

Abstract
With the development of single‐cell RNA sequencing technology (scRNA‐seq), we have the ability to study biological questions at the level of the individual cell transcriptome. Nowadays, many analysis tools, specifically suitable for single‐cell RNA sequencing data, have been developed. In this review, the currently commonly used scRNA‐seq protocols are discussed. The upstream processing flow pipeline of scRNA‐seq data, including goals and popular tools for reads mapping and expression quantification, quality control, normalization, imputation, and batch effect removal is also introduced. Finally, methods to evaluate these tools in both cellular and genetic dimensions, clustering and differential expression analysis are presented.
Keywords: bioinformatics, biomedical engineering, genomics, RNA
This review discussed the currently commonly used scRNA‐seq protocols. introduced the upstream processing flow pipeline of scRNA‐seq data, including goals and popular tools, and presented methods to evaluate these tools in both cellular and genetic dimensions.
1. INTRODUCTION
It has been possible to gain unprecedented insight into the cellular diversity of organismal tissues through high‐throughput single‐cell transcriptomics. The bulk RNA sequencing method can only quantify the average signal of many cells, ignores the stochastic nature of gene expression in different cells, and does not reveal cellular heterogeneity [1, 2, 3]. However, single‐cell transcriptomics can address the problems associated with the stochastic nature of gene expression. As of now, single‐cell transcriptome sequencing (scRNA‐seq) studies have shown great promise for discovering new cell types, reconstructing developmental trajectories, and the study of tumor heterogeneity [4]. A new understanding of biological development and disease has also been provided by single‐cell transcriptome sequencing technologies.
The main difference between bulk RNA seq and scRNA seq is whether each sequencing library represents a single cell or a group of cells. The main reasons for the difference between the two libraries are the extremely low number of transcripts in a single cell, the low efficiency of mRNA capture, the loss of technology in the process of reverse transcription (RT), and the amplification bias generated by a large number of cDNA amplification processes [5, 6]. For sequencing data alignment, the tools used for bulk RNA seq alignment are also applicable to scRNA seq data. The alignment methods dedicated to scRNA seq generally have advantages in computing resources and running speed [7]. In terms of quality control, compared with bulk RNA seq, scRNA seq needs to filter out low‐quality cells and multicells through various filtering indicators. For the drop‐based method, background noise can also be removed. For data normalization, the method applicable to bulk RNA seq may introduce technical artifacts [8]. In terms of imputation, the single‐cell RNA sequencing data usually contains many missing values, and imputation algorithms based on multiple models can be used to impute zero values generated by technical factors. For batch effect processing, scRNA seq needs to consider both technical deviation and biological difference, and retain the biological variation that researchers want to focus on by eliminating unwanted variation.
Despite the convenience that scRNA‐seq offers to biological research, the technique still has many drawbacks. Its gene expression matrices are extremely noisy, high‐dimensional, and sparse. Therefore, to make the best use of scRNA‐seq technology, computational tools specifically designed for scRNA sequence data are required. In recent years, the explosion of single‐cell analysis tools has increased the difficulty of selecting the right tool for a given dataset [9, 10]. Many of these tools exist as user‐friendly tools [11, 12, 13] to process and interpret scRNA sequence data, but they are somewhat of a “black box” for non‐expert users. Therefore, understanding the computational approach at each step will help researchers to select more appropriate processes and tools for their data, so a review of these tools and methods is needed.
In this review, some steps of the single‐cell transcriptome analysis process will be highlighted, starting with the currently available single‐cell transcriptome sequencing technologies and the single‐cell transcriptome sequencing data processing process, which describes the evaluation methods for single‐cell transcriptome sequencing data processing methods. Overall, we hope to provide some assistance to users when selecting methods and tools to process single‐cell transcriptome data.
2. scRNA‐seq TECHNOLOGY
Over the past few years, many scRNA‐seq techniques have been developed [14, 15, 16, 17, 18, 19, 20, 21, 22, 23]. In general, the process of scRNA‐seq includes the following steps: [1] first isolate individual cells from tissues; [2] perform cell lysis to obtain mRNA; [3] perform mRNA molecular capture; [5] reverse transcribe (RT) mRNA to cDNA; [6] amplify cDNA using PCR or IVT; and [7] perform library construction and sequencing. Table 1 shows some of the currently used single‐cell transcriptome sequencing protocols.
TABLE 1.
Current common single‐cell transcriptome sequencing protocols.
| Platforms | Isolation strategies | Cell scale | UMI | Amplification methods | Region |
|---|---|---|---|---|---|
| Smart‐seq2 | FACS | Small | NO | PCR | Full‐length |
| MATQ‐seq | FACS | Small | YES | PCR | Full‐length |
| Drop‐seq | Microdroplets | Large | YES | PCR | 3′ end |
| inDrop | Microdroplets | Large | YES | IVT | 3′ end |
| STRT‐seq | FACS | Small | YES | PCR | 5′ end |
| 10x Genomics | Microdroplets | Large | YES | PCR | 3′ end |
| CEL‐seq2 | FACS | Small | YES | IVT | 3′ end |
| Seq‐well | Micro‐fluidic | Large | YES | PCR | 3′ end |
| Fluidigm C1 | Micro‐fluidic | Small | NO | PCR | Full‐length |
| Quartz‐Seq2 | FACS | Small | YES | PCR | 3′ end |
Depending on the method of quantification, single‐cell sequencing technologies can be broadly divided into two categories. The first category is full‐length methods (e.g., Smart‐seq2 [14]), which provide reads of the entire gene length and improves overall sensitivity. Nevertheless, the count of shorter genes is often lost in full‐length library preparations due to their bias towards longer genes. The second category is tag‐based approaches that typically incorporate unique molecular identifiers (UMIs) to identify and quantify individual transcripts by capturing and sequencing only 3' end transcripts (e.g. inDrop [17] and Drop‐seq [16]) or 5′ end transcripts (e.g., STRT‐seq [18]). The sources of sequence information obtained by sequencing these protocols are different. The 3 'end protocol obtains a segment of sequence information after the amplification of the cDNA transcription start site, while the 5′ end protocol obtains a segment of sequence information after the amplification of the 5 'end of the cDNA. The role of UMI is to do absolute quantification and correct sequencing errors because the amplification efficiency of each mRNA is different. Even if the initial mRNA expression amount of two mRNA is consistent, after multiple rounds of amplification, it may be mistaken for their differential expression. Through the initial marker of UMI, the original gene expression can be known by counting the type of UMI after amplification. Moreover, UMI can eliminate the identified sequencing errors and report the unique read before alignment. In brief, full‐length methods covering more reads, tag‐based methods dominate quantification, especially in experiments with high cell counts.
