

Abstract
Direct, amplification-free detection of RNA has the potential to transform molecular diagnostics by enabling simple on-site analysis of human or environmental samples. CRISPR-Cas nucleases offer programmable RNA-guided recognition of RNA that triggers cleavage and release of a fluorescent reporter molecule1,2, but long reaction times hamper sensitivity and speed when applied to point-of-care testing. Here we show that unrelated CRISPR nucleases can be deployed in tandem to provide both direct RNA sensing and rapid signal generation, thus enabling robust detection of ~30 RNA copies/microliter in 20 minutes. Combining RNA-guided Cas13 and Csm6 with a chemically stabilized activator creates a one-step assay that can detect SARS-CoV-2 RNA extracted from nasopharyngeal samples with qRT-PCR-derived Ct values up to 33 in microfluidic chips, using a compact imaging system. This Fast Integrated Nuclease Detection In Tandem (FIND-IT) approach enables direct RNA detection in a simple format that is amenable to point-of-care infection diagnosis, as well as to a wide range of other diagnostic or research applications.
Introduction
Current strategies for RNA detection in clinical samples based on qRT-PCR (quantitative reverse-transcriptase-polymerase chain reaction) provide high sensitivity (limit of detection of ~1 molecule/μl) but are too complex to implement for rapid point-of-care testing, an essential component of pandemic control3–5. CRISPR-Cas proteins offer a simpler alternative to PCR-based methods due to their programmable, RNA-guided recognition of RNA sequences6–8. Detection takes advantage of the intrinsic enzymatic functions of CRISPR-Cas proteins from Type III and Type VI CRISPR-Cas systems, including the multisubunit effector Csm/Cmr, or the single-protein effector Cas131,2,9,10. Target RNA recognition by the effector triggers multiple-turnover trans-cleavage of single-stranded RNA (ssRNA) by either the same protein, in the case of Cas13, or by a separate protein called Csm6, in the case of Csm/Cmr1,2,11,12. Enzymatic RNA cleavage generates a fluorescent signal when directed towards a reporter oligonucleotide containing a quencher and dye pair1,2,13. Cas13 or Csm/Cmr is capable of reaching an RNA detection sensitivity in the attomolar range in under an hour when coupled to a target sequence amplification procedure, such as reverse transcription-recombinase polymerase amplification (RT-RPA) or reverse transcription-loop-mediated isothermal amplification (RT-LAMP)1,2,9,10,14,15. However, addition of RT-LAMP to CRISPR-based detection requires multiple liquid-handling steps and/or high-temperature incubation (55–65°C), procedures that are challenging for point-of-care testing9,10,16–18. RT-RPA can be combined with LbuCas13 detection into a single step, but exhibited a lower sensitivity than the two-step procedure on both purified RNA and unextracted clinical samples, possibly due to the enzymes from different steps not being fully compatible for integrated detection19,20. We reported that amplification-free RNA detection using L. buccalis Cas13 (LbuCas13) with three guide RNAs could rapidly identify SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) genomic RNA in patient samples with PCR-derived cycle threshold (Ct) values up to 22, corresponding to ~1600 copies/μl in the assay21. When tested in a mobile phone-based imaging device, this assay detects as low as ~100–200 cp/μl of target RNA within 30 min21. However, increased speed and sensitivity of one-pot detection chemistries are still needed to enable their widespread use for point-of-care diagnostics.
Csm6, a dimeric RNA endonuclease from Type III CRISPR-Cas systems, has the potential to boost RNA detection based on its endogenous function in signal amplification9,10,22. During CRISPR-Cas interference, activation of Csm or Cmr by viral RNA recognition triggers synthesis of cyclic tetra- or hexaadenylates (cA4 or cA6) that bind to the Csm6 CRISPR-associated Rossman fold (CARF) and activate its higher eukaryote/prokaryote nucleotide binding (HEPN) domain’s ribonuclease activity7,11,12,23. However, diagnostic methods using this cascade were limited to a detection sensitivity of ~500 fM to 1 nM RNA (~105-109 cp/μl) without inclusion of RT-LAMP to amplify the target sequence9,10. Use of E. italicus Csm6 (EiCsm6) in a reaction where Cas13’s target-triggered trans-ssRNA cleavage generated linear hexaadenylates with a 2´,3´-cyclic phosphate (A6>P, activating ligand of EiCsm6) resulted in low detection sensitivity (1 μM target RNA) and no reduction in detection time12,22. Thus, current methods using Csm6 have not resulted in a level of signal amplification that enables CRISPR proteins to directly detect RNA at a concentration relevant for diagnostics.
Here we describe a tandem nuclease assay using Cas13 and Csm6 to achieve both high sensitivity and fast signal generation, without requiring a preceding target amplification step. This involved the design of potent, chemically stabilized activators for Csm6, as well as use of eight different crRNA sequences for Cas13 detection. We also show that this assay can be implemented in a portable device, consisting of a microfluidic chip and a compact detector, to detect viral RNA extracted from patient samples. This indicates that the assay is robust and simple to adapt for use in point-of-care testing workflows. This also highlights the value of combining unrelated CRISPR-Cas effectors for sensitive, one-pot detection of RNA.
Results
The minimal signal generation observed with Csm6 in RNA detection assays has limited its use for diagnostics22. We hypothesized that this limitation could be explained by the ability of some Csm6 proteins to degrade cA6 or cA4 activators over time, causing auto-inactivation23–27. Although self-inactivation can be blocked by substituting all the 2´-hydroxyl (2´-OH) groups in the cyclic oligoadenylate with either a 2´-deoxy (2´-H) or 2´-fluoro (2´-F) group, these modifications either completely abolished or led to a 50-fold reduction in activation of their cognate Csm6 protein23,25. We wondered whether site-selective chemical modification might prevent degradation of the Csm6-activating oligonucleotide while maintaining high-level activation of Csm6. Using the Csm6 protein from the T. thermophilus Type III-A system (TtCsm6)12,28,29, which recognizes cA4 or A4>P for activation12, we first tested whether 5´-A3–6U6-3´ oligonucleotides would produce TtCsm6-activating ligands when cleaved by RNA target-bound LbuCas13 (Fig. 1A, B). The oligonucleotide A4-U6 stimulated robust reporter cleavage by TtCsm6 relative to oligonucleotides containing A3, A5, or A6 at the 5´-end (Fig. 1B), consistent with TtCsm6’s preference for a linear activator resembling its natural cA4 ligand11,12. Furthermore, the reaction requires the LbuCas13-crRNA complex, its target RNA, and the A4-U6 activator, confirming that TtCsm6 activation is tightly coupled to target RNA detection by LbuCas13 via A4-U6 cleavage (Fig. 1C).
Figure 1: Activation and inactivation of TtCsm6 in an LbuCas13-TtCsm6 assay.

a) Schematic of TtCsm6 activation by LbuCas13 assembled with a crRNA. Binding of a short RNA target sequence (blue) by LbuCas13 (pink) leads to activation of trans-ssRNA cleavage, which results in trimming of the polyU region (red) from the Csm6 activator. This liberates an oligoadenylate activator with a 2´,3´-cyclic phosphate (yellow) which binds to the Csm6 CARF domains (dark blue) and activates its HEPN (light blue) for cleavage of a fluorophore-quencher RNA reporter.
b) LbuCas13-TtCsm6 assay with 2 μM Csm6 activator added, containing different lengths of oligoadenylates (A3-A6 followed by U6). LbuCas13 is complexed with a single crRNA sequence derived from a spacer in the genome of a Lachnospiraceae bacterium, and all reactions contain 200 pM of a short target RNA oligonucleotide, which triggers LbuCas13 to trim off the U’s from the TtCsm6 activator. Normalized fluorescence is plotted (fluorescence value, F, at each timepoint divided by the initial fluorescence, F0). The bar graph (right) shows the mean normalized fluorescence and S.E.M. (n = 3) at the assay endpoint of 118 min.
c) LbuCas13-TtCsm6 assay with controls shown, in which one or more reagents are omitted. Cas13 refers to the Cas13 ribonucleoprotein complex containing both Cas13 protein and its crRNA. Reporter only refers to a reaction with only fluorescent reporter molecules in buffer supplemented with RNase inhibitor. Bar graphs show mean normalized fluorescence and S.E.M. (n = 3) at assay endpoint of 118 min.
d) As in b, but with varying concentrations of the A4-U6. oligonucleotide that is cleaved by LbuCas13 to generate A4>P, the activating ligand of TtCsm6.
e) Reactions were initiated as in b, but with 1 μM of A4-U6. The indicated reagents (A4-U6, Cas13’s target RNA, TtCsm6 protein, or buffer) were then added into the reaction at the time indicated by the arrow. A control reaction is shown without target RNA present in the reaction (no target RNA). Mean normalized fluorescence and S.E.M (n = 3) are plotted over the time course of the reaction.
Activation of TtCsm6 with varying concentrations of A4-U6 produced an initial burst of fluorescence followed by a plateau within ~20–30 minutes, with the final fluorescence level proportional to the amount of Csm6 activator present (Fig. 1D). Since equal amounts of reporter molecules were present in all reactions, the plateau in the fluorescence signal likely corresponds to a cessation of reporter cleavage by TtCsm6. Addition of A4-U6 activator to an LbuCas13-TtCsm6 reaction in which the fluorescence had plateaued was shown to rapidly increase reporter cleavage by TtCsm6, while addition of more target RNA or TtCsm6 had no effect relative to a buffer control (Fig. 1E). Taken together, these data suggest that Csm6’s A4>P ligand is depleted over time, thereby deactivating its RNase activity. Consistent with this conclusion, direct activation of Csm6 with 0.5–2 μM of A4>P in the absence of LbuCas13 exhibited a similar pattern of inactivation as that observed for the full LbuCas13-TtCsm6 reaction with A4-U6 (Extended Data Fig. 1A). Liquid chromatography-mass spectrometry (LC-MS) analysis also showed that A4>P degrades to A2>P following incubation with TtCsm6, suggesting that linear activators, like cyclic oligoadenylates24–26, are subject to Csm6-catalyzed cleavage (Supplementary Table 1). In the context of the natural cA4 ligand, A4>P may represent an intermediate on the pathway to full TtCsm6 inactivation (Fig. 2A)22,23.
Figure 2: Effect of single 2´-fluoro and 2´-deoxy activator modifications on Csm6-based signal amplification of RNA detection.
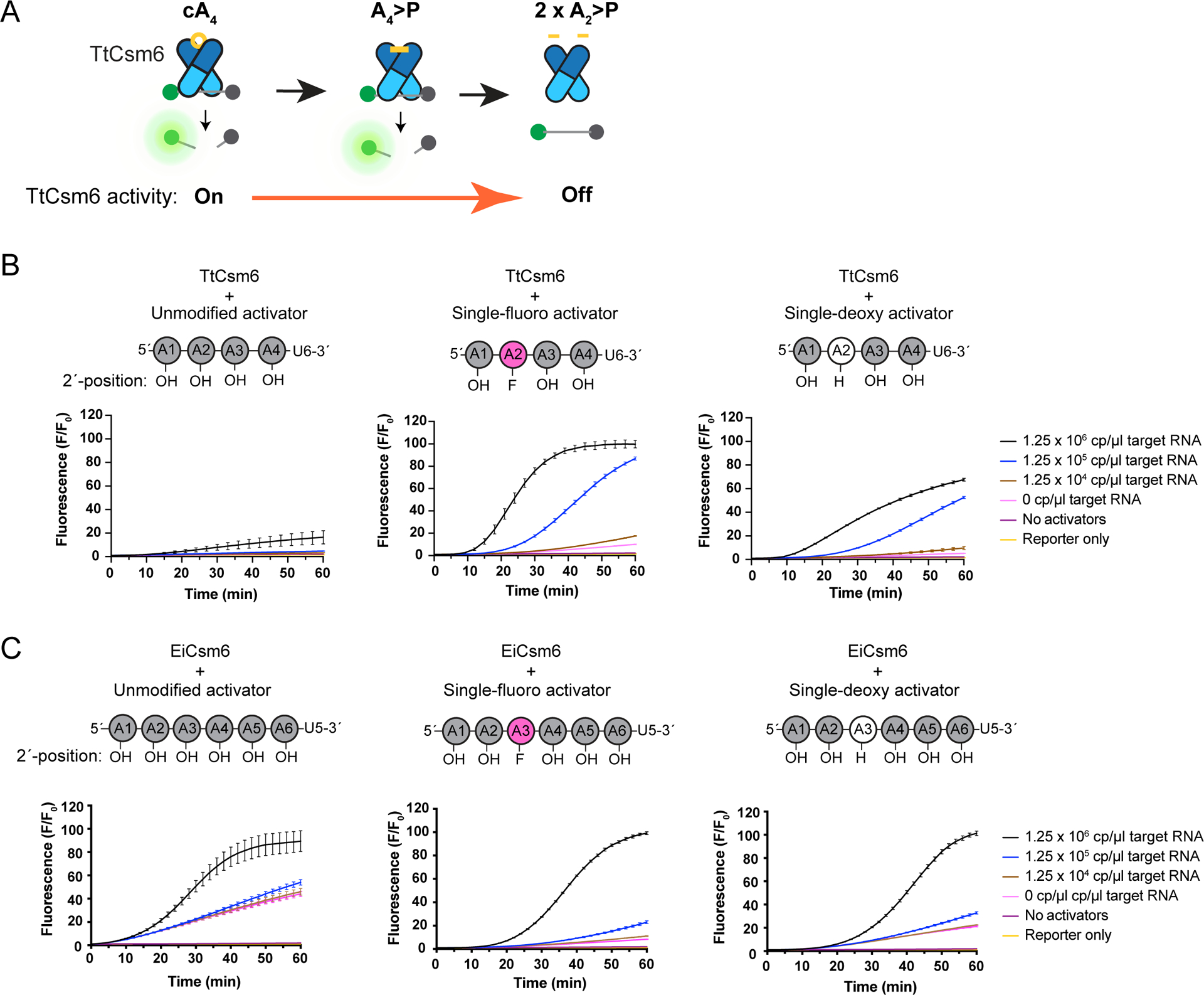
a) Schematic showing degradation of cA4 to A4>P and A2>P by Csm6’s CARF domain and its effect on RNA cleavage by the HEPN domain. Only binding of the cA4 or A4>P to the CARF domains (dark blue) of TtCsm6, can activate its HEPN domain (light blue) for ssRNA cleavage. Cleavage of the activator from cA4 to A4>P and A2>P is catalyzed by the CARF domain, and leads to inactivation of the HEPN domain.
b) LbuCas13-TtCsm6 reactions with 2 μM of unmodified A4-U6 (left), or A4-U6 bearing a single-fluoro modification (center) or single-deoxy modification (right) on the A2 nucleotide. LbuCas13 is loaded with crRNAs 604 and 612, which target an in vitro transcribed RNA transcript corresponding to a SARS-CoV-2 genome fragment. Target RNA was added at concentrations ranging from 0 cp/μl to 1.25 × 106 cp/μl, and control reactions lacking Csm6 activator and target RNA (“No activators”) or containing only reporter in buffer (Reporter only) are also shown. A schematic showing the position and type of modified nucleotide in the activator is shown above each graph. Adenosines with a 2´-OH, 2´-F, or 2´-H are shown in gray, pink, or white, respectively. Mean normalized fluorescence and S.E.M (n = 3) are plotted over 60 min as lines with error bars at the measurement timepoints.
c) As in b, but with an LbuCas13-EiCsm6 reaction with 0.5 μM of unmodified EiCsm6 activator, A6-U5 (left), a single-fluoro A6-U5 (center), and a single-deoxy A6-U5 (right). The 2´-modification is placed on the A3, rather than A2, to block degradation of A6>P to A3>P by EiCsm6’s CARF domain23.
