

Abstract
Alzheimer’s disease (AD) is one of the most common neurodegenerative diseases worldwide. The occult nature of the onset and the uncertainty of the etiology largely impede the development of therapeutic strategies for AD. Previous studies revealed that the disorder of energy metabolism in the brains of AD patients appears far earlier than the typical pathological features of AD, suggesting a tight association between energy crisis and the onset of AD. Energy crisis in the brain is known to be induced by the reductions in glucose uptake and utilization, which may be ascribed to the diminished expressions of cerebral glucose transporters (GLUTs), insulin resistance, mitochondrial dysfunctions, and lactate dysmetabolism. Notably, the energy sensors such as peroxisome proliferators-activated receptor (PPAR), transcription factor EB (TFEB), and AMP-activated protein kinase (AMPK) were shown to be the critical regulators of autophagy, which play important roles in regulating beta-amyloid (Aβ) metabolism, tau phosphorylation, neuroinflammation, iron dynamics, as well as ferroptosis. In this study, we summarized the current knowledge on the molecular mechanisms involved in the energy dysmetabolism of AD and discussed the interplays existing between energy crisis, autophagy, and ferroptosis. In addition, we highlighted the potential network in which autophagy may serve as a bridge between energy crisis and ferroptosis in the progression of AD. A deeper understanding of the relationship between energy dysmetabolism and AD may provide new insight into developing strategies for treating AD; meanwhile, the energy crisis in the progression of AD should gain more attention.
Keywords: Alzheimer’s disease, energy crisis, autophagy, ferroptosis, iron metabolism, beta-amyloid, tau protein
1. INTRODUCTION
Alzheimer’s disease (AD), characterized by progressive cognitive impairments and memory loss, is one of the most common dementias worldwide. Recent epidemic studies revealed that the number of Americans aged 65 and older with AD may grow to 13.8 million by the middle century [1]. Unfortunately, there is still a lack of effective strategies for the prevention or treatment of AD. Even worse, since the etiology of AD is also currently unknown, ectopic metabolism of beta-amyloid (Aβ), hyperphosphorylation of tau protein, chronic inflammation, oxidative damage as well as metal ion dysregulation are recognized as the potential risk factors of AD [2]. Among that, the Aβ cascade hypothesis is widely accepted as many AD patients exist the mutations of amyloid precursor protein (APP), presenilin 1 (PS1), presenilin 2 (PS2), and the epsilon 4 allele of apolipoprotein E (APOE4), which are highly associated with the metabolism of Aβ [3]. Interestingly, mounting clinic studies exerted that glucose hypometabolism is an invariant pathological perspective that precedes clinical symptoms and pathological changes even for decades in AD patients, suggesting that energy crisis may be an etiologic factor of AD [4, 5]. It is known that glucose hypometabolism paves the way to energy crisis, which is known to give rise to autophagy in normal and pathological conditions [6, 7]. In this paper, we reviewed the studies concerning energy metabolism in AD conditions, and further focused on the role of autophagy and ferroptosis in the progression of AD, aiming to unveil the potential networks between energy crisis, autophagy and ferroptosis in AD progression.
2. THE HALLMARKS OF ALZHEIMER’S DISEASE
One outstanding pathological hallmark of AD is the formation of senile plaques (SPs), which are predominately constituted by Aβ [8]. The production of Aβ depends on the successive cleavage of APP by β-secretase and γ-secretase [9]. Despite, the metabolism of Aβ is also modulated by the ability of Aβ clearance in the brain, such as neprilysin (NEP) and insulin-degrading enzyme (IDE), both of which possess the capacity to degrade Aβ [10]. Therefore, excessive generation of Aβ and impaired clearance of Aβ are the main causes of SPs formation in the brain [11]. Apart from the deposition of Aβ, the neurofibrillary tangles (NFTs) caused by the hyperphosphorylation of tau protein is another notable characteristic of AD. Tau protein is commonly found in the neurons of the central nervous system and has the function to stabilize microtubules and assist the transportation of cargo along axons [12]. Phosphorylation of tau controls its binding to microtubules and triggers the assembly of microtubules, but the hyperphosphorylation of tau results in the opposite effects [13]. Phosphorylation of tau is regulated by several enzymes such as cyclin-dependent kinase5 (CDK5), glycogen synthase kinase3α/β(GSK3α/β), and phosphatase 2A(PP2A), and these enzymes abnormalities lead to hyperphosphorylation of tau, thereby contributing to the formation of NFTs [14]. The abnormal accumulations of Aβ and NFTs in the brain trigger a series of events that may cause the occurrence of AD [15]. Furthermore, mounting evidence suggested that neuroinflammation is a contributor to the progression of AD. The inflammatory factors such as tumor necrosis factor α (TNFα), interleukin 1β (IL-1β), and interleukin 6 (IL-6) are increased in the AD brain, some of which are highly associated with Aβ production, tau phosphorylation, and blood-brain barrier (BBB) disruption. Nevertheless, it is still uncertain whether neuroinflammation is a cause or a consequence of AD. In addition, dysregulations of metal ions (such as iron, copper, and zinc) play attractive roles in the progress of AD. Especially, iron is accumulated in SPs and neurons in the AD brain, which in turn implicates the regulation of Aβ production, tau phosphorylation, and neuronal loss (Fig. 1) [16-18].
Fig. (1).
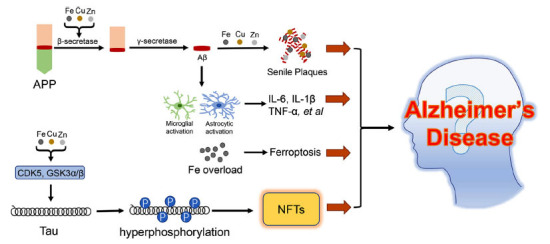
Singling pathways involved in the progression of AD. APP is successively cleaved by the β-secretase and γ-secretase to produce the Aβ, meanwhile, the metal ions (Fe, Cu, and Zn) could enhance the activity of β-secretase. With the help of metal ions, the Aβ is deposited to form senile plaques. On the other hand, the Aβ could induce microglial activation and astrocytic activation to produce inflammatory factors. Increased contents of metal ions lead to CDK5 and GSK3α/β activation, thus promoting tau hyperphosphorylation and NFTs formation. Iron overload is also observed in the cerebral neurons, which results in ferroptosis. All the aforementioned pathways participate in the progression of AD.
