

Abstract
Microbial experience fundamentally shapes immunity, particularly during the perinatal period when the immune system is underdeveloped, and novel microbial encounters are common. Most animal models are raised in specific pathogen free (SPF) conditions with relatively uniform microbial communities. How SPF housing conditions alter early life immune development relative to natural microbial exposure (NME) has not been thoroughly investigated. Here, we compare immune development in SPF-raised mice to mice born from immunologically experienced mothers in microbially diverse environments. NME induced broad immune cell expansion, including naïve cells suggesting mechanisms beside activation-induced proliferation contribute to the increase in immune cell numbers. We found NME conditions also expanded immune cell progenitor cell populations in the bone marrow, suggesting microbial experience enhances immune development at the earliest stages of immune cell differentiation. Multiple immune functions characteristically impaired in infants were also enhanced by NME, including T cell memory and Th1-polarization, B cell class-switching and antibody production, proinflammatory cytokine expression, and bacterial clearance following Listeria monocytogenes challenge. Collectively, our studies reveal numerous impairments in immune development in SPF conditions relative to natural immune development.
INTRODUCTION
At birth, newborns abruptly encounter a diverse community of previously unencountered microbes. The newborn immune system has the complex task of simultaneously defending the host against dangerous pathogens while ignoring innocuous signals and nurturing the formation of a healthy commensal microbiome. Failures of these processes can result in infection, sepsis, malnutrition, developmental delay, allergic reactions, or death. Immune development is a prolonged process whereby early life immune suppression is progressively reduced. This process can take months to years for some mammalian immune systems, during which the organism is susceptible to infections. While the factors that orchestrate immune development are not clearly defined, evidence suggests microbial experience can augment immune development (1–4). Moreover, the perinatal immune system may be more sensitive to modulation by microbial exposure than the mature immune system (5). The effects of microbial exposure on immune development are not limited to the postnatal period, as prenatal maternal microbial exposure also impacts immune development in offspring (6, 7). However, the field has been limited to single pathogen models and the use of relatively uniform conventional microbiota. While some recent naturalization models have elegantly incorporated physiological microbial exposures during pregnancy and preconception (4, 8, 9), the impact of perinatal and maternal microbial exposures on the trajectory of early immune development remains incompletely defined. Here, we use a mouse model that incorporates physiological microbial exposure beginning with the mother before conception and persisting through pregnancy and postpartum period. We define how natural microbial experience (NME) influences immune system development through characterization of immune cells and immune cell progenitor populations, cytokine/chemokine production, pattern recognition receptor (PRR) expression, and pathogen clearance.
MATERIALS AND METHODS
Mice
Male and female C57BL/6J (B6) (000664), and female B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J (B6.mTmG) (007676) mice were purchased from Jackson Laboratories (Bar Harbor, ME). B6.mTmG+/− mice were used as female breeders in some experiments to track immune cell origins. We observed no difference between mTmG and B6 mice in these experiments. Female pet store mice were purchased from local pet stores in the Minneapolis-Saint Paul, MN metro area. Serological testing has revealed pet store mice often carry some combination of: Rotavirus (EDIM), Mouse Hepatitus Virus (MHV), Murine Norovirus (MNV), Mouse Parvovirus NS1, Type 1/2 (MPV), Minute virus of mice (MVM), Theiler’s murine encephalomyelitis virus (TMEV), Sendai Virus (SEND), Lymphocytic Choriomeningitis (LCMV), Mouse Adenovirus 1/2 (MAV), Mouse Cytomegalo Virus (MCMV), Polyoma virus (POLY), Pneumonia virus of mice (PVM), Mycoplasma Pulmonis (MPUL), Clostridium Piliforme (CPIL), Pinworms, Furmites, and Enchephalitozoon cuniculi (ECUN). The pet store mice are also tested for Ectromelia virus (Mousepox), Reovirus (REO), and Cillia-Associated Respiratory Bacillus (CARB), but have never tested positive for these microbes. Our SPF colony is routinely tested to ensure the absence of the following pathogens: Mouse Parvovirus (MPV), Minute Virus of Mice (MVM), Mouse, Hepatitis Virus (MHV), Mouse Rotavirus-A (EDIM), Theiler’s Murine Encephalomyelitis Virus (TMEV), Sendai Virus (SEND), Pneumonia Virus of Mice (PVM), Reovirus (REO), Ectromelia (Mousepox), Mouse Adenovirus Type 1&2 (MAV), Polyoma Virus (POLY), Lymphocytic, Choriomeningitis Virus (LCMV), Mouse Cytomegalovirus (MCMV), Mycoplasma pulmonis (MPUL), Clostridium piliforme (CPIL, Tyzzers Disease), Cilia-Associated Respiratory Bacillus (CARB), Furmites (Myobia musculi, Radfordia affnis, Fadfordia ensifera, Myocoptes musculinus), Pinworms (aspicularis tetraptera, Syphacia abelata, Syphacia muris), and Encephalitozoon cuniculi (ECUN). Female laboratory mice were cohoused with pet store mice for >3 weeks in large rat cages to normalize microbial experience prior to introduction of male breeders. All mice were housed in AAALAC-approved animal facilities at the University of Minnesota (BSL-1/BSL-2 for SPF mice, and BSL-3 for cohoused mice). All animal use was performed per a University of Minnesota Institutional Animal Care and Use Committee-approved protocol (2106-39195A).
Flow Cytometry
Analysis was performed on the LSR Fortessa (BD Biosciences). Data were analyzed with FlowJo Software version 10 (BD Biosciences). Antibodies used for flow cytometry were purchased from BD Biosciences: CD3 (145-2C11), CD4 (GK1.5), CD8 (53-6.7), CD34 (RAM34), CD44 (IM7), CD45.2 (104), CD62L (MEL-14), CD64a/b (X54-5/7.1), Gata3 (L50-823), I-A/I-E (2G9), IgM (II/41), Ly6G (18A), RORγt (Q31-378); Invitrogen: B220 (RA3-682), CD11b (M1/70), CD11c (N418), FOXP3 (FJK-16s), IgD (11-26c(11-26)), Ki67 (SolA15), Ly6G (1AB-Ly6G), NK1.1 (PK136), PD-1 (J43), Ter119 (Ter119); Biolegend: CD16 (S17011E), CD19 (6D5), CD138 (281-2), c-kit (ACK2), CXCR5 (L138D7), Flt-3 (A2F10), Ly6C (HK1.4), Sca-1 (E13-161.7), T-bet (4B10), TCR γδ (GL3); and Tonbo: B220 (RA3-682), Ghost Dye Violet 510, Ghost Dye Red 780.
Plasma Cytokine Analysis
Blood was collected EDTA tubes and centrifuged at 1000g for 10 minutes. The plasma layer was removed, and sent to the University of Minnesota Cytokine Reference lab for cytokines/chemokines quantification by Luminex magnetic bead immunoassay using the MILLIPLEX mouse 32-plex (Millipore Sigma: G-CSF, GM-CSF, IFN-γ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17, CXCL10/IP-10, CXCL1/KC, LIF, CXCL5/LIX, CCL2/MCP-1, M-CSF, CxCL9/MIG, CCL3/MIP-1α, CCL4/MIP-1β, CXCL2/MIP-2, CCL/5RANTES, TNF-α, VEGF, CCL11/Eotaxin).
Listeria Infection and Bacterial Quantification
Listeria monocytogenes was grown to log phase in tryptic soy broth (TSB) supplemented with 50 μg/ml of streptomycin. Mice were infected with 1x105 – 2x105 CFU interperitoneally. Four days post-infection, the spleens of infected animals were homogenized in 4mL of 0.2% IGEPAL solution. Serial dilutions were plated on TSB agar supplemented with 50 μg/mL streptomycin for 24 h at 37°C, and the colonies were enumerated.
Generation of cDNA libraries and Quantitative real-time PCR (qPCR)
Splenocytes were cultured overnight in complete RPMI at 37°C 5% CO2 to permit the isolation of myeloid cells by plastic adherence (10, 11). Non-plastic adherent cells were washed away with 2 washes of 5 mL PBS. Total RNA was isolated from the adherent myeloid cells using TRIzol reagent (Invitrogen, Carlsbad, CA), and 1 μg RNA was reverse transcribed into cDNA using random hexamers and Superscript III (Invitrogen, Carlsbad, CA). The resulting cDNA was used as template for qPCR using TaqMan primer/probe sets for Tlr1, Tlr2, Tlr3, Tlr4, Tlr6, Cd14, Myd88, and 18s rRNA (Applied Biosystems).
Quantification and Statistical Analysis
Data were collected across multiple experiments performed over 2.5 years. GraphPad Prism 9 was used to perform statistical analysis. A two-tailed unpaired, nonparametric Mann-Whitney U test was used to compare two groups at a single time point. Simple linear regression analysis was used to test whether the slopes of cells or cytokines/chemokines over time were different between groups. * p < 0.05, ** p <0.01, *** p<0.001, **** p<0.0001.