Mathematical modeling suggested that blocking degradation of A4>P by Csm6 could dramatically improve fluorescent signal production in the Cas13-Csm6 detection assay (Extended Data Fig. 2). Mutations in Csm6 or replacement of the 2´-hydroxyl (2´-OH) in cyclic oligoadenylates with 2´-fluoro (2´-F) or 2´-deoxy (2´-H) were previously shown to inhibit activator degradation, but these strategies either drastically lowered or prevented activation of Csm6’s RNA cleavage activity21,22. To test whether modified linear activators can increase detection sensitivity and/or speed in a LbuCas13-TtCsm6 assay, we replaced the 2´-hydroxyls of A4-U6 with either a single or multiple 2´-deoxy, 2´-fluoro, or 2´-O-methyl groups to block cleavage within the A4 sequence (Fig. 2B, Extended Data Fig. 3A–C). Although inserting multiple modifications into the activators led to slow activation of TtCsm6, substitution of the 2´-hydroxyl of A2 with either a 2´-deoxy or 2´fluoro modification boosted the target RNA detection limit from 1.25 × 106 cp/μl (2 pM) to 1.25 × 104 cp/μl (20 fM), constituting a 100-fold increase in sensitivity over an unmodified A4-U6 (Fig. 2B, Extended Data Fig. 3A–C). In addition, these singly-modified activators enabled faster distinction between the sample fluorescence signal and background signal for reactions measured with 1.25 × 106 cp/μl (2 pM; above background at ~10 min), 1.25 × 105 cp/μl (200 fM; above background at ~20 min), and 1.25 × 104 cp/μl (20 fM; above background at ~40 min) of target RNA (Fig. 2B). The single-fluoro A4-U6 also exhibited improved signal-to-background over the single-deoxy A4-U6, indicating its superiority in maintaining TtCsm6 activation (Fig. 2B).
To confirm that the enhanced signal amplification provided by the singly-modified A4U6 activators was indeed due to better activation by the A4>P ligand, we showed that A4>P oligonucleotides containing a 2´-fluoro or 2´-deoxy at the A2 nucleotide could sustain TtCsm6’s activity for longer times compared to the unmodified A4>P (Extended Data Fig. 1A–C). In addition, incubation of TtCsm6 with the single-fluoro A4>P produced A3>P, indicating that Csm6-catalyzed degradation of A4>P to A2>P was blocked by the modification, but that cleavage could occur at other positions (Supplementary Tables 1, 2). Interestingly, we found that application of an analogous single-fluoro substitution strategy to EiCsm6 (EiCsm6), an ortholog that uses cA6/A6>P as an activating ligand12,23, did not lead to a significant improvement in the sensitivity or speed of an LbuCas13-EiCsm6 RNA detection assay (Fig. 2C; Extended Data Fig. 4A, B). It is possible that the longer A6>P activator of EiCsm6 is more susceptible to nucleolytic degradation, relative to the shorter A4>P activator of TtCsm6. Taken together, these results demonstrate that the single-fluoro A4-U6 activator of TtCsm6 improves signal amplification by 100-fold relative to an unmodified activator in a tandem nuclease detection assay with Cas13.
For TtCsm6 and its modified activator to be useful for enhancing RNA detection sensitivity and/or speed, the programmable nature of the CRISPR nuclease used for detection must be preserved. To test this in our Cas13-Csm6 assay, we added TtCsm6 and its single-fluoro activator to an LbuCas13 protein programmed with different crRNA sequences that target the RNA genome of SARS-CoV-2, the causative agent of COVID-19 (Fig. 3A). Detection using different crRNA sequences exhibited similar sensitivity and kinetics, showing that this one-step assay is programmable, and thus could potentially be adapted for detection of virtually any RNA sequence. To further improve detection sensitivity, we complexed LbuCas13 with a pool of eight crRNA sequences, which enables coverage of a wide range of SARS-CoV-2 variants (Supplementary Table 4) and exhibits improved signal-to-background compared to two crRNAs (Extended Data Fig. 5). Using this optimized set of crRNAs, LbuCas13 could detect as few as 63 RNA copies per microliter (cp/μl) of externally validated SARS-CoV-2 RNA in 2 hours (Fig. 3B). Inclusion of these eight guides in an LbuCas13-TtCsm6 tandem reaction enabled detection of 31 cp/μl of SARS-CoV-2 RNA in the same time frame (Fig. 3C). To determine whether addition of TtCsm6 could accelerate the time to detection, we compared the results of these two detection chemistries after a 20-minute reaction time. While LbuCas13 was unable to detect concentrations ranging from 31–125 cp/μl by 20 minutes, the assay containing both LbuCas13 and TtCsm6 could detect 31 cp/μl within 20 min (p-value < 0.05, Fig. 3B, C; Extended Data Figure 6). Taken together, these results demonstrate that, with optimized crRNAs for LbuCas13 and a chemically stabilized activator for TtCsm6, the tandem CRISPR nuclease assay can detect RNA sequences from an infectious pathogen with both high sensitivity and a fast time to detection that could be amenable for point-of-care testing workflows.
Figure 3: Programmability and benchmarking of the LbuCas13-TtCsm6 assay.
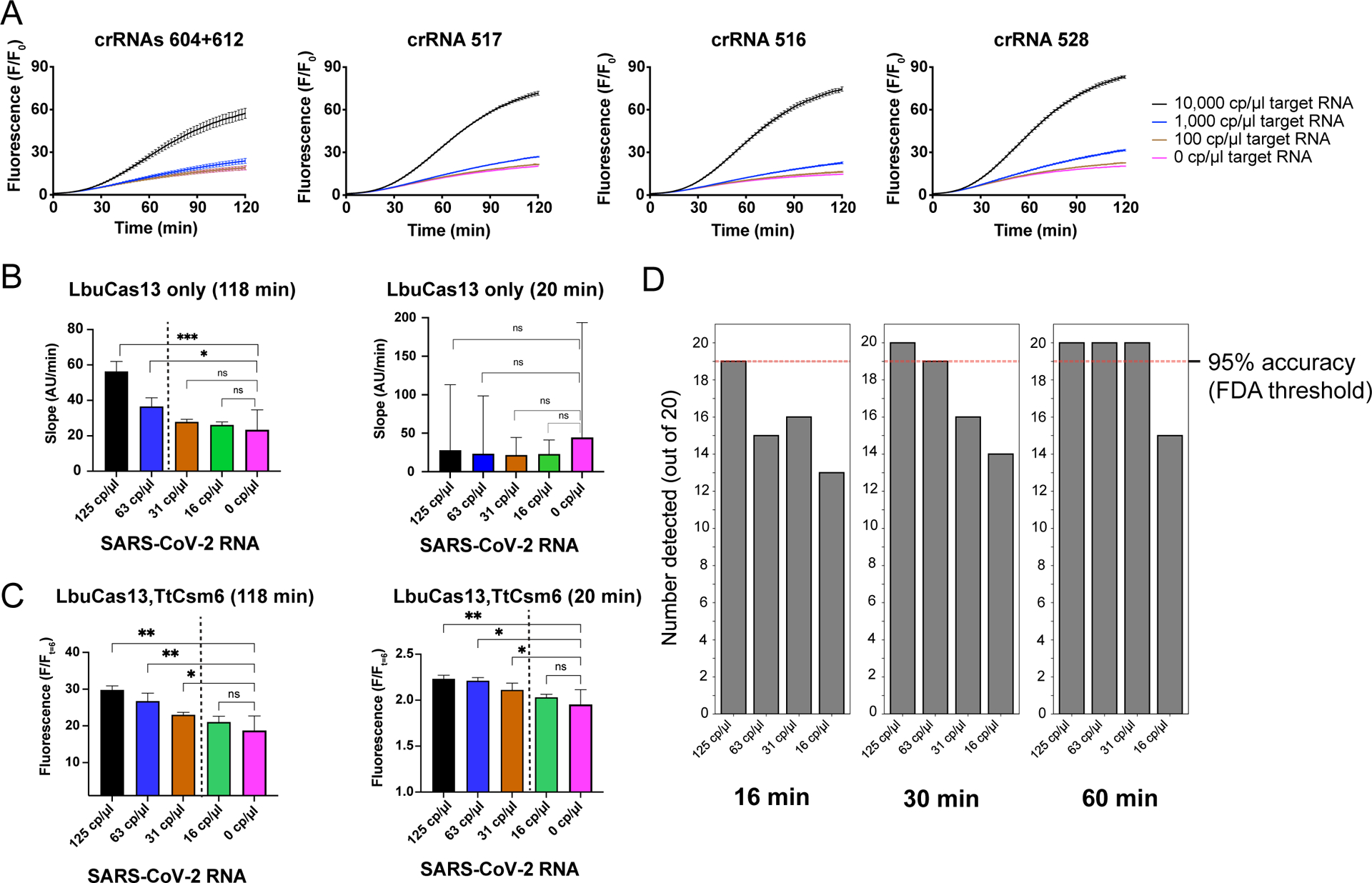
a) Testing the programmability of LbuCas13-TtCsm6 with different crRNAs targeting the SARS-CoV-2 genome. In the leftmost panel, LbuCas13 is loaded with two crRNAs, 604 and 612, and in the remaining panels, it is loaded with a single crRNA sequence (516, 517, or 528). Twist synthetic SARS-CoV-2 control RNA was used as the target. Mean normalized fluorescence and S.E.M (n = 3) is plotted over the two-hour time course of the reaction.
b) Direct detection of externally validated SARS-CoV-2 RNA control from BEI resources. The slopes of the fluorescent signal generated by reporter cleavage were analyzed at 118 mins and 20 min using LbuCas13 complexed with eight crRNAs targeting the SARS-CoV-2 genome. The mean rate of fluorescence increase, as determined by linear regression, are plotted, with the error bars showing the 95% confidence interval (n = 3). Pairwise comparisons of the slopes to 0 cp/μl by ANCOVA are shown as significant if the p-value < 0.05. Asterisks shown in the figure correspond to the following p-values: * for P ≤ 0.05, ** for P ≤ 0.01, and *** for P ≤ 0.001. Non-significant p-values (P > 0.05) are shown as “ns.” A dotted line indicates the boundary between detected and undetected target RNA concentrations.
c) LbuCas13-TtCsm6 detection of BEI SARS-CoV-2 RNA was carried out with eight crRNAs and the single-fluoro TtCsm6 activator. The mean normalized fluorescence (fluorescence at the endpoint divided by fluorescence at 6 min) of three technical replicates was determined at 118 min and 20 min, using t = 6 min as the initial data point for normalization (see Extended Data Figure 6). Error bars show the 95% confidence intervals. The significance of pairwise comparisons is represented as in b with asterisks to indicate the p-value or “ns” for non-significant p-values. A dotted line indicates the boundary between detected and undetected target RNA concentrations.
d) Accuracy of the LbuCas13-TtCsm6 assay assessed over 20 replicates in a plate reader. Individual positive replicates were compared to the 95th percentile of the control distribution to determine if their signal was higher; a difference between the positive replicate and the control distribution was considered significant when p-value of the comparison was < 0.05 (see Methods). The number detected out of 20 replicates is shown at 16, 30, and 60 min for 125 cp/μl, 63 cp/μl, 31 cp/μl and 16 cp/μl of BEI extracted SARS-CoV-2 RNA. The FDA threshold for limit of detection is shown as a dotted line in the bar graphs.
In addition to sensitivity and speed, diagnostic assays must meet the accuracy threshold set by the Food and Drug Administration (FDA) for emergency use authorization, which stipulates that 19 of 20 replicates (95% accuracy) are detected at ~1–2 times the limit of detection. To determine if the LbuCas13-TtCsm6 tandem nuclease assay would meet this validation criterion, we ran 20 replicates of our assay around the limit of detection and analyzed individual replicates by comparing them to the 95th percentile of the negative control distribution (Fig. 3D; Extended Data Figs. 7–8; see Methods). Our assay detected 125, 63, and 31 cp/μl of viral RNA with 100% accuracy (20/20 replicates), and 16 cp/μl with 75% accuracy (15/20 replicates) after 60 min, indicating that 31 cp/μl is the limit of detection as defined by the FDA (Fig. 3D). In addition, 125 cp/μl and 63 cp/μl of viral RNA could be detected with 95% accuracy (19/20 replicates) in as short as 16 min and 30 min, respectively. Taken together, the performance and simplicity of the LbuCas13-TtCsm6 tandem nuclease chemistry creates a Fast Integrated Nuclease Detection In Tandem (FIND-IT) assay that integrates the sensitivity of LbuCas13 with the accelerated time-to-detection of provided by the chemically stabilized TtCsm6 activator, while simultaneously achieving the accuracy required for diagnostic tests.