It is noteworthy that energy dysmetabolism is also a feature of the AD brain, such as glucose hypometabolism is prevalent in the brains of AD patients and AD animals, and it is prior to the appearance of the pathologic symptoms in the progress of AD [4, 5, 19, 20], suggesting that glucose hypometabolism may be a potential etiologic factor of AD. Unfortunately, there was pretty limited research conducted to uncover the roles of energy crisis in the progress of AD. We, therefore, summarized the studies regarding glucose hypometabolism in AD and hoped to find a better way to overcome this disease.
3. THE ENERGY DYSMETABOLISM IN ALZHEIMER’S DISEASE BRAIN
3.1. The Energy Metabolism in the Brain
The brain is a high-energy requirement organ that represents only 2% of the total body mass but consumes about 25% of the glucose from the body’s absorption [21]. Glucose is the main source of energy supplementation in the brain. Glucose crosses the BBB and enters the cells dependent on the glucose transporters (GLUTs) [22]. The brain mainly expresses glucose transporters GLUT1, GLUT2, GLUT3, GLUT4, GLUT5, GLUT6, and GLUT8, of which, GLUT1 locates in the blood vessel endothelium and astrocytic end feet, which is in charge of glucose entry from the peripheral circulation, whereas the GLUT3 and GLUT4 are abundantly expressed in the neuron [23], indicating the important roles of glucose in the regulation of neuronal activities.
The glucose in the cytoplasm is phosphorylated by hexokinase (HK) to produce glucose-6-phosphate, which may submit to three different metabolism pathways including glycolysis, pentose phosphate pathway (PPP), and glycogenesis (only in astrocytes) [21]. Glucose and lactic acid are involved in brain development and memory formation and are coupled with neuronal metabolism, meanwhile, astrocytes may play a regulatory role [24]. In neurons, glucose uptake is largely dependent on the GLUT3 and GLUT4, and subsequently, the glucose suffers glycolysis and tricarboxylic acid cycle (TAC cycle) to produce adenosine triphosphate(ATP) to maintain neuronal functions [23]. Glucose also has an important role in neuronal scavenging of ROS through the PPP to produce nicotinamide-adenine dinucleotide phosphate (NAPDH), which is required for the regeneration of reduced glutathione [25]. It is worth noting that the storage of glycogen in neurons is inhibited in normal conditions [26], however, several studies revealed that the storage of glycogen in neurons exists only in severe neurological diseases [27]. In addition, due to the high expression of lactate dehydrogenase 1 (LDH1) in neurons, the lactate can be easily converted to pyruvate to act as an energy substrate in neurons [28]. Lactic acid can also activate neuronal glutamate signals and play a protective effect on long-term memory [29-31]. Neurons have a stronger preference for the uptake of lactate than glucose [32], hence, the metabolic activity of lactate in the brain is critical for neuronal activity.
Astrocytes are identified to account for about 5%-15% of the brain’s energy expenditure, while their energy consumption becomes higher in non-resting states [21, 33, 34]. Astrocytes absorb glucose mainly through GLUT1 and convert it into lactic acid which is oxidized in mitochondria [28], and this action is related with the phosphorylation of pyruvate dehydrogenase (PH.D.) and the activity of cytochrome oxidase in astrocytes [35]. A previous study showed that repression of mitochondrial respiration in astrocytes (only aerobic glycolysis in astrocytes) did not affect astrocyte survival and memory performance in mice [36], suggesting that glycolysis is the main energy source of astrocytes. Another study from single-cell metabolic state analysis showed that lactic acid maintains mitochondrial length in radial glial progenitors [37], but it is not clear whether the effect is the same in mature individuals. Importantly, the neuron-absorbed lactate largely originates from astrocytic lactate. Lactate is secreted through monocarboxylate transporter 1/4 (MCT1/4) in astrocytes, and subsequently taken up by MCT2 in neurons [38] where it is converted into pyruvate as a direct source of energy [38]. Different from astrocytes, microglial energy metabolism depends primarily on oxidative phosphorylation [39], but converts to glycolysis upon activation [40]. This metabolic reprogramming is thought to be involved in the inflammatory response of microglia during the evolution of neurological diseases [41, 42]. Microglia possess the ability to produce and uptake lactate, and could rapidly change their metabolic pattern to select lactate as an energy donor in the presence of insufficient glucose supply [43]. This metabolic shift may accommodate the high ATP demand of microglia.
The brain owns the ability to sense and adapt to alternations in environmental energy and hormone modulation to survive periods of nutritional deficiency [44]. The hypothalamus and brainstem are the major brain regions responsible for energy regulation [45]. Due to the lack of a typical blood-brain barrier structure in the median eminence (ME) of the arcuate nucleus, it is considered to be a portal for sensing changes in hormones and nutrients in the circulatory system [45], by which, the brain can trigger a complementary effect to escape the energy crisis. Increasing evidence indicates that ketone bodies are substitution fuel for the brain during occasional periods of energy starvation caused by pathologic or physiologic factors [46]. Ketone bodies mainly contain β-hydroxybutyrate and acetoacetate, which are mainly produced by acetyl Coenzyme A(CoA) in the liver through the β-oxidation pathway [47]. However, the concentration of ketone bodies is low since the formation of ketone bodies is inhibited by insulin in the normal physiological state [48]. Several studies have suggested that the appropriate concentration of ketone bodies have protective effects on the neuronal system, the underlying mechanism may be due to the reduced metabolic pressure caused by alternative fuels [49], whereas the high concentration of ketone bodies have significant neurotoxicity via induction of plasma membrane depolarization [50]. A previous study provided evidence that the absence of the ketone transporter solute carrier family 5 member 8 (SLC5A8) contributed to the shortage of brain energy in mice, even though SLC5A8 was not expressed in the brain, suggesting that chronic ketone deficiency may affect the brain’s energy supply [51].
In addition to ketone bodies, insulin can also regulate brain development and energy metabolism. Insulin is known to be secreted by pancreatic beta-cell, which is critical to the systemic homeostasis of blood glucose. Insulin exerts its actions by binding to insulin receptors (IRs) present on most cells. Interestingly, IRs are widely expressed in all the cell types in the brain, especially in the cortex, hippocampus, striatum, and olfactory bulb [23]. GLUT4 is an insulin-sensitive glucose transporter in neurons, challenge with insulin induces GLUT4 translocation to the plasma membrane via an AKT-dependent manner [52]. Moreover, insulin also modulates glucose entry into the brain via promoting astrocytic GLUT1 translocation to the cell membrane by cooperation with insulin-like growth factor 1 (IGF1) [53]. It is notable to know that the insulin level in plasma is much higher than that in cerebrospinal fluid (CSF) [54], indicating that most of the insulin in the brain may stem from circulating pancreatic insulin.
