

Summary
Patients with short telomere syndromes (STS) are predisposed to developing cancer, believed to stem from chromosome instability in neoplastic cells. We evaluated this hypothesis in a large cohort of patients assembled over the last 20 years. We found that the only solid cancers to which patients with STS are predisposed were squamous cell carcinomas of the head and neck, anus or skin. Whole genome sequencing showed no increase in chromosome instability, such as translocations or chromothripsis. Moreover, the cancers acquired mutations in the TERT promoter. The spectrum of cancers in patients with STS was reminiscent of the cancers seen in patients with immunodeficiency. We therefore performed a detailed study of the immune status of patients with STS and found a striking T cell immunodeficiency at the time of cancer diagnosis. A similar immunodeficiency impaired tumor surveillance in mice with short telomeres. We conclude that STS patients’ predisposition to solid cancers is due to T cell exhaustion rather than autonomous defects in the neoplastic cells themselves.
Keywords: telomerase, aging, T cell aging, genome instability, head and neck cancer, senescence, immune aging
eTOC Blurb
Schratz et al. find a surprisingly low incidence of solid cancers in patients with short telomere syndromes except for squamous tumors. Tumors show no genome instability and arise in patients with short telomere-induced T cell immunodeficiency. T cell exhaustion in short telomere mice fails to survey and suppress tumors.
Graphical Abstract
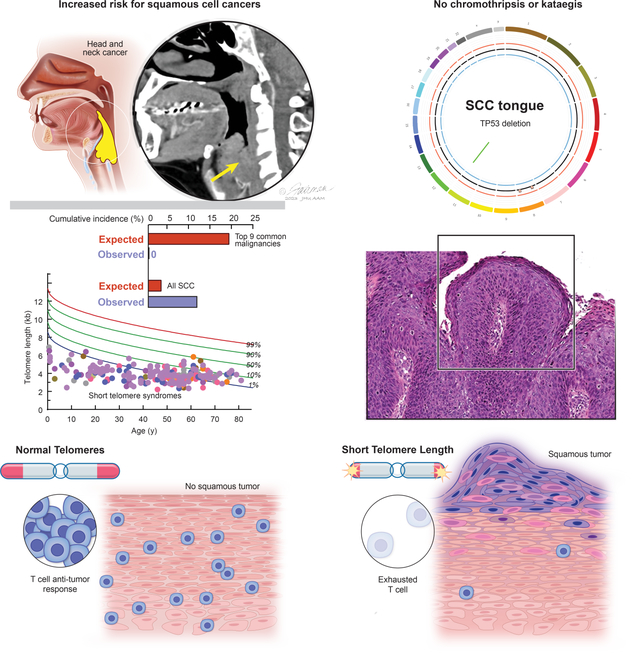
Epithelial cancers comprise nearly 90% of age-related malignancies1, but the biology that drives their development with aging is incompletely understood. Telomere shortening is a well-characterized mechanism of cellular aging. Telomeres protect chromosomes from fusion; their length is genetically determined and it predicts the onset of replicative senescence2. At critical thresholds, short telomere length signals a double strand DNA damage response and a TP53-dependent checkpoint that provokes apoptosis or cellular senescence3–5. This program inhibits tumorigenesis in vivo, and improves cancer-free survival in oncogene- as well as tumor suppressor-mediated animal models of cancer6–9. However, in mice where Tp53 loss was also engineered, short telomeres accelerated carcinogenesis and shifted the spectrum of cancers from sarcomas to epithelial tumors10. The tumors that formed had hallmarks of chromosome instability including non-reciprocal translocations10. In yeast that lack telomerase, chromosomal rearrangement and increased mutation rates have been observed11. Recent cell culture- based models of telomere dysfunction that was induced by telomere deprotection reported kataegis and chromothripsis12,13. These seemingly conflicting results as to whether short telomeres promote or protect against cancer have left open the question of how short telomeres influence human cancer risk and contribute to cancer development in humans with aging.
The short telomere syndromes (STS) are archetypal premature aging syndromes in that they capture a defect acquired universally with aging14. They are the most prevalent of premature aging syndromes because of their high incidence among adults with pulmonary fibrosis15. Loss-of-function mutations in the telomerase reverse transcriptase, TERT, is the most common cause of STS followed by mutations in the telomerase RNA component, TR16,17. Myelodysplastic syndrome and acute myeloid leukemia comprise the most common malignancies in STS; these cancers arise under selective pressures of telomere-mediated stem cell failure. In this context, somatic reversion mutations that offset the germline loss-of-function mutation in telomerase protect against myeloid malignant evolution16,18. Whether solid tumors that arise in the STS recapitulate the adenocarcinoma-predominant malignancy spectrum associated with aging has not been studied. Moreover, fundamentally, how these tumors overcome the inherited telomere maintenance defect to sustain their evolution is not known. Here we report a 20-year experience, analyzing for the first time the global somatic landscape of cancers derived from humans with STS. We uncover in this clinical context an unexpected mechanism where tumor-independent factors related to defective immune surveillance drive cancers that derive from the squamous epithelium.
Results
Solid tumors in the short telomere syndromes have a distinct spectrum from age-related malignancies
To understand the role of short telomeres in human carcinogenesis, we assessed the prevalence of invasive solid tumors among 226 individuals with STS who were recruited from 2003–2022 (Figure 1A and clinical characteristics in Table S1). Median age at enrollment was 50 (range 0.6–81 years, 10,890 lifetime-years) and 58% were male. TERT loss-of-function mutations were most common (44%), followed by TR, RTEL1 and DKC1 (Figure 1A and Table S1). Participants had very short telomeres relative to age-matched healthy controls with 93% falling below the 10th percentile (n=201) and 64% below the 1st percentile (n=138) (Figure 1A). Cumulative incidence of solid cancers increased with age, but was overall low [0.09 by age 70, 95% confidence interval (CI) 0.04–0.14, accounting for competing risks of mortality] and lower than the cumulative incidence of hematologic malignancies (0.18 at the same age, 95% CI 0.11–0.26) (Figure 1B and Table S2). Sixteen invasive solid tumors were diagnosed in 14 individuals and these comprised 40% of all the cancers with the hematologic malignancies being either myelodysplastic syndrome or acute myeloid leukemia (Figure 1B). Comparing the adjusted incidence to the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) data, there was a lower than expected number of the 9 most common cancers associated with mortality in adults (i.e. lung, colorectal, pancreatic, kidney, bladder, uterine, melanoma, thyroid and non-Hodgkin lymphoma1): 19.4 cases expected vs. none documented (Figure 1C). In contrast to age-related solid tumors, which are predominantly of adenocarcinoma histology, nearly all the solid tumors were derived from the squamous epithelium (88%, 14 of 16, Figure 1D). The most common sites were the oral cavity and other sites of the head and neck (44%, n=7), followed by anal squamous cell cancers (SCC) (19%, n=3) and skin SCC (13%, n=2) (Figure 1E). The risk of oral cavity SCC and all SCC was increased [Observed/Expected (O/E) ratio 16.2, 95% CI: 5.3–30.1 and O/E ratio 3.5, 95% CI 1.72–5.46, respectively] (Figure 1C). In examining the clinical features, nearly all the patients who developed solid tumors were male (93%, 13 of 14) (Figure 1E) even though hematologic malignancy diagnoses were evenly distributed among the sexes (Table S2). Males with X-linked DKC1 mutations had the highest risk [Hazard Ratio (HR) 6.93 relative to males with other mutations, 95% CI 1.87–25.68], and females had the lowest risk (HR 0.18, 95% CI 0.02–1.46, Figure 1F–G, Table S2 and S3). The data indicated that STS have a distinct solid tumor spectrum from what is seen with aging and it is largely limited to squamous carcinoma.
Figure 1. Cancer risk in the short telomere syndromes.
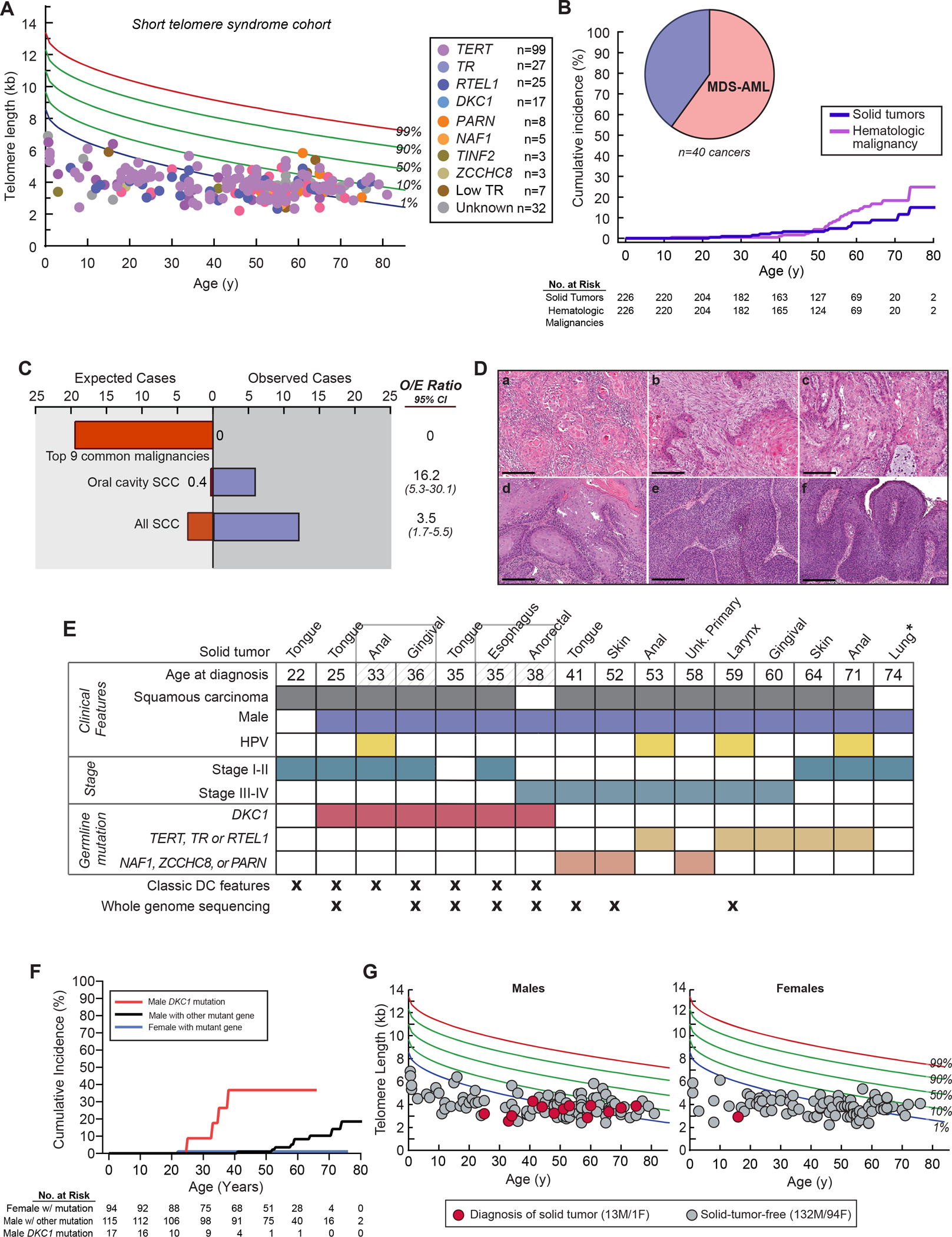
A. Telogram of individuals in the Johns Hopkins Telomere Syndrome Registry with each individual’s germline mutant gene (key) relative to the age-adjusted, clinically validated nomogram (reference 17). Data are shown for 217 of 226 individuals for whom lymphocyte telomere length data were available. B. Cumulative incidence of hematologic malignancies and solid tumors by age accounting for competing risk of death. Pie chart in left upper corner shows distribution of 40 cancers identified in 35 individuals. C. Observed cases of cancer in the Short Telomere Syndrome study participants relative to expected as derived from the Surveillance, Epidemiology, and End Results (SEER) Database and corrected for age and sex. The observed/expected (O/E) ratio is shown to the right. Lung cancer squamous cell cancer (SCC) was excluded from the all SCC comparison. D. Representative hematoxylin and eosin images (100x) of solid tumors: a. primary SCC of tongue, b. tongue SCC with involved local lymph node, c. lip invasive SCC with perineural invasion, d. gingival SCC, e. larynx SCC, and f. anal squamous carcinoma in situ of gluteal cleft which later progressed to anal SCC. Scale bars indicate 250 μm. E. Clinical characteristics of 14 individuals with 16 cancers (2 individuals with two malignancies are noted by brackets). HPV status was by determined by whole genome sequencing, and for anal cancers, was presumed positive given known association. *Denotes a donor-derived lung adenocarcinoma that developed after transplantation for pulmonary fibrosis. F. Cumulative incidence of cancers in male DKC1 mutation carriers relative to males and females with non-DKC1 mutations. G. Prevalence of all solid tumors in males (M) and females (F) in the Johns Hopkins study relative to age at the time of study enrollment and telomere length. Nine individuals who did not have telomere data are not included, but they had no invasive cancer diagnoses (7 M, 2 F). See also Table S1–S3.