Depending on how the cells are captured, single‐cell sequencing techniques can also be divided into plate‐based and droplet‐based approaches. Droplet‐based technologies (e.g., inDrop [17], Drop‐seq [16], and 10X Genomics [19]) have become the most popular methods for isolating single‐cell RNA. The droplet‐based Chromium protocol from 10X Genomics can capture thousands of cells per run [24]. Whereas Drop‐Seq is usually more affordable and corresponds to fewer transcript counts per cell with only a slight loss of sensitivity. And inDrop‐seq allows customisation of parameters, so this protocol may be ideal for detecting low levels of expressed genes. Droplet‐based approaches offer high throughput, but at the cost of low sequencing depth (which means that fewer transcripts are captured per cell) [25]. As a result, droplet‐based methods are more susceptible to technical noise compared to plate‐based methods, which are preferred when dealing with rare cell types.
3. scRNA‐seq DATA PROCESSING FLOW PIPELINE
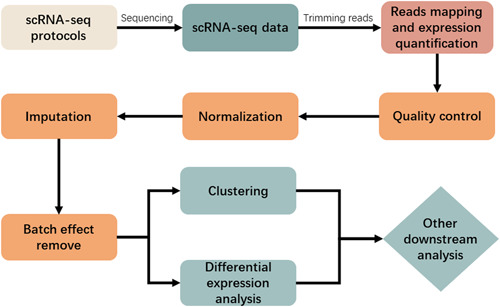
Figure 1 illustrates the current common single‐cell transcriptome sequencing data processing pipeline. After single‐cell transcriptome sequencing, the obtained scRNA‐seq data need to be analysed to generate a gene expression matrix, and in order to obtain the gene expression matrix, each sequenced reads needs to be mapped to a reference genome and quantified for expression. Before reads mapping, quality control (QC) of the FASTQ files obtained from sequencing is required. The most popular quality control tools include FastQC [26] and Cutadapt [27]. FastQC takes the sequenced FASTQ files obtained from the sequencing machine as input and returns a quality report of reads. After that, Cutadapt can be used to trim reads for quality improvement of alignment. For protocols that use UMIs, UMI‐tools [9] can also be executed to trim barcodes and perform demultiplexing because UMI sequences often have errors. UMI‐tools introduce a network‐based approach to interpret these errors when identifying PCR duplicates.
FIGURE 1.
Commonly used single‐cell transcriptome sequencing data processing pipeline.
The time and memory requirements for the mapping and expression quantification step are high. For example, CellRanger takes 22 h to process 784 million reads (equivalent to 50,000 cells when 15,000 reads per cell), and uses up to 30 GB of RAM. Other methods have significantly improved operation efficiency compared with CellRanger [28]. For the next steps, such as quality control, normalization, imputation, and batch effect removal, tools and methods based on statistical models or probabilistic models have certain advantages in running time and computing resource occupation, while tools and methods based on machine learning and deep learning models have high demand in running time and computing resource occupation. We recommend at least 32 threads, 128 GB of memory, and a computing environment with the Linux operating system installed.
3.1. Reads mapping and expression quantification
Figure 2 illustrates the basic process from sequencing data to the gene expression matrix. scRNA‐seq and bulk RNA‐seq technologies typically sequence transcripts into reads to generate raw data in fastq format, and since these two types of RNA‐seq data do not differ in sequence alignment, the original development for bulk RNA sequence mapping tools is also applicable to single‐cell RNA sequence data. Generally, reads mapping algorithms are divided into two main categories: those based on spacer seed indexing and those based on Burrows–Wheeler transform (BWT). The popular alignment tools are, TopHat2 [29], STAR [30], and HISAT [31], which perform well in terms of mapping speed and accuracy and can effectively map billions of reads to a reference genome or transcriptome. A number of alignment methods have been developed specifically for scRNA‐seq sequencing data, including CellRanger [19], dropEst [32], Kallisto‐BUStools [28], and Alevin [33]. CellRanger is generally used with the 10X genomics Chromium platform, is a fairly mature and complete pipeline, but CellRanger is slow and has high memory requirements. The dropEst process has faster runtimes and lower memory usage than CellRanger and yields more accurate gene expression estimates by correcting sequencing errors in cell barcodes and UMIs, and as development progresses, the latest packages support more and more sequencing protocols. The Kallisto pseudo‐alignment, used in combination with the bustools' method for storing and managing scRNA‐seq data, is an efficient alternative for alignment, and the combined K‐B method is tens of times faster than CellRanger, although the method does not remove empty cell barcodes and requires manual processing. Alevin is an extension of the pseudo‐alignment method that can be used to process scRNA‐seq data, running much faster than CellRanger.
FIGURE 2.
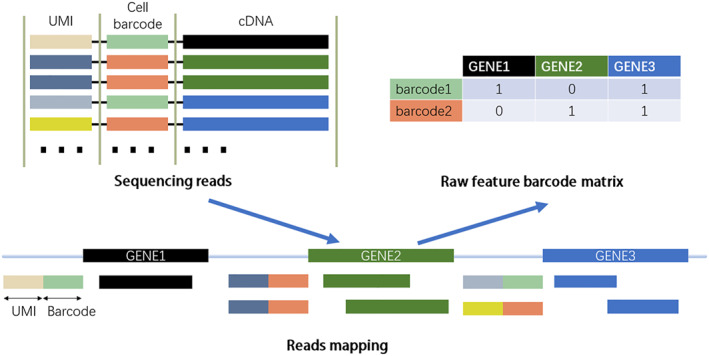
Common steps for processing scRNA‐seq data to gene expression matrix.
Depending on the range of transcript sequences captured by scRNA‐seq, different methods need to be selected for gene or transcript expression quantification. To quantify gene or transcript expression, the data generated by scRNA‐seq methods for full‐length transcripts (e.g. Smart‐seq2 and MATQ‐seq) can be analysed with bulk RNA sequencing software. An assembly without the reference genome is called de novo assembly (without a reference genome) and an assembly based on a reference genome or genome‐guided assembly is called genome‐guided assembly. Genome‐guided transcriptome assembly is usually more accurate than de novo transcriptome assembly, which is typically applied to organisms lacking a reference genome [34]. A wide range of single‐cell RNA sequence studies use Cufflinks [35], RSEM [36], and Stringtie [37] to obtain relative gene or transcript expression estimates for reads or fragments per kilobase per million mapped reads (RPKM or FPKM) or transcripts per million mapped reads (TPM). The StringTie method outperformed other genomic guidance methods in gene/transcript reconstruction and expression quantification, according to Pertea et al [37]. For 3′ end‐based, UMI‐based scRNA sequencing, such as CEL‐seq2, Drop‐seq, and InDrop‐seq, specific algorithms must be used in order to calculate gene expression. SAVER (Single Cell Analysis by Expression Recovery) is a recently proposed and effective UMI‐based tool for the accurate estimation of gene expression in single cells [38]. Theoretically, UMI‐based scRNA‐seq can greatly reduce technical noise [39], mainly to eliminate amplification bias generated by PCR, thus improving the accuracy of transcription counting. Table 2 shows the tools commonly used for reads mapping and expression quantification of scRNA‐seq data.