3.2. The Energy Crisis in Alzheimer’s Disease Brain
High energy is required to maintain synaptic activity, and neurons must maintain energy homeostasis to induce the formation of long-term memory [55]. Therefore, the brain is easy to be affected by low energy metabolism. Energy metabolism is different in different brain regions, while the areas of the brain with more connectivity certainly have higher energy requirements [56, 57]. Animal experiments showed that learning and memory increase glucose consumption in the hippocampus [58]. Minoshima S, et al. examined glucose metabolism levels in different brain regions of AD patients by autopsy and found decreased glucose metabolism in the parietotemporal association, posterior cingulate, and frontal association cortices [59]. Nowadays, several techniques (such as positron emission tomography (PET), functional magnetic resonance imaging (fMRI), and magnetic resonance spectroscopy (MRS) have been employed to measure the energy metabolism of the brain [60]. The fMRI aims to evaluate the brain activity and functional connectivity of different brain regions by monitoring blood oxygen level dependence (BOLD) [61]. An animal experiment showed diminished connectivity of the mesial temporal with other brain regions in the brain of aged rats, which may be associated with age-related cognitive impairment [62]. In clinical research, Wu YQ, et al. found that the bilateral inferior temporal gyrus signal was significantly lower in mild cognitive impairment (MCI) patients than that in healthy subjects [63], indicating that the decreased activity of the bilateral inferior temporal gyrus may be involved in cognitive impairment. The relationship between BOLD changes and AD risk genes (especially APOEε4) has been extensively studied. Subjects with the APOEε4 allele showed reduced functional connectivity in the hippocampus and middle temporal cortex [64]. Furthermore, female subjects with the APOEε4 allele also showed an age-dependent reduction in functional connectivity between the hippocampus and precuneus/posterior cingulate cortex [65]. Therefore, the fMRI functional connectivity can be used as an indicator for the early clinical diagnosis of AD and the judgment of AD progression.
MRS is a non-invasive, functional imaging technique that can be used to characterize changes in metabolite concentrations [66]. The function of MRS to detect metabolites depends on the different imaging modalities (MRS-active nuclei), such as 1H MRS quantifies N-acetylaspartate (NAA) and glutamate more efficiently, and 31P MRS is used to detect all phosphorylation products, especially for phosphatidyl inositol and ATP, while 13C MRS, for precise tracking of the metabolic fate of marked metabolites (for example, glucose and pyruvate) [67]. Using 1H MRS analysis, early AD patients showed low NAA/creatine (Cr) and late AD patients showed lower NAA/Cr and higher myo-Inositol/Cr in the posterior cingulate [68]. According to the 31P MRS analysis, part of the brain regions (retrosplenial cortex, hippocampi, and parieto-occipital) exhibited high energy phosphate metabolic activity in AD patients [69, 70]. The application of 13C MRS in AD patients is relatively limited at present [71], but 13C MRS has the function of being able to detect dynamic glucose transport, which will have a wider application in the future.
Glucose is converted to glucose-6-phosphate by HK, but deoxyglucose-6-phosphate cannot continue to be used as a substrate for glycolysis and therefore accumulates intracellularly [72]. Therefore, 18F-fluorodeoxyglucose (FDG) can be used with PET to monitor glucose metabolism in the brain [73]. FDG uptake in several brain regions decreases significantly with age, especially in the superior temporal pole [74]. Consistent with this result, Pezzoli S, et al. found that the decreased glucose metabolism in AD patients mainly occurred in the temporal lobe [75]. Other studies showed that decreased glucose metabolism also occurred in the posterior cingulate gyrus and precuneus in AD patients [76, 77]. Furthermore, according to the FDG uptake of PET studies in early AD brains, cognitively healthy older adult controls and MCI brains, the rates of glucose uptake were significantly reduced in partial regions of the AD brain, such as in frontal, parietal, temporal lobes, and cingulate gyrus, which were accompanied with brain volume reduction and cortical thinning [78]. In addition, by following up on FDG uptake in the brain of AD patients(range 2.0 - 4.0 years), Ossenkoppele R, et al. found reduced FDG uptake in the frontal, parietal, and lateral temporal lobes, which could effectively indicate the progress of AD [79]. The ventral arcuate nucleus of the hypothalamus locates at the medial edge of the third ventricle, contains neurons such as agouti-related peptide (AgRP) and pro-opiomelanocortin (POMC) neurons which are regulated by leptin, insulin and ghrelin to maintain the overall energy balance of the body [80]. Through PET/MR imaging and histopathological observation of the AD brains, it was found that the hypothalamus of AD patients had different degrees of atrophy, amyloid plaque formation, and NFTs [81]. Studies of PET using the FDG as a tracer also revealed a significant deficit in glucose metabolism in the hypothalamus in the AD subjects and MCI subjects as well as in “pre-symptomatic” transgenic mice overexpressing APP (Tg2576) [81, 82]. The enzyme creatine kinase (CK) reaction and kynurenine pathways are the keys to balancing brain energy metabolism. When local energy demand suddenly increases, CK reaction can quickly replenish ATP from energy buffer phosphocreatine (PCr) [83]. In the early degenerative regions of the brain in AD patients, PCr levels were elevated as assessed using phosphorus-31 MR spectroscopy (31P–MRS), further indicating changes in brain energy metabolism in the brains of AD patients [84]. However, whether changes in glucose uptake and utilization trigger the onset of AD or dysmetabolism of energy is due to the neurodegeneration in the AD brain, is still a matter of debate.
