

Background and Aims:
HDV infection leads to the most aggressive form of human viral hepatitis for which there is no FDA-approved therapy. PEG IFN-lambda-1a (Lambda) has previously demonstrated a good tolerability profile in HBV and HCV patients compared to PEG IFN-alfa. The goal of Phase 2 LIMT-1 trial was to evaluate the safety and efficacy of Lambda monotherapy in patients with HDV.
Approach and Results:
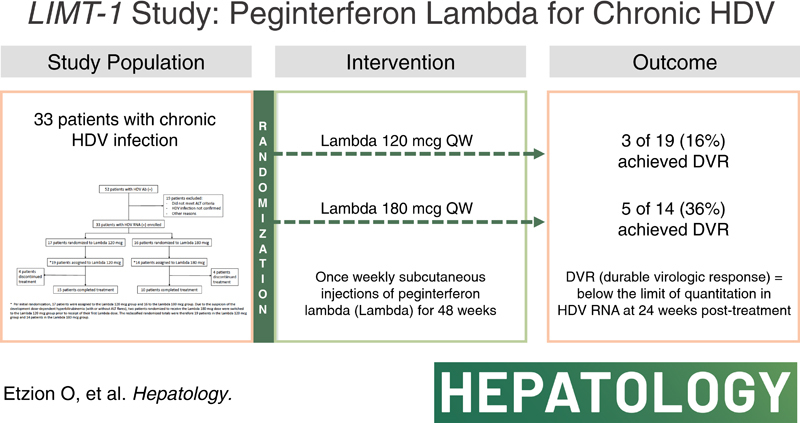
An open-label study of Lambda 120 or 180 mcg, administered once weekly by subcutaneous injections for 48 weeks, followed by 24 weeks of posttreatment follow-up. Thirty-three patients were allocated to Lambda 180 mcg (n=14) or 120 mcg (n=19). Baseline mean values: HDV RNA 4.1 log10 IU/mL (SD±1.4); ALT 106 IU/L (35–364); and bilirubin 0.5 mg/dL (0.2–1.2). Intention-to-treat rates of virologic response to Lambda 180 mcg and 120 mcg, 24 weeks following treatment cessation were 5 of 14(36%) and 3 of 19 (16%), respectively. The posttreatment response rate of 50% was seen in low BL viral load (≤4 log10) on 180 mcg. Common on-treatment adverse events included flu-like symptoms and elevated transaminase levels. Eight (24%) cases of hyperbilirubinemia with or without liver enzyme elevation, leading to drug discontinuation, were mainly observed in the Pakistani cohort. The clinical course was uneventful, and all responded favorably to dose reduction or discontinuation.
Conclusions:
Treatment with Lambda in patients with chronic HDV may result in virologic response during and following treatment cessation. Clinical phase 3 development of Lambda for this rare and serious disease is ongoing.
BACKGROUND
HDV is a satellite RNA virus that requires the surface antigen of HBV for its propagation. Consequently, HDV can only be acquired by individuals already infected with HBV (superinfection) or by those infected with HBV and HDV simultaneously (coinfection).1 Current estimates suggest that ~15–20 million people are chronically infected with HDV worldwide, with prevalence rates varying markedly between different countries and geographic regions.2,3
HDV infection is the most severe form of chronic viral hepatitis.4 At diagnosis, patients with chronic HDV tend to present at more advanced stages of liver fibrosis.5 Furthermore, progression to cirrhosis and HCC in chronic HDV occurs at rates that are 5 times higher than in HBV monoinfection. As of yet, there is no FDA-approved treatment for this severe disease, which presents an urgent unmet medical need. Peginterferon alfa (Alfa), a type I interferon, has been explored in small single-arm studies6–8 and in 2 randomized controlled clinical trials9,10 in HDV. In the HIDIT I study, the administration of Alfa 180 mcg for 48 weeks with or without adefovir led to undetectable HDV levels in 23% of patients at the end of treatment (EOT), while a durable response at 24 weeks after the treatment was achieved in 28%. In the HIDIT II study, treatment with Alfa 180 mcg was extended to 96 weeks and combined with either tenofovir or a placebo. HDV negativity at EOT was achieved in 40% of patients, but only 27% maintained HDV negative status at the end of the follow-up period. Importantly, in both studies, treatment with Alfa was associated with considerable rates of constitutional, flu-like, hematological, and psychiatric adverse events (AE) due to wide type I receptor distribution throughout the body.
Interferon Lambda is a first-in-class type III interferon. It shares a similar downstream signaling pathway and the capacity for broad induction of antiviral activity as type I interferons, but unlike interferon alfa type I receptors, type III receptors for interferon lambda are mainly expressed on the surface of epithelial cells of the gastrointestinal and respiratory tract. Consequently, it is expected that the exogenous administration of interferon lambda will be better tolerated than interferon alfa. Indeed, to date, the use of Lambda, a pegylated form of interferon lambda-1a, has been explored in over 3000 patients in HBV and HCV clinical trials, showing less frequent and less severe of the typical side effects observed with Alfa.11–14
Phase 2 LIMT-1 is a proof-of-concept study and the first demonstration of the safety and efficacy of Peginterferon Lambda (Lambda) treatment in patients infected with chronic HDV.
METHODS
Study design and participants
The LIMT-1 study was a Phase 2, open-label, 2 arm, controlled trial conducted at 4 hospitals in Pakistan, Israel, and New Zealand. Major inclusion criteria included age 18 or older, detectable and quantifiable HDV RNA by qPCR at study entry, ALT above the upper limit of normal (ULN) and below 10 times the ULN, and the presence of compensated liver disease. Patients were excluded from the study if they had other causes for chronic liver disease (viral and nonviral), were treated with interferon-based therapy in the 12 months before study enrollment, or had a contraindication for treatment with interferon. Full eligibility criteria are provided in the synopsis of the study protocol (Supplementary Material http://links.lww.com/HEP/D188).
The study protocol was approved by the institutional review boards of the participating hospitals and was conducted in compliance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulatory requirements. All patients provided written informed consent before the enrolment. This study is registered with ClinicalTrials.gov, number NCT02765802.
