

Abstract
Gene co‐expression network analysis is an efficient systems biology approach for the discovery of novel gene functions and trait‐associated gene modules. To identify clusters of functionally related genes involved in soybean nodule formation and development, we performed a weighted gene co‐expression network analysis. Two nodule‐specific modules (NSM‐1 and NSM‐2, containing 304 and 203 genes, respectively) were identified. The NSM‐1 gene promoters were significantly enriched in cis‐binding elements for ERF, MYB, and C2H2‐type zinc transcription factors, whereas NSM‐2 gene promoters were enriched in cis‐binding elements for TCP, bZIP, and bHLH transcription factors, suggesting a role of these regulatory factors in the transcriptional activation of nodule co‐expressed genes. The co‐expressed gene modules included genes with potential novel roles in nodulation, including those involved in xylem development, transmembrane transport, the ethylene signalling pathway, cytoskeleton organization, cytokinesis and regulation of the cell cycle, regulation of meristem initiation and growth, transcriptional regulation, DNA methylation, and histone modifications. Functional analysis of two co‐expressed genes using TILLING mutants provided novel insight into the involvement of unsaturated fatty acid biosynthesis and folate metabolism in nodule formation and development. The identified gene co‐expression modules provide valuable resources for further functional genomics studies to dissect the genetic basis of nodule formation and development in soybean.
Keywords: gene co‐expression network, nodule formation and development, soybean, TILLING
Two modules of highly co‐regulated soybean genes associated with nodule formation and development were identified using gene co‐expression network analysis.

Leguminous plants such as soybean (Glycine max), alfalfa (Medicago sativa), and common bean (Phaseolus vulgaris) form a symbiotic association with soil nitrogen‐fixing bacteria that leads to the formation of root nodules. The symbiotic association between leguminous plants and rhizobia is a host‐specific process initiated in the rhizosphere, in which bacterial host‐specific nodulation genes are activated by flavonoids released from plant roots as signal molecules (Roy et al., 2020). Transcriptional activation of host‐specific nodulation genes results in the synthesis of host‐specific lipo‐chitooligosaccharides known as Nod factors. Secretion of Nod factors is recognized and perceived by cognate host receptors, which induce deformation and curling of root hairs that result in entrapping bacterial cells in the host cell wall and penetrating root cells, leading to the formation of nitrogen‐fixing nodules (Roy et al., 2020). In the developing nodules, the bacteria differentiate into bacteroids, which use a nitrogenase enzyme complex to convert atmospheric nitrogen into ammonia, which can be readily absorbed by the host plant (Oldroyd & Downie, 2008; Roy et al., 2020).
In soybean, nodule organogenesis involves transcriptome programming of a significant number of plant genes implicated in a wide range of biological processes (Brechenimacher et al., 2008; Hayashi et al., 2008; Libault et al., 2009, 2010; Severin et al., 2010; Yuan et al., 2016, 2017). Our recent transcriptome analysis of 12‐, 22‐, and 36‐day‐old‐nodules, corresponding to formation, development, and senescence stages, implied that about half of soybean genes are transcriptionally regulated during nodule formation and development (Niyikiza et al., 2020). Of these, 9669 genes were similarly regulated across various nodule developmental stages and hence were considered as nodulation core genes (Niyikiza et al., 2020). However, this extended list of genes poses a major challenge for determining the causal gene‐to‐phenotype association. In this regard, gene co‐expression network analysis represents a powerful analytical tool to identify modules of highly co‐regulated genes that are trait‐correlated (Piya et al., 2022; Stuart et al., 2003). This approach is progressively used to prioritize candidate genes for further functional genomics studies in various plant species including, for example, tomato (Solanum lycopersicum) (Fukushima et al., 2012; Narise et al., 2017; Pan et al., 2013), maize (Zea mays) (Chen et al., 2014; Downs et al., 2013), rice (Oryza sativa) (Ficklin et al., 2010; Shaik & Ramakrishna, 2014), soybean (Liu et al., 2015; Piya et al., 2022; Zhu et al., 2022), and grapevine (Vitis vinifera) (Palumbo et al., 2014). With the recent expansion of RNA‐sequencing (RNA‐Seq) datasets spanning various plant tissues, organs, and developmental conditions, accurate identification of co‐regulated gene modules correlated with particular traits can be achieved.
