

ABSTRACT
Syndromic birth defects are rare diseases that can present with seemingly pleiotropic comorbidities. Prime examples are rare congenital heart and cardiovascular anomalies that can be accompanied by forelimb defects, kidney disorders and more. Whether such multi-organ defects share a developmental link remains a key question with relevance to the diagnosis, therapeutic intervention and long-term care of affected patients. The heart, endothelial and blood lineages develop together from the lateral plate mesoderm (LPM), which also harbors the progenitor cells for limb connective tissue, kidneys, mesothelia and smooth muscle. This developmental plasticity of the LPM, which founds on multi-lineage progenitor cells and shared transcription factor expression across different descendant lineages, has the potential to explain the seemingly disparate syndromic defects in rare congenital diseases. Combining patient genome-sequencing data with model organism studies has already provided a wealth of insights into complex LPM-associated birth defects, such as heart-hand syndromes. Here, we summarize developmental and known disease-causing mechanisms in early LPM patterning, address how defects in these processes drive multi-organ comorbidities, and outline how several cardiovascular and hematopoietic birth defects with complex comorbidities may be LPM-associated diseases. We also discuss strategies to integrate patient sequencing, data-aggregating resources and model organism studies to mechanistically decode congenital defects, including potentially LPM-associated orphan diseases. Eventually, linking complex congenital phenotypes to a common LPM origin provides a framework to discover developmental mechanisms and to anticipate comorbidities in congenital diseases affecting the cardiovascular system and beyond.
Keywords: Congenital heart disease, Heart development, Mesoderm, Lateral plate mesoderm, Rare diseases, Embryo patterning, Animal models, Genotype-phenotype correlation, Cell fate, Cardiopharyngeal field, Hematopoiesis
Summary: We conceptualize how phenotypes of syndromic congenital diseases affecting the cardiovascular system link to disrupted mechanisms in shared progenitor cells and developmental programs in the lateral plate mesoderm.
Introduction
Comparative observations in different model organisms guide our understanding of how the human body develops, functions and manifests congenital anomalies. Numerous rare congenital diseases present with complex syndromic phenotypes that affect seemingly disconnected organs or cell types. Studying such disease phenotypes in model organisms can (1) reveal new, clinically meaningful insights about a potentially shared embryonic origin or molecular mechanism, and (2) inform about possible additional phenotypes in affected patients. However, decoding the underlying developmental and molecular causes of complex syndromic birth defects remains challenging when we lack a unifying concept to connect the observed phenotypes.
Heart formation provides a powerful example for the close developmental and genetic relationship of embryonic lineages with seemingly distant locations in the final body plan (Bulatovic et al., 2016; Diogo et al., 2015; Meilhac and Buckingham, 2018; Stainier et al., 1993). The heart develops from the lateral plate mesoderm (LPM) that initially emerges at the edge of the forming vertebrate embryo (Meilhac and Buckingham, 2018; Prummel et al., 2020; Saint-Jeannet et al., 1992; Stainier et al., 1995) (Fig. 1). Curiously, the emerging heart field is part of a wider progenitor cell population called the cardiopharyngeal field (CPF) that includes progenitor cells forming various muscle groups of the neck and the head (Figs 1 and 2) (Chan et al., 2016; Diogo et al., 2015; Gopalakrishnan et al., 2015; Lescroart et al., 2015, 2021; Stolfi et al., 2010). In addition to the heart, the LPM also gives rise to several smooth muscle lineages, ventral dermis, mesothelia and connective tissue of the limbs, including long bones (Lane and Smith, 1999; Selleck and Stern, 1991; Warga and Nüsslein-Volhard, 1999) (Fig. 1). The kidneys, traditionally described as intermediate mesoderm in birds and mammals, share clonal and gene expression relationships with other LPM-derived progenitors across vertebrates (Fig. 1) (Davidson et al., 2019; Mansour et al., 2022; Mattonet et al., 2022; Takasato and Little, 2015; Takasato et al., 2014). This complex developmental and genetic architecture renders the LPM sensitive to defects in its patterning and cell fate specification, with broad consequences for several of its descendant organs and tissues. Case in point, heart-hand syndromes manifest as cardiovascular and forelimb anomalies (Camarata et al., 2010; Yin et al., 2018, 2020). Although these phenotypes present in seemingly distant organs, the progenitors for both the heart and forelimb skeleton share a close developmental connection as they emerge from adjacent cell territories within the LPM, which also have a considerable number of co-expressed genes (Figs 1 and 2) (Mori and Bruneau, 2004; Silverman and Hurst, 1968; Tamari and Goodman, 1974; Yin et al., 2020).
Fig. 1.
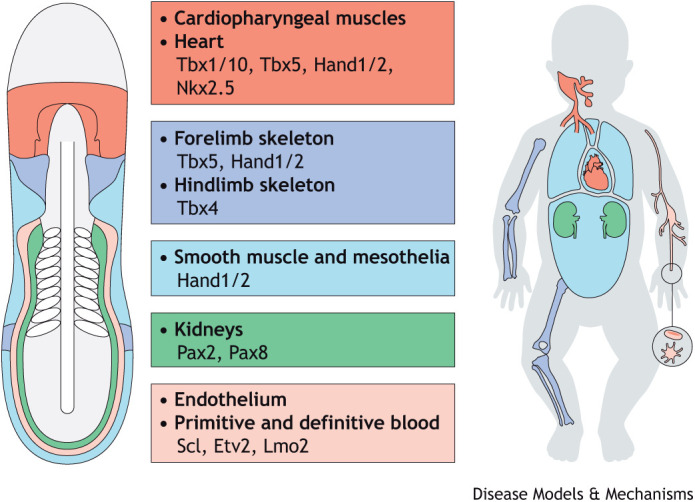
The lateral plate mesoderm (LPM) develops into a dispersed set of organs that share a common embryonic origin. Major LPM progenitor cell populations and their main transcription factors (middle), and their corresponding arrangement in a schematized vertebrate embryo during segmentation stages (left). The human infant (right) depicts the corresponding final organs and cell types with their distribution in the body.
Fig. 2.

Heart and limb develop from neighboring LPM populations with shared transcription factors. The heart and the forelimb progenitors emerge within the anterior LPM in close proximity to each other, as depicted in a schematic representing the vertebrate embryo (left). On the right, we depict heart and limb/fin development in the mammalian and zebrafish embryo; the developmental process in zebrafish is similar to that in mammals. The heart forms by medial progenitor migration and leftward jog, resulting in the primitive heart tube that then undergoes complex remodeling to form a multi-chambered heart (top). The limb buds for both forelimbs and hindlimbs emerge from dedicated territories within the LPM via a conserved cascade of signaling events; interaction with the apical ectodermal ridge through Fgf ligand secretion supports limb bud outgrowth followed by LPM-based formation of connective tissue and skeletal structures (bottom).
Overlaying complex syndromic disease phenotypes with developmental information provides a conceptual framework to discover underlying mechanisms and possible comorbidities relevant to long-term patient health. Here, we discuss the LPM as a developmental concept to connect seemingly disparate phenotypes in human birth defects affecting the heart and beyond with known and unknown causative genes. We further present as-of-yet orphan syndromes missing causative gene associations that manifest as potential LPM diseases due to their affecting two or more LPM-derived tissues. We lastly outline how clinical, computational and developmental biology laboratories can productively collaborate towards revealing the causes of complex congenital syndromes affecting the heart and comorbidities.
The LPM as context for cardiovascular system development
LPM formation and patterning is regulated by deeply conserved principles across chordates (Box 1) (Prummel et al., 2020). Its emergence is closely tied to dorsal-ventral patterning of the vertebrate embryo, which is driven by secreted Bmp and Nodal signaling molecules that control downstream Smad transcription factors (Graff et al., 1996; Liu et al., 1996; Müller et al., 1999; Savage et al., 1996; Schier and Talbot, 2005; Sekelsky et al., 1995; Shi et al., 1997; Zhang et al., 1996). While Bmp regulates the dorsal-ventral arrangement of the body axis, Nodal activity additionally supports ectoderm versus mesendoderm patterning (Conlon et al., 1991; Davidson et al., 2015; Gritsman et al., 1999; Hayes et al., 2021; Kattman et al., 2011; Martyn et al., 2019; Schier and Talbot, 2005; Zhou et al., 1993). During gastrulation, the LPM emerges as ventral and lateral mesoderm territory (Garcia-Martinez and Schoenwolf, 1993; Gurdon, 1995; Lane and Smith, 1999; Mead et al., 1996; Selleck and Stern, 1991). In zebrafish, Smad proteins cooperate with Eomesa, Foxh1 and Mixl1 to induce the LPM, yet whether this combination is necessary and sufficient across vertebrates remains to be determined (Bjornson et al., 2005; Prummel et al., 2019; Slagle et al., 2011). Nonetheless, the basic concept of ventral LPM-inducing activity seems to be conserved across chordates and sets up the emerging LPM as a dedicated progenitor field for its various downstream cell fates (Figs 1 and 2, Box 1) (Prummel et al., 2019, 2020).
Box 1. Evolutionary context of lateral plate mesoderm (LPM) and cardiovascular fate formation.
Despite a wealth of data on downstream fates, little is known about how the LPM partitions into its individual territories and how these relate to each other. Our current insights suggest that LPM-derived organs such as the heart evolved by integrating LPM-focused regulatory programs and cell fate mechanisms that became refined from pre-existing larger cell fields with as-of-yet unknown purpose (Kaplan et al., 2015; Monahan-Earley et al., 2013; Onimaru et al., 2011; Prummel et al., 2020; Tanaka, 2016) (Fig. 1). The seemingly disconnected functions and distributions of downstream fates emerging from the LPM have rendered the study of their joint origins challenging. Comparative studies involving distantly related chordate species now highlight the LPM as a fertile ground for evolutionary innovations that have contributed to critical aspects of vertebrates, including, but not limited to, head and neck muscles for feeding, appendages for locomotion and chambered hearts for increased metabolic needs (Ericsson et al., 2013; Onimaru et al., 2011; Prummel et al., 2019; Stephenson et al., 2017; Yong et al., 2021). In particular, the evolutionary context of heart development has advanced our understanding of cardiovascular development and connections to disease.
The forelimb and hindlimb skeleton are the most diverged, non-obvious LPM derivatives. Decades of phenotypic, genetic and molecular work across model systems and in human congenital disease have uncovered that both heart and forelimb progenitors express Tbx5, Hand2 and other transcription factors (Rallis et al., 2003; Steimle and Moskowitz, 2017). Further, both heart and limb progenitor fields form adjacent to each other in the LPM (Fig. 1 and 2). Zebrafish and mouse data propose that restriction of the cardiac mesoderm by retinoic acid is required for the initiation of the limb field (Tanaka, 2016; Waxman et al., 2008; Zhao et al., 2009). Zebrafish and mouse raldh2 (aldh1a2)/Raldh2 (Aldh1a2) mutants lack anterior paired appendages, and retinoic acid inhibition in zebrafish induces posterior expansion of the cardiac field correlating with failed fin bud initiation (Gibert et al., 2006; Grandel et al., 2002; Waxman et al., 2008). These phenotypes suggest that the regionalization of the LPM into cardiac mesoderm and more posterior derivatives contributed to forelimb field evolution. Analysis of Nkx2.5 expression dynamics documented that this regionalization occurs during lamprey development, but not in amphioxus, suggesting that this process evolved in the vertebrate family branch (Onimaru et al., 2011). The Tbx5 expression domain is defined by nested Hox expression, specifically at the anterior borders of the Hox5- and Hox6-expressing LPM (Minguillon et al., 2012). Although many questions regarding the evolutionary and developmental relationship of heart and forelimb mesoderm remain unresolved, experimental evidence suggests that the evolution of the forelimb field followed an anterior restriction of the cardiac mesoderm. However, the initial purpose of this restriction remains debated; forelimb origins have been suggested to have started as the seventh pharyngeal arch or as part of broader models of fin formation along the body axis, yet both possibilities await further data (Diogo, 2020; Sleight and Gillis, 2020; Tulenko et al., 2013).
The finding that second heart field progenitors share a clonal origin with craniofacial muscles has revealed another unanticipated developmental and evolutionary connection – the cardiopharyngeal field (CPF). The combined CPF is defined by expression of Tbx1, Nkx2.5 and others. Subsequent work across models, including the chordate Ciona, has revealed deeply conserved gene regulatory mechanisms within this intertwined progenitor field (Diogo et al., 2015; Stolfi et al., 2010; Wang et al., 2013) (Fig. 2). However, as with the heart and limb connection, the original purpose of an ancestral CPF remains a mystery. The cardiopharyngeal progenitors forming heart and head muscles are hypothesized to employ separately evolved, but functionally convergent, striated muscle programs (Diogo et al., 2015; Steinmetz et al., 2017). Over the course of chordate head evolution, cardiopharyngeal progenitor cells are thought to have acquired the ability to self-renew prior to differentiating into either heart or branchiomeric (head/neck) muscle groups and to respond to appropriate signals to specialize in different locations (Diogo et al., 2015; Kaplan et al., 2015; Tolkin and Christiaen, 2012). Evolutionary and comparative studies pointed out that the emergence of heterogeneous branchiomeric muscle groups coincides with the appearance of chambered hearts in stem vertebrates, suggesting a shared developmental and evolutionary history (Diogo et al., 2015; Moorman and Christoffels, 2003; Simões-Costa et al., 2005). In tunicates, trunk ventral cells are multipotent cardiopharyngeal progenitors that contribute to the heart and pharyngeal muscle groups (Kaplan et al., 2015; Stolfi et al., 2010; Wang et al., 2017b).
These LPM-associated evolutionary puzzles highlight the power of comparative, cross-species studies to start defining the cellular and molecular framework that govern the context of heart development. Combining developmental and evolutionary developmental biology (evo-devo) as comparative approaches with genetic insights from human disease has strong potential to advance our knowledge in all these disciplines.
A hallmark of LPM architecture is the shared expression of transcription factor genes across seemingly disparate lineage progenitors; whether this shared expression reflects a clonal origin of co-expressing progenitors or co-opted gene regulation for similar regulatory purposes remains a major open question. In the emerging heart field, Tbx5 synergizes with Nkx2.5 (Nkx2-5), Gata4, Hand1/2 and other cardiac transcription factors to regulate heart tube precursors that develop from the LPM-embedded first and second heart fields (Greulich et al., 2011; Hiroi et al., 2001; Pi-Roig et al., 2014; Steimle and Moskowitz, 2017). The first heart field contains early-differentiating progenitors that form the primitive heart tube; descendant cells of the second heart field differentiate subsequently at both poles of the emerging heart tube (Dyer and Kirby, 2009; Laura et al., 2020; Rochais et al., 2009) (Figs 1 and 2). In the LPM immediately posterior to the emerging heart field, forelimb/pectoral fin bud initiation requires Tbx5 expression in the fin/limb-specifying field to induce Fgf10, which then activates Fgf8 in the overlying apical ectodermal ridge (Cunningham and Duester, 2015; Garrity et al., 2002; Minguillon et al., 2009, 2012; Moreau et al., 2019; Parrie et al., 2013; Rodriguez-Esteban et al., 1999; Takeuchi et al., 1999) (Fig. 2). Hand2 establishes the anterior-posterior forelimb axis through activating Shh and antagonizing Gli3 (Charité et al., 2000; Galli et al., 2010; Osterwalder et al., 2014). In the hindlimb-specifying field, Pitx1 activates Tbx4, which in turn induces Fgf10, achieving an analogous feedback loop with Fgf8 in the apical ectoderm ridge. Although forelimbs and hindlimbs display unique morphologies, Tbx4 can function in place of Tbx5 when expressed in the forelimb-specifying region (Don et al., 2016; Minguillon et al., 2005, 2012; Rodriguez-Esteban et al., 1999; Takeuchi et al., 1999) (Fig. 2). Despite their physical proximity and the fact that both the heart and the forelimb fields express Tbx5 and Hand2, it remains unclear whether these two lineages share a common progenitor or whether they arise from disparate progenitor fields (Ahn et al., 2002; Charité et al., 2000; Fernandez-Teran et al., 2000; Garrity et al., 2002; Rodriguez-Esteban et al., 1999; Steimle and Moskowitz, 2017; Yelon et al., 2000).
Expanding the developmental context of heart formation within the LPM, emerging heart progenitors are part of the CPF that expresses Tbx1 and Nkx2.5/7 and contributes to craniofacial muscles beyond the heart (Comai et al., 2019; Diogo et al., 2015; Lescroart et al., 2010; Meilhac et al., 2004; Nomaru et al., 2021; Xu et al., 2004; Zhou et al., 2008). Whether the entire CPF is one lineage field or a composite of the LPM and other anterior mesodermal lineages remains to be clarified (Figs 1 and 2). Nonetheless, the activities of Mesp1, Nkx2.5 and Tbx1/10, as well as their related gene-regulatory networks, indicate a homology between tunicate trunk ventral cells and the vertebrate CPF (Stolfi et al., 2010; Tolkin and Christiaen, 2012; Wang et al., 2013) (see also Box 1).
During development, other LPM lineages intersect with cardiac precursors in origin, gene expression or both. The endocardium emerges from anterior Scl/Tal1- and Etv2-expressing progenitors that have a distinct morphology and migration path compared to that of other cranial endothelial cells (Bussmann et al., 2007; DeRuiter et al., 1992; Harris and Black, 2010; Ivanovitch et al., 2017; Misfeldt et al., 2009; Proulx et al., 2010) (Fig. 1). In the trunk, Scl/Tal1-expressing progenitors develop into trunk endothelium and first erythrocyte progenitors, which form the initial circulatory system components (De La Garza et al., 2019; Garcia-Martinez and Schoenwolf, 1992; Mead et al., 1996; Pardanaud et al., 1996; Stainier et al., 1996) (Figs 1 and 3). In parallel, Pax2- and Pax8-positive LPM progenitors form the kidney, as initially discovered by the contribution of Pax2/8 perturbation to kidney disease (Bouchard et al., 2002; Eccles et al., 2004; Keller et al., 1994; McMahon, 2016; Poleev et al., 1992; Torres et al., 1995) (Fig. 1). Recent work in zebrafish has documented that pax2a-expressing LPM progenitors contribute to endothelial and erythroid fates besides forming kidney lineages (Mattonet et al., 2022). The kidney precursors initially reside laterally to endothelial progenitors at least in the teleost LPM, contrary to in mammals, in which they reside between somites and endothelial precursors as ‘intermediate mesoderm’ (McMahon, 2016; Mullins et al., 1996; Perens et al., 2016; Takasato and Little, 2015; Terashima et al., 2014). These data suggest that kidney progenitors can be regarded as an LPM lineage. Lastly, the lateralmost LPM forms the Hand2-expressing mesothelium progenitors that surround internal organs and the body cavities, as well as different smooth muscle types (Ariza et al., 2016; Gays et al., 2017; Prummel et al., 2022; Shin et al., 2009). Work in chicken has documented interactions and stepwise separation between smooth muscles, blood and endothelial progenitors as driven by Hand2, Wnt, Bmp and Notch (Shin et al., 2009). Further underlining non-autonomous cross-talk between the stripes, loss of hand2 in zebrafish perturbs the balance between endothelial progenitors and kidney formation (Perens et al., 2016, 2021). Subsequent blood flow has a major impact on tissue remodeling and even the emergence of key cell fates, as, after the onset of circulation, mechanical forces influence the budding of Runx1-expressing definitive hematopoietic stem cell progenitors from the ventral wall of the dorsal aorta (Boisset et al., 2010; Downing et al., 1993; Kissa and Herbomel, 2010; Lancino et al., 2018; Müller et al., 1994; North et al., 2009; Traver et al., 2003).
Fig. 3.
