 777–822. [DOI] [PubMed] [Google Scholar]
- 47.Steitz T.A. (1998) A mechanism for all polymerases. Nature, 391, 231–232. [DOI] [PubMed] [Google Scholar]
- 48.Gardner A.F. and Jack,W.E. (1999) Determinants of nucleotide sugar recognition in an archaeon DNA polymerase. Nucleic Acids Res., 27, 2545–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Astatke M., Ng,K., Grindley,N.D. and Joyce,C.M. (1998) A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proc. Natl Acad. Sci. USA, 95, 3402–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonnin A., Lazaro,J.M., Blanco,L. and Salas,M. (1999) A single tyrosine prevents insertion of ribonucleotides in the eukaryotic-type phi29 DNA polymerase. J. Mol. Biol., 290, 241–251. [DOI] [PubMed] [Google Scholar]
- 51.Martin-Hernandez A.M., Domingo,E. and Menendez-Arias,L. (1996) Human immunodeficiency virus type 1 reverse transcriptase: role of Tyr115 in deoxynucleotide binding and misinsertion fidelity of DNA synthesis. EMBO J., 15, 4434–4442. [PMC free article] [PubMed] [Google Scholar]
- 52.Gao G. and Goff,S.P. (1998) Replication defect of moloney murine leukemia virus with a mutant reverse transcriptase that can incorporate ribonucleotides and deoxyribonucleotides. J. Virol., 72, 5905–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirsch R.C., Lavine,J.E., Chang,L.J., Varmus,H.E. and Ganem,D. (1990) Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as well as for reverse transcription. Nature, 344, 552–555. [DOI] [PubMed] [Google Scholar]
- 54.Olivares I., Sanchez-Merino,V., Martinez,M.A., Domingo,E., Lopez-Galindez,C. and Menendez-Arias,L. (1999) Second-site reversion of a human immunodeficiency virus type 1 reverse transcriptase mutant that restores enzyme function and replication capacity. J. Virol., 73, 6293–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]