Patient allocation
Patients were assigned 1:1 by the interactive web response system to receive either 120 or 180 mcg Lambda as a subcutaneous (SC) weekly injection.
Procedures
A full description of the study-related procedures is provided in Appendix 1 of the study protocol (Supplementary Material http://links.lww.com/HEP/D188).
Briefly, at baseline, all patients not previously treated with nucleos(t)ide analogues (NUCs) were initiated and maintained throughout the treatment and follow-up phases of the study, with either entecavir 0.5 mg/day or tenofovir 245 or 300 mg/day. Lambda 120 or 180 mcg was administered once weekly for 48 weeks, followed by a 24-week follow-up period. Per protocol, dose reductions of Lambda were allowed based on the prespecified dose adjustment rules. Patients’ clinical and laboratory assessments were done at baseline, week 1, week 2, week 4, and every 4 weeks thereafter, according to the clinical study protocol. Clinical assessment included a comprehensive physical examination at baseline and a brief, directed physical examination performed on all subsequent study visits. A 12-lead electrocardiogram was performed at screening, baseline, and weeks 4, 12, and 48. Retinal examinations were performed at screening unless the patient had a documented record of a retinal examination within 1 year before the screening and at week 48. Abdominal imaging (ultrasound, MRI, or CT) was performed at the screening in patients who did not have documentation of one of the above tests obtained≤6 months before screening.
Liver stiffness was evaluated by Fibroscan® (Echosense, Paris, France) at baseline, week 48, and week 72. Laboratory assessments included complete blood counts (CBC), routine chemistries, liver function tests, thyroid panels, coagulation studies, and urinalysis. For women of childbearing potential, a blood sample for serum hCG test was collected at the screening, and urine hCG testing was performed at all subsequent visits except for week 1.
Virologic studies
Viral serologic studies, including HBsAg, HBeAg, anti-HBe, and anti-HBs, were determined by the microparticle enzyme immunoassay method (Abbott Laboratories, North Chicago, IL); anti-HDV was determined by an enzyme immunoassay (Abbott Laboratories). HDV RNA was quantified in serum by real-time qPCR using the RoboGene® HDV RNA Quantification Kit (Analytik Jena, Jena, Germany, lower limit of quantitation [LLOQ] of 14 IU/mL, and lower limit of detection [LLOD] of 6 IU/mL). Serum HBV DNA quantification was performed by the Cobas TaqMan® HBV test (Roche Molecular Systems, Inc, Mannheim, Germany) and HBsAg quantification by the Architect HBsAg assay (Abbott Diagnostics, Germany).
Definitions of viral kinetic patterns
Viral decline patterns were categorized into groups based on the number of identified slopes (or phases) calculated by linear regression. A phase was defined as a 2-fold change in viral load between 2 consecutive time points (either decrease or increase). HDV RNA breakthrough (under therapy) was defined as a 2-fold increase from nadir viral load that was sustained for at least 2 consecutive time points.
Safety assessments
Safety was assessed by the investigators throughout the study based on physical examination, patient-reported outcomes diary, and routine review of laboratory and ancillary test results. All adverse events (AEs) were recorded and graded according to the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03, with modifications for leukocytes, platelets, prothrombin time, INR, ALT, AST, and bilirubin. Adherence with the prescribed regimen for Lambda and NUC was assessed using diary information.
DILISYM® model
DILIsym Services, Inc. (Research Triangle Park, NC) is focused on providing services with the DILIsym modeling system, a computational model of drug induced liver injury.15,16 The model estimates the degrees of hepatocyte loss associated with the drug of interest-based mainly on patterns of ALT and total bilirubin elevations. Modeling involves an initial performance of simulations in the baseline human to assess the magnitude of hepatocyte loss related to observed ALT or bilirubin elevations. Next, a human population sample (SimPops) is employed to determine the variability in the estimated hepatocyte loss. Subsequently, the estimated hepatocyte loss inferred from ALT elevations or bilirubin elevations are compared to assess the source of bilirubin elevation, which may result from hepatocyte loss and/or impaired function of hepatic enzyme/transporters involved in the bilirubin disposition.
Outcomes
The primary efficacy end points of the study were the change from the baseline in HDV viral load at week 48 (EOT) and week 72 (end of follow-up, EOFU). The secondary end points included the proportion of patients with HDV RNA below the limit of quantitation (BLQ) at EOT, week 12 after the treatment and EOFU, and changes from the baseline in HBV viral load, quantitative HBsAg (qHBsAg) levels, and Fibroscan® results at EOT and EOFU. Primary safety end points included treatment-emergent AEs, severe adverse events (SAEs), and the AEs leading to dose reduction or early discontinuation of the study drug.
Statistical analysis
Detailed information on the procedures used for the statistical analysis is provided in section 8 of the study protocol (Supplementary Material, http://links.lww.com/HEP/D188). Briefly, clinical and laboratory data were summarized using descriptive statistics (mean, SD, median, and range) for actual and change-from-baseline values at each timepoint. A sample size of 40 was chosen to allow the assessment of the safety, tolerability, and efficacy of Lambda at 120 versus 180 mcg/week. The sample size was not formally calculated using power analysis. Randomized (Intention-to-Treat [ITT]) Population consisted of all patients who were enrolled, randomized, and assigned to a treatment group using the IWRS. Patients were classified according to the randomized treatment group. An exception to this classification were 2 patients who were randomized to Lambda 180 mcg but were moved to the Lambda 120 mcg group before receiving their first dose of the study drug; these patients were included in the Lambda 120 mcg treatment group. Baseline and efficacy analyses were performed according to the ITT population.
Modified Intention-to-Treat (MITT) Population was defined as those patients in the randomized population who received at least 80% of the total study drug dose throughout the entire 48-week treatment period and for whom HDV viral load data are available for the day 1 (baseline) and end-of-treatment (week 48) study visits. Patients who received less than 80% of the dose on any given treatment day (eg, dose missed, interrupted, or reduced to dose reduction 1 [DR1] or dose reduction 2 [DR2]) were excluded. Sensitivity analyses of efficacy and safety were performed in patients in the MITT population.