Different type of cells has different changes in energy metabolism in the pathological process of AD. The expression levels of GLUT3 and GLUT4 in neurons were significantly decreased [85, 86], but the MCT levels were increased in AD brains [87]. These alternations may be associated with the increased susceptibility of neurons to take up lactate during energy crisis. Moreover, the astrocytic GLUT1 was also decreased, along with the activation of astrocytes in the progress of AD [88], suggesting the glucose hypometabolism in AD brains since GLUT1 acts as the “gatekeeper” for glucose entry into the brain. In addition, quantifications of FDG uptake by different cells in the brain of 5×FAD mice revealed a significant increase in microglial uptake compared to that in WT mice, while no significant differences were found in neurons and astrocytes [89]. This study further sequenced single cells of microglia and found that a subset of microglia with higher glucose transporters with defective glycolysis and oxidative phosphorylation was increased during the pathological progression in 5×FAD mice [89], indicating that a decreased glucose utilization happened in microglia. Previous studies revealed that glycolysis in microglia could attenuate the phagocytic activity of Aβ and increase the secretion of TNFα [90, 91]. In addition, lactate challenge could also promote the secretions of IL-6 and IL-1β in primary microglia [92]. A recent study further uncovered that lactate was increased in the microglia from 5×FAD mice, and lactate-induced lactylation of histone 4 lysine 12 lactonization (H4K12la) is highly enriched at the promoter of glycolysis-related genes and thereby increasing glycolytic activity in microglia, which subsequently exacerbates microglial dysfunction and promotes neuroinflammation [93]. Considering that oxidative phosphorylation is the main metabolic approach in microglia [39], remodeling the energy program in microglia may be a promising strategy to alleviate the progress of AD.
3.3. The Inducing Factors of Energy Dysmetabolism in the AD Brain
Since energy metabolism disorder is an outstanding pathological feature of AD, Doorduijn AS, et al. conducted a meta-analysis on the intake of energy and protein in normal people and AD patients [94]. The study found no sufficient evidence that the intake of energy or protein in AD patients was lower than that in the control group [94], suggesting that the energy metabolism disorder in the AD brain is not caused by insufficient intake, but may be associated with the uptake or utilization of cerebral glucose.
3.3.1. Ectopic Expressions of Glucose Transporters
AD patients are often associated with decreased expressions of glucose transporters (mainly GLUT1 and GLUT3), which may be regulated by hypoxia-inducible factor 1α (HIF-1α). Further studies also suggested that the expression level of GLUT1 in brain-derived endothelial cells (BDCECs) of the AD brain was significantly reduced than that in the normal brain [95], indicating that the uptake of glucose was diminished in the AD brain. Interestingly, GLUT1 deficiency in endothelium promoted the BBB breakdown and reduced the blood flow; Furthermore, GLUT1 deficiency in endothelium promoted the neuropathology and cognitive impairments in an animal model of AD [96], suggesting that energy crisis may be a potential causative factor for AD. Apart from GLUT1 and GLUT3, the translocation of GLUT4 to the cell membrane was also impaired in the AD brain, despite the fact that the GLUT4 expression level was unchanged in the condition of AD [97, 98]. The mechanism of mislocation of GLUT4 remains largely unknown, which may be highly associated with the dysregulation of insulin signaling in AD brains since GLUT4 is an insulin-regulated transporter [99].
3.3.2. Insulin Resistance
Insulin resistance results in a significant decrease in the uptake and utilization of glucose, which subsequently impairs the energy metabolism system in the body. Insulin resistance is a characteristic of type-2 diabetes, whereas a similar phenomenon has appeared in the AD brain, such as decreased insulin signaling activity, and abnormal binding of insulin with insulin receptors [23]. Furthermore, there is evidence to support an epidemiological linkage between AD and type-2 diabetes as ectopic inflammatory responses, insulin resistance, and mitochondrial dysfunction are observed in these diseases [100]. Therefore, several scholars also refer to AD as type-3 diabetes [101]. The depletion of insulin in diabetic rats activates the protein kinase A (PKA) pathway and induces tau phosphorylation [102]. While it was once thought that insulin resistance could worsen tau damage by disrupting the balance of tau kinases and phosphatases [103]. Interestingly, the brains of the tau transgenic mice showed an increased response to insulin without the expected insulin resistance, suggesting a complex linkage between insulin resistance and tau phosphorylation [104]. Of note, our previous study revealed that leptin knockout significantly worsens the Aβ pathology and cognitive deficits in APP/PS1 mice [105], suggesting that the chronic diabetic state is a contributor to AD. Interestingly, several other studies also suggested that insulin resistance can be induced by Aβ toxicity [106, 107], however, the mechanism underlying remains largely unknown. The insulin receptor substrate (IRS) protein family plays a critical role in the process of insulin signal transduction [108]. Insulin binds to its receptor IRs to recruit IRS and phosphatidylinositol 3-kinase (PI3K) to the plasma membrane for the production of phosphatidylinositol-3,4,5-trisphosphate (PIP3) from phosphatidylinositol-4,5-bisphos-phate [PI(4,5)P2], which subsequently activates AKT [108]. Further study revealed that insulin signaling is modulated by liquid-liquid phase separation (LLPS) by driving the formation of intracellular IRS1 condensates [109]. Insulin stimulation could promote the PIP3 production and recruitment of AKT in the IRS1 condensates, subsequently promoting insulin signal transduction from the plasma membrane into the cytoplasm [109]. Therefore, the failure of the IRS/PI3K/ AKT signal pathway is recognized as the main contributor to insulin resistance. GSK3β and rapamycin complex (mTOR) are the predominant downstream targets of the IRS/PI3K/ AKT signal pathway [110, 111]. The inactivation of PI3K activates GSK3β [112], which is directly related to tau hyperphosphorylation and glucose metabolism. On the other hand, the failure of the IRS/PI3K/AKT signal pathway could inhibit mTOR, which plays vital roles in the regulation of cell growth [113], glucose metabolism [114], amino acid metabolism [115], as well as autophagy [116]. Notably, the IRS1/2 levels and IRs were both significantly reduced in the brains of AD patients [117], suggesting that the disturbance of insulin signaling is involved in the progress of AD. Indeed, the translocations of insulin-dependent neuronal glucose transporters (such as GLUT4 and GLUT8) from intracellular compartments to the cell membranes were significantly inhibited possibly via the disturbance of insulin signaling, which may subsequently lead to energy crisis in neurons [99, 118].
3.3.3. Mitochondrial Dysfunction
Mitochondrial dysfunction is one of the pathological features of AD. In the brain of AD patients, the main manifestations of mitochondrial damage are included but are not limited to the decreased ATP production, the reduction of oxidative phosphorylation, and the impairment of antioxidant function [119]. Through proteomic analysis of triple transgenic mice (3xTg) and WT mice, Yu H, et al. found significant differences in the expressions of mitochondrial proteins in the entorhinal cortex, which mainly include glucose-regulating proteins and TCA-related proteins [120]. In addition, the methylation level in the d-loop region of the mitochondrial gene in the blood of AD patients was lower than that in normal people, but the methylation level in the inner olfactory cortex was increased [121], which was consistent with the decreased expression of complex I/IV subunit gene of mitochondrial electron transport chain in the brain of AD patients [122-124]. Dihydrolipoamide dehydrogenase (DLD) is an important mitochondrial enzyme related to energy metabolism, and its mutation and decreased activity are believed to be related to the genetic and pathological processes of AD [125].5-methoxyindole-2-carboxylic acid (MICA), an inhibitor of DLD, stimulated the expression of human Aβ in C. elegans and reduced the aggregation of Aβ [126]. However, the inhibition of DLD activity in C. elegans expressing tau resulted in the phosphorylation of tau protein and the increase of glucose in vivo [127].