TABLE 2.
Tools for reads mapping and expression quantification of scRNA‐seq data.
| Tools | Function | Language | URL |
|---|---|---|---|
| CellRanger | Analyse pipelines | Python | https://support.10xgenomics.com/single‐cell‐gene‐expression/software/pipelines/latest/what‐is‐cell‐ranger |
| DropEst | Analyse pipelines | C++ | https://github.com/hms‐dbmi/dropEst |
| Kallisto–BUStools | Reads mapping | Python,R | https://www.kallistobus.tools/getting_started |
| Alevin | Reads mapping | Rust | https://salmon.readthedocs.io/en/latest/alevin.html |
| Cufflinks | Expression quantification | Python | https://github.com/cole‐trapnell‐lab/cufflinks |
| RSEM | Expression quantification | C++ | https://github.com/deweylab/RSEM |
| Stringtie | Expression quantification | C++ | https://github.com/gpertea/stringtie |
| SAVER | Expression quantification | R | https://github.com/mohuangx/SAVER |
3.2. Quality control
scRNA‐seq may be affected by broken, dead, multiple cells mixed or contaminated by environmental RNA and generate part of the low‐quality data. Using low‐quality data may hinder downstream analysis and result in incorrect interpretations. Consequently, the second round of quality control should be performed using tools such as Seurat [11], scanpy [40] and Scater [41].
For broken or dead low‐quality cells, if exogenous echinocandins (e.g., ERCC) are used in the scRNA sequence, those cells with a very high ratio of reads corresponding to echinocandins, which may have been destroyed during cell capture, should be removed. Furthermore, fragmented cells usually lose cytoplasmic RNA but retain mitochondrial RNA, and the ratio of the mitochondrial genome to the cytoplasmic RNA can also be used to identify low‐quality cells in these cells. Detecting the number of genes or transcripts expressed in a particular cell is also useful. Only a small number of genes can be detected in a cell, indicating that the cell is damaged, dead, or suffering from RNA degradation. It is common practice to quantify expressed genes and subsequently filter low‐quality cells by using an FPKM/RPKM threshold. Microscopic imaging of each cell can also be performed, but this leads to a significant increase in cost, or cells can be stained using reactive dyes but may alter the transcriptional state of the cell, thus changing the outcome of the entire experiment. Machine learning methods can also be used to build models for isolating low‐quality cells in sequencing data by learning the biological and technical features in the reference data. Some quality control methods for scRNA‐seq, such as SinQC [42] and Scater [41], are tools that are useful for the quality control of scRNA sequence data.
For protocols using droplet sequencing technology, environmental mRNA expression profiles can be estimated from empty droplets by SoupX [43]. Then, estimate (or manually set) the contamination fraction in each cell data, that is, the UMI fraction from the background. Finally, the environmental mRNA expression profile is used to estimate contamination and correct the expression profile for each cell to remove an environmental RNA contamination.
Technical artifacts known as “doublets” frequently impact data with numerous cell mixtures, limiting cell throughput and producing erroneous biological results. Christopher S et al. mentioned that doublet detection could be performed on single‐cell RNA sequencing data by using an artificial nearest‐neighbour approach, and developed a tool called DoubletFinder [44] that uses gene expression features to predict doublets in scRNA‐seq data, thus improving the performance of differential gene expression analysis. Solo [45] performs better in more complex organisational data by semi‐supervised deep learning for doublets detection.
The principle of threshold selection is to have a deep biological understanding of target cells, so as to correctly interpret data and select appropriate quality control parameters. Quality Control (QC) filters out unqualified data (such as technical problems or cell quality problems) by using different indicators. The thresholds of QC parameters are not necessarily the same in different analyses, and the setting of thresholds depends on the cells or tissues sequenced.
When determining the QC threshold, the diversity of the analysed organisation must be considered. For example, when designing experiments to study metastatic cancer cells in the blood, the number of cancer cells is very low compared with the number of normal blood cells, so the counts of transcripts in QC indicators must be adjusted. In this tissue, blood cells are dominant cells, but compared with active cancer cells, their expression is considered to be in a relatively static state, with relatively low RNA content. Therefore, if the reference threshold is set to delete cells with transcripts more than twice the average, cancer cells may be mistaken for doubles because of their high transcriptional activity, and all of them will be removed [46]. Another common QC parameter is the number of mitochondrial genes. As with the number of transcripts, this parameter is highly dependent on the type of tissue and the problems studied. For example, due to the high energy demand of myocardial cells, 30% of the total mRNA in the heart is mitochondria, while the proportion of tissues with low energy demand is 5% or less. Therefore, 30% of mitochondrial mRNA is normal in myocardial cells, but abnormal in lymphocytes [47]. According to the purpose of the experiment, gene‐specific QC indicators can also be added. For genes with low expression in all cells and no statistical significance between cell types, we can consider setting a threshold to filter them out, so as to reduce the amount of calculation in the later stage.
By far, Seurat is one of the most popular tools with built‐in capabilities to perform low‐quality cell filtering, which requires manual setting of filtering thresholds that usually depend on the type of cells and tissues being analysed. Lambrechts et al. in their study filtered out cells expressing ≤100 or ≥6000 genes, UMI counts ≤200, and corresponding mitochondrial genes ≥10% [48], while Fan et al. retained high‐quality cells by using the following parameters: [1] 200< total number of expressed genes per cell (nGenes) <2500; [2] 300< total number of UMIs per cell (nUMIs) <15,000; and [3] percentage of UMIs mapping to mitochondrial genes (MT%) <10% [49]. Therefore, we recommend the flexibility to adjust the above QC thresholds according to specific disease states and tissue types, especially for mitochondrial gene‐based cellular filtering thresholds. Table 3 shows the current tools commonly used for scRNA‐seq data quality control.
TABLE 3.