In this study, we performed a weighted gene co‐expression network analysis (WGCNA; Langfelder & Horvath, 2008) to identify modules of highly co‐regulated soybean genes associated with nodule formation and development. The analysis was conducted using 138 RNA‐Seq datasets from 11 different soybean tissues (cotyledon, flower, lateral root, leaf, nodule, pod, root, root hair cell, seed, seedling, and stem; Table S1). It is important to mention that nine nodule RNA‐Seq datasets corresponding to the nodule formation, development, and senescence stages (Niyikiza et al., 2020) were selected because of the high sequencing depth and coverage. High‐quality reads from each dataset were mapped to the soybean reference genome (Wm82.a2.v1) using TopHat v. 2.0.14 (Trapnell et al., 2009) and the reads mapped to each gene were counted using HTSeq (Anders et al., 2015). Genes with fewer than 10 mapped reads in more than 95% of the samples were removed from the analysis. The data were then subjected to variance‐stabilizing transformation with the R package DESeq2 (Love et al., 2014) using the varianceStabilizingTransformation function. Genes showing zero variance across the samples were also removed. The batch effect of the remaining 27,749 genes was adjusted using R package ComBat (Johnson et al., 2007). To ensure scale‐free topology, the soft‐thresholding power of β = 9 was used. Then, we built the adjacency matrix and constructed the topological overlap matrix. Based on average hierarchical clustering and dynamic tree clipping, we identified a total of 34 colour‐coded modules (Figure 1a). Two upregulated modules were specific to nodules based on the module eigengenes across various tissues and the absence of any significant correlation with other tissues (Figure S1). Other modules with high correlation values in nodules were not considered because they showed high correlation values in other tissues. These two nodule‐specific modules (NSM) contained 304 (NSM‐1) and 203 genes (NSM‐2) (Figure 1b,c). These genes were then compared with the previously reported 9669 nodule core genes that were similarly regulated during various nodule developmental stages (Niyikiza et al., 2020). Notably, 187 NSM‐1 genes and 40 NSM‐2 genes overlapped with the nodule core genes, indicating that NSM‐1 and NSM‐2 include both core and stage‐specific genes.
FIGURE 1.

Identification of two nodule‐specific modules using weighted gene co‐expression network analysis (WGCNA). (a) Network heatmap plot showing the eigengenes of 34 modules. The gene dendrograms and module colours are displayed to the left and top of the heatmap. NSM‐1 and NSM‐2 are highlighted in dark grey and violet, respectively. (b and c) Gene networks showing co‐expression events between 304 genes in NSM‐1 (b) and 203 genes in NSM‐2 (c) The gene networks were visualized using Cytoscape v. 3.7.2. (d and e) Cis‐binding motifs enriched in the promoters of genes in NSM‐1 (d) or NSM‐2 (e).
Among the NSM‐1 genes, we found several with potential roles in nodulation, including glutamate synthase (Carvalho et al., 2003), Clavata3/ESR (CLE) (Hastwell et al., 2014; Lim et al., 2011), chalcone‐flavanone isomerases (Liu & Murray, 2016; Subramanian et al., 2007), pectin lyase‐like superfamily proteins (Xie et al., 2012), cysteine proteinase superfamily proteins (van Wyk et al., 2014), and several metabolite transporters (Udvardi & Poole, 2013). Similarly, NSM‐2 contained genes with possible function in nodulation, including haemoglobin 3, early nodulin‐related genes, SNARE‐like superfamily proteins, and hypoxia‐responsive family proteins (Larrainzar et al., 2020; Sogawa et al., 2019) Also, NSM‐2 contained several genes encoding wound‐responsive family proteins and ethylene signalling factors, suggesting that symbiosis signalling may share common components with the wound signal transduction pathway, particularly those associated with ethylene‐dependent defence responses.
Careful examination of the putative functions of NSM‐1 and NSM‐2 genes provided insight into the potential implication of a number of formerly unappreciated biological processes in nodule formation and development. For example, NSM‐1 included several genes involved in xylem and phloem pattern formation (Glyma.17G006600, Glyma.06g066200, Glyma.12g102600, and Glyma.14G087400), vesicle‐mediated transport (Glyma.04G029800, Glyma.15G192100, Glyma.08G135400, Glyma.10g243500, Glyma.14g147800, and Glyma.13g288600), plasmodesmata‐mediated intercellular transport (Glyma.10G190500 and Glyma.05G018200), cytokinesis (Glyma.13G310700, Glyma.03G211900, Glyma.13G025500, and Glyma.17G038700), sphingoid biosynthesis (Glyma.05G053300 and Glyma.02g106300), polyamine biosynthesis (Glyma.04G007700 and Glyma.12G185700), folate biosynthetic process (Glyma.02g217100, Glyma.12g226400, and Glyma.19G121600), and DNA methylation (Glyma.15G121800, Glyma.12G001300, Glyma.08G269000, Glyma.11G219200, and Glyma.18G038300) (Table S2). Similarly, NSM‐2 included several genes encoding functions involved in cysteine dioxygenase activity (Glyma.09g240100, Glyma.13g064300, Glyma.16g037600, Glyma.18g256400, Glyma.19g020500, Glyma.19g115300, and Glyma.19g115600), GTPase activity (Glyma.14G098000 and Glyma.17G226700), and anaerobic respiration and cellular response to hypoxia (Table S2).