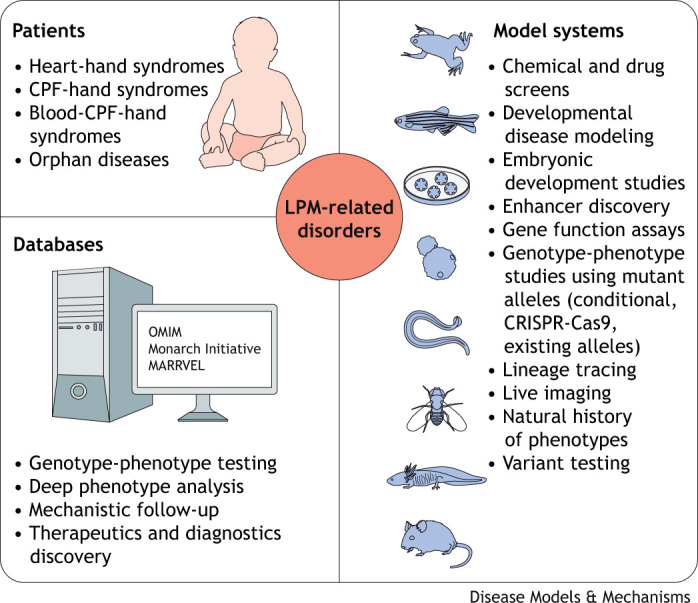
Discovery of disease-causing genes and mechanisms. Curated public databases assemble a wealth of patient-based sequencing data, genetic and phenotype information, and more. These data enable analyses of the intersection of diseases with related phenotypes and developmental similarities, such as LPM-centered anomalies. The developmental function of gene loss or gain of function, polymorphisms and other genetic changes are then testable in model systems, such as animal models that carry orthologs of human disease-associated human genes or patient-derived in vitro models. CPF, cardiopharyngeal field; LPM, lateral plate mesoderm; MARRVEL, Model organism Aggregated Resources for Rare Variant ExpLoration; OMIM, Online Mendelian Inheritance in Man.
Taken together, the LPM harbors progenitor populations for multiple organs, providing a model to link perturbations in shared precursors to causation of congenital defects affecting two or more LPM-derived organs (Gays et al., 2017; Nishimoto and Logan, 2016; Prummel et al., 2019, 2020, 2022; Tanaka, 2016). Yet, the LPM is not of single clonal origin, and major organ precursors segregate as early as during gastrulation. Consequently, alternative mechanisms contributing to comorbidities could involve perturbed organ progenitor positioning, signaling interactions and shared gene expression across LPM domains. For this Review, we consider LPM-affecting diseases as those that affect two or more organs or cell types of LPM origin and/or that share LPM-centered gene expression profiles. The developmental and evolutionary peculiarities of the LPM underline how mutations in genes affecting common progenitors or shared regulatory mechanisms could trigger syndromic defects with complex comorbidities in seemingly unrelated organ systems.
Syndromes with joint heart and limb anomalies
Joint heart and upper limb anomalies occur in several human congenital syndromes (Fig. 2). Affected patients share common clinical presentations that vary in their phenotypic expressivity, described under the umbrella term heart-hand or upper limb-cardiovascular syndromes. Although the genetic bases for several of these diseases have been uncovered (Table 1, Box 2), many remain without an assigned genetic cause (Table 2). These syndromes often present with additional respiratory, endocrine, metabolic, hematopoietic or neurologic symptoms for which clinical urgency can dominate over the heart and hand phenotypes, including symptoms that arise as complications from the primary cardiac defects (Metra et al., 2011; Tamari and Goodman, 1974) (Tables 1 and 2).
Table 1.
Simplified OMIM table of diseases with heart, forelimb and additional LPM-linked anomalies and with known genetic basis, including causative gene, pattern of inheritance and the top five phenotypic categories across syndromic diseases

Box 2. OMIM for diseases mentioned throughout the text resulting in a comparison table.
The table within the OMIM portal summarizes the causative genes and molecular bases of the syndrome as well as the inheritance pattern, if known, and several classes of phenotypes. This table was generated with the search query detailed in the supplementary information.
Table 2.
Simplified OMIM table of ‘orphan’ diseases with putative LPM association for which no causative genetic basis is known, including pattern of inheritance and the top five phenotypic categories across syndromic diseases

Holt-Oram syndrome is the prototypic heart-hand syndrome. It manifests as autosomal-dominant upper (anterior) limb and shoulder girdle abnormalities with a spectrum of congenital heart defects [Online Mendelian Inheritance in Man (OMIM) #142900] (Hurst et al., 1991; Temtamy, 1985). The most common presentation of Holt-Oram syndrome is triphalangeal thumbs and cardiac atrial septal defects, although the severities of limb and heart defects vary widely. In 1997, two separate groups confirmed that mutations in TBX5 were causative for Holt-Oram syndrome, resulting in predicted instances of haploinsufficiency (Basson et al., 1997; Yi et al., 1997). Ever since linking TBX5 mutations to the syndrome, a growing number of variant TBX5 alleles have been uncovered in affected patients. Loss-of-function mutations or TBX5-spanning large deletions cause the most severe phenotypes (Basson et al., 1997; Gruenauer-Kloevekorn and Froster, 2003) (Borozdin et al., 2006). Missense variants may cause distinct phenotypes depending on the position of the mutation (Yang et al., 2000). The human syndromic phenotypes are recapitulated in recessive Tbx5 loss-of-function mouse models (Bruneau et al., 2001; Rallis et al., 2003), and zebrafish tbx5a mutants and morphants have co-occurring heart and pectoral fin anomalies (Fig. 2, Table 3) (Ahn et al., 2002; Garrity et al., 2002). Select putative gain-of-function TBX5 alleles cause atypical Holt-Oram syndrome with mild skeletal defects and paroxysmal atrial fibrillation (Postma et al., 2008). A 48 kb duplication encompassing TBX5 exons 2 through 9 is associated with mild limb defects and non-septal cardiac defects (Patel et al., 2012).
Table 3.
Causative genes and existing animal models of syndromic diseases with putative LPM association

Loss of the Tbx5 paralog Tbx4 in mice (Naiche and Papaioannou, 2003; Rodriguez-Esteban et al., 1999), and tbx4 in zebrafish (Don et al., 2016), results in hindlimb or pelvic fin absence (Fig. 2, Table 3), although mutant mice do not survive embryonic stages due to fatal lung failure (Arora et al., 2012) (Table 3). This combination of developmental phenotypes links to Tbx4 expression in the hindlimb bud, as well as in the future lung mesothelium that is likely to be of LPM origin. Curiously, dominant TBX4 perturbations in humans largely cause cardiopulmonary defects, including pulmonary arterial hypertension and patent ductus arteriosus, in which the pulmonary circuit fails to remodel into a functional unit before birth (D'Alto and Mahadevan, 2012; Haarman et al., 2019; Haarman et al., 2020; Karolak et al., 2023; Welch and Chung, 2020; Yarboro et al., 2022). Patients with heterozygous TBX4 variants have mild limb anomalies affecting the patella or joints (Haarman et al., 2019, 2020; Karolak et al., 2023). Notably, the human phenotypes affecting hindlimbs are distinct from those arising in mouse loss-of-function models, which seem to stem from possible species-specific partial compensation of Tbx4 by Pitx1 and from Isl1/Ldb function in murine hindlimb development (Duboc et al., 2021; Itou et al., 2012; Narkis et al., 2012) (Fig. 2).
Mutations in HAND1 and HAND2 have been associated with cardiac anomalies such as tetralogy of Fallot, hypoplastic left ventricle and others (Lu et al., 2016; Reamon-Buettner et al., 2008, 2009). These basic helix-loop-helix (bHLH) transcription factors have widespread expression in the early LPM and in the subsequent cardiac, pharyngeal arch, limb and mesothelial progenitors (Charité et al., 2000; Gibert et al., 2006; Prummel et al., 2022). Hand1 and Hand2 have unique as well as redundant functions in cardiac and limb patterning in mice, controlling epithelium formation, extracellular matrix composition, cell migration and the expression of downstream transcription factor genes (Barnes and Firulli, 2009; Barnes et al., 2010; Conway et al., 2010; Firulli et al., 1998, 2010). Curiously, zebrafish only retained hand2 in their genome, and hand2 mutants develop cardiac and branchial arch defects, hypoplastic pectoral fins and reduced mesothelium that results in herniation (Garavito-Aguilar et al., 2010; Hu et al., 2011; Perens et al., 2016; Prummel et al., 2022; Yelon and Stainier, 2005; Yelon et al., 2000). Compared to genes encoding other cardiac transcription factors, HAND1/2 seem to be less frequently mutated in congenital disease, possibly due to their partial redundancy and potential to compensate for one another upon perturbation (Barnes et al., 2011; Charité et al., 2000; McFadden et al., 2005). Notably, partial trisomy of distal chromosome 4q (4q+; no OMIM reference), which contains the HAND2 locus, is associated with cardiac, digit, kidney and craniofacial defects, indicating broader LPM perturbation during development (Battaglia et al., 2005; Lundin et al., 2002; Lurie, 2005). Elegant studies have linked mouse Hand2 dosage to developmental phenotypes that recapitulate major aspects of human 4q+ syndrome (Table 3) (Tamura et al., 2013). Together, these shared heart and limb transcription factors provide clear examples for LPM-based comorbidities.
Congenital craniofacial anomalies with cardiac comorbidities
Heart-hand syndromes feature obvious LPM-linked comorbidities, yet additional heart-affecting syndromes with complex comorbidities seem also to be influenced by shared LPM progenitors and/or shared genetic programs (Figs 1 and 2, Tables 1 and 2, Box 2). DiGeorge syndrome (OMIM #188400) is caused by autosomal-dominant 22q11.2 deletions (Wilson et al., 1993). Varying widely between patients, the most consistent symptoms of DiGeorge syndrome are hypocalcemia from parathyroid hypoplasia, thymus hypoplasia and T-cell deficiencies, submucosal cleft palate and cardiac outflow tract defects. Patients with 22q11.2 deletion can also present with variable craniofacial defects, encompassing conditions known as Shprintzen (velocardiofacial) syndrome or Takao syndrome, associated with dysphagia and a broad range of heart defects (Karpinski et al., 2022; Motahari et al., 2019; Shprintzen, 1994; Yagi et al., 2003; Yamagishi et al., 2016). Nonetheless, 22q11.2 harbors several genes and as-of-yet incompletely charted regulatory elements (Fernández et al., 2005; Saitta et al., 2004), and no single gene has been linked to the full phenotype spectrum. Consequently, DiGeorge syndrome could comprise multiple syndromes caused by individual genes in the 22q11.2 locus (Burn, 1999; Wilson et al., 1993).
The major phenotypes of DiGeorge syndrome can potentially be explained by TBX1 haploinsufficiency, with other symptoms attributed to the cumulative effects from loss of other 22q11.2 genes (Jerome and Papaioannou, 2001; Lindsay et al., 2001; Paylor et al., 2006; Yagi et al., 2003). Notably, patients with point mutations in TBX1 display characteristic phenotypes of 22q11.2 deletion patients (Yagi et al., 2003), although they do not develop the full spectrum of DiGeorge syndrome. Mice hemizygous for the highly conserved 22q11.2 region display mild cardiac outflow tract defects, as phenocopied in heterozygous Tbx1 mutant mice (Lindsay et al., 1999, 2001). Homozygous Tbx1 mutant mice (Jerome and Papaioannou, 2001; Merscher et al., 2001) and tbx1 mutant zebrafish (van gogh, vgo) (Piotrowski et al., 2003) recapitulate the major structural defects observed in patients with severe DiGeorge syndrome, including heart, thymus and craniofacial defects (Table 3), but do not recapitulate all aspects of human DiGeorge syndrome. Notably, the expression pattern of Tbx1 is not single lineage or LPM specific, but rather a mix of different anterior/rostral cell types, including the LPM, neural crest and additional head mesoderm, that awaits concise definition. Consequently, untangling individual contributions of Tbx1 function in individual cell types, as well as in cell-cell crosstalk, in head and heart structures remains an ongoing challenge in the field.
In addition, patients with atypical 22q11.2 microdeletions that leave TBX1 unaffected still show craniofacial and cardiac defects, as well as velopharyngeal insufficiency (Shi and Wang, 2018; Verhagen et al., 2012). One possibility is that these patients lack cis-acting regulatory elements required for modulating TBX1 expression; alternatively, deletion of CRKL, another 22q11.2 gene that has also been linked to craniofacial and heart anomalies in mouse models (Guris et al., 2001, 2006; Motahari et al., 2019; Verhagen et al., 2012), triggers the craniofacial and cardiac defects of DiGeorge syndrome. Further, modifying alleles outside the 22q11.2 microdeletion may synergize with TBX1 to create unique, atypical phenotypic manifestations (Aggarwal and Morrow, 2008). Although DiGeorge syndrome is complex and incompletely understood, it provides a powerful example for the complex interplay of cell lineages associated with heart development across the LPM and beyond.
Hematological disorders with complex comorbidities
Several predominantly hematopoietic disorders feature seemingly unrelated structural comorbidities (Tables 1 and 2, Box 2). A classic example is thrombocytopenia-absent radius (TAR) syndrome (OMIM #274000), a rare congenital disease affecting less than 1:100,000 live births. In its most selective manifestation, TAR syndrome patients feature a severely reduced number of platelet-forming megakaryocytes (<30×109 platelets/l of blood), which correlates with hypo-megakaryocytic thrombocytopenia, and absent radius bones (Bonsi et al., 2009; Greenhalgh, 2002; Hall et al., 1969; Klopocki et al., 2007; Petit and Boussion, 2022; Thompson et al., 2001). The blood coagulation defect, which may be transient, is seen in all cases, and, in over 90% of patients, manifests as symptomatic within the first 4 months of life (Hall et al., 1969). Curiously, the coagulation defect resolves with increasing age, but it needs to be determined whether this is due to the migration of hematopoietic progenitors from the fetal liver to the fetal bone marrow. The non-hematopoietic syndromic phenotypes have variable expressivity, ranging from mild joint issues to absent radii, while several TAR syndrome patients also present with variable heart and kidney anomalies, intolerance to cow's milk presenting as persistent diarrhoea and failure to thrive, and can experience exacerbation of thrombocytopenia (Whitfield and Barr, 1976) later in life. More rarely, patients present with mild craniofacial anomalies. Genetically, TAR syndrome associates with a heterozygous 1q21.1 microdeletion removing at least ten genes, including the gene encoding the exon junction complex factor RBM8A/Y14 that ubiquitously contributes to the regulation of mRNA splicing, nonsense-mediated decay and mRNA localization (Gehring et al., 2005; Klopocki et al., 2007; Palacios et al., 2004). Hypomorphic perturbation of RBM8A is assumed to cause TAR syndrome, as the 1q21.1 microdeletion, combined with putative hypomorphic RBM8A/Y14 alleles, has been found in most sequenced patients (Albers et al., 2012, 2013). However, the mechanistic link between hypomorphic RBM8A and the peculiar phenotype combination of TAR syndrome remains to be elucidated. Notably, TAR syndrome affects predominantly LPM-derived cell lineages in the blood, forelimb skeleton, heart and kidneys. Considering TAR syndrome an LPM-associated pathology might provide a framework to investigate its mechanistic underpinnings (Kocere et al., 2023 preprint) (Figs 1 and 3).
Another ultra-rare syndrome, autosomal-dominant radioulnar synostosis with amegakaryocytic thrombocytopenia (RUSAT1; OMIM #605432), features severe coagulation defects with restricted pronation-supination of the forearm (Thompson and Nguyen, 2000; Thompson et al., 2001). Two RUSAT1-affected families have been reported to carry mutations in HOXA11, a Hox gene involved in regional bone, muscle and tendon patterning (Hawkins et al., 2021; Swinehart et al., 2013); how HOXA11 perturbation causes the hematopoietic defects remains unknown. By contrast, other RUSAT1 cases have been associated with mutations in the MECOM locus that spans the MDS1 and EVI1 genes (Niihori et al., 2015); the EVI1 transcription factor broadly controls cell cycle genes and is dynamically expressed in various tissues, including the forming limbs (Bard-Chapeau et al., 2012; Celá et al., 2013; Li et al., 2022; Nucifora, 1997). Notably, Evi1 is an upstream regulator of hematopoietic transcription factors, including the megakaryocyte-erythrocyte regulator Gfi1 (Ayoub et al., 2018; Chiba et al., 2020; Konantz et al., 2016).
In addition to TAR and RUSAT1, other rare hematopoietic disorders with co-occurring limb anomalies have been reported. Individuals with Fanconi anemia (OMIM #609054) can present with digit anomalies, such as unilateral radial anomalies that include duplicated thumbs (Bhandari et al., 2022; Tischkowitz and Hodgson, 2003). Other examples include WT limb-blood syndrome (OMIM #194350) (Gonzalez et al., 1977), Diamond-Blackfan anemia 3 (OMIM #610629) (Gazda et al., 2006; Landowski et al., 2013) and congenital hemidysplasia with ichthyosiform erythroderma and limb defects (OMIM #308050) (Happle et al., 1996). Notably, TAR, Fanconi anemia and Diamond-Blackfan anemia are seemingly caused by mutations in ubiquitous regulatory factors, suggesting an early developmental susceptibility to their activity levels that remains to be elucidated. Nonetheless, whether the root of these syndromic phenotypes is an earlier developmental LPM phenotype remains unclear, yet provides a working hypothesis to study their embryonic origins.
Orphan diseases with potential LPM connections
In the examples above, we outlined the rationale for considering a shared LPM origin for syndromic congenital heart and cardiovascular diseases as part of complex syndromes with comorbidities affecting LPM-associated structures. A conceptual framework to connect syndromic phenotypes to a joint developmental basis extends to linking structural birth defects with fetal exposure to environmental factors such as teratogenic compounds (Box 3). Ultimately, combining syndromes with similar phenotypes via shared molecular pathways or developmental processes may shed light on orphan diseases with as-of-yet unknown causes and, at times, no clear all-encompassing diagnoses. Orphan diseases (Table 2, Box 2) remain understudied, as they are exceedingly rare, at times affecting single families or, alternatively, may have complex polygenic origins (Chen and Altman, 2017).
Box 3. Environmental impact on heart and forelimb development.
Beyond syndromes with a trackable genetic basis, insights into developmental and genetic programs help explain the consequences of exposure to teratogenic agents. The most infamous example is thalidomide syndrome or embryopathy linked to the use of thalidomide (also known as α-phthalimidoglutarimide) to treat morning sickness in pregnancy (Donovan et al., 2018; Kim and Scialli, 2011; Rehman et al., 2011; Somers, 1960; Vargesson, 2015), which affected thousands of pregnancies in the late 1950s. Infants were born with severe limb defects (phocomelia), eye and ear deformities, and congenital heart defects, depending on the gestational age at exposure and duration of treatment (Donovan et al., 2018; Vargesson, 2015). We now know that thalidomide syndrome is caused by compound-promoted degradation of several zinc finger transcription factors, including SALL4 (Donovan et al., 2018). Consistent with this discovery, thalidomide syndrome phenocopies Duane-radial ray syndrome (also called Okihiro syndrome; OMIM #607323), which is caused by mutations in SALL4 (Asakawa and Kawakami, 2018; Cox et al., 2002; Kohlhase et al., 2005).
In murine cardiac and forelimb formation, Tbx5 regulates Sall4 expression, and coordinated positive and negative feed-forward loops between these two transcription factors are required for the correct patterning of the heart and limbs (Koshiba-Takeuchi et al., 2006). This relationship, at a molecular level, explains the shared phenotypes between patients with Duane-radial ray/Okihiro and thalidomide syndromes. Together with Holt-Oram syndrome phenotypes, these reflect the early developmental connection of the heart and forelimbs in the LPM.
