

Abstract
Acute lung injury (ALI) is still associated with high mortality. Growing evidence suggests that Club Cell Protein 16 (CC16) plays a protective role against ALI. However, the doses of recombinant CC16 (rCC16) used in preclinical studies are supraphysiological for clinical applications. Extracellular vesicles (EVs) are nanovesicles endogenously generated by mammalian cells. Our study demonstrated that CC16 is released via small EVs and EV-encapsulated CC16 (sEV-CC16) and has anti-inflammatory activities, which protect mice from lipopolysaccharide (LPS) or bacteria-induced ALI. Additionally, sEV-CC16 can activate the DNA damage repair signaling pathways. Consistent with this activity, we observed more severe DNA damage in lungs from Cc16 knockout (KO) than wild-type (WT) mice. Mechanistically, we elucidated that CC16 suppresses nuclear factor κB (NF-κB) signaling activation by binding to heat shock protein 60 (HSP60). We concluded that sEV-CC16 could be a potential therapeutic agent for ALI by inhibiting the inflammatory and DNA damage responses by reducing NF-κB signaling.
Keywords: SCGB1A1, acute respiratory distress syndrome, DNA repair, inflammation, NF-κB signaling, HSP60
Graphical abstract
The discovery of Club Cell Protein 16-containing extracellular vesicles provides a potential treatment to reduce morbidity and mortality in acute lung injury and acute respiratory distress syndrome.
Introduction
Acute lung injury (ALI) is a clinical syndrome associated with extensive lung inflammation, damage to the parenchyma, severe hypoxemia, and respiratory failure.1 ALI can deteriorate into a more severe form of acute respiratory distress syndrome (ARDS), which is difficult to treat and hence is associated with substantial morbidity and mortality.2 ARDS is often observed in infections, sepsis, aspiration of gastric contents, severe trauma, and patients in intensive care units.1,2,3 As ALI causes severe injury to the alveolar epithelium that often results in incomplete repair,4 long-term quality of life is poor in some ALI survivors.5,6 Primary pneumonia caused by viruses, bacteria, and fungi is the most common cause of ALI.7 Recently, the number of patients with pneumonia in coronavirus disease 2019 (COVID-19), caused by the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has dramatically increased, and ALI represents one of the most severe manifestations of COVID-19.8 Thus, ALI/ARDS is now receiving increased clinical and basic research interest.
Inflammation is the host immune response to ALI characterized by pulmonary infiltration of immune cells, such as neutrophils and macrophages.9,10 Although the inflammatory response is essential for host defense, excessive inflammation often causes collateral damage to the tissues.11 Excessive pro-inflammatory cytokines and chemokines produced by infiltrated inflammatory cells cause edema and deterioration of gas exchange.12 In addition, ARDS can lead to progressive multiple organ dysfunction caused by a cytokine storm in patients with severe infection, such as COVID-19.13,14 This phenomenon indicates that control of excessive inflammation is crucial for the treatment of ALI/ARDS.
Extracellular vesicles (EVs) are nanoparticles naturally released from cells.15 According to the International Society for Extracellular Vesicles, EVs can be classified as small EVs (sEVs; diameter < 200 nm) and medium/large EVs (m/lEVs; diameter > 200 nm).16 Recent studies have attempted to identify the biological functions of EVs after Raposo et al., reported that EVs carry antigens of their parent cells.17 EVs can enter cells by fusing with the membrane without the need for a specific receptor.18 EVs are a promising drug delivery vehicle because of their high stability, biocompatibility, low immunogenicity and toxicity, and the fact that diverse molecules (microRNA [miRNA], small interfering RNA [siRNA], DNA, proteins, and chemical drugs) can be loaded into them.19,20
Club cells are important multifunctional cells within the mammalian bronchial epithelium. Club cell protein (CC16) is the major secreted protein of club cells.21 CC16 exerts protective effects against oxidative stress and inflammation in the respiratory tract.22 CC16 suppresses pulmonary inflammation triggered by allergens, ozone, cigarette smoke (CS), and viruses.23 In addition, reduced bronchoalveolar lavage fluid (BALF) CC16 levels were observed in various lung diseases, including chronic obstructive pulmonary disease (COPD), asthma, and ALI/ARDS.23,24
In this study, we found that sEV-encapsulated CC16 (sEV-CC16) levels were decreased in BALF from patients with pneumonia compared with normal controls. To explore the role of sEV-CC16, we delivered sEV-CC16 obtained from BEAS-2B cells stably overexpressing CC16 to mice with lipopolysaccharide (LPS)-induced ALI. sEV-CC16 displayed therapeutic benefits in mice by protecting them from LPS-induced lung injury. Further, we attempted to elucidate the mechanism by which CC16 suppressed inflammation and DNA damage. Our data indicated that CC16 was able to suppress the activation of nuclear factor κB (NF-κB) signaling by binding to heat shock protein 60 (HSP60). Taken together, results from our study demonstrate the ability of sEV-CC16 to prevent ALI in a preclinical model by modulating the inflammatory and DNA damage responses via reducing NF-κB signaling and suggest that sEV-CC16 can be developed into a potential therapy for ALI/ARDS.
Results
sEV-derived CC16 is reduced in BALF from pneumonia patients
It has been reported that BALF-derived sEVs from patients with sarcoidosis contributed to inflammation by stimulating cytokine production from immune and epithelial cells.25 Although CC16 is a major secreted protein in epithelial lining fluid, it is unknown whether CC16 can be secreted within EVs. To address the question, we purified sEVs from human BALF (Table S1) and characterized them using nanoparticle tracking analysis (NTA). The size distributions and average diameters of purified BALF sEVs are shown in Figures 1A and 1B. The particle numbers of 200 μg of BALF sEV from normal human and pneumonia patients were approximately 2.86 × 1011 particles/mL and 2.90 × 1011 particles/mL, respectively (Figure 1C). No differences in size and concentration were observed between the two groups of sEVs. Transmission electron microscopy (TEM) and immunogold labeling with CC16 were conducted to show the morphological shape of sEVs and confirm the presence of CC16. CC16 was abundantly expressed in sEVs from healthy subjects (Figure 1D). However, CC16 protein levels were much lower in BALF m/lEVs from healthy subjects and BALF sEVs from pneumonia patients (Figure 1D). Flow cytometry analysis also showed that healthy subjects have more CC16-positive sEV particles compared with sEVs from pneumonia patients (Figures 1E and 1F). To further validate our results, a quantitative assessment of CC16 using mass spectrometry (MS) was performed, which is a mainstream molecular-level analysis technique.26 The number of CC16-derived amino acids within BALF sEVs was two orders of magnitude higher for normal subjects versus patients with pneumonia (4.9 × 106 versus 5.6 × 104, respectively) (Figures 1G and 1H). Considering the heterogeneity of BALF sEVs,27 we enriched CC16-positive sEVs (CC16+ sEVs) from total sEVs to elucidate the cell source of CC16-carrying sEVs. As shown in Figure S1, sEV membrane proteins (CD9, CD63, FLOT1) were detected in both total human BALF sEVs and CC16+ sEVs. Interestingly, the Pan Cytokeratin (pan-CK; epithelial cell marker) expression level was increased in CC16+ sEVs compared with the total sEVs. Meanwhile, protein levels of MPO (neutrophil marker), α-SMA (smooth muscle cell marker), and CD31 (endothelial cell marker) were decreased in CC16+ sEVs, indicating that CC16+ sEVs are mainly released from pulmonary epithelial cells. Furthermore, we confirmed our observations above using the LPS-induced mouse model of ALI. The murine BALF sEVs were characterized by NTA (Figures S2A–S2D). Consistent with the human data, the number of CC16-derived amino acids in sEVs from PBS- and LPS-treated mice were detected by MS, respectively (Figures S2E and S2F). These results indicate that CC16 is secreted through sEVs and that CC16 secretion in sEVs is reduced during ALI.
Figure 1.

sEV-derived CC16 is reduced in BALF from pneumonia patients
(A–D) Two-hundred micrograms of BALF sEVs from pooled normal human (n = 10) and pneumonia patients (n = 21) are randomly pooled as five pairs per group. sEVs samples are examined by NTA to analyze size distribution (A), average size (B), and particle numbers (C). Data are mean ± SD. Ns, not significant, p > 0.05. (D) Representative TEM images of BALF-derived sEVs are shown. sEV samples were stained using immunogold labeling with the antibody against CC16. Scale bar, 100 nm. (E and F) Detection of CC16-positive BALF sEVs from healthy (n = 10) and pneumonia patients (n = 21) using flow cytometry (E). Mean fluorescence intensity (MFI) is measured (F). The boxes in the boxplots show the medians with 25th and 75th percentiles, and the whiskers show the minimum and maximum values. ∗∗p < 0.01 versus the normal group. (G and H) Pooled BALF sEVs from normal human (n = 10) and pneumonia patients (n = 21) were purified. Twenty micrograms of pooled sEVs from each group are used for measuring the amino acid number of CC16 by MS.
Preparation and characterization of CC16-enriched sEVs
It has been shown that stable producer cells can be used to produce engineered sEVs.28 To generate CC16-enriched sEVs, BEAS-2B cells were transfected with a plasmid encoding human CC16 or vector. Stably transfected BEAS-2B cells were selected as described in Figure S3A. CC16 overexpression was confirmed using immunofluorescence staining (Figure S3B). sEVs released from BEAS-2B-Con (sEV-Con) and BEAS-2B-CC16 (sEV-CC16) were purified and characterized. The morphology of sEVs in both groups under TEM has a typical cup-like shape with a heterogeneous size range of less than 200 nm (Figure 2A). NTA results revealed that the average sizes of sEV-Con and sEV-CC16 were 141 and 149 nm, respectively (Figures 2B and 2C) and the particle numbers of sEV-Con and sEV-CC16 were 3.62 × 1011 and 3.23 × 1011, respectively (Figure 2D). Next, we verified sEV-Con and sEV-CC16 based on CC16 content, protein markers, CC16-positive particles, and the number of CC16-derived amino acids. First, ELISA showed that there was a significantly higher amount of CC16 in sEV-CC16 than in sEV-Con (Figure 2E). After calculating CC16 protein copy numbers in one single sEV particle, we found sEV-CC16 contained about 14 copies of CC16, whereas sEV-Con contained about one copy (Figure 2F). Results from the western blot analysis confirmed increased CC16 protein levels in sEV-CC16 versus sEV-Con; however, both types were positive for CD9, CD63, FLOT1, and β-actin and were negative for ribosomal protein SP-1 (Figure 2G). In addition, increased numbers of CC16-positive sEVs and CC16-derived amino acid numbers in sEV-CC16 were validated using flow cytometry analysis (Figures 2H and 2I) and MS (Figures 2J and 2K), respectively. These data demonstrated that the strategy for preparing sEV-CC16 is feasible and valid.
Figure 2.