Energy metabolism disorders may also be associated with oxidative stress and the unfolded protein response (UPR), primarily as a result of oxidative damage to enzymes involved in glycolysis, TCA, and ATP synthesis. These enzymes are damaged by free radicals not only in AD but also in down's syndrome, amnestic mild cognitive impairment (aMCI), type 2 diabetes, and other conditions [128, 129]. Studies using oxidized nucleoside 8-hydroxy-20-deoxyguanosine (8-OHdG) assay found that the oxidative damage of mitochondrial DNA was increased in the brain of AD patients, which may, in turn, aggravate the vicious cycle between mitochondrial dysfunction and oxidative stress [130]. The accumulation of proteins that are unfolded or misfolded in the endoplasmic reticulum (ER) can lead to stress. To alleviate this, cells activate an intracellular signaling network, collectively known as the UPR, that coordinates the recovery of ER function [131]. Mitochondria and ER stress are closely related to apoptosis in AD pathology. When ER stress occurs, UPR can feedback and control the stress, thus improving the folding efficiency. When the stress cannot be solved, UPR promotes cell apoptosis. It was verified by experiments that the overexpression of APP/Aβ or hyperphosphorylated tau can induce ER stress, which is manifested by the activation of UPR and the imbalance of mitochondria [132]. It should be noted that, from the current point of view, the pathogenesis of AD is unclear and multi-factorial. Similarly, the above energy metabolism factors usually do not occur independently in AD, and there is no clear evidence to indicate the order.
3.3.4. Lactate Dysmetabolism
Lactate from the astrocyte-to-neuron lactate shuttle is the main energy substrate for neurons [133]. Lactic acid could protect against glutamate toxicity and regulate neuronal signaling [134] and increase cellular respiration [135]. Production of lactic acid in astrocytes is largely dependent on AMPK activity, knockout of AMPK in astrocytes, but not neurons, leads to thioredoxin-interacting protein (TXNIP) hyperstability, GLUT1 misregulation, lactate reduction, and neuronal loss [136]. Notably, abnormal AMPK activity was reported in the postmortem AD patients and AD mouse models [137], suggesting that lactate homeostasis is impaired in the brains of AD patients. A previous study revealed that bilateral intrahippocampal injections of Aβ25-35 significantly decreased the lactate content and reduced the MCT2 expression in the brains of rats [138]. Interestingly, the lactate level was significantly elevated in the CSF of AD patients [139]. A similar result was also obtained in another study, which found that the high lactate level in the CSF of AD patients was positively associated with reduced glucose consumption in the left medial prefrontal cortex (mPFC), left orbitofrontal cortex (OFC), and left parahippocampal gyrus (PHG) [140]. This circumstance may be related to the loss of the ability to absorb lactate in neurons or neurons becoming more dependent on lactic acid metabolism for energy supply in the condition of AD. On the other hand, chronic inflammation was reported to increase the supply of lactic acid to neurons but impair the lactic acid oxidation in neurons [141], suggesting that neuroinflammation may also contribute to lactate dysmetabolism in AD pathology.
4. AUTOPHAGY MAY BE A BRIDGE BETWEEN AD AND ENERGY CRISIS
4.1. The Relationship between Autophagy and Energy Crisis
The causes of energy metabolism disorder in the brain are various, and the regulation of autophagy is also affected by various factors. In vivo, autophagy can be induced by a variety of cytokines, enzymes, and hormones, such as insulin [110], nitric oxide (NO) [142], interleukin, leptin, and angiogenin [143], and other conditions such as sleep (rapid eye movement sleep) [144], starvation [145] and nutritional deficiency. In the state of starvation, autophagy activity is enhanced which promotes the activation of peroxisome proliferators-activated receptor α (PPARα), regulates β-oxidation, and promotes the formation of ketones which are compensatory fuel for the organisms [145]. Furthermore, amino acid starvation could activate autophagy through activating amino acid starvation response (AAR), mechanisms that may involve activation of the mechanistic target of rapamycin complex 1 (mTORC1) and PKC [146].
Energy affects autophagic activity mainly through transcriptional and post-transcriptional regulations. At the transcriptional level, the energy-sensitive transcription factors PPAR and transcription factor EB (TFEB) induce autophagy by up-regulating the expressions of related genes, such as Beclin 1, LC3, autophagy-related protein 7 (Atg7), autophagy-related protein 9B (Atg9B), and UV Radiation Resistance-Associated Gene (UVRAG), WD repeat domain phosphoinositide-interacting protein 1 (WIPI1), etc. [147]. At the post-transcriptional level, AMPK signaling plays an important role in nutrition deprivation-mediated autophagy [148]. When the cells are suffering from the energy crisis, the AMPK is phosphorylated and activates and Unc-51-like autophagy-activating kinase 1 (ULK1/Atg1) at S317, and the activated ULK1 subsequently promotes the activation of Beclin 1 and then promotes LC3-I lipidation into LC3-II to promote the mature of autophagosomes (Fig. 2) [149]. It should be noted that the ULK1 is a vital regulator for autophagy, and several scholars believe that studying the post-translational modification of ULK1 will help to understand how energy and nutrient factors regulate autophagy [150].
Fig. (2).
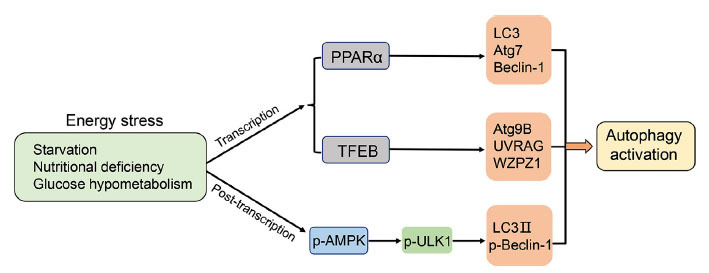
Energy crisis promotes autophagy activation. Energy crisis could be induced by starvation, nutritional deficiency, and glucose hypometabolism, which enhance the activity of PPARα, thus transcriptionally upregulates the expressions of LC3, Atg7 and Beclin-1 to induce autophagy; Energy crisis also activates TFEB, which subsequently transcriptionally increases the expressions of Atg9B, UVRAG, and WAPZ1 to induce autophagy. In addition, energy crisis enhances the phosphorylation of ULK1 via activation of AMPK, thus promoting the LC3 lipidation and Beclin-1 phosphorylation, resulting in autophagy activation.