To identify transcription factors with possible roles in regulating the expression of NSM‐1 and NSM‐2 genes, we examined the gene promoters of these two modules for transcription factor cis‐binding elements. The promoter regions, 2000 bp upstream of the translation initiation codon ATG, were analysed using the MEME database (Bailey et al., 2009). Genes in NSM‐1 were significantly enriched in cis‐binding elements for various ethylene response factors (ERFs), MYB, and C2H2‐type zinc finger factors (Figure 1d). The cis‐binding motifs for ERF, MYB, and C2H2‐type zinc transcription factors were detected in 33%, 38%, and 47% of the NSM‐1 genes, respectively. Notably, 74% (225 out of 304) of the NSM‐1 genes contained at least one of these three motifs in the promoter region. Similarly, the NSM‐2 gene promoters were significantly enriched in cis‐binding elements for TCP, bZIP, and bHLH, transcription factors, which were detected in 26%, 30%, and 40% of the genes (Figure 1e). At least one of these three binding motifs is present in 60% of the NSM‐2 gene promoters. Intriguingly, several genes encoding these six transcription factor family members are among the NSM‐1 and NSM‐2 gene modules. These findings suggest a possible role of these transcription factors in transcriptional activation of nodule co‐expressed genes. While few members of these transcription factor families have been reported to play key roles in rhizobium–legume symbiosis (reviewed in Chakraborty et al., 2022; Diédhiou & Diouf, 2018), our analysis extends the limited list of the regulatory factors likely controlling nodule formation and development, and provides a solid foundation for future studies aimed at defining the exact regulatory function of these transcription factors.
Heatmap analysis of normalized gene expression levels of NSM‐1 and NSM‐2 genes across 11 tissues revealed that the large majority of NSM‐1 and NSM‐2 are highly expressed in nodules. Notably, a number of NSM‐1 genes also exhibited high expression in lateral roots, seedlings, and developing seeds (Figure 2a). Similarly, genes in NSM‐2 exhibited high expression in root tissues (Figure 2b). These expression data suggest that the majority of genes in NSM‐1 and NSM‐2 are not nodule‐specific and most likely have other molecular functions beside their potential involvement in nodule organogenesis. Gene ontology (GO) enrichment analysis indicated that ontologies with functions related to the small molecule catabolic process, amine metabolic process, and cellular ketone metabolic process are significantly enriched among both the NSM‐1 and NSM‐2 genes (Figure 3a,b). Ontologies associated with negative regulation of macromolecule metabolic process and negative regulation of biological process are uniquely enriched among the NSM‐1 genes (Figure 3a). Ontologies with functions related to metabolic processes of carbohydrate, glucose, alcohol, monosaccharide, carboxylic acid, and organic acid are uniquely enriched among the NSM‐2 genes (Figure 3b). Also, the catabolic processes of cellular macromolecules and carbohydrate are specifically enriched among the NSM‐2 genes (Figure 3b). These data imply that genes involved in sugar‐related metabolic and catabolic processes play a key role in nodule formation and development. This is consistent with the fundamental role of sugar metabolism in nitrogen assimilation and transport during nodulation (Udvardi & Poole, 2013).
FIGURE 2.
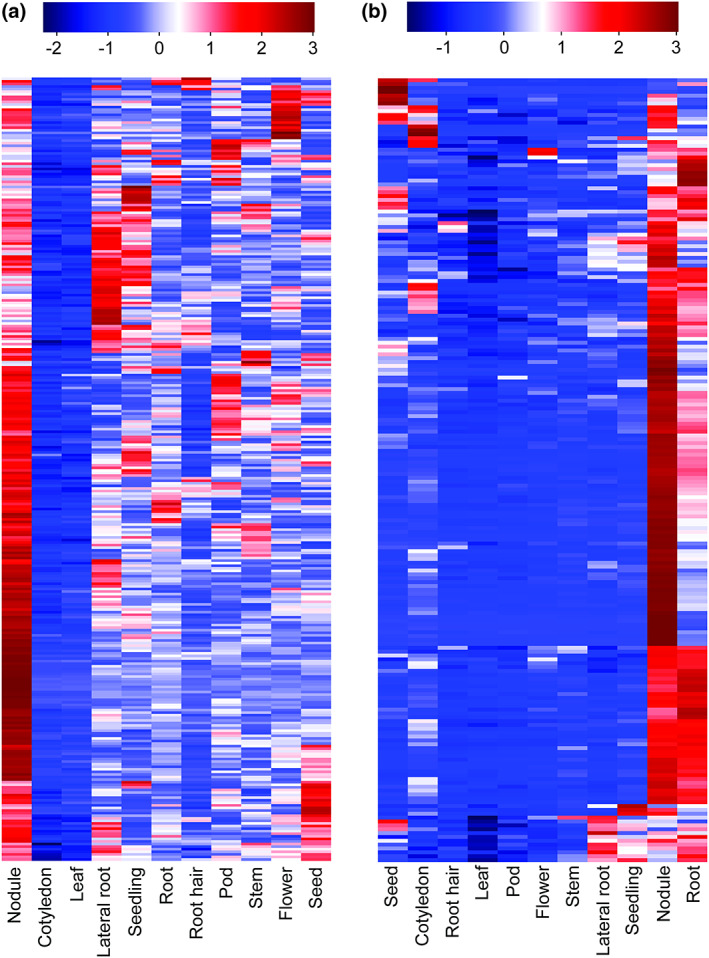
Heatmap demonstrating the expression patterns of NSM‐1 and NSM‐2 genes across 11 tissues. RNA‐Seq count data were normalized as RPKM and used to construct gene expression heatmaps of NSM‐1 (a) and NSM‐2 (b) genes. Colour scales on the top indicate log2 of normalized count data.
FIGURE 3.