RESULTS
NME induces broad expansion of immune cells
We cohoused female C57BL/6 background mice with female pet store mice for >3 weeks prior to breeding to ensure the dams acquired an experienced immune system and expose new litters to the diverse microbial communities of pet store mice from the earliest natural point (Fig. 1A). Breeding mice under NME conditions resulted in slight reductions in litter size and weight at weaning relative to litters born under conventional SPF conditions (Fig. 1B−C). We took a comprehensive and agnostic approach to measure the impact of NME on immune development using two flow cytometry panels to quantify 26 unique immune cell markers at three different ages: infancy (2 weeks of age), weaning (3 weeks of age), and adolescence (6 weeks of age). We observed a 4.4-fold expansion of leukocyte numbers (CD45+) in the lymph nodes of NME mice relative to SPF mice by 6 weeks of age (Fig. 2A). We subdivided this population into eight major immune cell types: CD4 T cells, CD8 T cells, B cells (CD19+), monocytes/macrophages (CD11b+), dendritic cells (DCs; CD11c+ MHC II+), neutrophils (Ly6G+), natural killer (NK) cells (NK1.1+), and Ɣδ T cells (Ɣδ TCR+). As expected, we observed a measurable expansion of most immune cell types in the lymph nodes (superficial cervical, axillary, brachial, inguinal, and mesenteric) of NME pups relative to SPF pups at 2 weeks of age and found this gap widened through 3 and 6 weeks of age (Fig. 2B−I). By 6 weeks, all eight major immune cell types were significantly more numerous in NME mice relative to SPF mice. Neutrophils and NK cells demonstrated the greatest expansion, achieving a >8-fold expansion in NME mice over SPF mice (Fig. 2D, G). This disproportionate expansion resulted in neutrophils, NK cells, and monocytes/macrophages making up a larger proportion of the immune cells in NME mice than SPF mice, while CD4 T cells were less frequent at certain timepoints (Fig. 2D, E, G, and H). In the spleen, the degree of NME-induced immune cell expansion was more modest than what was observed in the lymph nodes but followed similar trends (data not shown). These data reveal NME has a measurable impact on the immune system during early life, inducing global numerical expansion.
Figure 1: NME Breeding.
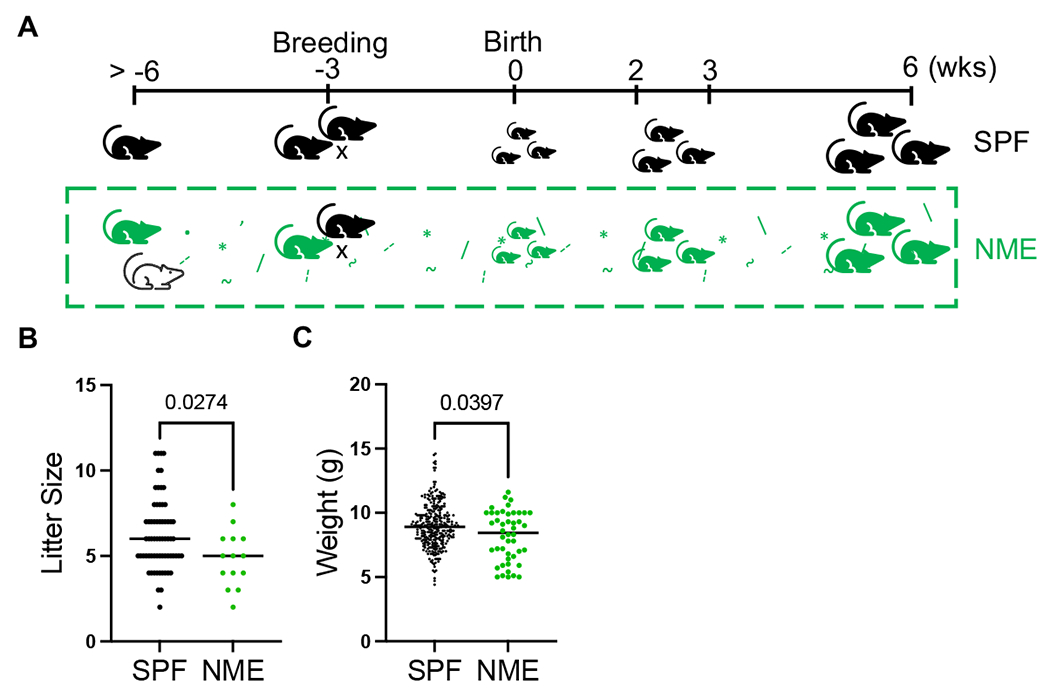
(A) Adult female C57BL/6 mice (green) are cohoused with pet store mice (white) for >3 weeks prior to breeding. Dam and litter remained with pet store mice for up to 6 wks after parturition. (B) Litter sizes (from 61 SPF litters and 14 NME litters) and (C) weight at weaning were measured (collected from 51 SPF litters and 10 NME litters). Data was collected over the course of one year of breeding. **p ≤ 0.01, ****p ≤ 0.0001.
Figure 2: NME induces broad immune cell expansion in the lymph nodes.
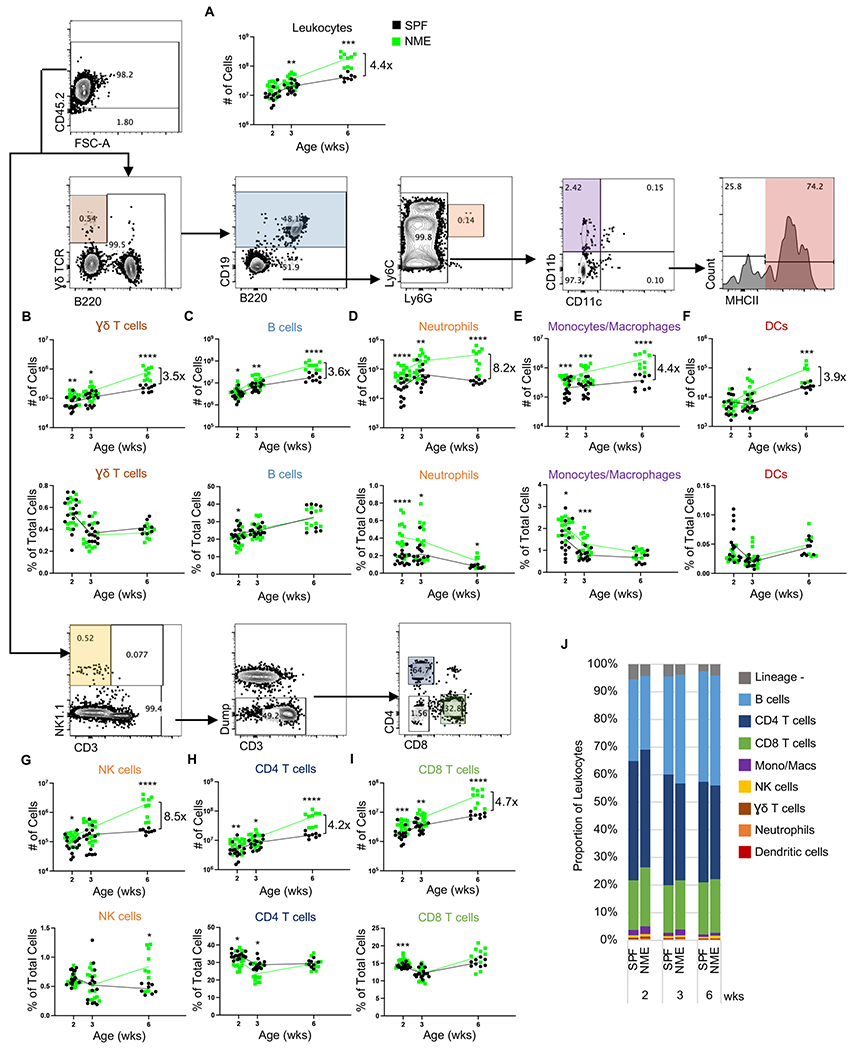
Lymph nodes were harvested from SPF (black) and NME (green) offspring at 2, 3, or 6 weeks of age and split in half for staining with two flow panels. (A) Leukocytes were identified by CD45.2 expression and enumerated. Number and frequency of eight immune cell types as indicated: (B) Ɣδ T cells, (C) B cells, (D) neutrophils, (E) monocytes/macrophages, (F) DCs, (G) NK cells, (H) CD4 T cells, and (I) CD8 T cells. (J) Leukocyte composition. 10-15 mice/group/time point. Data were collected from three individual experiments per group per age over two years. Example flow plots are from a 6-week-old NME mouse. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
NME pups exhibit increased T cell memory and effector T cell commitment
We next measured the effect of NME on CD8 T cell activation and memory commitment using a combination of standard murine markers of activation (CD44) and naïve or central memory (CD62L). In the lymph nodes of NME mice, we observed an increased number of central memory T (Tcm; CD44+ CD62L+) and effector (Teff)/effector memory (Tem; CD44+ CD62L−) CD8 T cells; however, we also observed an unexpected increase in the number of naïve (CD44− CD62L+) T cells (Fig. 3A−C). Compared to SPF mice, NME mice maintained higher proportions of activated CD8 T cells than SPF mice at each age (Fig. 3D). The lymph node-derived CD4 T cell population mirrored the CD8 T cell population with increased numbers and proportions of activated and memory committed CD4 T cells at each timepoint (Fig. 3E−H). Much like the CD8 T cells population, the naive CD4 population was larger in NME mice than SPF mice at 2 and 6 wks of age (Fig. 3E).
Figure 3: NME increases T cell memory commitment.
