

Abstract
The classical phosphatidylinositol 3-kinases (PI3Ks) are heterodimers of p110 and p85. PIK3CA, the gene encoding the catalytic p110α subunit, is one of the most frequently mutated oncogenes in human cancers with hot spot mutations occurring in the helical domain or in the kinase domain. Tumors with these two types of PIK3CA mutations show overlapping yet distinct phenotypes; however, the underlying mechanisms remain unclear. In a recent publication [1], Hao et al revealed exciting findings about the PI3K p85β regulatory subunit in promoting PIK3CA helical domain mutation-driven cancer progression. The authors found that p85β disassociated from the PI3K complex and translocated into the nucleus only in cancer cells harboring PIK3CA helical domain mutations. Disrupting nuclear localization of p85β suppressed mouse tumor growth of cancer cells with PIK3CA helical domain mutation. Mechanistically, they elegantly showed that nuclear p85β recruited the deubiquitinase USP7 to stabilize the histone methyltransferases EZH1/2, leading to enhanced H3K27 trimethylation and gene transcription. Combining an EZH inhibitor with a PI3K inhibitor specifically resulted in regression of mouse xenograft tumors with PIK3CA helical domain mutations. These findings illustrate a previously uncharacterized function of p85β in tumor development and suggest an effective approach to target tumors with PIK3CA helical mutations.
Keywords: PI3K, p110, p85, cancer mutation, nuclear translocation, colorectal cancer
The phosphatidylinositol 3-kinases (PI3Ks) are a group of highly conserved lipid kinases that play an important role in determining cellular fate. Although they are related to the protein kinases that phosphorylate proteins, PI3Ks phosphorylate lipids by adding a phosphate group to convert phosphatidylinositol-4,5-triphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3), the second messenger that recruits AKT to the plasma membrane through its interaction with the pleckstrin homolog (PH) domain of AKT [2], which then activates downstream pathways, including the mechanistic target of rapamycin 1 (mTORC1). PI3K can be activated by receptor tyrosine kinases (RTKs), cytokine/chemokine receptors, G-protein coupled receptors (GPCRs), and B/T-cell receptors. Therefore, the PI3K pathway regulates cell growth, nutrition and metabolism, cytoskeletal remodeling, cell proliferation and even cell death [3], controlling normal cell physiology and cellular transformation.
The PI3K family is divided into three classes (Class 1–3) based on structural differences, with Class 1 being the most widely studied. Class 1 is further subcategorized into 1A and 1B subtypes that transduce RTK and GPCR signaling, respectively. Type 1A PI3Ks are heterodimers that consist of a catalytic subunit (p110) and a regulatory subunit (p85 or p55). There are four distinct genes encoding PI3K catalytic subunits, p110α/β/γ/δ, whereas the regulatory subunit is encoded by three genes that translate into p85α, p85β, p55γ and splice variants. At rest or in the absence of stimuli, the p85 subunit binds to the p110 subunit and inhibits its catalytic activity [4]. Upon stimulation by, for instance, growth factors, the p110/p85 complex is recruited to the plasma membrane where p85 binds via its SH2 domains to tyrosine phosphorylated RTKs or adaptor proteins, such as insulin receptor substrate 1 (IRS1) [5], leading to the displacement of p85 from the PI3K complex followed by p110 activation. Hence, the expression level of p85 inside the cell is critical for regulating the activation of p110. In parallel, cells evolved a checkpoint system to turn off or limit the level of activation of PI3K-AKT, in which several lipid phosphatases, particularly phosphatase and tensin homolog (PTEN), convert PIP3 back to PIP2 by removing its 3’ phosphate [2].
The link between the PI3K pathway and cancer can be traced back to the original discovery of the first PI3K activity associated with oncogene-induced transformation, for instance, by Src and polyomavirus middle T antigen [6]. PIK3CA, which encodes the p110α catalytic subunit of PI3K, is among the top frequently mutated oncogenes in human cancers [7]. The negative regulator, the phosphatase PTEN, is also frequently mutationally inactivated in tumors, which thereby disrupts the normal kinase/phosphatase balance, resulting in constitutive elevation of PIP3 and activation of the PI3K-AKT pathway that drives transformation and promotes tumor progression [8]. While the p85α subunit is considered to be a tumor suppressor, the p85β subunit actually promotes tumor development [9]. PIK3R1, the gene encoding p85α, was found to be mutated in various cancers and such mutations disrupt its binding with p110. In contrast, PIK3R2, the gene encoding p85β, was found to be overexpressed in tumors [9]. Further, p85 can function in a p110-independent manner, largely by being an adaptor protein to facilitate the signaling transduction that regulates insulin response, senescence, unfolded protein stress, cytoskeleton re-arrangement, and endocytic trafficking, etc [10]. However, our understanding about how exactly these PI3K isoforms regulate tumor progression remained incomplete.