Preparation and characterization of sEV-Con and sEV-CC16
(A) TEM images of sEV-Con and sEV-CC16 isolated from culture media of BEAS-2B-Con and BEAS-2B-CC16. Scale bar, 100 μm. (B–D) Two-hundred micrograms of sEV-Con and sEV-CC16 are characterized using NTA for size distribution (B), diameter (C), and particle number (D). Results are represented as mean ± SD of a minimum of three independent experiments. ns, p > 0.05; ∗∗p < 0.01. (E) The CC16 protein amount in 200 μg of sEV-Con and sEV-CC16 is quantified using ELISA (n = 4). (F) Copy number of CC16 in single sEV-Con and sEV-CC16 is calculated (n = 4). Results are presented as mean ± SD. ∗∗p < 0.01. (G) sEV positive markers (CD9, CD63, and Flot1) and a negative marker (Sp1) are detected in 50 μg of protein from BEAS-2B-Con, BEAS-2B-CC16, sEV-Con, and sEV-CC16 using western blot (n = 3). (H and I) Detection of CC16-positive sEVs from sEV-Con and sEV-CC16 using flow cytometry (n = 5). Results are presented as mean ± SD. ∗∗p < 0.01. (J and K) sEV-Con and sEV-CC16 isolated from five independent sEV isolation processes were pooled and analyzed by MS. The number of CC16-derived amino acids in 20 μg of pooled sEV-Con and sEV-CC16 are quantified.
sEV-CC16 attenuates LPS and bacterial-induced lung injury in mice
Next, we evaluated whether sEV-CC16 could exert a protective effect against ALI. As shown in Figure 3A, 3 h after LPS intratracheal instillation (i.t.), sEV-Con or sEV-CC16 was also administered to the mice via the i.t. route. First, we assessed organ/tissue distributions of XenoLight DiR dye-labeled sEV-Con and sEV-CC16. Twenty-four hours after administration of sEVs, the obtained images showed lung-specific fluorescence intensity of DiR, indicating that sEV-Con and sEV-CC16 were effectively delivered into the lung rather than other major tissues/organs (Figure 3B). We next evaluated lung inflammation and injury, where rCC16 treatment was used as a positive control and to compare the treatment efficacy of sEV-CC16 and rCC16. BALF counts of macrophages (PBS, 2.11 ± 0.20 × 105/mL; sEV-Con, 2.18 ± 0.41 × 105/mL; sEV-CC16, 1.05 ± 0.25 × 105/mL; rCC16, 1.02 ± 0.15 × 105/mL) and neutrophils (PBS, 18.98 ± 1.78 × 105/mL; sEV-Con, 18.74 ± 3.19 × 105/mL; sEV-CC16, 9.44 ± 2.22 × 105/mL; rCC16, 9.16 ± 1.37 × 105/mL) were significantly reduced in sEV-CC16-treated mice and to a similar extent in rCC16-treated mice (Figures 3C–3E). Similarly, lung injury assessed by histopathology (Figures 3C and 3F) and lung wet/dry weight ratio (Figure 3G) were significantly decreased in sEV-CC16- and rCC16-treated groups. sEV-CC16 and rCC16 had comparable anti-inflammatory activities as assessed by reduced cytokines (interleukin [IL]-1β and tumor necrosis factor alpha [TNF-α]) and chemokines (CXCL1 and CXCL2) in BALF (Figures 3H–3K). In our experiment, sEV-Con did not show any immunomodulatory effect compared with the PBS-only-treated group, indicating that the therapeutic benefits of sEV-CC16 came from overexpressed CC16. Furthermore, both sEV-CC16 and rCC16 reduced the mRNA levels of cytokines (Il-1β, Il-6, and TNF-α) and chemokines (Cxcl1, Cxcl2, and Ccl2) in lung tissues and bronchoalveolar lavage (BAL) cells (Figures S3C–S3F). Besides LPS, we validated our data using a murine model of bacterial-induced ALI. As shown in Figure S4A, 3 h after Klebsiella pneumoniae infection, sEV-Con or sEV-CC16 was also administered to the mice via i.t. Like LPS, BALF counts of macrophages (sEV-Con, 0.87 ± 0.25 × 105/mL; sEV-CC16, 0.21 ± 0.05 × 105/mL; rCC16, 0.24 ± 0.12 × 105/mL) and neutrophils (sEV-Con, 7.84 ± 2.22 × 105/mL; sEV-CC16, 1.93 ± 0.43 × 105/mL; rCC16, 2.38 ± 1.26 × 105/mL) were significantly reduced in sEV-CC16-treated mice and rCC16-treated mice (Figures S4B–S4D). Corroborating this, lung injury score (Figures S4B and S4E) and lung wet/dry weight ratio (Figure S4F) were significantly reduced with sEV-CC16 and rCC16 treatments. In addition, inflammatory cytokines and chemokines were also significantly reduced by treatment with sEV-CC16 and rCC16 (Figures S4G–S4L). These results indicated that delivery of CC16-enriched sEVs can effectively attenuate endotoxin- and live bacterial-induced ALI.
Figure 3.

sEV-CC16 protects against LPS-induced lung injury in mice
Three hours after LPS, mice (five to eight mice per group) were given PBS, sEV-Con, sEV-CC16, or rCC16 and sacrificed 24 h after the indicated treatments. Schematic illustration of delivery of sEV-Con and sEV-CC16 into LPS-pretreated mice (A). In vivo imaging systems (IVIS) images of major organs obtained 24 h after the delivery of DiR-labeled sEVs (B). H&E staining of BAL cells and lung sections. M, macrophage; N, neutrophil; red arrows, neutrophils; blue arrows, alveolar disruption with hyaline membranes. Scale bar, 100 μm (C). The number of BALF macrophages (D) or neutrophils (E). Lung injury scored (F). Lung wet-to-dry weight ratios (G). Protein levels of IL-1β (H), TNF-α (I), CXCL-1 (J), and CXCL-2 (K) in BALF detected using ELISAs. The results presented as mean ± SD. In (D)–(I) and (K), the data were analyzed by a one-way ANOVA followed by Tukey’s HSD. In (J), the boxes in the boxplots show the medians with 25th and 75th percentiles, and the whiskers show the minimum and maximum values. Data are analyzed by the Kruskal-Wallis one-way analysis followed by pairwise comparisons using Mann-Whitney U tests. ns, p > 0.05; ∗p < 0.05; ∗∗p < 0.01.
sEV-CC16 attenuates LPS-induced lung injury in CC16 knockdown mice without complications
We confirmed that both cellular and released CC16 protein were decreased by LPS-induced lung injury (Figures S5A–S5D), which is in line with previous studies.29,30 To better address the importance of sEV-CC16 during ALI, we knocked down CC16 in wild-type (WT) mice using CC16 siRNA. The reduced CC16 protein expression was confirmed on day 3 and day 5 after delivering CC16 siRNA (Figures S6A and S6B). We also found that both secreted soluble CC16 and sEV-CC16 are decreased (Figures S6C and S6D). No adverse effects were observed in mice after the knockdown of CC16, such as body weight loss (Figure S6E). As illustrated in Figure S7A, we further explored whether CC16 knockdown can lead to more severe damage during ALI and tested the therapeutic effect of sEV-CC16, as well as its potential adverse effects. Twenty-four hours after LPS treatment, BALF cell counting and lung wet/dry ratio were significantly increased in the CC16 knockdown group, while administration of sEV-CC16 suppressed inflammatory response (Figures S7B–S7E). In addition to 24 h, we evaluated the long-term effect after sEV-CC16 administration. As shown in Figures S7F–S7H, LPS-induced body weight loss is recovered by administration of sEV-CC16 in both control and CC16 knockdown groups. sEV-CC16 administration also attenuates lung injury in LPS-treated mice on day 7 (Figures S7I–S7K). Furthermore, spleen enlargement was ameliorated on day 7 after sEV-CC16 treatment (Figure S8), indicating that sEV-CC16 administration did not cause specific complications.
Tracking sEV-CC16 recipient cells that mediate the anti-inflammatory effect
Our previous study showed that lung macrophages efficiently take up intratracheally instilled sEVs.31 Using the same methods, we labeled sEV-Con and sEV-CC16 with PKH26 to track their distribution in the lung. Co-localization of PKH26 (red) and CD68-positive cells (green, a marker of macrophage) were observed in lungs and BAL cells (Figures 4A and 4B), indicating that sEVs derived from BEAS-2B cells also can be taken up by lung macrophages. Next, we determined the uptake and anti-inflammatory effect of sEV-Con and sEV-CC16 using macrophage-like THP-1 cells. PKH67-labeled sEVs were efficiently taken up by THP-1 cells (Figure 4C). The anti-inflammatory role of sEV-CC16 was confirmed by decreased LPS-induced secretion of cytokines (IL-1β and TNF-α) and chemokines (CXCL1 and CXCL2) (Figures 4D–4G). The mRNA levels of cytokines (IL-1β, IL-6, and TNF-α) and chemokines (CXCL2 and CCL2) were reduced by sEV-CC16 compared with sEV-Con (Figures S9A–S9E). Similarly, sEV-CC16 inhibited cytokine gene expression induced by K. pneumoniae (Figures S9F–S9H). Further, sEV-CC16 also induced anti-inflammatory effects on murine bone marrow-derived primary macrophages (BMDMs) (Figures S9I–S9S).
Figure 4.

sEV-CC16 inhibits LPS-induced inflammation in macrophages
(A and B) Mice (n = 5 per group) received 1 μg of LPS (in 50 μL of PBS) via i.t. After 3 h, mice were given 7.5 × 1010 (in 50 μL of PBS) PKH26-labeled sEV-Con or PKH26-labeled EV-CC16 via the i.t. route. Mice were sacrificed 24 h after sEV treatment. Immunofluorescence staining was performed in lung sections (A) and BAL cells (B) using an antibody against CD68 (a macrophage marker) to track the uptake of sEV-Con or sEV-CC16. Scale bar, 100 μm. (C) EV-Con or EV-CC16 labeled with PKH67 was added to THP-1. After 24 h, cells were washed and the internalization of labeled sEV-Con or sEV-CC16 was detected using a fluorescence microscope. Scale bar, 100 μm. (D–G) Differentiated THP-1 cells were treated with 100 ng/mL LPS, 5 × 108/mL sEV-Con, 5 × 108/mL sEV-CC16, or 1 μg/mL rCC16 for 24 h. The amount of released IL-1β (D), TNF-α (E), CXCL-1 (F), and CXCL-2 (G) in culture media were detected using ELISAs. Results represent mean ± SD of three independent experiments and the data were analyzed by a one-way ANOVA followed by Tukey’s HSD. ∗∗p < 0.01.
Co-localization of PKH26 (red) and pan-CK (green, a marker of epithelial cells) was observed in mice with LPS-induced ALI (Figure 5A), which indicated that intratracheally delivered sEVs target airway epithelial cells as well as macrophages. In vitro, PKH26-labeled sEVs were internalized by normal human bronchial epithelial (NHBE) cells, as well as BEAS-2B cells (Figures 5B and 5C). Functionally, sEV-CC16 effectively reduced the cytokine and chemokine expression in BEAS-2B cells (Figures 5D–5F). Surprisingly, rCC16 failed to reduce the expression of these inflammation markers. Collectively, our results indicated that sEV-CC16 exerts an anti-inflammatory role by targeting macrophages and airway epithelial cells.
Figure 5.

sEV-CC16 inhibits bacterial-induced inflammation in lung epithelial cells
(A) Mice (n = 8 per group) received 1 μg of LPS (in 50 μL of PBS) via i.t. After 3 h, mice were given 7.5 × 1010 (in 50 μL of PBS) unlabeled or PKH26-labeled sEV-CC16. Mice were sacrificed 24 h after sEV-CC16 treatment. Immunofluorescence staining was performed in lung sections using an antibody against pan-CK (green, a marker of epithelial cell) to track the uptake of sEV-CC16. Scale bar, 100 μm. (B–F) NHBE and BEAS-2B cells were infected with K. pneumoniae (K. p) at an MOI of 1:5 ratio for 1 h. After washing, the cells were treated with 5 × 108/mL PKH26-labeled sEV-Con or sEV-CC16. The internalization of sEVs into NHBE (B) and BEAS-2B (C) is observed with a fluorescence microscope. Scale bar, 100 μm. (D–F) 5 × 108/mL sEV-Con, sEV-CC16 or 5 μg/mL rCC16 were added to K. pneumoniae (K. p)-infected BEAS-2B. mRNA levels of IL-1β (D), IL-6 (E), and IL-8 (F) were detected using qRT-PCR at 24 h after the sEV treatment. Results represent mean ± SD of three independent experiments and the data were analyzed by a one-way ANOVA followed by Tukey’s HSD. ns, p > 0.05; ∗∗p < 0.01.
