

Summary
Coronary artery disease (CAD) is a pandemic disease where up to half of the risk is explained by genetic factors. Advanced insights into the genetic basis of CAD require deeper understanding of the contributions of different cell types, molecular pathways, and genes to disease heritability. Here, we investigate the biological diversity of atherosclerosis-associated cell states and interrogate their contribution to the genetic risk of CAD by using single-cell and bulk RNA sequencing (RNA-seq) of mouse and human lesions. We identified 12 disease-associated cell states that we characterized further by gene set functional profiling, ligand-receptor prediction, and transcription factor inference. Importantly, Vcam1+ smooth muscle cell state genes contributed most to SNP-based heritability of CAD. In line with this, genetic variants near smooth muscle cell state genes and regulatory elements explained the largest fraction of CAD-risk variance between individuals. Using this information for variant prioritization, we derived a hybrid polygenic risk score (PRS) that demonstrated improved performance over a classical PRS. Our results provide insights into the biological mechanisms associated with CAD risk, which could make a promising contribution to precision medicine and tailored therapeutic interventions in the future.
Keywords: coronary artery disease, atherosclerosis, single cell, cell state, genome-wide association study, polygenic risk score, genetics, scRNA-seq, GWAS
Graphical abstract
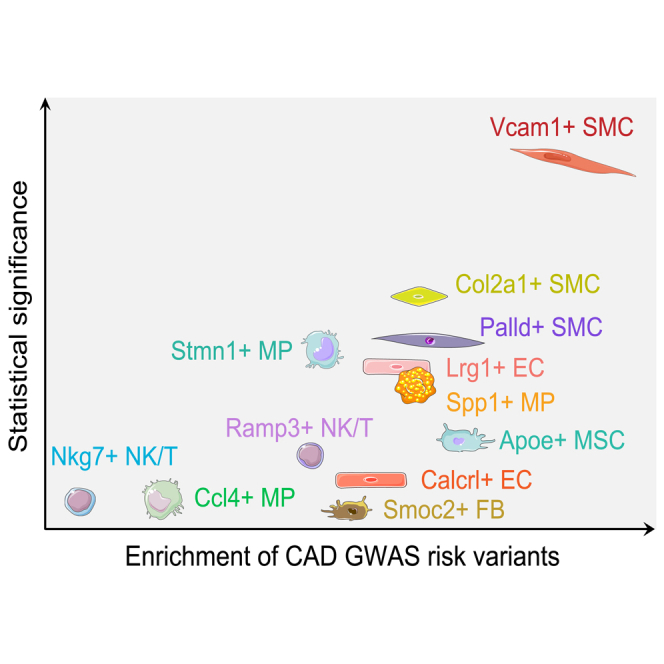
Using single-cell RNA sequencing, researchers identified 12 atherosclerosis-associated cell states and found that Vcam1+ smooth muscle cell state contributed the most to the genetic heritability of coronary artery disease. A hybrid polygenic risk score (PRS) that considers cell-type-specific regulatory elements demonstrated improved performance over a classical PRS.
Introduction
Cell cycle stages and cell types are well understood dimensions of cellular heterogeneity that are routinely applied to distinguish cell populations. Still, within a population of relatively homogeneous cells, significant variability in specific responses to an identical stimulus can be detected, also known as cell states. Efforts to determine the extent of such cell-to-cell heterogeneity in pathological conditions have been revolutionized by the emergence of single-cell RNA sequencing (scRNA-seq)-based studies. To this end, cardiovascular research has witnessed an explosion of studies describing an unprecedented degree of cell diversity in atherosclerotic lesions. scRNA-seq and cytometry by time of flight (CyTOF) methods have identified at least 19 types of leukocytes, consisting of 3–5 different macrophage subsets, 3–5 T cell subsets, two B cell subsets, two types of monocytes, two types of dendritic cells, one NK cell subset, neutrophils, and eosinophils in atherosclerotic mouse aortas.1 In addition, seven clusters of smooth muscle cells (SMCs) have been identified, demonstrating dedifferentiation of contractile SMCs into intermediate cells, termed “SEM” cells (stem cell, endothelial cell, monocyte) and further into fibromyocytes/fibrochondrocyte-like cells and to an osteogenic phenotype.2,3,4,5,6 Additionally, eight endothelial cell (EC) subpopulations have been identified as a result of diabetogenic high-fat diet or disturbed flow where proatherogenic conditions induced a dramatic transition of ECs into progenitor, proinflammatory, mesenchymal (EndMT), and immune cell-like phenotypes.7,8 Still, consistent definition and comparison of altered cell states in atherosclerosis is missing, complicating the understanding of mechanisms and pathways that contribute to pathological cell states.
An individual’s risk of atherosclerotic coronary artery disease (CAD) is determined by an interplay of environmental and genetic factors. The recent genome-wide association studies (GWASs) for CAD with over 1 million participants have identified over 300 risk loci.9,10 Despite extensive progress in statistical and experimental tools used to link regions of the genome to disease risk, it remains challenging to identify the causal genes underlying genetic associations and the cell types through which the effect is mediated. We have recently demonstrated that cell-type-specific chromatin accessibility and gene expression provide a means for predicting the cell type of action for CAD loci.11 To this end, we demonstrated that the cis-regulatory elements active in SMCs and ECs show the highest enrichment of GWAS SNPs for CAD and blood pressure among the lesional cell types. However, the contributions of specific disease-associated cell states or gene signatures to the disease risk and to CAD heritability remain unknown.
To shed light into these questions, we performed scRNA-seq of thoracic aortas of healthy and atherosclerotic Ldlr−/−/Apob100/100 mice during disease progression. Our results identify 12 cell states that are increased in response to atherogenic changes. We validate our findings in bulk RNA-sequencing (RNA-seq) studies in a separate mouse experiment and in human samples from affected individuals. We identify common pathways, transcription factors, and ligands that define the cell state changes. Finally, we link our findings to GWASs to prioritize the relevance of atherosclerosis-associated cell states in the biology and heritability of CAD and apply this knowledge in polygenic risk prediction.
Material and methods
Mouse model
To model a disease stage course of atherosclerosis in mouse, we performed four combinations of genotype and dietary protocol. We fed the atherosclerosis-prone low-density lipoprotein receptor-deficient mice expressing only apolipoprotein B100 (Ldlr−/−/Apob100/100)12,13 (The Jackson Laboratory strain #003000) a high-fat diet (HFD; Teklad TD.88137) for 0, 1, or 3 months to model prelesion, early, and late disease stages, respectively. We timed diet-starting age to equalize the age at sample collection between all groups (8 months old). Age-matched wild-type C57BL/6J mice fed chow diet were used as a healthy control. Male and female mice were used (mouse counts for each experiment are stated below). Throughout the study, mice were maintained on a 12-h light-dark cycle and had access to food and water ad libitum. All animal experiments were approved by the local ethics committee and carried out in compliance with European Union Directive 2010/63/EU.
Aorta single-cell RNA-seq library preparation
Mice were anesthetized with isoflurane and euthanized by cervical dislocation. The mice were perfused by cardiac puncture with 10 mL of ice-cold PBS supplemented with 20 U/mL heparin and placed on ice for dissection. The entire thoracic aorta (i.e., the ascending aorta, the aortic arch, and the descending aorta up to the diaphragm) was extracted and used in scRNA-seq sample preparation. The adventitia was mechanically removed under a dissection microscope and discarded. Each thoracic aorta minced with a scalpel and enzymatically dissociated into single-cell suspension in 0.8 mL of Multi Tissue Dissociation Kit 1 (Miltenyi Biotec #130-110-201) enzymatic mixture reconstituted in RPMI 1640 medium supplemented with 0.5% bovine serum albumin (BSA) and 20 mM HEPES buffer (pH = 7.2). The mixture was incubated at 37°C with end-over-end rotation. After 20, 40, and 60 min of incubation time, tissue pieces were left to settle for 10 s, and 0.8 mL of cell suspension supernatant was collected from the tube and placed on ice. To continue the digestion, we added 0.8 mL of fresh enzymatic digestion mixture. The collected cell suspension was filtered through a 30 μm cell strainer, centrifuged at 400 g for 8 min at 4°C, resuspended in PBS supplemented with 1% BSA, and placed on ice. The three cell suspension fractions for each aorta were subsequently pooled together and red blood cell lysis was carried out by adding 9 volumes of ice-cold 1X RBC Lysis Buffer, Multi-species (eBioscience #00-4300-54) and incubating on ice for 3 min. Subsequently, we added PBS to normalize the buffer and collected the cells by centrifugation (400 g for 8 min at 4°C). Magnetic removal of dead cells was carried out with the Dead Cell Removal Kit (Miltenyi Biotec #130-090-101) with Miltenyi MS magnetic columns following the manufacturer’s instructions. Cells were resuspended in PBS containing 0.04% BSA and counted by hemocytometry with trypan blue staining. Cell viability was between 74% and 85%. In each experiment group, aortic cell isolation was carried out separately from three male mice, the cells were stained with TotalSeq cell hashing antibodies (BioLegend) according to the manufacturers’ recommendations, and subsequently the cells from individual mice were pooled in equal proportions into one lane of the Chromium Controller microfluidics chip (10x Genomics). We only used male mice to minimize experimental variation and increase sensitivity of the experiment to detect disease-associated changes. This decision was justified on the basis of bulk RNA-seq results (below), which indicated that the effect of treatment on the major cell type proportions was stronger than the effect of individual factors such as sex (Figure S8C). We used the Chromium Single Cell 3′ Kit (v2 Chemistry; 10x Genomics) to prepare scRNA-seq libraries. Paired-end high-throughput sequencing was carried out on an Illumina NovaSeq instrument (read 1: 26 bp, read 2: 91 bp).