The safety population consisted of all patients who received at least 1 dose of the study drug. This population was used for the analysis of all safety end points. In each study population, patients were analyzed according to the dose group to which they were randomized. Associations between kinetic pattern categories and baseline levels of HDV RNA, ALT, HBsAg, and HBV DNA, and differences in phase durations and slopes were evaluated with the nonparametric Kruskal-Wallis test. Fisher exact tests were used for the comparison of proportions, to evaluate the associations between the kinetic categories and Lambda dosage and cirrhotic status, and to assess whether the different measures of treatment success were associated with the following: the absence of a breakthrough, HDV below limit of quantification (BLQ) at efficacy end points, and a decrease of more than 2 log in HDV RNA between the baseline and efficacy end points.
RESULTS
Patient characteristics
Between October 19, 2016 and July 12, 2017, a total of 52 patients underwent screening, of whom 33 were enrolled in the study (Figure 1). The original study design aimed to randomize patients 1:1 to 1 of the 2 treatment arms. Per initial randomization, 17 patients were assigned to the Lambda 120 mcg group and 16 to the Lambda 180 mcg group. However, due to an increased frequency of liver-related significant adverse events (hyperbilirubinemia with or without ALT flares) at the Pakistan site, starting April 27, 2017, two patients initially randomized to receive the 180 mcg/wk dose at this site were intentionally switched to the the Lambda 120 mcg group before the receipt of their first Lambda dose. Administration of Lambda 120 mcg in these patients was continued throughout the study (Figure 1).
FIGURE 1.
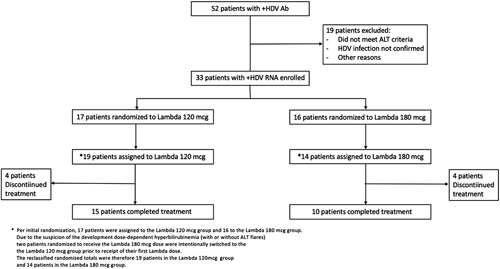
LIMT-1 study flow chart. Abbreviations: HDV, hepatitis delta virus; Lambda, PEG IFN-lambda-1a.
Overall, 19 patients were assigned to receive Lambda 120 mcg (group 1) and 14 patients to receive Lambda 180 mcg (group 2). Demographics and baseline disease characteristics of the patients enrolled are summarized in Table 1. The median age of participants was 36 (range 20–64). Twenty-two (66.7%) were male, and 9 (27%) were diagnosed with cirrhosis at baseline. All patients were infected with HDV genotype 1, with a mean HDV viral load of 4.03 log10 IU/mL (0.5, 7.3) and a mean ALT of 105.6 (35, 364). Most patients (64%) had previous exposure to interferon alfa therapy (either standard interferon or pegylated).
TABLE 1.
Baseline patient characteristics
| Characteristic | Overall (N=33) | 180 mcg (N=14) | 120 mcg (N=19) | p |
|---|---|---|---|---|
| Age, y (range) | 40 (20, 64) | 44.1 (23, 64) | 37.1 (20, 64) | 0.11 |
| Male, n (%) | 22 (66.7) | 15 (78.9) | 7 (50.0) | 0.14 |
| Race, n (%) | — | — | — | 0.19 |
| White | 13 (39.4) | 8 (57.1) | 5 (26.3) | — |
| Black | 1 (3.0) | 0 | 1 (5.3) | — |
| Pacific Islander | 4 (12.1) | 2 (14.3) | 2 (10.5) | — |
| Other | 15 (45.5) | 4 (28.6 | 11 (57.9) | — |
| BMI, kg/m2 (range) | 26.4 (14.4, 36.6) | 25.9 (14.4, 36.5) | 26.7 (20.4, 36.6) | 0.69 |
| Prior Interferon Use (%) | 16 (48.5) | 6 (42.9) | 10 (52.6) | 0.58 |
| Patients with Cirrhosis (%) | 3 (9%) | 2 (6%) | 1 (3%) | 0.71 |
| Liver stiffness, kPa (range) | 11.7 (4.6, 27.4) | 12.7 (4.6, 27.4) | 11.0 (7.1,17.3) | 0.32 |
| HDV RNA, log10 IU/mL (range)a | 4.03 (0.5, 7.3) | 3.86 (0.5, 7.3) | 4.15 (0.5, 6.1) | 0.66 |
| ALT, U/mL (range)b | 105.6 (35, 364) | 94.7 (35, 197) | 113.6 (47, 364) | 0.35 |
| Total Bilirubin, mcmol/L (range)b | 8.6 (3, 20) | 7.8 (4, 14) | 9.2 (3, 20) | 0.27 |
| Albumin, g/L (range)b | 43.8 (37, 52) | 44.2 (41, 49) | 43.5 (37, 52) | 0.47 |
| INR (range)b | 1.2 (1.0, 1.6) | 1.2 (1.1, 1.4) | 1.3 (1.0, 1.6) | 0.49 |
| Platelets, ×109/L (range)b | 183.2 (95, 303) | 168.9 (95, 303) | 193.7 (104, 281) | 0.23 |
RoboGene HDV RNA Quantification Kit 2.0 assay: lower limit of quantitation=14 IU/mL; lower limit of detection=6 IU/mL.
Normal ranges: ALT=10–35 U/mL (female); 10–50 U/mL (male); total bilirubin=1.71–20.5 mcmol/L; albumin=35–55 g/L; INR=≤1.1; platelets=150–400×109/L.
Abbreviations: ALT, alanine aminotransferase; BMI, body mass index; INR, international normalized ratio.
HDV RNA response
During 48 weeks of therapy and throughout the posttreatment follow-up, patients assigned to Lambda 180 mcg/wk demonstrated superior efficacy compared to the 120 mcg/wk dose, reaching a mean change from the baseline in HDV RNA of -2.14 log10 and -1.70 log10 at weeks 48 and 72, respectively. For the Lambda 120 mcg group, the mean change from the baseline in HDV RNA was -1.23 log10 at week 48 and -0.73 log10 at week 72 (Figure 2).