Cancers acquire telomere maintenance mechanisms and have no evidence of telomere crisis
To assess how solid tumors overcome the germline telomere defect, we performed paired tumor-normal high pass whole genome sequencing (mean 72x) on 8 cancers (7 SCC and 1 anorectal adenocarcinoma, Figure 2A). Nearly all the tumors carried at least one TP53 loss-of-function alteration (88%, 7 of 8), and 50% had two hits (4 of 8, Figure 2A, Table S4). Most tumors acquired telomere maintenance mechanisms with canonical TERT promoter mutations that upregulate TERT transcription being most common, but there was also amplification of the TERT and TR loci at 5p and 3q, respectively, as well as a POT1 mutation and MYC amplification detected (Figure 2A). Multiple telomere maintenance mechanisms co-existed in some tumors. No tumor had copy number loss or mutations in alternative lengthening of telomere (ALT)-associated genes, ATRX, DAXX or SMARCAL1. Relative to a comparably sequenced TCGA Head and Neck SCC dataset, the prevalence of TP53 alterations and TERT promoter mutations was higher (Figure 2B), but the tumor mutation burden was comparable and low (mean 8.7/Mb, range 1.0–22.6, Figure 2C). The predominant single base substitution signature (SBS) was that of replicative aging (C>T dominant, COSMIC SBS1) which was found in 5 of 8 cancers; it explained 22% of single nucleotide variants (range 13–42%, Figure S1A and S1B). Kataegis has been identified in some cell-based telomere deprotection models12,13, but we could not detect these patterns and the shortest inter-cluster distance identified was 515 kb, longer than the 2 kb inter-cluster threshold characteristic of kataegis (Figure S1C). Additionally, STS oral cavity SCC tumors had few deep focal copy number alterations (mean 26/genome, range 8–64, Table S4), a pattern that was distinct from oral cavity tumors derived from Fanconi anemia patients19. The data indicated that solid tumors arising in STS overcome the short telomere checkpoint through loss-of-function of TP53 while simultaneously acquiring telomere maintenance mechanisms.
Figure 2. Somatic landscape of solid tumors arising in the short telomere syndromes.
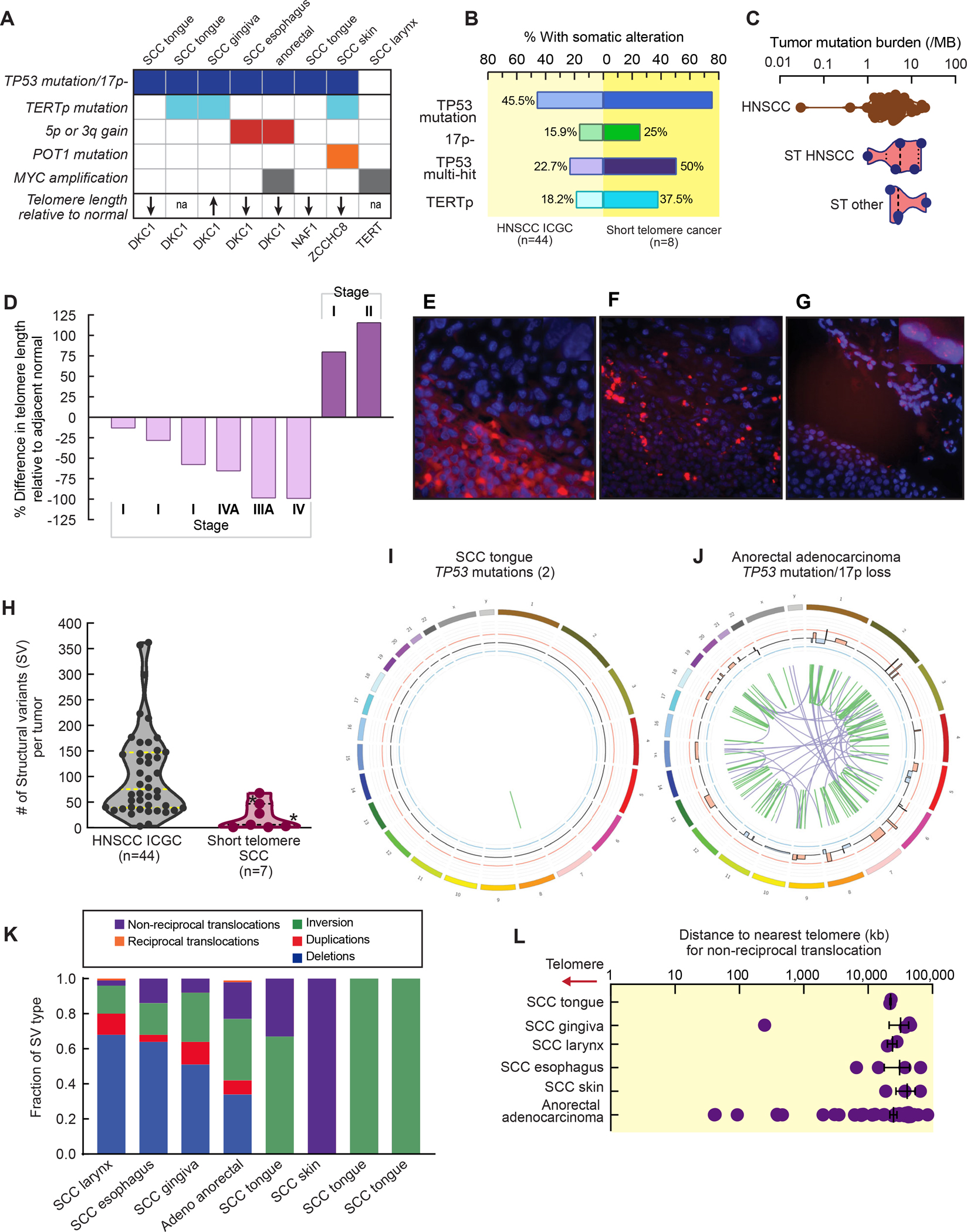
A. TP53 alterations and telomere maintenance mechanisms analyzed by whole genome sequencing. Bottom row shows relative telomere length differences for tumors available for measurement compared to normal as summarized in panel D. B. Somatic alteration frequencies relative to 44 Head and Neck Squamous Cell Carcinoma (HNSCC) from the International Cancer Genome Consortium (ICGC) (n=44 total, median age 57, 52% oral cavity SCC). C. Tumor mutation burden (TMB), calculated as the number of non-synonymous single nucleotide variants and indels per Mb of coding region, is plotted relative to HNSCC ICGC data. Plots showing distribution are truncated at the minimum and maximum values; dashed line marks the median with dotted lines marking the 25th and 75th percentiles. D. Relative change in telomere length as measured by quantitative fluorescence in situ hybridization (Q-FISH) of tumors and surrounding normal tissue. E-G. Representative fluorescence microscopy images (100x) with tumor (T) and normal (N) delineated respectively. Telomere fluorescent probe is in red and DAPI in blue. Scale bars indicate 50 μm. H. Number of structural variants (SVs) in short telomere syndrome SCC relative to HNSCC ICGC tumors with mean and 25th and 75th percentile lines marked. *Denotes the SCC of skin and esophagus. I and J. Two representative circos plots showing the spectrum of genomic alterations in specified short telomere solid tumors with listed TP53 status above. Remaining circos plots are in Figure S4. K. Proportion of SV types for each tumor are shown. L. Shortest distance from each translocation breakpoint to the start of the nearest telomere region plotted. Mean with standard error of the mean are shown. See also Fig. S1–S2 and Table S4.
Tumor telomere length and chromosome stability of tumors
In the context of the inherited telomere maintenance defect, we assessed the tumor telomere length by quantitative fluorescence situ hybridization (FISH). We found most cancers had somatic shortening relative to the adjacent normal tissues (6 of 8, 67%) (Figure 2D–F and Figure S2A and S2B). The extent of shortening was greatest in advanced stage cancers (Stage III-IV relative to Stage I-II) (Figure 2D). In two cases, the tumor telomere length was longer (Figure 2D,G and Figure S2C and S2D), but these cancers did not show ultra-bright telomere foci characteristic of ALT, and retained ATRX staining (Figure S2C and S2D). This absence of alterations associated with ALT genes (Table S4), and the telomere lengthening without ALT, is similar to what is seen in some epithelial cancers with yet-uncharacterized telomere maintenance mechanisms20.
Since short telomeres have been reported to drive genome instability in model systems10–13,21, we analyzed the structural variant (SV) burden, and found that the total number of SVs was in fact lower than sporadic head and neck SCC (Figure 2H). Moreover, using prediction algorithms which use contemporary and validated criteria that require co-occurrence of structural and copy number variants 22, there was no evidence of chromothripsis (Figure 2I,J and Figure S2E). Non-reciprocal translocations are the most common SVs associated with telomere dysfunction in yeast and mice10,23, but we found these events accounted for only 16% of all the structural variants (56 of 346 events, Table S4), and some tumors had no unbalanced translocations (Figure 2K and Table S4). To test if telomeres were involved in these translocations, we examined but did not identify telomere repeats around the breakpoint junctions of all the SVs including those involved in non-reciprocal translocation. The mean distance of each breakpoint to the closest telomere was 27.4 MB (Figure 2L), and there were no telomere end-to-end fusions in the unaligned whole genome sequence reads.