Aorta bulk RNA-seq library preparation
Mouse aortas were dissected as for scRNA-seq (described above), flash-frozen, and cryogenically pulverized with the Cellcrusher cryo-press cooled with liquid nitrogen. Total RNA was isolated and treated with DNaseI with the Absolutely RNA Nanoprep Kit (Agilent). RNA was quantified with the Bioanalyzer RNA 6000 pico assay (Agilent) and RNA-seq libraries were prepared with the SMARTer v2 Stranded Total RNA-Seq Pico Kit (Takara Bio). Sample size for bulk RNA-seq was six per group, consisting of four males and two females (except the 3-month HFD group, which had four males and one female). High-throughput Illumina sequencing (read length 75 bp, single-end) was carried out at EMBL GeneCore.
Single-cell RNA-seq data analysis
Sequencing reads were processed with the Cell Ranger pipeline (version 3.0.2; 10x Genomics) and the 10x Genomics mm10 reference transcriptome package (version 3.0.0). The Cell Ranger-filtered cell barcode count matrices were subsequently processed with Seurat (version 3.1.0)14 running in R version 3.5.3. The standard (log normalization-based) workflow recommended by Seurat authors was used. For each library, to remove low-quality barcodes, we visually evaluated cell quality metrics (genes per barcode, UMIs per barcode, and mitochondrial read fraction) by using violin plots and selected cutoffs, which resulted in retaining 87%–93% of barcodes. In total, 36,157 cells were retained with median 1,629 (mean 2,043) UMI counts per cell, median 875 (mean 965.4) genes per cell, and median 5.8% (mean 5.8%) mitochondrial counts. Cells from all libraries were integrated with the canonical correlation analysis (CCA) method implemented in Seurat version 314 with default parameters. After CCA, the standard count processing was rerun on the integrated assay with default parameters, except for the number of principle components used (set to 35) and the clustering resolution (set to 1.1). The resulting 23 clusters were manually annotated to a general cell type level with the following marker genes: macrophage (Csf1r, Cd68, Adgre1), smooth muscle (Myh11, Tagln, Cnn1), endothelial (Pecam1, Cdh5, Cldn5), pericyte (Rgs5), dedifferentiated SMC (Vcam1, Lgals3, Dcn), chondrocyte-like (Comp, Fmod), fibroblast (Fbln1, Pdgfra, Serpinf1), mesenchymal stromal (Ly6a, Pi16), NK/T (Cd3d, Cd8b1, Nkg7), and epithelial-like (Upk3b). Hashtag assignment was carried out with the HTODemux function of Seurat. As the Biolegend TotalSeq cell hashing staining reagent is a mixture of anti-CD45 and anti-MHC I antibodies, HTODemux was run separately for the immune and non-immune cells of each library. To remove ambient RNA contamination, we ran the DecontX function (celda R package version 1.1.6)15 by using the automated clusters (resolution 1.1) as the cell population labels and we used the decontaminated counts for subsequent analysis. Cells with an estimated ambient RNA contamination fraction > 0.3 were excluded from downstream analyses as possible doublets or otherwise low-quality cells.
To identify disease-associated cell states, we divided the Uniform Manifold Approximation and Projection (UMAP) plot from the integration of all libraries into a grid of 200 by 200 (for visualization) or 50 by 50 (for cell selection) subdivisions and counted the number of cells from each library falling into each sector. The cell occupancy was normalized to the total number of cells in each library, scaled to a library size of 10,000 cells, and log2-transformed. Subsequently, for each sector, the fold change in cell abundance was calculated between the late disease sample and the healthy control sample. UMAP regions displaying a more than 2-fold (>1 log2 fold change) increase in cell occupancy in response to disease were selected for further investigation.
Marker genes of disease-increased UMAP regions (cell states) were defined with the Wilcoxon rank-sum test (implemented in Seurat) comparing the cells located within the area of increased UMAP occupancy to the other cells of the same general cell type (i.e., cells not changed in abundance upon disease). Genes expressed in at least 10% of cells (in at least one of the cell populations of the comparison) were considered and the minimum required log fold change was 0.25. p values were corrected for multiple testing with false discovery rate (FDR) and padj < 0.05 was considered significant. One highly reliable marker gene was selected to name the cell state whenever possible. The full marker lists are available in Table S2.
Gene Ontology enrichment analysis for cell state marker gene lists was performed with the g:Profiler web tool (access date 2020-03-26).16 All cell state marker gene lists were tested for enrichment against all Gene Ontology Biological Process (GO:BP) categories. The cell state SMC-4 was not included in this analysis, as it had only three marker genes. To reduce redundant and overly general hits, we filtered the GO:BP enrichment results for each cell state marker gene list to remove categories with >1,000 genes in mouse and we further filtered for GO term semantic similarity by using the GOSemSim package (version 2.8.0)17 by ranking the GO terms on the basis enrichment p value and, starting from the top, removing any less significant terms with Rel similarity measure > 0.9. After the GO term filtering, the top four most significantly enriched GO:BP terms were selected from each cell state (provided the terms satisfy padj < 0.05) for a comparative enrichment heatmap.
To plot gene set enrichment at the single-cell level, we used the VISION package (version 2.0.0)18 with the GO:BP and MSigDB Hallmarks19 gene sets.
To predict which extracellular ligands may serve as upstream inducers of a cell state, we used the NicheNet package (version 0.1.0).20 For the query gene signature (i.e., the gene set to be explained by ligand-receptor interactions), an entire list of marker genes for a disease-associated cell state was used (described above). The cells of the disease-increased cell state were considered the “receiver” cell population (expressing receptor) and all other cell types in the dataset were considered potential “sender” cells (expressing ligands). A ligand or receptor was considered expressed in a cell population if detected in at least 10% of the cells. Predicted ligand activities were ranked with the Pearson correlation coefficient (default).
We used SCENIC (version 1.1.1)21 to predict transcriptional regulators, motifs, and regulons that are preferentially active in disease-associated cell states. SCENIC single-cell regulatory network inference was run following the published vignette for one cell type at a time. Cells from the maximally different disease stages (control and late disease) were used. For SMC-related cells, cells were further randomly subsampled with the automated clusters (resolution 1.1; described above), keeping a maximum of 200 cells per cluster. The cisTarget database was mouse mm9 transcriptional start site (TSS)-centered (±10 kb), v9 motifs, seven species (download date 2020-01-24). The activity of each predicted gene network was scored in each cell and binarized, as described by the SCENIC authors. We used the Wilcoxon rank-sum test to find regulons that are differentially active in one cell state compared to the other cells of the same cell type. For differential activity, the regulon was required to be active in at least 20% of the cells of one population and satisfy log fold change > 0.15 and FDR < 0.05.
Decomposition of cell states in mouse bulk RNA-seq
We trimmed mouse aorta RNA-seq reads by using Trim Galore (version 0.4.4; GitHub: https://github.com/FelixKrueger/TrimGalore) to remove adapters and bases with quality score below 20. We aligned reads to the mm9 mouse genome with STAR (version 2.5.4)22 and used HOMER (version 4.9)23 to generate a gene count matrix from uniquely mapped reads.
To evaluate relative changes in cell state abundance between atherosclerosis disease stages based on mouse aorta bulk RNA-seq profiles, we applied the Cell Population Mapping method (scBio R package; version 0.1.5).24 Aorta scRNA-seq cells and the integrated UMAP were used as the cell state space. The bulk RNA-seq count matrix was normalized for transcript length with reads per kilobase per million reads mapped (RPKM) and log-transformed. Cell Population Mapping was run in relative abundance mode (subtracting the mean of the reference group from all samples in the test group) with default parameters.
Cell state gene signature activity in human atherosclerosis study cohorts
Gene expression microarray data was obtained for human aortic plaque (n = 15; abdominal aorta), carotid plaque (n = 29), femoral plaque (n = 24), and non-atherosclerotic control artery (n = 24; left internal thoracic artery) from the Tampere Vascular Study25 and from human carotid artery segments classified as either advanced (n = 16) or early (n = 13) atherosclerotic plaque (GEO: GSE28829).26 Gene expression levels were quantile-normalized with the preprocessCore R package (version 1.52.1; GitHub: https://github.com/bmbolstad/preprocessCore). To calculate gene signature activity scores in these bulk RNA profiles, we used the approach of Tirosh et al.27 (implemented as the Seurat function AddModuleScore), wherein genes are binned on the basis of average log expression level across samples, and in each sample, a bin background level (calculated from random control genes in the same bin) is subtracted from the levels of the test genes. The full lists of marker genes of atherosclerosis-increased cell states were used as the tested gene sets (gene programs).
Prioritized genes at human CAD GWAS loci
To associate human CAD GWAS loci to their potential causal genes, we collected published gene prioritization results across multiple different prioritization approaches and GWASs.
We used the OpenTargets Genetics Portal (data release: version 5–21.06)28 to obtain data for the following published GWASs of CAD, myocardial infarction, and stroke29,30,31,32,33,34,35 and also for the biobank-based GWAS results36,37 for FinnGen coronary atherosclerosis and UK Biobank ischemic heart disease, myocardial infarction, and coronary atherosclerosis (OpenTargets study accessions FINNGEN_R5_I9_CORATHER, SAIGE_411, SAIGE_411_2, and SAIGE_411_4). Across the studies, the OpenTargets Locus2Gene algorithm, a machine learning method trained on a gold-standard curated result set, prioritized 238 unique genes, OpenTargets eQTL colocalization 164 genes, and OpenTargets “nearest gene” 387 genes. The recent very large CAD GWAS by Aragam et al.9 was additionally included. From this study, the per-association overall top-prioritized genes contributed 186 unique genes, polygenic priority score (PoPS) 386 genes, and “nearest gene” 216 genes, and for the GWAS 1% FDR threshold loci, “nearest gene” contributed 716 genes. Additionally, the transcriptome-wide association study (TWAS) of CAD by Li et al.38 prioritized 114 unique genes and the CAD GWAS review by Erdmann et al.39 listed 373 genes at CAD loci.
Partitioned heritability with linkage disequilibrium score regression (LDSC)
LDSC (version 1.0.1)40 and the van der Harst et al. CAD GWAS29 full summary statistics (downloaded from Mendeley Data: https://doi.org/10.17632/gbbsrpx6bs.1) were used for partitioned heritability analysis of CAD following the recommendations published by LDSC authors. The marker genes of atherosclerosis-associated cell states (described above) were used as the gene sets. The gene-transcribed regions in the hg19 human genome were padded with 100 kb upstream and downstream. The LD score window was 1 cM (default), the provided 1000 Genomes (1000G) Phase 3 EUR data files were used, and the 1000G Phase 3 EUR baseline models were included in the calculation.