FIGURE 2.
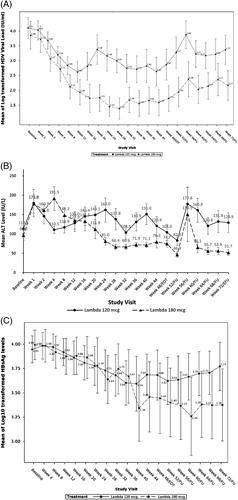
Virological, biochemical, and serological response to treatment, according to treatment group. Mean HDV RNA (A), ALT (B) and HBsAg (C) levels over time, in the two treatment groups. Abbreviations: ALT, alanine aminotransferase; HDV, hepatitis D virus; HBsAg, hepatitis B surface antigen.
At week 48, seven of the 14 patients (50%) on Lambda 180 mcg and 4 of the 19 patients (21%) treated on Lambda 120 mcg reached>2 log10 decline in HDV RNA levels compared to baseline values. HDV RNA BLQ was observed in 36% and 16% of patients in the Lambda 180 mcg and 120 mcg groups, respectively.
At week 72, all but 1 of the patients on Lambda 180 mcg/wk who reached HDV RNA BLQ at week 48, still maintained this status, while an additional patient with detectable HDV RNA at week 48, achieved HDV RNA BLQ (overall, 5 of 14 patients, 36%). Similarly, 1 additional patient in this group reached>2 log10 decline in HDV RNA level at week 72 and another patient that had reached>2 log10 decline at week 48 did not maintain this status (overall, 5 of 14 patients, 36%). In the Lambda 120 mcg/week group, all patients who reached HDV RNA BLQ at week 48 still maintained this status at week 72 (3 of 19, 16%), and 2 of 19 (10%) displayed an HDV RNA viral load decline of>2 log10 compared to baseline at this timepoint (Figure 3).
FIGURE 3.
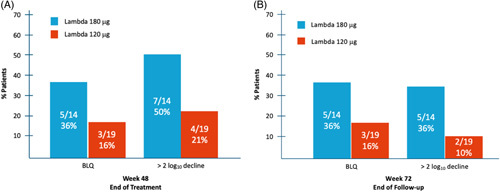
HDV RNA viral load levels at Weeks 48 and 72 by treatment group. The proportion of patients achieving viral load reduction > 2 log compared to baseline, or BLQ at Week 48 (A) and Week 72 (B). Abbreviations: BLQ, below limit of quantification; Lambda, PEG IFN-lambda-1a.
In a univariable analysis of the baseline patient characteristics, none was found to be associated with HDV RNA BLQ or>2 log HDV RNA decline at week 72 compared to baseline values (Supplementary Table 1, http://links.lww.com/HEP/D188).
Response rates at weeks 48 and 72 differed between patients with high (≥4 log10) versus low (<4 log10) baseline viral load (VL) in both treatment arms. In the 180 mcg arm, at week 48, 38% and 33% of patients with high versus low baseline viral loads, respectively, reached HDV RNA levels BLQ. At week 72, the difference between these 2 groups became more prominent, with 50% of patients in the low baseline VL reaching HDV RNA BLQ versus 25% in the high baseline VL meeting this endpoint. Similarly, in the 120 mcg arm, at week 48, 17% (high baseline viral load) and 14% (low baseline viral load) reached HDV RNA BLQ. At week 72, only 8% of patients with high baseline viral load reached HDV RNA BLQ, while 36% of patients in the low baseline viral load group reached this endpoint (Supplementary Table 2, http://links.lww.com/HEP/D188).
ALT normalization
Improvement in ALT levels is considered a surrogate marker for subsiding necroinflammation in the liver. At week 48, 2 of the 19 patients (11%) of Lambda 120 mcg and 2 of the 14 patients (14%) of Lambda 180 mcg group achieved ALT normalization. ALT levels continued to decline in 3 additional patients in each group throughout posttreatment follow-up, eventually leading to ALT normalization of 26% and 36% in Lambda 120 and 180 mcg, respectively, at week 72 (Figure 4).
FIGURE 4.
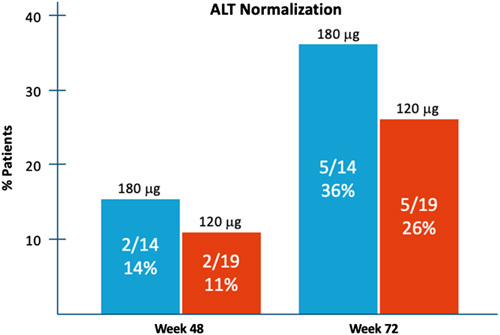
ALT normalization rates at Weeks 48 and 72 by treatment group. Abbreviation: ALT, alanine aminotransferase.
Combined ALT normalization and HDV viral load
To assess the combined biochemical and antiviral effect of Lambda in patients chronically infected with HDV, we conducted a post hoc analysis of the proportion of patients achieving a composite end point of ALT normalization and either HDV RNA decline>2 log10 compared to baseline or BLQ. At week 48, one of the 19 patients (5%) in the 120 mcg and 2 of the 14 patients (14%) in the Lambda 180 mcg reached this end point, while at week 72, response rates increased to 11% and 29% in the Lambda 120 mcg and 180 mcg, respectively (Figure 5).
FIGURE 5.
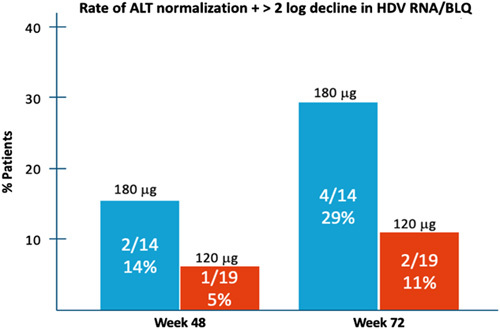
Composite response at Weeks 48 and 72 by treatment group. The rate of combined biochemical (ALT normalization) and virological (>2 log decline in viral load compared to baseline or HDV RNA BLQ) response, according to treatment group, at Weeks 48 and 72. Abbreviations: ALT, alanine aminotransferase; BLQ, below limit of quantification; HDV, hepatitis D virus.