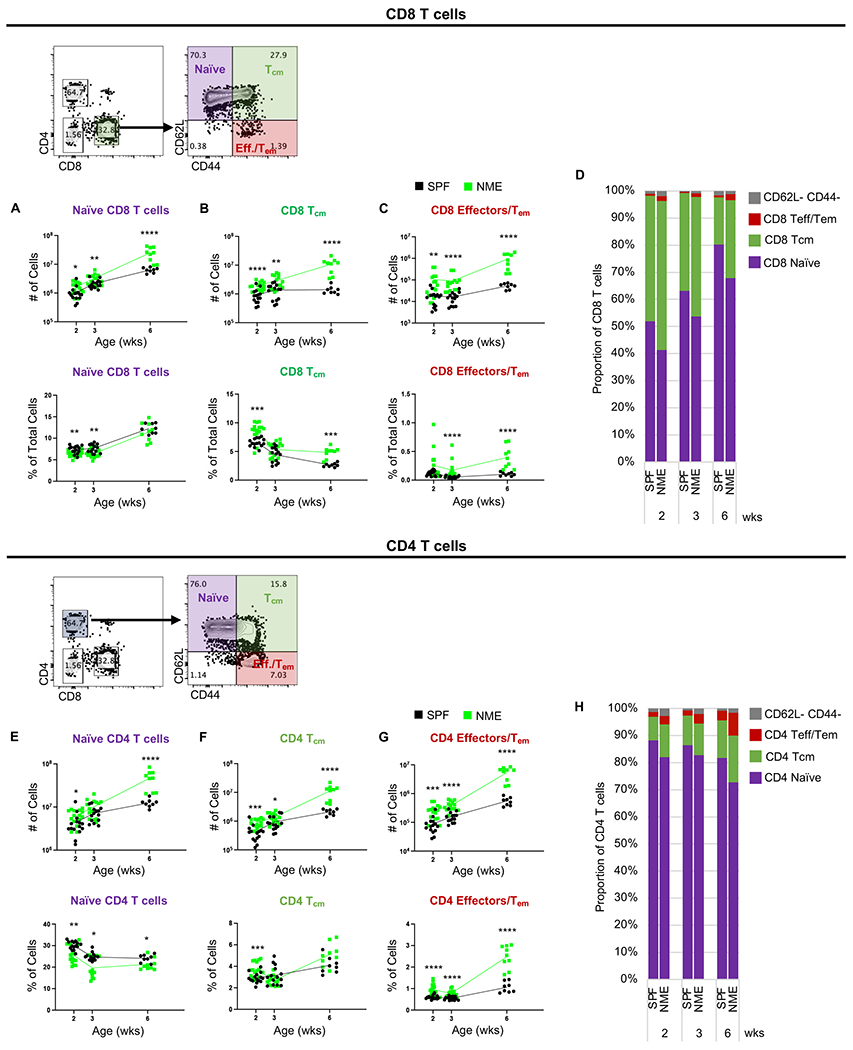
Number and frequency of (A) naïve, (B) Tcm, and (C) Tem/Teff CD8 T cells in the lymph nodes harvested from SPF (black) and NME (green) offspring at 2, 3, or 6 weeks of age. (D) Lymph node CD8 T cell composition. Number and frequency of (E) naïve, (F) Tcm, and (G) Tem/Teff CD4 T cells in the lymph node. (H) Lymph node CD4 T cell composition. 10-15 mice/group/time point. Data were collected from three individual experiments per group per age over two years. Example flow plots are from a 6-week-old NME mouse. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
In addition to examining the extent of CD4 T cell activation and memory commitment, we also investigated the impact of NME on CD4 T helper (Th) cell lineage commitment by assessing the expression of lineage defining transcription factors. SPF mice maintained a steady level of ~10% commitment of CD4 T cells to definable Th subsets in the lymph nodes throughout the study, while the proportion of polarized CD4 T cells in NME mice reached 15% by 6 weeks (Fig. 4F). Foxp3-expressing regulatory T (Treg) cells exhibited a slight increase in number in NME mice relative to SPF mice at 2 weeks but made up a similar proportion of total cells in the lymph node (Fig. 4A). However, by 6 weeks Treg cells in NME mice outnumbered those of SPF mice and were more frequent (Fig. 4A). We observed robust increases in the number and proportion of Th1 (Tbet+) cells in NME mice relative to SPF mice at all developmental stages (Fig. 4B). Th17 (RORγt+) and Th2 (Gata3+) cells were also significantly more numerous and made up a greater proportion of lymph node cells in NME mice than SPF mice at 2 and 6 weeks (Fig. 4C−D). Finally, follicular helper T (Tfh; PD-1+ CXCR5+) cells were more numerous in NME relative to SPF mice at each timepoint, though their relative proportion was not significantly different (Fig. 4E). In the spleen, the number and proportion of Th subsets were equivalent between NME and SPF mice through 3 weeks; however, by 6 weeks NME mice displayed a slight increase in the number of each Th subset over SPF mice (data not shown). CD4 T cell phenotypes are often described in simplified terms of the Th1 versus Th2 dichotomy, intending to contrast the cellular and humoral potential of the immune system, respectively (12, 13). Low Th1:Th2 ratios are often associated with tolerance, like those observed in neonates and pregnant women, while higher Th1:Th2 ratios suggest a more proinflammatory immune environment (12, 13). We found the CD4 T cell compartment in NME mice was skewed toward a more proinflammatory ratio (Th1 and Th17: Treg, Th2, and Tfh) than SPF mice early in both the lymph node (Fig. 4G) and spleen (data not shown). Collectively, these data demonstrate microbial exposure during early life enhances T cell commitment to mature effector and memory lineages; however, it unexpectedly also increased the number of naïve T cells.
Figure 4: NME enhances helper T cell polarization.
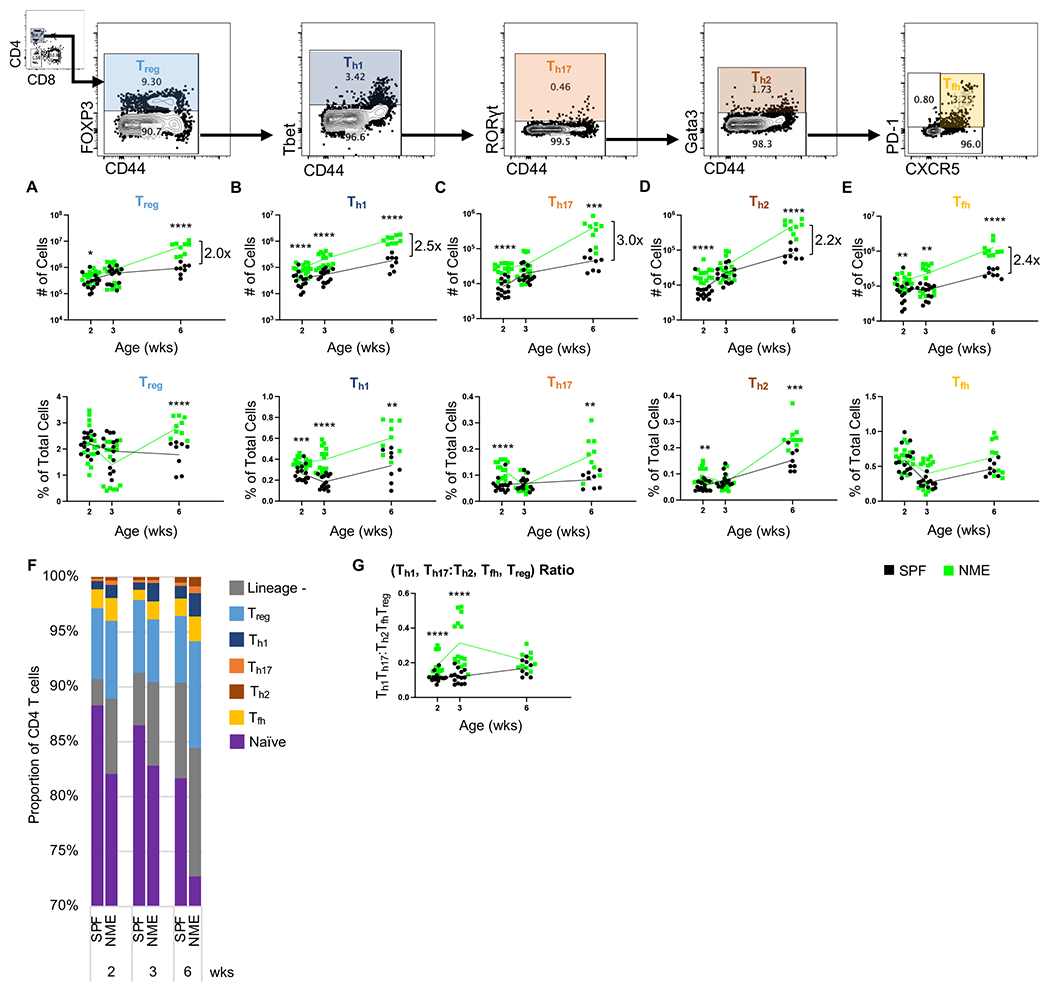
Number and frequency of (A) Treg, (B) Th1, (C) Th17, (D) Th2, (E) Tfh CD4 T cells in the lymph node harvested from SPF (black) and NME (green) offspring at 2, 3, or 6 weeks of age. (F) Lymph node CD4 T cell lineage composition. (G) Lymph node Th1 and Th17 to Treg, Th2, and Tfh ratio. 10-15 mice/group/time point. Data were collected from three individual experiments per group per age over two years. Example flow plots are from a 6-week-old NME mouse. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Macrophages display a more activated phenotype in NME pups.