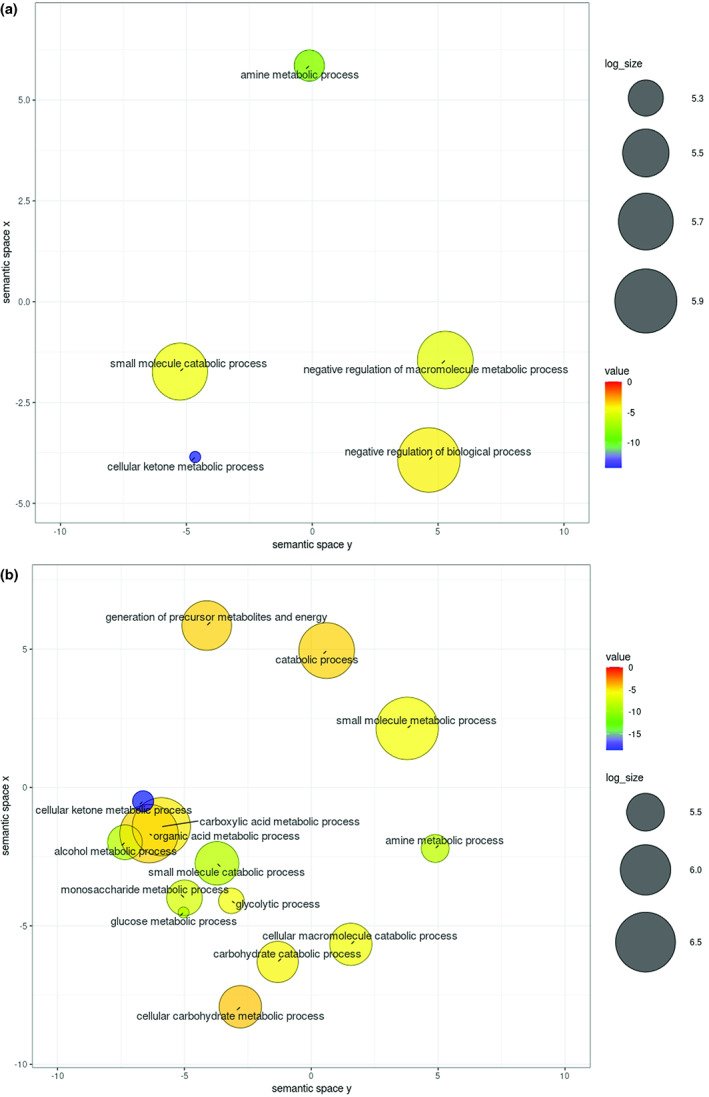
Gene ontology (GO) term enrichment analysis of nodule specific modules. (a and b) Ontology enrichment analyses of the NSM‐1 (a) and NSM‐2 (b) genes were conducted using the agriGO database with Fisher's exact test and Bonferroni multitest adjustment. Adjusted p value <0.01 was set as statistically significant. Redundant GO terms were grouped based on semantic similarity to other terms in the Uniprot database and visualized as a scatter plot using the REViGO Web server (Supek et al., 2011). The size of the bubbles reflects the abundance of the GO terms. The colour scales represent log10 of the GO term adjusted p values.
We evaluated the potential impact of two of the NSM‐1 and NSM‐2 genes (Glyma.19G121600 and Glyma.14G121400) on nodule formation and development using TILLING (targeting‐induced local lesions in genomes) mutant lines. These two genes were selected because they represent distinct connectivity and varied expression patterns. Glyma.19G121600 showed low connectivity with only two co‐expression events and is highly expressed in the developing nodules, roots, leaf buds, and flowers (Table S1). In contrast, Glyma.14G121400 showed high connectivity with 50 co‐expression events and is specifically highly expressed in the nodules (Table S1). In addition, the functions of these two genes in nodule formation and development remain largely unknown. Screening our TILLING mutants' collection generated in the soybean cv. Forrest background (Lakhssassi et al., 2020), we identified three mutants (FM3_1726, FM4_217, and FM2_1775) for Glyma.19G121600 and five mutants (FM4_1416, FM5_605_28, FM5_813, FM3_90, and FM2_111) for Glyma.14G121400 (Figure 4a). We determined nodule number and size in these eight TILLING mutant lines as well as the wild‐type Forrest in two independent experiments. Four‐day‐old seedlings were inoculated with Bradyrhizobium diazoefficiens (strain USDA110) as previously described (Niyikiza et al., 2020). Twenty‐two days after inoculation, the number of nodules per root system was counted and the nodules were then photographed for size measurements using ImageJ software. Each mutant plant was confirmed by sequencing PCR‐amplified products generated using mutant‐specific primers as described by Lakhssassi et al. (2020). Interestingly, the average numbers of nodules developed on the eight TILLING mutants were significantly reduced when compared with the wild‐type Forrest (Figure 4b). The average number of nodules formed on the Glyma.19G121600 and Glyma.14G121400 mutants ranged, respectively, from 6.5 to 8.4 and 5.9 to 22.3 compared with 39.4 nodules determined in wild‐type plants (Figure 4b). Notably, the Glyma.19G121600 mutants showed significant reduction in the nodule size as compared with wild‐type plants (Figure 4c). Only two TILLING mutant lines of Glyma.14G121400 developed nodules smaller than those formed on wild‐type plants (Figure 4c).