Changes in liver stiffness
Liver stiffness was measured at baseline and at weeks 48 and 72. We did not find statistically significant changes in the liver stiffness measurements between the different time points in both treatment arms and in responders versus nonresponders to the treatment at week 72 (Supplementary Table 3, http://links.lww.com/HEP/D188).
HDV RNA and HBsAg kinetic patterns
Six patients were excluded from the HDV kinetic analysis; 3 due to null response and 3 because the HDV levels were BLQ throughout the treatment. The remaining 27 patients each experienced dramatic drops in the serum HDV RNA level following a pharmacological delay, during which the HDV RNA remained at the pretreatment level. We identified 5 distinct viral kinetic patterns: 6 patients with a monophasic decline (either a single phase of viral decline or 2 viral decline slopes that differed by less than a factor of 2), 3 patients with a biphasic decline (a first phase decline 2-fold larger [or smaller] followed by a second phase decline rate), 5 patients with a flat-partial response (a first phase viral decline followed by an extremely slow [or flat] second phase), 10 patients with a triphasic decline (similar to a flat-partial response but with the resumption of viral decline before EOT), and 3 with a staircase decline (more than 3 distinct phases of viral decline). Supplementary Figure 1, (http://links.lww.com/HEP/D189) displays representative HDV RNA kinetics (along with HBV DNA, HBsAg, and ALT levels) for each of these patterns. During the treatment, 12 patients experienced viral breakthroughs, 2 patients in the monophasic, 2 patients in the biphasic, 1 patient in the flat-partial decline, and 7 patients in the triphasic groups. There were no significant associations between the viral kinetic pattern and the baseline levels of HDV RNA, ALT, HBsAg, or HBV DNA, nor was there any association of kinetic pattern with Lambda dosage or cirrhotic status. In addition, qHBsAg levels did not show statistically significant changes in either treatment group throughout the study compared to baseline, and no association was found between changes in the qHBsAg levels and either HDV RNA or ALT values.
There was a statistically significant association between the viral kinetic pattern category and response to the treatment at EOT and EOFU. The Viral kinetic pattern was associated with HDV RNA BLQ at EOT (p = 0.02), and>2 log10 IU/mL reduction in HDV RNA at EOT (p = 0.02), and EOFU (p = 0.04) compared to baseline. Meaningful treatment response was primarily associated with a monophasic kinetic pattern, with all 6 (100%) patients who exhibited a monophasic pattern of viral decline reaching VL BLQ by EOT and 4 of 6 (66%) maintaining VL BLQ at EOFU. This is in contrast to only 3 of 21 (14%), and 2 of 21(10%) patients with other (nonmonophasic) kinetic patterns achieving VL BLQ at EOT and EOFU, respectively.
Safety
Overall, 159 AEs were recorded in over 1300 weeks of the Lambda therapy. The majority of AEs were considered possibly related or related to the treatment, with most being resolved without additional treatment. The severity of AEs was classified as grade 1 in 88 (55%) cases, grade 2 in 51 (32%) cases, grade 3 in 17 (11%) cases, and grade 4 in 3 (2%) cases. The most common system organ classes with AE severity of ≥grade 2 were related to the gastrointestinal system or to investigations of the liver enzyme and/or bilirubin elevations (Supplementary Table 4, http://links.lww.com/HEP/D188). The most common (≥30%) AEs by the preferred term following the Lambda administration were headache (21 [63.6%]), pyrexia, and arthralgia (15 [45.5%]), dyspepsia (13 [39.4%]), myalgia (12 [36.4%]), fatigue (11 [33.3%]), and diarrhea (10 [30.3%]) (Supplementary Table 5, http://links.lww.com/HEP/D188).
AEs of special interest (ie, those historically observed with standard interferon or peginterferon alfa-2a or 2b treatment) were generally mild in nature with Lambda treatment. The incidence of AEs reported by the patients in the Lambda 180 mcg group was higher for constitutional (fatigue, asthenia) and neurological (headache, dizziness) AEs, but otherwise comparable for flu-like, musculoskeletal, and psychiatric AEs when compared with the Lambda 120 mcg group.
Throughout the treatment phase of the study, there was only 1 case of mild depression (grade 1) which did not require medication. Median values of hemoglobin, neutrophils, and platelets did not show significant changes in both treatment arms throughout the study, with only one case of neutropenia (grade 4), which resolved within 1 week of dose reduction. No events of thrombocytopenia were recorded, and there was no use of hematopoietic growth factors (Table 2 and Supplementary Tables 4 and 5, http://links.lww.com/HEP/D188, and Supplementary Figure 2, http://links.lww.com/HEP/D189).
TABLE 2.