Squamous cell carcinomas arise in individuals with T cell immunodeficiency
Although the squamous cancer spectrum did not overlap with the common cancers of aging, we recognized it included malignancies that have an increased risk in patients with T cell immunodeficiency including solid organ transplant recipients on immunosuppression and those with HIV/AIDS24,25. We examined and found nearly all the patients had a T cell immunodeficiency at the time of diagnosis (86%, 12 of 14, Figure 3A and Table S5). Individuals diagnosed prior to age 50 years had a primary unprovoked lymphopenia and severely low CD4+ T cell counts (mean 305/mm3 with 5 of 6 falling below the lower end of the normal range, Table S5). Older adults had a milder baseline T cell lymphopenia, but had additionally been exposed to T cell immunosuppressive medications prior to their cancer diagnosis (Table S5). In the latter group, SCC was diagnosed after lung/liver transplantation, or after T cell cytotoxic medications (e.g. azathioprine or mycophenolate) were prescribed for presumed autoimmune disease prior to the diagnosis of a STS (Figure 3A and Table S5). The median time to diagnosis of a solid tumor after initiation of immunosuppression was 4.25 years (range 2–11 years, n=6), and 4 of these older individuals died within one year from rapidly progressive cancer (Figure 3B–E). Supporting a potential link between primary T cell immunodeficiency and risk for cancer, none of the individuals who underwent hematopoietic stem cell transplantation with T cell engraftment developed solid tumors (0 of 16, 51 lifetime years). Since T cell immunity protects against virally-induced cancers, we examined more than 9000 viral genomes in the whole genome sequence in addition to those commonly assayed in tumors clinically, and found only a minority of the tumors were virally associated (4 of 14 cancers); all of them contained human papilloma virus (3 anal, 1 laryngeal SCC, Figure 1F and Table S5).
Figure 3. Short telomere patients with solid tumors have T cell immunodeficiency.
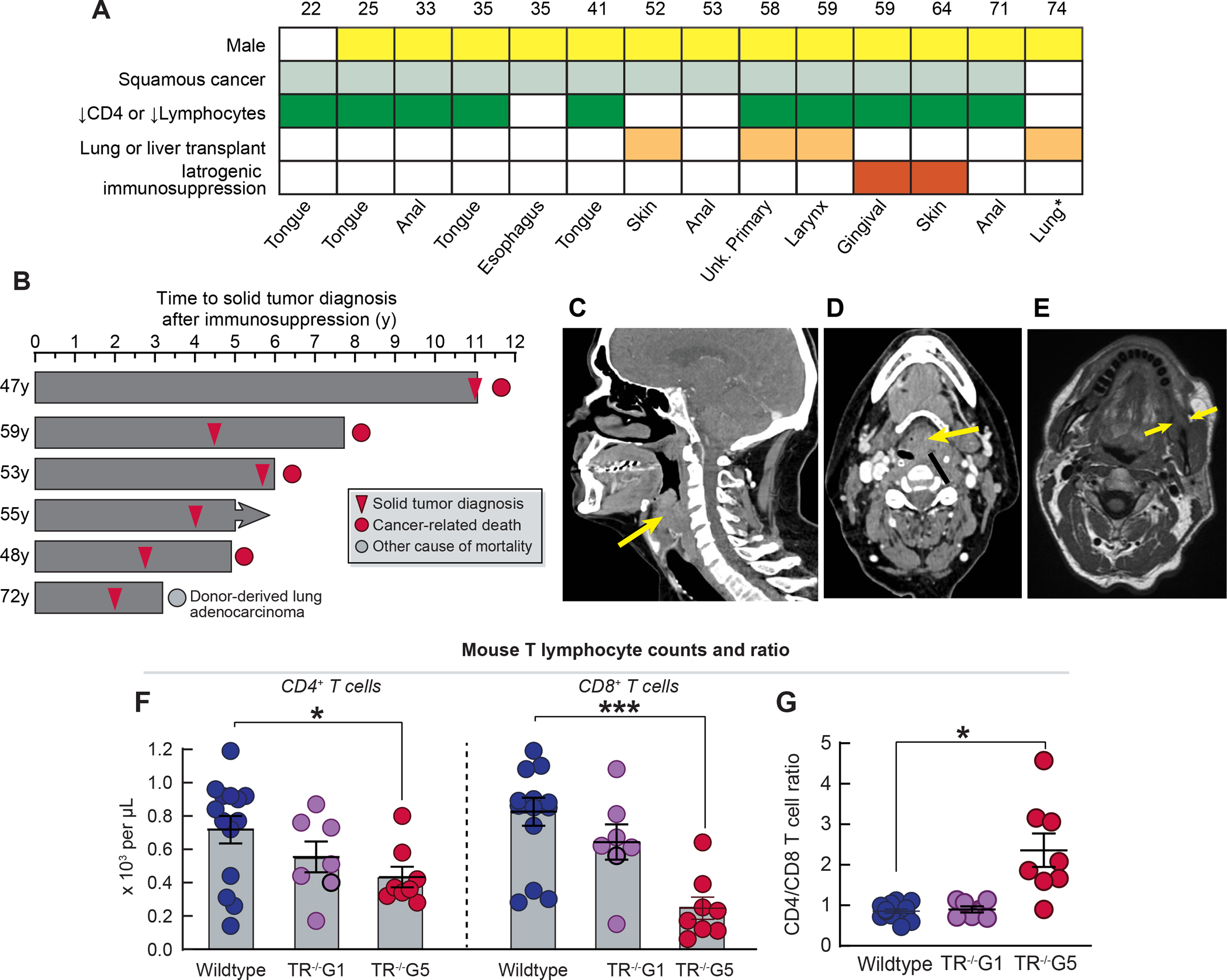
A. T cell immunodeficiency, both primary and secondary (in some cases both), in 14 individuals with germline mutations in telomere maintenance genes who developed solid tumors. B. Swimmer plot shows interval in years from initiation of immunosuppression to diagnosis of solid tumor in 6 individuals: 4 who underwent solid organ transplant, and 2 who were treated with immunosuppression for presumed autoimmune disease. C and D. Sagittal and axial computed tomography images (respectively) for a TERT mutation carrier who was diagnosed with a laryngeal squamous cancer (arrows) after lung transplantation for idiopathic pulmonary fibrosis. Images show rightward tracheal displacement due to mass effect. E. Sagittal magnetic resonance image of gingival squamous cell cancer (arrow) diagnosed after initiation of mycophenolate for hypersensitivity pneumonitis prior to short telomere syndrome diagnosis. F. Peripheral blood absolute CD4+ and CD8+ T cell counts in wildtype, TR−/−G1 and TR−/−G5 mice. G. CD4:CD8 ratio of mice in panel F. Mice were on average 16 weeks (range 5–23): Wildtype [7 males (M)/7 females (F)], TR−/−G1 (5M/2F), and TR−/−G5 (3M/5F). For F and G, mean and standard error of the mean are plotted. *P<0.05 and ***P<0.001 (Student’s t-test, two-sided). See also Table S5.
Mice with short telomeres have defective tumor immune surveillance
Intact replicative potential and telomere length are essential for T cell proliferation, expansion and sustained memory with aging26. In contrast to children with severe STS who have a complex immunodeficiency that contributes to early mortality27, adults with STS predominantly suffer from a T cell immunodeficiency of variable penetrance 26. The compromised T cell immunity is provoked by depleted hematopoietic stem cells and by the DNA damage response which provokes p53-dependent intrinsic apoptosis that leads to premature thymic involution5,26. With aging, the burden of T cell apoptosis and dropout manifests as a progressive T cell lymphopenia, restriction of T cell repertoire and a propensity for opportunistic infections including reactivation of latent DNA viruses26,28,29. The co-occurrence of T cell immunodeficiency with SCC led us to hypothesize that short telomere length impairs T cell-dependent cancer immunity. To test this, we studied telomerase RNA null mice (TR−/−), which have no phenotype in the first generation (G1), but after successive breeding, later generation mice (fifth generation, G5) acquire short telomere length30,31. These mice recapitulate the T cell defects seen in patients with STS5,26, including in their CD4+ and CD8+ lymphopenia (Figure 3F,G). To test the impact of short telomere length in T cell immune surveillance capacity, we implanted immunogenic tumor cells derived from KrasG12D/+Trp53R172H/+ mice that were engineered to express the human ovalbumin antigen. These cells grow tumors initially, but subsequently elicit a CD8-dependent host response that eradicates them32. Although this is not a model of SCC, it allowed us to test the effect of short telomere length on T cell cancer surveillance capacity in vivo. We performed a randomized study that was frequency-matched for age and sex with a prespecified endpoint of tumor growth. Cells implanted into wildtype and TR−/−G1 mice initially generated tumors, but they all became undetectable after 14 days (0 of 12 and 0 of 7 mice; 0 of 24 and 0 of 14 flanks with relapse, respectively, Figure 4A). In contrast, 41% of TR−/−G5 mice (13 of 32) had measurable tumors (P=0.0009, Fisher’s exact test, adjusted for age and sex), and 30% of implanted cells became tumorigenic (19 of 64 flanks, P<0.0001, Fisher’s exact test). In a subset of cases, tumors grew unchecked after implantation (8 of 19 flanks, 42%), but more commonly, tumors were initially suppressed, similar to controls, but then relapsed (11 of 19 flanks, Figure 4A). Male and female wildtype and TR−/−G1 mice showed comparable tumor sizes during the first 14 days, but male TR−/−G5 acquired larger tumors than females (81.8 mm3 vs. 51.8 mm3, P=0.03, Mann-Whitney test, Figure S3A). To understand the mechanism of impaired immune surveillance, we first assessed if TR−/−G5 tumors lost the immunogenic antigen, but found that ovalbumin was retained at the time of relapse (8 of 8 tumors analyzed, Figure S3B). When we analyzed the tumor infiltrating lymphocytes (TILs), we found there was a robust CD4 and CD8 T cell infiltrate initially at day 7 across all the groups (Figure 4B and 4C). However, by day 30, the tumor infiltrating lymphocytes had vanished in TR−/−G5 with essentially no CD4+ T cells visible and markedly fewer CD8+ T cells compared to three weeks earlier (Figure 4B and 4C).
Figure 4. Impaired immune surveillance of tumors in late generation telomerase null mice is associated with T cell exhaustion.