Other examples of embryopathies linked to fetal teratogenic agent exposure include lithium-associated congenital heart defects and fetal retinoid syndrome. As retinoid acid signaling is involved in second heart field patterning, perturbations in this pathway cause heart malformations. The most common cause of fetal retinoid syndrome is exposure to isotretinoin, a retinoid derived from vitamin A that is used to treat cystic acne. An analysis of 154 patients with fetal exposure to isotretinoin concluded that there is a 35% risk of embryopathy when isotretinoin is taken beyond the 15th day post-conception (Lammer et al., 2010; Mondal et al., 2017). Isotretinoin exposure-associated congenital abnormalities include craniofacial and cardiac defects, as well as altered central nervous system development and thymic malformations (Lammer et al., 2010; Mondal et al., 2017). Mechanistically, isotretinoin increases apoptosis in cells of the cranial neural crest due to upregulation of FoxO3a (FOXO3) and TRAIL (TNFSF10) (Draghici et al., 2021; Ej, 1992; Gudas and Wagner, 2011; Niederreither et al., 2001). Further studies are required to tease out additional signaling players in isotretinoin-induced cardiac and neural crest defects during fetal development, and how the LPM is affected in these syndromes.
Lithium is a common treatment for bipolar disorders, acting to slow or halt the progression of neuronal loss through targets including neurotrophins, GSK3B and additional mitochondrial/endoplasmic reticulum-associated proteins (Machado-Vieira et al., 2009). Fetal lithium exposure is associated with Ebstein's anomaly, a right ventricular outflow tract defect: a comprehensive study found that infants exposed to lithium have an adjusted risk ratio of 1.65% for cardiac malformations relative to unexposed infants (Patorno et al., 2017). GSK3B inhibits canonical Wnt signaling (Dominguez et al., 1995; He et al., 1995; Klein and Melton, 1996; Peifer et al., 1994; Pierce and Kimelman, 1995; Stambolic et al., 1996; Yost et al., 1996), hinting at a possible mechanistic connection to the heart defects, as Wnt signaling contributes to several steps in cardiac patterning and morphogenesis (Cohen et al., 2007; Mandal et al., 2017; Martin et al., 2010; Schneider and Mercola, 2001; Ueno et al., 2007; Verhoeven et al., 2011). Nonetheless, lithium still remains the drug of choice to treat bipolar pregnant mothers due to evidence of efficacy compared to other drugs, as well as evidence that other drugs may also show low levels of teratogenicity (Patorno et al., 2017; Poels et al., 2018).
These examples underscore the risks of undesired effects from fetal exposure to environmental teratogenic agents. The modes of action of these drugs affect complex signaling pathways in different tissues and organs, but are often incompletely understood. Further, susceptibility to these environmental effects may greatly vary between patients due to unique modifying genetic variants, highlighting the need to integrate these factors when studying the mechanisms of syndromic diseases. The intertwined organ functions emerging from the LPM as well as its shared and co-deployed regulatory processes during development might render this mesoderm territory particularly sensitive to environmental perturbations.
Holt-Oram syndrome and Duane-radial ray syndrome, which we discuss in Box 3, present similar phenotypes due to genetic perturbations in a shared molecular pathway (Harvey and Logan, 2006): Tbx5 acts directly upstream of Sall4 during heart and appendage development in mice (Koshiba-Takeuchi et al., 2006) and in zebrafish (Harvey and Logan, 2006). Other heart-hand syndromes may result from developmental perturbation of the same or interacting pathways and the progenitor cell populations they control. Heart-hand syndrome type 3 (also known as Spanish type; OMIM #140450) features the characteristic upper limb deformities and congenital heart defects. Affecting three individuals of a single Spanish family, no causative gene has been assigned to the condition to date (Ruiz de la Fuente and Prieto, 1980). Additional orphan diseases that primarily present as congenital heart and limb defects include patent ductus arteriosus and bicuspid aortic valve with hand anomalies (OMIM #604381) (Gelb et al., 1999) and long-thumb brachydactyly syndrome (OMIM #112430) (Gelb et al., 1999; Hollister and Hollister, 1981) (Table 2). Despite the overlapping phenotypes, a shared progenitor cell or genetic origin has not been established in these orphan diseases. Therefore, investigating potential perturbations of TBX5, SALL4 and other cardiac/forelimb transcription factors in the LPM-derived heart and limb mesoderm warrants further consideration (Fan et al., 2009; Harvey and Logan, 2006; Koshiba-Takeuchi et al., 2006).
Numerous orphan diseases outside the heart-hand syndromes feature symptoms that affect LPM-derived organs or tissues. Examples include the previously mentioned WT limb-blood syndrome, with patients presenting endothelial, limb and craniofacial defects (Gonzalez et al., 1977), and thoracoabdominal syndrome (OMIM #313850), which affects craniofacial and neck structures, the heart, blood and kidneys (Carmi et al., 1990; Gonzalez et al., 1977). These clearly complex phenotypes could result from early perturbations in LPM patterning yet could also result from more pleiotropic developmental issues. Considering the joint LPM origin of various affected organs as the developmental stencil to narrow down possible connecting causes could advance the investigation of underlying mutations for these orphan diseases. Although orphan diseases are exceedingly rare, collectively, affected patients make up a large percentage of genetic diseases and deserve diagnostic and therapeutic investigation (Orphanet).
Connecting and testing mechanisms of syndromic diseases using animal models
As outlined above, a LPM disease-associated gene may be expressed in multiple organ lineages. This is based on lineage connections or co-option of gene regulatory mechanisms during organ development in disparate cell types (McQueen and Rebeiz, 2020; Monteiro, 2012; Olson, 2006; Shubin et al., 2009). Throughout this Review, we have highlighted how model organisms have fundamentally contributed to our understanding of developmental birth defects, gene ontology and biological mechanisms (Fig. 3, Table 3). For example, the notion that head and neck muscles, together with the heart, are affected in DiGeorge syndrome indicates that they emerge from the Tbx1-expressing CPF (Diogo et al., 2015; Greulich et al., 2011; Lescroart et al., 2015; Swedlund and Lescroart, 2019). Lineage tracing experiments have established firm evidence for shared clonal origins of heart and head muscle lineages (Diogo et al., 2015; Lescroart et al., 2010, 2015; Mahadevan et al., 2004; Meilhac et al., 2004). Consequently, mutations in undifferentiated early CPF progenitors, such as mutations in TBX1, can potentially influence all downstream lineages. However, how Tbx1 is regulated in the CPF and whether its downstream expression in different muscle lineages uses shared or divergent mechanisms in each lineage remains to be clarified.
TBX5, as the causative gene in Holt-Oram syndrome, provides another potent example for joint expression in different LPM descendants. The recessive zebrafish mutant heartstrings carries a mutant tbx5a allele (Garrity et al., 2002). heartstrings mutants, as well as tbx5a morphants, display heart and pectoral fin defects, recapitulating the Tbx5 mutant mouse and human Holt-Oram phenotypes (Ahn et al., 2002; Garrity et al., 2002) (Table 3). Studies in zebrafish tbx5a mutants and morphants have provided new insights into how Tbx5 contributes to left-right laterality, cardiac looping and myocardial conductivity (Boyle Anderson and Ho, 2018; Mao et al., 2015; Mao et al., 2021; Mosimann et al., 2015; Parrie et al., 2013). These insights provide basic concepts of the cardiac manifestations that could arise in patients carrying TBX5 gene variants. Mice heterozygous for a Tbx5 deletion (Tbx5del/+) recapitulate the hallmarks of the human syndrome with congenital forelimb and heart defects, whereas mice with homozygous deletions (Tbx5del/del) are embryonic lethal (Bruneau et al., 2001; Mori and Bruneau, 2004; Zhou et al., 2005). Mechanistic work revealed that Tbx5 and Nk2.5 synergistically interact to promote the expression of the gap junction protein Cx40 (Gja5), reduction of which is assumed to contribute to the cardiac anomalies in Holt-Oram syndrome (Bruneau et al., 2001; Zhou et al., 2005). In the limbs, absence of Tbx5 abrogates the Fgf10-Fgf8 signaling loop, which is critical for limb bud outgrowth (Agarwal et al., 2003; Arora et al., 2012; Minguillon et al., 2012). However, as we discussed earlier, whether Tbx5 activity in both cardiac and forelimb progenitors reflects a joint clonal LPM origin of these organ precursors, or whether Tbx5 expression and function are co-opted by forelimb precursors in a separate gene-regulatory event, remains unresolved. The mouse Tbx5 locus harbors several regulatory elements, including a dedicated forelimb enhancer (Cunningham et al., 2018; Minguillon et al., 2012) and multiple cardiac enhancers (Richter et al., 2020; Smemo et al., 2012); more regulatory elements (and their upstream control) await discovery (Bickley and Logan, 2014; Bruneau et al., 1999). Similarly, zebrafish hand2 and mouse Hand2 mutants have provided key insights into the developmental mechanisms in heart, limbs and additional LPM organs influenced by this deeply conserved transcription factor (Barnes et al., 2011; Firulli et al., 1998; George et al., 2022; Hashimoto et al., 2019; Keegan et al., 2005; Laurent et al., 2017; McFadden et al., 2005; Osterwalder et al., 2014; Perens et al., 2016; Prummel et al., 2022; Vincentz et al., 2021; Yamagishi et al., 2000; Yelon and Stainier, 2005). The upstream regulation of Hand2 in mouse and zebrafish, beyond the pharyngeal arches (Charité et al., 2001; Iklé et al., 2012), remains vastly uncharacterized, as regulatory elements for broad early LPM expression or for heart and limb/pectoral fin activity have not been identified to date.
Multiple lineages may also develop in an interconnected manner, whereby perturbation of one may have consequences for another. Beyond Tbx5, the heart and the pectoral fin/forelimb also provide an example for inter-organ signaling interactions during development. In the absence of retinoic acid signaling in the prospective pectoral fin/forelimb field, the heart field expands in cell number (Cunningham and Duester, 2015; Duong et al., 2021; Gibert et al., 2006; Zhao et al., 2009). This interaction might reflect the evolutionary relationship between heart and forelimb, with models proposing that delimiting the heart field was a prerequisite to paired appendage evolution (Tanaka, 2013, 2016). Beyond the developmental impact of perturbing retinoic acid signaling and its upstream regulation, avoiding embryo or fetal exposure to retinoic acid analogs remains paramount (Box 3) (Cunningham and Duester, 2015; Duester, 2008; Lammer et al., 2010; Mondal et al., 2017; Niederreither et al., 2001). In the forming trunk, the kidney, hematopoietic and endothelial progenitors form in close proximity within the posterior LPM and are influenced by signaling interactions that include the lateralmost mesothelial progenitors (Fig. 1) (Chapman, 2019; Cheng et al., 2015; Dressler, 2009; Fleming et al., 2013; Perens et al., 2016; Shin et al., 2009; Wingert and Davidson, 2008). Consequently, perturbations of these developmental interactions could lead to multi-organ defects that manifest as complex syndromic phenotypes.
While work across animal models has discovered causes and consequences of congenital heart anomalies and their comorbidities, animal models also emphasize inter-species differences in genetic susceptibility to structural heart disorders. In humans, the majority of genetic congenital heart disease patients carry heterozygous mutations (Richards and Garg, 2010; Andersen et al., 2014; Nees and Chung, 2020; Pierpont et al., 2018; Williams et al., 2019; Yasuhara and Garg, 2021). By contrast, mouse and zebrafish models of these disorders frequently require bi-allelic loss of function of the orthologous gene(s) to reveal a phenotype, as documented for mutations in Tbx5, Hand2, Sall4, Gata family members and others (Bruneau et al., 2001; Garrity et al., 2002; Granados-Riveron et al., 2012; Harvey and Logan, 2006; Yelon et al., 2000) (Fig. 3, Table 3). Beyond complete loss of function, numerous mutations identified in patients result in amino acid substitutions or splicing and regulatory alterations with more subtle impact on gene function. Therefore, recapitulating the exact patient-derived mutation may be necessary to appropriately model a particular syndrome or patient cohort (Albadri et al., 2017; Kimura et al., 2014; Prykhozhij et al., 2018). Rather than reducing the validity of animal models, these nuances reveal the critical roles of gene dosage or of compensatory genetic mechanisms that await further elucidation (Rossi et al., 2015; Sztal and Stainier, 2020).
Taken together, the LPM provides a conceptual framework to connect seemingly disperse disease phenotypes, either by common clonal origin, close lineage connection during development or shared regulatory gene expression among progenitors. Considering developmental and molecular connections between cells and organs has vast potential in directing diagnostics for possible and plausible comorbidities, avoiding potential incomplete diagnoses, and facilitating the design of complex treatment strategies.
Outlook
Next-generation sequencing has advanced ground-breaking insights into mutations and gene variants associated with developmental disorders and invigorated association studies for disease susceptibility loci (Austin-Tse et al., 2022; Lightbody et al., 2019; Morash et al., 2018; Yang and Kim, 2018). Several publicly available databases curate information on disease genetics, such as the Monarch Initiative and OMIM (McMurry et al., 2016; Shefchek et al., 2020; Motulsky, 1993) (Fig. 3). Although they are exceedingly powerful in aggregating diverse data from clinical and functional studies, any developmental or mechanistic connections for LPM-associated diseases, such as joint LPM origin of affected organs, remains to be determined by the database user. Further, drawing functional or causative conclusions from individual mutations or genetic variation is heavily influenced by insights gained in model organism studies (Alghamdi et al., 2022), in particular, assessing the phenotypic impact of individual gene perturbations on structural birth defects. As nowhere near all orthologs and paralogs of human genes have been functionally tested in developmental model organisms (Table 3, Box 2), the community still faces a gap in gene ontology knowledge that restricts the sequence-based prediction of disease traits. CRISPR-mediated mutagenesis for basic functional testing of candidate genes is possible by microinjection in zebrafish and Xenopus: these CRISPR-induced somatic mutants, so-called crispants (Burger et al., 2016), can recapitulate major aspects of the candidate gene's function. Nonetheless, although crispants provide a powerful discovery tool for first functional insights before mouse knockout studies, they require stringent controls and germline mutant validation in species that enable this workflow (Burger et al., 2016; Naert and Vleminckx, 2018; Shah et al., 2015; Trubiroha et al., 2018; Wu et al., 2018) (Fig. 3).
The broad lack of interfaces linking patient genetic data to functional in vivo follow-up, such as missing cross-species integration with animal models in individual databases, remains a key gap. The Model organism Aggregated Resources for Rare Variant ExpLoration (MARRVEL) website curates available model organism resources for human disease-relevant genes, providing a powerful resource for overcoming this gap (Wang et al., 2017a). Additional information, such as genomic coordinates, or interconnection with databases mapping non-coding features of the genome, could significantly expand patient-derived sequence data. Further, the varied versions of the human genome assembly used across individual databases renders comparisons challenging. Although individual genes of interest can be cross-referenced manually, the systematic generation of candidate gene lists to validate in model organisms remains labor intensive and not broadly accessible. Augmenting databases towards driving experimental validation of mutations, variants and causal gene discovery overall would provide fertile grounds for collaborations between clinicians, bioinformaticians and developmental biologists to accelerate research on rare LPM disorders, as we started to define throughout this Review, and beyond.
Ultimately, a major hurdle to diagnose and fully care for syndromic congenital anomalies remains the sheer complexity of human phenotypes such as heart-hand disorders. Patients with a specific organ-affecting disease often require highly specialized care. Performing a full diagnostic phenotype assessment of potentially dozens of symptoms in multiple organs is frequently not possible for reasons involving time, finances, technology or lacking consent. Nonetheless, providing insights into the underlying biology of linked comorbidities of syndromic congenital disease has the potential to improve long-term care and consequently the quality of life of affected patients. Predicting or anticipating care-relevant congenital phenotypes that can co-occur based on embryonic connections gleaned from developmental biology is only the starting point towards this goal.
Supplementary Material
Acknowledgments
This article is part of a collection ‘Moving Heart Failure to Heart Success: Mechanisms, Regeneration & Therapy’, which was launched in a dedicated Special Issue guest edited by Jeroen Bakkers, Milena Bellin and Ravi Karra. See related articles in this collection at https://journals.biologists.com/collection/8169/Moving-Heart-Failure-to-Heart-Success.
Acknowledgements
We thank the Mosimann laboratory and the members of the TBX4Life initiative for constructive input on the manuscript.
Footnotes
Funding
This work has been supported by the National Institutes of Health (1R01DK129350-01A1) to A.B.; the Swiss Bridge Foundation and Anschutz Medical Campus, University of Colorado School of Medicine to A.B. and C.M.; and the Children's Hospital Colorado Foundation, Blood Transfusion Center Zurich and National Science Foundation (2203311) to C.M.
Contributor Information
Christian Mosimann, Email: christian.mosimann@cuanschutz.edu.
Alexa Burger, Email: alexa.burger@cuanschutz.edu.