Transcription profile altered by sEV-CC16 in LPS-treated macrophages
Macrophages play a key role in the innate immune system.32 To better understand the effects of sEV-CC16 on the inflammatory gene expression profile, a microarray analysis was conducted using Clariom S Assay. The expression of 21448 genes was compared after adding sEV-Con or sEV-CC16 to LPS-treated THP-1 cells. The regulated genes in the heatmap showed several distinct clustering areas between sEV-Con and sEV-CC16 (Figure 6A). We enriched this dataset of differentially expressed genes using wikiPathway analysis, and we confirmed that inflammation-related pathways were regulated by inhibiting the expression of pro-inflammatory genes (Table S2). Unexpectedly, we found that treatment of sEV-CC16 significantly activated multiple DNA damage repair-related pathways, including DNA replication, DNA mismatch repair, DNA damage and cellular response, and DNA repair pathways (Figure 6B). Moreover, several DNA repair genes, such as LIG1, PCNA, BRCA2, and EXO1, were significantly upregulated by sEV-CC16 treatment compared with the sEV-Con (Figure 6C). The upregulation of these DNA repair genes was confirmed by qRT-PCR in murine BAL cells after administration of sEV-CC16 (Figure S10A) and human THP-1 macrophages (Figure S10B). Then, we evaluated LPS-induced DNA damage using a Comet assay after treating THP-1 cells and BEAS-2B cells with sEV-Con, sEV-CC16, or rhCC16. sEV-CC16 suppressed LPS-induced DNA damage in both cell lines (Figures 6D–6G), as demonstrated by the decreased length of tail DNA. However, rCC16 only decreased DNA damage in macrophages, not the epithelial cells. To compare the effect of sEV-Con or sEV-CC16 on DNA repair-related gene expression in epithelial cells induced by K. pneumoniae exposure, we conducted TaqMan Array and identified four upregulated genes, POLQ, MBD4, MAPK9, and MGMT (Figure S10C). These genes were upregulated by sEV-CC16 in NHBE and BEAS-2B (Figures S10D and S10E), indicating that sEV-CC16 not only inhibits inflammation but also activates DNA repair pathways.
Figure 6.

Transcription profile altered by sEV-CC16 in LPS-treated macrophage-like cells
(A-C) 100 ng/mL LPS-treated THP-1 were incubated with 5 × 108/mL sEV-Con or sEV-CC16 for 24 h. Human Gene Clariom S assay was used to represent differential gene expression by a heatmap between sEV-Con- and sEV-CC16-treated THP-1 (n = 3 per group). Hierarchical clustering indicated individual gene expression into blue (reduced expression) and red (increased expression) (A). Transcriptome Analysis Console was conducted for pathway analysis (B). DNA repair pathway-related genes are depicted, and qRT-PCR-confirmed genes are indicated in the scatter plot. Significance was calculated using two-sided Fisher’s exact test (C). (D–G) 100 ng/mL LPS-treated THP-1 (D and E) or 1 μg/mL LPS-treated BEAS-2B (F and G) (n = 5 per group) were incubated with 5 × 108/mL sEV-Con, or 5 × 108/mL sEV-CC16, or 2 μg/mL rCC16 for 24 h. Images of representative Comet assay images were taken using a fluorescence microscope. Scale bar, 100 μm. The length of tail DNA was quantified using ImageJ software. Results represent mean ± SD and the data were analyzed by a one-way ANOVA followed by Tukey’s HSD. ns, p > 0.05; ∗∗p < 0.01.
sEV-CC16 protects against lung injury by modulating the DNA repair pathways
Based on our observations from in vitro analysis, Cc16 KO mice were used to further confirm the role of CC16 in DNA repair in vivo (Figure S11A). Lung sections from unchallenged 3- and 18-month-old WT mice and Cc16 KO mice were stained with γ-H2AX, a marker of DNA damage. As shown in Figures S11B and S11C, 18-month-old Cc16 KO mice showed a significantly increased number of γ-H2AX-positive cells in lung tissues compared with 18-month-old WT. We observed similar results using TUNEL assay, which also indicated enhanced DNA damage in Cc16 KO mouse lungs compared with WT (Figures S11D and S11E). Next, we evaluated the DNA damage in the LPS-treated mice. Double immunofluorescence staining of p-γ-H2AX and pan-CK suggests that sEV-CC16 but not rCC16 administration can strongly attenuate LPS-induced DNA damage in lung epithelial cells (Figures 7A and 7B), which is in line with the sEV-CC16 reduced apoptosis (Figures 7C and 7D). Consistently, sEV-CC16 at 5 × 108/mL (approximately 0.4 μg/mL) fully rescued the decrease of viability caused by LPS in BEAS-2B cells (Figure 7E). In contrast, rCC16 at 5 μg/mL shows no beneficial effect on LPS-reduced cell viability.
Figure 7.

sEV-CC16 protects against DNA damage induced by LPS
Mice received 1 μg of LPS in (50 μL of PBS) via the i.t. route. After 3 h, mice were treated with sEV-Con, sEV-CC16, or cCC16 via the i.t. route. Mice were sacrificed 24 h after sEV treatment. (A and B) Lung sections (n = 3 per group) were stained using an antibody against γ-H2AX (DNA damage marker) and pan-CK (epithelial cell marker). sEV-Con-treated mice were stained with a non-immune rabbit IgG. Scale bar, 100 μm (A). Relative fluorescence intensity is measured by ImageJ software (B). (C and D) TUNEL staining (n = 3 per group) was conducted to detect DNA damage in the lung (C). TUNEL-positive cells were calculated in the lung tissues (D). (E) BEAS-2B cells were treated with 100 μg/mL LPS, 5 μg/mL rCC16, 5 × 108/mL sEV-Con, or 5 × 108/mL sEV-CC16 as indicated for 24 h. Cell viability was detected by MTS Assay Kit. In (B) and (D), results are mean ± SD and analyzed by a one-way ANOVA followed by Tukey’s HSD. In (E), data are presented as mean ± SD. ns, p > 0.05; ∗∗p < 0.01.
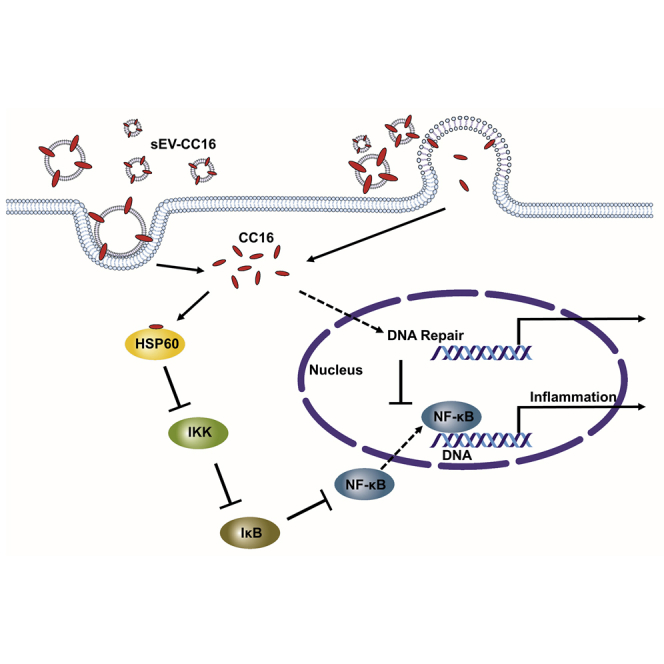
CC16 suppresses NF-κB activity through the interaction with HSP60
We aimed to identify the CC16-binding protein(s) to elucidate the molecular mechanism by which CC16 reduces inflammation and DNA damage upon lung injury. We conducted glutathione S-transferase (GST) pull-down combined with MS analysis and identified pyruvate kinase M1/2 (PKM1/2, also called PKM2) and HSP60 as potential CC16-binding proteins (Figure 8A). Next, we performed coimmunoprecipitation (coIP) to validate the interaction between CC16 and HSP60. However, we did not observe the interaction between CC16 and PKM1/2 using a coIP assay (Figure 8B). Chun et al. reported that Hsp60 activates NF-κB signaling by directly interacting with an inhibitor of κB kinase (IKK) complex.33 We also observed the same interaction between HSP60 and IKKα (Figure 8C). However, when we overexpressed CC16 in 293T cells, the interaction between HSP60 and IKKα was blocked (Figure 8C). Based on this finding, we hypothesized that intracellular CC16 could inhibit NF-κB activity by interacting with HSP60. Consequently, intracellular CC16 reduces the inflammatory response and enhances DNA repair processes. To verify this hypothesis, we measured NF-κB reporter activity and found sEV-CC16 significantly reduced NF-κB reporter activity in THP-1 and 293T cells (Figures 8D and 8E). We also conducted an NF-κB luciferase reporter assay and western blot after the overexpression of CC16 and HSP60. Overexpression of HSP60 induced NF-κB signaling in response to LPS treatment, while CC16 overexpression can rescue HSP60-induced NF-κB activation (Figure 8F). In addition, overexpression of HSP60 in LPS-activated BEAS-2B and NHBE cells increased the expression of pro-inflammation markers, including IL-1β, CXCL2, and IL-8 (Figures S12A and S12B). The pro-inflammatory effect induced by HSP60 overexpression was rescued when a CC16-expressing plasmid was co-transfected into NHBE cells (Figure S12C). As shown in Figure 8G, overexpression of CC16 can inhibit the phosphorylation and degradation of IκBα, which is induced by LPS and HSP60 overexpression. Furthermore, the protein expression of NF-κB p65 in the nuclear fraction was increased by LPS and HSP60, but CC16 can suppress the nuclear translocation of p65, indicating that CC16 is a negative regulator of NF-κB signaling by blocking the interaction between HSP60 and IKK.
Figure 8.

CC16 suppresses NF-κB activity through interaction with HSP60
(A) A representative image of affinity chromatography using GST and GST-tagged CC16. (B) pRP-CMV-CC16 was co-transfected with pcDNA-HSP60 or pEGFP-C1-PKM2 into 293T cells at 1:1 ratio. After 24 h, 1 μg/mL LPS was added to the cells. CoIP assay was conducted using lysates 48 h after transfection (n = 3). (C) pRP-CMV-CC16, pcDNA-HSP60, or empty vector were transfected into BEAS-2B cells at 1:1 ratio. Twenty-four hours later, 1 μg/mL LPS was added to the cells. CoIP assay was conducted using BEAS-2B cell lysates 48 h after transfection (n = 3). (D and E) sEV-Con or sEV-CC16 were added together with LPS after transfecting pNL3.2. NF-κB-RE plasmid. NF-κB reporter activity in THP-1 cells (D) and 293T cells (E). (F and G) pRP-CMV-CC16, pcDNA-HSP60, or empty vector was transfected into 293T cells (F) or THP-1 cell (G) at 1:1 ratio. Twenty-four hours after transfection, LPS was added to the cells for an additional 24 h. NF-κB reporter activity (F) and nuclear translocation of NF-κB sub-unit in cytoplasmic and nuclear fractions (G) were determined. β-Actin and H3 were used as a loading control for western blot. In (D) and (E), results are mean ± SD of three independent experiments and analyzed by two-tailed unpaired Student’s t test. In (F), results represent mean ± SD and were analyzed by one-way ANOVA followed by Tukey’s HSD. ns, p > 0.05; ∗p < 0.05; ∗∗p < 0.01.