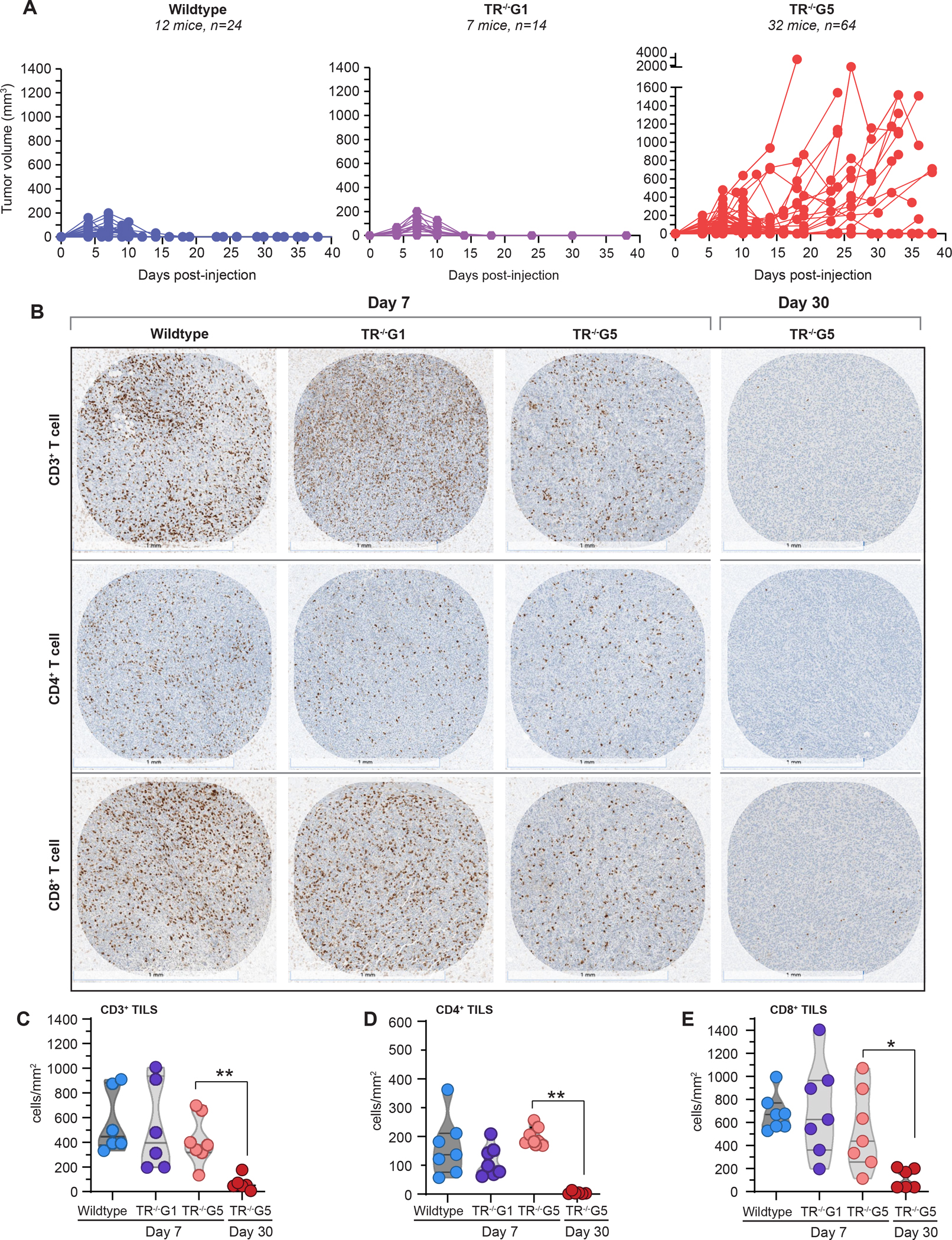
A. Tumor volume relative in mice injected with OvaB6 tumorigenic cells subcutaneously. Each line represents tumor size measured over the time course in one flank. Mice studied were on average 16 weeks old (range 6–24): Wildtype [24 flanks, 12 mice, 6 male (M)/6 female (F)], TR−/−G1 (14 flanks, 7 mice, 4M/3F), TR−/−G5 (64 flanks, 32 mice, 16M/16F). B. CD3, CD4 and CD8 immunohistochemistry in tumors harvested at each time point and for each genotype shown in panel A. At day 7, tumors showed comparable burdens of tumor infiltrating T lymphocytes (TILs) but TILs subsequently vanished by day 30 in TR−/−G5 tumors. Scale bar below each image refers to 1 mm. C-E. TIL quantification per tumor area at the timepoints shown in panel B. Violin plots with median and 25th/75th percentile lines marked. For day 7, wildtype (n=6–7 flanks, 4 mice, 2M/2F), TR−/−G1 (6–7 flanks, 7 mice, 5M/2F), and TR−/− G5 (7 flanks, 5 mice, 3M/2F) are shown. For day 30, data for TR−/−G5 (6 flanks, 4 mice, 2M/2F) are shown. **P<0.01 and ***P<0.001 (Mann-Whitney test, corrected for multiple comparisons). See also Fig. S3.
Discussion
In this 20-year study, we found that individuals with the shortest telomeres in the human population have an increased risk of SCC, while simultaneously having a lower risk of other cancers associated with aging. The data provide decisive interpretation of longstanding conflicting data from model organisms on the role of telomeres in human cancer development with aging. They support a paradigm wherein short telomere length generally protects against cancer development with aging and does not fuel chromosome instability in humans. Even in tumors with biallelic TP53 loss, there was no evidence of kataegis or chromothripsis and the STS cancers had fewer structural variants than comparable sporadic tumors. These observations align with an emerging body of genetic evidence that points to longer telomere length as a potent driver of cancer risk in the human population, including the majority of malignancies associated with aging33–35. In contrast to the STS, autosomal dominant-inherited mutations that predispose to long telomere length promote ubiquitous clonal evolution and increase the risk for a wide spectrum of cancers including those commonly associated with aging36.
We focused our studies on T cell immunity because the solid cancers we documented had a nearly identical spectrum to those seen in patients with acquired T cell immunodeficiency. Adults with STS suffer from T cell defects that are exacerbated over time with aging in the face of chronic antigen exposure26, and memory T cells also have the highest rate of telomere attrition relative to all other leukocytes37. Short, dysfunctional telomeres signal a T cell exhaustion program that is provoked by a p53-dependent apoptosis; this program is evident in patients with STS as well as in late generation telomerase knockout mice5,26. The cumulative effects of T cell dropout with aging leads to an immunodeficiency which can clinically manifest as reactivation of latent DNA viruses such as cytomegalovirus, especially when patients are additionally exposed to iatrogenic immune suppression26,29. Our data demonstrate that persistent exposure to a tumor antigen also leads to T cell exhaustion and loss of immune memory26,28. The combined clinical and animal model data therefore support that short telomere length, at certain extreme thresholds, is sufficient to impair cancer immune surveillance and increases the risk for subsets of cancers that rely on T cell competence for their suppression.
The SCC risk we documented was almost exclusively restricted to males, in contrast to the hematologic malignancies which affects the sexes equally. Other monogenic T cell immunodeficiencies also show more severe phenotypes in males38. Older adult males similarly have more severe T cell dysfunction than age-matched females38. The sex disparity has been proposed to be related to the hemizygous nature of the many X-linked immune genes, although the precise mechanisms remain poorly understood38. The highest risk of cancers we saw was among males with DKC1 mutations. DKC1 encodes dyskerin which plays ubiquitous roles in RNA modification and stability beyond telomerase. Our data do not rule out the possibility that other dyserkin functions may be playing a role in the increased SCC risk, however, DKC1 mutation carriers shared the same immunodeficiency as other STS patients consistent with a convergent mechanism. The increased SCC predilection, in contrast to the risk of myeloid cancers, supports that T cell immunity may be more compromised in these male DKC1 mutation carriers. Our findings have clinical implications for cancer screening and indicate that adult males with STS, especially those on immunosuppressive medications and with DKC1 mutations, have the highest risk. Targeted screening may be of high yield in this subset.
The dependence of squamous epithelial-lined mucosa on T cell competence for tumor surveillance is not well understood, but the same cancer spectrum we documented is observed in other T cell immunodeficient states including HIV/AIDS24,25,39. It also overlaps with what is seen in Fanconi anemia, another DNA repair syndrome, although Fanconi anemia patients have a higher risk of head and neck SCC (30% lifetime risk compared to the 9% in STS), and they have no known T cell compromise40. The genomic landscape of head and neck SCC derived from patients with Fanconi anemia was recently characterized and found to have high copy number alterations19. The contrast between the Fanconi anemia genomic landscape with STS cancers highlights the heterogeneity of pathways that provoke squamous carcinogenesis. An integrated view suggests that T cell competence, as well as intact interstrand crosslink DNA repair, are two non-overlapping tumor suppressive mechanisms of squamous carcinogenesis. Deeper understanding of the relevant drivers of squamous carcinogenesis is needed given the increasing incidence of oral cavity SCC especially among younger individuals41, and the yet-limited treatment options available for individuals who develop these cancers.
STAR Methods
RESOURCE AVAILABILITY
Lead contact.
Further information and requests for data or materials should be addressed to: Dr. Mary Armanios (marmani1@jhmi.edu)
Materials availability.
This study did not generate new unique reagents.
Data and code availability.