References
- Agarwal, P., Wylie, J. N., Galceran, J., Arkhitko, O., Li, C., Deng, C., Grosschedl, R. and Bruneau, B. G. (2003). Tbx5 is essential for forelimb bud initiation following patterning of the limb field in the mouse embryo. Development 130, 623-633. 10.1242/dev.00191 [DOI] [PubMed] [Google Scholar]
- Aggarwal, V. S. and Morrow, B. E. (2008). Genetic modifiers of Velo-cardio-facial syndrome/DiGeorge syndrome. Dev. Disabil. Res. Rev. 14, 19-25. 10.1002/ddrr.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn, D. G., Kourakis, M. J., Rohde, L. A., Silver, L. M. and Ho, R. K. (2002). T-box gene tbx5 is essential for formation of the pectoral limb bud. Nature 417, 754-758. 10.1038/nature00814 [DOI] [PubMed] [Google Scholar]
- Albadri, S., Del Bene, F. and Revenu, C. (2017). Genome editing using CRISPR/Cas9-based knock-in approaches in zebrafish. Methods 121-122, 77-85. 10.1016/j.ymeth.2017.03.005 [DOI] [PubMed] [Google Scholar]
- Albers, C. A., Paul, D. S., Schulze, H., Freson, K., Stephens, J. C., Smethurst, P. A., Jolley, J. D., Cvejic, A., Kostadima, M., Bertone, P.et al. (2012). Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat. Genet. 44, 435-439. S1-2. 10.1038/ng.1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers, C. A., Newbury-Ecob, R., Ouwehand, W. H. and Ghevaert, C. (2013). New insights into the genetic basis of TAR (thrombocytopenia-absent radii) syndrome. Curr. Opin. Genet. Dev. 23, 316-323. 10.1016/j.gde.2013.02.015 [DOI] [PubMed] [Google Scholar]
- Alghamdi, S. M., Schofield, P. N. and Hoehndorf, R. (2022). Contribution of model organism phenotypes to the computational identification of human disease genes. Dis. Model. Mech. 15, dmm049441. 10.1242/dmm.049441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen, T. A., Troelsen, K. D. L. L. and Larsen, L. A. (2014). Of mice and men: molecular genetics of congenital heart disease. Cell. Mol. Life Sci. 71, 1327-1352. 10.1007/s00018-013-1430-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariza, L., Carmona, R., Cañete, A., Cano, E. and Muñoz-Chápuli, R. (2016). Coelomic epithelium-derived cells in visceral morphogenesis: coelomic epithelium in morphogenesis. Dev. Dyn. 245, 307-322. 10.1002/dvdy.24373 [DOI] [PubMed] [Google Scholar]
- Arora, R., Metzger, R. J. and Papaioannou, V. E. (2012). Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet. 8, e1002866. 10.1371/journal.pgen.1002866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakawa, K. and Kawakami, K. (2018). Protocadherin-mediated cell repulsion controls the central topography and efferent projections of the abducens nucleus. Cell Rep. 24, 1562-1572. 10.1016/j.celrep.2018.07.024 [DOI] [PubMed] [Google Scholar]
- Austin-Tse, C. A., Jobanputra, V., Perry, D. L., Bick, D., Taft, R. J., Venner, E., Gibbs, R. A., Young, T., Barnett, S., Belmont, J. W.et al. (2022). Best practices for the interpretation and reporting of clinical whole genome sequencing. NPJ Genomic Med. 7, 27. 10.1038/s41525-022-00295-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub, E., Wilson, M. P., McGrath, K. E., Li, A. J., Frisch, B. J., Palis, J., Calvi, L. M., Zhang, Y. and Perkins, A. S. (2018). EVI1 overexpression reprograms hematopoiesis via upregulation of Spi1 transcription. Nat. Commun. 9, 4239. 10.1038/s41467-018-06208-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard-Chapeau, E. A., Jeyakani, J., Kok, C. H., Muller, J., Chua, B. Q., Gunaratne, J., Batagov, A., Jenjaroenpun, P., Kuznetsov, V. A., Wei, C. L.et al. (2012). Ecotopic viral integration site 1 (EVI1) regulates multiple cellular processes important for cancer and is a synergistic partner for FOS protein in invasive tumors. Proc. Natl. Acad. Sci. USA 109, 2168-2173. 10.1073/pnas.1119229109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes, R. M. and Firulli, A. B. (2009). A twist of insight - the role of Twist-family bHLH factors in development. Int. J. Dev. Biol. 53, 909-924. 10.1387/ijdb.082747rb [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes, R. M., Firulli, B. A., Conway, S. J., Vincentz, J. W. and Firulli, A. B. (2010). Analysis of the Hand1 cell lineage reveals novel contributions to cardiovascular, neural crest, extra-embryonic, and lateral mesoderm derivatives. Dev. Dyn. 239, 3086-3097. 10.1002/dvdy.22428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes, R. M., Firulli, B. A., VanDusen, N. J., Morikawa, Y., Conway, S. J., Cserjesi, P., Vincentz, J. W. and Firulli, A. B. (2011). Hand2 loss-of-function in Hand1-expressing cells reveals distinct roles in epicardial and coronary vessel development. Circ. Res. 108, 940-949. 10.1161/CIRCRESAHA.110.233171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basson, C. T., Bachinsky, D. R., Lin, R. C., Levi, T., Elkins, J. A., Soults, J., Grayzel, D., Kroumpouzou, E., Traill, T. A., Leblanc-Straceski, J.et al. (1997). Mutations in human cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 15, 30-35. 10.1038/ng0197-30 [DOI] [PubMed] [Google Scholar]
- Battaglia, A., Chen, Z., Brothman, A. R., Morelli, S., Palumbos, J. C., Carey, J. C., Hudgins, L. and Disteche, C. (2005). Karyotype/phenotype correlations in duplication 4q: evidence for a critical region within 4q27-28 for preaxial defects. Am. J. Med. Genet. A 134A, 334-337. 10.1002/ajmg.a.30644 [DOI] [PubMed] [Google Scholar]
- Bhandari, J., Thada, P. K. and Puckett, Y. (2022). Fanconi Anemia. StatPearls. [PubMed] [Google Scholar]
- Bickley, S. R. B. and Logan, M. P. O. (2014). Regulatory modulation of the T-box gene Tbx5 links development, evolution, and adaptation of the sternum. Proc. Natl. Acad. Sci. USA 111, 17917-17922. 10.1073/pnas.1409913111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornson, C. R. R., Griffin, K. J. P., Farr, G. H., Terashima, A., Himeda, C., Kikuchi, Y. and Kimelman, D. (2005). Eomesodermin is a localized maternal determinant required for endoderm induction in zebrafish. Dev. Cell 9, 523-533. 10.1016/j.devcel.2005.08.010 [DOI] [PubMed] [Google Scholar]
- Boisset, J.-C., van Cappellen, W., Andrieu-Soler, C., Galjart, N., Dzierzak, E. and Robin, C. (2010). In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature 464, 116-120. 10.1038/nature08764 [DOI] [PubMed] [Google Scholar]
- Bonsi, L., Marchionni, C., Alviano, F., Lanzoni, G., Franchina, M., Costa, R., Grossi, A. and Bagnara, G. P. (2009). Thrombocytopenia with absent radii (TAR) syndrome: from hemopoietic progenitor to mesenchymal stromal cell disease? Exp. Hematol. 37, 1-7. 10.1016/j.exphem.2008.09.004 [DOI] [PubMed] [Google Scholar]
- Borozdin, W., Bravo-Ferrer Acosta, A. M., Seemanova, E., Leipoldt, M., Bamshad, M. J., Unger, S. and Kohlhase, J. (2006). Contiguous hemizygous deletion ofTBX5,TBX3, andRBM19 resulting in a combined phenotype of Holt-Oram and ulnar-mammary syndromes. Am. J. Med. Genet. Part A 140A, 1880-1886. 10.1002/ajmg.a.31340 [DOI] [PubMed] [Google Scholar]
- Bosman, E. A., Penn, A. C., Ambrose, J. C., Kettleborough, R., Stemple, D. L. and Steel, K. P. (2005). Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum. Mol. Genet. 14, 3463-3476. 10.1093/hmg/ddi375 [DOI] [PubMed] [Google Scholar]
- Bouchard, M., Souabni, A., Mandler, M., Neubüser, A. and Busslinger, M. (2002). Nephric lineage specification by Pax2 and Pax8. Genes Dev. 16, 2958. 10.1101/gad.240102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle Anderson, E. A. T. and Ho, R. K. (2018). A transcriptomics analysis of the Tbx5 paralogues in zebrafish. PLoS One 13, e0208766. 10.1371/journal.pone.0208766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau, B. G., Logan, M., Davis, N., Levi, T., Tabin, C. J., Seidman, J. G. and Seidman, C. E. (1999). Chamber-specific cardiac expression of Tbx5 and heart defects in Holt- Oram syndrome. Dev. Biol. 211, 100-108. 10.1006/dbio.1999.9298 [DOI] [PubMed] [Google Scholar]
- Bruneau, B. G., Nemer, G., Schmitt, J. P., Charron, F., Robitaille, L., Caron, S., Conner, D. A., Gessler, M., Nemer, M., Seidman, C. E.et al. (2001). A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106, 709-721. 10.1016/S0092-8674(01)00493-7 [DOI] [PubMed] [Google Scholar]
- Bulatovic, I., Månsson-Broberg, A., Sylvén, C. and Grinnemo, K. H. (2016). Human fetal cardiac progenitors: the role of stem cells and progenitors in the fetal and adult heart. Best Pract. Res. Clin. Obstet. Gynaecol. 31, 58-68. 10.1016/j.bpobgyn.2015.08.008 [DOI] [PubMed] [Google Scholar]
- Burger, A., Lindsay, H., Felker, A., Hess, C., Anders, C., Chiavacci, E., Zaugg, J., Weber, L. M. L. M., Catena, R., Jinek, M.et al. (2016). Maximizing mutagenesis with solubilized CRISPR-Cas9 ribonucleoprotein complexes. Development 143, 2025-2037. 10.1242/dev.134809 [DOI] [PubMed] [Google Scholar]
- Burn, J. (1999). Closing time for CATCH22. J. Med. Genet. 36, 737-738. 10.1136/jmg.36.10.737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussmann, J., Bakkers, J. and Schulte-Merker, S. (2007). Early endocardial morphogenesis requires Scl/Tal1. PLoS Genet. 3, e140. 10.1371/journal.pgen.0030140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarata, T., Snyder, D., Schwend, T., Klosowiak, J., Holtrup, B. and Simon, H.-G. (2010). Pdlim7 is required for maintenance of the mesenchymal/epidermal Fgf signaling feedback loop during zebrafish pectoral fin development. BMC Dev. Biol. 10, 104. 10.1186/1471-213X-10-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmi, R., Barbash, A. and Mares, A. J. (1990). The thoracoabdominal syndrome (TAS): a new X-linked dominant disorder. Am. J. Med. Genet. 36, 109-114. 10.1002/ajmg.1320360122 [DOI] [PubMed] [Google Scholar]
- Celá, P., Balková, S. M., Bryjová, A., Horáková, D., Míšek, I., Richman, J. M. and Buchtová, M. (2013). Expression, function and regulation of Evi-1 during embryonic avian development. Gene Expr. Patterns 13, 343-353. 10.1016/j.gep.2013.06.002 [DOI] [PubMed] [Google Scholar]
- Chan, S. S.-K., Hagen, H. R., Swanson, S. A., Stewart, R., Boll, K. A., Aho, J., Thomson, J. A. and Kyba, M. (2016). Development of bipotent cardiac/skeletal myogenic progenitors from MESP1+ mesoderm. Stem Cell Rep. 6, 26-34. 10.1016/j.stemcr.2015.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, D. L. (2019). Impaired intermediate formation in mouse embryos expressing reduced levels of Tbx6. Genesis 57, e23270. 10.1002/dvg.23270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charité, J., Mcfadden, D. G. and Olson, E. N. (2000). The bHLH transcription factor dHAND controls Sonic hedgehog expression and establishment of the zone of polarizing activity during limb development. Development 127, 2461-2470. 10.1242/dev.127.11.2461 [DOI] [PubMed] [Google Scholar]
- Charité, J., McFadden, D. G., Merlo, G., Levi, G., Clouthier, D. E., Yanagisawa, M., Richardson, J. A. and Olson, E. N. (2001). Role of Dlx6 in regulation of an endothelin-1-dependent, dHAND branchial arch enhancer. Genes Dev. 15, 3039-3049. 10.1101/gad.931701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, B. and Altman, R. B. (2017). Opportunities for developing therapies for rare genetic diseases: focus on gain-of-function and allostery. Orphanet J. Rare Dis. 12, 61. 10.1186/s13023-017-0614-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H., Chang, C.-H., Perrin, E. and Perrin, J. (1982). A lethal, Larsen-like multiple joint dislocation syndrome. Am. J. Med. Genet. 13, 149-161. 10.1002/ajmg.1320130207 [DOI] [PubMed] [Google Scholar]
- Cheng, C. N., Verdun, V. A. and Wingert, R. A. (2015). Recent advances in elucidating the genetic mechanisms of nephrogenesis using zebrafish. Cells 4, 218-233. 10.3390/cells4020218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba, A., Masamoto, Y., Mizuno, H. and Kurokawa, M. (2020). GFI1 is a downstream target of EVI1 in normal hematopoiesis. Blood 136, 33. 10.1182/blood-2020-134071 [DOI] [Google Scholar]
- Clayton-Smith, J. and Donnai, D. (1988). A further patient with the lethal type of Larsen syndrome. J. Med. Genet. 25, 499-500. 10.1136/jmg.25.7.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, E. D., Wang, Z., Lepore, J. J., Lu, M. M., Taketo, M. M., Epstein, D. J. and Morrisey, E. E. (2007). Wnt/β-catenin signaling promotes expansion of Isl-1–positive cardiac progenitor cells through regulation of FGF signaling. J. Clin. Invest. 117, 1794-1804. 10.1172/JCI31731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai, G., Heude, E., Mella, S., Paisant, S., Pala, F., Gallardo, M., Langa, F., Kardon, G., Gopalakrishnan, S. and Tajbakhsh, S. (2019). A distinct cardiopharyngeal mesoderm genetic hierarchy establishes antero-posterior patterning of esophagus striated muscle. Elife 8, e47460. 10.7554/eLife.47460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon, F. L., Barth, K. S. and Robertson, E. J. (1991). A novel retrovirally induced embryonic lethal mutation in the mouse: assessment of the developmental fate of embryonic stem cells homozygous for the 413.d proviral integration. Development 111, 969-981. 10.1242/dev.111.4.969 [DOI] [PubMed] [Google Scholar]
- Conway, S. J., Firulli, B. and Firulli, A. B. (2010). A bHLH code for cardiac morphogenesis. Pediatr. Cardiol. 31, 318-324. 10.1007/s00246-009-9608-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, R., Bouzekri, N., Martin, S., Southam, L., Hugill, A., Golamaully, M., Cooper, R., Adeyemo, A., Soubrier, F., Ward, R.et al. (2002). Okihiro syndrome is caused by SALL4 mutations. Hum. Mol. Genet. 11, 2979-2987. 10.1093/hmg/11.23.2979 [DOI] [PubMed] [Google Scholar]
- Cunningham, T. J. and Duester, G. (2015). Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 16, 110-123. 10.1038/nrm3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, D., Swartzlander, D., Liyanarachchi, S., Davuluri, R. V. and Herman, G. E. (2005). Changes in gene expression associated with loss of function of the NSDHL sterol dehydrogenase in mouse embryonic fibroblasts. J. Lipid Res. 46, 1150-1162. 10.1194/jlr.M400462-JLR200 [DOI] [PubMed] [Google Scholar]
- Cunningham, T. J., Lancman, J. J., Berenguer, M., Dong, P. D. S. and Duester, G. (2018). Genomic knockout of two presumed forelimb Tbx5 enhancers reveals they are nonessential for limb development. Cell Rep. 23, 3146-3151. 10.1016/j.celrep.2018.05.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alto, M. and Mahadevan, V. S. (2012). Pulmonary arterial hypertension associated with congenital heart disease. Eur. Respir. Rev. 21, 328-337. 10.1183/09059180.00004712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson, K. C., Mason, E. A. and Pera, M. F. (2015). The pluripotent state in mouse and human. Development 142, 3090-3099. 10.1242/dev.116061 [DOI] [PubMed] [Google Scholar]
- Davidson, A. J., Lewis, P., Przepiorski, A. and Sander, V. (2019). Turning mesoderm into kidney. Semin. Cell Dev. Biol. 91, 86-93. 10.1016/j.semcdb.2018.08.016 [DOI] [PubMed] [Google Scholar]
- De La Garza, A., Cameron, R. C., Gupta, V., Fraint, E., Nik, S. and Bowman, T. V. (2019). The splicing factor Sf3b1 regulates erythroid maturation and proliferation via TGFb signaling in zebrafish. Blood Adv. 3, 2093-2104. 10.1182/bloodadvances.2018027714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deruiter, M. C., Poelmann, R. E., Vanderplas-De Vries, I., Mentink, M. M. T. and Gittenberger-de Groot, A. C. (1992). The development of the myocardium and endocardium in mouse embryos. Fusion of two heart tubes? Anat. Embryol. 185, 461-473. 10.1007/BF00174084 [DOI] [PubMed] [Google Scholar]
- Diogo, R. (2020). Cranial or postcranial—Dual origin of the pectoral appendage of vertebrates combining the fin-fold and gill-arch theories? Dev. Dyn. 249, 1182-1200. 10.1002/dvdy.192 [DOI] [PubMed] [Google Scholar]
- Diogo, R., Kelly, R. G., Christiaen, L., Levine, M., Ziermann, J. M., Molnar, J. L., Noden, D. M. and Tzahor, E. (2015). A new heart for a new head in vertebrate cardiopharyngeal evolution. Nature 520, 466-473. 10.1038/nature14435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez, I., Itoh, K. and Sokol, S. Y. (1995). Role of glycogen synthase kinase 3 beta as a negative regulator of dorsoventral axis formation in Xenopus embryos. Proc. Natl. Acad. Sci. USA 92, 8498-8502. 10.1073/pnas.92.18.8498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Don, E. K., De Jong-Curtain, T. A., Doggett, K., Hall, T. E., Heng, B., Badrock, A. P., Winnick, C., Nicholson, G. A., Guillemin, G. J., Currie, P. D.et al. (2016). Genetic basis of hindlimb loss in a naturally occurring vertebrate model. Biol. Open 5, 359-366. 10.1242/bio.016295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan, K. A., An, J., Nowak, R. P., Yuan, J. C., Fink, E. C., Berry, B. C., Ebert, B. L. and Fischer, E. S. (2018). Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. Elife 7, e38430. 10.7554/eLife.38430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing, J., Head, D., Curcio-Brint, A., Hulshof, M., Motroni, T., Raimondi, S., Carroll, A., Drabkin, H., Willman, C. and Theil, K. (1993). An AML1/ETO fusion transcript is consistently detected by RNA-based polymerase chain reaction in acute myelogenous leukemia containing the (8;21)(q22;q22) translocation. Blood 81, 2860-2865. 10.1182/blood.V81.11.2860.2860 [DOI] [PubMed] [Google Scholar]
- Draghici, C.-C., Miulescu, R.-G., Petca, R.-C., Petca, A., Dumitrașcu, M. C. and Șandru, F. (2021). Teratogenic effect of isotretinoin in both fertile females and males (Review). Exp. Ther. Med. 21, 534. 10.3892/etm.2021.9966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressler, G. R. (2009). Advances in early kidney specification, development and patterning. Development 136, 3863-3874. 10.1242/dev.034876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboc, V., Sulaiman, F. A., Feneck, E., Kucharska, A., Bell, D., Holder-Espinasse, M. and Logan, M. P. O. (2021). Tbx4 function during hindlimb development reveals a mechanism that explains the origins of proximal limb defects. Development 148, dev199580. 10.1242/dev.199580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duester, G. (2008). Retinoic acid synthesis and signaling during early organogenesis. Cell 134, 921-931. 10.1016/j.cell.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong, T. B., Holowiecki, A. and Waxman, J. S. (2021). Retinoic acid signaling restricts the size of the first heart field within the anterior lateral plate mesoderm. Dev. Biol. 473, 119-129. 10.1016/j.ydbio.2021.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer, L. A. and Kirby, M. L. (2009). The role of secondary heart field in cardiac development. Dev. Biol. 336, 137-144. 10.1016/j.ydbio.2009.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles, M. R., He, S., Legge, M., Kumar, R., Fox, J., Zhou, C., French, M. and Tsai, R. W. S. (2004). PAX genes in development and disease: the role of PAX2 in urogenital tract development. Int. J. Dev. Biol. 46, 535-544. 10.1111/j.1440-169x.2004.00770.x [DOI] [PubMed] [Google Scholar]