T cell activation and lineage commitment are mediated by interactions with antigen presenting cells. Therefore, we measured the influence of early life microbial exposure on the activation status of the antigen presenting cell populations. When we analyzed lymph node-derived monocyte/macrophage population (CD11b+ CD11c−) for the expression of activation markers Ly6C and MHC II, we identified five distinct populations: Ly6C+ MHC II− (P1), Ly6C+ MHC IIint (P2), Ly6Clow MHC IIint (P3a), Ly6Clow MHC IIhi (P3b), and Ly6C− MHC II− (P4) (Fig. 5A−E). These populations correspond with the “waterfall” developmental progression pattern described in multiple organs, where the Ly6C+ MHC II− (P1) population is similar to blood monocytes that begin to express MHC II after activation to become Ly6C+ MHC IIint (P2) (14–16). These cells then decrease Ly6C expression to extravasate and become P3 macrophages (14–16). We also observed distinct MHC IIint (P3a) and MHC IIhi (P3b) populations in the lymph node (Fig. 5D−E). The Ly6C− MHC II− population (P4) likely consists of mature macrophages and other rare immune cell lineages not effectively excluded by upstream gating (i.e., CD11b+ T cells and granulocytes) (14–16). Each of these five monocyte/macrophage subpopulations were more numerous in NME mice than SPF mice across all ages (Fig. 5A−E). Much like our observations with T cells, NME appeared to expand all macrophage subsets, even the least activated, P1 subset (Fig. 5A). When we considered the proportions of these populations in the lymph nodes, we observed several notable NME-induced alterations to the immune cell composition. Ly6C+ MHC II− (P1) cells made up a greater proportion of lymph cells early in NME pups but equaled SPF proportions by 6 weeks (Fig. 5A, C). The Ly6C− MHC II− (P4) and Ly6Clow MHC IIint (P3a) population made up a larger proportion of lymph cells in NME pups at only the 3-week time point (Fig. 5C−D), while the Ly6C+ MHC IIint (P2) and Ly6Clow MHC IIhi (P3b) populations trended toward a larger proportion of lymph cells in NME mice at every developmental stage (Fig. 5B, D). Together, these data demonstrate that NME conditions broadly expand and activate monocytes/macrophages relative to SPF mice, likely facilitating the observed maturation of the T cell compartment.
Figure 5: Monocyte/macrophage subsets.
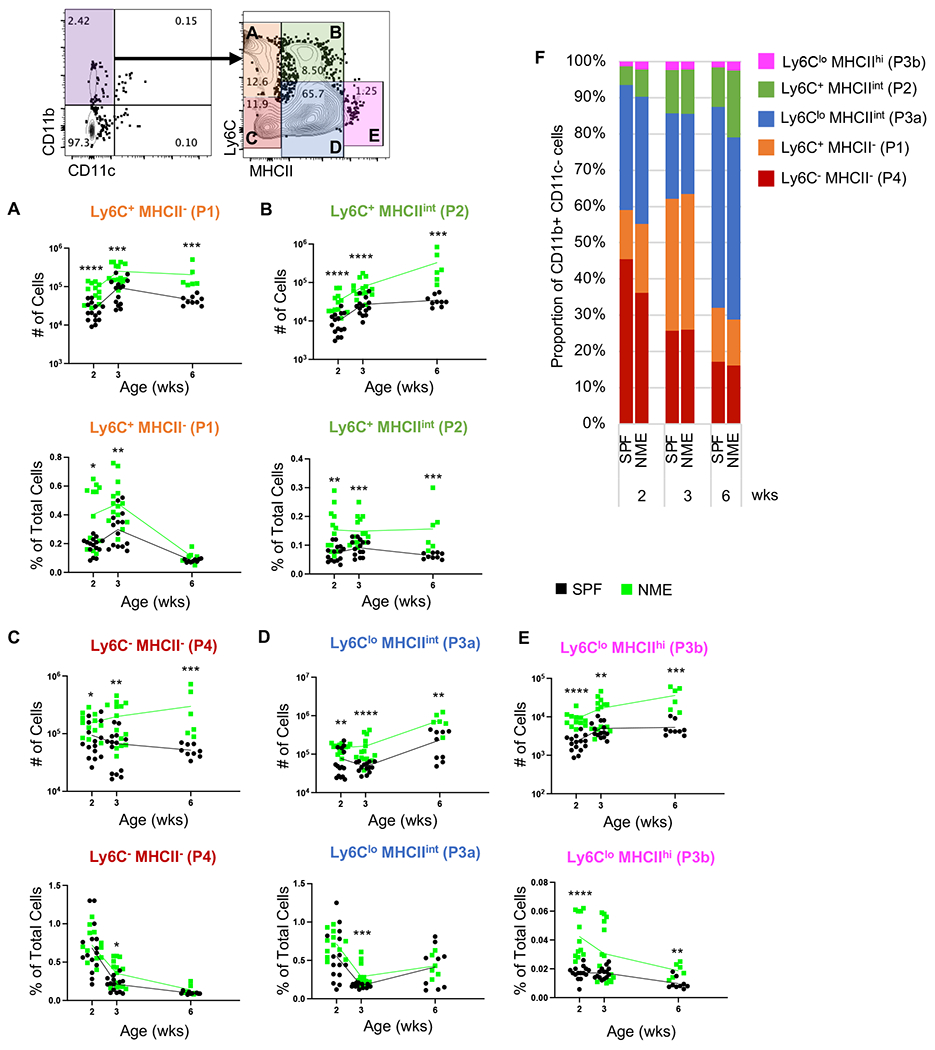
Number and frequency of (A) Ly6C+ MHCII− (P1), (B) Ly6C+ MHCIIint (P2), (C) Ly6C− MHCII− (P4), (D) Ly6Clo MHCint (P3a), (E) Ly6Clo MHCIIhi (P3b) in the lymph node harvested from SPF (black) and NME (green) offspring at 2, 3, or 6 weeks of age. (F) Lymph node monocyte/macrophage composition. 10-15 mice/group/time point. Data were collected from multiple experiments over two years. Example flow plots are from a 6-week-old NME mouse. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Physiological microbial experience enhances B cell class-switching, plasma cell commitment, and antibody production.
Infant B cells produce less immunoglobulin, undergo less class-switching, and produce fewer memory B cells and plasma cells in response to antigen than adult B cells. While cell intrinsic factors are involved, the relative paucity of Tfh cells and germinal centers are also contributing factors (17, 18). We observed a >2-fold increase in the quantity of Tfh cells in NME mice (Fig. 4E). To determine whether this NME-induced increase in Tfh cells correlated with enhanced B cell activation, we measured the expression of IgD and IgM among the CD19+ B220+ B cell population to define IgD+ IgM+ antigen inexperienced naïve B cells, IgD− IgM+ immature B cells, IgD+ IgM− mature activated B cells, and IgD− IgM− class-switched B cells. All B cell subsets expanded in NME mice relative to SPF mice in the lymph node, resulting in similar proportion at 2 and 3 weeks (Fig. 6A–D). By 6 weeks, class-switched (IgD− IgM−) B cells made up a larger proportion of lymph node cells in NME mice than SPF mice (Fig. 6C). Plasma cells are terminally differentiated B cells that secrete large amounts of antibody. While plasma cells typically reside in the bone marrow, they are generated in the germinal centers of the spleen and lymph nodes. We found NME mice had increased numbers of plasma cells (B220− CD19− CD138+) at 3 and 6 weeks of age in the lymph node (Fig. 6F) and spleen (data not shown) compared to SPF mice. Antibody production was also greater in NME mice than SPF mice at 3 weeks of age as measured by total IgG ELISA (Fig. 6G). Altogether, our observations suggest NME conditions expand all B cell populations and augment B cell activation and germinal center activity during early life.
Figure 6: NME increases B cell activation.
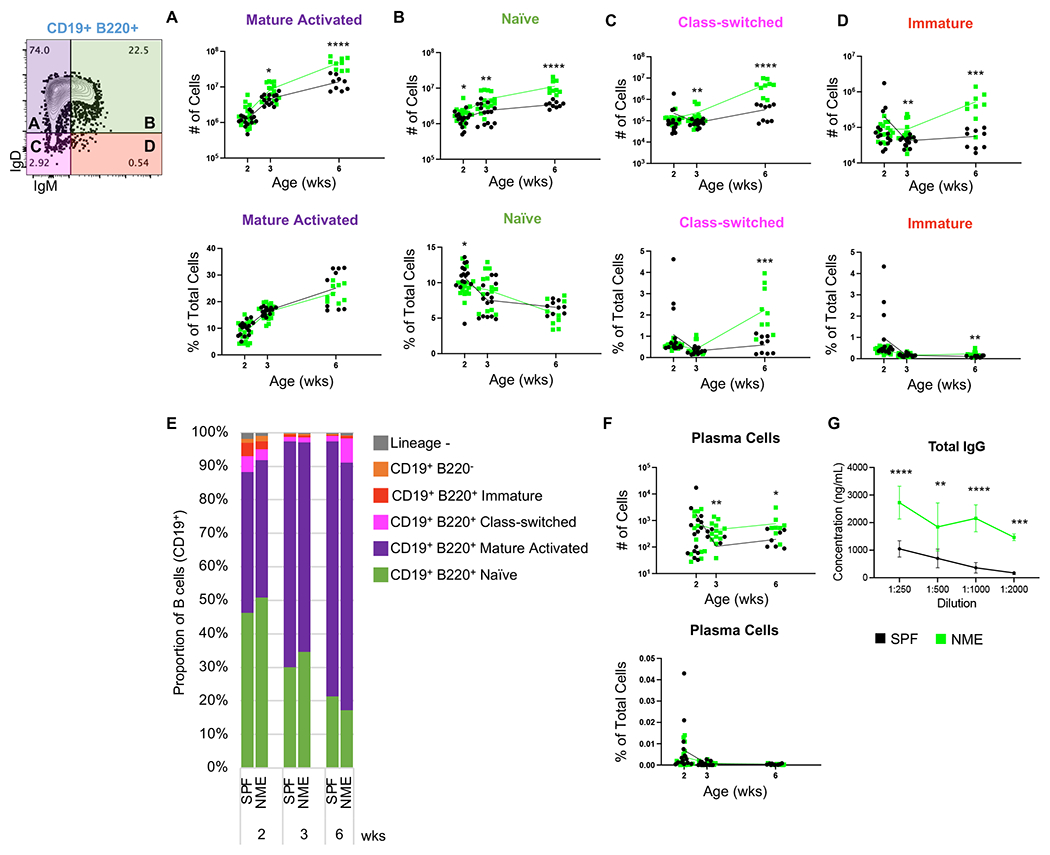
Number and frequency of (A) mature activated, (B) naïve, (C) class-switched, (D) and immature B cells in the lymph node harvested from SPF (black) and NME (green) offspring at 2, 3, or 6 weeks of age. (E) Lymph node B cell composition. (F) Number and frequency of plasma cells in the lymph node. 10-15 mice/group/time point. Data were collected from three individual experiments per group per age over two years. Example flow plots are from a 6-week-old NME mouse. (G) Total IgG concentration (ng/mL) in the plasma of 3-week-old SPF and NME mice measured by a single ELISA, samples were assayed in duplicate. 3-4 mice/group. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
NME increases the number of immune cell progenitors in perinatal bone marrow.