Discussion
An increasing number of high-profile studies on EVs have described their significant roles in the regulation of several molecular pathways related to aging, cancer, infectious diseases, and obesity.15 Although EV studies have attracted widespread interest following the publication of rigorously conducted EV studies, consensus still has not emerged on the classification of EV subtypes such as endosome-origin “exosomes” (size range 50–150 nm) and plasma membrane-derived “ectosomes” (size range 100–500 nm).34 Several studies recently tried to distinguish exosomes and ectosomes based on specific protein expression levels of CD6 and CD63 to separate exosomes and small ectosomes in the same size range.35 Nonetheless, these studies have only been conducted on certain cell lines. Therefore, we conducted this study using heterogeneous particles of less than 200 nm as sEVs (commonly known as exosomes) following the guideline Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV 2018).16 Since it has been reported that sEVs can efficiently deliver various therapeutic materials to target cells, sEV-based drug delivery has been considered a potential therapeutic system.36 sEVs are secreted by most types of cells and can be detected in various body fluids, including saliva, breast milk, synovial fluid, bile, blood, urine, and BALF.15 In this study, we used BALF, which is a bronchoalveolar washing fluid containing the lung immune cells; thus, important immunological information about the lung can be provided.37
CC16 is the major protein secreted by club cells and the most abundant protein in BALF, accounting for 2%–3% of the total protein.21 Decreases in levels of CC16 in the BALF are closely linked to lung injury, including ALI and its severe form, ARDS. Lung injury allows excess fluid to move into the lungs from circulation due to increased epithelial permeability induced by an inflammatory response, causing decreased lung compliance and severe hypoxemia associated with high mortality.38 Functionally, CC16 has an anti-inflammatory effect against various insults, such as allergens, viruses, bacteria, and CS.29,39 In Cc16 deficient condition, increased emphysema development and pulmonary inflammation were observed compared with age-matched WT mice. In particular, Cc16 KO mice showed increased apoptosis and inflammatory responses compared with WT mice in pathological conditions such as CS exposure40,41 and asthma.42 In addition, Cc16 KO mice showed spontaneous development of airspace enlargement, higher lung compliance, and lower lung resistance than WT mice over 18 months of age.41 Although CC16 showed a lot of beneficiary effects, alveolar barrier damage caused by inflammatory responses can trigger CC16 leakage from the respiratory tract into blood vessels,43 and circulating CC16 is eventually eliminated from the body by renal clearance.44 The expression level of CC16 has also been shown to be decreased during LPS-induced ALI.29 Thus, during ALI, reductions in lung CC16 levels may contribute to excessive inflammation and associated lung injury.
BALF-derived sEVs are mainly secreted from lung resident cells such as alveolar macrophages, epithelial cells, stem cells, and endothelial cells.45 Bourdonnay et al. reported that sEVs mediate the crosstalk between alveolar macrophages and epithelial cells in the inflammatory response.46 Furthermore, the study suggested that molecules packaged into sEV may exhibit physiological similarities to exogenously administered therapeutics.46 While studies have reported that administration of rCC16 ameliorates inflammation preparedness by lung macrophages and epithelial cells,39,47,48 our study has revealed that CC16 is naturally secreted by Club cells via sEVs; thus, CC16 delivered via sEVs has therapeutic potential to treat lung diseases. We attempted to develop an sEV-based nanotherapy to improve the pharmacokinetic properties of CC16. CC16 has a highly conserved N-terminal signal peptide among various species, which is likely to target CC16 not only in the secretory pathway but also secreted EVs. A previous study profiled the protein content of sEVs from mouse BALF and found that CC16 is one of the 1,140 high-confidence proteins identified in BAL EXOs.49 In this study, CC16 was efficiently encapsulated into the sEVs of BEAS-2B-CC16, a stable CC16-expressing cell encoding human CC16. In addition, sEV-CC16 was able to exert beneficial activities on lung macrophages and epithelial cells. Although the fluorescence intensity of PKH26-labeled sEV-CC16 is weaker than in macrophages (red dots in Figure 5A), it successfully confirmed that sEVs were delivered into epithelial cells. sEVs improved lung injury and the lung inflammatory response in mice with LPS-induced ALI. In addition, sEVs constrained the inflammatory response in LPS- and K. pneumoniae-stimulated macrophages and epithelial cells. Therefore, our study provides compelling evidence that sEV-CC16 has a marked potential as a therapeutic agent for inflammatory lung diseases such as ALI/ARDS.
The advantage of sEVs is that they can increase drug delivery efficiency. Compared with existing synthetic nanoparticles, a distinct advantage of sEVs is their natural origin,50 which enables them to fuse with the recipient cell without triggering immunogenicity and to promote the cytoplasmic release of cargos after endosomal uptake.51 A recent study by Johnson et al. reported that CC16 blocked the adhesion of macrophages to human endothelial cells by binding to α4β1 integrin, thereby reducing lung inflammation and damage.52 Although we did not confirm whether sEV-CC16 can exhibit anti-inflammatory effects by binding to α4β1 integrin, our results indicated that sEV-CC16 exerts its therapeutic function without binding to a receptor due to direct internalization into target cells, which is an advantage of sEVs over rCC16 protein.53 Another key finding of our study is that sEV-CC16 exerts similar beneficial effects at much lower concentrations than rCC16. Although rCC16 could be internalized to epithelial cells through phagocytosis and pinocytosis without receptor, the phagocyte capacity of macrophages is much stronger.54,55 So, only a small amount of rCC16 may be able to go into cells other than macrophages due to the lack of CC16-specific membrane receptors. However, sEVs can go into the cell with their therapeutic cargos by fusion and/or endocytosis.56 In our study, sEV-CC16 was effective in experimental models such as the bacterial infection cellular model, where rCC16 was not found to be therapeutically beneficial.39 Several studies have reported that sufficient therapeutic effects of rCC16 are achieved only at very high concentrations (of up to 2 μg/mL in macrophages and 200 μg/mL in epithelial cells).47,48 In our study, sEV-CC16 and rCC16 showed a similar therapeutic effect on lung injury in mice with LPS-induced ALI, the CC16 concentration in sEV-CC16 (40 ng/kg body weight) was much lower than needed for rCC16 (2 mg/kg body weight). Similar results were also observed in in vitro experiments. Thus, sEV-CC16 has advantages over CC16 as a therapeutic approach by reducing the amount of protein that needs to be delivered to achieve a therapeutic response.
Another novel finding from our study is that sEV-CC16 promotes DNA repair-related pathways in stressed lung epithelial cells and macrophages. Inflammation and DNA damage are closely linked pathologies.57 Also, dysfunction of lung epithelial cells and lung macrophages can be triggered by reactive oxygen species (ROS)-induced-mitochondrial DNA damage, which in turn evokes inflammatory and immune responses.58 ROS produced by LPS at inflammatory sites often trigger DNA damage response.59,60 Several studies suggest that DNA damage causes an inflammatory response by activating NF-κB.61,62 By investigating the Cc16 KO mice, we identified a novel protective function for CC16 by preventing DNA damage in lung cells of unchallenged animals. The protective effect of sEV-CC16 against DNA damage was also confirmed in lung epithelial cells and macrophages in vitro. Previously, we reported that CC16 inhibited ROS generation and cytokine secretion in bacterial-infected lung epithelial cells,39 supporting that CC16 is capable of several different functions, including anti-inflammatory effects, reducing DNA damage and ROS. However, the molecular mechanism by which CC16 activates the DNA repair pathway was discovered in our study.
The anti-inflammatory effect of CC16 has been investigated not only in response to LPS treatment but also in cells or animals exposed to bacteria, ozone, allergens, viruses, and CS.39,40,41,47,48 CC16 has been proposed to inhibit two pro-inflammatory pathways, phospholipase A2 and NF-κB.41,63 However, the direct binding protein of CC16 is still unclear. NF-κB signaling is a central mediator of pro-inflammatory gene induction and functions in innate and adaptive immune cells. Although several studies observed that CC16 can inhibit NF-κB activation,39,40,41,47 the molecular mechanism is still unclear. Lesur et al. first reported that CC16 regulated inflammation by inhibiting phospholipase A2 (PLA2) activity, which can modulate NF-κB translocation.63 However, Laucho-Contreras et al. reported that there was no relationship between secretory PLA2 (sPLA2) and CC16 in the Cc16 KO mice model.41 Another of our principal findings is the identification of the molecular target of CC16 using an affinity purification approach. With this approach, we identified that HSP60 is a direct binding target of CC16 and regulates the affinity for NF-kB signaling. HSP60 activated NF-κB signaling by inducing phosphorylation and nuclear localization in several cell lines, such as cardiomyocytes and microglia.64,65 We also observed that overexpression of HSP60 in lung epithelial cells initiated an inflammatory response. It has been reported that HSP60 induces phosphorylation of IKKα/β complex by binding with IKKα and IKKβ.33 Activated IKKα/β induces IκB phosphorylation, which in turn degrades the NF-κB complex and permits translocation of NF-κB dimers into the nucleus to trigger transcription of pro-inflammatory genes.66,67 Interestingly, the interaction of HSP60 with IKKα was blocked when we overexpressed CC16, suggesting that CC16 may compete with IKKα for the interaction of HSP60 to inhibit inflammatory signaling. As a result, inactivated IKKα by binding of CC16 to HSP60 blocked NF-κB signaling by inhibiting phosphorylation and degradation of IκBα and localization of NF-κB p65 into the nuclear (Figures 8 and S12).
Currently, technical limitations do not allow us to completely separate exosomes from small ectosomes. Some studies separated exosomes and small ectosomes using beads,35,68 but it is still difficult to obtain sufficient amounts of exosomes for in vivo experiments.69 Another limitation in our study is that, although sEV-CC16 are secreted from the CC16-overexpressed stable cell line, CC16 may not be the only component that differs from the EVs from control cells. Several studies have reported that stem cell-derived sEVs have bioactivities, including immunoregulation, angiogenesis, and tissue regeneration.70 Nevertheless, the therapeutic benefits of EV-CC16 remain unchallenged by this limitation. Also, sEV-Con derived from epithelial cells did not show bioactivity compared with the PBS group (Figures S7 and S8). In addition, sEV-CC16 treatment did not show any adverse reactions compared with the rCC16 treatment group, suggesting that the therapeutic function of sEV-CC16 mainly came from the CC16 protein itself. However, further studies are needed to fully explore the other proteins or RNA components that are differentially present in sEV-CC16. Even though the sEV-CC16 treatment did not cause any significant side effects, weight loss, or mortality in animals 1 week after treatment, potential untoward/toxic effects of sEV-CC16 need to be further evaluated before clinical use. Moreover, several limitations present a major obstacle to the clinical application of sEV-CC16. First, sEV-CC16 used in our study is derived from BEAS-2B-CC16 stable cells. Given that BEAS-2B is an immortalized cell line by SV40-containing adenovirus, the safety concern may limit the usage of sEV-CC16 in translational studies. Second, roughly 400 mL of culture medium from thirty 10-cm dishes are required to collect enough sEV-CC16 for treating one single mouse due to the low yield of sEV-CC16 from cultured cells. Last, sEV-CC16 contains a low CC16 amount, approximately 14 copies per sEV, as shown in Figure 2F. Despite these limitations, we successfully proposed a new platform for targeted delivery of CC16 using sEVs and highlighted the potential of sEV-CC16 as a promising nanotherapeutic for the treatment of ALI. In summary, our work demonstrates that sEV-CC16 can be secreted naturally and have therapeutic potential for ALI/ARDS and potentially other lung diseases associated with inflammation by inhibiting the NF-κB pathway through its direct interaction with HSP60.