FIGURE 4.
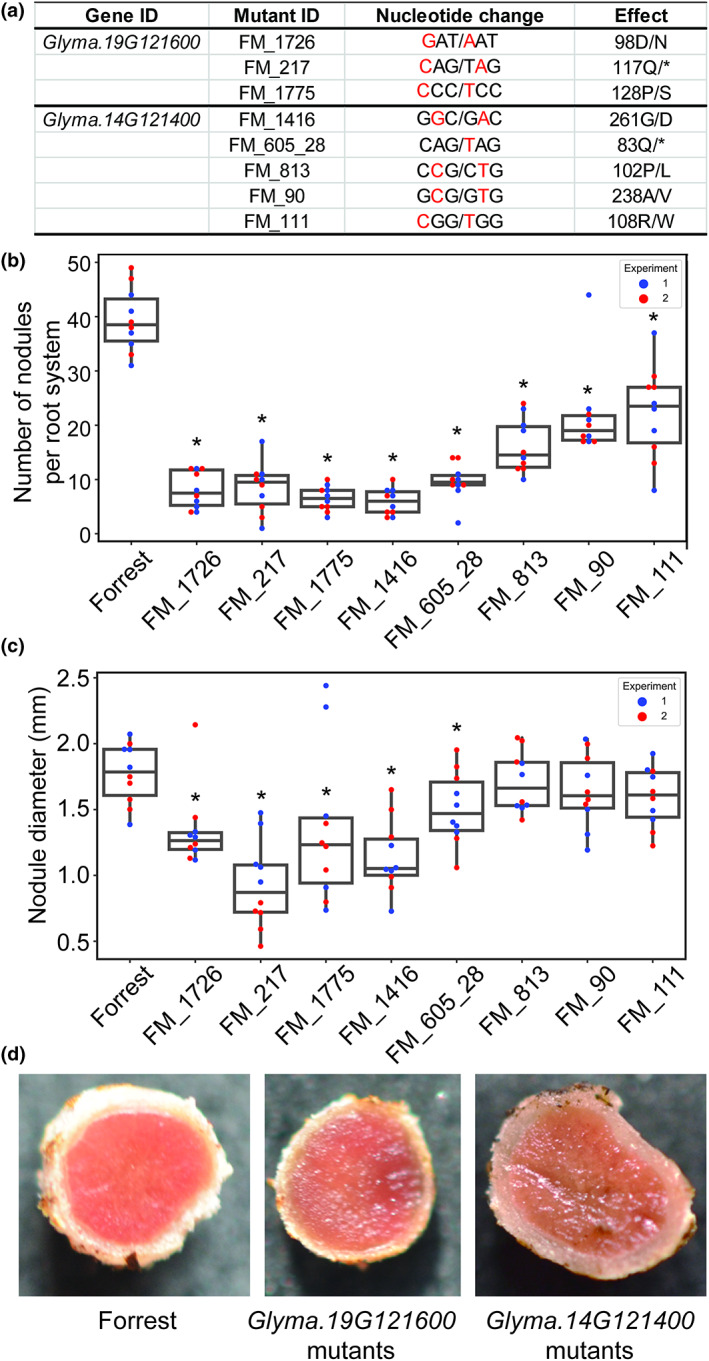
TILLING mutations of Glyma.19G121600 and Glyma.14G121400 impact nodule formation and development. (a) Description of TILLING mutations of Glyma.19G121600 and Glyma.14G121400, and the corresponding amino acid substitutions. (b and c) Impact of TILLING mutations in Glyma.19G121600 and Glyma.14G121400 on nodule formation and development. Nodule number (a) and size (b) were determined 22 days after inoculation with Bradyrhizobium diazoefficiens (strain USDA110). Data were obtained from at least 10 plants from two independent experiments, which are indicated in blue and red. Dots in the graphs represent values from individual plants. Asterisks indicate statistically significant differences from the wild‐type cv. Forrest as determined by analysis of variance (p < 0.05). (d) Cross‐sections of 22‐day‐old nodules formed on wild‐type Forrest and TILLING mutants of Glyma.19G121600 and Glyma.14G121400. The pink colour in the wild‐type nodule reflects a high concentration of leghaemoglobin associated with active nitrogen fixation. In contrast, the brown colour in the nodules formed in TILLING mutants of Glyma.14G121400 and Glyma.19G121600 reflects a low concentration of leghaemoglobin associated with reduced nitrogen fixation.
To gain more insight into the function of Glyma.14G121400 and Glyma.19G121600 in nitrogen fixation, we performed cross‐sections of the nodules formed on these eight mutants and wild‐type plants, and examined nodule structure and colour using light microscopy. No obvious differences in nodule structure between the mutant lines and wild‐type plants were observed. Interestingly, the root nodules of wild‐type plants were bright pink in colour due to the presence of a high concentration of leghaemoglobin associated with high nitrogen fixation (Figure 4d). The root nodules of the TILLING mutants of both genes were brown, indicative of reduced nitrogen fixation activity (Figure 4d). Together, these data imply a role of Glyma.14G121400 and Glyma.19G121600 in nodule formation, development, and nitrogen fixation.