Unreported germline variants were submitted to the Telomerase Database (http://telomerase.asu.edu) and ClinVar Annotation Database (https://www.ncbi.nlm.nih.gov/clinvar/). Processed and verified somatic variants, copy number and structural variant calls are included in Table S4. TCGA HNSCC data is accessible from the International Cancer Genome Consortium (ICGC)-25K Data Portal. Additional information required to reanalyze the data is available upon request provided they fall within the limits of the available consent and with approval from the Johns Hopkins Medicine Institutional Review Board. To request access, contact Dr. Mary Armanios at Johns Hopkins School of Medicine as aforementioned. The Key Resources Table includes the links under “Deposited Data”.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CD3 | Abcam | Cat# ab5690; RRID:AB_305055 |
| Rabbit monoclonal anti-CD4 | Abcam | Cat# ab183685; RRID:AB_2686917 |
| Mouse monoclonal anti-CD8a | ThermoFisher Scientific | Cat# 14-0195-82; RRID:AB_2637159 |
| Rabbit polyclonal anti-Ovalbumin | Abcam | Cat# ab181688 |
| Rabbit monoclonal anti-rat IgG | Vector Labs | Cat# AI-4001; RRID:AB_2336209 |
| Goat polyclonal anti-rabbit IgG | Abcam | Cat# ab205718; RRID:AB_2819160 |
| Biological samples | ||
| Human tumor and paired germline samples | This manuscript | N/A |
| Mouse tumor samples | This manuscript | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Citrate unmasking solution | Vector Labs | Cat# H-3300-250 |
| ImmPACT DAB Peroxidase (HRP) Substrate | Vector Labs | Cat# SK-4103 |
| Mayer’s hematoxylin | Sigma-Aldrich | Cat# MHS1 |
| Critical commercial assays | ||
| Qiagen Puregene Blood Kit B | Qiagen | Cat# 158389 |
| Siemens Tissue Preparation System | Siemens Healthineers | N/A |
| TruSeq Nano DNA Library Prep kit | Illumina | Cat # 20015965 |
| Deposited data | ||
| Previously unreported germline variants | This manuscript | ClinVar annotation database: https://www.ncbi.nlm.nih.gov/clinvar/ Telomerase database: http://telomerase.asu.edu |
| Processed genome sequencing data | This manuscript | Table S4 |
| The Cancer Genome Atlas (TCGA) HNSCC genomic sequencing | International Cancer Genome Consortium-25K Data Portal | https://dcc.icgc.org/ |
| Experimental models: Cell lines | ||
| Mouse: Ova-albumin-antigen expressing cells (OvaB6) derived from KrasLSL-G12D/+, Tp53LSL-R172H/+, Pdx1-Cre (KPC) mice | Dr. Robert Vonderheide’s laboratory, University of Pennsylvania | Ref 32 |
| Experimental models: Organisms/strains | ||
| Mouse: B6.Cg-Terctm1RdP/J (mTR-/-) | Jackson Lab | Cat# 004132; RRID:IMSR_JAX:0041 |
| Mouse: C57BL/6 | Jackson Lab | Cat# 000664 |
| Oligonucleotides | ||
| Alexa 488-labeled telomere (AATCCC)3 probe | Panagene | http://www.panagene.com/_ENG/html/dh_product/prod_view/32/?cate_no=3 |
| Software and algorithms | ||
| SeerStat version 8.4.0 | National Cancer Institute | https://seer.cancer.gov/seerstat/ |
| Telometer, ImageJ plugin, v. 3.0.05 | Meeker et al., 2002 | demarzolab.pathology.jhmi.edu/telometer |
| Nikon NIS-Elements v. 5.21.02 | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| Burrows-Wheeler Aligner (v0.7.7) | Li, 2013 | https://github.com/lh3/bwa |
| Picard-tools1.119 | Broad Institute | https://broadinstitute.github.io/picard/ |
| GATK v3.6.0 | Broad Institute | https://gatk.broadinstitute.org/hc/enus |
| MuTect2 v3.6.0 | Broad Institute | https://gatk.broadinstitute.org/hc/enus/articles/360037593851-Mutect2 |
| snpEFF v4.1 | Microsoft Genomics | http://pcingola.github.io/SnpEff/ |
| Variant Effect Predictor Ensembl API | Ensembl | https://useast.ensembl.org/info/docs/tools/vep/index.html |
| Bambino | Edmonson et al., 2011 | https://cgwb.nci.nih.gov/goldenPath/bamview/documentation/index.html |
| Mutalisk mutation analysis toolkit | Lee et al., 2018 | http://mutalisk.org |
| SigProfilerSingleSample v1.3 | Alexandrov et al., 2020 | https://github.com/AlexandrovLab/SigProfilerExtractor |
| CNVkit v0.9.4 | Talevich et al., 2016 | https://cnvkit.readthedocs.io/en/stable/ |
| CONSERTING | Chen et al., 2015 | http://www.stiuderesearch.org/site/lab/zhang |
| Manta v0.29.6 | Chen et al., 2016 | https://github.com/Illumina/manta |
| Lumpy v0.2.11 | Layer et al., 2014 | https://github.com/arq5x/lumpy-sv |
| bedTools | Quinlan Lab, University of Utah | https://bedtools.readthedocs.io/en/latest/ |
| circos v0.69 | Krzywinski et al., 2009 | http://circos.ca/software/download/circos/ |
| ShatterSeek | Cortes-Ciriano et al. | https://github.com/parklab/ShatterSeek |
| kallisto v0.43.1 | Bray et al. | https://pachterlab.github.io/kallisto/about |
| FACSDiva v.8.0.1 | BD Biosciences | https://www.bdbiosciences.com/enus/products/software/instrument-software/bd-facsdiva-software |
| FlowJo v.10.1 | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads/ |
| Halo image analysis software | Indica Labs | https://indicalab.com/halo/ |
| GraphPad Prism v.7.04 | GraphPad Software | https://www.graphpad.com/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human participants.
Study participants were recruited to the Johns Hopkins Telomere Syndrome Registry 16 from July 2003 to July 2022. Participants were assigned the short telomere syndrome diagnosis if they 1) carried a pathogenic mutation in telomerase or other telomere maintenance gene and short telomere length, or 2) had short telomere length and classic features of a familial short telomere syndrome as defined 42, or 3) had short telomere syndrome clinical features with abnormally low telomerase RNA (TR) levels, as defined previously 43,44. Dyskeratosis congenita was assigned if an individual had one of the three mucocutaneous features. Cancer diagnoses were adjudicated by original pathology reports by two of the authors and surgical pathology was centrally reviewed for tumors that were sequenced and/or examined for telomere studies. The study was approved by the Johns Hopkins Medicine Institutional Review Board (IRB) and participants gave written informed consent.
Vertebrate Animal study approval and mice studied.
Mice were housed in the Johns Hopkins School of Medicine East Baltimore campus. The study was approved by the Johns Hopkins Animal Care and Use Committee. mTR−/− mice were maintained and bred as described (founders deposited at JAX lab) 30 with heterozygous mice interbred to generate the first generation and knockout mice successively bred to generate later generation mice with short telomeres. Wildtype mice were maintained in a separate dedicated colony, and both wildtype and mTR−/− mice were on the C57BL6/J background. Mice were 16 weeks on average (range 6–24) at the time of injection and male and female mice were studied.
Mouse cell lines.
Ova-albumin-antigen expressing cells (V6.Ova) were generated from a mouse pancreatic adenocarcinoma cell line isolated from a spontaneous pancreatic tumor in a KrasLSL-G12D/+, Trp53LSL-R172H/+, Pdx1-Cre (KPC) mouse 32. Tumors were excised from the whole pancreata and single cell suspensions were generated then cells were retrovirally transduced with ovalbumin-expressing construct labeled with a Td-tomato marker to create cell lines that express full-length OVA; cells were then sorted for expression of Td-tomato and a single representative clone was selected for these studies (Clone V6.Ova) 32. Cells were grown in complete medium in normoxic (5% CO2) conditions until injections.
METHOD DETAILS
Leukocyte telomere length measurement, germline DNA sequencing, and quantitative real-time PCR for telomerase RNA (TR).
Peripheral blood leukocyte telomere length was measured by flow cytometry and fluorescence in situ hybridization (flowFISH) at the Johns Hopkins Pathology Laboratories17. In this assay, a greater deviation from the median in children and young adults signifies more severe disease17. Germline mutations were identified by genome 43,44, whole exome45, or panel-based sequencing17, and where a variant was known in a family, by targeted Sanger sequencing42 using peripheral blood isolated genomic DNA. Germline mutations were reported previously16–18, and additional germline variants not previously reported are listed in Table S5 (included in the Telomerase Database telomerase.asu.edu 46). TR levels were measured in EBV-transformed lymphoblastoid cell lines using quantitative real time PCR as previously described 43,47, and “low TR” individuals were assigned if TR levels fell near 50% or lower levels relative to a group of healthy controls.
Tumor telomere quantitative fluorescence in situ hybridization (FISH).
Tumor telomere length was measured by quantitative fluorescence in situ hybridization (FISH) using an Alexa 488-labeled telomere (AATCCC)3 probe (Panagene) on unstained 5 μm sections from tumor-containing paraffin-embedded tissue blocks 48,49. Briefly, tumor and normal regions were delineated by a pathologist (LMR) then imaged (Nikon ECLIPSE Ni with Intensilight C-HGFI illuminator and fluorescence excitation/emission filters). For each normal and tumor area, 5–7 high power (x100/1.4 Oil) images from the same slide were captured in DAPI and TRITC channels at a fixed exposure time to avoid signal saturation (Nikon NIS-Elements v. 5.21.02). Grayscale images were analyzed using Telometer (ImageJ plugin, v. 3.0.05, available at demarzolab.pathology.jhmi.edu/telometer/50 kind gift from Dr. Angelo DeMarzo and Dr. Alan Meeker). Average telomere length per cell was measured as the sum of fluorescence signals in a DAPI-defined nuclear area, and 20–30 nuclei were analyzed for each of the normal and tumor areas.
Whole genome sequencing and analysis
Microdissection and DNA extraction.
Tumor area was demarcated on hematoxylin and eosin-slides and 5 μm unstained paraffin-embedded sections (3–6 per tumor) were micro-dissected manually. Tumor DNA was purified using an automated Siemens Tissue Preparation System in a clinically-validated pipeline (Siemens Healthineers). DNA extracted from peripheral blood [Qiagen Gentra Puregene kit (Qiagen, Blood Core Kit B)] was used as a germline source. DNA concentrations were quantified using Quibit (Thermo Fisher Scientific).
Paired tumor-normal whole genome sequencing and alignment.
DNA libraries were constructed using TruSeq DNA Nano kit (Illumina) with 200 ng of genomic DNA. Sequencing was performed on an Ilumina NovaSeq6000 S4 using paired-end 150 bp chemistry with 60x target coverage. The actual mean coverage was 72x (range 26–129x) across the normal and tumor genomes. llumina’s bcl2fastq v2.15.0 was used to convert BCL files to demultiplexed FASTQ files. Demultiplexed reads were aligned to GRCh38 using BWA-MEM v0.7.7 with default parameters. Piccard-tools1.119 was used to add read groups as well as remove duplicate reads. GATK v3.6.0 base call recalibration steps were used to create a final alignment file.
Somatic variant calling and analysis.
Somatic single nucleotide variants and insertions/deletions between tumor-normal pairs were called from the BAM file using MuTect2 v3.6.0 with default parameters. Passed calls were annotated with snpEFF v4.1 and variant call files were analyzed for non-synonymous mutations in candidate telomere lengthening and DNA damage response genes listed (Table S4). The pathogenicity of each identified mutation was manually assessed using in COSMIC v94 and CADD score (Table S5). Tumor mutation burden was defined as non-synonymous somatic mutations per Mb of coding region and calculated using Variant Effect Predictor Ensembl API 51.
Mutational Signature analysis.
For mutational signature analysis, variants were called using Bambino52 and passed somatic variant calls were analyzed with the Mutalisk mutation analysis toolkit53. For one sample with higher background, variant calls were generated from Mutect2. Program input was the VCF file, and the multinomial Maximum Likelihood Estimate method was used to identify all COSMIC signatures and generate localized hypermutation rainfall plots relative to distance between mutations. The mutational signature results with more than 2% variants associated with a single cosmic signature were considered for interpretation. Additionally, high quality variants (SNVs) were run through SigProfilerSingleSample v1.354 to determine the presence of known COSMIC signatures within samples. Mutational signature analysis resulted in cosine score values above 0.90. Kataegis was examined manually using the rainfall plots, and defined using standard criteria as the presence of distinct vertical lines of SNV ‘pile up’ that have short inter-mutation cluster distance less than 2 kb, as had also been defined previously 12.
Copy number analysis.
Copy number analyses were performed using both CNVkit v0.9.455 and CONSERTING56 algorithms with default parameters. Passed calls were further analyzed for focal, gene-level deep amplifications (log2 ≥1) or deletions (log2 ≤−1) in the candidate genes (Table S4). Due to the high sequencing coverage and corresponding noise, larger segment and whole chromosome copy number alterations and regions of loss of heterozygosity were assessed by manual inspection of allelic imbalance plots created using high quality CONSERTING calls for each paired tumor-normal comparison.
Analysis of structural variants.