Materials and methods
Reagents
LPSs from Escherichia coli O111:B4, PMA, and PEG 6000 were purchased from Sigma-Aldrich. PBS, TBS, penicillin-streptomycin (10,000 U/mL), FBS, EV-depleted FBS, DMEM, and RPMI-1640 were purchased from Gibco. Lipofectamine 2000 reagent, protease inhibitor tablet, and BSA were purchased from Thermo Fisher Scientific. The K. pneumoniae strain was purchased from ATCC (#43186, Manassas, VA). Recombinant human CC16 was purchased from Novus Biologicals (NBP2-35126, Centennial, CO).
Cell culture
BEAS-2B, THP-1, and HEK293T cells were purchased from ATCC (Manassas, VA). Primary NHBE cells were purchased from Lonza Bioscience. BEAS-2B and NHBE cells were cultured using Bronchial Epithelial Cell Growth Medium (BEGM) Bullet Kit (Lonza). HEK293T cells were cultured in 10% FBS/DMEM with 1% penicillin/streptomycin. THP-1 cells maintained in 10% FBS/RPMI-1640 were differentiated into macrophage-like cells using 40 nM PMA treatment for 48 h. BMDMs were isolated as previously described71 and maintained with 30% L929 cells conditioned medium in DMEM complete medium for 7 days before any further experimental procedure. All cells were cultured in an incubator at 37°C supplied with 5% CO2.
In vitro pro-inflammatory stimulation
For LPS treatment, 100 ng/mL was used to activate THP-1 and BMDM. A concentration of 1 μg/mL was used for NHBE, BEAS-2B, and 293T cells. For bacterial stimulation, K. pneumoniae were grown in Luria-Bertani medium at 37˚C in a rotator at 250 rpm overnight and then sub-cultured using fresh medium until they reached mid-log phase. Bacterial concentrations were assessed by optical density 600 (OD600), K. pneumoniae was diluted to the desired colony-forming units (CFU), and 5 × 106 CFU of K. pneumoniae were added to 106 cultured cells for 1 h. After the incubation, bacteria were removed by washing, and 50 μg/mL gentamicin was added to kill the remaining bacteria as described before.72 Cells were then maintained with rCC16, sEV-Con, and sEV-CC16 for 24 h.
Plasmids
Plasmid pRP-Puro-CMV-CC16 and its vector pRP-Puro-CMV were purchased from VectorBuilder (Chicago, IL). Human HSP60 cDNA amplified by RT-PCR was subcloned into the Kpn I and Xho I sites of pcDNA3.1 vector (CMV promoter; Invitrogen) to obtain pcDNA-HSP60, and the primer sequences used for cloning are provided in Table S3. pEGFP-C1-PKM2 (Addgene plasmid #64698) was a gift from Axel Ullrich.73
Generation of stable cell line and sEV-CC16
To produce sEV-CC16, we generated stable CC16 producer cells, as described in Figure S3A. Briefly, plasmid pRP-Puro-CMV-CC16 or vector pRP-Puro-CMV was transfected into BEAS-2B through electroporation (1,290 V, 20 ms, two pulses) using the Neon Transfection System (Thermo Fisher Scientific). At 48 h post transfection, 2 μg/mL puromycin was added to select stably transfected cells. The puromycin-added media were changed every 2–3 days until colonies formed. Then, a single colony was expanded and maintained for 24 h more in EV-depleted medium to purify sEV-CC16 or sEV-Con.
Purification of sEVs
sEVs were prepared using sequential centrifugation protocol with slight modifications.74,75 BALF samples and conditioned media were collected and centrifuged at 500 × g for 5 min to remove cells. The supernatant was centrifuged at 2000 × g for 20 min to eliminate apoptotic bodies. The resulting supernatants were mixed with an equal volume of a 2 × PEG (16% PEG with 1 M NaCl) solution and were incubated at 4°C overnight, followed by centrifugation at 3,000 × g for 1 h to pellet total EVs. To remove m/lEVs, pellet EVs were resuspended with PBS followed by centrifugation at 16,000 × g for 1 h. The resulting supernatants were filtered using a 0.22-μm filter and ultracentrifuged at 120,000 × g for 1 h to pellet sEVs. To wash sEVs, PBS was added and sEVs were ultracentrifuged at 120,000 × g for 1 h. To enrich CC16+ sEVs from human BALF sEVs, we used Pierce Protein A/G Magnetic Beads (Thermo Fisher Scientific) coated with CC16 antibody (NovusBio, NBP2-75705, clone JU34-03) as previously described.76 Briefly, we coated a 1-mg magnetic bead with 10 μg of CC16 antibody for 1 h at room temperature in immunoprecipitation (IP) buffer (Pierce IP Lysis Buffer, Thermo Fisher Scientific). After washing the bead using IP buffer, coated beads were incubated with human BALF sEVs at 4°C overnight and were washed three times using IP buffer. CC16+ sEVs were eluted for western blot analysis.
Animal studies
WT C57BL/6 mice of both genders (8 weeks old) were purchased from the Jackson Laboratory. To induce lung injury, mice were anesthetized and intratracheally intubated by a blunt-ended feeding needle. Then 1 μg of LPS or 5 × 103 CFU of K. pneumoniae in 50 μL of PBS was delivered through the needle. After 3 h, mice were given 50 μL of PBS, sEV-Con (7.5 × 1010 in 50 μL of PBS), sEV-CC16 (7.5 × 1010 in 50 μL of PBS), or rCC16 (2 mg/kg body weight) via the i.t. route. Mice were euthanized by an aerosolized isoflurane overdose at the indicated time point. To perform the knockdown of CC16 in the lung, we loaded 400 pmol of CC16 siRNA into 20 μL of Lipofectamine 2000 reagent according to the manufacturer’s protocol. Pre-designed control and CC16 siRNA (TriFECTa RNAi Kit) were purchased from Integrated DNA Technologies. siRNA was delivered intratracheally 5 days before LPS or sEV administration. A total of 15 to 21 mice per group were used and three to eight mice as indicated in the dot plot were used per group in each experiment. After euthanasia, we isolated BALF as previously described.77 Briefly, mice (five to eight mice per group) were used to lavage total mouse lungs and washed with 1.8 mL (0.6 mL × 3) of PBS. BALF was then cytocentrifuged at 500 × g for 5 min to obtain BAL cells. Total inflammatory cell counts were measured using a hemocytometer as previously described.78 BAL cells were air-dried and stained with PROTOCOL Hema 3 fixative and solutions (Fisher Scientific). Images of the stained BAL cells were visualized and captured using a microscope (Zeiss Observer Z1, Carl Zeiss, Oberkochen, Germany). To measure the weight ratio, we dissected the inferior lobe of the right lung from mice. The wet weight was measured immediately after its excision and dried at 60°C for 24 h and re-weighed for dry weight. The wet/dry weight ratio was calculated by dividing the wet by the dry weight. The remaining part of the lung was used for RNA or protein extraction. To observe histologic images of lung sections, we obtained the whole lungs from five mice per group and made cryosections for immunofluorescence and H&E staining. All the animal studies were approved by Charlie Norwood Veterans Affairs Medical Center Institutional Animal Care and Use Committee and followed the ARRIVE guidelines.
Cc16 KO murine model
The formalin-fixed paraffin-embedded blocks of lung tissues from both WT and Cc16 KO mice were kindly provided by Dr. Caroline A. Owen. Animal experiments were conducted following ARRIVE guidelines. Cc16 KO mice were generated as described previously,79 and C57BL/6 WT and Cc16 KO mice of both genders were housed in the same room in a barrier facility under specific pathogen-free conditions for 3 months (n = 3 per group) and 18 months (n = 8 for WT; n = 9 for Cc16 KO). The Brigham and Women’s Hospital Institutional Animal Care and Use Committee approved all procedures performed on mice.
Characterization of sEVs
As previously described,80 the negative-staining TEM images and immunogold-labeled TEM images with monoclonal anti-CC16 (R&D Systems, MAB4218, clone #394324) and non-immune rat immunoglobulin G (IgG) (R&D Systems, MAB005, Clone # 43414) were taken using JEM 1230 TEM (JEOL USA, Peabody, MA) at the Electron Microscopy Core Laboratory at Augusta University. NTA data were obtained using Nanosight NS500 at the Nanomedicines Characterization Core Facility (The University of North Carolina at Chapel Hill). To track the sEV distributions, PKH26 (red dye) and PKH67 (green dye) Fluorescent Cell Linker Kits for General Cell Membrane Labeling (Sigma-Aldrich) were used according to the manufacturer’s protocol. For in vivo study, PKH26-labeled sEVs were administered into mice as described above. Lung sections and BAL cells were incubated with polyclonal anti-CD68 (Abcam, ab125047) and monoclonal pan-CK (Sigma-Aldrich, c2562, clone C-11+PCK-26+CY-90+KS-1A3+M20 + A53-B/A2) overnight at 4°C. After washing, polyclonal anti-rabbit Alexa 488-conjugated (Thermo Fisher Scientific, A11070) and polyclonal anti-mouse Alexa 488-conjugated (Thermo Fisher Scientific, A11001) secondary antibodies were applied. For in vitro experiments, after treating with PKH26- or PKH67-labeled sEVs, THP-1, BEAS-2B, and NHBE cells were washed and fixed. Nuclei were counterstained with DAPI (Abcam). Fluorescence images were visualized and captured using a Zeiss Observer Z1 microscope (Carl Zeiss). To measure the expression levels of CC16 in sEVs, flow cytometric analysis was performed as described previously.81 Purified 2-μg sEVs were coupled with 10 μL of aldehyde/sulfate latex beads (Thermo Fisher Scientific) for 2 h. After blocking in 4% BSA for 1 h, the bead-bound sEVs were permeabilized for 5 min with 0.2% Triton X-100 and fixed for 5 min with 2% formaldehyde. For analysis, bead-bound sEVs were incubated with monoclonal anti-CC16 (NovusBio, NBP2-75705, clone JU34-03), followed by incubation with polyclonal anti-rabbit Alexa 488-conjugated secondary antibodies (Thermo Fisher Scientific, A11070) antibody. Flow cytometric analysis was performed using Cytoflex Flow Cytometer (Beckman Coulter).
Human samples
Demographic information of human samples was included in Table S1. Normal human BAL samples were purchased from BioIVT (Westbury, NY). For BAL samples from pneumonia patients, nine were purchased from Discovery Life Sciences (Huntsville, AL) and 12 were collected from patients enrolled in the Albuterol to Treat Acute Lung Injury (ALTA) Trial82 and provided by the National Heart, Lung, and Blood Institute (NHLBI) Biologic Specimen and Data Repository Information Coordinating Center (BioLINCC). Public-use data and plasma samples were collected from a previously conducted study, the ALTA trial, and provided via BioLINCC. All human procedures in the ALTA trial were approved by the institutional review boards, and written informed consents were obtained from patients or surrogates at each participating hospital. Plasma specimens were measured at Augusta University laboratories and this study was approved by the Augusta University Institutional Review Board (IRB number 1128838-13) prior to its initiation.