Glyma.19G121600 encodes a dihydrofolate reductase, which catalyses the conversion of dihydrofolate into tetrahydrofolates, multifunctional cofactors that play a key role in the cell division and biosynthesis of nucleic acids and amino acids (Banuelos et al., 2021). Several transcriptomic and proteomic studies suggest a role of folates in nodule formation and development, but direct evidence remains elusive (reviewed in Banuelos et al., 2021). In this context, our data provide genetic evidence that potential changes in folate metabolism or level as a result of Glyma.19G121600 knockout impacted both nodule number and size as well as nitrogen fixation. The observed defects may be linked to the role of folates and their derivatives in biological processes fundamental for symbiosis, including biosynthesis of purines, DNA methylation, endoreduplication, nitrogen translocation, and hormone biosynthesis.
Glyma.14G121400 encodes a stearoyl‐acyl carrier protein desaturase, which catalyses the conversion of stearic acid to oleic acid, and hence the total content of unsaturated fatty acids. Stearoyl‐acyl carrier protein desaturases have been shown to control various aspects of nodule development in soybean (Gillman et al., 2014; Krishnan et al., 2016; Lakhssassi et al., 2017, 2020). Although the soybean genome contains five genes coding for stearoyl‐acyl carrier protein desaturase (Lakhssassi et al., 2020), only Glyma.14G121400 is expressed in soybean nodules (Niyikiza et al., 2020). This finding may suggest a role of Glyma.14G121400 in nodule development via controlling the levels of unsaturated fatty acids. This suggestion is further supported by a recent study documenting the essential role of fatty acids and lipid biosynthesis for nodule formation and development in soybean (Zhang et al., 2020). Because primary metabolic pathways are highly interconnected, it not surprising that mutations in Glyma.14G121400 could impact a broad range of nodule phenotypic and physiological characteristics. In this context, future metabolic profiling of these mutants would reveal the exact metabolites and pathways impacted by these mutations.
In conclusion, our gene co‐expression analysis resulted in identifying not only genes with known functions in nodulation but, more crucially, also pointed to novel genes with potential roles in nodulation. For example, NSM‐1 and/or NSM‐2 included genes involved in response to wounding, xylem development, transmembrane transport, ethylene signalling pathway, cytoskeleton organization, cytokinesis, regulation of cell cycle, regulation of meristem initiation and growth, transcriptional regulation, DNA methylation, and histone modifications with potential novel roles in regulating nodule development in soybean. Functional characterization of the genes coding these functions is expected to improve our understanding of nodule organogenesis and identify targets for improving nodule formation and development to reduce synthetic fertilizer use.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest.
Supporting information
Figure S1 Module‐trait heatmap showing the correlations between the module eigengenes and various tissues and organs. The nodule‐specific modules NSM‐1 and NSM‐2 are highlighted in dark grey and violet, respectively. The colour bar in the right represents the correlation value
Table S1 Read count data of 138 RNA‐Seq samples used for gene co‐expression analysis
Table S2 Identification numbers and functional annotations of the NSM‐1 and NSM‐2 genes
ACKNOWLEDGEMENTS
This project was supported by funds from the Tennessee Soybean Promotion Board to Tarek Hewezi and Vince Pantalone.
Piya, S. , Pantalone, V. , Zadegan, S.B. , Shipp, S. , Lakhssassi, N. , Knizia, D. et al. (2023) Soybean gene co‐expression network analysis identifies two co‐regulated gene modules associated with nodule formation and development. Molecular Plant Pathology, 24, 628–636. Available from: 10.1111/mpp.13327
DATA AVAILABILITY STATEMENT
The data supporting the findings of this study are provided in the manuscript and supporting information.