To demonstrate the feasibility of incorporating FIND-IT into a point-of-care testing workflow, we developed a detector consisting of a microfluidic chip with reaction chambers, a heating module that maintains the reactions at 37°C, and a compact fluorescence imaging system (Fig. 4A–C). The detector, which simultaneously monitors signal from two reaction chambers, is capable of sensing changes in signal as small as 1%, corresponding to a difference of 3 times the Root-Mean-Square-Error (RMSE) in the mean signal. We applied LbuCas13-TtCsm6 reactions containing target RNA (SARS-CoV-2 genomic RNA) or no target RNA to the reaction chambers and monitored the fluorescence signal for one hour (Fig. 4D). The signal in a reaction containing 400 cp/μl of extracted SARS-CoV-2 genomic RNA increased non-linearly, resulting in a ~4.7-fold increase in one hour, while a negative control lacking target RNA exhibited a ~1.7-fold change (Fig. 4D, left). This ~270% increase in fluorescence of the 400 cp/μl reaction compared to the background control contrasts with the ~3% variation between two negative controls run side-by-side (Fig. 4D, right). Comparison of samples to a control reaction run in parallel on the same chip could thus be used to normalize baseline signal fluctuations between runs. Combined with the substantial difference in output between positive and negative samples, this suggests that the LbuCas13-TtCsm6 reaction is both chemically compatible with the microfluidic chip, and, when implemented in a compact detector, is suitable for use as a point-of-care diagnostic.
Figure 4: Implementation of FIND-IT on a compact fluorescence detector for testing of COVID-19 patient samples.
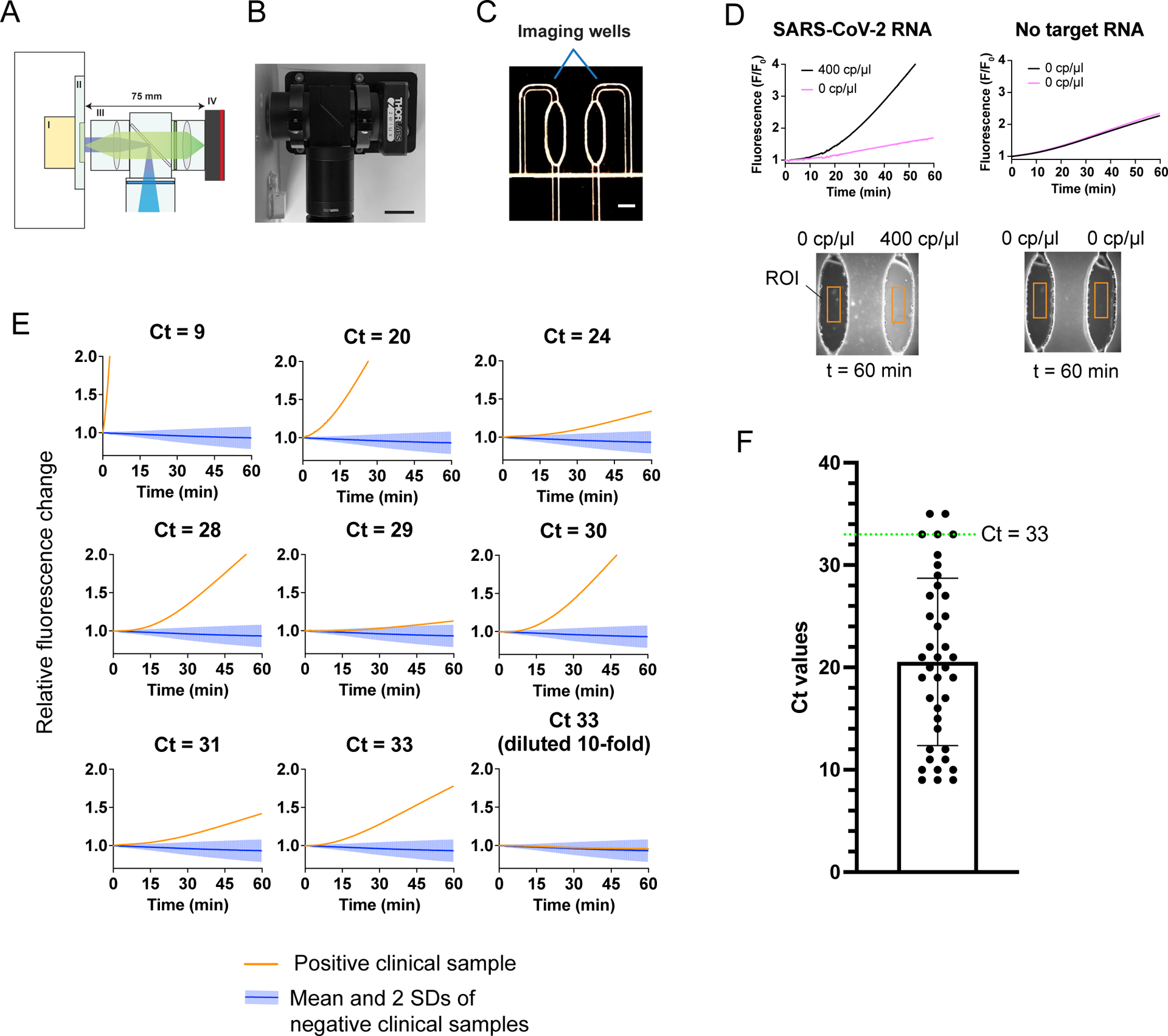
a) A compact and sensitive fluorescence detector for the LbuCas13-TtCsm6 assay. The heating module (I), sample cartridge (II), imaging optics (III), and camera (IV) are indicated by roman numerals. The size of the imaging optical train is shown above in millimeters (mm).
b) Photograph showing the compact fluorescence imaging system. Scale bar is 20 mm.
c) Photograph showing the two imaging chambers on the custom microfluidic chip, with a scale bar of 1.75 mm.
d) Detection of BEI SARS-CoV-2 genomic RNA on the compact fluorescence detector. Either 400 cp/μl of target RNA or water (0 cp/μl) was added to the reactions, and normalized fluorescence signal is shown over 1 hour. Images below each graph show the final image collected of the imaging chambers. Typical examples of ROIs (regions of interest) selected for integrating fluorescence intensity are shown as orange boxes.
e) Detection of SARS-CoV-2 RNA in total RNA extracted from nasopharyngeal swab samples obtained from the IGI testing laboratory30. Each sample was analyzed in parallel on the microfluidic chip with a no-target control (0 cp/μl), in which water instead of extracted RNA was added to the reaction. The fluorescence change of positive samples relative to the control are plotted as red-orange lines, with its corresponding qRT-PCR-derived Ct value above the graph. Positive samples with Ct values ranging from 9 to 33 are shown. The mean fluorescence change of six negative patient samples (Ct > 37) relative to their controls is plotted as a blue line on each graph, with the shaded region representing two standard deviations. Reactions were run for 60 min, with images collected every 10 s. Individual plots for samples and controls that were run in parallel can be found in Extended Data Fig. 9.
f) Scatter dot plot showing the distribution of Ct values of 39 positive patient samples identified in 296 samples total, obtained from the IGI testing laboratory30. A green dotted line indicates a Ct value of 33. Samples with Ct values at and below the green dotted line were detected by the FIND-IT assay in the integrated fluorescence detector.
To test whether FIND-IT could detect SARS-CoV-2 genomic sequences in total RNA extracted from nasopharyngeal patient samples, we tested nine positive samples with qRT-PCR-derived Ct values ranging from 9–33 (Supplementary Table 5), as well as six negative patient samples (Ct > 37), on the fluorescence detector (Fig. 4E, Extended Data Fig. 9)30. To account for fluorescence variations from cartridge to cartridge, we determined the fluorescence change of the samples relative to a no-target control that was run in parallel, as in Fig. 4D (See Methods). The negative samples ran slightly below or close to the background control, possibly due to the presence of human RNA competing with Cas13- or Csm6-mediated cleavage in the FIND-IT assay (Extended Data Fig. 9). To set a threshold for positive signal, we required that the relative fluorescence change be larger than the mean and two standard deviations of the clinical negative distribution (n = 6). Using this criterion, FIND-IT detected samples with Ct values of 9 and 20 in <5 min, and samples with Ct values of 24–28 by ~15–20 min. For samples with Ct values 29–33, the time to detection and signal increase was more variable (15–45 min). This could be due to either incomplete conversion of RNA to DNA during the reverse transcriptase step during qRT-PCR or variable levels of total human RNA in the clinical samples, which could compete with the fluorogenic reporter for TtCsm6-mediated RNA cleavage. It could also be due to partial sample degradation due to prior freeze-thaw cycles and handling, since these samples were obtained and tested after the initial qRT-PCR analysis by the IGI testing lab. Notably, dilution of the Ct 33 sample by 10-fold (equivalent to ~3 Ct values; Extended Data Fig. 10) led to a decrease in signal to background levels, as expected for lower concentrations of viral RNA. Based on the Ct values of all COVID-19-positive samples (n = 39) identified in patient samples from the IGI testing laboratory (n = 296), our assay could detect ~95% of all positives in less than an hour (Fig. 4F)30. Combined with upstream RNA extraction, this assay could be deployed as part of a point-of-care testing workflow to capture most individuals at the early, more infectious stages of COVID-19, when Ct values range between 20–3031.
Discussion
CRISPR-Cas nucleases are attractive tools for diagnostic applications given their programmability and ability to directly detect sequences in viral genomes. However, increases in the sensitivity and speed of CRISPR-based detection are needed to enable widespread use of these technologies as diagnostics. In this study, we demonstrate that LbuCas13 can be coupled with TtCsm6 using chemically modified RNA activators, leading to a 100-fold increase in sensitivity over unmodified activators. Combining TtCsm6 with an LbuCas13 effector containing eight different crRNAs also enabled detection of as low as 31 copies per μl, with detection accuracy reaching 100% by 60 min over 20 replicates. Higher viral RNA concentrations can be detected with similar accuracy at earlier timepoints, with 125 cp/μl and 63 cp/μl detected in 16 and 30 min, respectively. We also show that the intrinsic programmability of LbuCas13 is preserved in this assay with different crRNAs targeting the SARS-CoV-2 viral genome. The assay can be readily adapted to a compact device comprising a microfluidic cartridge integrated with a compact LED camera. It also detected SARS-CoV-2 RNA in clinical samples with Ct values ranging between 9 to 33, indicating its ability to potentially capture individuals at highly infectious stages of disease31. Full concordance of positive samples identified previously by qRT-PCR was also observed. The signal of all negative samples we tested could also be distinguished from the positive samples by comparison to a background control. Implementation of a full point-of-care testing workflow using FIND-IT will require additional development of suitable sample intake and processing procedures, as well as a robust data analysis and management pipeline. Future studies with greater numbers of patient samples will also be required for clinical validation of diagnostic testing strategies that utilize FIND-IT for RNA detection. The simplicity and accelerated response of this detection chemistry could have broad use for detection of SARS-CoV-2 and other viral or cellular RNA sequences in the future.
This approach is one of the few technologies that does not use target amplification to increase sensitivity and time to detection, relying instead on tandem nucleases, Cas13 and Csm6, to achieve both high sensitivity and fast signal generation in a single-pot reaction. It achieves a sensitivity of ~30 cp/μl, which is below the 100 cp/μl that is considered useful for point-of-care diagnostics in pandemic control3. Importantly, the FIND-IT chemistry achieves this detection sensitivity in the context of a single combined reaction containing both nucleases and the activator oligonucleotide, a workflow that is simple and reduces cost. Integration of TtCsm6 and its potent modified activator with other Cas13-based detection assays could lead to further improvements in time to detection and/or sensitivity of current methods for RNA detection21,32.
These results establish FIND-IT as a one-step assay that enables rapid RNA detection with sensitivity, accuracy, and adaptability that is amenable to point-of-care detection of virtually any RNA sequence. FIND-IT demonstrates the value of stabilizing Csm6 nuclease activation and enabling its tandem use with RNA-cleaving CRISPR-Cas proteins for direct and rapid RNA detection33–36. This technology could enable practical on-site detection of viral or human RNA in clinical samples, or plant, fungal, or microbial RNA in environmental samples.
Methods
Protein purification
The DNA construct encoding Leptotricia buccalis Cas13a (LbuCas13a) was codon-optimized for expression in E. coli13. LbuCas13a expression and purification were carried out as previously described13 with modifications, summarized here: A construct encoding for N-terminal His6-MBP-TEV-tagged LbuCas13a (used for production of protein used in Fig. 1 and Extended Data Fig. 3) or His6-SUMO-tagged LbuCas13a (used for production of protein used in Figs. 2–4 and all other Extended Data Figures), was transformed into E. coli Rosetta 2 (DE3) pLysS cells and cultured in terrific broth at 37°C. At an OD600 of 0.6–0.8, cultures were cooled on ice for 15 min prior to induction with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and expression overnight at 16°C. Compact cell pellets measuring ~10 mL in a 50 mL Falcon tube were resuspended in 100 mL of lysis buffer (50 mM HEPES pH 7.0, 500 mM NaCl, 1 mM TCEP, 10% glycerol, supplemented with 0.5 mM PMSF and 4 EDTA-free protease inhibitor tablets, Roche) and lysed by sonication. Soluble His6-MBP-TEV or His6-SUMO LbuCas13a were clarified by centrifugation at 35,000xg, the supernatant was applied to a 5 mL HiTrap NiNTA column (GE Healthcare) and eluted over a linear imidazole (0.01–0.3 M) gradient via FPLC (ÄKTA Pure). Following overnight dialysis of the peak fractions with TEV or SUMO protease, LbuCas13a was further purified by HiTrap SP (GE Healthcare) ion exchange and, in the case of the MBP-tagged construct, removal of MBP using MBP trap HP columns (GE Healthcare). Finally, size-exclusion chromatography was carried out using a Superdex 200 16/600 column (GE Healthcare) with 1 mM TCEP supplemented into the gel filtration buffer. Peak fractions were pooled, concentrated, and aliquoted into PCR strip tubes before snap freezing in liquid nitrogen.
The codon-optimized sequence encoding amino acid residues 2–467 of TtCsm6 (also known as TTHB152) was cloned into an expression vector that also encodes an N-terminal His6-SUMO tag followed by a TEV (Tobacco Etch Virus) cleavage site. The protein was expressed in BL21(DE3) cells, as described previously, and pellets were stored at −20°C29. Thawed pellets were resuspended in lysis buffer (20 mM HEPES pH 8.0, 500 mM KCl, 5 mM imidazole, 1 mM TCEP) supplemented with EDTA-free protease inhibitors (Roche) and lysed by sonication. Following clarification of the lysate, the His-SUMO-TtCsm6 protein was immobilized on HIS-Select Nickel Affinity Gel (Sigma) at 4°C, washed with lysis buffer, and eluted using the same buffer supplemented with 250 mM imidazole. TEV protease was added to remove the His-SUMO tag, and the protein was dialyzed overnight at 4°C against buffer containing 25 mM HEPES pH 7.5, 150 mM NaCl, 5% (v/v) glycerol, and 1 mM TCEP in a 10,000 MWCO dialysis cassette. The protein solution was then centrifuged to remove aggregates, concentrated, and subjected to size-exclusion chromatography purification on a Superdex 200 16/600 column (GE Healthcare). Peak fractions were pooled, concentrated to ~600 μM, and flash-frozen in liquid nitrogen. Working stocks were prepared by diluting the protein to 50 μM in buffer containing 20 mM HEPES (pH 7.5), 200 mM KCl, 5% (v/v) glycerol, 1 mM TCEP and flash-freezing in 5 μl aliquots using liquid nitrogen.