Accessible chromatin by cell type in human atherosclerotic plaques
We used human endarterectomy single-cell assay for transposase-accessible chromatin with sequencing (scATAC-seq)11 transposase cut site coordinates and cell annotations (available at FigShare: https://doi.org/10.6084/m9.figshare.14501985.v2) to aggregate cut sites to pseudobulk at the level of cell lineages (SMC, EC, macrophage, T/NK, and B/plasma) and we called peaks in each cell lineage separately by using MACS241 with fragment extension 200 bp and shift −100 bp. Where equal-width peaks are stated, we resized peaks centering on the MACS2-called peak summit coordinate (cell type specific).
Gene set-based PRSs
We used the PRSice-2 (version 2.3.3)42 feature PRSet,43 which performs region-aware (coordinate set-based) SNP clumping, to generate clumping and thresholding PRSs based on sets of cell state marker genes, scATAC-seq peaks, or genes of a biological pathway. In PRSet, SNPs falling within regions of interest are preferentially retained for each LD clump of SNPs. The clumping distance was 500 kb to either side of the index SNP and the LD r2 threshold was 0.2.
The base data for all PRSs were the additive model summary statistics of the CARDIoGRAMplusC4D 1000 Genomes-based CAD GWAS,44 a study that does not have significant sample overlap with the UK Biobank.29 We filtered base data genetic variants to keep variants with INFO score ≥ 0.8 and to exclude variants with strand-ambiguous alleles.
The UK Biobank45 was used as the target cohort for all PRS scoring. We filtered the imputed genotype data to keep autosomal variants with MAF ≥ 0.01 and INFO score ≥ 0.8 and exclude variants with Hardy-Weinberg equilibrium test p < 1e−25 or genotype missingness rate > 0.1 by using PLINK (version 2.00).46 The final variant set across base and target data consisted of 4,958,173 variants. We included participants of self-reported White ancestry in the precomputed UK Biobank PCA calculation (unrelated samples) and selected available imputed genotype data and filtered to exclude participants with sex chromosome aneuploidy, heterozygosity or genotype missingness outliers, participants with excess relatives, kinship inference analysis exclusions, those more than seven standard deviations away from mean in the first six principal components (PCs) (GitHub: https://github.com/Nealelab/UK_Biobank_GWAS), and participants who had withdrawn consent. CAD phenotype status in the UK Biobank was defined on the basis of the example of Choi et al.43 (specifically, GitLab: https://gitlab.com/choishingwan/prset_analyses/-/blob/master/script/sql/generate_cad.sql), which defined CAD cases by using primary or secondary ICD10 or ICD9 codes for hospital inpatient records or cause of death and additionally OPSC4 operation codes and self-reporting by the participant. In total, there were 21,600 cases and 359,254 controls.
For gene set-based PRSs generated from gene coordinates alone (i.e., not using scATAC-seq regulatory region information), we extended the transcript regions to include 35 kb upstream and 10 kb downstream. To utilize scATAC-seq peaks to inform cell state marker gene PRSs, we only considered the scATAC-seq peaks called in the corresponding cell type and peaks within ±500 kb of the gene TSS were retained for PRS generation.
PRS performance was evaluated as set out in the PRSet method.43 We evaluated variance explained by the PRS (PRS.R2) by subtracting the pseudo R2 of the full model (CAD ∼ PRS + covariates) from the pseudo R2 of the null model (CAD ∼ covariates). The covariates used in PRS modeling were sex, age (earliest age of CAD for cases, oldest age at attending UK Biobank center for controls), UK Biobank assessment center, genotype batch, and the first ten genetic PCs. We used the PRSet competitive p value calculation,43 based on permutation testing (10,000 permutations), to test for signal enrichment compared to identically clumped SNPs in regions of the genome considered background (defined as either all genes or all scATAC peaks).
Results
Identification of atherosclerosis-associated cell states
To provide an unbiased, enrichment free, analysis of all the cell types of the healthy and atherosclerotic vascular wall, we performed scRNA-seq from single cell suspensions extracted from thoracic aorta. Cells from three mice were labeled with hashtag antibodies and pooled from each of four conditions, including healthy control mice and Ldlr−/−/Apob100/100 mice with pre-lesioned, early, and late atherosclerotic lesions, and prepared into libraries with the 10× Chromium system (Figure 1A). The scRNA-seq profiles of 36,157 cells passed quality control and were selected for downstream analysis (Figure S1). Joint analysis of atherosclerotic and control samples with automated clustering identified 23 cell clusters that corresponded to nine major cell types, including endothelial cells (ECs), NK/T cells, monocyte/macrophages (MPs), smooth muscle cells (SMCs), pericytes, dedifferentiated SMCs, chondrocyte-like cells, mesenchymal stromal cells (MSCs), and fibroblasts (FBs) according to marker gene-based curation (Figures 1B and S2 and Table S1).
Figure 1.

Clustering and identification of atherosclerosis-associated cell states
(A) Schematic overview of the experimental setup.
(B and C) (B) UMAP projection of the scRNA-seq profiles represented as eleven manually annotated clusters. (C) UMAP regional occupancy analysis demonstrating relative changes in cell density comparing atherosclerotic vascular wall to healthy controls. Atherosclerosis-associated cell states are revealed by increased local abundance of cells in regions of the UMAP plot (log2FC, log2 fold change).
(D) UMAP plot depicting the 12 disease-associated cell states and the selected top marker genes.
(E) Relative changes in the cell state proportions during different stages of atherosclerosis shown for each of the three biological replicates. Diamond represents the average of the three replicates.
Extensive variability in the regional occupancy of the Uniform Manifold Approximation and Projection (UMAP) was observed across different disease stages after data integration, indicating a spectrum of cellular states associated with atherosclerosis. To define discrete cell states (within broader cell types) that are atherosclerosis associated, we selected regions of the UMAP plot where the local cell density was increased >2-fold in late disease compared to control. (Figures 1C and S3). Differential neighborhood abundance analysis based on the k-nearest neighbor graph (Milo method47) largely confirmed the regions of altered abundance (Figure S4). The marker genes of disease-increased cell states were defined by comparing the cells located within the area of increased UMAP occupancy to the other cells of the same cell type that did not change in abundance (full marker lists in Table S2). Therefore, it should be noted that the cell state markers represent a distinct signature from the general cell type markers that are obtained by comparison between cell types (Tables S1 versus S2). Using the marker gene sets to score gene program activity revealed that the cell state signatures tend to be upregulated gradually, and the cells within the defined cell state showed >1 SD unit of elevated expression relative to the expression variation in the cell type as a whole (Figure S5).
Our analysis identified 12 cell states that were increased in at least one of the diseased conditions (Figure 1D), which were named on the basis of one of the top marker genes. These included three SMC cell states, including Vcam1+ SMC, Col2a1+ SMCs, and Palld+ SMCs, three macrophage-derived cell states called Spp1+ MPs, Ccl4+ MPs, and Stmn1+ MPs, two EC cell states corresponding to Lrg1+ ECs and Calcrl+ ECs, and two NK/T cell states, Nkg7+ NK/Ts and Ramp3+ NK/Ts, as well as Smoc2+ FB state and Apoe+ MSC state. We noted a further candidate population of disease-increased cells among the contractile (Myh11+) SMCs (Figures S2B, S4, and 1D). However, it appeared to be a very mild perturbation, showing only three marker genes (Slc22a1, Rbp4, Ndufa3), and therefore was not included in subsequent analyses. Importantly, the cell states demonstrated temporal patterns corresponding to the progression of the disease. For example, the proportion of cells associated with EC and MP states as well as Vcam1+ SMC and Palld+ SMC state cells exhibited a gradual increase toward late disease stage, whereas NK/T states, Col2a1+ SMCs, and Smoc2+ FBs were mostly detected at the late disease stage (Figure 1E). This highlights the importance of temporal resolution in scRNA-seq-based analysis of disease progression. Among the cell state markers, we selected LRG1, VCAM1, and PALLD for validation with immunohistochemistry (Figure S6) and Lrg1 and Palld for confirmation by using spatial transcriptomics with the Molecular Cartography platform by Resolve Biosciences (Figure S7). In addition, we used alternative marker genes to visualize the Vcam1+ SMCs (Col6a3), Col2a1+ SMCs (Col6a3 and Sox9), Ccl4+ MPs (Il1b), Spp1+ MPs (Abca1), and Stmn1+ MPs (Top2a) because the primary marker genes of these states exceeded the maximum expression level limit and had to be excluded at the panel design stage (Figure S7). Altogether, this analysis confirmed the distinct location and identities of several of the cell states.
Validation of the atherosclerosis-associated cell states in mouse and human lesions
To confirm that the identification of atherosclerosis-associated cell states is not confounded by technical issues related to scRNA-seq, we further sough to analyze the presence of the gene signatures in bulk RNA-seq. In particular, cell dissociation could alter the relative proportions of cell populations in scRNA-seq experiments, an effect that would not occur with bulk RNA-seq.48 We therefore performed a parallel bulk RNA-seq experiment with 5–6 replicates and estimated the performance of these two methods in capturing cell type proportions during atherosclerosis progression (Figure 1A). Altogether 1,049 unique genes were upregulated and 851 downregulated during the course of disease development (Table S3). Notably, a large majority of the bulk RNA disease-induced genes were expressed in immune cells, in line with expected infiltration of these cells to the vascular wall during disease progression and indicating that changes in cell type proportion are a major driver of bulk RNA-seq differential expression (Figures S8A and S8B). Deconvolution of bulk RNA-seq data with the scRNA-seq data as in Newman et al.49 confirmed the gradual increase of macrophage and NK/T cells relative to other cell types (Figure S8C). This was also evident from the scRNA-seq data itself, although the relative proportion of leukocytes was smaller compared to bulk RNA deconvolution results, possibly reflecting selective cell loss due to tissue dissociation (Figure S8D). To further leverage the bulk RNA-seq data to validate the atherosclerosis-associated cell states, we estimated the relative cell state abundance changes by using cell population mapping,24 where scRNA-seq profiles are used to infer the composition of cell states from bulk transcriptome data. This analysis supported the increase in cell state abundance for majority (9/12) of the cell states but failed to recapitulate Palld+ SMCs, Smoc2+ FBs, and Apoe+ MSC state changes, possibly because of the low amount of cells in these populations (Figure 2A). Altogether, this analysis supports the reliability of our workflow for cell state identification from single cell expression data.