REFERENCES
- Anders, S. , Pyl, P.T. & Huber, W. (2015) HTSeq‐a python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, T.L. , Boden, M. , Buske, F.A. , Frith, M. , Grant, C.E. , Clementi, L. et al. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Research, 37(suppl_2), W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banuelos, J. , Martínez‐Romero, E. , Montaño, N.M. & Camargo‐Ricalde, S.L. (2021) Folates in legume root nodules. Physiologia Plantarum, 171, 447–452. [DOI] [PubMed] [Google Scholar]
- Brechenimacher, L. , Kim, M.Y. , Benitez, M. , Li, M. , Joshi, T. , Calla, B. et al. (2008) Transcription profiling of soybean nodulation by Bradyrhizobium japonicum . Molecular Plant‐Microbe Interactions, 21, 631–645. [DOI] [PubMed] [Google Scholar]
- Carvalho, H.G. , Lopes‐Cardoso, I.A. , Lima, L.M. , Melo, P.M. & Cullimore, J.V. (2003) Nodule‐specific modulation of glutamine synthetase in transgenic Medicago truncatula leads to inverse alterations in asparagine synthetase expression. Plant Physiology, 133, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, S. , Valdés‐López, O. , Stonoha‐Arther, C. & Ané, J.M. (2022) Transcription factors controlling the Rhizobium–legume symbiosis: integrating infection, organogenesis, and the abiotic environment. Plant and Cell Physiology, 63, 1326–1343. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Zeng, B. , Zhang, M. , Xie, S. , Wang, G. , Hauck, A. et al. (2014) Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiology, 166, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diédhiou, I. & Diouf, D. (2018) Transcription factors network in root endosymbiosis establishment and development. World Journal of Microbiology and Biotechnology, 34, 37. [DOI] [PubMed] [Google Scholar]
- Downs, G.S. , Bi, Y.M. , Colasanti, J. , Wu, W. , Chen, X. , Zhu, T. et al. (2013) A developmental transcriptional network for maize defines coexpression modules. Plant Physiology, 161, 1830–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficklin, S.P. , Luo, F. & Feltus, F.A. (2010) The association of multiple interacting genes with specific phenotypes in rice using gene coexpression networks. Plant Physiology, 154, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima, A. , Nishizawa, T. , Hayakumo, M. , Hikosaka, S. , Saito, K. , Goto, E. et al. (2012) Exploring tomato gene functions based on coexpression modules using graph clustering and differential coexpression approaches. Plant Physiology, 158, 1487–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillman, J.D. , Stacey, M.G. , Cui, Y. , Berg, H.R. & Stacey, G. (2014) Deletions of the SACPD‐C locus elevate seed stearic acid levels but also result in fatty acid and morphological alterations in nitrogen fixing nodules. BMC Plant Biology, 14, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastwell, A.H. , Gresshoff, P.M. & Ferguson, B.J. (2014) The structure and activity of nodulation‐suppressing CLE peptide hormones of legumes. Functional Plant Biology, 42, 229–238. [DOI] [PubMed] [Google Scholar]
- Hayashi, S. , Gresshoff, P.M. & Kinkema, M. (2008) Molecular analysis of lipoxygenases associated with nodule development in soybean. Molecular Plant‐Microbe Interactions, 21, 843–853. [DOI] [PubMed] [Google Scholar]
- Johnson, W.E. , Li, C. & Rabinovic, A. (2007) Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Krishnan, H.B. , Alaswad, A.A. , Oehrle, N.W. & Gillman, J.D. (2016) Deletion of the SACPD‐C locus alters the symbiotic relationship between Bradyrhizobium japonicum USDA110 and soybean, resulting in elicitation of plant defense response and nodulation defects. Molecular Plant‐Microbe Interactions, 29, 862–877. [DOI] [PubMed] [Google Scholar]
- Lakhssassi, N. , Colantonio, V. , Flowers, N.D. , Zhou, Z. , Henry, J. , Liu, S. et al. (2017) Stearoyl‐acyl carrier protein desaturase mutations uncover an impact of stearic acid in leaf and nodule structure. Plant Physiology, 174, 1531–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhssassi, N. , Zhou, Z. , Liu, S. , Piya, S. , Cullen, M.A. , El Baze, A. et al. (2020) Soybean TILLING‐by‐sequencing+ reveals the role of novel GmSACPD members in unsaturated fatty acid biosynthesis while maintaining healthy nodules. Journal of Experimental Botany, 71, 6969–6987. [DOI] [PubMed] [Google Scholar]
- Langfelder, P. & Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrainzar, E. , Villar, I. , Rubio, M.C. , Pérez‐Rontomé, C. , Huertas, R. , Sato, S. et al. (2020) Hemoglobins in the legume–Rhizobium symbiosis. New Phytologist, 228, 472–484. [DOI] [PubMed] [Google Scholar]
- Libault, M. , Farmer, A. , Brechenmacher, L. , Drnevich, J. , Langley, R.J. , Bilgin, D.D. et al. (2010) Complete transcriptome of the soybean root hair cell, a single‐cell model, and its alteration in response to Bradyrhizobium japonicum infection. Plant Physiology, 152, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libault, M. , Joshi, T. , Takahashi, K. , Hurley‐Sommer, A. , Puricelli, K. , Blake, S. et al. (2009) Large‐scale analysis of putative soybean regulatory gene expression identifies a Myb gene involved in soybean nodule development. Plant Physiology, 151, 1207–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, C.W. , Lee, Y.W. & Hwang, C.H. (2011) Soybean nodule‐enhanced CLE peptides in roots act as signals in GmNARK‐mediated nodulation suppression. Plant Cell Physiology, 52, 1613–1627. [DOI] [PubMed] [Google Scholar]