4.2. Autophagy is Impaired in Alzheimer’s Disease Brain
The prevailing view is that autophagy is a protective mechanism in cells, and its imbalance is closely related to the occurrence of neurodegenerative diseases [151]. Autophagy is mainly divided into three types: macro-autophagy, micro-autophagy, and chaperone-mediated autophagy, meanwhile, macro-and chaperone-mediated autophagy have gained considerable attention in the research of AD and other neurodegenerative diseases [152]. In terms of macro-autophagy, it refers to the transport of “cargo” (such as damaged mitochondria and misfolding proteins) to lysosomes for degradation [153]. The formation of autophagic complexes is a key step in the initiation of autophagy. The mechanisms in the formations of autophagic complexes are still under investigation and may be divided into several units by their functions: Atg9 vesicles, the Atg2-Atg18/WIPI complex, the Atg8/LC3 conjugation system, the class III phosphatidyl inositol 3-kinase(PI3K) complex, the Atg12 conjugation system, and the Atg1/ULK complex [154]. Macro-autophagy is mainly characterized by autophagosomes formed by extending a double-layer membrane structure to surround substances that need to be swallowed and degraded, which can be induced by cell signals including energy crisis [155]. Recently, researchers employed single population laser capture microdissection coupled with custom-designed microarray profiling to study postmortem brain tissue samples of precuneus cathepsin D positive (a marker susceptible to NFTs) and found that the autophagosome-related genes were misexpressed in MCI and AD samples [156]. Interestingly, the AD-related gene PS1 plays essential role in the acidification of lysosomes and activation of cathepsins, and PS1 mutations produce a significant autophagic deficiency in the fibroblasts from AD patients [157]. The previous study also uncovered that high brain cholesterol promoted the formation of autophagosomes while blocking its fusion with the endosomal-lysosomal system, these actions, therefore, reduced the Aβ degradation but promoted Aβ secreted out of the cells [158].
Different from macro-autophagy, the chaperone-mediated autophagy does not depend on the formation of autophagosomes but relies on the chaperone heat shock cognate 70 (HSC70) targeting proteins containing KFERQ-like motifs, and subsequently enters into the lysosomal lumen with the help of lysosomal complexes containing lysosome-associated membrane protein 2 (LAMP2A) and heat shock protein 90 (HSP90) [159]. It is noteworthy that tau and Aβ both contain KFERQ-like motifs, raising the possibility that tau and Aβ can be targeted by HSC70 and enter lysosomes for degradation [160, 161]. A previous study revealed that the mRNA of HSC70 was significantly decreased in the postmortem brain of AD patients compared to the controls, indicating a potential role of HSC70 in the progression of AD [162]. Surely, inhibition of HSC70 by its inhibitor VER-155008 effectively reduced the Aβ burden and paired helical filament tau accumulation in 5XFAD mice [163]. However, HSC70 was shown to tightly interact with tau fibrils and avoid their aggregation by capping the nucleating ends [164]. Moreover, the Hyperphosphorylated tau also interacts with the HSC70-carboxyl terminus of the HSC70-interacting protein (CHIP) complex which promoted the ubiquitination of tau, therefore, ubiquitinated tau was removed and subsequently inhibited the tau-mediated cell death [165].In addition, the scaffolding protein Ran-binding protein 9 (RanBP9) was increased in the brains of AD patients and AD mouse models, which could enhance the abilities of HSP90-HSC70 binding to ATP and increase their ATPase activities. However, genetic reduction of RanBP9 effectively improved the synaptic plasticity and tau pathology in P301S mice [166]. These studies suggest that the functions of CMA may be context-dependent, and the regulation of CMA should be taken into consideration in the treatment of AD.
Mitophagy is a specific autophagy phenomenon that selectively degrades and recycles damaged mitochondria [167]. Mitophagy-mediated removal of damaged mitochondria by PTEN-induced putative kinase 1 (PINK1) is a vital step in the maintenance of mitochondrial homeostasis. PINK1 is recruited to the damaged mitochondria because of decreased mitochondrial membrane potential and depolarization, and subsequently, Parkin, an E3 ubiquitin ligase, is also recruited to the mitochondria and mediates the formation of two distinct poly-ubiquitin chains, linked through Lys 63 and Lys 27 [168, 169]. The mitochondrial voltage-dependent anion channel 1 (VDAC1) is the target of Parkin-mediated Lys 27 polyubiquitination [169]. The autophagic adaptor p62 then binds to mitochondria via ubiquitin labeling and then promotes mitochondrial degradation [170]. The mitophagy pathway was extensively damaged in the brains of AD patients and AD animal models [171], suggesting that the damaged mitochondria may continuously be accumulated, which results in energy crisis and ROS overproduction in the pathological process of AD. A previous study showed that energy crisis could trigger mitochondrial fission [172], which may exacerbate the deterioration of the tissue energy environment and cause cell death [167]. Of note, Aβ could decrease mitophagy by reducing the expression of PINK1 and worsening the cognitive ability in mice [173]. Moreover, the aggregation of Aβ protein could limit the autophagic pathway of neurons [174], leading to intracellular calcium overload and mitochondrial damage [175], and subsequently contributing to the progression of AD. Furthermore, overexpression of human tau could impair mitophagy via elevating mitochondrial membrane potential, reducing Parkin expression, and inhibiting Parkin translocation to mitochondria in neurons [176, 177]. Together, these results indicate that mitophagy is impaired under the condition of AD, and targeting mitophagy may be a new strategy for combating AD.