Treatment emergent adverse events of special interest
| Number of patients experiencing adverse event | |||
|---|---|---|---|
| Classification and preferred term | 120 mcg (n=19), n (%) | 180 mcg (n=14), n (%) | Total (N=33), n (%) |
| Flu-like | 13 (68.4) | 9 (64.3) | 22 (66.7) |
| Pyrexia | 8 (42.1) | 7 (50.0) | 15 (45.5) |
| Chills | 5 (26.3) | 3 (21.4) | 8 (24.2) |
| Arthralgia | 8 (42.1) | 7 (50.0) | 15 (45.5) |
| Myalgia | 5 (26.3) | 7 (50.0) | 12 (36.4) |
| Musculoskeletal | 10 (52.6) | 10 (71.4) | 20 (60.6) |
| Arthralgia | 8 (42.1) | 7 (50.0) | 15 (45.5) |
| Myalgia | 5 (26.3) | 7 (50.0) | 12 (36.4) |
| Back pain | 6 (31.6) | 3 (21.4) | 9 (27.3) |
| Constitutional | 4 (21.1) | 9 (64.3) | 13 (39.4) |
| Fatigue | 3 (15.8) | 8 (57.1) | 11 (33.3) |
| Asthenia | 1 (5.3) | 3 (21.4) | 4 (12.1) |
| Neurological | 9 (47.4) | 12 (85.7) | 21 (63.6) |
| Headache | 9 (47.4) | 12 (85.7) | 21 (63.6) |
| Dizziness | 1 (5.3) | 4 (28.6) | 5 (15.2) |
| Psychiatric | 0 | 1 (7.1) | 1 (3.0) |
| Depression | 0 | 1 (7.1) | 1 (3.0) |
| Irritability | 0 | 0 | 0 |
| Insomnia | 0 | 0 | 0 |
A total of 17 (51.5%) patients experienced a significant AE (ie, an event leading to dose interruption, dose reduction, and/or study drug discontinuation). There were 13 cases of dose reduction, 6 in the Lambda 120 mcg and 7 in the Lambda 180 mcg groups, 2 cases of dose interruption (1 in each group), and 8 cases of drug discontinuation, 4 in each treatment group. No statistically significant differences were seen in the frequency of dose reductions, interruptions, or discontinuations between the 2 groups (Supplementary Table 6, http://links.lww.com/HEP/D188). The criteria for dose interruption/reduction or drug discontinuation in response to the development of significant AE are described in the Supplementary material (Table 4 of the study protocol, http://links.lww.com/HEP/D188).
Of note, the event rate for significant adverse events was approximately the same for the 2 Lambda doses employed in this study: (Lambda 120 mcg [n=10; 52.3%]; and Lambda 180 mcg [n=8; 57.1%]). Similarly, the rate of events requiring study drug discontinuation was also comparable across the 2 treatment groups: (Lambda 120 mcg [n=5; 26.3%]; and Lambda 180 mcg [n=3; 21.4%]).
All treatment discontinuations occurred due to the development of hepatobiliary laboratory abnormalities (hyperbilirubinemia with or without liver enzyme elevations). Age, gender, and BMI distribution did not differ between patients who discontinued treatment prematurely compared to the rest of the cohort. A higher rate of hyperbilirubinemia mandating treatment discontinuation was observed in the group of patients recruited in Karachi, Pakistan (5/15 [33%], compared to all other sites combined [3/18, 17%)]. Four additional patients developed grade 1 or 2 hyperbilirubinemia that was managed with a dose reduction of Lambda. The development of hyperbilirubinemia was not associated with high baseline liver stiffness (>13 kPa) or low (<160) baseline platelet count (p = 0.240 and p = 0.701, respectively). In all the patients experiencing hyperbilirubinemia, it was not associated with symptoms, such as weakness, nausea, vomiting, or pruritus, or with the development of clinical or laboratory signs of liver decompensation. Following the discontinuation of Lambda, bilirubin levels gradually subsided, returning to baseline values by the end of the study in all but 1 patient (See supplementary Table 7, [http://links.lww.com/HEP/D188] for detailed description of the baseline, peak, and follow-up values of biochemical indices of patients who discontinued treatment) Extensive DILIsym® modeling of the transaminase and total bilirubin levels suggested that most patients with hyperbilirubinemia did not suffer more than 25% hepatocyte loss. These results are consistent with a transporter-based mechanism rather than direct hepatotoxicity.
No deaths were reported. Eleven SAEs, all considered related or possibly related to the Lambda treatment, occurred in 7 (21.2%) patients, and all were hepatobiliary in nature. Of the 19 patients in the Lambda 120 mcg group, 5 (26.3%) experienced a total of 8 SAEs. In the Lambda 180 mcg group, 2 of the 14 (14.3%) patients experienced a total of 3 SAEs. With the exception of 1 grade 2 SAE, and 1 grade 3 SAE, these events were grade 1 in severity. All SAEs were considered possibly related or related to treatment with Lambda and were hepatobiliary in nature (Supplementary Table 8, http://links.lww.com/HEP/D188).
DISCUSSION
Our results showed that the treatment with Lambda for 48 weeks is associated with a viral response at EOT and EOFU, in both treatment arms. In patients assigned to Lambda180 mcg, HDV RNA levels were significantly reduced, with 36% of patients showing HDV RNA undetectable or BLQ at the end of treatment and 50% reaching more than a 2 log10 reduction in the viral load compared to baseline values. More importantly, the effect of Lambda 180 mcg was durable 24 weeks after stopping treatment, with 36% of patients reaching HDV RNA BLQ at the end of 24 weeks of follow-up. Viral responses at EOT and EOFU were also observed in patients assigned to the Lambda 120 mcg, although to a lesser extent. The durable effect of Lambda (both 180 mcg and 120 mcg) was even more prominent in patients with low versus high baseline viral load, although this observation is based on a small number of patients in each group.