Figure 2.

Atherosclerosis-associated cell state signatures are activated in mouse and human lesions based on bulk RNA-seq
(A–C) (A) Cell population mapping using the scRNA-seq cell state space to plot the differential cell abundance estimated from the mouse bulk RNA-seq data. The gene set activity scores for each cell state were further investigated in the (B) Tampere Vascular Study15 representing 68 advanced atherosclerotic plaques (15 aortic, 29 carotid, and 24 femoral plaques) and 28 controls (left internal thoracic artery) and (C) the Maastricht Pathology Tissue Collection16 representing atherosclerotic carotid artery segments from 13 early intimal thickening/xanthoma lesions and from 16 advanced fibrous cap atheroma lesions.
We next sought to investigate the reproducibility of the atherosclerosis-associated cell states in public datasets of mouse and human scRNA-seq. To achieve this, we compared the enrichment of gene set modules in the cell type of interest in our data, in different mouse models of atherosclerosis4 and in human coronary lesions3 (Figures S9–S14). In the Pan et al.4 dataset including Ldlr−/− (0, 8, 16, and 26 weeks of HFD) and Apoe−/− (8, 16, and 22 weeks of HFD) mouse models, the cell state markers were enriched in a specific subpopulation of cells, suggesting that they represent distinct phenotypes (Figure S11). The fraction of gene set positive cells were increased upon disease progression for all other cell types except the Palld+ SMC and Calcrl+ EC states, which demonstrated a relative decrease from the earliest timepoints in both mouse models (Figures S12 and S13). In addition, FB and MSC states were only increased between 8 and 16 weeks of HFD in the Ldlr−/− mouse model, whereas NK/T states exhibited a decrease in cell fraction between 16 and 26 weeks of HFD, suggesting mouse-model- and time-point-specific differences.
In human lesions,3 all the cell state markers with the exception of Calcrl+ ECs had a distinct localization within the cell type cluster (Figure S14), supporting that the same cell states are present. However, the available human scRNA-seq datasets do not allow comparison between healthy and diseased vasculature, which is why we further extended our analysis to bulk RNA-seq datasets to investigate whether the cell state signatures were associated with disease progression. To achieve this, we studied the expression of the cell state markers in two human clinical cohorts, the Tampere Vascular Study,25 representing 68 advanced atherosclerotic plaques (15 aortic, 29 carotid, and 24 femoral plaques) and 28 controls (left internal thoracic artery), and the Maastricht Pathology Tissue Collection,26 representing atherosclerotic carotid artery segments from 13 lesions at the early intimal thickening/xanthoma stage and from 16 advanced fibrous cap atheroma lesions. Altogether, 10 out of the 13 disease state gene signatures separated the diseased samples from the controls (Figures 2B and 2C). The Palld+ SMC and Calcrl+ EC markers were only induced in the aortic plaques, compared to femoral or carotid plaques or control arteries, suggesting vascular-bed-specific differences. These results demonstrate similarities between the atherosclerotic cell states in mouse and human and support a potential role for these genes in disease etiology.
Identification of cell-state-specific and shared pathways among gene signatures
The maintenance and transition of cellular states are controlled by environmental signals that translate into gene regulatory mechanisms and biological processes. To investigate the similarity of the biological process activities of the atherosclerosis-associated cell states, we next evaluated the uniqueness of the cell state markers and their associated gene ontologies. Comparison of the marker gene sets of each disease-associated cell state demonstrated that the large majority (71%; 1,516/2,122) of the signature genes were unique to one cell state (Figures 3A and 3B). Still, 12 genes were common to five or more cell states, as exemplified by matrix Gla protein (Mgp), stem cell antigen-1 (Sca1; also called Ly6A) and legumain (Lgmn), syndecan 4 (Sdc4), and insulin-like growth factor binding protein 4 (Igfbp4), and could thus represent global markers of atherosclerosis.
Figure 3.

Characterization of atherosclerosis-associated cell states and key biological pathways
(A) Marker gene counts for the 12 most abundant disease-associated cell states.
(B) Common markers between disease-increased cell states.
(C) UpSet plot showing the gene overlaps between cell state signatures and the gene ontologies enriched (log10 of adjusted p value) in the intersections.
(D) Single-cell gene set enrichment scoring for selected biological processes. Enrichment score is shown from the lowest 5% (Q5) to highest 95% (Q95).
Comparison of marker gene lists at the level of gene ontology allowed to further shed light into the phenotypes of these cell states (Figures 3C and 3D; Table S4). For example, Vcam1 itself and the other associated cell state markers participating in ECM remodeling have been previously identified as hallmarks of SMC-derived intermediate cells,4 also called fibromyocytes.3 These cells have been suggested to differentiate into chondrocyte-like cells,2,5 i.e., fibrochondrocytes,4 which correspond to the Col2a1+ SMC state. This gave us a unique opportunity to model a three-state continuum for the SMCs (Myh11+ - Vcam1+ - Col2a1+) not evident for the other cell types. The hypothesis proposing that contractile SMCs can undergo transdifferentiation into Vcam1+ SMCs and Col2a1+ SMCs was further corroborated by the results of pseudotime trajectory analysis (Figure S15).
Among ECs, the Lrg1+ ECs expressed markers of endothelial-to-mesenchymal transition, such as Sox4,50 Tubb3,51 and Fbln5,52 in line with a recent report.8 On the other hand, Calcrl+ EC state genes were indicative of regulation of endothelial function by mitochondrial reactive oxygen species (electron-transport chain) and shear stress (e.g., Calcrl, Klf4, Pecam1, Tek [Tie2], Jun, and Fos).7 Our analysis also uncovered the phenotypes of the five atherosclerosis-associated immune cell states as similar to lipid-associated Trem2+ MPs (Spp1+ MP1), proinflammatory MPs (Ccl4+ MPs), proliferating MPs (Stmn1+ MPs),1 Nkg7+ Cytotoxic CD8 T cells, and Ramp3+ antigen-specific CD4 T cells.53,54
Despite extensive cell state specificity of the marker gene sets, we also identified shared biological processes between some cell states. Gene ontologies related to ECM organization, blood vessel development, endodermal cell differentiation, and angiogenesis were enriched among the shared genes of Vcam1+ SMC, Col2a1+ SMC, and Lrg1+ EC states, whereas enrichment for cholesterol transport and homeostasis genes was shared between Vcam1+ SMC, Col2a1+ SMC, and Spp1+ MP states (Figures 3C and 3D). Taken together, these results provide evidence that the disease-associated cell states largely contribute to specific cellular functions in the atherosclerotic niche with, however, important overlapping activities related to vascular development, angiogenesis, and lipid metabolism.
Disease states share similar upstream regulators but differ in predicted TF activities
We next sought to investigate how the gene signatures of disease-associated cell states could be defined by the cellular microenvironment by inferring intercellular communications and intracellular signaling networks. We first looked for expressed ligands and receptors and modeled their gene regulatory effects in putative ligand-receiving cells for the five most abundant disease states by using the NicheNet tool20 (Figure 4A). Ligands belonging to the transforming growth factor β (TGFβ), inflammation (TNF, IL-1α/β), angiogenesis (VEGFA, FGF1/2), and cholesterol pathways (ApoE) were identified as the most probable ligands to give rise to the gene sets, suggesting extensive similarities in the microenvironmental signals underlying cell-state-specific gene expression (Figures 4A and 4B). Among them, Tnf and Il1a/b were mostly expressed by the lipid-associated Spp1+ and proinflammatory Ccl4+ MPs, suggesting extensive auto- and paracrine proinflammatory signals originating from myeloid cells (Figure 4C). Importantly, these proinflammatory ligands were predicted to regulate several cell state genes specific for Vcam1+ and Col2a1+ SMCs (Figure 4B). To test the validity of these computational predictions, we stimulated mouse SMCs in vitro by using the top-ranked ligand IL-1β for 24 and 48 h and modeled the pseudotemporal trajectory of the gene expression response on the basis of scRNA-seq. Our results demonstrated that the genes upregulated along the in vitro SMC IL-1β response trajectory were induced along trajectory of in vivo SMC disease-associated transition and vice versa, supporting the predictions (Figure S16). Finally, Col2a1+ chondrocyte-like cells were predicted to be the main producers of TGFβ1, VEGFA, and BMP2, highlighting their potential role in a cell-cell signaling network of autocrine osteochondrogenic signaling and paracrine signaling driving the fibrogenic and angiogenic gene expression programs in Vcam1+ SMCs and Lrg1+ ECs (Figures 4D and S17).
Figure 4.

Modeling intercellular communication between cell states
Ligand–receptor–target gene analysis was carried out with NicheNet.
(A) Top 10 prioritized upstream ligands for cell state signature gene sets.
(B) Ligand target gene networks presented for Vcam1+ SMCs, Col2a1+ SMCs, and Lrg1+ ECs.
(C) Expression of the prioritized ligands by the different cell states and types. Row normalized gene expression (TPM = transcripts per million) is shown.
(D) Schematic of predicted ligand-mediated signaling between Vcam1+ and Col2a1+ SMC states involving autocrine and paracrine signaling.