Tools for quality control of scRNA‐seq data.
| Tools | Function | Language | URL |
|---|---|---|---|
| Seurat | Analyse pipelines | R | https://github.com/satijalab/seurat |
| Scanpy | Analyse pipelines | Python | https://github.com/scverse/scanpy |
| Scater | Analyse pipelines | R | https://github.com/jimhester/scater |
| SoupX | Background contamination removal | R | https://github.com/constantAmateur/SoupX |
| DoubletFinder | Doublet removal | R | https://github.com/ddiez/DoubletFinder |
| Solo | Doublet removal | Python | https://github.com/calico/solo |
3.3. Normalization
Normalization is a significant step to take interest signals of relevance from scRNA‐seq sequencing data and is often accomplished by correcting for unintended biases caused by capture efficiency, sequencing depth, deletions, and other technical factors. Normalization only considers technical deviation and has nothing to do with whether there is batch effect; normalization methods based on probabilistic models usually include the function of batch effect removal. The technical noise of scRNA‐seq is significant due to the small number of starting RNAs and the inherent limitations of different experimental protocols. scRNA‐seq data normalization is highly beneficial for downstream analysis, such as cell subpopulation identification and differential expression analysis. Generally, within‐sample normalization and between‐sample normalization are the two categories of normalization [50]. The goal of within‐sample normalization is to remove gene‐specific biases (such as those related to the GC content and gene length) so that measures of gene expression within a sample (such as RPKM/FPKM and TPM) can be compared. In contrast, between‐sample normalization is used to correct for sample‐specific variations (such as sequencing depth and capture efficiency) so that gene expression levels between samples may be compared. Simple normalization techniques frequently rely on the upper quartile or sequencing depth. The spike‐ins or UMIs can be used to improve the normalization performance of a scRNA‐seq protocol. [51].
For within‐sample normalization of bulk RNA sequencing data, a number of techniques have been developed, including DESeq2 [52] and trimmed mean of M values (TMM) [53]. While TMM gets rid of extreme logarithmic difference multiples, DEseq2 produces scaling factors depending on read counts of various samples. However, batch‐based normalization methods may not apply to data from single‐cell transcriptomics. Due to the technical nature of scRNA‐seq, which generates a large number of zero expression values and has more technical variation than batch RNA‐seq, the use of batch RNA‐seq normalization methods may lead to overcorrection of scRNA‐seq for low‐expressed genes [50] and under‐correction for highly expressed genes, so there is a need to develop scRNA‐seq specifically applicable to normalization methods. Zhijin Wu et al. mentioned that system technology noise is not only cell‐specific but also affects genes differently, so a simple cell size factor adjustment for normalization is not appropriate, and they proposed a non‐linear normalization method ‘SC2P’ for each gene in each cell. The cell‐ and gene‐specific normalization factors can reduce technical variation more than other methods without reducing biological variation, and SC2P normalization also eliminates bias due to uneven technical noise when technical effects, like sequencing depth, are unbalanced between cell populations. However, since this method is only applicable to scRNA‐seq experiments that do not use unique molecular identifiers (UMIs), amplification bias is still present in these analyses [54]. In contrast, “Dino” is a normalization method based on a flexible negative binomial mixed gene expression model, also a non‐linear normalization method, which is robust to shallow sequencing, sample heterogeneity, and varying zero ratios, thus improving the performance of downstream analysis in many settings [55]. There are also inverse fold product methods introduced in recent years that use total expression values in cell clusters for normalization, such as SCnorm based on quantile regression, and Bacher et al. noted that SCnorm successfully normalises scRNA seq data and enhances the performance of principal component analysis (PCA) and differentially expressed gene identification results. Conversely, typical normalization approaches intended for bulk RNA seq may introduce artifacts [56]. Table 4 shows some of the tools currently available specifically for scRNA‐seq data normalization, which generally have better performance than methods designed for batch RNA‐seq data normalization.
TABLE 4.
Tools for normalization of scRNA‐seq data.
| Tools | Language | URL |
|---|---|---|
| SC2P | R | https://github.com/haowulab/SC2P |
| Dino | R | https://github.com/JBrownBiostat/Dino |
| SCnorm | R | https://github.com/rhondabacher/SCnorm |
| scTransform | R | https://github.com/satijalab/sctransform |
| scran | R | https://github.com/marionilab/scran |
3.4. Imputation
Single‐cell RNA sequencing data often contain many missing values or deletions, as evidenced by the fact that many genes have zero expression values. Some of these zero values are biologically significant, representing that the gene is not expressed in the cell or that the mRNA has been degraded after expression. And more zeros may arise because of the scRNA‐seq technology itself, such as technical zeros due to reverse transcription failure of mRNA due to reverse transcription bias or the low number of corresponding mRNA, sampling zeros due to low amplification efficiency or limited sequencing depth, and lack of reads or UMIs of the gene in the cell. These non‐biological zeros increase intercellular variability, blur the detection of gene‐to‐gene relationships, and may largely affect downstream analysis. In contrast, imputation is an effective strategy to replace missing data (zeros) with alternative values.
Although some methods have been proposed to impute bulk RNA‐seq data, they are not directly applicable to scRNA‐seq data. In recent years, several impute methods have been developed for scRNA‐seq technology, including SAVER [38], SAVERX [57], WEDGE [58], MAGIC [59], mcImpute [60], scVI [61], scGNN [62], scIGANs [63], and AutoImpute [64]. SAVER is a Bayesian‐based model for recovering the true expression levels of all genes in UMI‐based scRNA‐seq data. SAVERX may extract migrable gene‐gene connections from data from various laboratories, circumstances, and species by combining a deep auto‐encoder with a Bayesian model to filter out noise from new target datasets. WEDGE uses biased matrix decomposition to interpolate gene expression values from single‐cell RNA sequence datasets. MAGIC estimates gene expression by constructing Markov affinity‐based graphs. Mclmpute uses a low‐rank matrix‐based complementation technique to estimate deletions in single‐cell expression data. scVI is a comprehensive analysis tool for single‐cell data based on hierarchical Bayesian models with variational inference. scGNN is a method based on graph neural networks with an autoencoder that can be used for imputation and clustering. It performs well for noisy large‐scale datasets. scIGAN is a generative adversarial network‐based method that performs well on noisy small‐scale datasets. AutoImpute is an autoencoder‐based technique for imputing missing values that discovers the natural distribution of scRNA sequence data.