Immunofluorescence and H&E staining
Immunofluorescence staining was performed as previously described.31 Briefly, lung sections were incubated overnight at 4°C with monoclonal anti-CC16 (NovusBio, NBP2-75705, clone JU34-03), polyclonal anti-CD68 (Abcam, ab125047), monoclonal pan-CK (Sigma-Aldrich, c2562, clone C-11+PCK-26+CY-90+KS-1A3+M20 + A53-B/A2), and monoclonal anti-γ-H2AX (Abcam, ab81299, clone EP854(2)Y). After washing, polyclonal anti-rabbit Alexa 488-conjugated (Thermo Fisher Scientific, A11070), polyclonal anti-mouse Alexa 488-conjugated (Thermo Fisher Scientific, A11001), and polyclonal anti-rabbit Alexa 546-conjugated (Thermo Fisher Scientific, A11071) secondary antibodies were applied. Nuclei were counterstained with DAPI (Sigma-Aldrich). Images of the stained lung sections were visualized using a Zeiss Observer Z1 microscope (Carl Zeiss). The BAL cells were cytocentrifuged at 500 × g for 5 min using a Shandon Cytospin 4 (Thermo Fisher Scientific) and concentrated onto a microscope slide. BAL cells were air-dried and stained with PROTOCOL Hema 3 fixative and solutions (Thermo Fisher Scientific). Total inflammatory cell counts in the BALF were determined using a hemocytometer. H&E staining of lung sections was conducted in the Histology Core Laboratory at Augusta University. Histologic images of lung sections were captured using a Zeiss Observer Z1 microscope (Carl Zeiss). The histologic lung injury score was assessed in the double-blinded condition as previously described.83 Briefly, lung injury was assessed on the following criteria: (1) the number of neutrophils in the alveolar space, (2) the number of neutrophils in the interstitial space, (3) the number of hyaline membranes, (4) proteinaceous debris filling the airspaces, and (5) alveolar septal thickening.
TUNEL assay
A TUNEL Assay Kit - BrdU-Red (Abcam, ab66110) was used according to the manufacturer’s instructions. In brief, lung sections were fixed using 4% formaldehyde/PBS for 15 min. Fixed tissues were treated with 20 μg/mL proteinase K solution for 5 min to expose the DNA for end-labeling with the TdT enzyme. Then, DNA labeling solution containing TdT enzyme and Br-dUTP were treated for 1 h at 37°C in the dark, following treatment of anti-BrdU-Red antibody for 30 min and DAPI for nuclei counterstaining. Images of the stained lung sections were captured using a Zeiss Observer Z1 microscope (Carl Zeiss). To measure the percentage of TUNEL-positive cells, 100 DAPI stained cells were counted from 10 random microscopic fields per sample.
In vivo imaging systems
To track biodistribution, we labeled 100 μg of sEVs using DiR near-infrared (NIR) fluorescence dye (XenoLight, 125964), according to the manufacturer’s instructions. Briefly, 8.3 mg/mL DiR dissolved in ethanol was prepared and diluted to 640 μg/mL using PBS, and 100 μg of sEVs in 25 μL was incubated with 25 μL of 640 μg/mL DiR for 30 min at room temperature. DiR-labeled sEVs were washed and purified using ultracentrifugation to remove unbound DiR dye. LPS-treated mice were given DiR-labeled sEV-Con or sEV-CC16 intratracheally. Major organs/tissues were collected for ex vivo imaging and visualized using LI-COR Pearl Impulse Imaging System at 24 h after EV administration.
Western blot analysis, coIP assay, and ELISA
Western blot analysis was performed as described previously.84 In brief, cells or sEVs were lysed in RIPA buffer with protease inhibitor. Cytoplasmic and nuclear fractions were extracted by Nuclear Extraction Kit (Abcam, ab113474) according to the manufacturer’s protocol. Protein lysates were resolved on SDS-PAGE gels and transferred to the polyvinylidene fluoride (PVDF) membrane. Membranes were blocked in 5% BSA/TBST incubated with primary antibodies with monoclonal anti-CD9 (Abcam, ab92726, clone EPR2949), polyclonal anti-CD63 (Abcam, ab216130), monoclonal anti-Flotillin 1 (Abcam, ab133497, clone EPR6041), polyclonal anti-Sp1 (Abcam, ab227383), monoclonal anti-CC16 (NovusBio, NBP2-75705, clone JU34-03), monoclonal HSP60 (Abcam, ab59457, clone LK-1), monoclonal anti-IKKα (Abcam, ab32041, clone Y463), monoclonal pan-CK (Sigma-Aldrich, c2562, clone C-11+PCK-26+CY-90+KS-1A3+M20 + A53-B/A2), monoclonal CD31 (BD Pharmingen, 553369, clone MEC 13.3), polyclonal MPO (R&D Systems, AF3667), monoclonal α-SMA (Sigma-Aldrich, A5691, clone 1A4), monoclonal IκBα (Cell Signaling Technology, #4814, clone L35A5), monoclonal p-IκBα (Cell Signaling Technology, #2859, clone 14D4), monoclonal NF-κB p65 (Cell Signaling Technology, #8242, clone D14E12), monoclonal histone H3 (Cell Signaling Technology, #4499, clone D1H2), and monoclonal anti-β-actin (Sigma-Aldrich, A5441, clone AC-15) overnight at 4°C. They were then incubated with polyclonal anti-rabbit horseradish peroxidase (HRP) secondary antibody (R&D Systems, HAF008), polyclonal anti-mouse HRP secondary antibody (R&D Systems, HAF007), polyclonal anti-rat HRP secondary antibody (R&D Systems, HAF005), and polyclonal anti-goat HRP secondary antibody (R&D Systems, HAF017). Images were obtained by X-ray film and Chemidoc image system (Bio-Rad). For the coIP assay, samples were lysed in IP Lysis Buffer (Thermo Fisher Scientific) and incubated with Pierce Protein A/G Magnetic Beads (Thermo Fisher Scientific), followed by incubation with primary antibody at 4°C overnight. IgG was used as a negative control. The precipitated samples were directly used for western blot analysis. Input was used as a positive control. DuoSet ELISA Kits for human proteins (CC16, IL-1β, TNF-α, CXCL-1, and CXCL-2) and mouse proteins (IL-1β, TNF-α, CXCL-1, and CXCL-2) were purchased from R&D Systems. The mouse CC16 ELISA kit was purchased from MyBioSource (MyBioSource, MBS2708332). ELISA was conducted according to the manufacturer’s protocol.
RNA preparation, reverse transcription, qRT-PCR, and PCR array
Total RNA was isolated using PureLink RNA Mini Kit (Invitrogen). First-strand cDNA was prepared from an RNA template using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qRT-PCR was performed by using the Power SYBR Green PCR Master Mix and StepOne Plus Real-time PCR System (Applied Biosystems). Tbp was used as a normalization control. The sequences of the primers are summarized in Table S3. For analysis of human DNA repair mechanism, qRT-PCR was performed using TaqMan Array Human DNA Repair Mechanism (Thermo Fisher Scientific). Pooled RNA from three independent samples was converted into cDNA in a reverse-transcription reaction. Microarray heatmap was then generated using GraphPad Prism version 9 (GraphPad Software).
The GST pull-down assay
GST pull-down assay (Thermo Fisher Scientific) was conducted using GST (Proteintech), GST-tagged CC16 (Proteintech), and THP-1 cells. Briefly, 108 differentiated THP-1 cells were treated with 100 ng/mL LPS for 24 h and then homogenized with 1 mL of pull-down lysis buffer. Then 1 mL of cell lysate was mixed with 150 μg of GST or GST-tagged CC16 and separated by glutathione agarose affinity resin, which can capture the GST-tagged protein. Samples were concentrated and separated by SDS-PAGE and visualized by silver staining. The samples eluted from glutathione agarose affinity resin and visualized band by silver staining were analyzed by Orbitrap Fusion Tribrid MS (Thermo Scientific) coupled with an Ultimate 3000 nano-UPLC system (Thermo Scientific) in the Proteomics Core Laboratory at Augusta University.
NF-κB reporter assay
Transfection of the plasmid pNL3.2. NF-κB-RE (Promega, N1111) into THP-1 cells and HEK293T cells was conducted by using Lipofectamine 2000. At 24 h after transfection, a luciferase assay was performed using the Nano-Glo Luciferase Assay System (Promega) according to the manufacturer’s protocol. Luciferase activities were measured and analyzed after adding LPS (100 ng/mL) to THP-1 cells or LPS (1 μg/mL) to HEK293T cells for 24 h with sEV-Con (5 × 108/mL), sEV-CC16 (5 × 108/mL), or plasmid transfection as indicated.
Comet assay
Comet assays were performed using Comet assay kit (Abcam) following the manufacturer’s instructions. In brief, THP-1 cells and BEAS-2B cells were suspended at 105 cells/mL in ice-cold PBS and were combined with Comet agarose in a 1/10 ratio. The mixture was immediately transferred onto the Comet slide. After incubating with prechilled lysis buffer, the slides were subjected to electrophoresis using Tris-borate-EDTA (TBE) electrophoresis solution. After electrophoresis, Vista Green DNA dye was added to Comet slides. Images were captured by a Zeiss Observer Z1 microscope (Carl Zeiss). Ten pictures were randomly taken, and the tail length and tail DNA (%) were calculated using ImageJ software.
Clariom S assay and data analysis
Total RNA was isolated from THP-1 cells treated with sEV-Con or sEV-CC16 in 100 ng/mL LPS condition using RNeasy Plus Kits (Qiagen). For confirming RNA quality, RNA integrity number (RIN) was assessed by the Agilent 2200 TapeStation (Agilent Technologies). Clariom S assay and microarray were performed in Georgia Cancer Center Integrated Genomics and Bioinformatics Core at Augusta University. Further analysis for the scatter plot, heatmap, and the signaling pathway was carried out using the Transcriptome Analysis Console 4.0 (TAC 4.0) (Applied Biosystems)
Copy number of CC16 calculation
CC16 protein copies per single sEV-Con and sEV-CC16 were calculated as described by Davidson et al.85 Briefly, we measured the amount in 200 μg of sEV-Con or sEV-CC16. The number of CC16 was then calculated using this equation: protein number = mass of proteins/molecular weight of CC16 (15.8) × Avogadro’s constant (6.02 × 1023). The protein number was then divided by particle number from 200 μg of sEV-Con or sEV-CC16, respectively.
Cell viability assay
BEAS-2B cells were seeded in a 96-well plate with 100 μL of culture media per well. After the indicated treatment, 20 μL of CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) (Promega, G3582, Madison, WI) was added to each well and incubated for 2 h at 37°C, in 5% CO2 atmosphere (following the manufacturer’s protocol). The absorbance at 490 nm was measured.
Statistical analysis
SigmaPlot (Systat Software) was used for data analysis. For comparing two groups, the two-tailed unpaired Student’s t test for variables with a normal distribution or the Mann-Whitney U test for variables without a normal distribution were used. For comparing three or more groups, a one-way analysis of variance with a Tukey’s honestly significant difference (HSD) test for variables with normal distribution or the Kruskal-Wallis one-way analysis followed by pairwise comparisons using Mann-Whitney U tests for those without a normal distribution were selected. Parametric data are shown as mean ± SD. Non-parametric data are shown as boxplots showing medians and 25th and 75th percentiles and whisker plots showing minimum and maximum values. A p value ≤0.05 was considered to be significant. Data without normal distribution were visualized by box and whisker plots or data with normal distribution were visualized by scattering dot plots using GraphPad Prism (GraphPad Software).
Acknowledgments
We thank the Integrated Genomics Shared Resource in the Georgia Cancer Center at Augusta University for help with microarray. We thank Katherine Hardwick (Clinical and Administrative Pharmacy, College of Pharmacy, University of Georgia) for administrative support. This work was supported by National Institutes of Health grant R00 HL141685 (D.Z.) and National Institutes of Health grant R03 AI152003 (D.Z.).
Author contributions
D.Z., Y.H., and P.R.S. designed the research. Y.H., Y.Z., and X.W. performed the experiments. Y.Z., S.A., D.Z., and X.W. collected, analyzed, and interpreted data. Y.H., D.Z., P.R.S., and C.A.O. wrote the manuscript.