The predicted similarities in the microenvironmental signals suggest that the differences in disease-associated gene signatures could arise as a result of differential response of the cell types to the same stimulus and thus cell-state-specific transcription factor (TF) activity. To investigate this, we inferred TF regulatory networks by single-cell regulatory network inference and clustering (SCENIC) analysis for the three major cell lineages (SMCs, ECs, and MPs; Figure 5A).21 First, we predicted the transcription factors for the Palld+, Vcam1+, and Col2a1+ SMC-derived cell states (Figure 5B). The Palld+ cell state gene expression profile was predicted to be driven by proinflammatory TFs such as the NF-κB, AP-1, and Maf family members. In line with the predicted differentiation trajectory of the Vcam1+ and Col2a1+ cell states from contractile SMCs, the SCENIC predictions supported a gradient of TF activity (Figures 5E and S15C) and evolution of gene networks. CEBPD and RUNX1 were identified as the key regulators for both atherosclerosis-associated cell states despite the higher expression of Cebpd in Vcam1+ SMCs and Runx1 in Col2a1+ SMCs. In addition, the transition from Vcam1+ cell state to Col2a1+ state was predicted to be driven by the ER-Golgi stress transducer CREB3 family members55 as well as EndMT associated HAND2 and SNAI1 TFs.56 TFs induced by inflammation and metabolic stress, including XBP1, ATF4, RARG, and NFATC1/2, were identified as key drivers of the Col2a1+ state.
Figure 5.

Prediction of cell-state-specific transcription factor activities
(A–D) (A) The most abundant atherosclerosis-associated cell states were selected for SCENIC analysis21 along with disease-unperturbed cells of the same cell type. Differentially active gene regulatory networks identified for (B) smooth muscle cell (SMC), (C) endothelial cell (EC), and (D) macrophage (MP) cell states. The predicted regulon activity and transcription factor gene expression (row normalized TPM) are shown.
(E) Selected examples of regulon activities and transcription factor gene expression plotted on UMAP.
In line with marker gene expression, SOX4 was predicted as the main driver of the Lrg1+ EC state, followed by XBP1, TCF4, CEBPB, IRF3, CREB3L2, and ELK3 (Figures 5C and 5E). The analysis was unable to identify candidate regulators for Calcrl+ EC state. Interestingly, XBP1, TCF4, CEBPB, CREB3L2, and ELK3 were also identified as key regulators of the Vcam1+ and Col2a1+ SMC states, suggesting a key role of these TFs across cell types. As expected, the inflammatory Ccl4+ MPs showed enrichment of proinflammatory TF motifs, such as STAT, IRF, and AP-1 factors, whereas the Spp1+ lipid-associated MPs were enriched for SREBF1, USF1, NR1H3, and the ER stress response TFs DDIT3 and XBP1, which all have been implicated in lipid homeostasis57 (Figures 5D and 5E). The proliferating Stmn1+ MPs were enriched for TF motifs implicated in cell cycle regulation, as exemplified by the members of the E2F TFs and EZH2. Among the TFs identified, XBP1, MAFB, MLX, and NFE2L2 (also called NRF2) were also identified as drivers of the Vcam1+ and Col2a1+ SMC states. Overall, we conclude that the majority of the single-cell gene regulatory networks are cell state specific and could provide critical insights into essential factors driving the progression of atherosclerosis.
Interrogation of GWAS loci and partitioning the heritability of CAD highlights the importance of smooth muscle cell states
GWASs have identified over 300 risk loci for CAD.9,10,29 Nonetheless, for a vast majority of the loci, the causal gene(s) underlying the association are not known with certainty. In the last few years, however, several new methods have emerged that, on the basis of genomic proximity, protein-coding variants, variant association to gene expression (QTL, TWAS), enhancer-gene maps, or similarity in gene functions or pathways, establish links between risk loci and genes,9,28,38,39 providing hundreds of candidate causal genes. We made use of these candidate causal gene lists to compute their enrichment among the cell state gene sets by using the hypergeometric test with expressed genes (>1 TPM in any aortic cell type or state; 14,902 genes) as the background. Our results demonstrate that 11 of the 12 cell states included genes prioritized as candidate causal genes, with the highest enrichment detected for the three SMC state (Vcam1+, Col2a1+ and Palld+) and Lrg1+ EC state genes (Figures 6A and S18). This enrichment trend was also evident when the marker gene lists were truncated to a specific number of top genes (Figure S19). In a further analysis, we equalized the number of marker genes for each cell state across a larger range (top 25 to 500 genes) by also including sub-threshold marker genes where needed. These results (Figure S20) also confirmed SMC (Vcam1+, Col2a1+, and Palld+) and EC (Lrg1+) states as the most enriched for CAD-GWAS-prioritized genes, including when measured by overlap ratio.
Figure 6.

The contribution of cell states to CAD heritability
(A) Overlap of the CAD GWAS candidate causal gene lists from nine different sources (different colors) with the cell state markers. NA indicates no overlapping genes.
(B) Results from LD score regression (LDSC)40 applied to cell state marker genes (using 100 kb flanking regions) to partition CAD heritability within the genome.
To further investigate the contribution of atherosclerosis-associated cell state signatures to the SNP-based heritability of CAD, we applied the LDSC tool40 to partition heritability to gene sets by using CAD GWAS summary statistics. In our analysis, CAD heritability was most significantly enriched in regions surrounding Vcam1+ SMC state genes, followed by Col2a1+ and Palld+ SMC states, Spp1+ and Stmn1+ MPs, and Lrg1+ ECs (Figure 6B). Our results demonstrate significant enrichment of SMC cell state genes over all other cell types of the lesions, which could help in functionally interpreting the GWAS signal.
Cell-state- and pathway-specific PRSs provide insight into the biological mechanisms of CAD risk
An emerging body of evidence has shown that aggregation and weighing of CAD-associated variants into PRSs can improve an individual’s risk prediction beyond traditional risk factors and provide an opportunity to identify novel mechanisms influencing CAD risk.58 GWAS results could be considered a composite of signals corresponding to CAD-relevant processes encoded by different genomic regions and biological pathways. We therefore sought to test how much polygenic risk for CAD is influenced by the cell state, cell type, and gene ontology pathways by aggregating the risk alleles across the respective gene sets. To study this, we employed the PRSice-2 extension PRSet,43 which performs LD clumping in a region set-aware manner, generating PRSs that preferentially include SNPs falling into regions of interest. We used the GWAS summary statistics from the CARDIoGRAMplusC4D44 study as the basis for PRS generation, and we used the UK Biobank cohort (21,600 CAD cases and 359,254 controls) to evaluate PRS performance.
We first applied PRSet to cell state marker gene sets (gene body with −35 kb and +10 kb flanks). To evaluate PRS performance, PRSet computes the amount of phenotypic variance explained by the PRS, defined as the increase in model R2 when PRS is included, compared to a null model consisting of covariates only. Further, permutation-based significance testing is carried out, comparing the performance for a PRS to identically clumped SNP sets from background regions.43 We observed that PRS derived from the Vcam1+ SMC state genes explained greatest CAD risk variance and was strongly enriched in the predictive power relative to background, followed by Col2a1+ SMCs and Lrg1+ ECs (Figure 7A). Importantly, the cell state genes outperformed the corresponding cell type marker genes in predictive performance (Figure S21). Truncating the cell state marker gene sets to a specific number of top genes revealed that performance does tend to increase with the number of genes; however, the increase appears more rapid for some cell states than others, and permutation-based p value (i.e., predictive power relative to background SNP sets) tends to plateau for several cell states (Figure S22).
Figure 7.

Pathway- and cell-state-specific polygenic risk scores shed light into the genetic basis of CAD
(A) Cell-state-specific PRS was constructed with (A) gene coordinates (−35 kb upstream to 10 kb downstream) using PRSet.43 To obtain the empirical p value, random SNP sets containing the same number of post-clumping SNPs were selected from background regions of the genome, selected from all genic regions.
(B) Explained variance of each pathway-specific PRS to polygenic risk of CAD calculated for the gene sets listed in Figure 3C.
(C) Cell-state-specific PRS analysis constructed with plaque scATAC-seq peak coordinates that were found within ±500 kb of the TSS.
(D) Proportion of variance of CAD explained by PRS in genome-wide analysis. The values represent PRS calculated for all cell-type-specific scATAC-seq peaks at different p value thresholds, which are compared to the classical genome-wide clumping and thresholding PRS.
To investigate how marker genes of non-plaque cell types perform in a similar analysis, we derived marker sets (Table S5) for 79 human cell types across the body by using the gene expression profiles compiled from scRNA-seq datasets.59 As an evaluation of the marker sets, related cell types showed more pairwise shared markers (Figure S23), as expected, and testing the 79 cell type marker sets (top 500 genes each) for overlap with CAD-GWAS-prioritized genes revealed the strongest enrichment in SMC and EC, followed by FB and adipocytes, while most cell types showed no significant enrichment (Figure S24). Next, we constructed gene set-based PRSs for CAD by using between 20 and 500 top markers for each cell type (gene body regions + flanks) and evaluated PRS R2. Out of the 79 cell types, hepatocytes ranked highest, followed by SMCs, with adipocytes and ECs also among the top ten cell types (Figure S25). Several other cell types in the top ten tend to be ones that share markers with SMCs (Figure S23).
Pathway-based PRSs have been suggested to better inform disease biology compared to classical PRSs.43,58 We therefore also performed PRS analysis on the gene ontologies that were significantly enriched among the cell state signature genes (Figures 2C and 2D). For each of the significant ontology categories (Figures 2C and 2D), we collected all human genes assigned to the ontology and used these to create a CAD PRS. Interestingly, the PRSs derived from functional categories that were shared among several cell states (Figures 2C and 2D), including cholesterol transport, lipid localization, blood vessel development, and angiogenesis gene sets, were among the top in predictive performance and enrichment significance (Figure 7B). In addition, enzyme-linked and growth factor receptor signaling, cell division, leukocyte differentiation, and cell cycle, were among the top ten categories associated with CAD risk. Altogether, our analysis suggests pivotal roles for disease-associated cell states and pathways as mediators of the genetic risk for CAD.