As of yet, no FDA-approved therapy is available for chronic HDV. Bulevirtide, which recently received conditional authorization from the European Medicinal Agency,17 is still being evaluated for long-term efficacy in Phase 3 clinical trials. Alfa, for the treatment of chronic HDV, has been modestly used for chronic HDV over the past several decades due to its poor tolerability and limited efficacy. Lambda, a first-in-class type III interferon, has a limited receptor distribution throughout the body and has demonstrated better tolerability versus Alfa. Indeed, in our study, Lambda resulted in only 159 AEs after administration for over 1300 weeks. This amounts to a very low event rate of 6 per 48-week course of therapy. Most AEs were mild to moderate in nature and did not compromise patients’ quality of life or occupational capacity. Unlike treatment with Alfa, which is associated with bone marrow suppression and psychiatric manifestations in a substantial proportion of treated patients, in the current study, only 1 patient developed significant neutropenia that resolved spontaneously within 1 week of Lambda discontinuation and with no need for hematopoietic growth factor administration. In addition, a single patient-reported symptoms suggestive of mild-grade depression that resolved during the study, with no need for drug intervention. As a comparison, in the HIDIT I and II trials, a significant proportion of patients experienced psychiatric and hematologic side effects. In HIDIT I, psychiatric AEs and neutropenia or thrombocytopenia were reported in 12% and 20% of patients treated with Alfa, respectively. In HIDIT II, psychiatric disorders and neutropenia or thrombocytopenia were reported in 24% and 28% of patients treated with Alfa, respectively.9,10
In addition, two-thirds of patients in LIMT-1 were previously treated with Alfa, where they were unable to complete 48 weeks of Alfa treatment due to the debilitating side effects. All these patients tolerated Lambda well and completed 48 weeks of Lambda treatment, aside from those who terminated therapy as required by per-protocol stopping rules due to significant hyperbilirubinemia. Significant hyperbilirubinemia occurred in 8 of the 33 patients. Most of these events (63%) occurred in patients from Pakistan. The rate of significant hyperbilirubinemia in patients from the other sites was comparable to those reported in the previous studies of Lambda in HBV and HCV patients. To better understand the mechanism underlying this phenomenon, we employed the DILISYM® platform, a computational model used for the investigation of suspected drug-related toxicity in clinical trials. The DILISYM analysis suggested that a transporter-based mechanism is the most plausible explanation for the development of hyperbilirubinemia due to Lambda. These findings were also consistent with the observation that none of the patients experiencing the total bilirubin elevations manifested any clinical signs or symptoms and/or laboratory abnormalities indicative of hepatic decompensation. Further, molecular and genetic studies are planned to investigate this hypothesis.
Analysis of the HDV viral kinetics under the Lambda treatment identified 5 distinct patterns of viral decline, with the monophasic pattern showing a strong association with durable viral response. Interestingly, our previous analysis of viral kinetics in a cohort of 10 patients with chronic HDV infection treated with Alfa for 28 weeks found only 2 response patterns (biphasic and flat-partial).18 Another notable difference between the kinetic patterns observed in patients treated with Alfa versus Lambda was in the association of HDV viral decline with qHBsAg levels. While HBsAg kinetics paralleled the second phase slope of HDV RNA in all 10 patients treated with Alfa, no association between HDV RNA and HBsAg kinetics was observed with Lambda. Further experimental and theoretical efforts are needed to determine whether the differences in kinetics reflect differences in the mechanisms of action of Alfa and Lambda in the treatment of HDV.
This study had several limitations, including the relatively small cohort size and the fact that patients were not evenly randomized between the 2 treatment groups. Nevertheless, results from this study encourage further investigation of Lambda as a potential therapeutic for chronic HDV infection. Indeed, the Phase 3 LIMT-2 randomized, open-label multicenter trial assessing the safety and efficacy of Lambda 180 mcg for 48 weeks in patients with chronic HDV infection was initiated in December 2022 and is enrolling and dosing.
After a long-awaited period, several novel drugs with distinct mechanisms of action for chronic HDV are currently being explored in the ongoing late-stage clinical trials. These include bulevirtide (entry inhibitor), lonafarnib (prenylation inhibitor), and Lambda (immunomodulator). While these drugs show promise, it is not yet clear whether the cure for HDV can be achieved with the monotherapy of these agents. It is reasonable to assume that combinations may be needed to maximize clinical benefits. Results from the Phase 2 LIMT-1 study are promising as Lambda is shown to provide comparable efficacy to Alfa but with improved tolerability and may therefore substitute Alfa as the immunomodulator of choice in combination studies for HDV therapy. Recently, the results of the combination of Lambda with lonafarnib 50 mg bid boosted with ritonavir 100 mg bid for 24 weeks were reported in the Phase 2 LIFT-1 trial. Results from this study show that 77% of patients achieved>2 log10 reduction in HDV RNA, and 50% were BLQ or virus undetectable at the end of treatment compared to baseline, with most having only mild to moderate side effects. Twenty-four weeks after treatment, 23% of patients demonstrated a durable virologic response, and 50% of patients achieved a>2-point reduction in histological activity index (HAI).19 A follow-on study of Lambda in combination with lonafarnib boosted with ritonavir for 48 weeks of treatment is planned to initiate this year. Findings from these studies, together with our results further suggest that Lambda may be utilized for the treatment of chronic HDV infection either as a standalone therapy or in combination with other drugs with different mechanisms of action.
In conclusion, the LIMT-1 study shows that the treatment of chronic HDV with Lambda for 48 weeks leads to a durable virologic response and is associated with a favorable side effect profile. Further studies are needed to explore the utility of this drug as monotherapy or in combination with other therapeutics for the treatment of chronic HDV.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Yang K and Longo D of DILISym for their contribution to the study, and Gorstein E for statistical analysis.
CONFLICTS OF INTEREST
Ohad Etzion consults for Eiger Biopharmaceuticals. Edward J. Gane advises Assembly, Arbutus, Aligos, Roche and Vir. He is on the speakers’ bureau for Abbvie. Ingrid Choong owns stock in, is employed by, and owns intellectual property rights in Eiger Biopharmaceuticals. Jeffrey Glenn owns stock in, is employed by, and owns intellectual property rights in Eiger Biopharmaceuticals. The remaining authors have no conflicts to report.
Footnotes
Funding Information NIH, Grant/Award Number: R01AI144112 and R01AI146917.
Abbreviations: AEs, adverse events; Alpha, peginterferon-alpha; BLQ, below limit of quantification; CBC, complete blood count; DR1, dose reduction 1; DR2, dose reduction 2; EOFU, end of follow-up; EOT, end of treatment; HDV, hepatitis delta virus; ITT, intention-to-treat; IWRS, interactive web response system; Lambda, pEG IFN-lambda-1a; LLOD, lower limit of detection; LLOQ, lower limit of quantification; MITT, modified intention-to-treat; NUCs, nucleos(t)ide analogues; qHBsAg, quantitative HBsAg levels; SAEs, severe adverse events; SC, subcutaneous; VL, viral load.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal's website, www.hepjournal.com
Contributor Information
Ohad Etzion, Email: ohadet34@yahoo.com.
Saeed Hamid, Email: saeed.hamid@aku.edu.
Yoav Lurie, Email: yoavtalitami@gmail.com.
Edward J. Gane, Email: EdGane@adhb.govt.nz.