Manta v0.29.657 was used to call somatic structural variants (SV) and indels between the tumor-normal pairs using default parameters. Lumpy v0.2.11 58 was used to call SVs independently on tumor samples. SV calls that were called by both platforms (Manta and Lumpy) with overlapping SV breakpoints (assessed by bedTools pairToPair) were considered to ensure high quality data. Variants were annotated according to UCSC refseq annotations using a custom script and classified as deletions, insertion, inversions or translocations. As the mean whole genome sequencing coverage was 72x, we retained high confidence variants by requiring SV supports from both discordantly mapped read pairs and junction reads (3 in each category), or at least 10% of the reads containing discordantly mapped read pairs (i.e. 7 discordantly mapped read pairs). Each SV that passed this threshold was further examined by manual inspection in IGV to exclude additional variants caused by mapping error, and only high quality SVs with no mismapping issues were included in the analyses. To determine if any translocation breakpoint was located near a telomeric region, we used bedtools to inspect if the start and end position of the SV region fell within 500 bp upstream or downstream of the telomeric region as defined by the UCSC Genome Browser (August 1, 2022). We also searched for telomere repeat sequences (TTAGGG)3–20 at structural variant junctions (2 mismatches allowed), including at breakpoints of non-reciprocal translocations.
Circos plot generation.
To visualize data, scaled circos plots of the entire genome, excluding chromosome Y, were created using circos v0.6959 for each tumor. Copy number alterations detected by CNVkit containing megabase pairs with log2 >0.35 (amplification/gains) or log2 < −0.35 (deletions/loss) and covering more than 3 Mb of the genome were included in the plots. Overlapping SVs called by both Manta and Lumpy, as above, were included in the circos plots (Table S4).
Chromothripsis analysis.
Curated copy number and SV calls for each sample were visually inspected and analyzed using ShatterSeek22, an R package prediction program for identifying chromothripsis patterns. The ShatterSeek software specifically assigns each SV as having no, low or high level of evidence for chromothripsis based on statistical criteria as described in Cortes-Ciriano et al.22 ShatterSeek assesses the entire set of structural rearrangements for each chromosome, including the clustering of breakpoints, oscillatory pattern of copy number states and relative order and orientation of rearranged segments, to identify alterations consistent with chromothripsis.
Viral integration.
Human papilloma virus (HPV) status was inferred clinically using immunohistochemistry by standard methods. Anal cancers were presumed to be HPV positive given the known association 60. To detect viral DNA in short telomere individual cancer genomes, we constructed a custom viral reference database from eukaryotic viral genomes retrieved from NCBI RefSeq (> 9,000 genomes)61. Unmapped and soft-clipped genome sequencing reads (aligned to GRCh37-lite) were mapped to the constructed viral reference database using kallisto v0.43.1 62 at 25 basepair sequences at a time. The final virus-like reads were mapped by blastn 63 against both the unmasked custom viral reference database (using e-value cutoff of 10−10) and against a NCBI RefSeq genomes of viruses, representative bacteria, archaea, and fungi, and UCSC genomes of human, chimp, mouse, chicken, and fruit fly, and NCBI VecScreen vectors. A read was considered virus-specific if its top hit with the lowest e-value was a virus from the reference database64. Viral integration status was determined by the presence of chimeric/split and soft-clipped reads and required a minimum of two supporting reads to report a viral integration event.
Comparison with published squamous cell cancer data.
Somatic mutation data for the Head and neck squamous cell cancers (HNSCC) of The Cancer Genome Atlas (TCGA) cohort were obtained through International Cancer Genome Consortium (ICGC)-25K Data Portal65 (data release 28, n=44 cancers) using somatic variant calls obtained from whole genome sequence data. Multiple variant call pipelines were cross-referenced to create a final consensus set of variants66. Tumor mutation burden was calculated in the coding regions, and the frequency of TERT promoter mutations was assessed at 3 hotspots: c.−146C>T, c.−124C>T/A and c.−57A>C. TP53 loss-of-function was assessed by curating coding variants and 17p encompassing deletions.
Mouse Studies
Flank injections and animal monitoring.
Two days prior to injections, age- and sex-matched mice were anesthetized with inhaled isoflurane, and flank fur was removed using an electric razor or Nair Hair Remover Lotion. On the day of injections, OvaB6 cells were harvested (90% confluence), washed and suspended in sterile DMEM, and 5 million cells in 100 μl volume were injected subcutaneously (two flanks for each mouse). For each experiment, 2–3 wildtype mice were injected with 100 μl media only to ensure sterility of the media. Mice were monitored every 3 days and tumors were measured using a digital caliper. Tumor volume was calculated using the longest measurement [length (L)] and shortest [width (w)], as described32. Mice were euthanized if the animal appeared distressed or if there were tumor-associated morbidities such as ascites, muscle invasion, ulcers, or if tumor size was greater than 1,500 mm3. Harvested tumors were dissected from euthanized mice, preserved in 10% formalin, and then embedded in paraffin and sectioned using standard procedures.
Mouse peripheral T cell subset analysis.
To quantify peripheral lymphocyte counts, mice were euthanized and 300 μl from a cardiac puncture were collected in capillary K3 EDTA Microvette tubes (Sarstedt). Complete blood counts were performed on 100 μl on the Procyte Dx Hematology Analyzer (IDEXX Laboratories), and remaining volume was analyzed for T cell subset quantification by flow cytometry26. Briefly, after red blood cells lysis using 1x RBC Lysis Buffer (eBioscience), cells were stained using antibodies against the cell surface markers listed below. Single-color controls were used for each antibody. Flow cytometry data were acquired using FACSDiva (v.8.0.1, BD Biosciences) on an LSR II (BD Biosciences), and analyzed using FlowJo (v.10.1, BD Biosciences). After a lymphocyte gate was created on forward-side-scatter plots, doublet exclusion gating strategies were used and CD3+ CD4+ and CD3+ CD8+ subpopulations were analyzed. At least 20,000 viable events were collected for each animal, and absolute T cell counts were calculated based on the total leukocyte count in peripheral blood.
| Marker | Fluorochrome | Clone # | Source |
|---|---|---|---|
| CD3e | PE | 145-2C11 | eBioscience |
| CD4 | FITC | RM4–5 | Biolegend |
| CD8a | APC | 53–6.7 | Biolegend |
| CD19 | PECY7 | 6D5 | Biolegend |
Tumor infiltrating lymphocyte and ovalbumin immunohistochemistry.
CD3+, CD4+ and CD8+ staining was performed on formalin-fixed, paraffin-embedded 5 micron sections using standard procedures (Johns Hopkins Oncology Tissue Services, Ventana Discovery Ultra autostainer, Roche Diagnostics). For ovalbumin detection, slides were deparaffinized and rehydrated, incubated in Citrate Unmasking solution at 100°C for 1h (Vector Labs, 100X dilution), blocked in 10% donkey serum for 1.5h, incubated in the anti-Ovalbumin primary antibody overnight at room temperature, then blocked in H2O2, washed and incubated in the Goat Anti-Rabbit IgG H&L (HRP) secondary antibody for 1h at room temperature. Signal was detected using ImmPACT DAB Peroxidase (HRP) Substrate (Vector Laboratories), counterstained in Mayer’s hematoxylin (Sigma-Aldrich, catalog # MHS1), dehydrated, and mounted using DPX (Sigma-Aldrich). The following primary antibodies were utilized: anti-CD3 (rabbit polyclonal, ab5690, 1:200; abcam), anti-CD4 (rabbit monoclonal, ab183685, 1:1000; abcam), anti- CD8a [(monoclonal, clone 4SM16, 1:125, ThermoFisher Scientific), followed by rabbit anti-rat linker antibody (1:500 dilution; AI4001; Vector Labs)] and anti-Ovalbumin (rabbit polyclonal, ab181688, 1:500; abcam). Primary antibodies were detected using an anti-rabbit HQ detection system followed by Chromomab DAB IHC detection kit (Roche Diagnostics) for CD3, CD4 and CD8. Goat Anti-Rabbit IgG H&L (HRP) (goat polyclonal, ab205718, 1:5000; abcam) was used to detect the primary ovalbumin antibody.
Quantification of tumor infiltrating lymphocytes.
Stained slides were digitally scanned at 20x resolution on the Aperio ScanScope CS system and lymphocyte infiltration within tumors were identified by a board certified pathologist. Areas of tumor were identified and the density of total CD3, CD4 and CD8 T cells were quantified using Halo image analysis software (Indica Labs, Corrales, NM). Image acquisition and analysis were done blinded to mouse genotype or timepoint
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Methods of clinical data analyses.
Cumulative incidence of myelodysplastic syndrome and acute myeloid leukemia (MDS/AML) or a solid tumor diagnosis was estimated using Fine and Gray’s method67, using the participant’s age at diagnosis. Participants who were cancer-free were censored at the last age at follow-up. Death before a cancer diagnosis was analyzed as a competing event. Hazard ratios for comparison of subgroups, defined by sex and DKC1 mutation status, were estimated using proportional sub-distribution hazards regression.
To assess the observed cancer incidence relative to age- and sex-matched cohorts, we compared the observed events relative to the number in the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) Program data (www.seer.cancer.gov) during the same time of followup. We performed two analyses. The first was for the cancers identified in the short telomere syndrome patients: i.e. MDS/AML and squamous cell cancers (SCC). The latter was performed for oral cavity SCC (the largest subset of solid tumors) as well as all SCC excluding lung SCC. In a second analysis, we compared the relative cancer incidence of the top 9 most common malignancies listed in the 2021 American Cancer Society Cancer Facts and Figures1. This encompassed any of lung/bronchus, colorectal, bladder, pancreas, kidney/renal pelvis, melanoma, thyroid, non-Hodgkin lymphoma, and endometrial cancers. Breast and prostate cancers were excluded from this analysis as these were not documented with primary pathology reports in the participants of the Telomere Syndrome study.
The analyses followed a similar algorithm as described previously16. Specifically, we used SeerStat version 8.4.0 software to calculate age-adjusted incidence rates of each malignancy separately for men and women, according to race (white, black, other), and by year from 1975 through 2019; rates for the 2020 to 2021 period were extrapolated using a regression model. For each individual, we calculated the cumulative sum of the yearly incidence rate beginning with either the individual’s birth year or 1975 (whichever was later), and up through the year of last known follow-up or year of diagnosis, if affected. For each cancer, we took the sum of these values across all individuals as the expected number of diagnoses for this cohort. The observed-to-expected (O/E) ratio was calculated as the sum of the observed diagnoses in this cohort divided by the expected number in SEER. The 95% confidence interval (CI) was calculated using a bootstrap approach with 10,000 simulations.
Statistical analyses for mouse studies.
For the mouse studies, pilot data were used to calculate sample size and mice were age- and sex-matched mice. Analysis for T cell infiltrates was done blinded to the recipient animal’s genotype. Prism v.7.04 (GraphPad Software) was used to generate the figures. R version 4.2.1 was used to calculate the P-values for the mouse experiments. The proportion of flanks that showed tumor growth across genotypes was compared using logistic regression, adjusting for age and sex of the mice. Peak tumor volume for each flank in the first 10 days was also compared between males and females using generalized estimating equations (GEE) to control for repeated measures in the same mice. All P-values shown are two-sided, and P-values less than 0.05 were considered to be significant. For comparisons of CD3, CD4 and CD8 TILs across genotypes, P-values less than 0.0167 was considered statistically significant to account for multiple comparisons.