The observation that all immune cell types were expanded in NME mice, including multiple naive immune cell populations (Figs. 3A, 3E, 5A, and 6B), led us to investigate whether NME conditions influence the production of hematopoietic stem cells and progenitors (HSCPs). LSK cells, defined by the lack of expression of mature immune cell markers (Lin−) and the co-expression of Sca-1 and c-Kit, include multi-potent progenitor (MPP) cell subsets 1-4, which give rise to oligopotent progenitors: common lymphoid progenitors (CLPs), granulocyte-monocyte progenitors (GMPs), and megakaryocyte-erythroid progenitors (MEP) (Fig. 7A)(19). We enumerated LSK cells in the bone marrow of 3-day old NME and SPF pups and found NME pups had more LSK cells than SPF pups, indicating NME conditions stimulated expansion of HSPCs (Fig.7 B). Enumeration of Lin−, Sca-1+, c-kit+, CD34−, Flt3− cells, which fit the profile of hematopoietic stem cells (HSCs) thought to have the broadest differentiation potential, were not as robustly expanded, but did trend toward increased numbers in NME mice compared to SPF mice (Fig. 7B). Given that NME broadly induced expansion of many leukocyte subsets we investigated whether oligopotent progenitor subsets were also expanded. We identified a population of Lin−, Sca-1+, c-kit+, CD34+, Flt3hi expressing cells enriched for MPP4 cells, also referred to as Lympho-myeloid primed progenitors (LMPPs) (49), which are thought to give rise to oligopotent CLPs (enriched for here by Lin−, Sca-1lo, c-kitlo, Flt3+) that differentiate into lymphocytes. Both the MPP4/LMPP and CLP populations were expanded in NME mice relative to SPF mice (Fig. 7D, G). Likewise, GMP (Lin−, Sca-1−, c-kit+, CD16+, CD34+) cells, which differentiate into granulocytes and monocytes were also expanded in NME mice relative to SPF mice (Fig. 7E). Conversely, the number of MEP cells, which differentiate into platelets and red blood cells, were not altered by NME conditions, indicating a bias toward leukocyte expansion (Fig. 7F). Altogether, these observations suggest that physiological microbial experience during early life stimulates production of the earliest immune cell progenitors, which may promote the expansion of both innate and adaptive immune cells.
Figure 7. NME enhances hematopoiesis.
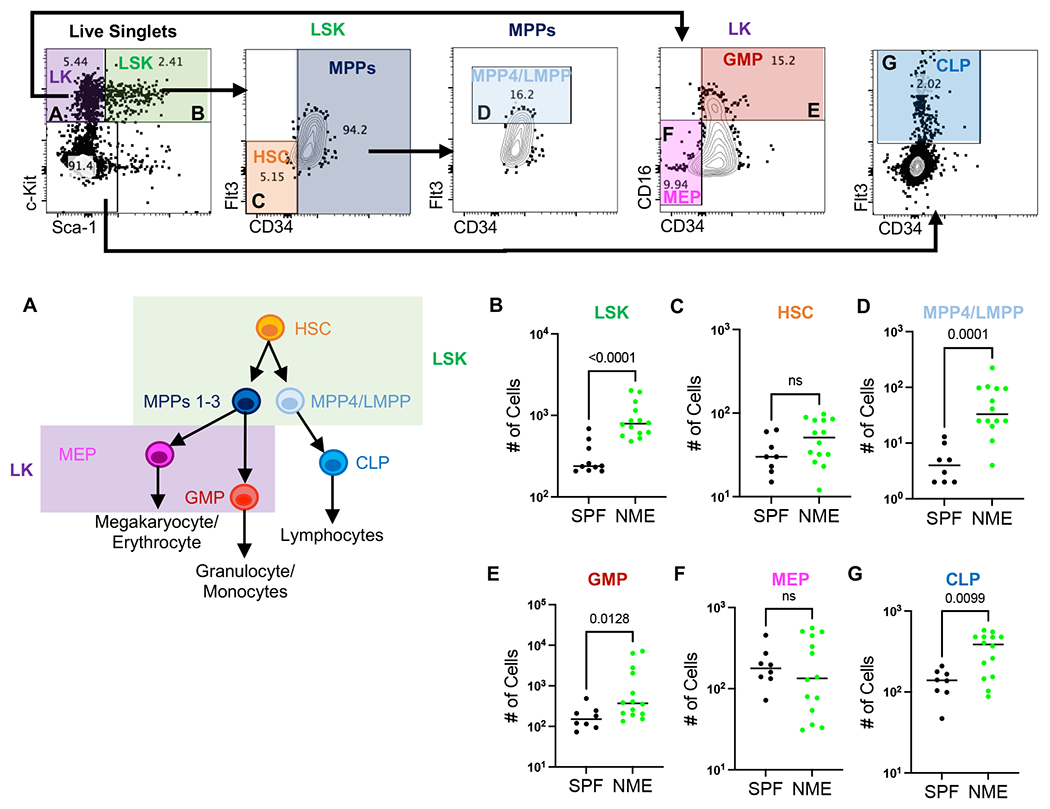
(A) Hierarchy of differentiation for immune cell progenitors. Bone marrow was harvested from SPF (black) and NME (green) pups at day of life (DOL) 3 and (B) LSK (Lin− Sca-1+ c-Kit+), (C) HSC (Lin− Sca-1+ c-Kit+ Flt-3− CD34−), (D) MPP4/LMPP (Lin− Sca-1+ c-Kit+ Flt-3hi), (E) GMP (Lin− Sca-1− c-Kit+ CD16+ CD34+), (F) MEP (Lin− Sca-1− c-Kit+ CD16− CD34−), (F) CLP (Lin− Sca-1− c-Kit− Flt3+) were enumerated flow cytometrically. 8-14 mice/group. Data was collected from 2-3 separate experiments. Example flow plots are from a DOL 3 NME mouse. Statistical analyses were determined using Mann-Whitney Test.
NME pups demonstrate more robust and precocious expression of multiple cytokines
To gain insight into the possible signaling pathways orchestrating immune cell expansion and activation in NME mice, we measured the concentrations of 32 cytokines and chemokines in the plasma from NME and SPF mice at 3 and 6 weeks of age. At 3 weeks, NME mice displayed elevated plasma levels of IL-6, IL-17, G-CSF, and CCL5 over SPF mice. By 6 weeks, 22 of 32 cytokines/chemokines tested were elevated in NME mice over SPF mice (Fig. 8A and Supplemental Fig. 1). CCL11, an eosinophil chemoattractant, was the only chemokine that demonstrated decreased plasma levels in NME mice relative to SPF mice (Fig. 8A and Supplemental Fig. 1). Individual cytokines and chemokines followed similar trends in NME and SPF mice from 3 to 6 weeks, although NME mice displayed more exaggerated slopes for IL-4, VEGF, IL-12p70, LIF, IL-17, IL-10, M-CSF, GM-CSF, CCL3, IL-15, IL-9, and CCL11 (Supplemental Fig. 1). Notably, plasma levels of TNFα in 3-week-old NME pups were equivalent to those of 6-week-old SPF mice, suggesting NME induced earlier expression of TNFα than SPF conditions (Supplemental Fig. 1). Age-associated increases in IFNγ are also well documented (20–24). Our analysis revealed 15% of 3-week-old NME pups displayed unusually high levels of plasma IFNγ, while no SPF pups had detectable serum levels of IFNγ (Supplemental Fig. 1). Furthermore, IFNγ was never detected in 6-week-old SPF pups, whereas two-thirds of 6-week-old NME mice registered detectable plasma IFNγ levels (Supplemental Fig. 1).
Figure 8: NME enhances cytokine, PRR expression, and host defense.