David Yardeni, Email: yardeda@gmail.com.
Sarah Duehren, Email: sduehren@luc.edu.
Nimrah Bader, Email: n.bader@aku.edu.
Anat Nevo-Shor, Email: anatnev@gmail.com.
Saleh Muhammad Channa, Email: sm.chann2@aku.edu.
Scott J. Cotler, Email: scotler@luc.edu.
Minaz Mawani, Email: m.mawani@aku.edu.
Om Parkash, Email: drom73@yahoo.com.
Harel Dahari, Email: hdahari@luc.edu.
Ingrid Choong, Email: ichoong@eigerbio.com.
Jeffrey S. Glenn, Email: jsglenn@stanford.edu.
REFERENCES
- 1.Bonino F, Heermann KH, Rizzetto M, Gerlich WH. Hepatitis delta virus: protein composition of delta antigen and its hepatitis B virus-derived envelope. J Virol. 1986;58:945–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lempp FA, Ni Y, Urban S. Hepatitis delta virus: insights into a peculiar pathogen and novel treatment options. Nat Rev Gastroenterol Hepatol. 2016;13:580–589. [DOI] [PubMed] [Google Scholar]
- 3.Wranke A, Pinheiro Borzacov LM, Parana R, Lobato C, Hamid S, Ceausu E, et al. Clinical and virological heterogeneity of hepatitis delta in different regions world-wide: The Hepatitis Delta International Network (HDIN). Liver Int. 2018;38:842–850. [DOI] [PubMed] [Google Scholar]
- 4.Yurdaydin C, Idilman R, Bozkaya H, Bozdayi AM. Natural history and treatment of chronic delta hepatitis. J Viral Hepat. 2010;17:749–756. [DOI] [PubMed] [Google Scholar]
- 5.Fattovich G. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. Gut. 2000;46:420–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erhardt A, Gerlich W, Starke C, Wend U, Donner A, Sagir A, et al. Treatment of chronic hepatitis delta with pegylated interferon-α2b. Liver Int. 2006;26:805–810. [DOI] [PubMed] [Google Scholar]
- 7.Castelnau C, Le Gal F, Ripault MP, Gordien E, Martinot-Peignoux M, Boyer N, et al. Efficacy of peginterferon alpha-2b in chronic hepatitis delta: Relevance of quantitative RT-PCR for follow-up. Hepatology. 2006;44:728–735. [DOI] [PubMed] [Google Scholar]
- 8.Niro GA, Ciancio A, Gaeta GB, Smedile A, Marrone A, Olivero A, et al. Pegylated interferon alpha-2b as monotherapy or in combination with ribavirin in chronic hepatitis delta. Hepatology. 2006;44:713–720. [DOI] [PubMed] [Google Scholar]
- 9.Wedemeyer H, Yurdaydìn C, Dalekos GN, Erhardt A, Çakaloğlu Y, Değertekin H, et al. Peginterferon plus Adefovir versus either drug alone for hepatitis delta. N Engl J Med. 2011;364:322–331. [DOI] [PubMed] [Google Scholar]
- 10.Wedemeyer H, Yurdaydin C, Hardtke S, Caruntu FA, Curescu MG, Yalcin K, et al. Peginterferon alfa-2a plus tenofovir disoproxil fumarate for hepatitis D (HIDIT-II): a randomised, placebo controlled, phase 2 trial. Lancet Infect Dis. 2019;19:275–286. [DOI] [PubMed] [Google Scholar]
- 11.Ramos EL. Preclinical and clinical development of pegylated interferon-Lambda 1 in chronic hepatitis C. J Interf Cytokine Res. 2010;30:591–595. [DOI] [PubMed] [Google Scholar]
- 12.Muir AJ, Shiffman ML, Zaman A, Yoffe B, de la Torre A, Flamm S, et al. Phase 1b study of pegylated interferon lambda 1 with or without ribavirin in patients with chronic genotype 1 hepatitis C virus infection. Hepatology. 2010;52:822–832. [DOI] [PubMed] [Google Scholar]
- 13.Muir AJ, Arora S, Everson G, Flisiak R, George J, Ghalib R, et al. A randomized phase 2b study of peginterferon lambda-1a for the treatment of chronic HCV infection. J Hepatol. 2014;61:1238–1246. [DOI] [PubMed] [Google Scholar]
- 14.Chan HLY, Ahn SH, Chang TT, Peng CY, Wong D, Coffin CS, et al. Peginterferon lambda for the treatment of HBeAg-positive chronic hepatitis B: A randomized phase 2b study (LIRA-B). J Hepatol. 2016;64:1011–1019. [DOI] [PubMed] [Google Scholar]
- 15.Howell BA, Yang Y, Kumar R, Woodhead JL, Harrill AH, Clewell HJ, et al. In vitro to in vivo extrapolation and species response comparisons for drug-induced liver injury (DILI) using DILIsymTM: a mechanistic, mathematical model of DILI. J Pharmacokinet Pharmacodyn. 2012;39:527–541. [DOI] [PubMed] [Google Scholar]
- 16.Howell BA, Siler SQ, Watkins PB. Use of a systems model of drug-induced liver injury (DILIsym®) to elucidate the mechanistic differences between acetaminophen and its less-toxic isomer, AMAP, in mice. Toxicol Lett. 2014;226:163–172. [DOI] [PubMed] [Google Scholar]
- 17.Human medicine European public assessment report (EPAR): Hepcludex. Accessed February 15, 2022. https://www.ema.europa.eu/en/medicines/human/EPAR/hepcludex
- 18.Guedj J, Rotman Y, Cotler SJ, Koh C, Schmid P, Albrecht J, et al. Understanding early serum hepatitis D virus and hepatitis B surface antigen kinetics during pegylated interferon-alpha therapy via mathematical modeling. Hepatology. 2014;60:1902–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koh C, et al. A Phase 2 Study of Peginterferon Lambda Lonafarnib and Ritonavir for 24 Weeks: End of Study Results from the LIFT HDV study. The Digital International Liver Congress™. 2020. [Google Scholar]