- Ej, L. (1992). Malformations of hindbrain structures among humans exposed to isotretinoin (13-cis-retinoic acid) during early embryogenesis. In Retinoids in Normal Development and Teratogenesis (ed. Morriss-Kay G.), pp. 281-295. Oxford University Press. [Google Scholar]
- Ericsson, R., Knight, R. and Johanson, Z. (2013). Evolution and development of the vertebrate neck. J. Anat. 222, 67-78. 10.1111/j.1469-7580.2012.01530.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, C., Chen, Q. and Wang, Q. K. (2009). Functional role of transcriptional factor TBX5 in pre-mRNA splicing and holt-oram syndrome via association with SC35. J. Biol. Chem. 284, 25653-25663. 10.1074/jbc.M109.041368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fath, M. A., Mullins, R. F., Searby, C., Nishimura, D. Y., Wei, J., Rahmouni, K., Davis, R. E., Tayeh, M. K., Andrews, M., Yang, B.et al. (2005). Mkks-null mice have a phenotype resembling Bardet-Biedl syndrome. Hum. Mol. Genet. 14, 1109-1118. 10.1093/hmg/ddi123 [DOI] [PubMed] [Google Scholar]
- Fernández, L., Lapunzina, P., Pajares, I. L., Criado, G. R., García-Guereta, L., Pérez, J., Quero, J. and Delicado, A. (2005). Higher frequency of uncommon 1.5-2 Mb deletions found in familial cases of 22q11.2 deletion syndrome. Am. J. Med. Genet. Part A 136A, 71-75. 10.1002/ajmg.a.30756 [DOI] [PubMed] [Google Scholar]
- Fernandez-Teran, M., Piedra, M. E., Kathiriya, I. S., Srivastava, D., Rodriguez-Rey, J. C. and Ros, M. A. (2000). Role of dHAND in the anterior-posterior polarization of the limb bud: Implications for the Sonic hedgehog pathway. Development 127, 2133-2142. 10.1242/dev.127.10.2133 [DOI] [PubMed] [Google Scholar]
- Firulli, A. B., McFadden, D. G., Lin, Q., Srivastava, D. and Olson, E. N. (1998). Heart and extra-embryonic mesodermal defects in mouse embryos lacking the bHLH transcription factor Hand1. Nat. Genet. 18, 266-270. 10.1038/ng0398-266 [DOI] [PubMed] [Google Scholar]
- Firulli, B. A., McConville, D. P., Byers, J. S., III, Vincentz, J. W., Barnes, R. M. and Firulli, A. B. (2010). Analysis of a Hand1 hypomorphic allele reveals a critical threshold for embryonic viability. Dev. Dyn. 239, 2748-2760. 10.1002/dvdy.22402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming, B. M. M., Yelin, R., James, R. G. G. and Schultheiss, T. M. M. (2013). A role for Vg1/Nodal signaling in specification of the intermediate mesoderm. Development 140, 1819-1829. 10.1242/dev.093740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock, R. L., Kudlow, B. A., Evans, A. M., Jameson, S. A., Hauschka, S. D. and Kennedy, B. K. (2006). Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 20, 486-500. 10.1101/gad.1364906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli, A., Robay, D., Osterwalder, M., Bao, X., Bénazet, J. D., Tariq, M., Paro, R., Mackem, S. and Zeller, R. (2010). Distinct roles of Hand2 in initiating polarity and posterior Shh expression during the onset of mouse limb bud development. PLoS Genet. 6, e1000901. 10.1371/journal.pgen.1000901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangras, P., Gallagher, T. L., Parthun, M. A., Yi, Z., Patton, R. D., Tietz, K. T., Deans, N. C., Bundschuh, R., Amacher, S. L. and Singh, G. (2020). Zebrafish rbm8a and magoh mutants reveal EJC developmental functions and new 3′UTR intron-containing NMD targets. PLoS Genet. 16, e1008830. 10.1371/journal.pgen.1008830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavito-Aguilar, Z. V., Riley, H. E. and Yelon, D. (2010). Hand2 ensures an appropriate environment for cardiac fusion by limiting Fibronectin function. Development 137, 3215-3220. 10.1242/dev.052225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Martinez, V. and Schoenwolf, G. C. (1992). Positional control of mesoderm movement and fate during avian gastrulation and neurulation. Dev. Dyn. 193, 249-256. 10.1002/aja.1001930305 [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez, V. and Schoenwolf, G. C. (1993). Primitive-streak origin of the cardiovascular system in avian embryos. Dev. Biol. 159, 706-719. 10.1006/dbio.1993.1276 [DOI] [PubMed] [Google Scholar]
- Garrity, D. M., Childs, S. and Fishman, M. C. (2002). The heartstrings mutation in zebrafish causes heart/fin Tbx5 deficiency syndrome. Development 129, 4635-4645. 10.1242/dev.129.19.4635 [DOI] [PubMed] [Google Scholar]
- Gays, D., Hess, C., Camporeale, A., Ala, U., Provero, P., Mosimann, C. and Santoro, M. M. (2017). An exclusive cellular and molecular network governs intestinal smooth muscle cell differentiation in vertebrates. Development 144, 464-478. 10.1242/dev.133926 [DOI] [PubMed] [Google Scholar]
- Gazda, H. T., Grabowska, A., Merida-Long, L. B., Latawiec, E., Schneider, H. E., Lipton, J. M., Vlachos, A., Atsidaftos, E., Ball, S. E., Orfali, K. A.et al. (2006). Ribosomal protein S24 gene is mutated in diamond-blackfan anemia. Am. J. Hum. Genet. 79, 1110-1118. 10.1086/510020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring, N. H., Kunz, J. B., Neu-Yilik, G., Breit, S., Viegas, M. H., Hentze, M. W. and Kulozik, A. E. (2005). Exon-junction complex components specify distinct routes of nonsense-mediated mRNA decay with differential cofactor requirements. Mol. Cell 20, 65-75. 10.1016/j.molcel.2005.08.012 [DOI] [PubMed] [Google Scholar]
- Gelb, B., Zhang, J., Sommer, R., Wasserman, J., Reitman, M. and Willner, J. (1999). Familial patent ductus arteriosus and bicuspid aortic valve with hand anomalies: a novel heart-hand syndrome. Am. J. Med. Genet. 87, 175-179. [DOI] [PubMed] [Google Scholar]
- George, R. M., Guo, S., Firulli, B. A., Rubart, M. and Firulli, A. B. (2022). Neonatal deletion of Hand1 and Hand2 within murine cardiac conduction system reveals a novel role for HAND2 in rhythm homeostasis. J. Cardiovasc. Dev. Dis. 9, 214. 10.3390/jcdd9070214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibert, Y., Gajewski, A., Meyer, A. and Begemann, G. (2006). Induction and prepatterning of the zebrafish pectoral fin bud requires axial retinoic acid signaling. Development 133, 2649-2659. 10.1242/dev.02438 [DOI] [PubMed] [Google Scholar]
- Gonzalez, C., Durkin-Stamm, M. V., Geimer, N. F., Shahidi, N., Schilling, R. F., Rubira, F. and Opitz, J. (1977). The WT syndrome—a “new” autosomal dominant pleiotropic trait of radial/ulnar hypoplasia with high risk of bone marrow failure and/or leukemia. Birth Defects Orig. Artic. Ser. 13, 31-38. [PubMed] [Google Scholar]
- Gopalakrishnan, S., Comai, G., Sambasivan, R., Francou, A., Kelly, R. G. and Tajbakhsh, S. (2015). A Cranial Mesoderm Origin for Esophagus Striated Muscles. Dev. Cell 34, 694-704. 10.1016/j.devcel.2015.07.003 [DOI] [PubMed] [Google Scholar]
- Graff, J. M., Bansal, A. and Melton, D. A. (1996). Xenopus Mad proteins transduce distinct subsets of signals for the TGF beta superfamily. Cell 85, 479-487. 10.1016/S0092-8674(00)81249-0 [DOI] [PubMed] [Google Scholar]
- Granados-Riveron, J. T., Pope, M., Bu'Lock, F. A., Thornborough, C., Eason, J., Setchfield, K., Ketley, A., Kirk, E. P., Fatkin, D., Feneley, M. P.et al. (2012). Combined mutation screening of NKX2-5, GATA4, and TBX5 in congenital heart disease: multiple heterozygosity and novel mutations. Congenit. Heart Dis. 7, 151. 10.1111/j.1747-0803.2011.00573.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandel, H., Lun, K., Rauch, G.-J., Rhinn, M., Piotrowski, T., Houart, C., Sordino, P., Küchler, A. M., Schulte-Merker, S. and Geisler, R.et al. (2002). Retinoic acid signalling in the zebrafish embryo is necessary during pre-segmentation stages to pattern the anterior-posterior axis of the CNS and to induce a pectoral fin bud. Development 129, 2851-2865. 10.1242/dev.129.12.2851 [DOI] [PubMed] [Google Scholar]
- Greenhalgh, K. L. (2002). Thrombocytopenia-absent radius syndrome: a clinical genetic study. J. Med. Genet. 39, 876-881. 10.1136/jmg.39.12.876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greulich, F., Rudat, C. and Kispert, A. (2011). Mechanisms of T-box gene function in the developing heart. Cardiovasc. Res. 91, 212-222. 10.1093/cvr/cvr112 [DOI] [PubMed] [Google Scholar]
- Gritsman, K., Zhang, J., Cheng, S., Heckscher, E., Talbot, W. S. and Schier, A. F. (1999). The EGF-CFC protein one-eyed pinhead is essential for nodal signaling. Cell 97, 121-132. 10.1016/S0092-8674(00)80720-5 [DOI] [PubMed] [Google Scholar]
- Gruenauer-Kloevekorn, C. and Froster, U. G. (2003). Holt-Oram syndrome: a new mutation in the TBX5 gene in two unrelated families. Ann. Genet. 46, 19-23. 10.1016/S0003-3995(03)00006-6 [DOI] [PubMed] [Google Scholar]
- Gudas, L. J. and Wagner, J. A. (2011). Retinoids regulate stem cell differentiation. J. Cell. Physiol. 226, 322-330. 10.1002/jcp.22417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdon, J. B. (1995). The formation of mesoderm and muscle in xenopus. In Organization of the Early Vertebrate Embryo (ed. N. Zagris, A. M. Duprat, A. Durston), pp. 51-59. Boston: Springer. [Google Scholar]
- Guris, D. L., Fantes, J., Tara, D., Druker, B. J. and Imamoto, A. (2001). Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat. Genet. 27, 293-298. 10.1038/85855 [DOI] [PubMed] [Google Scholar]
- Guris, D. L., Duester, G., Papaioannou, V. E. and Imamoto, A. (2006). Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev. Cell 10, 81-92. 10.1016/j.devcel.2005.12.002 [DOI] [PubMed] [Google Scholar]
- Haarman, M. G., Kerstjens-Frederikse, W. S. and Berger, R. M. F. (2019). The ever-expanding phenotypical spectrum of human TBX4 mutations: from toe to lung. Eur. Respir. J. 54, 1901504. 10.1183/13993003.01504-2019 [DOI] [PubMed] [Google Scholar]
- Haarman, M. G., Kerstjens-Frederikse, W. S. and Berger, R. M. F. (2020). TBX4 variants and pulmonary diseases: getting out of the “Box.”. Curr. Opin. Pulm. Med. 26, 277-284. 10.1097/MCP.0000000000000678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, J. G., Levin, J., Kuhn, J. P., Ottenheimer, E. J., van Berkum, K. A. P. and McKusick, V. A. (1969). Thrombocytopenia with absent radius (TAR). Medicine 48, 411-440. 10.1097/00005792-196948060-00001 [DOI] [PubMed] [Google Scholar]
- Happle, R., Effendy, I., Megahed, M., Orlow, S. J. and Kiister, W. (1996). CHILD syndrome in a boy. Am. J. Med. Genet. 62, 19-194. [DOI] [PubMed] [Google Scholar]
- Harris, I. S. and Black, B. L. (2010). Development of the endocardium. Pediatr. Cardiol. 31, 391-399. 10.1007/s00246-010-9642-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey, S. A. and Logan, M. P. O. (2006). sall4 acts downstream of tbx5 and is required for pectoral fin outgrowth. Development 133, 1165-1173. 10.1242/dev.02259 [DOI] [PubMed] [Google Scholar]
- Hashimoto, H., Wang, Z., Garry, G. A., Malladi, V. S., Botten, G. A., Ye, W., Zhou, H., Osterwalder, M., Dickel, D. E., Visel, A.et al. (2019). Cardiac reprogramming factors synergistically activate genome-wide cardiogenic stage-specific enhancers. Cell Stem Cell 25, 69-86.e5. 10.1016/j.stem.2019.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins, M. B., Henke, K., Correspondence, M. P. H. and Harris, M. P. (2021). Latent developmental potential to form limb-like skeletal structures in zebrafish. Cell 184, 899-911.e13. 10.1016/j.cell.2021.01.003 [DOI] [PubMed] [Google Scholar]
- Hayes, K., Kim, Y. K. and Pera, M. F. (2021). A case for revisiting Nodal signaling in human pluripotent stem cells. Stem Cells 39, 1137-1144. 10.1002/stem.3383 [DOI] [PubMed] [Google Scholar]
- He, X., Saint-Jeannet, J. P., Woodgett, J. R., Varmus, H. E. and Dawid, I. B. (1995). Glycogen synthase kinase-3 and dorsoventral patterning in Xenopus embryos. Nature 374, 617-622. 10.1038/374617a0 [DOI] [PubMed] [Google Scholar]
- Hiroi, Y., Kudoh, S., Monzen, K., Ikeda, Y., Yazaki, Y., Nagai, R. and Komuro, I. (2001). Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat. Genet. 28, 276-280. 10.1038/90123 [DOI] [PubMed] [Google Scholar]
- Hollister, D. W. and Hollister, W. G. (1981). The “long-thumb” brachydactyly syndrome. Am. J. Med. Genet. 8, 5-16. 10.1002/ajmg.1320080103 [DOI] [PubMed] [Google Scholar]
- Hu, Y. C., Newman, C. B., Porter, R. W. and Albuquerque, F. C. (2011). Transarterial Onyx embolization of sacral chordoma: case report and review of the literature. J. Neurointerv. Surg. 3, 85-87. 10.1136/jnis.2010.003020 [DOI] [PubMed] [Google Scholar]
- Hurst, J. A., Hall, C. M. and Baraitser, M. (1991). The Holt-Oram syndrome. J. Med. Genet. 28, 406-410. 10.1136/jmg.28.6.406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iklé, J. M., Artinger, K. B. and Clouthier, D. E. (2012). Identification and characterization of the zebrafish pharyngeal arch-specific enhancer for the basic helix-loop-helix transcription factor Hand2. Dev. Biol. 368, 118-126. 10.1016/j.ydbio.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itou, J., Kawakami, H., Quach, T., Osterwalder, M., Evans, S. M., Zeller, R. and Kawakami, Y. (2012). Islet1 regulates establishment of the posterior hindlimb field upstream of the Hand2-Shh morphoregulatory gene network in mouse embryos. Development 139, 1620-1629. 10.1242/dev.073056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovitch, K., Temiño, S. and Torres, M. (2017). Live imaging of heart tube development in mouse reveals alternating phases of cardiac differentiation and morphogenesis. Elife 6, e30668. 10.7554/eLife.30668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerome, L. A. and Papaioannou, V. E. (2001). DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 27, 286-291. 10.1038/85845 [DOI] [PubMed] [Google Scholar]
- Kaplan, N., Razy-Krajka, F. and Christiaen, L. (2015). Regulation and evolution of cardiopharyngeal cell identity and behavior: insights from simple chordates. Curr. Opin. Genet. Dev. 32, 119-128. 10.1016/j.gde.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolak, J. A., Welch, C. L., Mosimann, C., Bzdega, K., West, J. D., Montani, D., Eyries, M., Mullen, M. P., Abman, S. H., Prapa, M.et al. (2023). Molecular function and contribution of TBX4 in development and disease. Am. J. Respir. Crit. Care. Med. 207, 855-864. 10.1164/rccm.202206-1039TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinski, B. A., Maynard, T. M., Bryan, C. A., Yitsege, G., Horvath, A., Lee, N. H., Moody, S. A. and Lamantia, A. S. (2022). Selective disruption of trigeminal sensory neurogenesis and differentiation in a mouse model of 22q11.2 deletion syndrome. Dis. Model. Mech. 15, dmm047357. 10.1242/dmm.047357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattman, S. J., Witty, A. D., Gagliardi, M., Dubois, N. C., Niapour, M., Hotta, A., Ellis, J. and Keller, G. (2011). Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 8, 228-240. 10.1016/j.stem.2010.12.008 [DOI] [PubMed] [Google Scholar]
- Keegan, B. R., Feldman, J. L., Begemann, G., Ingham, P. W. and Yelon, D. (2005). Retinoic acid signaling restricts the cardiac progenitor pool. Science 307, 247-249. 10.1126/science.1101573 [DOI] [PubMed] [Google Scholar]
- Keller, S. A., Jones, J. M., Boyle, A., Barrow, L. L., Killen, P. D., Green, D. G., Kapousta, N. V., Hitchcock, P. F., Swank, R. T. and Meisler, M. H. (1994). Kidney and retinal defects (Krd), a transgene-induced mutation with a deletion of mouse chromosome 19 that includes the Pax2 locus. Genomics 23, 309-320. 10.1006/geno.1994.1506 [DOI] [PubMed] [Google Scholar]
- Kim, J. H. and Scialli, A. R. (2011). Thalidomide: the tragedy of birth defects and the effective treatment of disease. Toxicol. Sci. 122, 1-6. 10.1093/toxsci/kfr088 [DOI] [PubMed] [Google Scholar]
- Kimura, Y., Hisano, Y., Kawahara, A. and Higashijima, S. (2014). Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Sci. Rep. 4, 6545. 10.1038/srep06545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissa, K. and Herbomel, P. (2010). Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature 464, 112-115. 10.1038/nature08761 [DOI] [PubMed] [Google Scholar]
- Klein, P. S. and Melton, D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 93, 8455-8459. 10.1073/pnas.93.16.8455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopocki, E., Schulze, H., Strauss, G., Ott, C.-E. E., Hall, J., Trotier, F., Fleischhauer, S., Greenhalgh, L., Newbury-Ecob, R. A., Neumann, L. M.et al. (2007). Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am. J. Hum. Genet. 80, 232-240. 10.1086/510919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocere, A., Chiavacci, E., Mendez-Acevedo, K. M., Soneson, C., Hiltabidle, M. S., MacGowan, J. S., Panakova, D., Williams, M. L. K., Robinson, M. D., Mosimann, C.et al. (2023). TAR Syndrome-associated Rbm8a deficiency causes hematopoietic defects and attenuates Wnt/PCP signaling. bioRxiv 2023.04.12.536513. 10.1101/2023.04.12.536513 [DOI] [Google Scholar]
- Kohlhase, J., Chitayat, D., Kotzot, D., Ceylaner, S., Froster, U. G., Fuchs, S., Montgomery, T. and Rösler, B. (2005). SALL4 mutations in Okihiro syndrome (Duane-radial ray syndrome), acro-renal-ocular syndrome, and related disorders. Hum. Mutat. 26, 176-183. 10.1002/humu.20215 [DOI] [PubMed] [Google Scholar]