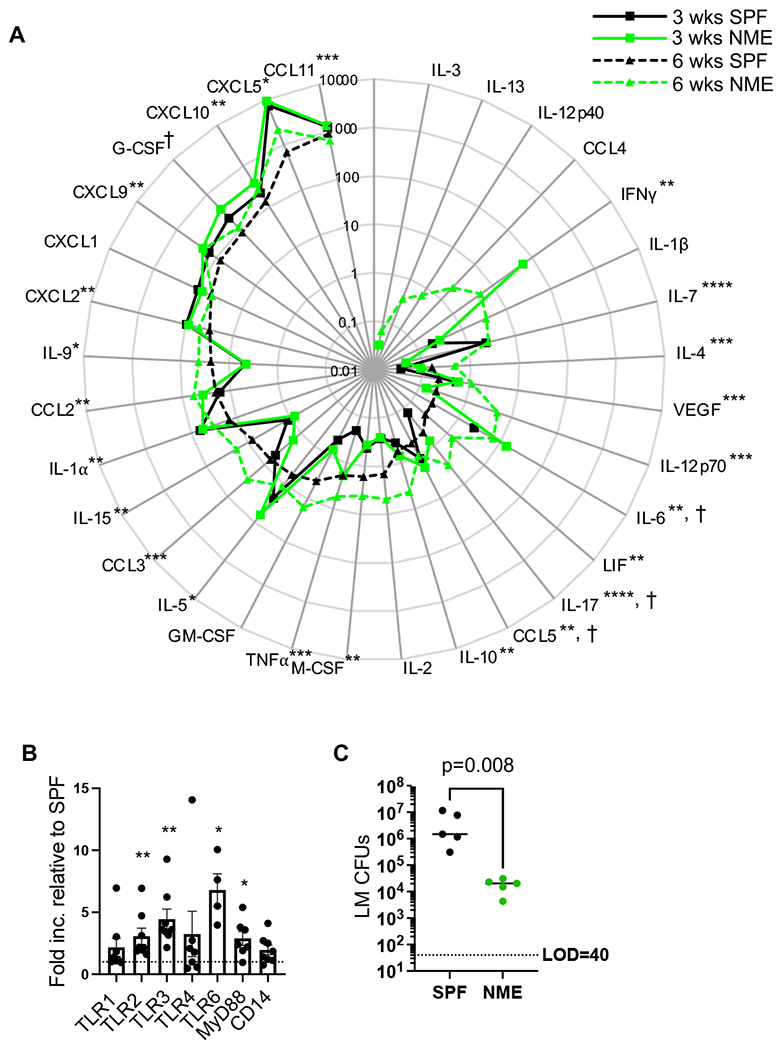
(A) Cytokine and chemokine levels in plasma harvested from 3-week-old SPF (solid black line), 3-week-old NME (solid green line), 6-week-old SPF (dotted black line), and 6-week-old NME (dotted green line) mice. Data depicts the average of 13 mice and two experimental replicates. (B) qPCR analysis of TLRs, MyD88, and CD14 in adherent myeloid cells isolated from spleens of SPF (n = 4) and NME (n = 8) mice. Data are representative of 2 technical replicates. (C) LM CFU in spleen 4 days post inoculation in SPF (black) and NME (green) mice. 9 mice/group were analyzed, representative of 2 technical replicates. †p < 0.05, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
PRR expression increases under NME conditions
Pattern recognition receptors (PRRs) recognize conserved pathogen associated molecular patterns (PAMPs). PRRs are expressed by a wide variety of mammalian cells but are most often associated with innate immune cells. PRRs play a crucial role in early detection of pathogens and the initiation immune responses by directing the expression of cytokines and transcription factors. PAMP stimulation is known to induce upregulation of PRRs, although the extent to which diverse microbial exposure alters PRR profiles during early life has not been described. We harvested cDNA from adherent splenocytes and measured the expression of a panel of PRRs and downstream signaling proteins by qPCR. We found multiple toll-like receptors (TLRs) were upregulated including membrane bound TLR2, which can heterodimerize with TLR1 and TLR6 to recognize a wide variety of PAMPs. TLR2 binding partner TLR6 and TLR signal adapter protein MyD88 were also significantly upregulated. TLR3, an endosomal TLR that recognized dsRNA and signals through TRAM/TRIF instead of MyD88, also demonstrated increased expression in NME mice than SPF mice (Fig. 8B). These data suggest NME conditions increased the sensitivity of myeloid cells to some PAMPs, which might begin to explain the differences between NME and SPF cytokine profiles and downstream activation of immune cells.
NME improves host defense in 3-week-old pups
In prior studies, cohousing adult laboratory mice with pet store mice improved bacterial clearance following challenge with a virulent strain of Listeria monocytogenes (LM), a bacterial strain commonly used to assess immune function in mouse models as well as a pathogen in human infants (25–27). Importantly, LM has not been observed to be a part of the natural ecology of pet store mice in our colony; therefore LM challenge represents a novel pathogen under these conditions (26). We challenged 3-week-old NME and SPF pups with LM and measured bacterial burden in the spleen 4 days post infection (dpi). Much like the adult cohoused mice, 3-week-old NME pups demonstrated a >2 log reduction in bacterial burden compared to their age-matched SPF counterparts (Fig. 8C). These data show microbial experience during early life exerts not only a quantitative impact on basal immunity but also improves protective immune function.
DISCUSSION
NME broadly and dramatically increased the quantity of immune cells. As expected, we observed NME-induced increases in the number of activated macrophages and lymphocytes suggesting proliferation in response to microbial exposure expands the number of immune cells. However, we also observed an increased number of naïve lymphocytes and undifferentiated macrophages/monocytes, implying some other mechanism must contribute to the observed expansion of immune cells. Enumeration of HSCPs in the bone marrow indicated NME also increased the size of multiple immune cell progenitor subsets, including LSK (Inclusive of HSCs and MPPs) and oligopotent progenitors, GMP and CLP, demonstrating NME broadly expanded immune cell progenitors. It is well established that inflammation can modulate hematopoiesis (28–31). Multiple pro-inflammatory cytokines augment hematopoiesis in vivo, including cytokines that we observed to be induced by NME: IL-6 (3 and 6 weeks), IL-17 (3 and 6 weeks), G-CSF (3 and 6 weeks), TNFα (6 weeks), M-CSF (6 weeks), GM-CSF (6 weeks), and IFNγ (6 weeks) (28–31). HSCPs have also been demonstrated to express TLRs and may therefore sense PAMPs directly (30, 32). It has been hypothesized that microbial sensing by HSPCs helps to replenish depleted innate cells – which lack the proliferative capacity of many adaptive cells – during system infections (32). However, our observations suggest NME induces the production of HSPCs with broad lineage commitment potential, including both innate and adaptive cell lineages. In line with our observations in NME mice, recently published research found that administering PolyI:C to the mother during gestation to mimic viral infection induced inflammatory cytokine mediated expansion of fetal lymphoid progenitors (33).
The newborn immune system is characterized by broad immunosuppression (34, 35). Compared to the immune systems of mice raised in low microbial burden conditions, the immune systems of mice exposed to microbially diverse environments from conception demonstrated improved immune responsiveness. Indeed, when we challenged recently weaned NME and SPF mice with a previously unencountered bacterial pathogen (i.e., virulent LM), NME mice cleared the infection more rapidly. Because LM has not been observed in our NME colony, it is less likely that persistent passively transferred LM-specific maternal antibody contributed to bacterial clearance. A hallmark of infant immunity is a reduced ability to produce Th1 and memory T cell responses. Our data show T cells from NME mice commit to Th1 and memory more often than those of SPF mice, making up a greater proportion of the T cell compartment throughout development. Likewise, germinal center function is considered deficient in infants. However, NME conditions enhanced class-switching, plasma cell differentiation, and antibody production. Infant cytokine profiles also differ from those of adults, favoring anti-inflammatory over proinflammatory cytokines (35–37). NME conditions stimulated more robust, and in some cases earlier expression of proinflammatory cytokines compared to SPF counterparts. One interpretation of these observations is that diverse microbial exposure during early life accelerates immune maturation, alternatively microbial experience may be inducing trained immunity, whereby immune stimuli enhance innate immunity heightening immune responsiveness and supporting adaptive responses. While the two concepts share many features, trained immunity is not thought to be permanent, whereas immune maturation would be expected to result in enduring and progressive change. Continued tracking, manipulation of the timing of microbial exposure, and comparisons of epigenetic profiles might provide insights into whether NME induces trained immunity, accelerates immune maturation, or both. We noted multiple parallels between immunological changes induced by Mycobacterium bovis bacillus Calmette-Guérin (BCG) vaccination in human infants and those of mice raised in NME conditions. Like NME, BCG induces Th1-memory responses, proinflammatory cytokines, and provides immunity to non-mycobacterial pathogens (17, 38, 39). The BCG vaccine is thought to invoke superior long-term innate immune function because it stimulates five different TLRs, perhaps triggering multiple microbial sensing pathways in a manner similar to NME conditions (39). The BCG vaccine might thus be a real-world example of the impact of microbial stimulation on infant immunity and a precedent for applying this concept to improve infant immunity. However, the BCG vaccine consists of a singular weakened strain of tuberculosis bacteria and is only administered once, typically between 4-6 weeks of age; therefore, it may serve to supplement the diverse microbes naturally encountered by human infants.