- Liu, J.Y. , Chen, N.N. , Grant, J.N. , Cheng, Z.M. , Stewart, C.N. & Hewezi, T. (2015) Soybean kinome: functional classification and gene expression patterns. Journal of Experimental Botany, 66, 1919–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C.W. & Murray, J.D. (2016) The role of flavonoids in nodulation host‐range specificity: an update. Plants, 5, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. & Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narise, T. , Sakurai, N. , Obayashi, T. , Ohta, H. & Shibata, D. (2017) Co‐expressed pathways DataBase for tomato: a database to predict pathways relevant to a query gene. BMC Genomics, 18, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niyikiza, D. , Piya, S. , Routray, P. , Miao, L. , Kim, W.S. , Burch‐Smith, T. et al. (2020) Interactions of gene expression, alternative splicing, and DNA methylation in determining nodule identity. The Plant Journal, 103, 1744–1766. [DOI] [PubMed] [Google Scholar]
- Oldroyd, G.E. & Downie, J.A. (2008) Coordinating nodule morphogenesis with rhizobial infection in legumes. Annual Review of Plant Biology, 59, 519–546. [DOI] [PubMed] [Google Scholar]
- Palumbo, M.C. , Zenoni, S. , Fasoli, M. , Massonnet, M. , Farina, L. , Castiglione, F. et al. (2014) Integrated network analysis identifies fight‐club nodes as a class of hubs encompassing key putative switch genes that induce major transcriptome reprogramming during grapevine development. The Plant Cell, 26, 4617–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Y. , Bradley, G. , Pyke, K. , Ball, G. , Lu, C. , Fray, R. et al. (2013) Network inference analysis identifies an APRR2‐like gene linked to pigment accumulation in tomato and pepper fruits. Plant Physiology, 161, 1476–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piya, S. , Hawk, T. , Patel, B. , Baldwin, L. , Rice, J.H. , Stewart, C.N., Jr. et al. (2022) Kinase‐dead mutation: a novel strategy for improving soybean resistance to soybean cyst nematode Heterodera glycines . Molecular Plant Pathology, 23, 417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, S. , Liu, W. , Nandety, R.S. , Crook, A. , Mysore, K.S. , Pislariu, C.I. et al. (2020) Celebrating 20 years of genetic discoveries in legume nodulation and symbiotic nitrogen fixation. The Plant Cell, 32, 15–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severin, A.J. , Woody, J.L. , Bolon, Y.T. , Joseph, B. , Diers, B.W. , Farmer, A.D. et al. (2010) RNA‐Seq atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biology, 10, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik, R. & Ramakrishna, W. (2014) Machine learning approaches distinguish multiple stress conditions using stress‐responsive genes and identify candidate genes for broad resistance in rice. Plant Physiology, 164, 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogawa, A. , Yamazaki, A. , Yamasaki, H. , Komi, M. , Manabe, T. , Tajima, S. et al. (2019) SNARE proteins LjVAMP72a and LjVAMP72b are required for root symbiosis and root hair formation in Lotus japonicus . Frontiers in Plant Science, 9, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart, J.M. , Segal, E. , Koller, D. & Kim, S.K. (2003) A gene‐coexpression network for global discovery of conserved genetic modules. Science, 302, 249–255. [DOI] [PubMed] [Google Scholar]
- Subramanian, S. , Stacey, G. & Yu, O. (2007) Distinct, crucial roles of flavonoids during legume nodulation. Trends in Plant Science, 12, 282–285. [DOI] [PubMed] [Google Scholar]
- Supek, F. , Bošnjak, M. , Škunca, N. & Šmuc, T. (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One, 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. & Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udvardi, M. & Poole, P.S. (2013) Transport and metabolism in legume–rhizobia symbioses. Annual Review of Plant Biology, 64, 781–805. [DOI] [PubMed] [Google Scholar]
- van Wyk, S.G. , Du Plessis, M. , Cullis, C.A. , Kunert, K.J. & Vorster, B.J. (2014) Cysteine protease and cystatin expression and activity during soybean nodule development and senescence. BMC Plant Biology, 14, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, F. , Murray, J.D. , Kim, J. , Heckmann, A.B. , Edwards, A. , Oldroyd, G.E. et al. (2012) Pectate lyase required for root infection by rhizobia. Proceedings of the National Academy of Sciences of the United States of America, 109, 633–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, S. , Li, R. , Chen, S. , Chen, H. , Zhang, C. , Chen, L. et al. (2016) RNA‐Seq analysis of differential gene expression responding to different Rhizobium strains in soybean (Glycine max) roots. Frontiers in Plant Science, 7, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, S.L. , Li, R. , Chen, H.F. , Zhang, C.J. , Chen, L.M. , Hao, Q.N. et al. (2017) RNA‐Seq analysis of nodule development at five different developmental stages of soybean (Glycine max) inoculated with Bradyrhizobium japonicum strain 113–2. Scientific Reports, 7, 42248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G. , Ahmad, M.Z. , Chen, B. , Manan, S. , Zhang, Y. , Jin, H. et al. (2020) Lipidomic and transcriptomic profiling of developing nodules reveals the essential roles of active glycolysis and fatty acid and membrane lipid biosynthesis in soybean nodulation. The Plant Journal, 103, 1351–1371. [DOI] [PubMed] [Google Scholar]
- Zhu, X. , Zhang, B. , Gao, F. , Huang, F. , Zhang, H. & Huang, J. (2022) A soybean non‐coding RNA mining and co‐expression resource based on 1,596 RNA‐seq and small RNA‐seq libraries. Plant Physiology, 189, 1911–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Module‐trait heatmap showing the correlations between the module eigengenes and various tissues and organs. The nodule‐specific modules NSM‐1 and NSM‐2 are highlighted in dark grey and violet, respectively. The colour bar in the right represents the correlation value
Table S1 Read count data of 138 RNA‐Seq samples used for gene co‐expression analysis
Table S2 Identification numbers and functional annotations of the NSM‐1 and NSM‐2 genes
Data Availability Statement
The data supporting the findings of this study are provided in the manuscript and supporting information.