Supplementary Material
Table S4. List of somatic mutations, copy number alterations and structural variants (supplementary to Figure 2)
Table S5. Clinical characterization of immune deficiency of short telomere syndrome participants with solid tumors and germline mutations not previously reported in the Johns Hopkins Telomere Syndrome Registry (supplementary to Figure 3 and STAR Methods)
Highlights.
Patients with short telomere syndromes (STS) have lower incidence of most solid cancers
Squamous cancers that arise lack hallmarks of chromosome instability
Cancers arise primarily due to short telomere-mediated T cell immunodeficiency
T cell exhaustion in STS mice fails to survey and suppress immunogenic tumors
Acknowledgements.
The authors are grateful for all the participants and to their referring providers. We are thankful for help from Margaret Strong and Dr. Carol Greider for providing mTR−/− mice, for helpful comments on the manuscript from Dr. Bert Vogelstein, and discussion of ALT mechanisms with Dr. Alan Meeker. We appreciate help from Sheila Iyer with computational analyses and acknowledge support from these Johns Hopkins Core Facilities and their staff: Genetics Research Core Facility, Bloomberg School of Public Health Flow Cytometry Core, Phenotyping and Pathology Core, and the Sidney Kimmel Comprehensive Cancer Center (SKCCC) Oncology Tissue Services and Experimental and Computational Genomics Core Shared Resources. This work was funded by National Institutes of Health (R01CA225027, R01HL119476), the S&R, Gary Williams, and Commonwealth Foundations (MA). MA would like to acknowledge a gift in the name of Mrs. P. Godrej and a gift from the Harrington family. KES was supported by K08HL163468, the ASH Scholar Award and an award from the Dresner Foundation, DLG by T32GM007309, PLV by T32CA009071 and VG by F32HL142207. KES and DLG received support from the Turock Scholars Fund to the Telomere Center at Johns Hopkins. RVH received support from R01CA229803. SKCCC Shared Resources are supported by P30CA006973.
Inclusion and diversity statement.
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of interests. Dr. Vonderheide is an inventor on licensed patents related to cancer cellular immunotherapy and cancer vaccines, receives royalties from Boston Children’s Hospital for a licensed research-only monoclonal antibody, and has received consulting fees from Bristol Myers Squibb.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Fuchs HE, and Jemal A (2021). Cancer Statistics, 2021. CA Cancer J Clin 71, 7–33. 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, Greider CW, and Harley CB (1992). Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A 89, 10114–10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, and DePinho RA (1999). p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97, 527–538. S0092–8674(00)80762-X [pii]. [DOI] [PubMed] [Google Scholar]
- 4.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, and Jackson SP (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198. [DOI] [PubMed] [Google Scholar]
- 5.Lee HW, Blasco MA, Gottlieb GJ, Horner JW 2nd, Greider CW, and DePinho RA (1998). Essential role of mouse telomerase in highly proliferative organs. Nature 392, 569–574. [DOI] [PubMed] [Google Scholar]
- 6.Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, and DePinho RA (1999). Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell 97, 515–525. [DOI] [PubMed] [Google Scholar]
- 7.Feldser DM, and Greider CW (2007). Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 11, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perera SA, Maser RS, Xia H, McNamara K, Protopopov A, Chen L, Hezel AF, Kim CF, Bronson RT, Castrillon DH, et al. (2008). Telomere dysfunction promotes genome instability and metastatic potential in a K-ras p53 mouse model of lung cancer. Carcinogenesis 29, 747–753. [DOI] [PubMed] [Google Scholar]
- 9.Rudolph KL, Millard M, Bosenberg MW, and DePinho RA (2001). Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet 28, 155–159. [DOI] [PubMed] [Google Scholar]
- 10.Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, and DePinho RA (2000). Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406, 641–645. [DOI] [PubMed] [Google Scholar]
- 11.Hackett JA, Feldser DM, and Greider CW (2001). Telomere dysfunction increases mutation rate and genomic instability. Cell 106, 275–286. [DOI] [PubMed] [Google Scholar]
- 12.Maciejowski J, Li Y, Bosco N, Campbell PJ, and de Lange T (2015). Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163, 1641–1654. 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maciejowski J, Chatzipli A, Dananberg A, Chu K, Toufektchan E, Klimczak LJ, Gordenin DA, Campbell PJ, and de Lange T (2020). APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat Genet 52, 884–890. 10.1038/s41588-020-0667-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armanios M, and Blackburn EH (2012). The telomere syndromes. Nature reviews. Genetics 13, 693–704. 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alder JK, and Armanios M (2022). Telomere-mediated lung disease. Physiol Rev 102, 1703–1720. 10.1152/physrev.00046.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schratz KE, Haley L, Danoff SK, Blackford A, DeZern A, Gocke CD, Duffield AS, and Armanios M (2020). Cancer spectrum and outcomes in the Mendelian short telomere syndromes. Blood May 28 135, 1946–1956. 10.1182/blood.2019003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alder JK, Hanumanthu VS, Strong MA, DeZern AE, Stanley SE, Takemoto CM, Danilova L, Applegate CD, Bolton SG, Mohr DW, et al. (2018). Diagnostic utility of telomere length testing in a hospital-based setting. Proc Natl Acad Sci U S A 115, E2358–e2365. 10.1073/pnas.1720427115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schratz KE, Gaysinskaya V, Cosner ZL, DeBoy EA, Xiang Z, Kasch-Semenza L, Florea L, Shah PD, and Armanios M (2021). Somatic reversion impacts myelodysplastic syndromes and acute myeloid leukemia evolution in the short telomere disorders. J Clin Invest 131. 10.1172/JCI147598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Webster ALH, Sanders MA, Patel K, Dietrich R, Noonan RJ, Lach FP, White RR, Goldfarb A, Hadi K, Edwards MM, et al. (2022). Genomic signature of Fanconi anaemia DNA repair pathway deficiency in cancer. Nature 612, 495–502. 10.1038/s41586-022-05253-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meeker AK, Hicks JL, Gabrielson E, Strauss WM, De Marzo AM, and Argani P (2004). Telomere shortening occurs in subsets of normal breast epithelium as well as in situ and invasive carcinoma. Am J Pathol 164, 925–935. 10.1016/S0002-9440(10)63180-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maciejowski J, and de Lange T (2017). Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 18, 175–186. 10.1038/nrm.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cortes-Ciriano I, Lee JJ, Xi R, Jain D, Jung YL, Yang L, Gordenin D, Klimczak LJ, Zhang CZ, Pellman DS, et al. (2020). Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet 52, 331–341. 10.1038/s41588-019-0576-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hackett JA, and Greider CW (2003). End resection initiates genomic instability in the absence of telomerase. Mol Cell Biol 23, 8450–8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grulich AE, van Leeuwen MT, Falster MO, and Vajdic CM (2007). Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet 370, 59–67. 10.1016/S0140-6736(07)61050-2. [DOI] [PubMed] [Google Scholar]
- 25.Tota JE, Engels EA, Madeleine MM, Clarke CA, Lynch CF, Ortiz AP, Hernandez BY, and Chaturvedi AK (2018). Risk of oral tongue cancer among immunocompromised transplant recipients and human immunodeficiency virus-infected individuals in the United States. Cancer 124, 2515–2522. 10.1002/cncr.31359. [DOI] [PubMed] [Google Scholar]
- 26.Wagner CL, Hanumanthu VS, Talbot CC Jr., Abraham RS, Hamm D, Gable DL, Kanakry CG, Applegate CD, Siliciano J, Jackson JB, et al. (2018). Short telomere syndromes cause a primary T cell immunodeficiency. J Clin Invest 128, 5222–5234. 10.1172/JCI120216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jonassaint NL, Guo N, Califano JA, Montgomery EA, and Armanios M (2013). The gastrointestinal manifestations of telomere-mediated disease. Aging cell 12, 319–323. 10.1111/acel.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Popescu I, Mannem H, Winters SA, Hoji A, Silveira F, McNally E, Pipeling MR, Lendermon EA, Morrell MR, Pilewski JM, et al. (2019). Impaired Cytomegalovirus Immunity in Idiopathic Pulmonary Fibrosis Lung Transplant Recipients with Short Telomeres. Am J Respir Crit Care Med 199, 362–376. 10.1164/rccm.201805-0825OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silhan LL, Shah PD, Chambers DC, Snyder LD, Riise GC, Wagner CL, Hellstrom-Lindberg E, Orens JB, Mewton JF, Danoff SK, et al. (2014). Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J 44, 178–187. 10.1183/09031936.00060014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, and Greider CW (1997). Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91, 25–34. [DOI] [PubMed] [Google Scholar]
- 31.Alder JK, Barkauskas CE, Limjunyawong N, Stanley SE, Kembou F, Tuder RM, Hogan BL, Mitzner W, and Armanios M (2015). Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci U S A 112, 5099–5104. 10.1073/pnas.1504780112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans RA, Diamond MS, Rech AJ, Chao T, Richardson MW, Lin JH, Bajor DL, Byrne KT, Stanger BZ, Riley JL, et al. (2016). Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight 1. 10.1172/jci.insight.88328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNally EJ, Luncsford PJ, and Armanios M (2019). Long telomeres and cancer risk: the price of cellular immortality. J Clin Invest 130. 10.1172/JCI120851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armanios M (2022). The Role of Telomeres in Human Disease. Annu Rev Genomics Hum Genet. 10.1146/annurev-genom-010422-091101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haycock PC, Burgess S, Nounu A, Zheng J, Okoli GN, Bowden J, Wade KH, Timpson NJ, Evans DM, Willeit P, et al. (2017). Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol. 10.1001/jamaoncol.2016.5945 2604820 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeBoy EA, Tassia MG, Schratz KE, Yan SM, Cosner ZL, McNally EJ, Gable DL, Xiang Z, Lombard DB, Antonarakis ES, et al. (provisionally accepted). A long telomere syndrome’s pronearia predisposes to clonal hematopoiesis. [DOI] [PMC free article] [PubMed]
- 37.Aubert G, Baerlocher GM, Vulto I, Poon SS, and Lansdorp PM (2012). Collapse of telomere homeostasis in hematopoietic cells caused by heterozygous mutations in telomerase genes. PLoS Genet 8, e1002696. 10.1371/journal.pgen.1002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klein SL, and Flanagan KL (2016). Sex differences in immune responses. Nat Rev Immunol 16, 626–638. 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- 39.Engels EA, Pfeiffer RM, Fraumeni JF Jr., Kasiske BL, Israni AK, Snyder JJ, Wolfe RA, Goodrich NP, Bayakly AR, Clarke CA, et al. (2011). Spectrum of cancer risk among US solid organ transplant recipients. JAMA 306, 1891–1901. 10.1001/jama.2011.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kutler DI, Patel KR, Auerbach AD, Kennedy J, Lach FP, Sanborn E, Cohen MA, Kuhel WI, and Smogorzewska A (2016). Natural history and management of Fanconi anemia patients with head and neck cancer: A 10-year follow-up. Laryngoscope 126, 870–879. 10.1002/lary.25726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Database SS (2021). Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence - SEER 12 Research Data 1975–2019, National Cancer Institute, DCCPS, Surveillance Research Program, based on the November 2021 submission.