Jiacheng Wang et al. mentioned that scGNN and scIGANs significantly outperform other methods in terms of imputation effectiveness and improvement on downstream analysis tasks. However, due to their complex model structure, a large amount of computing time is required to obtain excellent performance [65]. Wenpin Hou et al. mentioned that researchers can choose the appropriate imputation method depending on the single‐cell protocol and the downstream analysis to be performed, for example, when the dataset used is based on UMIs tags, the best performing method for differential expression analysis is mcimpute, and the best performing method for clustering analysis is MAGIC. When the dataset used is based on the plate method, the best performing method for differential representation analysis is scVI, and the best performing method for clustering analysis is MAGIC. kNN smoothing and MAGIC methods have the best computational performance when researchers focus more on computation time, memory consumption, and large‐scale computation of the dataset [66]. Ruochen Jiang mentioned the applicability of imputation for single‐cell data, and they pointed out that UMIs‐based single‐cell sequencing data are considered non‐zero‐inflated, so methods based on the zero‐inflated model for imputation are never used. For UMI data, cell‐level analysis such as cell dimensionality reduction or cell clustering can be performed without imputation. However, when performing DE analysis, the best performance can be obtained by choosing the appropriate imputation method. For non‐UMI data, imputation using a non‐zero inflation model is better for cell‐level analysis such as cell downscaling or cell clustering. When performing DE analysis, all imputation methods perform better than no imputation or binarisation processing. Finally, when the cell library is large enough (the sequencing depth is most adequate), imputation can be left out [67]. Table 5 shows the commonly used scRNA‐seq data imputation tools, and users can choose the appropriate imputation tool according to their own analysis needs.
TABLE 5.
Tools for imputation of scRNA‐seq data.
| Tools | Language | URL |
|---|---|---|
| SAVER | R | https://github.com/mohuangx/SAVER |
| SAVERX | R | https://github.com/mohuangx/SAVERX |
| WEDGE | C++ | https://github.com/QuKunLab/WEDGE |
| MAGIC | Python | https://github.com/KrishnaswamyLab/magic |
| mcImpute | Matlab | https://github.com/aanchalMongia/McImpute_scRNAseq |
| scVI | Python | https://github.com/scverse/scvi‐tools |
| scGNN | Python | https://github.com/juexinwang/scGNN |
| scIGANs | Python | https://github.com/xuyungang/scIGANs |
| AutoImpute | Python | https://github.com/divyanshu‐talwar/AutoImpute |
3.5. Batch effect remove
Batch effects are a common source of technical variation in high‐throughput sequencing experiments. Large‐scale scRNA sequence datasets may be generated by different operators at different times or in multiple laboratories by using different cell isolation methods, library preparation methods, or sequencing platforms. The gene expression patterns in one batch of data may be systematically different from those in another as a result of these factors' potential to cause systematic mistakes. Batch effect removal takes into account both technical deviation and unwanted biological differences and retains the biological variation that researchers want to focus on.
Several methods have been developed specifically to deal with batch effects in single‐cell transcriptome sequencing data. For example, Batchelor [68] is an R package for batch effect correction based on the concept of mutual nearest neighbours (MNN), which defines highly variable gene (HVG) sets to identify mutually nearest neighbouring cells and corrects for batch effects by estimating a correction vector for HVG sets and query gene sets and applying Gaussian kernel weights to all cells within the same batch MNN. scVI [61] is a tool implemented using a variational autoencoder for normalising latent variables from different sources of variation (e.g., batch effects and the library size), and the distribution of each latent factor can be calculated to infer standard deviations. Seurat [11] is an R package for single‐cell transcriptome sequencing data analysis that allows various analyses. For batch effect correction, Seurat is similar to Batchelor, based on MNN to identify ensembles (similar cells in different batches). In contrast, Beer [69] determined the pairs of cells that were physically closest to one another, combined datasets from several batches, used tSNE to reduce the dimensionality of each batch dataset to one, and used Kendall's tau to calculate the distance between cells across batches. Then eliminated PCA subspaces with significant batch effects to normalise the batch effects. MMD‐ResNet [70] minimises the maximum mean discrepancy between the multivariate distributions of two replicates measured in different batches by training a residual neural network. scScope [71] is a deep learning‐based approach that uses correcting layer learning modules to eliminate batch effects.
Beer, Seurat, and Batchelor all used MNN identification in the batch effect correction process, using it for batch effect removal. Tian Lan et al. mentioned that Beer and Batchelor outperformed the other tools. In particular, Beer could distinguish batch effects from population heterogeneity in most cases while normalising most simulated batch effects. This is mainly attributed to the fact that Beer directly identifies PCA subspaces with high batch effects and simply removes these subspaces; information on cell population heterogeneity can still be retained [72]. Sometimes the identified MNNs may be incorrect MNN pairs, especially if the batch effect is not small enough compared to the biological effect (population heterogeneity), which may lead to the population heterogeneity being incorrectly corrected. In contrast, MMD‐ResNet, scVI and Seurat methods may have some performance advantages when there are more cell types and data from different batches. Table 6 shows the tools currently available for batch effect processing of scRNA‐seq data.
TABLE 6.
Tools for batch effect removal of scRNA‐seq data.
| Tools | Language | URL |
|---|---|---|
| Batchelor | Python | https://www.cnpython.com/pypi/batchelor |
| Beer | R | https://github.com/jumphone/BEER |
| Seurat | R | https://github.com/satijalab/seurat |
| scVI | Python | https://github.com/scverse/scvi‐tools |
| MMD‐ResNet | Python | https://github.com/ushaham/BatchEffectRemoval |
| scScope | Python | https://github.com/AltschulerWu‐Lab/scScope |
4. EVALUATION OF DATA PROCESSING METHODS FOR scRNA‐seq
Single‐cell transcriptome sequencing data processing methods can usually be evaluated in two aspects, such as cell clustering, which can assess the performance of data processing methods at the cellular level and secondly differential expression analysis, which can evaluate the data processing methods at the gene level.
4.1. Clustering
Clustering is a necessary step to determine the structure of cell types and cell subpopulations in single‐cell sequencing data, but the performance of clustering analysis is mainly influenced by several factors. First, technical noise and bias can be caused by cell‐specific traits (such as the cell cycle stage or cell size) as well as by technology or data sources (like low capture efficiency, amplification bias, and sequencing depth). Second, when different single‐cell populations in a collection of samples are examined simultaneously, technical biases and biological variations between populations take precedence in the clustering of single cells, leading to clustering with the original sample rather than with cells of a similar type. Since these constraints on the clustering performance of single‐cell sequencing data can be mitigated by many of the data processing methods now being developed, we can evaluate the method or combination of methods by corresponding to the downstream clustering performance of the preprocessing method.
The metrics commonly used to evaluate the clustering performance are the adjusted Rand index (ARI) to assess the correctness of clustering using a priori labels, normalised mutual information (NMI) and Jaccard index to measure the similarity of two clustering results, and Silhouette Coefficient and Dunn index to assess the compactness and separateness of clusters. Researchers can use these metrics to assess clustering performance and thus reflect the suitability of upstream data processing methods.
4.2. Differential expression analysis
Finding significantly differentially expressed genes (DEGs) between various subpopulations or cell types is possible with the use of differential expression analysis. For an explanation of biological variations between two control groups, DEGs are essential. But differential expression analysis of scRNA‐seq data is hampered by technological variability, high noise, and huge sample sizes. And there may be multiple cell states in the cell population, resulting in different gene expression patterns in different cells. Similarly, data processing methods have a significant impact on differential expression analysis, so we can use the results of differential expression analysis to evaluate the performance of the corresponding processing methods.