The authors previously reported that the p110α helical domain hotspot mutations lead to its direct association with IRS1, independently of p85, thus recruiting the mutant p110α to the plasma membrane to convert PIP2 to PIP3 and promoting tumor growth [11]. However, tumors with PIK3CA helical domain mutations are believed to be less responsive to inhibitors targeting PI3K, AKT or mTOR than hotspot mutations in the catalytic domain [12]. These results also indicate there must be as-yet to be identified mechanisms driving the distinct phenotypes of the p110 helical domain mutants. In the current study, Hao et al. made a surprising yet intriguing observation about the role of the p85β, but not p85α subunit, in specifically promoting tumor development of cancer cells with p110 helical domain mutations, but not p110 wild type or with kinase domain mutations. They started by demonstrating a reduced interaction between the p85β, but not the p85α subunit, and p110 with helical domain mutations, but not wild type or with kinase mutations. By aligning the amino acid sequences of p85β and p85α, the authors found that the N-terminal regions of the two isoforms are less highly conserved than the C-terminal regions that contain the two SH2 domains, and by swapping the N-terminal regions, which include the SH3 and GAP domains, between p85β and p85α, they confirmed that this region of p85β is indeed responsible for the lower affinity of p85β to p110, leading to higher levels of the free form of p85β.
In determining the significance of p85β in cancer, the authors analyzed TCGA data in colon, bladder, endometrial and breast cancers and found that high expression of PIK3R2 (encoding p85β) correlated with a worse prognosis only in cancers bearing p110 helical domain mutations, indicating a tumor promoting role of p85β. Consistently, knockdown of p85β specifically inhibited the growth of tumors derived from colon cancer cells harboring p110 helical domain mutations, while having no effect on the tumors from cancer cells with p110 wild type or kinase domain mutations.
This leads to an important question about how p85β promotes the growth of tumors harboring a p110 helical domain mutation? The authors ruled out the involvement of canonical PI3K downstream factors such as AKT and mTOR in this process, pointing to a previously unknown mechanism. During their analysis, the authors spotted an increase in the nuclear signal of p85β in cancer cells expressing the p110 helical domain mutations, suggesting free p85β can translocate into the nucleus. They indeed found a non-canonical nuclear localization signal (NLS, amino acids 474–484 with a sequence of LQMKRTAIEAF) in p85β that mediates its nuclear translocation.
To understand how nuclear p85β drives tumor progression, the authors conducted RNA sequencing in isogenic cell lines expressing wild type p85β or an NLS-disrupted mutant (K477R478 to A477A478). The results reveal that many tumor-suppressing genes, including ATM, BRCA1/2, and APC, were upregulated in p85β NLS mutant cells, indicating that p85β wild type in the nucleus downregulates tumor suppressing genes, which may contribute to its tumor-promoting role. Further analysis showed that p85β specifically upregulated the level of trimethylated H3K27, but not that of H3K9, H3K36 or H3K76, leading to a global gene transcriptional regulation. Consistently, p85β increased the protein levels of the EZH1/2 methyltransferases, but not the other subunits of the PRC2 repressor complex. Even though p85β worked as a global transcriptional regulator, its effect on the level of EZH1/2 was not through transcriptional upregulation, but instead via protein stability regulation. Again, these changes were only found in cancer cells with p110 helical domain mutations. The authors then showed that p85β stabilized EZH1/2 through recruiting the deubiquitinase USP7 (also called HAUSP) to remove ubiquitin from EZH1/2, leading to the stabilization of these two histone methyltransferases.
The authors did not stop here, but went on to test if combining an EZH1/2 inhibitor with a PI3K inhibitor could specifically target colon cancers with p110 helical mutations. Sure enough, they found that the combination suppressed the xenograft tumor growth of human colon cancer cell lines bearing helical domain mutations of p110 more strongly than that of cell lines with kinase domain mutations or the wild type p110, which was confirmed in colorectal PDX tumors with p110 helical domain mutation.
In conclusion, Hao et al [1] presented compelling and exciting findings about a novel function of p85β in the nucleus, filling a knowledge gap in our understanding about how the PI3K pathway drives cancer progression specifically in the genetic background of p110 helical domain mutations (Figure 1). These findings also suggest an effective approach to treat tumors with p110α helical domain mutations by combining an EZH1/2 inhibitor with a PI3K inhibitor.
Figure 1.