The codon-optimized EiCsm6 gene was cloned into a pET expression vector encoding His6-tagged EiCsm6 with an intervening TEV cleavage site (ENLYFQG) by Genscript. The protein was expressed and purified by Shanghai ChemPartner using the following steps. The plasmid encoding His-TEV-EiCsm6 was transformed into BL21(DE3) Star cells and grown in LB broth at 37°C to an OD of ~0.6. Expression was induced with 0.5 mM IPTG and cultures were transferred to 18°C for 16 h. Cells were resuspended in lysis buffer (20 mM HEPES pH 7.5, 500 mM KCl, 5 mM imidazole) supplemented with EDTA-free protease inhibitors (Roche) and lysed by sonication. The protein was immobilized on a 5-ml Ni2+-NTA fast-flow column, washed with lysis buffer, and then eluted with a step gradient of imidazole with fractions at 16%, 50%, and 100% of elution buffer (20 mM HEPES pH 7.5, 500 mM KCl, 500 mM imidazole) collected for further purification. The protein was incubated with TEV protease for 32 h at 25°C to remove the His6-tag. Following this, the protein was re-applied to the Ni2+-NTA column, washed with 20 mM HEPES pH 7.5, 500 mM KCl and untagged protein was eluted with 20 mM imidazole. Then, the protein was subjected to size-exclusion chromatography in buffer containing 20 mM HEPES pH 7.5, 500 mM KCl, and 5% (v/v) glycerol. Peak fractions were collected and concentrated to 15 mg/ml, and flash-frozen in small aliquots using liquid nitrogen.
Oligonucleotides
Reporter oligonucleotides, which comprised either a polycytosine sequence for Csm6 assays (C5) or a polyuridine sequence for Cas13 assays (U5) were labeled with a 5´-fluorescein and 3´-Iowa Black moiety. They were ordered HPLC-purified from IDT in bulk (250 nmole to 1 μmole) to avoid batch-to-batch variation in basal fluorescence. For direct comparisons of Csm6 reactions done with different activators or target concentrations, the same batch of reporter was always used. The crRNA guides GJK_073 and R004 were ordered as desalted RNA oligonucleotides from IDT at a scale of 25 nanomoles. Cas13 crRNAs 516, 517, 528, 604, 612, 542, 546, 564, 569, 588, and 596 targeting different regions of the SARS-CoV-2 genome were ordered as desalted oligonucleotides from Synthego at a scale of 5 nanomoles. Unmodified and 2´-modified A4>P oligonucleotides were synthesized by IDT at a 250 nmole scale. Sequences of oligonucleotides are listed in Supplementary Table 3. The set of eight crRNAs for LbuCas13 (604, 612, 542, 546, 564, 569, 588, and 596) used in Fig. 3B–D are able to detect all published SARS-CoV-2 strains in silico (51631 complete SARS-CoV-2 complete genome sequences were downloaded from NCBI under the taxonomy ID 2697049 as of 03/04/2021). The inclusivity for each guide is listed in Supplementary Table 4.
Preparation of SARS-CoV-2 RNA targets
An in vitro transcribed RNA corresponding to a fragment of the SARS-CoV-2 genome was used as a target in Fig. 2 and Extended Fig 4. A gene fragment (gBlock) corresponding to nucleotides 27222–29890 of the SARS-CoV-2 Wuhan-Hu-1 variant genome (MN908947.2) was obtained as a gift from Emily Connelly in the lab of Charles Craik at UCSF. The gBlock was PCR amplified with a 5’ primer bearing an extended T7 promoter sequence (GTCGAAATTAATACGACTCACTATAGG) before separating and extracting the template on an agarose (2% w/v) gel (0.5X TAE buffer) and further purification by phenol-chloroform extraction (pH 8.0) of the purified template and ethanol precipitation. The highly purified and RNase-free template was used in a high yield in vitro transcription reaction containing 1X transcription buffer (30 mM Tris-Cl, 25 mM MgCl2, 0.01% (v/v) Triton X-100 and 2 mM spermidine, pH 8.1), 5 mM of each NTP, 10 mM DTT, 1 μg/mL pyrophosphatase (Roche), and 100 μg/mL T7 RNA polymerase (purified in house) incubated at 37°C for 4 hours. The reaction was quenched with the addition of 25 units of RNase-free DNase (Promega) at 37°C for 30 mins before the addition of 2 volumes of acidic phenol and subsequent phenol-chloroform extraction. The RNA was then ethanol precipitated and snap-frozen for storage at −80°C.
Twist synthetic SARS-CoV-2 RNA control 2 (Catalog # SKU 102024) was used as a target in Fig. 3A. An externally validated genomic SARS-CoV-2 RNA control from BEI resources (NIAID/NIH) was used in Fig. 3B–D, Fig. 4D, and Extended Data Fig. 6 for better comparability to previous studies using this reagent (Lot numbers 70034085 and 70034826). This reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: Genomic RNA from SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52285. BEI control RNA was handled in accordance with environmental health and safety regulations for biosafety level 2 materials.
LbuCas13-TtCsm6 plate reader assays
In Fig. 1 and Extended Data Fig. 3, LbuCas13-TtCsm6 reactions contained 40 nM LbuCas13, 20 nM crRNA (GJK073 or R004), 100 nM TtCsm6, 100–200 pM RNA target (GJK075 or R010), and 200 nM C5 reporter, with either Csm6 activator (A3–6U6) or DEPC-treated water added for the no-activator control. The reactions were carried out at 37°C in buffer containing 20 mM HEPES (pH 6.8), 50 mM KCl, 5 mM MgCl2, 100 μg/ml BSA, 0.01% Igepal CA-630, and 2% glycerol.
For experiments in Fig. 1E, the same conditions were used as above except 1 μM of the A4-U6 activator was added. The reaction was initiated and allowed to proceed until it reached a plateau, at which point 1 μl of either 20 μM A4-U6, 4 nM target RNA, or 2 μM TtCsm6 protein was added to double the amount of each reagent that was in the reaction. For buffer controls, 1 μl of the reaction buffer was added instead. LbuCas13-crRNA was assembled at a concentration of 1 μM LbuCas13 and 500 nM crRNA for 15 minutes at room temperature. 20-μl plate reader assays were started by mixing 15 μl of a mastermix containing the LbuCas13-crRNA complex and TtCsm6 in buffer with 5 μl of a second mastermix containing RNA target, reporter, and the Csm6 activator in buffer. The protein mastermix was also equilibrated to room temperature for ~15–20 min prior to starting the reaction. Fluorescence measurements were made every 2 minutes on a Tecan Spark plate reader set with an excitation wavelength of 485 nm, emission wavelength of 535 nm, and z-height optimized to a well containing the reporter molecule in buffer.
In Figs. 2 and 3, LbuCas13-TtCsm6 reactions contained 50 nM LbuCas13, 50 nM crRNA, 100 nM TtCsm6, 1 U/μl of Murine RNase inhibitor (New England Biolabs), 2 μM of TtCsm6 activator, and 200 nM C5 reporter, with either target RNA or DEPC water added. The reaction buffer used for these assays contained 20 mM HEPES pH 6.8, 50 mM KCl, 5 mM MgCl2, and 5% (v/v) glycerol in DEPC-treated, nuclease-free water (Fisher Scientific or Invitrogen) supplemented with 1 U/μl Murine RNase inhibitor, as previously described21. Reactions were started by mixing 15 μl of the protein mastermix, containing LbuCas13, crRNA, TtCsm6, with 5 μl of the activator/reporter mastermix, containing TtCsm6 activator, target RNA, and a fluorescent C5 reporter. Measurements were taken in a plate reader every 2 min at 37°C. In Figs. 2 and 3A, measurements were taken on a Biotek plate reader with excitation of 485 nm and emission of 528 nm with z-height of 10.0 mm, and in Figs. 3C–D, a Tecan Spark plate reader was used with excitation wavelength of 485 nm and emission wavelength of 535 nm and z-position optimized to a well containing the reporter molecule in buffer. For the 20-replicate experiments in Fig. 3D, the z-position setting on the Tecan Spark was closely matched between experiments (z-position of 18962–18966 μm used across 125, 63, 31, and 16 cp/μl datasets) to ensure consistency across experiments for comparison.
For LbuCas13-EiCsm6 assays in Fig. 2 and Extended Data Fig. 4, the same buffer and concentrations of LbuCas13, crRNA, and reporter were used as for the LbuCas13-TtCsm6 assays, except 10 nM EiCsm6 and 0.5 μM EiCsm6 activator were added instead of TtCsm6 and its activator. 20 μl reactions were started by addition of a 15-μl LbuCas13/EiCsm6 mastermix to a 5 μl activator/reporter mastermix. Murine RNase inhibitor (NEB) was also included in both mastermixes at 1 U/μl. Measurements were taken every 2 or 3 min at 37°C in a Biotek plate reader, with excitation wavelength of 485 nm, emission wavelength of 528 nm, gain of 43, and z-height of 10.0 mm.
All plate reader assays were carried out in low-volume, flat-bottom, black 384-well plates with a non-binding surface treatment (Corning #3820). The data in Extended Data Figure 4 was collected using the same plate type, but without a non-binding surface (NBS) treatment (Corning #3821), due to a shortage of the NBS-treated plates during the COVID-19 pandemic. Fluorescence of Csm6 reactions were normalized where shown by dividing all values by the initial value at t = 0 (F/F0), except in Figs. 3C–D, where they were normalized to t = 6 min (F/Ft=6), to allow for ~5 min of temperature equilibration to 37°C at the start of the assay (see Extended Data Fig. 8). Optically clear ABsolute qPCR Plate Seals (Thermofisher Scientific #AB1170) were used to cover the 384-well plate when doing assays with extracted B.E.I. SARS-CoV-2 genomic RNA. All LbuCas13-Csm6 plate reader experiments in Figs. 1, 2, 3A, 3C and Extended Data Figs. 1, 3, 4 were performed in triplicate. The mean and standard error of the mean (S.E.M.) are plotted in graphs showing the reaction time course, where indicated in the legends.
Direct activation of TtCsm6 by A4>P oligonucleotides
For direct activation experiments, 100 nM TtCsm6 in 1X reaction buffer (20 nM HEPES pH 6.8, 50 mM KCl, 5 mM MgCl2, and 5% (v/v) glycerol in DEPC-treated, nuclease-free water) supplemented with 1 U/μl Murine RNase inhibitor was mixed with varying concentrations (0.5–2 μM) of an A4>P oligonucleotide. Measurements were taken at 37°C every 2 min in a Tecan Spark plate reader, using an excitation wavelength of 485 nm, emission wavelength of 535 nm, and z-height optimized to a well containing the reporter molecule in buffer. All reactions were performed in triplicate. The mean and standard error of the mean (S.E.M.) are plotted in graphs showing the reaction time course.
Modeling of the Cas13-Csm6 detection reaction
A kinetic scheme of chemical reactions was created and populated with known kinetic rates and equilibrium constants. Kinetic rates were used where they were known, where only equilibrium rates were known, the forward rates were assumed to be 1 nM−1 × sec−1 and a reverse rate was chosen to produce the known equilibrium constant. Rates and equilibrium constants were similar to those reported in previous publications1,12,23,37,38. For the CARF domain, a Km was known instead of a Kd, so the CARF kcat was subtracted from the reverse rate to produce an approximate Kd. The Cas13 background cleavage rate was selected to match the background rate observed in purified LbuCas13 preparations. The full kinetic scheme can be found in the Supplementary Information (SI). This kinetic scheme was then converted into a system of ordinary differential equations modeling the rate of change in the concentration of each reaction component as a function of time and the concentrations of the components using Mathematica. A numerical solution to the system of ordinary differential equations was created with a time step of 0.001 seconds and a total time of 2000 seconds using the Mathematica’s NDSolve at different concentrations of viral target and at different values for the CARF kcat. The full system of ordinary differential equations and starting conditions can be found in SI.
LbuCas13 direct detection experiments and analysis
These assays were performed and analyzed as described previously21, with the following modifications. Data were collected on a Tecan Spark plate reader at 37°C with a gain of 87 and a z-position optimized to a well in the plate containing reporter molecule in buffer. Fluorescence was measured every 2 minutes up to 118 min using an excitation wavelength of 485 nm (10 nm bandwidth) and 535 nm emission (20 nm bandwidth). Linear regression and pairwise comparison of slopes using ANCOVA was done using GraphPad Prism 9 as described, considering replicate y-values as individual data points21. For determination of the limit of detection at 20 min, we used only data collected in first 20 minutes of the assay. All assays were done in triplicate.
LC-MS Analysis
The degradation of cA4, A4>P or fA4>P was examined by incubating 5 μM TtCsm6 with 25 μM RNA activator at 37°C for 30 min in a total volume of 15 μl. The reaction was quenched by adding 1 μL 0.5 M EDTA, incubating the sample at 95°C for 5 min and finally centrifuging at 16,000 × g for 5 min to pellet and separate denatured Csm6 protein from its RNA activator. Oligonucleotide samples were then analyzed using a Synapt G2-Si mass spectrometer that was equipped with an electrospray ionization (ESI) source and a BEH C18 ionKey (length: 50 mm, inner diameter: 150 μm, particle size: 1.7 μm, pore size: 130 Å), and connected in line with an Acquity M-class ultra-performance liquid chromatography system (UPLC; Waters, Milford, MA). Acetonitrile, formic acid (Fisher Optima grade, 99.9%), and water purified to a resistivity of 18.2 MΩ·cm (at 25°C) using a Milli-Q Gradient ultrapure water purification system (Millipore, Billerica, MA) were used to prepare mobile phase solvents. Solvent A was 99.9% water/0.1% formic acid and solvent B was 99.9% acetonitrile/0.1% formic acid (volume/volume). The elution program consisted of isocratic flow at 1% B for 2 min, a linear gradient from 1% to 99% B over 2 min, isocratic flow at 99% B for 5 min, a linear gradient from 99% to 1% B over 2 min, and isocratic flow at 1% B for 19 min, at a flow rate of 1.5 μL/min. Mass spectra were acquired in the negative ion mode and continuum format, operating the time-of-flight mass analyzer in resolution mode, with a scan time of 1 s, over the range of mass-to-charge ratio (m/z) = 100 to 5000. Mass spectrometry data acquisition and processing were performed using MassLynx software (version 4.1, Waters).