Cytokines are key regulators of immune responses. Cohousing adult laboratory mice with pet store mice induced greater expression of numerous cytokines/chemokines (26). Conversely, when we analyzed 3-week-old NME mice we observed increased expression of only 4 out of 32 cytokines/chemokines measured. However, by 6 weeks of age most of the cytokines/chemokines were augmented by NME conditions, demonstrating age-restricted effects of NME on plasma cytokine expression. Multiple infant immune cells have reduced capacity to produce pro-inflammatory cytokines leading to a cytokine profile skewed toward Th2 associated and anti-inflammatory cytokines, which could explain the limited effect of NME at 3 weeks of age (20, 22, 34–36, 40). Age-dependent expression of several cytokines have been described, most notably, TNFα, IFNγ, and IL-12 increase with age, while IL-6 and IL-10 display a bi-modal expression pattern (20–24, 34, 40). In line with this literature, we observed an increase in TNFα expression between 3 and 6 weeks of age. Interestingly, TNFα levels in 3-week-old NME mice were equivalent to that of 6-week-old SPF mice suggesting NME conditions induce precocious expression of TNFα. IL-6, an anti-inflammatory and Th2 polarizing cytokine, appeared to decrease in NME and SPF mice from 3 to 6 weeks. The opposing functional, temporal relationships, and the pleotropic effects of IL-6 and TNFα could signify important roles for these cytokines in immune development (24). Indeed, the tumor necrosis factor superfamily was recently identified as an important modulator of early life immune development (41).
Altogether, we found exposure to diverse microorganisms from conception has a prodigious impact on mammalian immune development. The combined effect broadly activated the innate and adaptive arms of immunity and improved immune function. Microbial experience is a major contributor to immune development; however, the multiple mechanisms that orchestrate immune development are still unresolved. Many questions about the complex immunology of healthy pregnancy, infection, preeclampsia, and premature birth have been difficult to answer with conventional animal models. NME could provide a more faithful model of normal pregnancy immunology, vertical transfer of immunity, and maternal vaccination strategies. Furthermore, some features of newborn mice are underdeveloped relative to newborn humans such that they more closely resemble preterm infants, who are 10-19% more likely to suffer severe complications from infection than term infants (42–46). NME conditions may better model the complex microbial ecology of preterm infant infections. Better understanding of the deficiencies and strengths of infant immunity could uncover novel developmental stage-specific treatments and preventions to reduce infant hospitalization and deaths from infection. The ability to evaluate and appropriately support immune development could help close the gap between premature and term infant immune function. The data herein provide the foundation and improved model for these future studies. While vital differences still exist between the NME model and human immune development, NME provides a unified model of natural immune development that complements SPF research and could prove invaluable as a preclinical model.
LIMITATIONS
NME is inherently variable, and while serology identifies the presence of many pathogens including bacteria, viruses, and parasites, many more cannot be identified making it difficult to interrogate microbe-specific effects. Ongoing research is investigating how NME conditions alter the formation of the infant microbiome. We view microbial variability as a key feature of the model that better replicates natural development; however, it can complicate interpretation. Importantly, despite this variability, many of the metrics tested here display similar variance among NME mice as was observed in SPF mice.
Advancements in transcriptomics and proteomics have led to changes in how HSCPs are subcategorized and defined (19). The HSCP flow cytometry panel we used here is limited. As such, some populations, while enriched for the cells of interest may not be pure or may be referred to by different names in the literature. Additionally, inflammation can induce the expression of Sca-1 in otherwise Sca-1− progenitor populations, including GMPs. These points raise the possibility that the LSK population of NME mice, while it may be more representative of physiological conditions, could contain GMP cells, confounding interpretation (48).
Our experiments were designed to capture the levels of cytokines/chemokines induced by NME conditions; we did not assess the total capacity of immune cells to produce individual cytokine/chemokine. Ex vivo stimulation or cytokine reporter mice might demonstrate age-related deficiencies in cytokine/chemokine production more clearly and could reveal whether NME-induced increases in cytokine/chemokine expression represent augmented capacity or reflect increased stimulation within the same limits of SPF conditions. Plasma cytokine/chemokine profiles are helpful for understanding systemic characteristics of the immune system, but they do not necessarily correspond to the cytokine/chemokine profiles of bone marrow or sites of infection. Deeper analyses will have to be performed to investigate how NME alters the profiles of specific microenvironments.
The “hygiene hypothesis” and “neonatal window of opportunity” posit that a perinatal period exists during which microbial exposures have the greatest impact on immune development (50–52). Increasingly clean human environments are thought to limit immune activation during childhood preventing appropriate immune development which leads to future immunopathology in the form of asthma, allergy, and/or autoimmunity (50–53). Our data clearly demonstrate a potent effect of diverse microbial experience on early life immune development, but the current research does not address the impact on future immune health or immunopathology. While previous studies investigated the effects of cohousing adult laboratory mice with pet store mice, we do not directly compare the two models here (25, 26, 54). It is possible the unique function of the early life immune system could establish a very different baseline in response to diverse microbial experience that cannot be achieved by microbial experience later in life. Multiple studies support the idea that early life exposures can have long term impacts on immunity, some of which cannot be replicated by similar exposures later in life (4–9, 55, 56). However, the period of optimum effect in the context of diverse microbial exposure and normal immune development has yet to be defined. The NME model could provide a system in which the timing of diverse microbial exposure can be manipulated to determine if a window of optimum microbial experience exists during development.
Supplementary Material
Key Points:
Natural microbial exposure expands immune cell progenitors in neonatal mice.
Natural microbial exposure broadly expands mature immune cells in young mice.
Natural microbial exposure enhances early life host defense.
ACKNOWLEDGEMENTS
We would like to extend our gratitude to the University of Minnesota Flow Cytometry Core, Research Animal Resources (RAR), Cytokine Reference Lab, and the Center for Immunology for their support. Special thanks to Dr. Bryce Binstadt for critical review and suggestions regarding the manuscript.
This study was generously supported by awards from the University of Minnesota Department of Pediatrics grants: The Masonic Cross-Departmental Grant in Children’s Health Research (to N.J.S and S.E.H) and The Department of Pediatrics “R” Award (to N.J.S), by the University of Minnesota Undergraduate Research Opportunities Program (UROP) (to T.S), by NIH Grants GM140881 (T.S.G.), NIH R01AI155468 (S.E.H), and a Department of Veterans Affairs Merit Review Award I01BX001324 (T.S.G.). T.S.G. is the recipient of a Research Career Scientist award (IK6BX006192) from the Department of Veterans Affairs.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, Umesaki Y, Mathis D, Benoist C, Relman DA, and Kasper DL. 2012. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149: 1578–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith K, McCoy KD, and Macpherson AJ. 2007. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol 19: 59–69. [DOI] [PubMed] [Google Scholar]
- 3.Levy M, Thaiss CA, and Elinav E. 2016. Metabolites: messengers between the microbiota and the immune system. Genes & Development 30: 1589–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabilas C, Iu DS, Daly CWP, Yee Mon KJ, Reynaldi A, Wesnak SP, Grenier JK, Davenport MP, Smith NL, Grimson A, and Rudd BD. 2022. Early microbial exposure shapes adult immunity by altering CD8+ T cell development. Proc Natl Acad Sci U S A 119: e2212548119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, and Blumberg RS. 2012. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336: 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomez de Agüero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, Steinert A, Heikenwalder M, Hapfelmeier S, Sauer U, McCoy KD, and Macpherson AJ. 2016. The maternal microbiota drives early postnatal innate immune development. Science 351: 1296–1302. [DOI] [PubMed] [Google Scholar]
- 7.Lim AI, McFadden T, Link VM, Han SJ, Karlsson RM, Stacy A, Farley TK, Lima-Junior DS, Harrison OJ, Desai JV, Lionakis MS, Shih HY, Cameron HA, and Belkaid Y. 2021. Prenatal maternal infection promotes tissue-specific immunity and inflammation in offspring. Science 373. [DOI] [PubMed] [Google Scholar]
- 8.Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, Hickman HD, McCulloch JA, Badger JH, Ajami NJ, Trinchieri G, Pardo-Manuel de Villena F, Yewdell JW, and Rehermann B. 2017. Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell 171: 1015–1028.e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosshart SP, Herz J, Vassallo BG, Hunter A, Wall MK, Badger JH, McCulloch JA, Anastasakis DG, Sarshad AA, Leonardi I, Collins N, Blatter JA, Han SJ, Tamoutounour S, Potapova S, Foster St Claire MB, Yuan W, Sen SK, Dreier MS, Hild B, Hafner M, Wang D, Iliev ID, Belkaid Y, Trinchieri G, and Rehermann B. 2019. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inaba K, Swiggard WJ, Steinman RM, Romani N, Schuler G, and Brinster C. 2009. Isolation of dendritic cells. Curr. Protoc. Immunol Chapter 3: Unit 3 7. [DOI] [PubMed] [Google Scholar]