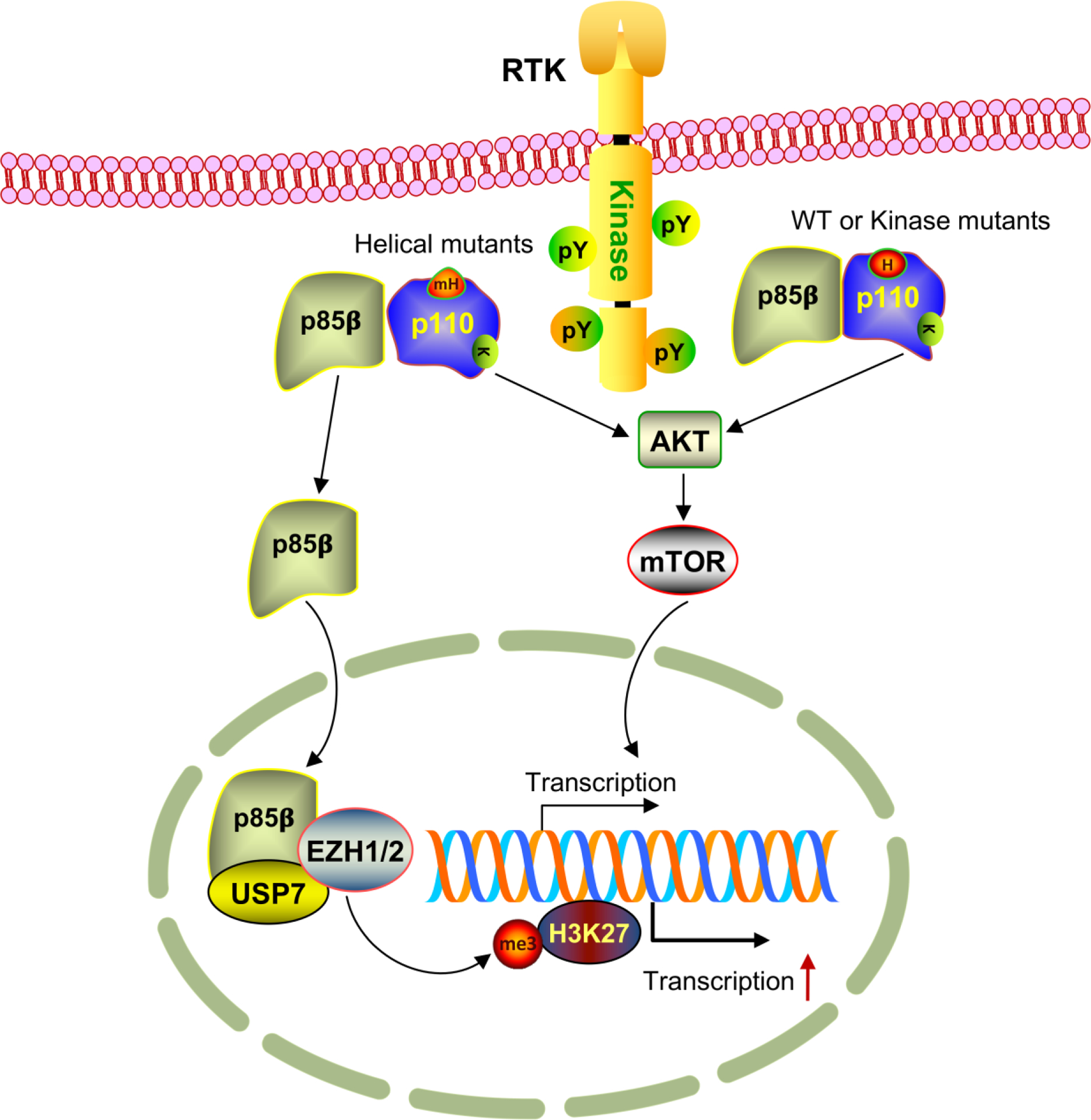
Model for the novel nuclear function of p85β in promoting tumor growth. Receptor tyrosine kinase (RTK) activates the p110/p85 complex, which can go through two routes to stimulate cell growth. For wild type (WT) or kinase domain mutant p110, p85β binds p110 as the canonical PI3K complex, which then activates downstream AKT and mTOR to promote gene expression and cell growth. However, when p110 has mutations in the helical domain (mH), p85β disassociates from p110 to become free forms that will be translocated into the nucleus though its built-in nuclear localization signal. In the nucleus, p85β recruits USP7 to EZH1/2 and stabilizes the latter, allowing H3K27 trimethylation and global transcriptional gene expression.