Declaration of interests
All authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.01.009.
Supplemental information
Data and materials availability
All data are available in the main text or the supplemental materials.
References
- 1.Raghavendran K., Napolitano L.M. Definition of ALI/ARDS. Crit. Care Clin. 2011;27:429–437. doi: 10.1016/j.ccc.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubenfeld G.D., Caldwell E., Peabody E., Weaver J., Martin D.P., Neff M., Stern E.J., Hudson L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 3.Matthay M.A., Zemans R.L., Zimmerman G.A., Arabi Y.M., Beitler J.R., Mercat A., Herridge M., Randolph A.G., Calfee C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers. 2019;5:18. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spadaro S., Park M., Turrini C., Tunstall T., Thwaites R., Mauri T., Ragazzi R., Ruggeri P., Hansel T.T., Caramori G., Volta C.A. Biomarkers for Acute Respiratory Distress syndrome and prospects for personalised medicine. J. Inflamm. 2019;16:1. doi: 10.1186/s12950-018-0202-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dowdy D.W., Eid M.P., Dennison C.R., Mendez-Tellez P.A., Herridge M.S., Guallar E., Pronovost P.J., Needham D.M. Quality of life after acute respiratory distress syndrome: a meta-analysis. Intensive Care Med. 2006;32:1115–1124. doi: 10.1007/s00134-006-0217-3. [DOI] [PubMed] [Google Scholar]
- 6.Bein T., Weber-Carstens S., Apfelbacher C. Long-term outcome after the acute respiratory distress syndrome: different from general critical illness? Curr. Opin. Crit. Care. 2018;24:35–40. doi: 10.1097/MCC.0000000000000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matthay M.A., Zemans R.L. The acute respiratory distress syndrome: pathogenesis and treatment. Annu. Rev. Pathol. 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallelli L., Zhang L., Wang T., Fu F. Severe acute lung injury related to COVID-19 infection: a review and the possible role for escin. J. Clin. Pharmacol. 2020;60:815–825. doi: 10.1002/jcph.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polidoro R.B., Hagan R.S., de Santis Santiago R., Schmidt N.W. Overview: systemic inflammatory response derived from lung injury caused by SARS-CoV-2 infection explains severe outcomes in COVID-19. Front. Immunol. 2020;11:1626. doi: 10.3389/fimmu.2020.01626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front. Immunol. 2020;11:1722. doi: 10.3389/fimmu.2020.01722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang D., Lee H., Wang X., Groot M., Sharma L., Dela Cruz C.S., Jin Y. A potential role of microvesicle-containing miR-223/142 in lung inflammation. Thorax. 2019;74:865–874. doi: 10.1136/thoraxjnl-2018-212994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moldoveanu B., Otmishi P., Jani P., Walker J., Sarmiento X., Guardiola J., Saad M., Yu J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009;2:1–11. [PMC free article] [PubMed] [Google Scholar]
- 13.Li L., Huang Q., Wang D.C., Ingbar D.H., Wang X. Acute lung injury in patients with COVID-19 infection. Clin. Transl. Med. 2020;10:20–27. doi: 10.1002/ctm2.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu B., Bao L., Wang L., Li F., Wen M., Li H., Deng W., Zhang X., Cao B. Anti-IFN-gamma therapy alleviates acute lung injury induced by severe influenza A (H1N1) pdm09 infection in mice. J. Microbiol. Immunol. Infect. 2021;54:396–403. doi: 10.1016/j.jmii.2019.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Yáñez-Mó M., Siljander P.R.M., Andreu Z., Zavec A.B., Borràs F.E., Buzas E.I., Buzas K., Casal E., Cappello F., Carvalho J., et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles. 2015;4 doi: 10.3402/jev.v4.27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poupardin R., Wolf M., Strunk D. Adherence to minimal experimental requirements for defining extracellular vesicles and their functions. Adv. Drug Deliv. Rev. 2021;176 doi: 10.1016/j.addr.2021.113872. [DOI] [PubMed] [Google Scholar]
- 17.Raposo G., Nijman H.W., Stoorvogel W., Liejendekker R., Harding C.V., Melief C.J., Geuze H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996;183:1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russell A.E., Sneider A., Witwer K.W., Bergese P., Bhattacharyya S.N., Cocks A., Cocucci E., Erdbrügger U., Falcon-Perez J.M., Freeman D.W., et al. Biological membranes in EV biogenesis, stability, uptake, and cargo transfer: an ISEV position paper arising from the ISEV membranes and EVs workshop. J. Extracell. Vesicles. 2019;8 doi: 10.1080/20013078.2019.1684862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mir B., Goettsch C. Extracellular vesicles as delivery vehicles of specific cellular cargo. Cells. 2020;9 doi: 10.3390/cells9071601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sutaria D.S., Badawi M., Phelps M.A., Schmittgen T.D. Achieving the promise of therapeutic extracellular vesicles: the devil is in details of therapeutic loading. Pharm. Res. 2017;34:1053–1066. doi: 10.1007/s11095-017-2123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laucho-Contreras M.E., Polverino F., Tesfaigzi Y., Pilon A., Celli B.R., Owen C.A. Club cell protein 16 (CC16) augmentation: a potential disease-modifying approach for chronic obstructive pulmonary disease (COPD) Expert Opin. Ther. Targets. 2016;20:869–883. doi: 10.1517/14728222.2016.1139084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Broeckaert F., Bernard A. Clara cell secretory protein (CC16): characteristics and perspectives as lung peripheral biomarker. Clin. Exp. Allergy. 2000;30:469–475. doi: 10.1046/j.1365-2222.2000.00760.x. [DOI] [PubMed] [Google Scholar]
- 23.Almuntashiri S., Zhu Y., Han Y., Wang X., Somanath P.R., Zhang D. Club cell secreted protein CC16: potential applications in prognosis and therapy for pulmonary diseases. J. Clin. Med. 2020;9 doi: 10.3390/jcm9124039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kropski J.A., Fremont R.D., Calfee C.S., Ware L.B. Clara cell protein (CC16), a marker of lung epithelial injury, is decreased in plasma and pulmonary edema fluid from patients with acute lung injury. Chest. 2009;135:1440–1447. doi: 10.1378/chest.08-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qazi K.R., Torregrosa Paredes P., Dahlberg B., Grunewald J., Eklund A., Gabrielsson S. Proinflammatory exosomes in bronchoalveolar lavage fluid of patients with sarcoidosis. Thorax. 2010;65:1016–1024. doi: 10.1136/thx.2009.132027. [DOI] [PubMed] [Google Scholar]
- 26.Urban P.L. Quantitative mass spectrometry: an overview. Philos. Trans. A. Math. Phys. Eng. Sci. 2016;374 doi: 10.1098/rsta.2015.0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee H., Groot M., Pinilla-Vera M., Fredenburgh L.E., Jin Y. Identification of miRNA-rich vesicles in bronchoalveolar lavage fluid: insights into the function and heterogeneity of extracellular vesicles. J. Control Release. 2019;294:43–52. doi: 10.1016/j.jconrel.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu C., Su C. Design strategies and application progress of therapeutic exosomes. Theranostics. 2019;9:1015–1028. doi: 10.7150/thno.30853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arsalane K., Broeckaert F., Knoops B., Wiedig M., Toubeau G., Bernard A. Clara cell specific protein (CC16) expression after acute lung inflammation induced by intratracheal lipopolysaccharide administration. Am. J. Respir. Crit. Care Med. 2000;161:1624–1630. doi: 10.1164/ajrccm.161.5.9812157. [DOI] [PubMed] [Google Scholar]
- 30.Bolton S.J., Pinnion K., Marshall C.V., Wilson E., Barker J.E., Oreffo V., Foster M.L. Changes in Clara cell 10 kDa protein (CC10)-positive cell distribution in acute lung injury following repeated lipopolysaccharide challenge in the rat. Toxicol. Pathol. 2008;36:440–448. doi: 10.1177/0192623308315357. [DOI] [PubMed] [Google Scholar]
- 31.Zhang D., Lee H., Wang X., Rai A., Groot M., Jin Y. Exosome-Mediated small RNA delivery: a novel therapeutic approach for inflammatory lung responses. Mol. Ther. 2018;26:2119–2130. doi: 10.1016/j.ymthe.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe S., Alexander M., Misharin A.V., Budinger G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Invest. 2019;129:2619–2628. doi: 10.1172/JCI124615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chun J.N., Choi B., Lee K.W., Lee D.J., Kang D.H., Lee J.Y., Song I.S., Kim H.I., Lee S.H., Kim H.S., et al. Cytosolic Hsp60 is involved in the NF-kappaB-dependent survival of cancer cells via IKK regulation. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meldolesi J. Exosomes and ectosomes in intercellular communication. Curr. Biol. 2018;28:R435–R444. doi: 10.1016/j.cub.2018.01.059. [DOI] [PubMed] [Google Scholar]
- 35.Mathieu M., Névo N., Jouve M., Valenzuela J.I., Maurin M., Verweij F.J., Palmulli R., Lankar D., Dingli F., Loew D., et al. Specificities of exosome versus small ectosome secretion revealed by live intracellular tracking of CD63 and CD9. Nat. Commun. 2021;12:4389. doi: 10.1038/s41467-021-24384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pitt J.M., Kroemer G., Zitvogel L. Extracellular vesicles: masters of intercellular communication and potential clinical interventions. J. Clin. Invest. 2016;126:1139–1143. doi: 10.1172/JCI87316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Hoecke L., Job E.R., Saelens X., Roose K. Bronchoalveolar lavage of murine lungs to analyze inflammatory cell infiltration. J. Vis. Exp. 2017;10 doi: 10.3791/55398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ware L.B., Matthay M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 39.Almuntashiri S., Han Y., Zhu Y., Dutta S., Niazi S., Wang X., Siddiqui B., Zhang D. CC16 regulates inflammation, ROS generation and apoptosis in bronchial epithelial cells during Klebsiella pneumoniae infection. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms222111459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laucho-Contreras M.E., Polverino F., Gupta K., Taylor K.L., Kelly E., Pinto-Plata V., Divo M., Ashfaq N., Petersen H., Stripp B., et al. Protective role for club cell secretory protein-16 (CC16) in the development of COPD. Eur. Respir. J. 2015;45:1544–1556. doi: 10.1183/09031936.00134214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laucho-Contreras M.E., Polverino F., Rojas-Quintero J., Wang X., Owen C.A. Club cell protein 16 (Cc16) deficiency increases inflamm-aging in the lungs of mice. Physiol. Rep. 2018;6 doi: 10.14814/phy2.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu L., An L., Ran D., Lizarraga R., Bondy C., Zhou X., Harper R.W., Liao S.Y., Chen Y. The club cell marker SCGB1A1 downstream of FOXA2 is reduced in asthma. Am. J. Respir. Cell Mol. Biol. 2019;60:695–704. doi: 10.1165/rcmb.2018-0199OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hermans C., Bernard A. Lung epithelium-specific proteins: characteristics and potential applications as markers. Am. J. Respir. Crit. Care Med. 1999;159:646–678. doi: 10.1164/ajrccm.159.2.9806064. [DOI] [PubMed] [Google Scholar]
- 44.Doyle I.R., Hermans C., Bernard A., Nicholas T.E., Bersten A.D. Clearance of Clara cell secretory protein 16 (CC16) and surfactant proteins A and B from blood in acute respiratory failure. Am. J. Respir. Crit. Care Med. 1998;158:1528–1535. doi: 10.1164/ajrccm.158.5.9712097. [DOI] [PubMed] [Google Scholar]