Limit-of-detection analysis for LbuCas13-TtCsm6
For determination of limit of detection using the triplicate data in Fig. 3C, a one-tailed t-test with Welch’s correction was used to compare mean fluorescence fold-change of a positive reaction (with initial point being t = 6 min) to that of the control without RNA target, using GraphPad Prism 9. The limit of detection was determined using the triplicate data collected at 20 min and 118 min as timepoints for comparison to LbuCas13 detection assays. Asterisks shown in the figure correspond to the following p-values: * for P ≤ 0.05, ** for P ≤ 0.01, and *** for P ≤ 0.001. The sequences of the crRNAs used are provided in Supplementary Table 3.
Limit-of-detection analysis for 20-replicate experiments
For all time-series experiments, data were collected at 2 min intervals and normalized by dividing each replicate by its value at t = 6 min to account for initial signal fluctuations during temperature equilibration (Extended Data Fig. 6). Samples were prepared and analyzed in batches of 10 with five experimental samples and five 0 cp/μl negative controls started simultaneously by a multichannel pipette. Reactions were set up as described in the section above (LbuCas13-TtCsm6 plate reader assays). Analysis was performed at the 16-, 30-, and 60-minute time points. At each of these time points the mean and standard deviation of the 0 cp/μl negative controls within each batch were determined and used to fit a normal distribution. The distribution of negative results was then used to calculate the probability of seeing a point equal to or higher than each experimental sample using the survival function of the normal distribution. This value is equivalent to a one-tailed p-value associated with a null hypothesis in which the experimental value is not greater than the value of the 0 cp/μl negative control. The results are displayed as the fraction of experimental samples out of the total 20 that could be distinguished from the negative control using a. p-value cutoff of 0.05. These cutoffs represent a predicted 5% false positive rate. We did not perform multiple hypothesis correction on this analysis to mirror the analysis that would be performed in a clinical setting, where an individual’s sample would not be corrected based on the number of samples run but where type 1 error (false positive rate) would instead be controlled by setting an appropriate p-value cutoff. The full time-course of the data used in this analysis are shown in Extended Data Fig. 7 and the improved signal-to-noise for normalizing data to t = 6 min for plate reader measurements is shown for part of the 125 cp/μl dataset in Extended Data Fig. 8.
Experiments using the compact LED-based fluorescence detector
The system camera was focused on and aligned with the imaging wells using an empty cartridge. Then, premixed 60 μl reactions of the LbuCas13-TtCsm6 reaction containing the single-fluoro A4-U6 activator were loaded directly into inlets leading to the ~15 μl imaging chambers of a microfluidic chip using a pipette, until the chambers were completely filled. The custom chips were fabricated from poly(methyl methacrylate) and pressure-sensitive adhesive. Reactions were assembled using the same conditions and reagent concentrations as the reactions in Fig. 3C and 3D, except a total reaction volume of 60 μl was used. The target RNA volume constituted 10% of the total reaction volume in all reactions testing clinical samples and BEI control SARS-CoV-2 RNA. The reaction chamber temperature was maintained at 37°C with ~1–2°C variation between runs. Reactions were then imaged every 10 s for 30–60 min using the system camera gain 2 dB and exposure setting of either 150 ms or 100 ms. LED excitation was strobed in synchrony with the camera exposure to avoid photobleaching between data points. ROIs used for signal integration were 150 pixels wide and 400 pixels long, centered in each chamber. For imaging the ~15 μl sample chambers, we require a fairly large field of view (FOV) and a modest numerical aperture (NA) enabling significant depth of focus without images of adjacent sample wells overlapping. To accomplish this in a relatively low-cost, compact device, we designed a custom system using a pair of eyepieces (Edmund Optics, PN#66–208 and 66–210), yielding a system with NA 0.09, FOV diameter 12.0mm, and magnification (M) of 0.54 (chosen to match the sensor size of the Thorlabs CS165MU1 camera to the FOV; sampling at ≥ Nyquist is unnecessary in this “light-bucket” application). The overall system is compact, with nominal track length (sample to camera) of ~75mm (Fig. 4A, B). Fluorescence filters were from Chroma Technologies, ET470/40x, T495lpxr, and ET535/70m, with excitation provided by a 965mW, 470nm LED (Thorlabs M470L4), providing a maximum of ~ 225mW into the 12 mm dia. sample FOV in an epi-illumination Kohler geometry. Custom control of the imaging hardware was implemented in MATLAB (2020a), using Thorlabs drivers and SDK (ThorCam) to control the camera acquisition, and serial communication to an Arduino Bluefruit Feather board to electronically trigger the LED illumination.
To accurately compare signals between two samples chambers, we used a prospective correction method to account for nonuniform illumination and background effects39. Briefly, the measured pixel intensity of reaction chambers is related to actual signals through
| (1) |
where S is a linear scaling factor that models distortions to an image due to illumination non-uniformity; B is an illumination-dependent background signal that accounts for scattering and background fluorescence from the reaction chambers; and D is an additive zero-light term, which is a result of camera offset and fixed pattern thermal (dark) signal. Prior to actual measurements, we determined S, B, and D by acquiring images of blank reaction chambers filled with 1X reaction buffer (20 mM HEPES pH 6.8, 50 mM KCl, 5 mM MgCl2, and 5% (v/v) glycerol in DEPC-treated, nuclease-free water) and a fluorescent slide, respectively. Experimental images were processed according to equation (1) to retrieve actual signals from each channel.
Detection of positive signal in clinical samples
First, illumination-corrected fluorescence values (described in the previous section) were normalized by dividing by the first measurement (F/F0), so that sample and controls curves start at the same value. This avoids signal from the sample well spuriously appearing to be above the negative control signal due to slight offsets in the initial fluorescence. Then, the normalized fluorescence of the clinical sample was divided by the normalized fluorescence of the paired background control (no-target) at every timepoint of the assay (every 10 s). This is henceforth referred to as the “relative fluorescence change” of a sample compared to its corresponding control curve. This analysis was performed for every positive and negative clinical sample run on the detector. Then, to determine the threshold for detection, we determined the mean and standard deviation of the relative fluorescence change for the negative samples (n = 6). We then considered positive signal in a reaction as being over two standard deviations above the mean. This corresponds to signal that would be higher than ~97.5% of the negative clinical sample distribution, as we consider all signal at or below two standard deviations above the mean to be negative.
Determination of Ct values for patient samples
Human nasopharyngeal swabs were collected, processed, and subjected to RNA extraction and qRT-PCR using primers for SARS-CoV-2 in the IGI testing laboratory, as described previously30. Briefly, individual Ct values were determined using primers for the N gene, S gene, and Orf1ab of the SARS-CoV-2 genome for each sample. Samples were considered positive if two out of the three SARS-CoV-2 targets exhibited Ct values of 37 or below. MS2 phage RNA was spiked into the sample and used as an RNA extraction process control. The Ct values reported in this study are the average of the individual Ct values obtained for N and S genes, and Orf1ab. The individual Ct values for all samples and MS2 phage RNA can be found in Supplementary Table 5.
Determination of a standard curve for Ct values using known concentrations of RNA
Two-fold dilutions of TaqPath COVID-19 Combo Kit positive control (ThermoFisher Scientific) were made in nuclease-free water to generate the indicated copies/μl concentrations. 5 μl of diluted control was added to the multiplexed COVID-19 real-time PCR assay (ThermoFisher Scientific), with a final reaction volume of 12.5 μl. Samples were amplified on a QuantStudio6 per the manufacturer’s protocol and analyzed using the Design & Analysis software, v4.2.3 (ThermoFisher Scientific).
Extended Data
Extended Data Figure 1: Direct activation of TtCsm6 by A4>P oligonucleotides.
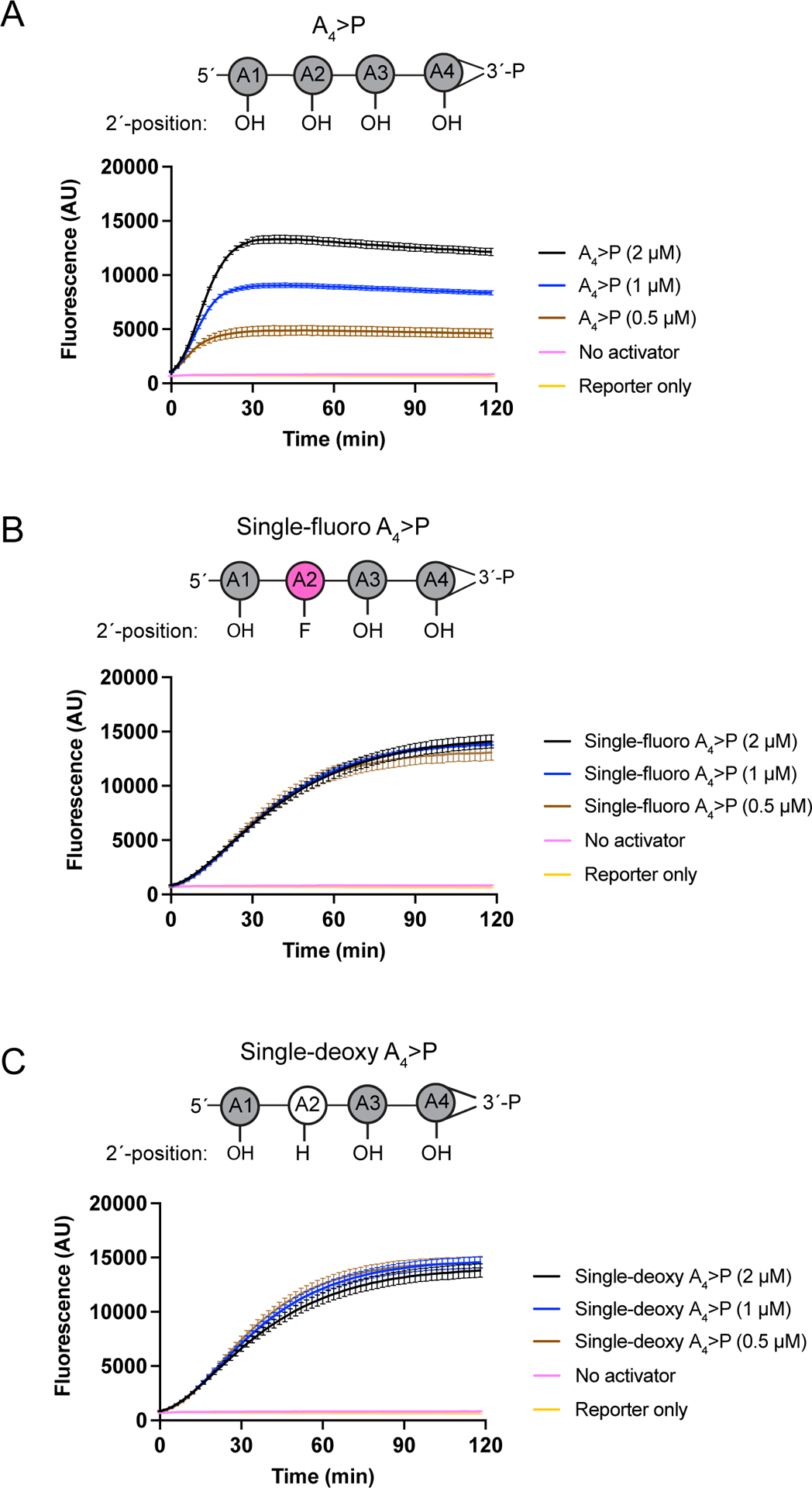
a) Activation of TtCsm6 by A4>P added at varying concentrations. Mean raw fluorescence intensity with error bars indicating S.E.M. (n = 3) is shown in arbitrary units (AU). The schematic on the right is a representation of the A4>P, with adenosines numbered A1-A4 from 5´ to 3´.
b) As in a, but showing activation of TtCsm6 by single fluoro-modified A4>P at varying concentrations. The schematic on the right is as in a, but with the modified nucleotide colored pink.
c) As in a, but showing activation of TtCsm6 by single deoxy-modified A4>P. The schematic on the right is as in a, but with the modified nucleotide colored white.
Extended Data Figure 2: Modeling of the Cas13-Csm6 detection reaction in the presence and absence of activator degradation.
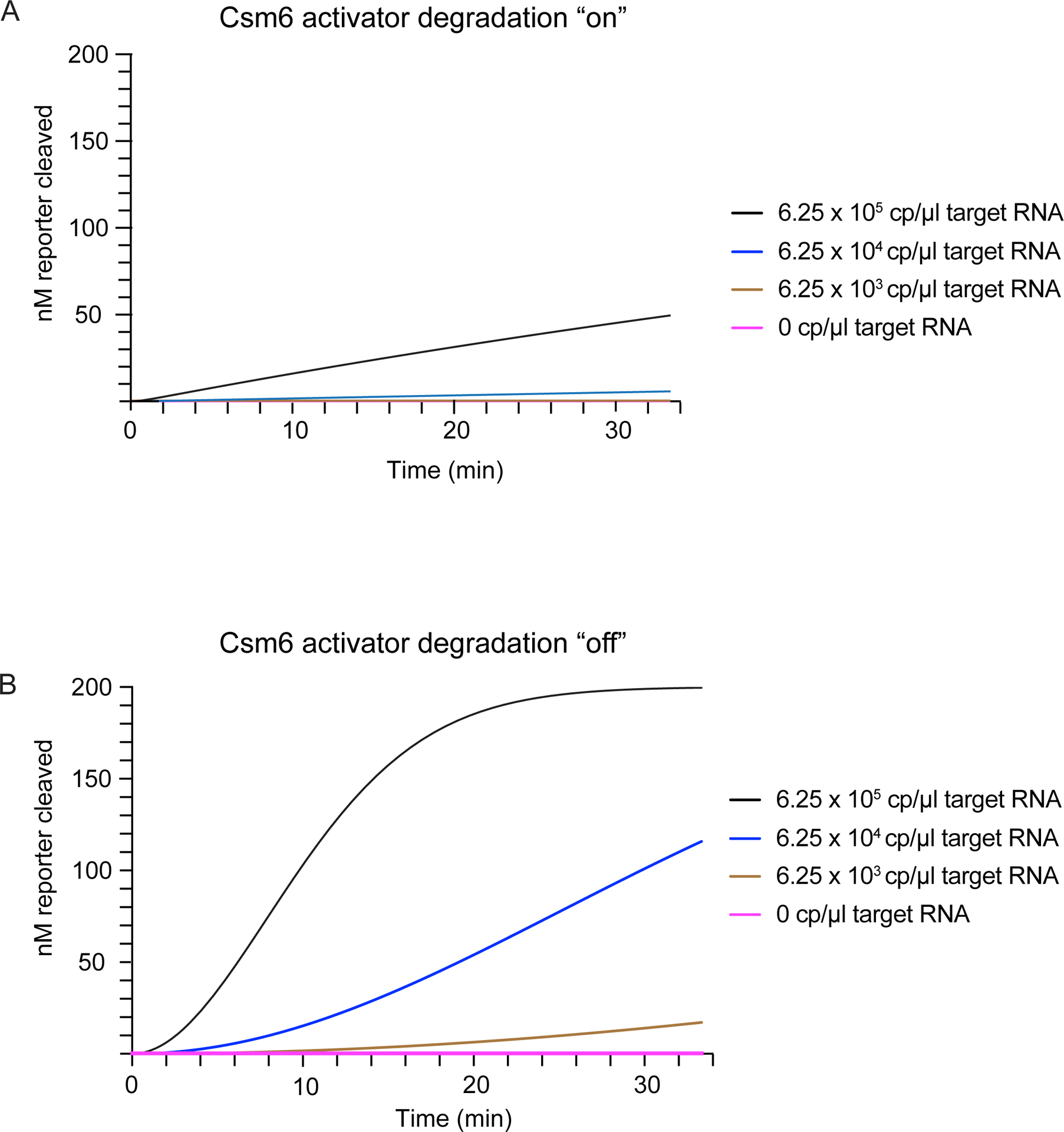
a) A graph showing the modeled kinetics of reporter cleavage by the HEPN domain of Csm6 when 0 cp/μl to 6.25 × 105 cp/μl (1 pM) target RNA is present and the CARF domain is active for cleavage of the A4>P fragment generated by Cas13 cleavage of the A4-U6 activator (CARF kcat = 0.05).
b) As in a, but in a situation where the CARF domain is unable to cleave the A4>P fragment generated by Cas13 cleavage of the A4-U6 activator (CARF kcat = 0).