A great majority of the genetic variants associated with CAD are located within non-coding elements of the genome where they are thought to play a role in gene expression regulation. In line with this, inclusion of functional annotations of genomic and epigenomic elements has been shown to improve the prediction accuracy of PRSs.60 Therefore, we derived a next set of PRSs prioritizing for variants in plaque cell type regulatory elements, represented by scATAC-seq peaks.11 Using scATAC peaks in range TSS ± 500 kb for cell state marker genes resulted in PRSs consisting of similar numbers of variants as the gene coordinate-based PRSs above, allowing direct comparison of power. Notably, PRS performance was considerably improved when we used the regulatory elements as functional priors, with ∼2-fold increase in R2 (Figures 7A versus 7C). In line with gene coordinate-based analysis, Vcam1+ SMC state gene continued to outperform other cell states.
As the regulatory element-based analysis appeared to inform SNP selection, we further tested scATAC peak-based PRSs genome wide (i.e., without gene set limitations). Out of the cell types studied, SMC scATAC-based PRS performed the best, although the combined PRS from all plaque scATAC cell types outperformed it (Figure 7C). This was not dependent on the total peak counts, as selecting the strongest 10,000 cell type unique peaks reproduced similar results (Figure S26). At its optimal p value threshold, the standard (classical) genome-wide PRS outperformed the scATAC PRSs. However, the p value threshold curves for scATAC and standard PRSs were differently shaped, and the scATAC PRS continued to gain power even at weaker p value thresholds whereas the standard PRS lost power (Figure 7D). Based on this observation, we constructed a hybrid PRS where all strong p value SNPs were included irrespective of scATAC data, and scATAC was only used for weaker p value variants. This hybrid PRS outperformed the classical PRS at its optimal p value threshold (Figure 7D).
Because other tissues such as liver and adipose tissue have been associated with the risk of CAD, we also generated genome region-based PRSs for CAD by using the recent scATAC atlas of 30 adult human tissues, consisting of 111 cell types and approximately 890,000 peaks.61 To confirm our cell type marker peak selection, we generated a pairwise marker sharing matrix, which revealed the expected similarity patterns between related cell types (Figure S27). In line with our plaque scATAC-based PRS analysis, SMC-related cell types explained the greatest CAD risk variance, followed by adipocytes, ECs, and fibroblasts. Notably, cardiomyocytes, hepatocytes, and immune cells scored considerably lower in R2-based ranking irrespective of the total peak count used (peaks selected by ATAC signal strength; Figures S28 and 29). The cell type ranking was similar for peaks selected by cell type specificity (Figure S30).
Discussion
By performing a cell-type-unbiased time-course analysis of single cell transcriptomes in mouse atherosclerotic aorta, we identified 12 disease-associated cell states. Majority of these states are concordant with the previous studies focusing on specific subtypes of cells and lineage-tracing experiments.1,2,3,4,5,6,7,8,53,54 In addition, we identified four less abundant disease states corresponding to Calcrl+ ECs, Smoc2+ FBs, Apoe+ MSCs, and Palld+ SMCs that require further experimental validation. Specifically, these cell states were not activated in Ldlr−/− and Apoe−/− mouse models to the same extent and could also represent states that are specific to certain aortic vascular beds. Still, our enrichment-free setup allowed us to provide the first comparison of shared and differential gene regulatory mechanisms underlying the atherosclerosis-associated cell states. Despite that the majority of marker genes were specific to a disease state, a handful of genes were shared between five or more states and thus represent potential candidates for global biomarkers of CAD. To this end, circulating levels of LGMN, MGP, SDC4, and IGFBP4 have been associated with atherosclerosis and acute cardiovascular events with potential prognostic or risk stratification value.62,63,64,65,66 For example, levels of MGP and IGFBP4 could reflect differential vascular calcification burden and highlight differences in plaque pathobiology between ST-segment-elevation myocardial infarction (STEMI) and non-ST segment elevation myocardial infarction (NSTEMI).66
Signals from the microenvironment can be transmitted into the intracellular gene expression programs through multiple layers of signal propagation including ligand-receptor interactions, signaling molecules, and transcription factors. Based on the predicted ligand-receptor activities and downstream gene expression changes, our analysis strongly suggests that TGFβ, IL-1α/β, TNF, VEGFA, FGF1/2, APOE, and SPP1 signaling pathways dominate in the atherosclerotic microenvironment and are shared as upstream inducers of many cell states. This suggests that the distinct gene content of the disease-associated gene signatures is a result of cell-type-specific responses to the same environmental stimuli. This is in line with the recent report by us and others where proatherogenic stimulus-induced gene expression responses in ECs, SMCs, and MPs appeared very cell type specific.67,68 Supporting this, we demonstrate that cell states are largely governed by specific TFs that form intricate gene regulatory networks. Still, a few TF modules were shared between the SMC and EC cell states (XBP1, TCF4, CEBPB, CREB3L2, and ELK3) and between SMC and MP cell states (XBP1, MAFB, MLX, and NRF2), identifying them as potential candidates driving the expression of the genes in shared ontology categories related to ECM organization, blood vessel development, angiogenesis, cholesterol transport, and lipid localization. Indeed, XBP1, a key modulator of unfolded protein response, has been demonstrated to play an important role in the regulation of lipid metabolism and angiogenesis and the inhibition of this pathway alleviates atherosclerosis.57,69,70
Analysis of the atherosclerotic cell state signatures in human lesions supported the relevance of the majority of the cell states in separating healthy from diseased samples or early disease from advanced disease samples. But in cases where the cell state gene sets were short, the power of sample classification was not apparent. Supporting the relevance of the disease signatures, we further demonstrated that many of the cell state marker genes were predicted target genes of CAD GWAS variants and the genome regions where the marker genes reside contribute significantly to the heritability of CAD. Importantly, the genetic variation in the Vcam1+ SMCs and Col2a1+ SMCs contributed to CAD heritability beyond other cell states, suggesting that these cell states are particularly important in understanding the pathobiology of atherosclerosis. This is also supported by other recent studies looking into the cell-type-specific expression of predicted GWAS target genes.71,72 Importantly, our analysis demonstrated that atherosclerosis-associated cell state markers explain a larger proportion of CAD risk variance compared to cell type markers, suggesting a more prominent role for genes that participate in pathological changes compared to those that maintain healthy cell identity.
Substantial ongoing efforts are looking into applying CAD PRS in the clinical practice including risk stratification and prediction of treatment response. By enhancing early prediction of CAD beyond traditional risk factors, PRS could also guide new treatment strategies. This is exemplified by several landmark studies that demonstrated that the individuals at high genetic risk for CAD experience the greatest benefit from lipid-lowering treatments.73,74,75 However, the standard PRS sums an individual’s genetic profile to a single estimate that may fail to identify more nuanced phenotypes that are necessary for risk stratification, prediction of treatment response, or identification of pathways leading to novel treatment.43 Here, we evaluated the performance of PRS, which accounts for genomic substructure/regional functional heterogeneity by aggregating risk alleles across cell states/types and biological pathways. Our results demonstrate that while the regular (genome-wide) PRS outperforms the cell state gene set-based PRSs in absolute accuracy, regulatory element-based PRS effectively quality filters the SNPs with weaker GWAS p value. This allowed us to construct a hybrid PRS that achieved higher power than the regular PRS at its optimal p value threshold. On the other hand, pathway-based PRS identified biological processes conferring higher genetic risk, providing pathway-level processes to target in drug design. In particular, the pathways shared between several cell states related to cholesterol transport, lipid localization, extracellular matrix, blood vessel development, and angiogenesis could provide actionable targets. While our analysis provides the first steps toward cell-state-, cell-type-, and pathway-level understanding of the genetic risk, future studies are needed to evaluate whether incorporation of such functional prior information improves polygenic prediction accuracy in individual stratification or prediction of treatment response.
In summary, we provide in depth characterization of atherosclerosis-associated cell states and demonstrate the value of cell-state-specific markers in understanding the genetic basis of CAD. Substantial work still needs to be done to functionally validate the role of these genes in the pathophysiology of atherosclerosis. Defining the mechanisms that contribute to distinct cell states in pathological conditions could provide a basis for applying precision medicine and targeted therapies in the future.
Data and code availability
The accession number for the mouse bulk RNA-seq and scRNA-seq reported in this paper is GEO: GSE205929 and GSE205930. The MOVAS count matrices are available at FigShare DOI: https://doi.org/10.6084/m9.figshare.20059649.v1. The Maastricht Pathology Tissue Collection data were downloaded under GEO accession number GEO: GSE28829. Previously published mouse and human atherosclerosis scRNA-seq datasets were obtained from GEO accessions GEO: GSE155513 and GSE131778, respectively.
Acknowledgments
We acknowledge Single Cell Genomics Core, Genome Center and Cell and Tissue Imaging Unit at University of Eastern Finland, Finnish Functional Genomics Centre, and Biocenter Finland for infrastructure support. This research has been conducted with the UK Biobank Resource (https://www.ukbiobank.ac.uk/) under application number 58990. We are grateful to Resolve Biosciences for their excellent service in optimizing and performing the Molecular Cartography experiments. This study was funded by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant no. 802825 to M.U.K.), Academy of Finland (grant no. 333021 and 335973 to M.U.K.; 311081 and 314557 to T. Lönnberg; 349708 to P.P.M.; 314556 and 335975 to A. Roivainen; 321535 and 328835 to J.P.L.), InFLAMES Flagship Programme of the Academy of Finland (337530 to T. Lönnberg), Finnish Foundation for Cardiovascular Research (M.U.K., P.R.M., and J.P.L.), Instrumentarium Science Foundation (P.R.M.), Ida Montinin Foundation (P.R.M.), Sigrid Jusélius Foundation (M.U.K.), GenomMed doctoral program (740264 to A. Ravindran), and Jane and Aatos Erkko Foundation (A. Roivainen). The Tampere Vascular Study (TVS) was supported with grants from the Competitive Research Funding of the Tampere University Hospital (grants 9M048 and 9N035 to T. Lehtimäki), the Emil Aaltonen Foundation (to T. Lehtimäki), the Pirkanmaa Regional Fund of the Finnish Cultural Foundation, the Research Foundation of Orion Corporation, the Jenny and Antti Wihuri Foundation, and the Academy of Finland (grants 322098 and 104821), the Finnish Foundation for Cardiovascular Research, the Yrjö Jahnsson Foundation, European Union 7th Framework Program (grant 201668 for AtheroRemo), EU Horizon 2020 (grant 755320 for TAXINOMISIS and grant 848146 for To Aition), and the Academy of Finland grant 322098 and Tampere University Hospital Supporting Foundation.