4.3. Autophagy Involves in Aβ Metabolism
Mounting evidence has suggested a bidirectional relationship between autophagy and AD [178]. Autophagy plays important role in the regulation of the Aβ generation. Activation of Atg5-dependent autophagy can promote the degradation of APP, which subsequently reduces the accumulation of Aβ in the AD animal models [179]. Furthermore, a previous study revealed that a significant portion of BACE1 is recruited to the autophagic vacuoles along axons in the condition of AD [180], and inhibition of the mTOR pathway triggered autophagy could effectively reduce the production of Aβ by reducing the expression of BACE1 [181]. Despite, autophagy is also involved in the secretion of Aβ, evidenced by that conditional knockout of Atg7 induced autophagic deficiency increased the content of Aβ in neurons but reduced the extracellular Aβ deposition in APP23 mice [182]. The underlying mechanism of autophagy-modulated Aβ secretion remains largely unknown. Cholesterol dysmetabolism should be considered since it widely exists in AD brains and plays a vital role in the regulation of Aβ clearance and secretion [158]. On the other hand, the ectopic metabolism of Aβ also has a role in the regulation of autophagy. Aβ can trigger oxidative stress by increasing the expression of NADPH oxidase 4 (NOX4), which results in neuronal death via over-activation of autophagy [183].
It is noteworthy that AMPK pathways and mTOR signaling can sense the energy and glucose states, but different activation modes of AMPK have the opposite effect on the clearance of Aβ [184]. It has been reported that the cognitive function of APP/PS1 mice is negatively correlated with the Aβ plaque load, the activation of mTOR, and the activation of autophagy [185]. Based on the method of computer simulation, several scholars believe that the formation of soluble Aβ, the activation of mTOR, and the activation of autophagy are connected in a vicious cycle of mutual influence: the increase of soluble Aβ will activate mTOR, while the activation of mTOR will inhibit the activation of autophagy, thus affecting the clearance of Aβ [186].
4.4. The Relationship Between Autophagy and Tau Metabolism
The tau phosphorylation is modulated by autophagic bioactivity [178]. The hyperphosphorylation of tau can be reduced by the activation of autophagy. Metformin, an activator of the AMPK pathway, effectively decreased the phosphorylation of tau via enhancing the autophagic activity in db/db mice [187]. Similar results were observed in APP/PS1 transgenic mice. Nitazoxanide is an autophagy activator that has been demonstrated in vivo and in vitro to enhance autophagy and inhibit tau phosphorylation [188]. Another research also showed that loss of β-interferon can lead to the accumulation of tau in mice, which can be alleviated by stimulating macro-autophagy [189]. Notably, hyperphosphorylated tau can damage the mitochondria and autophagic function possibly via increasing the number of autophagosomes and affecting the permeability of the lysosomal membrane [190, 191], thus in return, exacerbate the tau pathology.
Increasing evidence suggested that the autophagy-lysosome pathway plays a vital role in tau clearance, both pathological and physiological tau secretion from neurons is in an autophagy-dependent manner. Autophagy inhibitors or knockout of Beclin1 in SH-SY5Y cells that overexpress human tau protein promote the expression of tau protein and the secretion of phosphorylated tau [192]. Activation of autophagy promoted the degradation of insoluble tau and protected against its toxicity in Drosophila [193]. mTOR-independent activation of autophagy also facilitated neuronal survival by reducing the aggregation of tau in the brains of human tauopathy model mice [194]. Furthermore, cathepsin D, a lysosomal protease, owns the ability to degrade tau protein, and inhibition of cathepsins effectively delayed tau degradation and promoted the formation of high molecular weight species of tau [195]. The clearance of tau can also be regulated by its acetylation since a total of 15 acetylation sites were identified in tau protein using tandem mass spectrometry assay [196]. p300 is a kind of lysine acetyltransferase, p300 and its homologous CBP can acetylate tau and reduce tau toxicity [197]. p300 can regulate tubulin acetylation through the role of sirtuin 2 (SIRT2), and the decrease in acetylated tau or the increase in tubulin acetylation is conducive to the phosphorylation of tau and its clearance through autophagy [198]. Another study showed that hyperactivation of p300/CBP can block autophagic flux and increase the secretion of tau in neurons [199]. Meanwhile, the intracellular role of p300 is thought to be a factor that integrates misfolded protein stress, but this role is thought to be achieved by the highly disordered variable spliceosome in the p300 molecule independent of the acetyltransferase domain [200]. Thus, p300 also has the function to overcome the intracellular misfolded proteins.
4.5. Autophagy Interacts with Neuroinflammation
An increasing number of studies showed that neuroinflammation contributes to the progression of AD [201]. The elevated biomarkers of inflammation were found in the blood and peripheral tissues of AD patients [202]. Neuroinflammation is the chronic activation of the immune response of the central nervous system, which is the body’s response to harmful stimuli. Autophagy and neuroinflammation crosstalk each other through certain inflammatory factors and signaling pathways. NLRP3 inflammasomes activation leads to IL-1β and TNFα production which are highly associated with the aggregation of tau and Aβ. NLRP3 inflammasomes activation regulates tau kinases and phosphatases to promote tau protein phosphorylation and aggregation, and increases the deposition of Aβ by reducing the phagocytic process [203]. The NLRP3 inflammasomes activation could be inhibited by the activation of autophagy [204], in return, the autolysosome functions could also be impaired by NLRP3 inflammasomes mediated inflammatory response in the condition of AD [205]. The PI3K-Akt signaling pathway is an important regulatory pathway in inflammatory stress and autophagy disorders in neurodegenerative diseases. Activation of the PI3K-Akt signaling pathway results in the inhibition of autophagy by the activation of mTOR, and also enhances the nuclear location of nuclear factor kappa-B (NF-κB) to promote the production of inflammatory factors in the M1 microglia [206], suggesting the beneficial roles of PI3K-Akt signaling pathway inhibition of AD. A high level of glucocorticoid (GC) exposure is thought to be associated with age-related cognitive decline in humans. Reduction of GC in senescence-accelerated mouse-prone 8 (SAMP8) mice enhanced the hippocampal autophagy, which was accompanied by decreased expressions of inflammatory markers (heme oxygenase 1 (HMOX1), aldehyde dehydrogenase 2 (ALDH2), IL-1β and C-C Motif Chemokine Ligand 3 (CCL3) [207], suggesting that the inflammatory and autophagic processes occurring in nerve cells are antagonistic to each other. Of note, cell selection for inflammation or autophagy is related to the changes in the environmental energy of the cell. A study evaluated the acute alternation in ambient glucose on microglia and found that inflammation was activated when the cellular environment changed from normal glucose to high glucose, whereas autophagy was activated on the contrary [208].