Extended Data Figure 3: Triple-modified TtCsm6 activators lead to slow kinetics of reporter cleavage in a LbuCas13-TtCsm6 detection assay.
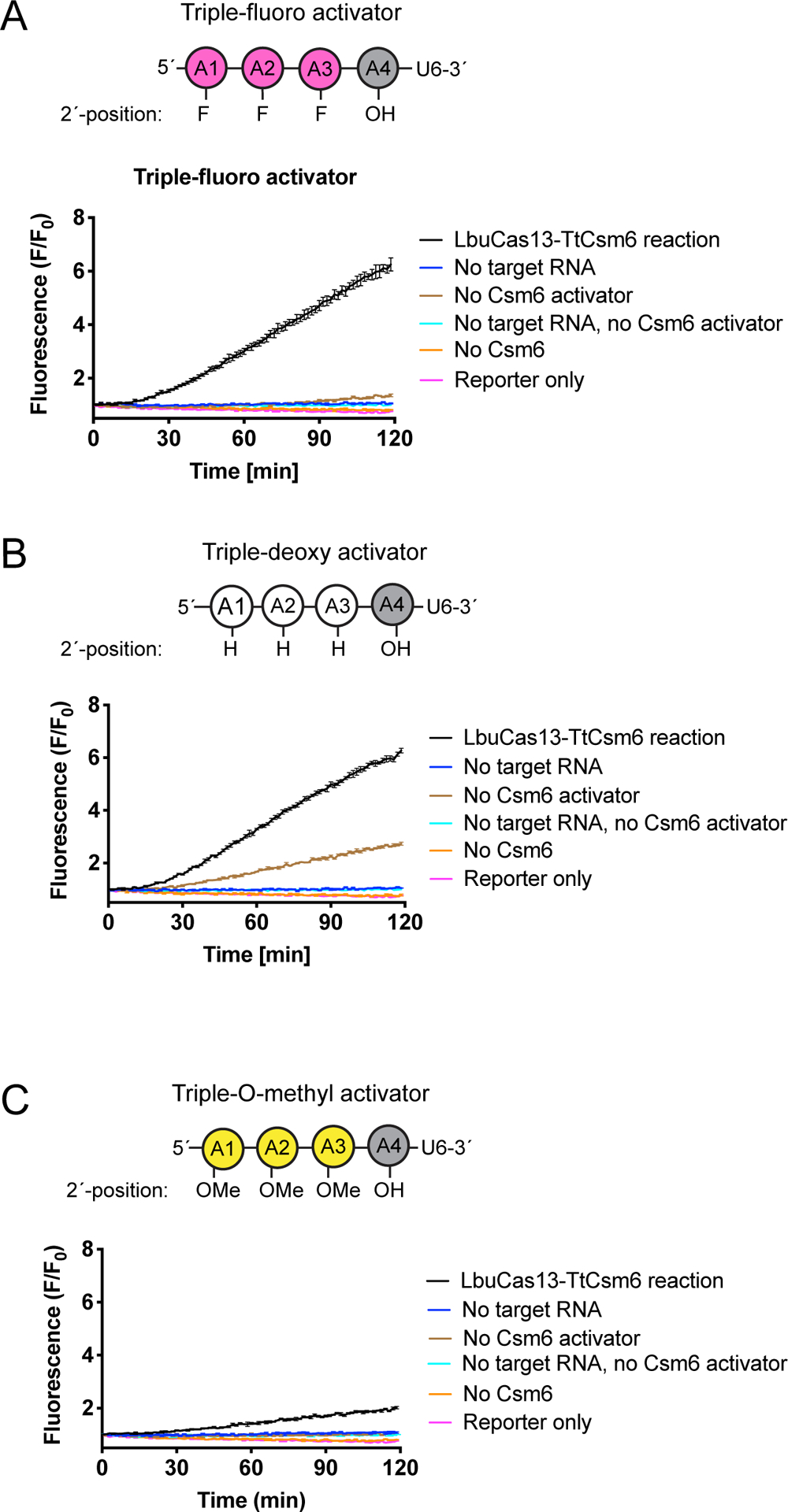
a) LbuCas13-TtCsm6 reaction with crRNA R004, 100 pM of a complementary target RNA (R010) and 4 μM of the triple-fluoro A4-U6 activator. Mean normalized fluorescence and S.E.M (n = 3) are plotted over 120 min. Controls without target RNA, TtCsm6 activator, and TtCsm6 protein are shown, as well as a reaction showing the reporter in buffer (Reporter only). Positions of the 2´-fluoro modifications in the activator are shown in the schematic above the graph.
b) As in a but with 4 μM triple-deoxy A4-U6 to activate TtCsm6. Positions of the 2´-deoxy modifications in the activator are shown above the graph.
c) As in a but with 4 μM triple-O-methyl A4-U6 to activate TtCsm6. Positions of the 2´-O-methyl (O-Me) modifications in the activator are shown in the schematic above the graph.
Extended Data Figure 4, related to Fig 2: LbuCas13-EiCsm6 detection with unmodified and single 2´-fluoro modified activators at high target RNA concentrations.
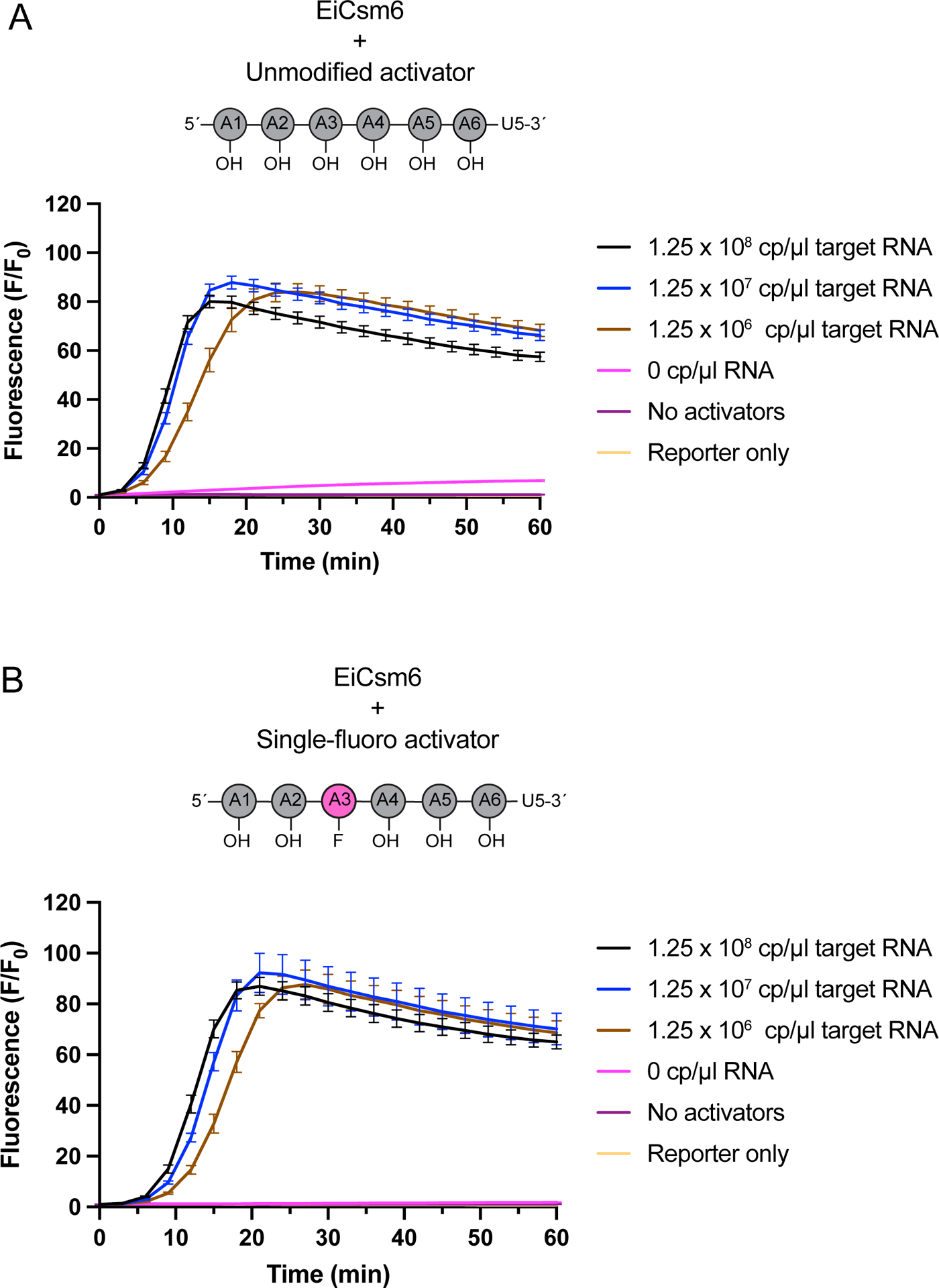
a) Detection assay using LbuCas13 complexed with crRNAs 604 and 612, and EiCsm6 with 0.5 μM of a A6-U5 activator22. An in vitro transcribed RNA corresponding to a fragment of the SARS-CoV-2 genome was added at target concentrations ranging from 2 pM (1.25 × 106 cp/μl) to 200 pM (1.25 × 108 cp/μl). Control reactions with target RNA and Csm6 activator omitted (No activators) or with the reporter in buffer (Reporter only). Mean normalized fluorescence and S.E.M (n = 3) are plotted over 60 min. The position of the modification in the EiCsm6 activator is shown in the schematic above the graph.
b) As in a but with a single-fluoro A6-U5 activator. The position of the modification in the activator is shown in the schematic above the graph.
Extended Data Figure 5, related to Fig. 3: Comparison of LbuCas13 detection assay with two and eight crRNA guides targeting SARS-CoV-2 RNA.

a) Direct detection of 10–10,000 cp/μl of Twist synthetic SARS-CoV-2 RNA control using 100 nM LbuCas13 loaded with two different crRNAs, 604 and 612 and a polyU FQ reporter. The mean slope of fluorescence increase over 2 hours and the 95% confidence interval (CI) of the mean are plotted (n = 3).
b) As in a, but with LbuCas13 complexed with eight crRNAs (604, 612, 542, 546, 564, 569, 588, 596).
c) Full 2-hour timecourse of the direct detection assay in a, with normalization to the first fluorescence measurement (F/F0). This accounts for slight variations in the initial fluorescence of each replicate. Data shown are the mean F/F0 values of three replicates and S.E.M.
d) Full 2-hour timecourse of the direct detection assay in b, with normalization to the first fluorescence measurement (F/F0). Data shown are the mean F/F0 values of three replicates and the S.E.M.
Extended Data Figure 6, related to Fig. 3: Full timecourses of LbuCas13 and LbuCas13-TtCsm6 detection data in Fig. 3B–C.