Declaration of interests
The authors declare no competing interests.
Published: April 14, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.03.013.
Contributor Information
Tiit Örd, Email: tiit.ord@uef.fi.
Minna U. Kaikkonen, Email: minna.kaikkonen@uef.fi.
Supplemental information
References
- 1.Zernecke A., Winkels H., Cochain C., Williams J.W., Wolf D., Soehnlein O., Robbins C.S., Monaco C., Park I., McNamara C.A., et al. Meta-Analysis of Leukocyte Diversity in Atherosclerotic Mouse Aortas. Circ. Res. 2020;127:402–426. doi: 10.1161/CIRCRESAHA.120.316903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dobnikar L., Taylor A.L., Chappell J., Oldach P., Harman J.L., Oerton E., Dzierzak E., Bennett M.R., Spivakov M., Jørgensen H.F. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat. Commun. 2018;9:4567. doi: 10.1038/s41467-018-06891-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wirka R.C., Wagh D., Paik D.T., Pjanic M., Nguyen T., Miller C.L., Kundu R., Nagao M., Coller J., Koyano T.K., et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019;25:1280–1289. doi: 10.1038/s41591-019-0512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan H., Xue C., Auerbach B.J., Fan J., Bashore A.C., Cui J., Yang D.Y., Trignano S.B., Liu W., Shi J., et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation. 2020;142:2060–2075. doi: 10.1161/CIRCULATIONAHA.120.048378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alencar G.F., Owsiany K.M., Karnewar S., Sukhavasi K., Mocci G., Nguyen A.T., Williams C.M., Shamsuzzaman S., Mokry M., Henderson C.A., et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation. 2020;142:2045–2059. doi: 10.1161/CIRCULATIONAHA.120.046672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim J.B., Zhao Q., Nguyen T., Pjanic M., Cheng P., Wirka R., Travisano S., Nagao M., Kundu R., Quertermous T. Environment-Sensing Aryl Hydrocarbon Receptor Inhibits the Chondrogenic Fate of Modulated Smooth Muscle Cells in Atherosclerotic Lesions. Circulation. 2020;142:575–590. doi: 10.1161/CIRCULATIONAHA.120.045981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andueza A., Kumar S., Kim J., Kang D.W., Mumme H.L., Perez J.I., Villa-Roel N., Jo H. Endothelial Reprogramming by Disturbed Flow Revealed by Single-Cell RNA and Chromatin Accessibility Study. Cell Rep. 2020;33:108491. doi: 10.1016/j.celrep.2020.108491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao G., Lu H., Liu Y., Zhao Y., Zhu T., Garcia-Barrio M.T., Chen Y.E., Zhang J. Single-Cell Transcriptomics Reveals Endothelial Plasticity During Diabetic Atherogenesis. Front. Cell Dev. Biol. 2021;9:689469. doi: 10.3389/fcell.2021.689469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aragam K.G., Jiang T., Goel A., Kanoni S., Wolford B.N., Atri D.S., Weeks E.M., Wang M., Hindy G., Zhou W., et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet. 2022;54:1803–1815. doi: 10.1038/s41588-022-01233-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tcheandjieu C., Zhu X., Hilliard A.T., Clarke S.L., Napolioni V., Ma S., Lee K.M., Fang H., Chen F., Lu Y., et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat. Med. 2022;28:1679–1692. doi: 10.1038/s41591-022-01891-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Örd T., Õunap K., Stolze L.K., Aherrahrou R., Nurminen V., Toropainen A., Selvarajan I., Lönnberg T., Aavik E., Ylä-Herttuala S., et al. Single-Cell Epigenomics and Functional Fine-Mapping of Atherosclerosis GWAS Loci. Circ. Res. 2021;129:240–258. doi: 10.1161/CIRCRESAHA.121.318971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powell-Braxton L., Véniant M., Latvala R.D., Hirano K.I., Won W.B., Ross J., Dybdal N., Zlot C.H., Young S.G., Davidson N.O. A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat. Med. 1998;4:934–938. doi: 10.1038/nm0898-934. [DOI] [PubMed] [Google Scholar]
- 13.Farese R.V., Jr., Véniant M.M., Cham C.M., Flynn L.M., Pierotti V., Loring J.F., Traber M., Ruland S., Stokowski R.S., Huszar D., Young S.G. Phenotypic analysis of mice expressing exclusively apolipoprotein B48 or apolipoprotein B100. Proc. Natl. Acad. Sci. USA. 1996;93:6393–6398. doi: 10.1073/pnas.93.13.6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang S., Corbett S.E., Koga Y., Wang Z., Johnson W.E., Yajima M., Campbell J.D. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol. 2020;21:57. doi: 10.1186/s13059-020-1950-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu G. Gene Ontology Semantic Similarity Analysis Using GOSemSim. Methods Mol. Biol. 2020;2117:207–215. doi: 10.1007/978-1-0716-0301-7_11. [DOI] [PubMed] [Google Scholar]
- 18.DeTomaso D., Jones M.G., Subramaniam M., Ashuach T., Ye C.J., Yosef N. Functional interpretation of single cell similarity maps. Nat. Commun. 2019;10:4376. doi: 10.1038/s41467-019-12235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Browaeys R., Saelens W., Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods. 2020;17:159–162. doi: 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- 21.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frishberg A., Peshes-Yaloz N., Cohn O., Rosentul D., Steuerman Y., Valadarsky L., Yankovitz G., Mandelboim M., Iraqi F.A., Amit I., et al. Cell composition analysis of bulk genomics using single-cell data. Nat. Methods. 2019;16:327–332. doi: 10.1038/s41592-019-0355-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sulkava M., Raitoharju E., Levula M., Seppälä I., Lyytikäinen L.P., Mennander A., Järvinen O., Zeitlin R., Salenius J.P., Illig T., et al. Differentially expressed genes and canonical pathway expression in human atherosclerotic plaques - Tampere Vascular Study. Sci. Rep. 2017;7:41483. doi: 10.1038/srep41483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Döring Y., Manthey H.D., Drechsler M., Lievens D., Megens R.T.A., Soehnlein O., Busch M., Manca M., Koenen R.R., Pelisek J., et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation. 2012;125:1673–1683. doi: 10.1161/CIRCULATIONAHA.111.046755. [DOI] [PubMed] [Google Scholar]
- 27.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mountjoy E., Schmidt E.M., Carmona M., Schwartzentruber J., Peat G., Miranda A., Fumis L., Hayhurst J., Buniello A., Karim M.A., et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet. 2021;53:1527–1533. doi: 10.1038/s41588-021-00945-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Harst P., Verweij N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018;122:433–443. doi: 10.1161/CIRCRESAHA.117.312086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson C.P., Goel A., Butterworth A.S., Kanoni S., Webb T.R., Marouli E., Zeng L., Ntalla I., Lai F.Y., Hopewell J.C., et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 2017;49:1385–1391. doi: 10.1038/ng.3913. [DOI] [PubMed] [Google Scholar]
- 31.Malik R., Chauhan G., Traylor M., Sargurupremraj M., Okada Y., Mishra A., Rutten-Jacobs L., Giese A.K., van der Laan S.W., Gretarsdottir S., et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 2018;50:524–537. doi: 10.1038/s41588-018-0058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kichaev G., Bhatia G., Loh P.R., Gazal S., Burch K., Freund M.K., Schoech A., Pasaniuc B., Price A.L. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am. J. Hum. Genet. 2019;104:65–75. doi: 10.1016/j.ajhg.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsunaga H., Ito K., Akiyama M., Takahashi A., Koyama S., Nomura S., Ieki H., Ozaki K., Onouchi Y., Sakaue S., et al. Transethnic Meta-Analysis of Genome-Wide Association Studies Identifies Three New Loci and Characterizes Population-Specific Differences for Coronary Artery Disease. Circ. Genom. Precis. Med. 2020;13:e002670. doi: 10.1161/CIRCGEN.119.002670. [DOI] [PubMed] [Google Scholar]
- 34.Koyama S., Ito K., Terao C., Akiyama M., Horikoshi M., Momozawa Y., Matsunaga H., Ieki H., Ozaki K., Onouchi Y., et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet. 2020;52:1169–1177. doi: 10.1038/s41588-020-0705-3. [DOI] [PubMed] [Google Scholar]
- 35.Hartiala J.A., Han Y., Jia Q., Hilser J.R., Huang P., Gukasyan J., Schwartzman W.S., Cai Z., Biswas S., Trégouët D.A., et al. Genome-wide analysis identifies novel susceptibility loci for myocardial infarction. Eur. Heart J. 2021;42:919–933. doi: 10.1093/eurheartj/ehaa1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou W., Nielsen J.B., Fritsche L.G., Dey R., Gabrielsen M.E., Wolford B.N., LeFaive J., VandeHaar P., Gagliano S.A., Gifford A., et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 2018;50:1335–1341. doi: 10.1038/s41588-018-0184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurki M.I., Karjalainen J., Palta P., Sipilä T.P., Kristiansson K., Donner K., Reeve M.P., Laivuori H., Aavikko M., Kaunisto M.A., et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613:508–518. doi: 10.1038/s41586-022-05473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L., Chen Z., von Scheidt M., Li S., Steiner A., Güldener U., Koplev S., Ma A., Hao K., Pan C., et al. Transcriptome-wide association study of coronary artery disease identifies novel susceptibility genes. Basic Res. Cardiol. 2022;117:6. doi: 10.1007/s00395-022-00917-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erdmann J., Kessler T., Munoz Venegas L., Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc. Res. 2018;114:1241–1257. doi: 10.1093/cvr/cvy084. [DOI] [PubMed] [Google Scholar]