- 11.Steinman RM, Kaplan G, Witmer MD, and Cohn ZA. 1979. Identification of a novel cell type in peripheral lymphoid organs of mice. V. Purification of spleen dendritic cells, new surface markers, and maintenance in vitro. J Exp Med 149: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kidd P 2003. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev 8: 223–246. [PubMed] [Google Scholar]
- 13.Chaouat G 2007. The Th1/Th2 paradigm: still important in pregnancy? Semin Immunopathol 29: 95–113. [DOI] [PubMed] [Google Scholar]
- 14.Mysore V, Tahir S, Furuhashi K, Arora J, Rosetti F, Cullere X, Yazbeck P, Sekulic M, Lemieux ME, Raychaudhuri S, Horwitz BH, and Mayadas TN. 2022. Monocytes transition to macrophages within the inflamed vasculature via monocyte CCR2 and endothelial TNFR2. J Exp Med 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bain CC, and Schridde A. 2018. Origin, Differentiation, and Function of Intestinal Macrophages. Front Immunol 9: 2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, Guilliams M, Malissen B, Agace WW, and Mowat AM. 2013. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol 6: 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Semmes EC, Chen JL, Goswami R, Burt TD, Permar SR, and Fouda GG. 2020. Understanding Early-Life Adaptive Immunity to Guide Interventions for Pediatric Health. Front Immunol 11: 595297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen SCA, and Boyd SD. 2019. New technologies and applications in infant B cell immunology. Current Opinion in Immunology 57: 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng H, Zheng Z, and Cheng T. 2020. New paradigms on hematopoietic stem cell differentiation. Protein & Cell 11: 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sack U, Burkhardt U, Borte M, Schädlich H, Berg K, and Emmrich F. 1998. Age-dependent levels of select immunological mediators in sera of healthy children. Clin Diagn Lab Immunol 5: 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Decker ML, Grobusch MP, and Ritz N. 2017. Influence of Age and Other Factors on Cytokine Expression Profiles in Healthy Children-A Systematic Review. Front Pediatr 5: 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Georgountzou A, and Papadopoulos NG. 2017. Postnatal Innate Immune Development: From Birth to Adulthood. Front Immunol 8: 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diehl S, and Rincón M. 2002. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol 39: 531–536. [DOI] [PubMed] [Google Scholar]
- 24.Angelone DF, Wessels MR, Coughlin M, Suter EE, Valentini P, Kalish LA, and Levy O. 2006. Innate Immunity of the Human Newborn Is Polarized Toward a High Ratio of IL-6/TNF-α Production In Vitro and In Vivo. Pediatric Research 60: 205–209. [DOI] [PubMed] [Google Scholar]
- 25.Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, Sekaly RP, Jenkins MK, Vezys V, Haining WN, Jameson SC, and Masopust D. 2016. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532: 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huggins MA, Sjaastad FV, Pierson M, Kucaba TA, Swanson W, Staley C, Weingarden AR, Jensen IJ, Danahy DB, Badovinac VP, Jameson SC, Vezys V, Masopust D, Khoruts A, Griffith TS, and Hamilton SE. 2019. Microbial Exposure Enhances Immunity to Pathogens Recognized by TLR2 but Increases Susceptibility to Cytokine Storm through TLR4 Sensitization. Cell Rep 28: 1729–1743.e1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang N, Strugnell RA, Wijburg OL, and Brodnicki TC. 2013. Systemic infection of Mice with Listeria monocytogenes to characterize host immune responses. Methods Mol Biol 1031: 125–144. [DOI] [PubMed] [Google Scholar]
- 28.King KY, and Goodell MA. 2011. Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nature Reviews Immunology 11: 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuettpelz LG, and Link DC. 2013. Regulation of hematopoietic stem cell activity by inflammation. Front Immunol 4: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mirantes C, Passegué E, and Pietras EM. 2014. Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp Cell Res 329: 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pietras EM 2017. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood 130: 1693–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takizawa H, Boettcher S, and Manz MG. 2012. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood 119: 2991–3002. [DOI] [PubMed] [Google Scholar]
- 33.López DA, Apostol AC, Lebish EJ, Valencia CH, Romero-Mulero MC, Pavlovich PV, Hernandez GE, Forsberg EC, Cabezas-Wallscheid N, and Beaudin AE. 2022. Prenatal inflammation perturbs murine fetal hematopoietic development and causes persistent changes to postnatal immunity. Cell Reports 41: 111677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsafaras GP, Ntontsi P, and Xanthou G. 2020. Advantages and Limitations of the Neonatal Immune System. Front Pediatr 8: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simon AK, Hollander GA, and McMichael A. 2015. Evolution of the immune system in humans from infancy to old age. Proc Biol Sci 282: 20143085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dowling DJ, and Levy O. 2014. Ontogeny of early life immunity. Trends Immunol 35: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saso A, and Kampmann B. 2017. Vaccine responses in newborns. Semin Immunopathol 39: 627–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marchant A, Goetghebuer T, Ota MO, Wolfe I, Ceesay SJ, De Groote D, Corrah T, Bennett S, Wheeler J, Huygen K, Aaby P, McAdam KP, and Newport MJ. 1999. Newborns develop a Th1-type immune response to Mycobacterium bovis bacillus Calmette-Guérin vaccination. J Immunol 163: 2249–2255. [PubMed] [Google Scholar]
- 39.Ramos L, Lunney JK, and Gonzalez-Juarrero M. 2020. Neonatal and infant immunity for tuberculosis vaccine development: importance of age-matched animal models. Disease Models & Mechanisms 13: dmm045740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, and Levy O. 2017. Protecting the Newborn and Young Infant from Infectious Diseases: Lessons from Immune Ontogeny. Immunity 46: 350–363. [DOI] [PubMed] [Google Scholar]
- 41.Gutierrez MJ, Nino G, Hong X, and Wang X. 2020. Epigenetic Dynamics of the Infant Immune System Reveals a Tumor Necrosis Factor Superfamily Signature in Early Human Life. Epigenomes 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davidesko S, Wainstock T, Sheiner E, and Pariente G. 2020. Long-Term Infectious Morbidity of Premature Infants: Is There a Critical Threshold? Journal of Clinical Medicine 9: 3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singer JR, Blosser EG, Zindl CL, Silberger DJ, Conlan S, Laufer VA, DiToro D, Deming C, Kumar R, Morrow CD, Segre JA, Gray MJ, Randolph DA, and Weaver CT. 2019. Preventing dysbiosis of the neonatal mouse intestinal microbiome protects against late-onset sepsis. Nat Med 25: 1772–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torow N, and Hornef MW. 2017. The Neonatal Window of Opportunity: Setting the Stage for Life-Long Host-Microbial Interaction and Immune Homeostasis. The Journal of Immunology 198: 557–563. [DOI] [PubMed] [Google Scholar]
- 45.Mold JE, and McCune JM. 2012. Immunological tolerance during fetal development: from mouse to man. Adv Immunol 115: 73–111. [DOI] [PubMed] [Google Scholar]
- 46.Adkins B, Leclerc C, and Marshall-Clarke S. 2004. Neonatal adaptive immunity comes of age. Nat Rev Immunol 4: 553–564. [DOI] [PubMed] [Google Scholar]
- 47.Kumar R, Fossati V, Israel M, and Snoeck HW. 2008. Lin-Sca1+kit- bone marrow cells contain early lymphoid-committed precursors that are distinct from common lymphoid progenitors. J Immunol 181: 7507–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pietras EM, Lakshminarasimhan R, Techner J-M, Fong S, Flach J, Binnewies M, and Passegué E. 2014. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. Journal of Experimental Medicine 211: 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buza-Vidas N, Woll P, Hultquist A, Duarte S, Lutteropp M, Bouriez-Jones T, Ferry H, Luc S, and Jacobsen SE. 2011. FLT3 expression initiates in fully multipotent mouse hematopoietic progenitor cells. Blood 118: 1544–1548. [DOI] [PubMed] [Google Scholar]
- 50.Apostol AC, Jensen KDC, and Beaudin AE. 2020. Training the Fetal Immune System Through Maternal Inflammation-A Layered Hygiene Hypothesis. Front Immunol 11: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hornef MW, and Torow N. 2020. ‘Layered immunity’ and the ‘neonatal window of opportunity’ – timed succession of non‐redundant phases to establish mucosal host–microbial homeostasis after birth. Immunology 159: 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Renz H, Adkins BD, Bartfeld S, Blumberg RS, Farber DL, Garssen J, Ghazal P, Hackam DJ, Marsland BJ, Mccoy KD, Penders J, Prinz I, Verhasselt V, Von Mutius E, Weiser JN, Wesemann DR, and Hornef MW. 2018. The neonatal window of opportunity—early priming for life. Journal of Allergy and Clinical Immunology 141: 1212–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okada H, Kuhn C, Feillet H, and Bach J-F. 2010. The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clinical and Experimental Immunology 160: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamilton SE, Badovinac VP, Beura LK, Pierson M, Jameson SC, Masopust D, and Griffith TS. 2020. New Insights into the Immune System Using Dirty Mice. J Immunol 205: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henrick BM, Rodriguez L, Lakshmikanth T, Pou C, Henckel E, Arzoomand A, Olin A, Wang J, Mikes J, Tan Z, Chen Y, Ehrlich AM, Bernhardsson AK, Mugabo CH, Ambrosiani Y, Gustafsson A, Chew S, Brown HK, Prambs J, Bohlin K, Mitchell RD, Underwood MA, Smilowitz JT, German JB, Frese SA, and Brodin P. 2021. Bifidobacteria-mediated immune system imprinting early in life. Cell 184: 3884–3898.e3811. [DOI] [PubMed] [Google Scholar]
- 56.Arnesen H, Hitch TCA, Steppeler C, Müller MHB, Knutsen LE, Gunnes G, Angell IL, Ormaasen I, Rudi K, Paulsen JE, Clavel T, Carlsen H, and Boysen P. 2021. Naturalizing laboratory mice by housing in a farmyard-type habitat confers protection against colorectal carcinogenesis. Gut Microbes 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.