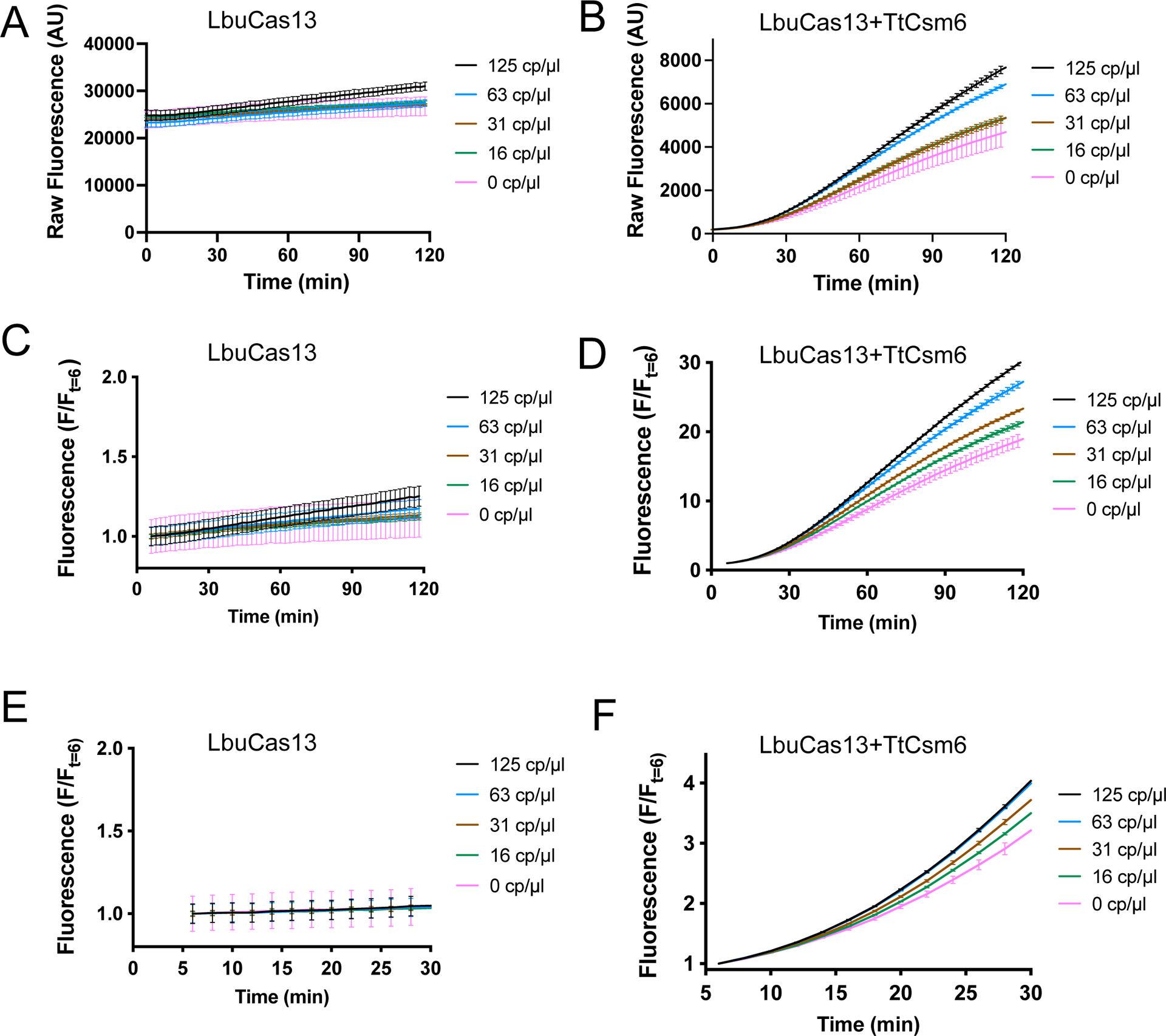
a) Raw fluorescence measurements of an LbuCas13 detection assay using an externally validated B.E.I. extracted SARS-CoV-2 RNA control as the target over 2 hours. Measurements are shown as mean fluorescence and S.E.M. (n = 3). These data were used for slope determination and comparison in Fig. 3B after 20 min and 118 min.
b) Raw fluorescence measuremements for LbuCas13-TtCsm6 detection assay using an externally validated B.E.I. extracted SARS-CoV-2 RNA control as the target over 2 hours. Measurements are shown as mean fluorescence and S.E.M. (n = 3).
c) Graph of the data in a, normalized to the fluorescence at t = 6 min, which was done to account for small well-to-well variations in the starting fluorescence and allow a short period of temperature equilibration. Measurements are shown as mean F/Ft=6 and S.E.M. (n = 3).
d) Graph of the data in b, normalized to the fluorescence at t = 6 min, which was done to account for small well-to-well variations in the starting fluorescence and allow for a short period of temperature equilibration. Measurements are shown as mean F/Ft=6 and S.E.M. (n = 3). These data were used for endpoint fluorescence comparison in Fig. 3C after 20 min and 118 min.
e) Same dataset shown in c, but zoomed to the first 30 min of the LbuCas13 detection assay.
f) Same dataset shown in d, but zoomed to the first 30 min of the LbuCas13-TtCsm6 assay.
Extended Data Figure 7, related to Fig 3D: Full time course of 20-replicate LbuCas13-TtCsm6 datasets for 125 cp/μl and 63 cp/μl target RNA concentrations shown in Fig. 3D.
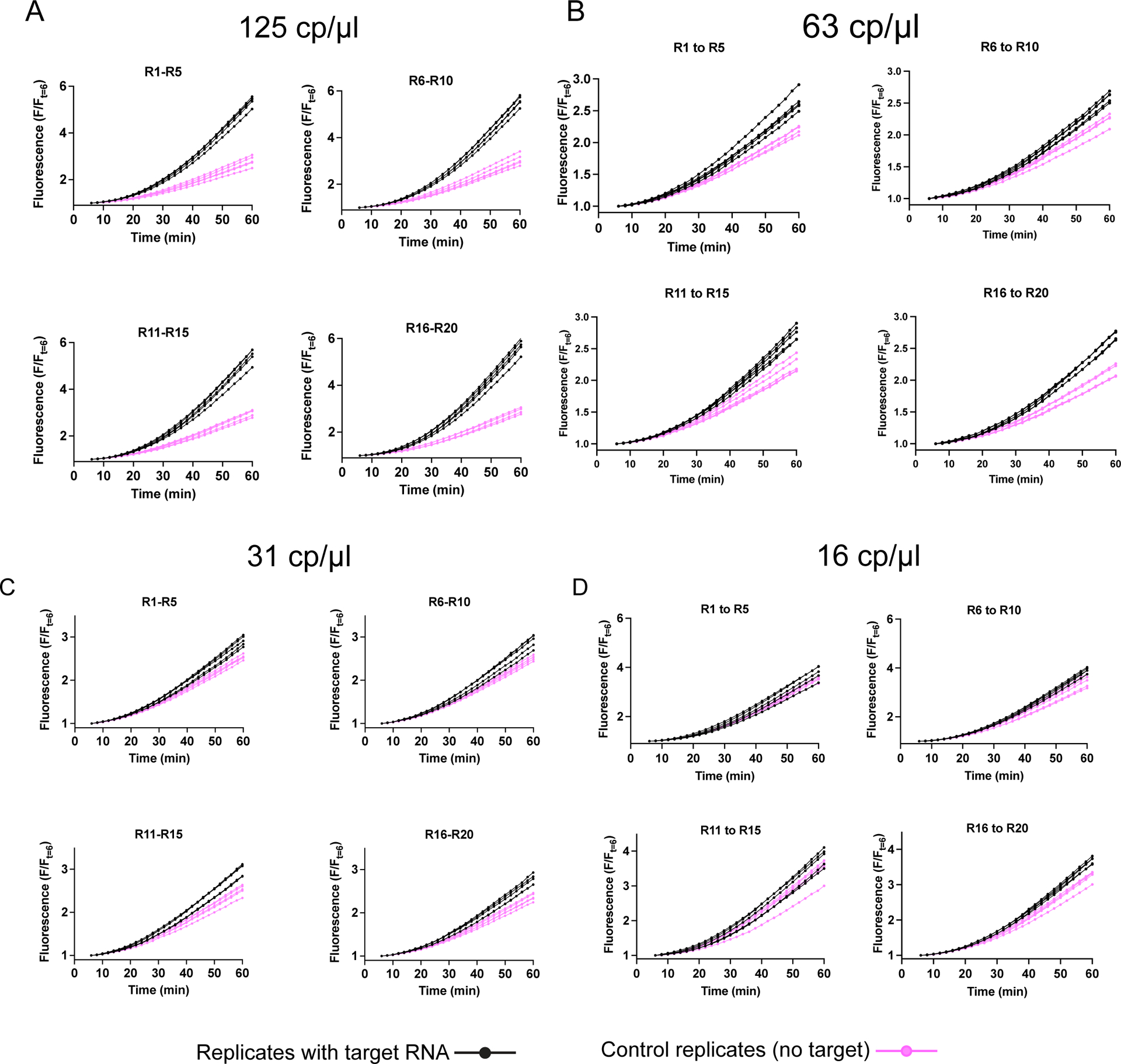
a) 1-hour time course data for 20 replicates of FIND-IT reactions containing 125 cp/μl B.E.I. extracted SARS-CoV-2 RNA. Each set of 5 sample and 5 control (no target RNA) replicates were started together (R1-R5, R6-R10, R11-R15, R16-R20). Replicates with target RNA are plotted in black and control replicates lacking target RNA (controls) are plotted in magenta. Fluorescence was normalized to the fluorescence value at 6 min (F/Ft=6) to allow for temperature equilibration to 37°C (See Extended Data Fig. 8)
b) As in a, but for sample reactions containing 63 cp/μl B.E.I. extracted SARS-CoV-2 RNA.
c) 20-replicate dataset for 31 cp/μl B.E.I. extracted SARS-CoV-2 RNA is shown over an hour. Each set of 10 replicate reactions (5 samples containing target RNA and 5 controls per graph) were started simultaneously. The normal distribution around the mean of the negative controls for each set of 10 reactions was used to determine a 95th percentile threshold for detection of the samples containing target RNA (see Methods for details). Replicates with target RNA are plotted in black and control replicates lacking target RNA (controls) replicates are plotted in magenta.
d) As in a, but for sample reactions containing 16 cp/μl B.E.I. extracted SARS-CoV-2 RNA.
Extended Data Figure 8, related to Fig. 3C-D: Effect of normalizing fluorescence relative to different timepoints in a plate reader assay.
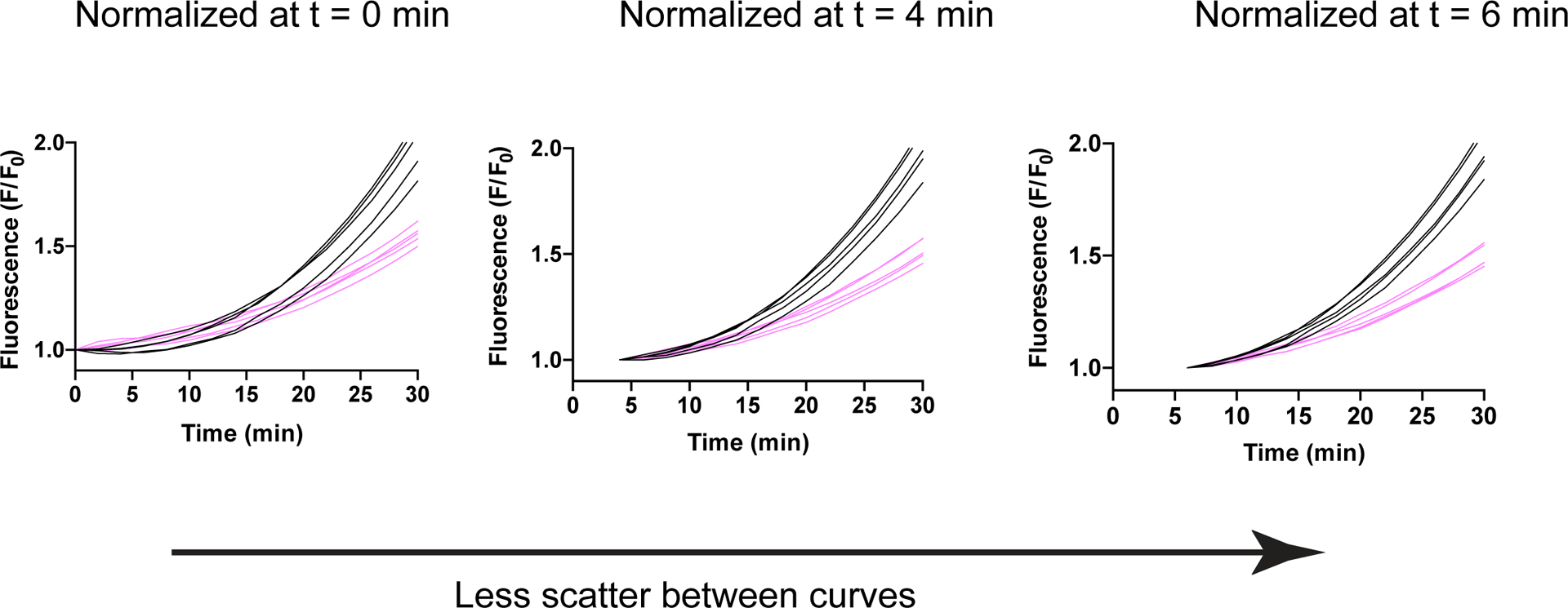
Graphs of 10 individual replicates from the LbuCas13-TtCsm6 assay in Fig. 3D using eight crRNAs targeting the SARS-CoV-2 genome, the single-fluoro TtCsm6 activator, and 125 cp/μl of BEI SARS-CoV-2 RNA as the target. Black curves indicate replicates with target RNA present, and pink curves indicate control replicates without target RNA. Comparison of the normalization at 0, 4, and 6 min (t = 0, 4, 6) shows that normalizing a few minutes after the start of the plate reader assay reduces the effect of initial signal fluctuations on the normalization.
Extended Data Figure 9, related to Fig. 4: Testing of clinical samples with FIND-IT assay in a compact fluorescence detector.