- 45.Admyre C., Grunewald J., Thyberg J., Gripenbäck S., Tornling G., Eklund A., Scheynius A., Gabrielsson S. Exosomes with major histocompatibility complex class II and co-stimulatory molecules are present in human BAL fluid. Eur. Respir. J. 2003;22:578–583. doi: 10.1183/09031936.03.00041703. [DOI] [PubMed] [Google Scholar]
- 46.Bourdonnay E., Zasłona Z., Penke L.R.K., Speth J.M., Schneider D.J., Przybranowski S., Swanson J.A., Mancuso P., Freeman C.M., Curtis J.L., Peters-Golden M. Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J. Exp. Med. 2015;212:729–742. doi: 10.1084/jem.20141675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pang M., Yuan Y., Wang D., Li T., Wang D., Shi X., Guo M., Wang C., Zhang X., Zheng G., et al. Recombinant CC16 protein inhibits the production of pro-inflammatory cytokines via NF-kappaB and p38 MAPK pathways in LPS-activated RAW264.7 macrophages. Acta Biochim. Biophys. Sin. 2017;49:435–443. doi: 10.1093/abbs/gmx020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin J., Li J., Shu M., Wu W., Zhang W., Dou Q., Wu J., Zeng X. The rCC16 protein protects against LPS-induced cell apoptosis and inflammatory responses in human lung pneumocytes. Front. Pharmacol. 2020;11:1060. doi: 10.3389/fphar.2020.01060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maroto R., Zhao Y., Jamaluddin M., Popov V.L., Wang H., Kalubowilage M., Zhang Y., Luisi J., Sun H., Culbertson C.T., et al. Effects of storage temperature on airway exosome integrity for diagnostic and functional analyses. J. Extracell. Vesicles. 2017;6 doi: 10.1080/20013078.2017.1359478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han Y., Jones T.W., Dutta S., Zhu Y., Wang X., Narayanan S.P., Fagan S.C., Zhang D. Overview and update on methods for cargo loading into extracellular vesicles. Processes (Basel) 2021;9 doi: 10.3390/pr9020356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wiklander O.P.B., Brennan M.Á., Lötvall J., Breakefield X.O., El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aav8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson M.D.L., Younis U.S., Menghani S.V., Addison K.J., Whalen M., Pilon A.L., Cress A.E., Polverino F., Romanoski C.E., Kraft M., et al. CC16 binding to alpha4beta1 integrin protects against mycoplasma pneumoniae infection. Am. J. Respir. Crit. Care Med. 2021;203:1410–1418. doi: 10.1164/rccm.202006-2576OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gurung S., Perocheau D., Touramanidou L., Baruteau J. The exosome journey: from biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021;19:47. doi: 10.1186/s12964-021-00730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharma L., Feng J., Britto C.J., Dela Cruz C.S. Mechanisms of epithelial immunity evasion by respiratory bacterial pathogens. Front. Immunol. 2020;11:91. doi: 10.3389/fimmu.2020.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kruth H.S. Receptor-independent fluid-phase pinocytosis mechanisms for induction of foam cell formation with native low-density lipoprotein particles. Curr. Opin. Lipidol. 2011;22:386–393. doi: 10.1097/MOL.0b013e32834adadb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gonda A., Kabagwira J., Senthil G.N., Wall N.R. Internalization of exosomes through receptor-mediated endocytosis. Mol. Cancer Res. 2019;17:337–347. doi: 10.1158/1541-7786.MCR-18-0891. [DOI] [PubMed] [Google Scholar]
- 57.Kirolikar S., Prasannan P., Raghuram G.V., Pancholi N., Saha T., Tidke P., Chaudhari P., Shaikh A., Rane B., Pandey R., et al. Prevention of radiation-induced bystander effects by agents that inactivate cell-free chromatin released from irradiated dying cells. Cell Death Dis. 2018;9:1142. doi: 10.1038/s41419-018-1181-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu X., Chen Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J. Transl. Med. 2017;15:207. doi: 10.1186/s12967-017-1306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qiao W., Huang Y., Bian Z., Sun X., Wang X., Gao Q., Peng Y., Meng L. Lipopolysaccharide-induced DNA damage response activates nuclear factor kappaB signalling pathway via GATA4 in dental pulp cells. Int. Endod. J. 2019;52:1704–1715. doi: 10.1111/iej.13180. [DOI] [PubMed] [Google Scholar]
- 60.Chan T.K., Tan W.S.D., Peh H.Y., Wong W.S.F. Aeroallergens induce reactive oxygen species production and DNA damage and dampen antioxidant responses in bronchial epithelial cells. J. Immunol. 2017;199:39–47. doi: 10.4049/jimmunol.1600657. [DOI] [PubMed] [Google Scholar]
- 61.McCool K.W., Miyamoto S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012;246:311–326. doi: 10.1111/j.1600-065X.2012.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Georgakilas A.G., Kotsinas A. Editorial: DNA damage and inflammation under stress. Front. Genet. 2017;8:152. doi: 10.3389/fgene.2017.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lesur O., Bernard A., Arsalane K., Lauwerys R., Bégin R., Cantin A., Lane D. Clara cell protein (CC-16) induces a phospholipase A2-mediated inhibition of fibroblast migration in vitro. Am. J. Respir. Crit. Care Med. 1995;152:290–297. doi: 10.1164/ajrccm.152.1.7541278. [DOI] [PubMed] [Google Scholar]
- 64.Tian J., Guo X., Liu X.M., Liu L., Weng Q.F., Dong S.J., Knowlton A.A., Yuan W.J., Lin L. Extracellular HSP60 induces inflammation through activating and up-regulating TLRs in cardiomyocytes. Cardiovasc. Res. 2013;98:391–401. doi: 10.1093/cvr/cvt047. [DOI] [PubMed] [Google Scholar]
- 65.Swaroop S., Mahadevan A., Shankar S.K., Adlakha Y.K., Basu A. HSP60 critically regulates endogenous IL-1beta production in activated microglia by stimulating NLRP3 inflammasome pathway. J. Neuroinflammation. 2018;15:177. doi: 10.1186/s12974-018-1214-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Israël A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang W.C., Hung M.C. Beyond NF-kappaB activation: nuclear functions of IkappaB kinase alpha. J. Biomed. Sci. 2013;20:3. doi: 10.1186/1423-0127-20-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jeppesen D.K., Fenix A.M., Franklin J.L., Higginbotham J.N., Zhang Q., Zimmerman L.J., Liebler D.C., Ping J., Liu Q., Evans R., et al. Reassessment of exosome composition. Cell. 2019;177:428–445.e18. doi: 10.1016/j.cell.2019.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Onódi Z., Pelyhe C., Terézia Nagy C., Brenner G.B., Almási L., Kittel Á., Manček-Keber M., Ferdinandy P., Buzás E.I., Giricz Z. Isolation of high-purity extracellular vesicles by the combination of iodixanol density gradient ultracentrifugation and bind-elute chromatography from blood plasma. Front. Physiol. 2018;9:1479. doi: 10.3389/fphys.2018.01479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wei W., Ao Q., Wang X., Cao Y., Liu Y., Zheng S.G., Tian X. Mesenchymal stem cell-derived exosomes: a promising biological tool in nanomedicine. Front. Pharmacol. 2020;11 doi: 10.3389/fphar.2020.590470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee H., Zhang D., Zhu Z., Dela Cruz C.S., Jin Y. Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci. Rep. 2016;6 doi: 10.1038/srep35250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Happel K.I., Zheng M., Young E., Quinton L.J., Lockhart E., Ramsay A.J., Shellito J.E., Schurr J.R., Bagby G.J., Nelson S., Kolls J.K. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J. Immunol. 2003;170:4432–4436. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Steták A., Veress R., Ovádi J., Csermely P., Kéri G., Ullrich A. Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer Res. 2007;67:1602–1608. doi: 10.1158/0008-5472.CAN-06-2870. [DOI] [PubMed] [Google Scholar]
- 74.Lee H., Zhang D., Wu J., Otterbein L.E., Jin Y. Lung epithelial cell-derived microvesicles regulate macrophage migration via MicroRNA-17/221-induced integrin beta1 recycling. J. Immunol. 2017;199:1453–1464. doi: 10.4049/jimmunol.1700165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rider M.A., Hurwitz S.N., Meckes D.G., Jr. ExtraPEG: a polyethylene glycol-based method for enrichment of extracellular vesicles. Sci. Rep. 2016;6 doi: 10.1038/srep23978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oksvold M.P., Neurauter A., Pedersen K.W. Magnetic bead-based isolation of exosomes. Methods Mol. Biol. 2015;1218:465–481. doi: 10.1007/978-1-4939-1538-5_27. [DOI] [PubMed] [Google Scholar]
- 77.Han Y., Zhu Y., Youngblood H.A., Almuntashiri S., Jones T.W., Wang X., Liu Y., Somanath P.R., Zhang D. Nebulization of extracellular vesicles: a promising small RNA delivery approach for lung diseases. J. Control Release. 2022;352:556–569. doi: 10.1016/j.jconrel.2022.10.052. [DOI] [PubMed] [Google Scholar]
- 78.Wang X., Polverino F., Rojas-Quintero J., Zhang D., Sánchez J., Yambayev I., Lindqvist E., Virtala R., Djukanovic R., Davies D.E., et al. A disintegrin and A metalloproteinase-9 (ADAM9): a novel proteinase culprit with multifarious contributions to COPD. Am. J. Respir. Crit. Care Med. 2018;198:1500–1518. doi: 10.1164/rccm.201711-2300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stripp B.R., Reynolds S.D., Boe I.M., Lund J., Power J.H.T., Coppens J.T., Wong V., Reynolds P.R., Plopper C.G. Clara cell secretory protein deficiency alters clara cell secretory apparatus and the protein composition of airway lining fluid. Am. J. Respir. Cell Mol. Biol. 2002;27:170–178. doi: 10.1165/ajrcmb.27.2.200200270c. [DOI] [PubMed] [Google Scholar]
- 80.Helwa I., Cai J., Drewry M.D., Zimmerman A., Dinkins M.B., Khaled M.L., Seremwe M., Dismuke W.M., Bieberich E., Stamer W.D., et al. A comparative study of serum exosome isolation using differential ultracentrifugation and three commercial reagents. PLoS One. 2017;12 doi: 10.1371/journal.pone.0170628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee H., Zhang D., Laskin D.L., Jin Y. Functional evidence of pulmonary extracellular vesicles in infectious and noninfectious lung inflammation. J. Immunol. 2018;201:1500–1509. doi: 10.4049/jimmunol.1800264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.National Heart, Lung and Blood Institute Acute Respiratory Distress Syndrome Clinical Trials Network. Matthay M.A., Brower R.G., Carson S., Douglas I.S., Eisner M., Hite D., Holets S., Kallet R.H., Liu K.D., et al. Randomized, placebo-controlled clinical trial of an aerosolized beta(2)-agonist for treatment of acute lung injury. Am. J. Respir. Crit. Care Med. 2011;184:561–568. doi: 10.1164/rccm.201012-2090OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matute-Bello G., Downey G., Moore B.B., Groshong S.D., Matthay M.A., Slutsky A.S., Kuebler W.M., Acute Lung Injury in Animals Study Group An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 2011;44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang D., Lee H., Haspel J.A., Jin Y. Long noncoding RNA FOXD3-AS1 regulates oxidative stress-induced apoptosis via sponging microRNA-150. FASEB J. 2017;31:4472–4481. doi: 10.1096/fj.201700091R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Davidson S.M., Takov K., Yellon D.M. Exosomes and cardiovascular protection. Cardiovasc. Drugs Ther. 2017;31:77–86. doi: 10.1007/s10557-016-6698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplemental materials.