- 40.Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.R., Anttila V., Xu H., Zang C., Farh K., et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi S.W., O'Reilly P.F. PRSice-2: Polygenic Risk Score software for biobank-scale data. GigaScience. 2019;8:giz082. doi: 10.1093/gigascience/giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi S.W., Garcia-Gonzalez J., Ruan Y., Wu H.M., Porras C., Johnson J., Bipolar Disorder Working group of the Psychiatric Genomics Consortium. Hoggart C.J., O'Reilly P.F. PRSet: Pathway-based polygenic risk score analyses and software. PLoS Genet. 2023;19:e1010624. doi: 10.1371/journal.pgen.1010624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nikpay M., Goel A., Won H.H., Hall L.M., Willenborg C., Kanoni S., Saleheen D., Kyriakou T., Nelson C.P., Hopewell J.C., et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O'Connell J., et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dann E., Henderson N.C., Teichmann S.A., Morgan M.D., Marioni J.C. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 2022;40:245–253. doi: 10.1038/s41587-021-01033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Denisenko E., Guo B.B., Jones M., Hou R., de Kock L., Lassmann T., Poppe D., Clément O., Simmons R.K., Lister R., Forrest A.R.R. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 2020;21:130. doi: 10.1186/s13059-020-02048-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng C.K., Lin X., Pu Y., Tse J.K.Y., Wang Y., Zhang C.-L., Cao X., Lau C.W., Huang J., He L., et al. SOX4 is a novel phenotypic regulator of endothelial cells in atherosclerosis revealed by single-cell analysis. J. Adv. Res. 2022;43:187–203. doi: 10.1016/j.jare.2022.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sobierajska K., Wawro M.E., Ciszewski W.M., Niewiarowska J. Transforming Growth Factor-beta Receptor Internalization via Caveolae Is Regulated by Tubulin-beta2 and Tubulin-beta3 during Endothelial-Mesenchymal Transition. Am. J. Pathol. 2019;189:2531–2546. doi: 10.1016/j.ajpath.2019.08.004. [DOI] [PubMed] [Google Scholar]
- 52.Lee Y.H., Albig A.R., Regner M., Schiemann B.J., Schiemann W.P. Fibulin-5 initiates epithelial-mesenchymal transition (EMT) and enhances EMT induced by TGF-beta in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis. 2008;29:2243–2251. doi: 10.1093/carcin/bgn199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Winkels H., Wolf D. Heterogeneity of T Cells in Atherosclerosis Defined by Single-Cell RNA-Sequencing and Cytometry by Time of Flight. Arterioscler. Thromb. Vasc. Biol. 2021;41:549–563. doi: 10.1161/ATVBAHA.120.312137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernandez D.M., Rahman A.H., Fernandez N.F., Chudnovskiy A., Amir E.A.D., Amadori L., Khan N.S., Wong C.K., Shamailova R., Hill C.A., et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019;25:1576–1588. doi: 10.1038/s41591-019-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sampieri L., Di Giusto P., Alvarez C. CREB3 Transcription Factors: ER-Golgi Stress Transducers as Hubs for Cellular Homeostasis. Front. Cell Dev. Biol. 2019;7:123. doi: 10.3389/fcell.2019.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laurent F., Girdziusaite A., Gamart J., Barozzi I., Osterwalder M., Akiyama J.A., Lincoln J., Lopez-Rios J., Visel A., Zuniga A., Zeller R. HAND2 Target Gene Regulatory Networks Control Atrioventricular Canal and Cardiac Valve Development. Cell Rep. 2017;19:1602–1613. doi: 10.1016/j.celrep.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moncan M., Mnich K., Blomme A., Almanza A., Samali A., Gorman A.M. Regulation of lipid metabolism by the unfolded protein response. J. Cell Mol. Med. 2021;25:1359–1370. doi: 10.1111/jcmm.16255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klarin D., Natarajan P. Clinical utility of polygenic risk scores for coronary artery disease. Nat. Rev. Cardiol. 2022;19:291–301. doi: 10.1038/s41569-021-00638-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karlsson M., Zhang C., Méar L., Zhong W., Digre A., Katona B., Sjöstedt E., Butler L., Odeberg J., Dusart P., et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021;7:eabh2169. doi: 10.1126/sciadv.abh2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu Y., Lu Q., Powles R., Yao X., Yang C., Fang F., Xu X., Zhao H. Leveraging functional annotations in genetic risk prediction for human complex diseases. PLoS Comput. Biol. 2017;13:e1005589. doi: 10.1371/journal.pcbi.1005589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang K., Hocker J.D., Miller M., Hou X., Chiou J., Poirion O.B., Qiu Y., Li Y.E., Gaulton K.J., Wang A., et al. A single-cell atlas of chromatin accessibility in the human genome. Cell. 2021;184:5985–6001.e19. doi: 10.1016/j.cell.2021.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lunde N.N., Holm S., Dahl T.B., Elyouncha I., Sporsheim B., Gregersen I., Abbas A., Skjelland M., Espevik T., Solberg R., et al. Increased levels of legumain in plasma and plaques from patients with carotid atherosclerosis. Atherosclerosis. 2017;257:216–223. doi: 10.1016/j.atherosclerosis.2016.11.026. [DOI] [PubMed] [Google Scholar]
- 63.Lunde N.N., Gregersen I., Ueland T., Shetelig C., Holm S., Kong X.Y., Michelsen A.E., Otterdal K., Yndestad A., Broch K., et al. Legumain is upregulated in acute cardiovascular events and associated with improved outcome - potentially related to anti-inflammatory effects on macrophages. Atherosclerosis. 2020;296:74–82. doi: 10.1016/j.atherosclerosis.2019.12.008. [DOI] [PubMed] [Google Scholar]
- 64.Kumric M., Borovac J.A., Ticinovic Kurir T., Martinovic D., Frka Separovic I., Baric L., Bozic J. Role of Matrix Gla Protein in the Complex Network of Coronary Artery Disease: A Comprehensive Review. Life. 2021;11:737. doi: 10.3390/life11080737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Solbu M.D., Kolset S.O., Jenssen T.G., Wilsgaard T., Løchen M.L., Mathiesen E.B., Melsom T., Eriksen B.O., Reine T.M. Gender differences in the association of syndecan-4 with myocardial infarction: The population-based Tromso Study. Atherosclerosis. 2018;278:166–173. doi: 10.1016/j.atherosclerosis.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 66.Cediel G., Rueda F., Oxvig C., Oliveras T., Labata C., de Diego O., Ferrer M., Aranda-Nevado M.C., Serra-Gregori J., Núñez J., et al. Prognostic value of the Stanniocalcin-2/PAPP-A/IGFBP-4 axis in ST-segment elevation myocardial infarction. Cardiovasc. Diabetol. 2018;17:63. doi: 10.1186/s12933-018-0710-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen P.Y., Qin L., Li G., Wang Z., Dahlman J.E., Malagon-Lopez J., Gujja S., Cilfone N.A., Kauffman K.J., Sun L., et al. Endothelial TGF-beta signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 2019;1:912–926. doi: 10.1038/s42255-019-0102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moreau P.R., Tomas Bosch V., Bouvy-Liivrand M., Õunap K., Örd T., Pulkkinen H.H., Pölönen P., Heinäniemi M., Ylä-Herttuala S., Laakkonen J.P., et al. Profiling of Primary and Mature miRNA Expression in Atherosclerosis-Associated Cell Types. Arterioscler. Thromb. Vasc. Biol. 2021;41:2149–2167. doi: 10.1161/ATVBAHA.121.315579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Binet F., Sapieha P. ER Stress and Angiogenesis. Cell Metab. 2015;22:560–575. doi: 10.1016/j.cmet.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 70.Tufanli O., Telkoparan Akillilar P., Acosta-Alvear D., Kocaturk B., Onat U.I., Hamid S.M., Çimen I., Walter P., Weber C., Erbay E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA. 2017;114:E1395–E1404. doi: 10.1073/pnas.1621188114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Slenders L., Landsmeer L.P.L., Cui K., Depuydt M.A.C., Verwer M., Mekke J., Timmerman N., van den Dungen N.A.M., Kuiper J., de Winther M.P.J., et al. Intersecting single-cell transcriptomics and genome-wide association studies identifies crucial cell populations and candidate genes for atherosclerosis. Eur. Heart J. Open. 2022;2:oeab043. doi: 10.1093/ehjopen/oeab043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Depuydt M.A.C., Prange K.H.M., Slenders L., Örd T., Elbersen D., Boltjes A., de Jager S.C.A., Asselbergs F.W., de Borst G.J., Aavik E., et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 2020;127:1437–1455. doi: 10.1161/CIRCRESAHA.120.316770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marston N.A., Kamanu F.K., Nordio F., Gurmu Y., Roselli C., Sever P.S., Pedersen T.R., Keech A.C., Wang H., Lira Pineda A., et al. Predicting Benefit From Evolocumab Therapy in Patients With Atherosclerotic Disease Using a Genetic Risk Score: Results From the FOURIER Trial. Circulation. 2020;141:616–623. doi: 10.1161/CIRCULATIONAHA.119.043805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Damask A., Steg P.G., Schwartz G.G., Szarek M., Hagström E., Badimon L., Chapman M.J., Boileau C., Tsimikas S., Ginsberg H.N., et al. Patients With High Genome-Wide Polygenic Risk Scores for Coronary Artery Disease May Receive Greater Clinical Benefit From Alirocumab Treatment in the ODYSSEY OUTCOMES Trial. Circulation. 2020;141:624–636. doi: 10.1161/CIRCULATIONAHA.119.044434. [DOI] [PubMed] [Google Scholar]
- 75.Mega J.L., Stitziel N.O., Smith J.G., Chasman D.I., Caulfield M., Devlin J.J., Nordio F., Hyde C., Cannon C.P., Sacks F., et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet. 2015;385:2264–2271. doi: 10.1016/S0140-6736(14)61730-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the mouse bulk RNA-seq and scRNA-seq reported in this paper is GEO: GSE205929 and GSE205930. The MOVAS count matrices are available at FigShare DOI: https://doi.org/10.6084/m9.figshare.20059649.v1. The Maastricht Pathology Tissue Collection data were downloaded under GEO accession number GEO: GSE28829. Previously published mouse and human atherosclerosis scRNA-seq datasets were obtained from GEO accessions GEO: GSE155513 and GSE131778, respectively.