Numerous single‐cell research on DEG have employed tools created for bulk RNA‐seq data, but it is still unknown how well these techniques perform, why they work, and whether they can be used to discover DEGs in scRNA‐seq data. Recently, certain particular techniques for scRNA‐seq data‐based differential expression calling have been developed, such as MAST [73], DEsingle [74], Census [75], BCseq [76], edgeR‐LRT [77], and DESeq2‐Wald [52], among others.
MAST is based on linear model fitting and likelihood ratio tests. DEsingle uses a zero‐inflated negative binomial model to estimate deletion rates and actual zeros. Census uses an algorithm to convert relative RNA‐seq expression levels to relative transcript counts, and analysis of changes in relative transcript counts significantly improves the accuracy of differential expression analysis. BCseq mitigates technical noise in a data‐adaptive manner. EdgeR‐LRT uses likelihood ratio tests, while DESeq2‐Wald uses Wald tests for negative binomial model coefficients, both tools for a given type of cell, first aggregating the counts of biological repeats and then converting the cell‐gene expression matrix to a biological repeat‐gene expression matrix using matrix multiplication. Jordan W. Squair et al. mentioned that the currently widely used single‐cell differentially expressed gene identification methods can find hundreds of differentially expressed genes in the absence of biological differences. It is recommended to use tools that first integrate biological duplicates to form pseudobulks, such as edgeR‐LRT, DESeq2‐Wald, etc., for differential expression analysis [78]. Table 7 shows the current common tools for differential expression analysis of scRNA‐seq data. Choosing the right tool for differential expression analysis will greatly reduce the risk of finding incorrectly differentially expressed genes.
TABLE 7.
Tools for differential expression analysis of scRNA‐seq data.
| Tools | Language | URL |
|---|---|---|
| MAST | R | https://github.com/RGLab/MAST |
| DEsingle | R | https://bioconductor.org/packages/DEsingle |
| Census | R | http://cole‐trapnell‐lab.github.io/monocle‐release/ |
| BCseq | R | http://www‐rcf.usc.edu/~liangche/software.html |
| edgeR‐LRT | R | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| DESeq2‐Wald | R | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
5. DISCUSSION
In the past few years, scRNA‐seq technology has made tremendous progress and a variety of scRNA‐seq protocols have been developed. scRNA‐seq protocol development and innovation have largely facilitated single‐cell transcriptomics studies, yielding deeper insights into studying the heterogeneity of gene expression in individual cells in large cell populations and determining the kinetic basis of tissue and organism development discoveries. However, the continuous development or iterative updating of scRNA‐seq protocols and the large amount of sparse and noisy data generated by scRNA‐seq pose new challenges to the accompanying analytical tools.
In this review, we discuss the analysis of different sequencing protocols and data processing steps for noisy sequencing data. Specifically, we review the goals and popular tools for reads mapping and expression quantification, quality control, normalization, interpolation, and batch effect removal applied to scRNA sequence data analysis. Also, we present methods to evaluate these tools in both cellular and genetic dimensions, namely clustering and differential expression analysis. We hope that this review will help researchers to choose the appropriate processing method to analyse scRNA‐seq data.
The current state of affairs is that there is a lack of standardised processes for analysis in this field due to the rapid iteration of newer single‐cell sequencing technologies. Therefore, more comparisons of processing work tools are needed to evaluate the available methods to address the complexity of scRNA sequence data. In addition, data integration and analysis methods will become increasingly important as more and more scRNA‐seq datasets become available. While many computational tools and workflows are already available for analysing scRNA‐seq data, a thorough comparison of different tools and hands‐on workflows is still needed to better find the best analysis methods to take full advantage of scRNA‐seq.
AUTHOR CONTRIBUTION
Lu Junru: Methodology, Writing – original draft, Writing – review & editing. Sheng Yuqi: Investigation, Writing – review & editing. Qian Weiheng: Resources, Writing – review & editing. Pan Min: Writing – review & editing. Zhao Xiangwei: Supervision, Writing – review & editing. Ge (GE) Qinyu: Conceptualization, Resources, Supervision, Writing – review & editing.
CONFLICT OF INTEREST STATEMENT
We declare that we have no conflict of interest.
ACKNOWLEDGEMENTS
This work was supported by the grant from National Key Research and Development Program of China (2022YFF0710800), the National Natural Science Foundation of China No.81827901 & No.61801108 and the Natural Science Foundation of Jiangsu Province (BK20201148 and BK20211166).
Lu, J. , et al.: scRNA‐seq data analysis method to improve analysis performance. IET Nanobiotechnol. 17(3), 246–256 (2023). 10.1049/nbt2.12115
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Raj, A. , van Oudenaarden, A. : Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135(2), 216–26 (2008). 10.1016/j.cell.2008.09.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Munsky, B. , Neuert, G. , van Oudenaarden, A. : Using gene expression noise to understand gene regulation. Science 336(6078), 183–7 (2012). 10.1126/science.1216379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stegle, O. , Teichmann, S.A. , Marioni, J.C. : Computational and analytical challenges in single‐cell transcriptomics. Nat. Rev. Genet. 16(3), 133–45 (2015). 10.1038/nrg3833 [DOI] [PubMed] [Google Scholar]
- 4. Liu, S. , Trapnell, C. : Single‐cell transcriptome sequencing: recent advances and remaining challenges. F1000Research 5 (2016). 10.12688/f1000research.7223.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kiselev, V.Y. , Andrews, T.S. , Hemberg, M. : Challenges in unsupervised clustering of single‐cell RNA‐seq data. Nat. Rev. Genet. 20(5), 273–82 (2019). 10.1038/s41576-018-0088-9 [DOI] [PubMed] [Google Scholar]
- 6. Hicks, S.C. , et al.: Missing data and technical variability in single‐cell RNA‐sequencing experiments. Biostatistics 19(4), 562–78 (2018). 10.1093/biostatistics/kxx053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kharchenko, P.V. : The triumphs and limitations of computational methods for scRNA‐seq. Nat. Methods 18(7), 723–32 (2021). 10.1038/s41592-021-01171-x [DOI] [PubMed] [Google Scholar]