- 42.Parry EM, Alder JK, Qi X, Chen JJ, and Armanios M (2011). Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood 117, 5607–5611. 10.1182/blood-2010-11-322149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gable DL, Gaysinskaya V, Atik CC, Talbot CC Jr., Kang B, Stanley SE, Pugh EW, Amat-Codina N, Schenk KM, Arcasoy MO, et al. (2019). ZCCHC8, the nuclear exosome targeting component, is mutated in familial pulmonary fibrosis and is required for telomerase RNA maturation. Genes Dev 33, 1381–1396. 10.1101/gad.326785.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanley SE, Gable DL, Wagner CL, Carlile TM, Hanumanthu VS, Podlevsky JD, Khalil SE, DeZern AE, Rojas-Duran MF, Applegate CD, et al. (2016). Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Science translational medicine 8, 351ra107. 10.1126/scitranslmed.aaf7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alder JK, Stanley SE, Wagner CL, Hamilton M, Hanumanthu VS, and Armanios M (2015). Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest 147, 1361–1368. 10.1378/chest.14-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Podlevsky JD, Bley CJ, Omana RV, Qi X, and Chen JJ (2008). The telomerase database. Nucleic Acids Res 36, D339–343. gkm700 [pii] 10.1093/nar/gkm700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaysinskaya V, Stanley SE, Adam S, and Armanios M (2020). Synonymous Mutation in DKC1 Causes Telomerase RNA Insufficiency Manifesting as Familial Pulmonary Fibrosis. Chest Dec, 2449–2457. 10.1016/j.chest.2020.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meeker AK, Gage WR, Hicks JL, Simon I, Coffman JR, Platz EA, March GE, and De Marzo AM (2002). Telomere length assessment in human archival tissues: combined telomere fluorescence in situ hybridization and immunostaining. Am J Pathol 160, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alder JK, Guo N, Kembou F, Parry EM, Anderson CJ, Gorgy AI, Walsh MF, Sussan T, Biswal S, Mitzner W, et al. (2011). Telomere length is a determinant of emphysema susceptibility. Am J Respir Crit Care Med 184, 904–912. 10.1164/rccm.201103-0520OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, et al. (2008). Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A 105, 13051–13056. 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biol 17, 122. 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edmonson MN, Zhang J, Yan C, Finney RP, Meerzaman DM, and Buetow KH (2011). Bambino: a variant detector and alignment viewer for next-generation sequencing data in the SAM/BAM format. Bioinformatics 27, 865–866. 10.1093/bioinformatics/btr032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee J, Lee AJ, Lee JK, Park J, Kwon Y, Park S, Chun H, Ju YS, and Hong D (2018). Mutalisk: a web-based somatic MUTation AnaLyIS toolKit for genomic, transcriptional and epigenomic signatures. Nucleic Acids Res 46, W102–W108. 10.1093/nar/gky406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Talevich E, Shain AH, Botton T, and Bastian BC (2016). CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 12, e1004873. 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X, Gupta P, Wang J, Nakitandwe J, Roberts K, Dalton JD, Parker M, Patel S, Holmfeldt L, Payne D, et al. (2015). CONSERTING: integrating copy-number analysis with structural-variation detection. Nat Methods 12, 527–530. 10.1038/nmeth.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 58.Layer RM, Chiang C, Quinlan AR, and Hall IM (2014). LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15, R84. 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, and Marra MA (2009). Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645. 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ouhoummane N, Steben M, Coutlee F, Vuong T, Forest P, Rodier C, Louchini R, Duarte E, and Brassard P (2013). Squamous anal cancer: patient characteristics and HPV type distribution. Cancer epidemiology 37, 807–812. 10.1016/j.canep.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 61.Brister JR, Ako-Adjei D, Bao Y, and Blinkova O (2015). NCBI viral genomes resource. Nucleic Acids Res 43, D571–577. 10.1093/nar/gku1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nature biotechnology 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 63.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. Journal of molecular biology 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 64.Moustafa A, Xie C, Kirkness E, Biggs W, Wong E, Turpaz Y, Bloom K, Delwart E, Nelson KE, Venter JC, and Telenti A (2017). The blood DNA virome in 8,000 humans. PLoS Pathog 13, e1006292. 10.1371/journal.ppat.1006292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, Bajari R, Andric D, Gerthoffert F, Lepsa A, Nahal-Bose H, Stein LD, and Ferretti V (2019). The International Cancer Genome Consortium Data Portal. Nature biotechnology 37, 367–369. 10.1038/s41587-019-0055-9. [DOI] [PubMed] [Google Scholar]
- 66.Consortium I.T.P.-C.A.o.W.G. (2020). Pan-cancer analysis of whole genomes. Nature 578, 82–93. 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fine JP, and Gray RJ (1999). A Proportional Hazards Model for the Subdistribution of a Competing Risk. Journal of the American Statistical Association 94, 496–509. 10.1080/01621459.1999.10474144. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S4. List of somatic mutations, copy number alterations and structural variants (supplementary to Figure 2)
Table S5. Clinical characterization of immune deficiency of short telomere syndrome participants with solid tumors and germline mutations not previously reported in the Johns Hopkins Telomere Syndrome Registry (supplementary to Figure 3 and STAR Methods)
Data Availability Statement
Unreported germline variants were submitted to the Telomerase Database (http://telomerase.asu.edu) and ClinVar Annotation Database (https://www.ncbi.nlm.nih.gov/clinvar/). Processed and verified somatic variants, copy number and structural variant calls are included in Table S4. TCGA HNSCC data is accessible from the International Cancer Genome Consortium (ICGC)-25K Data Portal. Additional information required to reanalyze the data is available upon request provided they fall within the limits of the available consent and with approval from the Johns Hopkins Medicine Institutional Review Board. To request access, contact Dr. Mary Armanios at Johns Hopkins School of Medicine as aforementioned. The Key Resources Table includes the links under “Deposited Data”.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CD3 | Abcam | Cat# ab5690; RRID:AB_305055 |
| Rabbit monoclonal anti-CD4 | Abcam | Cat# ab183685; RRID:AB_2686917 |
| Mouse monoclonal anti-CD8a | ThermoFisher Scientific | Cat# 14-0195-82; RRID:AB_2637159 |
| Rabbit polyclonal anti-Ovalbumin | Abcam | Cat# ab181688 |
| Rabbit monoclonal anti-rat IgG | Vector Labs | Cat# AI-4001; RRID:AB_2336209 |
| Goat polyclonal anti-rabbit IgG | Abcam | Cat# ab205718; RRID:AB_2819160 |
| Biological samples | ||
| Human tumor and paired germline samples | This manuscript | N/A |
| Mouse tumor samples | This manuscript | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Citrate unmasking solution | Vector Labs | Cat# H-3300-250 |
| ImmPACT DAB Peroxidase (HRP) Substrate | Vector Labs | Cat# SK-4103 |
| Mayer’s hematoxylin | Sigma-Aldrich | Cat# MHS1 |
| Critical commercial assays | ||
| Qiagen Puregene Blood Kit B | Qiagen | Cat# 158389 |
| Siemens Tissue Preparation System | Siemens Healthineers | N/A |
| TruSeq Nano DNA Library Prep kit | Illumina | Cat # 20015965 |
| Deposited data | ||
| Previously unreported germline variants | This manuscript | ClinVar annotation database: https://www.ncbi.nlm.nih.gov/clinvar/ Telomerase database: http://telomerase.asu.edu |
| Processed genome sequencing data | This manuscript | Table S4 |
| The Cancer Genome Atlas (TCGA) HNSCC genomic sequencing | International Cancer Genome Consortium-25K Data Portal | https://dcc.icgc.org/ |
| Experimental models: Cell lines | ||
| Mouse: Ova-albumin-antigen expressing cells (OvaB6) derived from KrasLSL-G12D/+, Tp53LSL-R172H/+, Pdx1-Cre (KPC) mice | Dr. Robert Vonderheide’s laboratory, University of Pennsylvania | Ref 32 |
| Experimental models: Organisms/strains | ||
| Mouse: B6.Cg-Terctm1RdP/J (mTR-/-) | Jackson Lab | Cat# 004132; RRID:IMSR_JAX:0041 |
| Mouse: C57BL/6 | Jackson Lab | Cat# 000664 |
| Oligonucleotides | ||
| Alexa 488-labeled telomere (AATCCC)3 probe | Panagene | http://www.panagene.com/_ENG/html/dh_product/prod_view/32/?cate_no=3 |
| Software and algorithms | ||
| SeerStat version 8.4.0 | National Cancer Institute | https://seer.cancer.gov/seerstat/ |
| Telometer, ImageJ plugin, v. 3.0.05 | Meeker et al., 2002 | demarzolab.pathology.jhmi.edu/telometer |
| Nikon NIS-Elements v. 5.21.02 | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| Burrows-Wheeler Aligner (v0.7.7) | Li, 2013 | https://github.com/lh3/bwa |
| Picard-tools1.119 | Broad Institute | https://broadinstitute.github.io/picard/ |
| GATK v3.6.0 | Broad Institute | https://gatk.broadinstitute.org/hc/enus |
| MuTect2 v3.6.0 | Broad Institute | https://gatk.broadinstitute.org/hc/enus/articles/360037593851-Mutect2 |
| snpEFF v4.1 | Microsoft Genomics | http://pcingola.github.io/SnpEff/ |
| Variant Effect Predictor Ensembl API | Ensembl | https://useast.ensembl.org/info/docs/tools/vep/index.html |
| Bambino | Edmonson et al., 2011 | https://cgwb.nci.nih.gov/goldenPath/bamview/documentation/index.html |
| Mutalisk mutation analysis toolkit | Lee et al., 2018 | http://mutalisk.org |
| SigProfilerSingleSample v1.3 | Alexandrov et al., 2020 | https://github.com/AlexandrovLab/SigProfilerExtractor |
| CNVkit v0.9.4 | Talevich et al., 2016 | https://cnvkit.readthedocs.io/en/stable/ |
| CONSERTING | Chen et al., 2015 | http://www.stiuderesearch.org/site/lab/zhang |
| Manta v0.29.6 | Chen et al., 2016 | https://github.com/Illumina/manta |
| Lumpy v0.2.11 | Layer et al., 2014 | https://github.com/arq5x/lumpy-sv |
| bedTools | Quinlan Lab, University of Utah | https://bedtools.readthedocs.io/en/latest/ |
| circos v0.69 | Krzywinski et al., 2009 | http://circos.ca/software/download/circos/ |
| ShatterSeek | Cortes-Ciriano et al. | https://github.com/parklab/ShatterSeek |
| kallisto v0.43.1 | Bray et al. | https://pachterlab.github.io/kallisto/about |
| FACSDiva v.8.0.1 | BD Biosciences | https://www.bdbiosciences.com/enus/products/software/instrument-software/bd-facsdiva-software |
| FlowJo v.10.1 | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads/ |
| Halo image analysis software | Indica Labs | https://indicalab.com/halo/ |
| GraphPad Prism v.7.04 | GraphPad Software | https://www.graphpad.com/ |