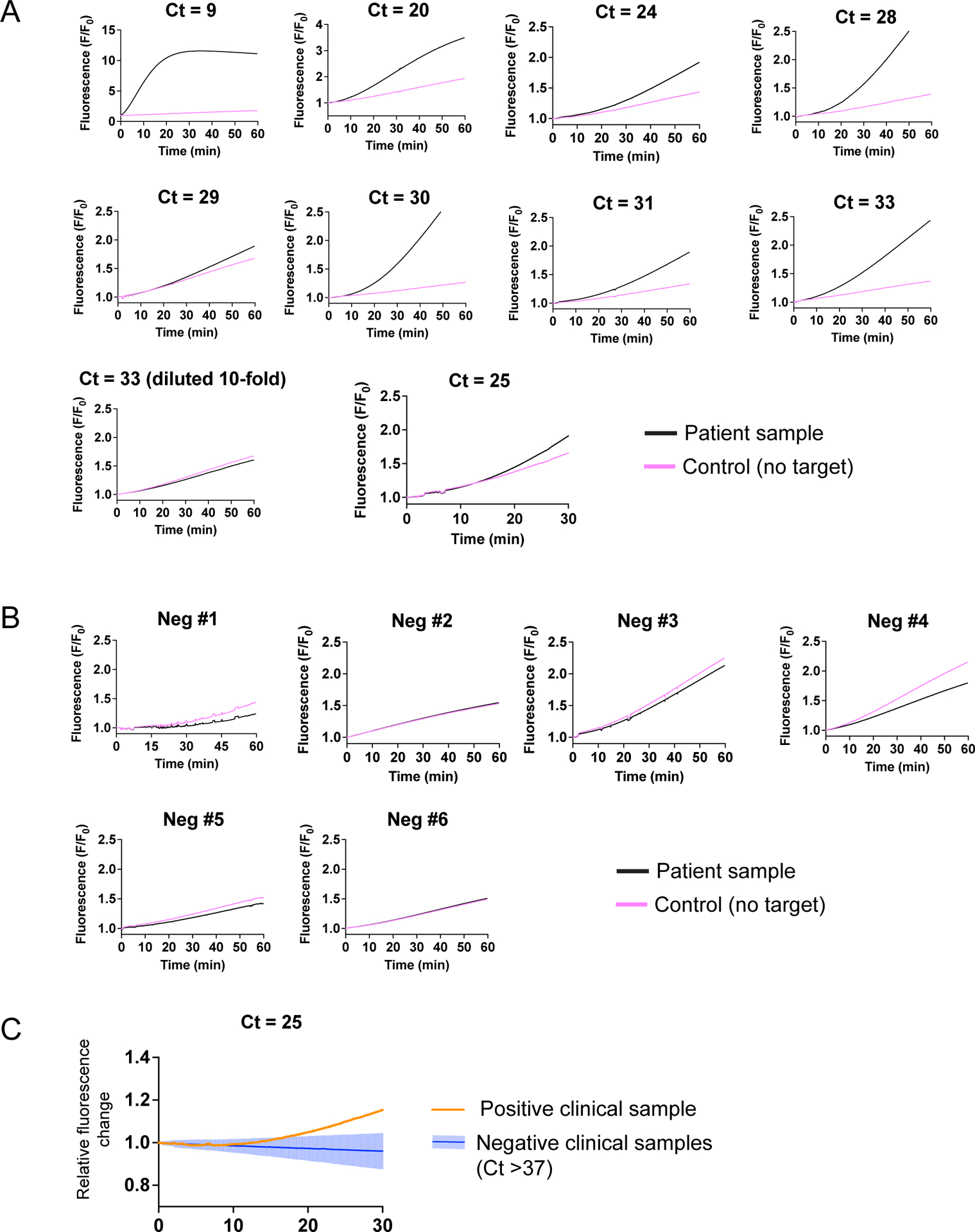
a) Testing of SARS-CoV-2-positive clinical samples with FIND-IT using a compact detector. Images of reaction wells were taken every 10 s by the detector for either 30 min (Ct 25) or 60 min (all other samples). Fluorescence (F/F0, where F0 is the initial fluorescence intensity) of a sample reaction (black lines) and a control reaction run in parallel (magenta lines) are plotted. Ct values of samples were previously determined by qRT-PCR by the IGI testing lab.
b) As in b, but for samples with Ct values >37, which are considered negative for SARS-CoV-2, based on the IGI testing laboratory’s qRT-PCR assay. These data were used to determine the mean fluorescence change of negative samples (black) relative to their no-target controls (magenta), as shown in Fig. 4 and in c.
c) The relative fluorescence change of an additional positive sample (Ct = 25) measured for 30 min instead of 60 min, is plotted alongside the relative fluorescence change of negative clinical samples (mean and two standard deviations plotted; n = 6).
Extended Data Figure 10, related to Fig. 4: Standard curve of Ct values versus target RNA concentration.
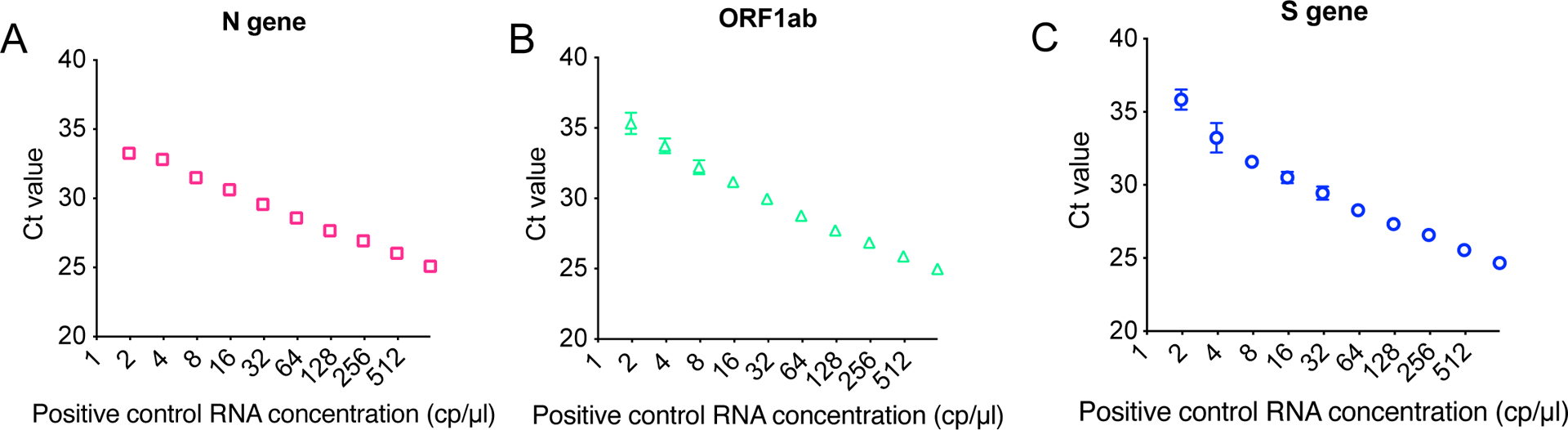
a) Ct values obtained from qRT-PCR for a dilution series of the positive control RNA provided by the Thermo TaqPath combo kit. The highest concentration of sample added to the reaction is 1000 cp/μl and other sample RNA concentrations were obtained by diluting the concentrated sample two-fold down to ~2 cp/μl. Primers for the N gene of SARS-CoV-2 were used for amplification. Mean Ct value and S.D. (n = 2) are graphed for each concentration of control RNA tested.
b) As in a but with primers for the Orf1ab gene of SARS-CoV-2.
c) As in a but with primers for the S gene of SARS-CoV-2.
Supplementary Material
Acknowledgments
This work was supported by DARPA under award N66001-20-2-4033. The views, opinions and/or findings expressed are those of the authors and should not be interpreted as representing the official views or policies of the Department of Defense or the U.S. Government. The work was also supported by the Howard Hughes Medical Institute, and the National Institutes of Health (NIH) (R01GM131073 and DP5OD021369 to P.D.H., R01GM127463 to D.F.S., and grant 5R61AI140465-03 to J.A.D., D.A.F., and M.O). A mass spectrometer was also purchased with NIH support (grant 1S10OD020062-01). This work was made possible by a generous gift from an anonymous private donor in support of the ANCeR diagnostics consortium. We also thank the David & Lucile Packard Foundation and the Shurl and Kay Curci Foundation for their generous support of this project. We thank Integrated DNA Technologies and Synthego Corporation for support with oligonucleotide modifications and synthesis, and Shanghai ChemPartner for expression and purification of the EiCsm6 protein. We thank QB3 MacroLabs for subcloning TtCsm6, A. J. Aditham for assistance purifying TtCsm6, and D. Colognori for helpful discussions. J.A.D. is an HHMI investigator. G.J.K. acknowledges support from the NHMRC (Investigator Grant, EL1, 1175568). B.W.T. is supported by a National Science Foundation (NSF) Graduate Fellowship. P.F. was supported by the NIH/NIAID (F30AI143401). M.D.d.L.D. was supported by the UC MEXUS-CONACYT Doctoral Fellowship.
Competing Interests
D.F.S. is a co-founder of Scribe Therapeutics and a scientific advisory board member of Scribe Therapeutics and Mammoth Biosciences. P.D.H. is a cofounder of Spotlight Therapeutics and serves on the board of directors and scientific advisory board, and is a scientific advisory board member to Vial Health and Serotiny. The Regents of the University of California have patents issued and/or pending for CRISPR technologies on which J.A.D., T.Y.L., P.D.H., N.P., D.F.S., D.C.J.S., S.E.K., B.W.T., E.J.C., S.J., and G.J.K. are inventors. M.O., P.F., G.R.K., D.F., S.S., and N.A.S. have also filed patent applications related to this work. J.A.D. is a cofounder of Caribou Biosciences, Editas Medicine, Scribe Therapeutics, Intellia Therapeutics and Mammoth Biosciences. J.A.D. is a scientific advisory board member of Caribou Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Mammoth Biosciences, Synthego, Algen Biotechnologies, Felix Biosciences and Inari. J.A.D. is a Director at Johnson & Johnson and has research projects sponsored by Biogen, Pfizer, AppleTree Partners and Roche.
Data and code availability statement
Data that support the findings of this study are available from the corresponding author upon request. Plasmids used in this study will be made available through Addgene. All custom code used for analysis will be made publicly accessible on Github or upon request. Sequences of oligonucleotides used in this study are available in Supplementary Table 3.
References
- 1.East-Seletsky A et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gootenberg JS et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larremore DB et al. Test sensitivity is secondary to frequency and turnaround time for COVID-19 screening. Science Advances 7, 1–11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vogels CBF et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT–qPCR primer–probe sets. Nature Microbiology 5, 1299–1305 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mina MJ, Parker R & Larremore DB Rethinking Covid-19 Test Sensitivity — A Strategy for Containment. New England Journal of Medicine 383, e120 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Knott GJ & Doudna JA CRISPR-Cas guides the future of genetic engineering. Science (New York, N.Y.) 361, 866–869 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu TY & Doudna JA Chemistry of Class 1 CRISPR-Cas effectors: Binding, editing, and regulation. Journal of Biological Chemistry 295, 14473–14487 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makarova KS et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nature Reviews Microbiology 18, 67–83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steens JA et al. SCOPE: Flexible targeting and stringent CARF activation enables type III CRISPR-Cas diagnostics. bioRxiv 2021.02.01.429135 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santiago-Frangos A et al. Intrinsic signal amplification by type-III CRISPR-Cas systems provides a sequence-specific viral diagnostic. medRxiv (2020). doi: 10.1101/2020.10.14.20212670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kazlauskiene M, Kostiuk G, Venclovas Č, Tamulaitis G & Siksnys V A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science 357, 605–609 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Niewoehner O et al. Type III CRISPR–Cas systems produce cyclic oligoadenylate second messengers. Nature 548, 543–548 (2017). [DOI] [PubMed] [Google Scholar]
- 13.East-Seletsky A, O’Connell MR, Burstein D, Knott GJ & Doudna JA RNA Targeting by Functionally Orthogonal Type VI-A CRISPR-Cas Enzymes. Molecular Cell 66, 373–383.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagamine K, Hase T & Notomi T Accelerated reaction by loop-mediated isothermal amplification using loop primers. Molecular and Cellular Probes 16, 223–229 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Piepenburg O, Williams CH, Stemple DL & Armes NA DNA Detection Using Recombination Proteins. PLoS Biology 4, e204 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joung J et al. Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. New England Journal of Medicine 383, 1492–1494 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broughton JP et al. CRISPR–Cas12-based detection of SARS-CoV-2. Nature Biotechnology 38, 870–874 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joung J et al. Point-of-care testing for COVID-19 using SHERLOCK diagnostics. medRxiv (2020). doi: 10.1101/2020.05.04.20091231 [DOI] [Google Scholar]
- 19.Arizti-Sanz J et al. Streamlined inactivation, amplification, and Cas13-based detection of SARS-CoV-2. Nature Communications 11, 5921 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang F, Abudayyeh OO, Gootenberg JS, Sciences C & Mathers L A protocol for detection of COVID-19 using CRISPR diagnostics. Bioarchive 1–8 (2020). [Google Scholar]
- 21.Fozouni P et al. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 184, 323–333.e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gootenberg JS et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Doval C et al. Activation and self-inactivation mechanisms of the cyclic oligoadenylate-dependent CRISPR ribonuclease Csm6. Nature Communications 11, 1596 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Athukoralage JS, Rouillon C, Graham S, Grüschow S & White MF Ring nucleases deactivate type III CRISPR ribonucleases by degrading cyclic oligoadenylate. Nature 562, 277–280 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia N, Jones R, Yang G, Ouerfelli O & Patel DJ CRISPR-Cas III-A Csm6 CARF Domain Is a Ring Nuclease Triggering Stepwise cA4 Cleavage with ApA>p Formation Terminating RNase Activity. Molecular Cell 75, 944–956.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Athukoralage JS, Graham S, Grüschow S, Rouillon C & White MF A Type III CRISPR Ancillary Ribonuclease Degrades Its Cyclic Oligoadenylate Activator. Journal of Molecular Biology 431, 2894–2899 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smalakyte D et al. Type III-A CRISPR-associated protein Csm6 degrades cyclic hexa-adenylate activator using both CARF and HEPN domains. Nucleic Acids Research 48, 9204–9217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu TY, Liu J-J, Aditham AJ, Nogales E & Doudna JA Target preference of Type III-A CRISPR-Cas complexes at the transcription bubble. Nature Communications 10, 3001 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niewoehner O & Jinek M Structural basis for the endoribonuclease activity of the type III-A CRISPR-associated protein Csm6. RNA 22, 318–329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Consortium I. testing.. Blueprint for a pop-up SARS-CoV-2 testing lab. Nature Biotechnology 38, 791–797 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Bullard J et al. Predicting Infectious Severe Acute Respiratory Syndrome Coronavirus 2 From Diagnostic Samples. Clinical Infectious Diseases 71, 2663–2666 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ackerman CM et al. Massively multiplexed nucleic acid detection using Cas13. Nature (2020). doi: 10.1038/s41586-020-2279-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McMahon SA et al. Structure and mechanism of a Type III CRISPR defence DNA nuclease activated by cyclic oligoadenylate. Nature Communications 11, 500 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lau RK et al. Structure and Mechanism of a Cyclic Trinucleotide-Activated Bacterial Endonuclease Mediating Bacteriophage Immunity. Molecular Cell 77, 723–733.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rostøl JT et al. The Card1 nuclease provides defence during type III CRISPR immunity. Nature 590, 624–629 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molina R et al. Structure of Csx1-cOA4 complex reveals the basis of RNA decay in Type III-B CRISPR-Cas. Nature Communications 10, 4302 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Athukoralage JS et al. The dynamic interplay of host and viral enzymes in type III CRISPR-mediated cyclic nucleotide signalling. eLife 9, 1–16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou T et al. CRISPR/Cas13a Powered Portable Electrochemiluminescence Chip for Ultrasensitive and Specific MiRNA Detection. Advanced Science 7, 1903661 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith K et al. CIDRE: an illumination-correction method for optical microscopy. Nature Methods 12, 404–406 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data that support the findings of this study are available from the corresponding author upon request. Plasmids used in this study will be made available through Addgene. All custom code used for analysis will be made publicly accessible on Github or upon request. Sequences of oligonucleotides used in this study are available in Supplementary Table 3.