- Konantz, M., Alghisi, E., Müller, J. S., Lenard, A., Esain, V., Carroll, K. J., Kanz, L., North, T. E., Lengerke, C., Adamo, L.et al. (2016). Evi1 regulates Notch activation to induce zebrafish hematopoietic stem cell emergence. EMBO J. 459, 1131-1135. 10.15252/embj.201593454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba-Takeuchi, K., Takeuchi, J. K., Arruda, E. P., Kathiriya, I. S., Mo, R., Hui, C. C., Srivastava, D. and Bruneau, B. G. (2006). Cooperative and antagonistic interactions between Sall4 and Tbx5 pattern the mouse limb and heart. Nat. Genet. 38, 175-183. 10.1038/ng1707 [DOI] [PubMed] [Google Scholar]
- Lammer, E. J., Chen, D. T., Hoar, R. M., Agnish, N. D., Benke, P. J., Braun, J. T., Curry, C. J., Fernhoff, P. M., Grix, A. W. J., Lott, I. T.et al. (2010). Retinoic acid embryopathy. N. Engl. J. Med. 313, 837-841. 10.1056/NEJM198510033131401 [DOI] [PubMed] [Google Scholar]
- Lancino, M., Majello, S., Herbert, S., De Chaumont, F., Tinevez, J.-Y., Olivo-Marin, J.-C., Herbomel, P. and Schmidt, A. (2018). Anisotropic organization of circumferential actomyosin characterizes hematopoietic stem cells emergence in the zebrafish. Elife 7, e37355. 10.7554/eLife.37355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landowski, M., O'donohue, M. F., Buros, C., Ghazvinian, R., Montel-Lehry, N., Vlachos, A., Sieff, C. A., Newburger, P. E., Niewiadomska, E., Matysiak, M.et al. (2013). Novel deletion of RPL15 identified by array-comparative genomic hybridization in Diamond-Blackfan anemia. Hum. Genet. 132, 1265-1274. 10.1007/s00439-013-1326-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane, M. C. and Smith, W. C. (1999). The origins of primitive blood in Xenopus: implications for axial patterning. Development 126, 423-434. 10.1242/dev.126.3.423 [DOI] [PubMed] [Google Scholar]
- Laura, V. G., Marcela, S. G., Ricardo, J. C., Roberto, L., Filiberto, T.-T. and Sánchez Gómez, C. (2020). Incorporation of the first and second heart fields and prospective fate of the straight heart tube via in vivo labeling of chicken embryos. PLoS One 15, e0234069. 10.1371/journal.pone.0234069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent, F., Girdziusaite, A., Gamart, J., Barozzi, I., Osterwalder, M., Akiyama, J. A., Lincoln, J., Lopez-Rios, J., Visel, A., Zuniga, A.et al. (2017). HAND2 target gene regulatory networks control atrioventricular canal and cardiac valve development. Cell Rep. 19, 1602-1613. 10.1016/j.celrep.2017.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescroart, F., Kelly, R. G., Le Garrec, J.-F., Nicolas, J.-F., Meilhac, S. M. and Buckingham, M. (2010). Clonal analysis reveals common lineage relationships between head muscles and second heart field derivatives in the mouse embryo. Development 137, 3269-3279. 10.1242/dev.050674 [DOI] [PubMed] [Google Scholar]
- Lescroart, F., Hamou, W., Francou, A., Théveniau-Ruissy, M., Kelly, R. G. and Buckingham, M. (2015). Clonal analysis reveals a common origin between nonsomite-derived neck muscles and heart myocardium. Proc. Natl. Acad. Sci. USA 112, 1446-1451. 10.1073/pnas.1424538112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescroart, F., Dumas, C. E., Adachi, N. and Kelly, R. Get al. (2021). Emergence of heart and branchiomeric muscles in cardiopharyngeal mesoderm. Exp. Cell Res. 410, 112931. 10.1016/j.yexcr.2021.112931 [DOI] [PubMed] [Google Scholar]
- Li, J., Glover, J. D., Zhang, H., Peng, M., Tan, J., Mallick, C. B., Hou, D., Yang, Y., Wu, S., Liu, Y.et al. (2022). Limb development genes underlie variation in human fingerprint patterns. Cell 185, 95-112.e18. 10.1016/j.cell.2021.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightbody, G., Haberland, V., Browne, F., Taggart, L., Zheng, H., Parkes, E. and Blayney, J. K. (2019). Review of applications of high-throughput sequencing in personalized medicine: barriers and facilitators of future progress in research and clinical application. Brief. Bioinform. 20, 1795. 10.1093/bib/bby051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay, E. A., Botta, A., Jurecic, V., Carattini-Rivera, S., Cheah, Y.-C., Rosenblatt, H. M., Bradley, A. and Baldini, A. (1999). Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 401, 379-383. 10.1038/43900 [DOI] [PubMed] [Google Scholar]
- Lindsay, E. A., Vitelli, F., Su, H., Morishima, M., Huynh, T., Pramparo, T., Jurecic, V., Ogunrinu, G., Sutherland, H. F., Scambler, P. J.et al. (2001). Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 410, 97-101. 10.1038/35065105 [DOI] [PubMed] [Google Scholar]
- Liu, F., Hata, A., Baker, J. C., Doody, J., Carcamo, J., Harland, R. M. and Massague, J. (1996). A human Mad protein acting as a BMP-regulated transcriptional activator. Nature 381, 620-623. 10.1038/381620a0 [DOI] [PubMed] [Google Scholar]
- Lu, J., Lian, G., Lenkinski, R., De Grand, A., Vaid, R. R., Bryce, T., Stasenko, M., Boskey, A., Walsh, C. and Sheen, V. (2007). Filamin B mutations cause chondrocyte defects in skeletal development. Hum. Mol. Genet. 16, 1661-1675. 10.1093/hmg/ddm114 [DOI] [PubMed] [Google Scholar]
- Lu, C. X., Gong, H. R., Liu, X. Y., Wang, J., Zhao, C. M., Huang, R. T., Xue, S. and Yang, Y. Q. (2016). A novel HAND2 loss-of-function mutation responsible for tetralogy of Fallot. Int. J. Mol. Med. 37, 445-451. 10.3892/ijmm.2015.2436 [DOI] [PubMed] [Google Scholar]
- Lundin, C., Zech, L., Sjörs, K., Wadelius, C. and Annerén, G. (2002). Trisomy 4q syndrome: presentation of a new case and review of the literature. Ann. Genet. 45, 53-57. 10.1016/S0003-3995(02)01117-6 [DOI] [PubMed] [Google Scholar]
- Lurie, I. W. (2005). Partial trisomy 4q and preaxial limb defects. Am. J. Med. Genet. Part A 138A, 304-305. 10.1002/ajmg.a.30960 [DOI] [PubMed] [Google Scholar]
- Machado-Vieira, R., Manji, H. K. and Zarate, C. A. (2009). The role of lithium in the treatment of bipolar disorder: convergent evidence for neurotrophic effects as a unifying hypothesis. Bipolar Disord. 11, 92. 10.1111/j.1399-5618.2009.00714.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan, N. R., Horton, A. C. and Gibson-Brown, J. J. (2004). Developmental expression of the amphioxus Tbx1/10 gene illuminates the evolution of vertebrate branchial arches and sclerotome. Dev. Genes Evol. 214, 559-566. 10.1007/s00427-004-0433-1 [DOI] [PubMed] [Google Scholar]
- Mandal, A., Holowiecki, A., Song, Y. C. and Waxman, J. S. (2017). Wnt signaling balances specification of the cardiac and pharyngeal muscle fields. Mech. Dev. 143, 32-41. 10.1016/j.mod.2017.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour, F., Hinze, C., Telugu, N. S., Kresoja, J., Shaheed, I. B., Mosimann, C., Diecke, S. and Schmidt-Ott, K. M. (2022). The centrosomal protein 83 (CEP83) regulates human pluripotent stem cell differentiation toward the kidney lineage. Elife 11, e80165. 10.7554/eLife.80165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, Q., Stinnett, H. K. and Ho, R. K. (2015). Asymmetric cell convergence-driven zebrafish fin bud initiation and pre-pattern requires Tbx5a control of a mesenchymal Fgf signal. Development 142, 4329-4339. 10.1242/dev.124750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, L. M. F., Boyle Anderson, E. A. T. and Ho, R. K. (2021). Anterior lateral plate mesoderm gives rise to multiple tissues and requires tbx5a function in left-right asymmetry, migration dynamics, and cell specification of late-addition cardiac cells. Dev. Biol. 472, 52-66. 10.1016/j.ydbio.2021.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, J., Afouda, B. A. and Hoppler, S. (2010). Wnt/beta-catenin signalling regulates cardiomyogenesis via GATA transcription factors. J. Anat. 216, 92-107. 10.1111/j.1469-7580.2009.01171.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyn, I., Siggia, E. D. and Brivanlou, A. H. (2019). Mapping cell migrations and fates in a gastruloid model to the human primitive streak. Development 146, dev179564. 10.1242/dev.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsson, H., Davey, E. J., Draptchinskaia, N., Hamaguchi, I., Ooka, A., Levéen, P., Forsberg, E., Karlsson, S. and Dahl, N. (2004). Targeted disruption of the ribosomal protein S19 gene is lethal prior to implantation. Mol. Cell. Biol. 24, 4032-4037. 10.1128/MCB.24.9.4032-4037.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattonet, K., Riemslagh, F. W., Guenther, S., Prummel, K. D., Kesavan, G., Hans, S., Ebersberger, I., Brand, M., Burger, A., Reischauer, S.et al. (2022). Endothelial versus pronephron fate decision is modulated by the transcription factors Cloche/Npas4l, Tal1, and Lmo2. Sci. Adv. 8, 31. 10.1126/sciadv.abn2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccright, B., Lozier, J. and Gridley, T. (2006). Generation of new Notch2 mutant alleles. Genesis 44, 29-33. 10.1002/gene.20181 [DOI] [PubMed] [Google Scholar]
- Mcfadden, D. G., Barbosa, A. C., Richardson, J. A., Schneider, M. D., Srivastava, D. and Olson, E. N. (2005). The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development 132, 189-201. 10.1242/dev.01562 [DOI] [PubMed] [Google Scholar]
- Mcmahon, A. P. (2016). Development of the mammalian kidney. Curr. Top. Dev. Biol. 117, 31-64. 10.1016/bs.ctdb.2015.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcmurry, J. A., Köhler, S., Washington, N. L., Balhoff, J. P., Borromeo, C., Brush, M., Carbon, S., Conlin, T., Dunn, N., Engelstad, M.et al. (2016). Navigating the phenotype frontier: the monarch initiative. Genetics 203, 1491. 10.1534/genetics.116.188870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcqueen, E. and Rebeiz, M. (2020). On the individuality of gene regulatory networks: how does network re-use affect subsequent evolution? Curr. Top. Dev. Biol. 139, 375. 10.1016/bs.ctdb.2020.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcsweeney, C., Dong, F., Chen, M., Vitale, J., Xu, L., Crowley, N., Luscher, B., Zou, D. and Mao, Y. (2020). Full function of exon junction complex factor, Rbm8a, is critical for interneuron development. Transl. Psychiatry 10, 379. 10.1038/s41398-020-01065-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead, P. E., Brivanlou, I. H., Kelley, C. M. and Zon, L. I. (1996). BMP-4-responsive regulation of dorsal–ventral patterning by the homeobox protein Mix.1. Nature 382, 357-360. 10.1038/382357a0 [DOI] [PubMed] [Google Scholar]
- Meilhac, S. M. and Buckingham, M. E. (2018). The deployment of cell lineages that form the mammalian heart. Nat. Rev. Cardiol. 15, 705-724. 10.1038/s41569-018-0086-9 [DOI] [PubMed] [Google Scholar]
- Meilhac, S. M., Esner, M., Kelly, R. G., Nicolas, J. F. and Buckingham, M. E. (2004). The clonal origin of myocardial cells in different regions of the embryonic mouse heart. Dev. Cell 6, 685-698. 10.1016/S1534-5807(04)00133-9 [DOI] [PubMed] [Google Scholar]
- Merscher, S., Funke, B., Epstein, J. A., Heyer, J., Puech, A., Lu, M. M., Xavier, R. J., Demay, M. B., Russell, R. G., Factor, S.et al. (2001). TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 104, 619-629. 10.1016/S0092-8674(01)00247-1 [DOI] [PubMed] [Google Scholar]
- Metra, M., Zacà, V., Parati, G., Agostoni, P., Bonadies, M., Ciccone, M., Cas, A. D., Iacoviello, M., Lagioia, R., Lombardi, C.et al. (2011). Cardiovascular and noncardiovascular comorbidities in patients with chronic heart failure. J. Cardiovasc. Med. 12, 76-84. 10.2459/JCM.0b013e32834058d1 [DOI] [PubMed] [Google Scholar]
- Minguillon, C., Del Buono, J. and Logan, M. P. (2005). Tbx5 and Tbx4 are not sufficient to determine limb-specific morphologies but have common roles in initiating limb outgrowth. Dev. Cell 8, 75-84. 10.1016/j.devcel.2004.11.013 [DOI] [PubMed] [Google Scholar]
- Minguillon, C., Gibson-Brown, J. J. and Logan, M. P. (2009). Tbx4/5 gene duplication and the origin of vertebrate paired appendages. Proc. Natl. Acad. Sci. USA 106, 21726-21730. 10.1073/pnas.0910153106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minguillon, C., Nishimoto, S., Wood, S., Vendrell, E., Gibson-Brown, J. J. and Logan, M. P. O. (2012). Hox genes regulate the onset of Tbx5 expression in the forelimb. Development 139, 3180-3188. 10.1242/dev.084814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misfeldt, A. M., Boyle, S. C., Tompkins, K. L., Bautch, V. L., Labosky, P. A. and Baldwin, H. S. (2009). Endocardial cells are a distinct endothelial lineage derived from Flk1+ multipotent cardiovascular progenitors. Dev. Biol. 333, 78-89. 10.1016/j.ydbio.2009.06.033 [DOI] [PubMed] [Google Scholar]
- Monahan-Earley, R., Dvorak, A. M. and Aird, W. C. (2013). Evolutionary origins of the blood vascular system and endothelium. J. Thromb. Haemost. 11 Suppl 1, 46-66. 10.1111/jth.12253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal, D., Shenoy, S. R. and Mishra, S. (2017). Retinoic acid embryopathy. Int. J. Appl. Basic Med. Res. 7, 264. 10.4103/ijabmr.IJABMR_469_16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro, A. (2012). Gene regulatory networks reused to build novel traits: co-option of an eye-related gene regulatory network in eye-like organs and red wing patches on insect wings is suggested by optix expression. BioEssays 34, 181-186. 10.1002/bies.201100160 [DOI] [PubMed] [Google Scholar]
- Moorman, A. F. M. and Christoffels, V. M. (2003). Cardiac chamber formation: development, genes, and evolution. Physiol. Rev. 83, 1223-1267. 10.1152/physrev.00006.2003 [DOI] [PubMed] [Google Scholar]
- Morash, M., Mitchell, H., Beltran, H., Elemento, O. and Pathak, J. (2018). The role of next-generation sequencing in precision medicine: a review of outcomes in oncology. J. Pers. Med. 8, 30. 10.3390/jpm8030030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau, C., Caldarelli, P., Rocancourt, D., Roussel, J., Denans, N., Pourquie, O. and Gros, J. (2019). Timed collinear activation of Hox genes during gastrulation controls the avian forelimb position. Curr. Biol. 29, 35-50.e4. 10.1016/j.cub.2018.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, A. D. and Bruneau, B. G. (2004). TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealed. Curr. Opin. Cardiol. 19, 211-215. 10.1097/00001573-200405000-00004 [DOI] [PubMed] [Google Scholar]
- Mosimann, C., Panáková, D., Werdich, A. A., Musso, G., Burger, A., Lawson, K. L., Carr, L. A., Nevis, K. R., Sabeh, M. K., Zhou, Y.et al. (2015). Chamber identity programs drive early functional partitioning of the heart. Nat. Commun 6, 8146. 10.1038/ncomms9146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostello, D., Hoechstetter, L., Bendon, R. W., Dignan, P. S. J., Oestreich, A. E. and Siddiqi, T. A. (1991). Prenatal diagnosis of recurrent Larsen syndrome: further definition of a lethal variant. Prenat. Diagn. 11, 215-225. 10.1002/pd.1970110403 [DOI] [PubMed] [Google Scholar]
- Motahari, Z., Moody, S. A., Maynard, T. M. and Lamantia, A. S. (2019). In the line-up: deleted genes associated with DiGeorge/22q11.2 deletion syndrome: are they all suspects? J. Neurodev. Disord. 11, 7. 10.1186/s11689-019-9267-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motulsky, A. G. (1993). Mendelian inheritance in man: catalogs of autosomal dominant, autosomal recessive, and X-linked phenotypes. JAMA 269, 2003-2004. 10.1001/jama.1993.03500150115041 [DOI] [Google Scholar]
- Müller, A. M., Medvinsky, A., Strouboulis, J., Grosveld, F. and Dzierzakt, E. (1994). Development of hematopoietic stem cell activity in the mouse embryo. Immunity 1, 291-301. 10.1016/1074-7613(94)90081-7 [DOI] [PubMed] [Google Scholar]
- Müller, F., Blader, P., Rastegar, S., Fischer, N., Knöchel, W. and Strähle, U. (1999). Characterization of zebrafish smad1, smad2 and smad5: the amino-terminus of smad1 and smad5 is required for specific function in the embryo. Mech. Dev. 88, 73-88. 10.1016/S0925-4773(99)00173-2 [DOI] [PubMed] [Google Scholar]
- Mullins, M. C., Hammerschmidt, M., Kane, D. A., Odenthal, J., Brand, M., Van Eeden, F. J., Furutani-Seiki, M., Granato, M., Haffter, P., Heisenberg, C. P.et al. (1996). Genes establishing dorsoventral pattern formation in the zebrafish embryo: the ventral specifying genes. Development 123, 81-93. 10.1242/dev.123.1.81 [DOI] [PubMed] [Google Scholar]
- Naert, T. and Vleminckx, K. (2018). CRISPR/Cas9 disease models in zebrafish and Xenopus: the genetic renaissance of fish and frogs. Drug Discov. Today Technol. 28, 41-52. 10.1016/j.ddtec.2018.07.001 [DOI] [PubMed] [Google Scholar]
- Naiche, L. A. and Papaioannou, V. E. (2003). Loss of Tbx4 blocks hindlimb development and affects vascularization and fusion of the allantois. Development 130, 2681-2693. 10.1242/dev.00504 [DOI] [PubMed] [Google Scholar]
- Narkis, G., Tzchori, I., Cohen, T., Holtz, A., Wier, E. and Westphal, H. (2012). Isl1 and Ldb co-regulators of transcription are essential early determinants of mouse limb development. Dev. Dyn. 241, 787. 10.1002/dvdy.23761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nees, S. N. and Chung, W. K. (2020). Genetic basis of human congenital heart disease. Cold Spring Harb. Perspect. Biol. 12, a036749. 10.1101/cshperspect.a036749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, L. T., Rakshit, S., Sun, H. and Wellik, D. M. (2008). The generation and expression of a Hoxa11eGFP targeted allele in mice. Dev. Dyn. 237, 3410. 10.1002/dvdy.21756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas, H. A., Hua, K., Quigley, H., Ivare, J., Tesson, F. and Akimenko, M. A. (2022). A CRISPR/Cas9 zebrafish lamin A/C mutant model of muscular laminopathy. Dev. Dyn. 251, 645-661. 10.1002/dvdy.427 [DOI] [PubMed] [Google Scholar]
- Niederreither, K., Vermot, J., Messaddeq, N., Schuhbaur, B., Chambon, P. and Dollé, P. (2001). Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 128, 1019-1031. 10.1242/dev.128.7.1019 [DOI] [PubMed] [Google Scholar]
- Niihori, T., Ouchi-Uchiyama, M., Sasahara, Y., Kaneko, T., Hashii, Y., Irie, M., Sato, A., Saito-Nanjo, Y., Funayama, R., Nagashima, T.et al. (2015). Mutations in MECOM, encoding oncoprotein EVI1, cause radioulnar synostosis with amegakaryocytic thrombocytopenia. Am. J. Hum. Genet. 97, 848-854. 10.1016/j.ajhg.2015.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto, S. and Logan, M. P. O. (2016). Subdivision of the lateral plate mesoderm and specification of the forelimb and hindlimb forming domains. Semin. Cell Dev. Biol. 49, 102-108. 10.1016/j.semcdb.2015.11.011 [DOI] [PubMed] [Google Scholar]