While these findings offer innovative insights into the PI3K signaling and its impact on tumor progression, they also raised several questions that are worthy of further investigation. First and foremost, why does p85β only demonstrate these effects in cancer cells harboring helical domain mutations of p110? For the nuclear translocation, is there a greatly reduced binding affinity between p85β and the helical domain mutated p110? X-ray crystallography and cryo-EM studies showed that the N-terminal ABD domain of p110α binds to the internal nSH2 and iSH2 domains of p85α [13, 14]. However, the ABD domain in p110 and the SH3 domain in p85 that are considered to interact with each other tend to be flexible, failing to produce high resolution structures. Yet, addition of a specific p110 inhibitor stabilized the structure of these domains, demonstrating that N-terminal SH3 domains of p85α, which is also the domain that is responsible for the observed different binding with p110 in the current study, points towards the catalytic domain, but not the helical domain of p110α [13]. Hence, it remains unknown if the amino terminus of p85β bind to the helical domain of p110. Future structural study is important to answer these questions. The identified NLS (LQMKRTAIEAF) in p85β only meets the minimum requirement (i.e., KR) for an NLS; hence, whether post-translational modifications such as phosphorylation on T479, methylation on R478 or acetylation on K477 is involved in promoting p85β nuclear shuttling is an interesting question that is worthy of investigating. It also remains unknown as to why p85β does not interact with USP7 or EZH1/2 in the nucleus in non-helical p110 mutant cancer cells, as the p110 subunit should be irrelevant for the function of p85β in the nucleus. Additionally, USP7 is a widely studied de-ubiquitin enzyme that regulates tumor through stabilizing a large number of important proteins that drive/promote or suppress tumor progression, such as CHK1, MDM2, c-MYC, P53, PTEN, etc., most likely in a context dependent manner [15]. This study adds another protein, EZH1/2, to the long list of USP7 substrates to promote colon tumor. Hence, combining a USP7 inhibitor with a PI3K inhibitor may produce a similar anti-tumor effect as seen by the combination of EZH1/2 inhibitor with PI3K inhibitor reported here. Last but not least, if the nuclear signaling is specific to the helical domain mutations of p110, an inhibitor that can specifically target helical domain mutations, without acting as a general p110 kinase inhibitor, might produce an even better tumor reduction effect when combined with EZH1/2 inhibitors.
Funding
Youwei Zhang is supported in part by NIH/NCI (R01CA230453) and the Case Comprehensive Cancer Center grant (P30CA043703).
Footnotes
Conflict of Interest: We declare no potential conflicts of interest
References:
- 1.Hao Y, et al. , Nuclear translocation of p85beta promotes tumorigenesis of PIK3CA helical domain mutant cancer. Nat Commun, 2022. 13(1): p. 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cantley LC, The phosphoinositide 3-kinase pathway. Science, 2002. 296(5573): p. 1655–7. [DOI] [PubMed] [Google Scholar]
- 3.Ghigo A, et al. , Phosphoinositide 3-kinases in health and disease. Subcell Biochem, 2012. 58: p. 183–213. [DOI] [PubMed] [Google Scholar]
- 4.Vadas O, et al. , Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Science signaling, 2011. 4(195): p. re2. [DOI] [PubMed] [Google Scholar]
- 5.Yu J, et al. , Regulation of the p85/p110 phosphatidylinositol 3’-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol, 1998. 18(3): p. 1379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitman M, et al. , Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature, 1985. 315(6016): p. 239–42. [DOI] [PubMed] [Google Scholar]
- 7.Kandoth C, et al. , Mutational landscape and significance across 12 major cancer types. Nature, 2013. 502(7471): p. 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantley LC and Neel BG, New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A, 1999. 96(8): p. 4240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vallejo-Diaz J, et al. , The Opposing Roles of PIK3R1/p85alpha and PIK3R2/p85beta in Cancer. Trends Cancer, 2019. 5(4): p. 233–244. [DOI] [PubMed] [Google Scholar]
- 10.Fox M, Mott HR, and Owen D, Class IA PI3K regulatory subunits: p110-independent roles and structures. Biochem Soc Trans, 2020. 48(4): p. 1397–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hao Y, et al. , Gain of interaction with IRS1 by p110alpha-helical domain mutants is crucial for their oncogenic functions. Cancer Cell, 2013. 23(5): p. 583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janku F, et al. , PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res, 2013. 73(1): p. 276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, et al. , Cryo-EM structures of PI3Kalpha reveal conformational changes during inhibition and activation. Proc Natl Acad Sci U S A, 2021. 118(45). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller MS, et al. , Structural basis of nSH2 regulation and lipid binding in PI3Kalpha. Oncotarget, 2014. 5(14): p. 5198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeasmin Khusbu F, Chen FZ, and Chen HC, Targeting ubiquitin specific protease 7 in cancer: A deubiquitinase with great prospects. Cell Biochem Funct, 2018. 36(5): p. 244–254. [DOI] [PubMed] [Google Scholar]