- 8. Cole, M.B. , et al.: Performance assessment and selection of normalization procedures for single‐cell RNA‐seq. Cell Syst 8(4), 315–28e8 (2019). 10.1016/j.cels.2019.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith, T. , Heger, A. , Sudbery, I. : UMI‐tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27(3), 491–9 (2017). 10.1101/gr.209601.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu, Y. , Zhang, K. : Tools for the analysis of high‐dimensional single‐cell RNA sequencing data. Nat. Rev. Nephrol. 16(7), 408–21 (2020). 10.1038/s41581-020-0262-0 [DOI] [PubMed] [Google Scholar]
- 11. Butler, A. , et al.: Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36(5), 411–20 (2018). 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lun, A.T. , McCarthy, D.J. , Marioni, J.C. : A step‐by‐step workflow for low‐level analysis of single‐cell RNA‐seq data with Bioconductor. F1000Res. 5, 2122 (2016). 10.12688/f1000research.9501.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Trapnell, C. , et al.: The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32(4), 381–6 (2014). 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Picelli, S. , et al.: Smart‐seq2 for sensitive full‐length transcriptome profiling in single cells. Nat. Methods 10(11), 1096–8 (2013). 10.1038/nmeth.2639 [DOI] [PubMed] [Google Scholar]
- 15. Sheng, K. , et al.: Effective detection of variation in single‐cell transcriptomes using MATQ‐seq. Nat. Methods 14(3), 267–70 (2017). 10.1038/nmeth.4145 [DOI] [PubMed] [Google Scholar]
- 16. Macosko, E.Z. , et al.: Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell 161(5), 1202–14 (2015). 10.1016/j.cell.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klein, A.M. , et al.: Droplet barcoding for single‐cell transcriptomics applied to embryonic stem cells. Cell 161(5), 1187–201 (2015). 10.1016/j.cell.2015.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Natarajan, K.N. : Single‐cell tagged reverse transcription (STRT‐Seq). Methods Mol. Biol. 1979, 133–53 (2019) [DOI] [PubMed] [Google Scholar]
- 19. Zheng, G.X. , et al.: Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8(1), 14049 (2017). 10.1038/ncomms14049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hashimshony, T. , et al.: CEL‐Seq2: sensitive highly‐multiplexed single‐cell RNA‐Seq. Genome Biol. 17(1), 77 (2016). 10.1186/s13059-016-0938-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gierahn, T.M. , et al.: Seq‐Well: portable, low‐cost RNA sequencing of single cells at high throughput. Nat. Methods 14(4), 395–8 (2017). 10.1038/nmeth.4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xin, Y. , et al.: Use of the Fluidigm C1 platform for RNA sequencing of single mouse pancreatic islet cells. Proc. Natl. Acad. Sci. U. S. A. 113(12), 3293–8 (2016). 10.1073/pnas.1602306113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sasagawa, Y. , et al.: Quartz‐Seq2: a high‐throughput single‐cell RNA‐sequencing method that effectively uses limited sequence reads. Genome Biol. 19(1), 29 (2018). 10.1186/s13059-018-1407-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dal Molin, A. , Di Camillo, B. : How to design a single‐cell RNA‐sequencing experiment: pitfalls, challenges and perspectives. Briefings Bioinf. 20(4), 1384–94 (2019). 10.1093/bib/bby007 [DOI] [PubMed] [Google Scholar]
- 25. Dillies, M.A. , et al.: A comprehensive evaluation of normalization methods for Illumina high‐throughput RNA sequencing data analysis. Briefings Bioinf. 14(6), 671–83 (2013). 10.1093/bib/bbs046 [DOI] [PubMed] [Google Scholar]
- 26. Andrews, S. : FastQC A Quality Control Tool for High Throughput Sequence Data (2014)
- 27. Martin, M. , Martin, M. : Cut adapt removes adapter sequences from high‐throughput sequencing reads, EMBnet. EMBnet 17:10‐12. Embnet Journal 17(1), 10 (2011). 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 28. Melsted, P. , et al.: Modular, efficient and constant‐memory single‐cell RNA‐seq preprocessing. Nat. Biotechnol. 39(7), 813–8 (2021). 10.1038/s41587-021-00870-2 [DOI] [PubMed] [Google Scholar]
- 29. Kim, D. , et al.: TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14(4), R36 (2013). 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dobin, A. , Gingeras, T.R. : Mapping RNA‐seq reads with STAR. Curr. Protoc. Bioinformatics 51(11), 4.1–4.9 (2015). 10.1002/0471250953.bi1114s51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim, D. , Langmead, B. , Salzberg, S.L. : HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12(4), 357–60 (2015). 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Petukhov, V. , et al.: dropEst: pipeline for accurate estimation of molecular counts in droplet‐based single‐cell RNA‐seq experiments. Genome Biol. 19(1), 78 (2018). 10.1186/s13059-018-1449-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patro, R. , et al.: Salmon provides fast and bias‐aware quantification of transcript expression. Nat. Methods 14(4), 417–9 (2017). 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garber, M. , et al.: Computational methods for transcriptome annotation and quantification using RNA‐seq. Nat. Methods 8(6), 469–77 (2011). 10.1038/nmeth.1613 [DOI] [PubMed] [Google Scholar]
- 35. Trapnell, C. , et al.: Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28(5), 511–5 (2010). 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li, B. , Dewey, C.N. : RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinf. 12(1), 323 (2011). 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pertea, M. , et al.: StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat. Biotechnol. 33(3), 290–5 (2015). 10.1038/nbt.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang, M. , et al.: SAVER: gene expression recovery for single‐cell RNA sequencing. Nat. Methods 15(7), 539–42 (2018). 10.1038/s41592-018-0033-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Islam, S. , et al.: Quantitative single‐cell RNA‐seq with unique molecular identifiers. Nat. Methods 11(2), 163–6 (2014). 10.1038/nmeth.2772 [DOI] [PubMed] [Google Scholar]
- 40. Wolf, F.A. , Angerer, P. , Theis, F.J. : SCANPY: large‐scale single‐cell gene expression data analysis. Genome Biol. 19(1), 15 (2018). 10.1186/s13059-017-1382-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCarthy, D.J. , et al.: Scater: pre‐processing, quality control, normalization and visualization of single‐cell RNA‐seq data in R. Bioinformatics 33(8), 1179–86 (2017). 10.1093/bioinformatics/btw777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jiang, P. , Thomson, J.A. , Stewart, R. : Quality control of single‐cell RNA‐seq by SinQC. Bioinformatics 32(16), 2514–6 (2016). 10.1093/bioinformatics/btw176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Young, M.D. , Behjati, S. : SoupX removes ambient RNA contamination from droplet‐based single‐cell RNA sequencing data. GigaScience 9(12) (2020). 10.1093/gigascience/giaa151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McGinnis, C.S. , Murrow, L.M. , Gartner, Z.J. : DoubletFinder: doublet detection in single‐cell RNA sequencing data using artificial nearest neighbors. Cell Syst 8(4), 329–37e4 (2019). 10.1016/j.cels.2019.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bernstein, N.J. , et al.: Doublet identification in single‐cell RNA‐seq via semi‐supervised deep learning. Cell Syst 11(1), 95–101e5 (2020). 10.1016/j.cels.2020.05.010 [DOI] [PubMed] [Google Scholar]