- Nomaru, H., Liu, Y., De Bono, C., Righelli, D., Cirino, A., Wang, W., Song, H., Racedo, S. E., Dantas, A. G., Zhang, L.et al. (2021). Single cell multi-omic analysis identifies a Tbx1-dependent multilineage primed population in murine cardiopharyngeal mesoderm. Nat. Commun. 12, 6645. 10.1038/s41467-021-26966-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord, H., Kahsay, A., Dennhag, N., Pedrosa Domellöf, F., Domellöf, P. and Von Hofsten, J. (2021). Genetic compensation between Pax3 and Pax7 in zebrafish appendicular muscle formation. Dev. Dyn. 251, 1423-1438. 10.1002/dvdy.415 [DOI] [PubMed] [Google Scholar]
- North, T. E., Goessling, W., Peeters, M., Li, P., Ceol, C., Lord, A. M., Weber, G. J., Harris, J., Cutting, C. C., Huang, P.et al. (2009). Hematopoietic stem cell development is dependent on blood flow. Cell 137, 736-748. 10.1016/j.cell.2009.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucifora, G. (1997). The EVI1 gene in myeloid leukemia. Leukemia 11, 2022-2031. 10.1038/sj.leu.2400880 [DOI] [PubMed] [Google Scholar]
- Ohnishi, T., Miura, I., Ohba, H., Shimamoto, C., Iwayama, Y., Wakana, S. and Yoshikawa, T. (2017). A spontaneous and novel Pax3 mutant mouse that models Waardenburg syndrome and neural tube defects. Gene 607, 16-22. 10.1016/j.gene.2016.12.037 [DOI] [PubMed] [Google Scholar]
- Olson, E. N. (2006). Gene regulatory networks in the evolution and development of the heart. Science 313, 1922-1927. 10.1126/science.1132292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omran, H., Häffner, K., Burth, S., Fernandez, C., Fargier, B., Villaquiran, A., Nothwang, H. G., Schnittger, S., Lehrach, H., Woo, D.et al. (2001). Human adolescent nephronophthisis: gene locus synteny with polycystic kidney disease in Pcy mice. J. Am. Soc. Nephrol. 12, 107-113. 10.1681/ASN.V121107 [DOI] [PubMed] [Google Scholar]
- Onimaru, K., Shoguchi, E., Kuratani, S. and Tanaka, M. (2011). Development and evolution of the lateral plate mesoderm: comparative analysis of amphioxus and lamprey with implications for the acquisition of paired fins. Dev. Biol. 359, 124-136. 10.1016/j.ydbio.2011.08.003 [DOI] [PubMed] [Google Scholar]
- Osterwalder, M., Speziale, D., Shoukry, M., Mohan, R., Ivanek, R., Kohler, M., Beisel, C., Wen, X., Scales, S. J., Christoffels, V. M.et al. (2014). HAND2 targets define a network of transcriptional regulators that compartmentalize the early limb bud mesenchyme . Dev. Cell 31, 345-357. 10.1016/j.devcel.2014.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios, I. M., Gatfield, D., St Johnston, D. and Izaurralde, E. (2004). An eIF4AIII-containing complex required for mRNA localization and nonsense-mediated mRNA decay. Nature 427, 753-757. 10.1038/nature02351 [DOI] [PubMed] [Google Scholar]
- Pardanaud, L., Luton, D., Prigent, M., Bourcheix, L. M., Catala, M. and Dieterlen-Lievre, F. (1996). Two distinct endothelial lineages in ontogeny, one of them related to hemopoiesis. Development 122, 1363-1371. 10.1242/dev.122.5.1363 [DOI] [PubMed] [Google Scholar]
- Parmar, K., D'andrea, A. and Niedernhofer, L. J. (2009). Mouse models of Fanconi anemia. Mutat. Res. 668, 133-140. 10.1016/j.mrfmmm.2009.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrie, L. E., Renfrew, E. M., Vander Wal, A., Mueller, R. L. and Garrity, D. M. (2013). Zebrafish tbx5 paralogs demonstrate independent essential requirements in cardiac and pectoral fin development. Dev. Dyn. 242, 485-502. 10.1002/dvdy.23953 [DOI] [PubMed] [Google Scholar]
- Parvari, R., Weinstein, Y., Ehrlich, S., Steinitz, M. and Carmi, R. (1994). Linkage localization of the thoraco-abdominal syndrome (TAS) gene to Xq25-26. Am. J. Med. Genet. 49, 431-434. 10.1002/ajmg.1320490416 [DOI] [PubMed] [Google Scholar]
- Patel, C., Silcock, L., Mcmullan, D., Brueton, L. and Cox, H. (2012). TBX5 intragenic duplication: a family with an atypical Holt-Oram syndrome phenotype. Eur. J. Hum. Genet. 20, 863-869. 10.1038/ejhg.2012.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patorno, E., Huybrechts, K. F., Bateman, B. T., Cohen, J. M., Desai, R. J., Mogun, H., Cohen, L. S. and Hernandez-Diaz, S. (2017). Lithium use in pregnancy and the risk of cardiac malformations. N. Engl. J. Med. 376, 2245. 10.1056/NEJMoa1612222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten, S. A., Jacobs-Mcdaniels, N. L., Zaouter, C., Drapeau, P., Albertson, R. C. and Moldovan, F. (2012). Role of Chd7 in zebrafish: a model for CHARGE syndrome. PLoS One 7, e31650. 10.1371/journal.pone.0031650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paylor, R., Glaser, B., Mupo, A., Ataliotis, P., Spencer, C., Sobotka, A., Sparks, C., Choi, C. H., Oghalai, J., Curran, S.et al. (2006). Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc. Natl. Acad. Sci. USA 103, 7729-7734. 10.1073/pnas.0600206103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer, M., Pai, L. M. and Casey, M. (1994). Phosphorylation of the Drosophila adherens junction protein Armadillo: roles for wingless signal and zeste-white 3 kinase. Dev. Biol. 166, 543-556. 10.1006/dbio.1994.1336 [DOI] [PubMed] [Google Scholar]
- Perens, E. A., Garavito-Aguilar, Z. V., Guio-Vega, G. P., Peña, K. T., Schindler, Y. L. and Yelon, D. (2016). Hand2 inhibits kidney specification while promoting vein formation within the posterior mesoderm. Elife 5, e19941. 10.7554/eLife.19941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perens, E. A., Diaz, J. T., Quesnel, A., Askary, A., Crump, J. G. and Yelon, D. (2021). osr1 couples intermediate mesoderm cell fate with temporal dynamics of vessel progenitor cell differentiation. Development 148, dev198408. 10.1242/dev.198408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit, F. and Boussion, S. (2022). Thrombocytopenia absent radius syndrome. In GeneReviews® [Internet] (ed. Adam M. P., Mirzaa G. M., Pagon R. A.et al.). Seattle: University of Washington, Seattle. [Google Scholar]
- Pi-Roig, A., Martin-Blanco, E. and Minguillon, C. (2014). Distinct tissue-specific requirements for the zebrafish tbx5 genes during heart, retina and pectoral fin development. Open Biol. 4, 140014. 10.1098/rsob.140014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce, S. B. and Kimelman, D. (1995). Regulation of Spemann organizer formation by the intracellular kinase Xgsk-3. Development 121, 755-765. 10.1242/dev.121.3.755 [DOI] [PubMed] [Google Scholar]
- Pierpont, M. E., Brueckner, M., Chung, W. K., Garg, V., Lacro, R. V., Mcguire, A. L., Mital, S., Priest, J. R., Pu, W. T., Roberts, A.et al. (2018). Genetic basis for congenital heart disease: revisited: a scientific statement from the American Heart Association. Circulation 138, e653-e711. 10.1161/CIR.0000000000000606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski, T., Ahn, D., Schilling, T. F., Nair, S., Ruvinsky, I., Geisler, R., Rauch, G.-J., Haffter, P., Zon, L. I., Zhou, Y.et al. (2003). The zebrafish van gogh mutation disrupts tbx1, which is involved in the DiGeorge deletion syndrome in humans. Development 130, 5043-5052. 10.1242/dev.00704 [DOI] [PubMed] [Google Scholar]
- Poels, E. M. P., Bijma, H. H., Galbally, M. and Bergink, V. (2018). Lithium during pregnancy and after delivery: a review. Int. J. Bipolar Disord. 6, 26. 10.1186/s40345-018-0135-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleev, A., Fickenscher, H., Mundlos, S., Winterpacht, A., Zabel, B., Fidler, A., Gruss, P. and Plachov, D. (1992). PAX8, a human paired box gene: isolation and expression in developing thyroid, kidney and Wilms’ tumors. Development 116, 611-623. 10.1242/dev.116.3.611 [DOI] [PubMed] [Google Scholar]
- Postma, A. V., Van De Meerakker, J. B. A., Mathijssen, I. B., Barnett, P., Christoffels, V. M., Ilgun, A., Lam, J., Wilde, A. A. M., Deprez, R. H. L. and Moorman, A. F. M. (2008). A gain-of-function TBX5 mutation is associated with atypical Holt-Oram syndrome and paroxysmal atrial fibrillation. Circ. Res. 102, 1433-1442. 10.1161/CIRCRESAHA.107.168294 [DOI] [PubMed] [Google Scholar]
- Proulx, K., Lu, A. and Sumanas, S. (2010). Cranial vasculature in zebrafish forms by angioblast cluster-derived angiogenesis. Dev. Biol. 348, 34-46. 10.1016/j.ydbio.2010.08.036 [DOI] [PubMed] [Google Scholar]
- Prummel, K. D., Hess, C., Nieuwenhuize, S., Parker, H. J., Rogers, K. W., Kozmikova, I., Racioppi, C., Brombacher, E. C., Czarkwiani, A., Knapp, D.et al. (2019). A conserved regulatory program initiates lateral plate mesoderm emergence across chordates. Nat. Commun. 10, 3857. 10.1038/s41467-019-11561-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prummel, K. D., Nieuwenhuize, S. and Mosimann, C. (2020). The lateral plate mesoderm. Development 147, dev175059. 10.1242/dev.175059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prummel, K. D., Crowell, H. L., Nieuwenhuize, S., Brombacher, E. C., Daetwyler, S., Soneson, C., Kresoja-Rakic, J., Kocere, A., Ronner, M., Ernst, A.et al. (2022). Hand2 delineates mesothelium progenitors and is reactivated in mesothelioma. Nat. Commun. 13, 1677. 10.1038/s41467-022-29311-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prykhozhij, S. V., Fuller, C., Steele, S. L., Veinotte, C. J., Razaghi, B., Robitaille, J. M., Mcmaster, C. R., Shlien, A., Malkin, D. and Berman, J. N. (2018). Optimized knock-in of point mutations in zebrafish using CRISPR/Cas9. Nucleic Acids Res. 46, e102-e102. 10.1093/nar/gky512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rallis, C., Bruneau, B. G., Del Buono, J., Seidman, C. E., Seidman, J. G., Nissim, S., Tabin, C. J. J., Logan, M. P. O. P., Ashby, P. R., Coutelle, O.et al. (2003). Tbx5 is required for forelimb bud formation and continued outgrowth. Development 130, 2741-2751. 10.1242/dev.00473 [DOI] [PubMed] [Google Scholar]
- Raman, R., Ramanagoudr-Bhojappa, R., Dhinoja, S., Ramaswami, M., Carrington, B., Jagadeeswaran, P. and Chandrasekharappa, S. C. (2022). Pancytopenia and thrombosis defects in zebrafish mutants of Fanconi anemia genes. Blood Cells. Mol. Dis. 93, 102640. 10.1016/j.bcmd.2021.102640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reamon-Buettner, S. M., Ciribilli, Y., Inga, A. and Borlak, J. (2008). A loss-of-function mutation in the binding domain of HAND1 predicts hypoplasia of the human hearts. Hum. Mol. Genet. 17, 1397-1405. 10.1093/hmg/ddn027 [DOI] [PubMed] [Google Scholar]
- Reamon-Buettner, S. M., Ciribilli, Y., Traverso, I., Kuhls, B., Inga, A. and Borlak, J. (2009). A functional genetic study identifies HAND1 mutations in septation defects of the human heart. Hum. Mol. Genet. 18, 3567-3578. 10.1093/hmg/ddp305 [DOI] [PubMed] [Google Scholar]
- Rehman, W., Arfons, L. M. and Lazarus, H. M. (2011). The rise, fall and subsequent triumph of thalidomide: lessons learned in drug development. Ther. Adv. Hematol. 2, 291. 10.1177/2040620711413165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, A. A. and Garg, V. (2010). Genetics of congenital heart disease. Curr. Cardiol. Rev. 6, 91. 10.2174/157340310791162703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter, F., Morton, S. U., Kim, S. W., Kitaygorodsky, A., Wasson, L. K., Chen, K. M., Zhou, J., Qi, H., Patel, N., Depalma, S. R.et al. (2020). Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat. Genet. 52, 769-777. 10.1038/s41588-020-0652-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochais, F., Mesbah, K. and Kelly, R. G. (2009). Signaling pathways controlling second heart field development. Circ. Res. 104, 933-942. 10.1161/CIRCRESAHA.109.194464 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Esteban, C., Tsukul, T., Yonel, S., Magallon, J., Tamura, K. and Izpisua Belmonte, J. C. (1999). The T-box genes Tbx4 and Tbx5 regulate limb outgrowth and identity. Nature 398, 814-818. 10.1038/19769 [DOI] [PubMed] [Google Scholar]
- Rossi, A., Kontarakis, Z., Gerri, C., Nolte, H., Hölper, S., Krüger, M. and Stainier, D. Y. R. R. (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230-233. 10.1038/nature14580 [DOI] [PubMed] [Google Scholar]
- Ruiz De La Fuente, S. and Prieto, F. (1980). Heart-hand syndrome. III. A new syndrome in three generations. Hum. Genet. 55, 43-47. 10.1007/BF00329125 [DOI] [PubMed] [Google Scholar]
- Saint-Jeannet, J. P., Levi, G., Girault, J. M., Koteliansky, V. and Thiery, J. P. (1992). Ventrolateral regionalization of Xenopus laevis mesoderm is characterized by the expression of alpha-smooth muscle actin. Development 115, 1165-1173. 10.1242/dev.115.4.1165 [DOI] [PubMed] [Google Scholar]
- Saitta, S. C., Harris, S. E., Gaeth, A. P., Driscoll, D. A., Mcdonald-Mcginn, D. M., Maisenbacher, M. K., Yersak, J. M., Chakraborty, P. K., Hacker, A. M., Zackai, E. H.et al. (2004). Aberrant interchromosomal exchanges are the predominant cause of the 22q11.2 deletion. Hum. Mol. Genet. 13, 417-428. 10.1093/hmg/ddh041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage, C., Das, P., Finelli, A. L., Townsend, S. R., Sun, C. Y., Baird, S. E. and Padgett, R. W. (1996). Caenorhabditis elegans genes sma-2, sma-3, and sma-4 define a conserved family of transforming growth factor beta pathway components. Proc. Natl. Acad. Sci. USA 93, 790. 10.1073/pnas.93.2.790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schier, A. F. and Talbot, W. S. (2005). Molecular genetics of axis formation in zebrafish. Annu. Rev. Genet. 39, 561-613. 10.1146/annurev.genet.37.110801.143752 [DOI] [PubMed] [Google Scholar]
- Schneider, V. A. and Mercola, M. (2001). Wnt antagonism initiates cardiogenesis in Xenopus laevis. Genes Dev. 15, 304-315. 10.1101/gad.855601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekelsky, J. J., Newfeld, S. J., Raftery, L. A., Chartoff, E. H. and Gelbart, W. M. (1995). Genetic characterization and cloning of mothers against dpp, a gene required for decapentaplegic function in Drosophila melanogaster. Genetics 139, 1347-1358. 10.1093/genetics/139.3.1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selleck, M. A. and Stern, C. D. (1991). Fate mapping and cell lineage analysis of Hensen's node in the chick embryo. Development 112, 615-626. 10.1242/dev.112.2.615 [DOI] [PubMed] [Google Scholar]
- Sen, R., Lencer, E., Geiger, E. A., Jones, K., Shaikh, T. H. and Artinger, K. B. (2021). The role of Kabuki Syndrome genes KMT2D and KDM6A in development: analysis in Human sequencing data and compared to mice and zebrafish. bioRxiv 2020.04.03.024646. 10.1101/2020.04.03.024646 [DOI] [Google Scholar]
- Shah, A. N., Davey, C. F., Whitebirch, A. C., Miller, A. C. and Moens, C. B. (2015). Rapid reverse genetic screening using CRISPR in zebrafish. Nat. Methods 12, 535-540. 10.1038/nmeth.3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidi, N. T. (1987). Fanconi anemia, dyskeratosis congenita, and WT syndrome. Am. J. Med. Genet. Suppl. 3, 263-278. 10.1002/ajmg.1320280531 [DOI] [PubMed] [Google Scholar]
- Shefchek, K. A., Harris, N. L., Gargano, M., Matentzoglu, N., Unni, D., Brush, M., Keith, D., Conlin, T., Vasilevsky, N., Zhang, X. A.et al. (2020). The Monarch Initiative in 2019: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res. 48, D704-D715. 10.1093/nar/gkz997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, H. and Wang, Z. (2018). Atypical microdeletion in 22q11 deletion syndrome reveals new candidate causative genes: a case report and literature review. Medicine 97, e9936. 10.1097/MD.0000000000009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Y., Hata, A., Lo, R. S., Massagué, J. and Pavletich, N. P. (1997). A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature 388, 87-93. 10.1038/40431 [DOI] [PubMed] [Google Scholar]
- Shin, M., Nagai, H. and Sheng, G. (2009). Notch mediates Wnt and BMP signals in the early separation of smooth muscle progenitors and blood/endothelial common progenitors. Development 136, 595-603. 10.1242/dev.026906 [DOI] [PubMed] [Google Scholar]
- Shprintzen, R. J. (1994). Velocardiofacial syndrome and DiGeorge sequence [3]. J. Med. Genet. 31, 423-424. 10.1136/jmg.31.5.423-b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubin, N., Tabin, C. and Carroll, S. (2009). Deep homology and the origins of evolutionary novelty. Nature 457, 818-823. 10.1038/nature07891 [DOI] [PubMed] [Google Scholar]
- Silverman, M. E. and Hurst, J. W. (1968). The hand and the heart. Am. J. Cardiol. 22, 718-728. 10.1016/0002-9149(68)90211-7 [DOI] [PubMed] [Google Scholar]
- Simões-Costa, M. S., Vasconcelos, M., Sampaio, A. C., Cravo, R. M., Linhares, V. L., Hochgreb, T., Yan, C. Y. I., Davidson, B. and Xavier-Neto, J. (2005). The evolutionary origin of cardiac chambers. Dev. Biol. 277, 1-15. 10.1016/j.ydbio.2004.09.026 [DOI] [PubMed] [Google Scholar]
- Slagle, C. E., Aoki, T. and Burdine, R. D. (2011). Nodal-dependent mesendoderm specification requires the combinatorial activities of FoxH1 and Eomesodermin. PLoS Genet. 7, e1002072. 10.1371/journal.pgen.1002072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleight, V. A. and Gillis, J. A. (2020). Embryonic origin and serial homology of gill arches and paired fins in the Skate, Leucoraja Erinacea. Elife 9, e60635. 10.7554/eLife.60635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smemo, S., Campos, L. C., Moskowitz, I. P., Krieger, J. E., Pereira, A. C. and Nobrega, M. A. (2012). Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 21, 3255-3263. 10.1093/hmg/dds165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers, G. F. (1960). Pharmacological properties of thalidomide (α-phthalimido glutarimide), a new sedative hypnotic drug. Br. J. Pharmacol. Chemother. 15, 111. 10.1111/j.1476-5381.1960.tb01217.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southgate, L., Machado, R. D., Snape, K. M., Primeau, M., Dafou, D., Ruddy, D. M., Branney, P. A., Fisher, M., Lee, G. J., Simpson, M. A.et al. (2011). Gain-of-Function Mutations of ARHGAP31, a Cdc42/Rac1 GTPase Regulator, Cause Syndromic Cutis Aplasia and Limb Anomalies. Am. J. Hum. Genet. 88, 574. 10.1016/j.ajhg.2011.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stainier, D. Y., Lee, R. K. and Fishman, M. C. (1993). Cardiovascular development in the zebrafish. I. Myocardial fate map and heart tube formation. Development 119, 31-40. 10.1242/dev.119.1.31 [DOI] [PubMed] [Google Scholar]