- 46. AlJanahi, A.A. , Danielsen, M. , Dunbar, C.E. : An introduction to the analysis of single‐cell RNA‐sequencing data. Mol Ther Methods Clin Dev 10, 189–96 (2018). 10.1016/j.omtm.2018.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Galow, A.M. , et al.: Quality control in scRNA‐Seq can discriminate pacemaker cells: the mtRNA bias. Cell. Mol. Life Sci. 78(19‐20), 6585–92 (2021). 10.1007/s00018-021-03916-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lambrechts, D. , et al.: Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 24(8), 1277–89 (2018). 10.1038/s41591-018-0096-5 [DOI] [PubMed] [Google Scholar]
- 49. Fan, X. , et al.: Single‐cell reconstruction of follicular remodeling in the human adult ovary. Nat. Commun. 10(1), 3164 (2019). 10.1038/s41467-019-11036-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vallejos, C.A. , et al.: Normalizing single‐cell RNA sequencing data: challenges and opportunities. Nat. Methods 14(6), 565–71 (2017). 10.1038/nmeth.4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bacher, R. , Kendziorski, C. : Design and computational analysis of single‐cell RNA‐sequencing experiments. Genome Biol. 17(1), 63 (2016). 10.1186/s13059-016-0927-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Love, M.I. , Huber, W. , Anders, S. : Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 15(12), 550 (2014). 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Robinson, M.D. , Oshlack, A. : A scaling normalization method for differential expression analysis of RNA‐seq data. Genome Biol. 11(3), R25 (2010). 10.1186/gb-2010-11-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu, Z. , Su, K. , Wu, H. : Non‐linear normalization for non‐UMI single cell RNA‐seq. Front. Genet. 12, 612670 (2021). 10.3389/fgene.2021.612670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brown, J. , et al.: Normalization by distributional resampling of high throughput single‐cell RNA‐sequencing data. Bioinformatics 37(22), 4123–4128 (2021). 10.1093/bioinformatics/btab450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bacher, R. , et al.: SCnorm: robust normalization of single‐cell RNA‐seq data. Nat. Methods 14(6), 584–6 (2017). 10.1038/nmeth.4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang, J. , et al.: Data denoising with transfer learning in single‐cell transcriptomics. Nat. Methods 16(9), 875–8 (2019). 10.1038/s41592-019-0537-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hu, Y. , et al.: WEDGE: imputation of gene expression values from single‐cell RNA‐seq datasets using biased matrix decomposition. Briefings Bioinf. 22(5) (2021) [DOI] [PubMed] [Google Scholar]
- 59. van Dijk, D. , et al.: Recovering gene interactions from single‐cell data using data diffusion. Cell 174(3), 716–29e27 (2018). 10.1016/j.cell.2018.05.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mongia, A. , Sengupta, D. , Majumdar, A. : McImpute: matrix completion based imputation for single cell RNA‐seq data. Front. Genet. 10, 9 (2019). 10.3389/fgene.2019.00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lopez, R. , et al.: Deep generative modeling for single‐cell transcriptomics. Nat. Methods 15(12), 1053–8 (2018). 10.1038/s41592-018-0229-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang, J. , et al.: scGNN is a novel graph neural network framework for single‐cell RNA‐Seq analyses. Nat. Commun. 12(1), 1882 (2021). 10.1038/s41467-021-22197-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xu, Y. , et al.: scIGANs: single‐cell RNA‐seq imputation using generative adversarial networks. Nucleic Acids Res. 48(15), e85 (2020). 10.1093/nar/gkaa506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Talwar, D. , et al.: AutoImpute: autoencoder based imputation of single‐cell RNA‐seq data. Sci. Rep. 8(1), 16329 (2018). 10.1038/s41598-018-34688-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang, J. , Zou, Q. , Lin, C. : A comparison of deep learning‐based pre‐processing and clustering approaches for single‐cell RNA sequencing data. Briefings Bioinf. 23(1) (2022). 10.1093/bib/bbab345 [DOI] [PubMed] [Google Scholar]
- 66. Hou, W. , et al.: A systematic evaluation of single‐cell RNA‐sequencing imputation methods. Genome Biol. 21(1), 218 (2020). 10.1186/s13059-020-02132-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jiang, R. , et al.: Statistics or biology: the zero‐inflation controversy about scRNA‐seq data. Genome Biol. 23(1), 31 (2022). 10.1186/s13059-022-02601-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Haghverdi, L. , et al.: Batch effects in single‐cell RNA‐sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36(5), 421–7 (2018). 10.1038/nbt.4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang, F. , Wu, Y. , Tian, W. : A novel approach to remove the batch effect of single‐cell data. Cell Discov. 5(1), 46 (2019). 10.1038/s41421-019-0114-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shaham, U. , et al.: Removal of batch effects using distribution‐matching residual networks. Bioinformatics 33(16), 2539–46 (2017). 10.1093/bioinformatics/btx196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Deng, Y. , et al.: Scalable analysis of cell‐type composition from single‐cell transcriptomics using deep recurrent learning. Nat. Methods 16(4), 311–4 (2019). 10.1038/s41592-019-0353-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lan, T. , et al.: Sequencing dropout‐and‐batch effect normalization for single‐cell mRNA profiles: a survey and comparative analysis. Briefings Bioinf. 22(4) (2021). 10.1093/bib/bbaa248 [DOI] [PubMed] [Google Scholar]
- 73. Finak, G. , et al.: MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single‐cell RNA sequencing data. Genome Biol. 16(1), 278 (2015). 10.1186/s13059-015-0844-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Miao, Z. , et al.: DEsingle for detecting three types of differential expression in single‐cell RNA‐seq data. Bioinformatics 34(18), 3223–4 (2018). 10.1093/bioinformatics/bty332 [DOI] [PubMed] [Google Scholar]
- 75. Qiu, X. , et al.: Single‐cell mRNA quantification and differential analysis with Census. Nat. Methods 14(3), 309–15 (2017). 10.1038/nmeth.4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen, L. , Zheng, S. : BCseq: accurate single cell RNA‐seq quantification with bias correction. Nucleic Acids Res. 46(14), e82 (2018). 10.1093/nar/gky308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Robinson, M.D. , McCarthy, D.J. , Smyth, G.K. : edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1), 139–40 (2010). 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Squair, J.W. , et al.: Confronting false discoveries in single‐cell differential expression. Nat. Commun. 12(1), 5692 (2021). 10.1038/s41467-021-25960-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.