- Stainier, D. Y., Weinstein, B. M., Detrich, H. W., Zon, L. I., Fishman, M. C., Detrich, H. W., 3rd., Zon, L. I. and Fishman, M. C. (1995). Cloche, an early acting zebrafish gene, is required by both the endothelial and hematopoietic lineages. Development 121, 3141-3150. 10.1242/dev.121.10.3141 [DOI] [PubMed] [Google Scholar]
- Stainier, D. Y., Fouquet, B., Chen, J. N., Warren, K. S., Weinstein, B. M., Meiler, S. E., Mohideen, M. A., Neuhauss, S. C., Solnica-Krezel, L., Schier, A. F.et al. (1996). Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development 123, 285-292. 10.1242/dev.123.1.285 [DOI] [PubMed] [Google Scholar]
- Stambolic, V., Ruel, L. and Woodgett, J. R. (1996). Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 6, 1664-1669. 10.1016/S0960-9822(02)70790-2 [DOI] [PubMed] [Google Scholar]
- Stawicki, T. M., Hernande, L., Esterberg, R., Linbo, T., Owens, K. N., Shah, A. N., Thapa, N., Roberts, B., Moens, C. B., Rubel, E. W.et al. (2016). Cilia-associated genes play differing roles in aminoglycoside-induced hair cell death in zebrafish. G3 6, 2225-2235. 10.1534/g3.116.030080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steimle, J. D. and Moskowitz, I. P. (2017). TBX5: a key regulator of heart development. Curr. Top. Dev. Biol. 122, 195-221. 10.1016/bs.ctdb.2016.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz, P. R. H., Aman, A., Kraus, J. E. M. and Technau, U. (2017). Gut-like ectodermal tissue in a sea anemone challenges germ layer homology. Nat. Ecol. Evol. 1, 1535-1542. 10.1038/s41559-017-0285-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson, A., Adams, J. W. and Vaccarezza, M. (2017). The vertebrate heart: an evolutionary perspective. J. Anat. 231, 787-797. 10.1111/joa.12687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolfi, A., Gainous, T. B., Young, J. J., Mori, A., Levine, M. and Christiaen, L. (2010). Early chordate origins of the vertebrate second heart field. Science 329, 565-568. 10.1126/science.1190181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, T., Escalante-Alcalde, D., Bhatt, H., Anver, M., Bhat, N., Nagashima, K., Stewart, C. L. and Burke, B. (1999). Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147, 913-919. 10.1083/jcb.147.5.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedlund, B. and Lescroart, F. (2019). Cardiopharyngeal progenitor specification: multiple roads to the heart and head muscles. Cold Spring Harb. Perspect. Biol. 12, a036731. 10.1101/cshperspect.a036731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinehart, I. T., Schlientz, A. J., Quintanilla, C. A., Mortlock, D. P. and Wellik, D. M. (2013). Hox11 genes are required for regional patterning and integration of muscle, tendon and bone. Development 140, 4574-4582. 10.1242/dev.096693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztal, T. E. and Stainier, Y. R. (2020). Transcriptional adaptation: a mechanism underlying genetic robustness. Development 147, dev186452. 10.1242/dev.186452. [DOI] [PubMed] [Google Scholar]
- Takasato, M. and Little, M. H. (2015). The origin of the mammalian kidney: implications for recreating the kidney in vitro. Development 142, 1937-1947. 10.1242/dev.104802 [DOI] [PubMed] [Google Scholar]
- Takasato, M., Er, P. X., Becroft, M., Vanslambrouck, J. M., Stanley, E. G., Elefanty, A. G. and Little, M. H. (2014). Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat. Cell Biol. 16, 118-126. 10.1038/ncb2894 [DOI] [PubMed] [Google Scholar]
- Takeuchi, J. K., Koshiba-Takeuchi, K., Matsumoto, K., Vogel-Höpker, A., Naitoh-Matsuo, M., Ogura, K., Takahashi, N., Yasuda, K. and Ogura, T. (1999). Tbx5 and Tbx4 genes determine the wing/leg identity of limb buds. Nature 398, 810-814. 10.1038/19762 [DOI] [PubMed] [Google Scholar]
- Tamari, I. and Goodman, R. M. (1974). Upper limb-cardiovascular syndromes: a description of two new disorders with a classification. Chest 65, 632-639. 10.1378/chest.65.6.632 [DOI] [PubMed] [Google Scholar]
- Tamura, M., Hosoya, M., Fujita, M., Lida, T., Amano, T., Maeno, A., Kataoka, T., Otsuka, T., Tanaka, S., Tomizawa, S.et al. (2013). Overdosage of Hand2 causes limb and heart defects in the human chromosomal disorder partial trisomy distal 4q. Hum. Mol. Genet. 22, 2471-2481. 10.1093/hmg/ddt099 [DOI] [PubMed] [Google Scholar]
- Tanaka, M. (2013). Molecular and evolutionary basis of limb field specification and limb initiation. Dev. Growth Differ. 55, 149-163. 10.1111/dgd.12017 [DOI] [PubMed] [Google Scholar]
- Tanaka, M. (2016). Developmental mechanism of limb field specification along the anterior–posterior axis during vertebrate evolution. J. Dev. Biol. 4, 18. 10.3390/jdb4020018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassabehji, M. (2003). Williams-Beuren syndrome: a challenge for genotype-phenotype correlations. Hum. Mol. Genet. 12, R229-R237. 10.1093/hmg/ddg299. [DOI] [PubMed] [Google Scholar]
- Temtamy, S. A. (1985). The genetics of hand malformations: updated. Congenit. Anom. 25, 73-92. 10.1111/j.1741-4520.1985.tb00636.x [DOI] [Google Scholar]
- Terashima, A. V., Mudumana, S. P. and Drummond, I. A. (2014). Odd skipped related 1 is a negative feedback regulator of nodal-induced endoderm development. Dev. Dyn. 243, 1571-1580. 10.1002/dvdy.24191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, A. A. and Nguyen, L. T. (2000). Amegakaryocytic thrombocytopenia and radio-ulnar synostosis are associated with HOXA11 mutation. Nat. Genet. 26, 397-398. 10.1038/82511 [DOI] [PubMed] [Google Scholar]
- Thompson, A. A., Woodruff, K., Feig, S. A., Nguyen, L. T. and Carolyn Schanen, N. (2001). Congenital thrombocytopenia and radio-ulnar synostosis: a new familial syndrome. Br. J. Haematol. 113, 866-870. 10.1046/j.1365-2141.2001.02834.x [DOI] [PubMed] [Google Scholar]
- Tischkowitz, M. D. and Hodgson, S. V. (2003). Fanconi anaemia. J. Med. Genet. 40, 1-10. 10.1136/jmg.40.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolkin, T. and Christiaen, L. (2012). Development and evolution of the ascidian cardiogenic mesoderm. Curr. Top. Dev. Biol. 100, 107-142. 10.1016/B978-0-12-387786-4.00011-7 [DOI] [PubMed] [Google Scholar]
- Torres, M., Gómez-Pardo, E., Dressler, G. R. and Gruss, P. (1995). Pax-2 controls multiple steps of urogenital development. Development 121, 4057-4065. 10.1242/dev.121.12.4057 [DOI] [PubMed] [Google Scholar]
- Traver, D., Paw, B. H., Poss, K. D., Penberthy, W. T., Lin, S. and Zon, L. I. (2003). Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat. Immunol. 4, 1238-1246. 10.1038/ni1007 [DOI] [PubMed] [Google Scholar]
- Trubiroha, A., Gillotay, P., Giusti, N., Gacquer, D., Libert, F., Lefort, A., Haerlingen, B., De Deken, X., Opitz, R., Costagliola, S.et al. (2018). A rapid CRISPR/Cas-based mutagenesis assay in zebrafish for identification of genes involved in thyroid morphogenesis and function. Sci. Rep. 8, 5647. 10.1038/s41598-018-24036-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulenko, F. J., Mccauley, D. W., Mackenzie, E. L., Mazan, S., Kuratani, S., Sugahara, F., Kusakabe, R. and Burke, A. C. (2013). Body wall development in lamprey and a new perspective on the origin of vertebrate paired fins. Proc. Natl. Acad. Sci. USA 110, 11899-11904. 10.1073/pnas.1304210110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno, S., Weidinger, G., Osugi, T., Kohn, A. D., Golob, J. L., Pabon, L., Reinecke, H., Moon, R. T. and Murry, C. E. (2007). Biphasic role for Wnt/β-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc. Natl. Acad. Sci. USA 104, 9685-9690. 10.1073/pnas.0702859104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargesson, N. (2015). Thalidomide–induced teratogenesis: history and mechanisms. Birth Defects Res. Part C Embryo Today Rev. 105, 140-156. 10.1002/bdrc.21096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney, G. K., Pei, W., Lafave, M. C., Idol, J., Xu, L., Gallardo, V., Carrington, B., Bishop, K., Jones, M., Li, M.et al. (2015). High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 25, 1030-1042. 10.1101/gr.186379.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen, J. M. A., Diderich, K. E. M., Oudesluijs, G., Mancini, G. M. S., Eggink, A. J., Verkleij-Hagoort, A. C., Groenenberg, I. A. L., Willems, P. J., Du Plessis, F. A., De Man, S. A.et al. (2012). Phenotypic variability of atypical 22q11.2 deletions not including TBX1. Am. J. Med. Genet. Part A 158A, 2412-2420. 10.1002/ajmg.a.35517 [DOI] [PubMed] [Google Scholar]
- Verhoeven, M. C., Haase, C., Christoffels, V. M., Weidinger, G. and Bakkers, J. (2011). Wnt signaling regulates atrioventricular canal formation upstream of BMP and Tbx2. Birth Defects Res. Part A - Clin. Mol. Teratol. 91, 435-440. 10.1002/bdra.20804 [DOI] [PubMed] [Google Scholar]
- Vincentz, J. W., Firulli, B. A., Toolan, K. P., Osterwalder, M., Pennacchio, L. A. and Firulli, A. B. (2021). HAND transcription factors cooperatively specify the aorta and pulmonary trunk. Dev. Biol. 476, 1-10. 10.1016/j.ydbio.2021.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W., Razy-Krajka, F., Siu, E., Ketcham, A. and Christiaen, L. (2013). NK4 antagonizes Tbx1/10 to promote cardiac versus pharyngeal muscle fate in the ascidian second heart field. PLoS Biol. 11, e1001725. 10.1371/journal.pbio.1001725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., Al-Ouran, R., Hu, Y., Kim, S. Y., Wan, Y. W., Wangler, M. F., Yamamoto, S., Chao, H. T., Comjean, A., Mohr, S. E.et al. (2017a). MARRVEL: integration of human and model organism genetic resources to facilitate functional annotation of the human genome. Am. J. Hum. Genet. 100, 843-853. 10.1016/j.ajhg.2017.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W., Niu, X., Jullian, E., Kelly, R. G., Satija, R. and Christiaen, L. (2017b). A single cell transcriptional roadmap for cardiopharyngeal fate diversification. Nat. Cell Biol. 21, 674-686. 10.1038/s41556-019-0336-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warga, R. M. and Nüsslein-Volhard, C. (1999). Origin and development of the zebrafish endoderm. Development 126, 827-838. 10.1242/dev.126.4.827 [DOI] [PubMed] [Google Scholar]
- Warren, M., Wang, W., Spiden, S., Chen-Murchie, D., Tannahill, D., Steel, K. P. and Bradley, A. (2007). A Sall4 mutant mouse model useful for studying the role of Sall4 in early embryonic development and organogenesis. Genesis 45, 51-58. 10.1002/dvg.20264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman, J. S., Keegan, B. R., Roberts, R. W., Poss, K. D. and Yelon, D. (2008). Hoxb5b acts downstream of retinoic acid signaling in the forelimb field to restrict heart field potential in zebrafish. Dev. Cell 15, 923. 10.1016/j.devcel.2008.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherbee, S. D., Niswander, L. A. and Anderson, K. V. (2009). A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum. Mol. Genet. 18, 4565. 10.1093/hmg/ddp422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, C. L. and Chung, W. K. (2020). Genetics and other omics in pediatric pulmonary arterial hypertension. Chest 157, 1287. 10.1016/j.chest.2020.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield, M. F. and Barr, D. G. D. (1976). Cows’ milk allergy in the syndrome of thrombocytopenia with absent radius. Arch. Dis. Child. 51, 337-343. 10.1136/adc.51.5.337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, K., Carson, J. and Lo, C. (2019). Genetics of congenital heart disease. Biomolecules 9, 879. 10.3390/biom9120879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, D. I., Burn, J., Scambler, P. and Goodship, J. (1993). DiGeorge syndrome: part of CATCH 22. J. Med. Genet. 30, 852-856. 10.1136/jmg.30.10.852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingert, R. A. and Davidson, A. J. (2008). The zebrafish pronephros: a model to study nephron segmentation. Kidney Int. 73, 1120-1127. 10.1038/ki.2008.37 [DOI] [PubMed] [Google Scholar]
- Wu, R. S., Lam, I. I., Clay, H., Duong, D. N., Deo, R. C. and Coughlin, S. R. (2018). A rapid method for directed gene knockout for screening in G0 Zebrafish. Dev. Cell 46, 112-125.e4. 10.1016/j.devcel.2018.06.003 [DOI] [PubMed] [Google Scholar]
- Xu, H., Morishima, M., Wylie, J. N., Schwartz, R. J., Bruneau, B. G., Lindsay, E. A. and Baldini, A. (2004). Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development 131, 3217-3227. 10.1242/dev.01174 [DOI] [PubMed] [Google Scholar]
- Yagi, H., Furutani, Y., Hamada, H., Sasaki, T., Asakawa, S., Minoshima, S., Ichida, F., Joo, K., Kimura, M., Imamura, S.et al. (2003). Role of TBX1 in human del22q11.2 syndrome. Lancet 362, 1366-1373. 10.1016/S0140-6736(03)14632-6 [DOI] [PubMed] [Google Scholar]
- Yamagishi, H., Olson, E. N. and Srivastava, D. (2000). The basic helix-loop-helix transcription factor, dHAND, is required for vascular development. J. Clin. Invest. 105, 261-270. 10.1172/JCI8856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi, H., Kodo, K., Maeda, J., Uchida, K., Tsuchihashi, T., Shibata, A., Ishizaki, R., Yamagishi, C. and Srivastava, D. (2016). A history and interaction of outflow progenitor cells implicated in “Takao syndrome”. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology [Internet] (ed. T. Nakanishi, R. R. Markwald, H. S. Baldwin, B. B. Keller, D. Srivastava, H. Yamagishi), pp. 201-209. Tokyo: Springer. [Google Scholar]
- Yamamoto, P. K., De Souza, T. A., Antiorio, A. T. F. B., Zanatto, D. A., Garcia-Gomes, M. S. A., Alexandre-Ribeiro, S. R., Oliveira, N. S., Menck, C. F. M., Bernardi, M. M., Massironi, S. M. G.et al. (2019). Genetic and behavioral characterization of a Kmt2d mouse mutant, a new model for Kabuki Syndrome. Genes. Brain. Behav. 18, e12568. 10.1111/gbb.12568 [DOI] [PubMed] [Google Scholar]
- Yang, M. and Kim, J.-W. (2018). Principles of genetic counseling in the era of next-generation sequencing. Ann. Lab. Med. 38, 291. 10.3343/alm.2018.38.4.291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J., Hu, D., Xia, J., Yang, Y., Ying, B., Hu, J. and Zhou, X. (2000). Three novel TBX5 mutations in Chinese patients with Holt-Oram syndrome. Am. J. Med. Genet. 92, 237-240. [DOI] [PubMed] [Google Scholar]
- Yarboro, M. T., Gopal, S. H., Su, R. L., Morgan, T. M. and Reese, J. (2022). Mouse models of patent ductus arteriosus (PDA) and their relevance for human PDA. Dev. Dyn. 251, 424-443. 10.1002/dvdy.408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara, J. and Garg, V. (2021). Genetics of congenital heart disease: a narrative review of recent advances and clinical implications. Transl. Pediatr. 10, 2366. 10.21037/tp-21-297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelon, D. and Stainier, D. Y. R. (2005). Hand2 regulates epithelial formation during myocardial differentiation. Curr. Biol. 15, 441-446. 10.1016/j.cub.2004.12.083 [DOI] [PubMed] [Google Scholar]
- Yelon, D., Ticho, B., Halpern, M. E., Ruvinsky, I., Ho, R. K., Silver, L. M. and Stainier, D. Y. R. (2000). The bHLH transcription factor hand2 plays parallel roles in zebrafish heart and pectoral fin development. Development 127, 2573-2582. 10.1242/dev.127.12.2573 [DOI] [PubMed] [Google Scholar]
- Yi, L. Q., Newbury-Ecob, R. A., Terrett, J. A., Wilson, D. I., Curtis, A. R. J., Ho Yi, C., Gebuhr, T., Bullen, P. J., Robson, S. C., Strachan, T.et al. (1997). Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 15, 21-29. 10.1038/ng0197-21 [DOI] [PubMed] [Google Scholar]
- Yin, Y., Ji, J., Borné, Y., Wang, Y., Zhao, J., Chen, S. and Tian, W. (2018). Clinical and epidemiological features of Heart-Hand Syndrome: a hospital-based study in China. Sci. Rep. 8, 8469. 10.1038/s41598-018-26727-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, Y., Ji, J., Zhao, J., Chen, S. and Tian, W. (2020). Clinical and epidemiological features of heart-hand syndrome, an updated analysis in China. BMC Musculoskelet. Disord. 21, 777. 10.1186/s12891-019-3017-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong, L. W., Lu, T.-M., Tung, C.-H., Chiou, R.-J., Li, K.-L. and Yu, J.-K. (2021). Somite compartments in amphioxus and its implications on the evolution of the vertebrate skeletal tissues. Front. Cell Dev. Biol. 9, 1010. 10.3389/fcell.2021.607057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost, C., Torres, M., Miller, J. R., Huang, E., Kimelman, D. and Moon, R. T. (1996). The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 10, 1443-1454. 10.1101/gad.10.12.1443 [DOI] [PubMed] [Google Scholar]
- Zhang, Y., Feng, X. H., Wu, R. Y. and Derynck, R. (1996). Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature 383, 168-172. 10.1038/383168a0 [DOI] [PubMed] [Google Scholar]
- Zhang, Y., Ear, J., Yang, Z., Morimoto, K., Zhang, B. and Lin, S. (2014). Defects of protein production in erythroid cells revealed in a zebrafish Diamond-Blackfan anemia model for mutation in RPS19. Cell Death Dis. 5, e1352. 10.1038/cddis.2014.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X., Sirbu, I. O., Mic, F. A., Molotkova, N., Molotkov, A., Kumar, S. and Duester, G. (2009). Retinoic acid promotes limb induction through effects on body axis extension but is unnecessary for limb patterning. Curr. Biol. 19, 1050-1057. 10.1016/j.cub.2009.04.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X., Sasaki, H., Lowe, L., Hogan, B. L. M. and Kuehn, M. R. (1993). Nodal is a novel TGF-β-like gene expressed in the mouse node during gastrulation. Nature 361, 543-547. 10.1038/361543a0 [DOI] [PubMed] [Google Scholar]
- Zhou, Y. Q., Zhu, Y., Bishop, J., Davidson, L., Henkelman, R. M., Bruneau, B. G. and Foster, F. S. (2005). Abnormal cardiac inflow patterns during postnatal development in a mouse model of Holt-Oram syndrome. Am. J. Physiol. - Hear. Circ. Physiol. 289, 992-1001. 10.1152/ajpheart.00027.2005 [DOI] [PubMed] [Google Scholar]
- Zhou, B., Ma, Q., Rajagopal, S., Wu, S. M., Domian, I., Rivera-Feliciano, J., Jiang, D., Von Gise, A., Ikeda, S., Chien, K. R.et al. (2008). Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454, 109-113. 10.1038/nature07060 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.