4.6. Autophagy Involves in Iron Metabolism and Ferroptosis
Iron is the most abundant metal ion in the brain [209]. There are two forms of iron in the organizations as ferrous iron and ferric iron, ferrous iron is a reduced form of iron, while ferric iron is an oxidation form. The ferrous iron forms alkoxy chloride with lipid peroxides and the release of ferrous iron is realized by the fusion of ferritin autophagosome and lysosome [210]. The regulations of iron in normal brains and AD brains have been specifically introduced in our previous reviews [17, 211]. Iron is an important element involved in the REDOX reaction of cells, but the excessive accumulation of iron will lead to oxidative damage and cell death,which has been observed in the brains of AD patients and animal models [212, 213]. Mounting evidence suggested that abnormal iron deposition was observed at the heart of neurological disorders (the cortex in AD, the substantia nigra in Parkinson’s disease, and the mitochondria in Huntington’s disease) [214]. Abnormal iron homeostasis in the brain can increase the production of harmful free radicals, resulting in damage to sensitive cells and subcellular structures [209]. It has been reported that autophagy plays important role in the modulation of iron homeostasis. Iron is stored in ferritin during times of iron excess and must be released under periods of iron demand. By interacting with ferritin to move to the lysosome lumen where the ferritin is destroyed and the iron is liberated, nuclear receptor coactivator 4 (NCOA4) can mediate the selective autophagy of ferritin [215]. Furthermore, activation of CMA could also promotes iron release by degrading ferritin [216]. It is noteworthy that deficiency of autophagy could give rise to impairment of ferritinophagy, thus leading to accumulation of ferritin and iron overload [215]. Therefore, dysregulation of the autophagy system may directly lead to intracellular iron accumulation in AD brains.
Ferroptosis, a new kind of cell death, is different from other forms of cell death (necrosis, apoptosis, pyroptosis), which is characterized by iron-dependent lipid peroxide, reduced or disappeared mitochondrial cristae, mitochondrial membrane concentration, and mitochondrial volume reduction [217]. Regulation of ferroptosis involves multiple signaling pathways, including the Nrf2-keap1 pathway, RAS/MAPK signal pathway, AMPK pathway, etc. [218]. Damages to the antioxidant system such as the dysfunctions of the plasma membrane system xc- (xCT) and peroxidase 4 (GPX4) could accelerate the progress of ferroptosis [219]. Notably, autophagy and ferroptosis take similar regulatory pathways, evidenced by that AMPK-mediated phosphorylation of Beclin1 at Ser90/93/96 is necessary for lipid peroxidation [220, 221], suggesting a potential connection between autophagy and ferroptosis. Surely, recent studies revealed that autophagy can promote cell ferroptosis via p62 selective degradation of aryl hydrocarbon receptor nuclear translocator-like (ARNTL) [222]; In addition, GPX4 is a target of HSC70, and degradation of GPX4 via activation of CMA could give rise to ferroptosis (Fig. 3) [223].
Fig. (3).
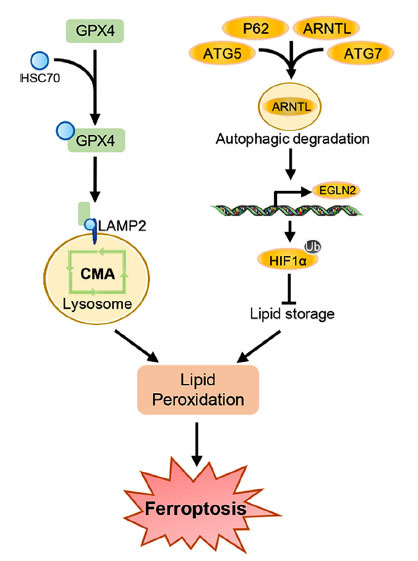
Ferroptosis is modulated by autophagy. Enhanced GPX4 degradation mediated by CMA activation gives rise to lipid peroxidation and ferroptosis. The macro-autophagy activation induces ARNTL degradation, thus transcriptionally promotes the EGLN2 expression, and subsequently inhibits HIF1α-mediated lipid storage, finally leading to lipid peroxidation and ferroptosis.
Ferroptosis occurs in a variety of neurodegenerative diseases such as AD and Parkinson’s disease [224], and could also be observed in vascular cognitive impairment, stroke, and traumatic brain injury [225]. The relationship between ferroptosis and AD has been noted. Metabolomics analyses showed that iron homeostasis, lipid peroxidation, and xCT are impaired in the AD brain [226]. Chronic iron exposure induced apoptosis, autophagy, and ferroptosis in mouse neurons, leading to loss of neurons and increasing the risk of AD [227]. Our previous study revealed that excess iron deposition was observed in the neurons of the P301S transgenic mice, and administration of iron chelator α-lipoic acid significantly reduced the phosphorylated tau and neuronal loss possibly by upregulation of GPX4 and xCT [18]. Moreover, mice with specific knockout of GPX4 in the cortex and hippocampal neurons exerted degeneration of neurons and impaired cognitive ability, while administration of vitamin E diet or ferroptosis inhibitor liproxstatin-1 effectively reversed these effects [228], suggesting that targeting ferroptosis may be a promising strategy for the treatment of AD. It is worth noting that mitochondrial dysfunction and abnormal glucose metabolism are hallmarks of AD, which suggest a potential association between energy crisis and ferroptosis. Indeed, several studies showed that mitochondrial dysfunction could induce lipid peroxidation, leading to ferroptosis [229]; Other studies also suggested that in the existence of mitochondrial DNA stress, cells were sensitive to ferroptosis inducers [230]. The relationship between ferroptosis and energy crisis is an interesting topic that should be further elucidated.
5. STRATEGIES TO FUEL THE BRAIN
In addition to targeting Aβ, tau, and neuroinflammation, improving the brain’s energy supply has recently gained great attention in the treatment of AD. The strategies are mainly divided into the following directions:
5.1. Energy-Replacement Therapy
Ketones are the important compensatory fuel for the brain in the condition of glucose deficiency. Medium-chain triglyceride (MCT), a ketone supplement, was used in patients with mild to moderate AD. The brain ketone and brain glucose intake were measured using PET, a study revealed that the total energy metabolism of the brain was increased without changing glucose utilization after ketone supplementation [231]. Similar to the aforementioned study, ketogenic drink treatment in patients with MCI showed an increase (about 230%) in ketone metabolism in the brain, which was accompanied by enhanced cognitive ability [232]. By comparing dietary interventions at different ages, increased intake of ketones and exogenous ketones increased the stability of the brain neural network [233]. Moreover, studies in different animal models have found that a ketogenic diet can also improve the symptoms of other neurological diseases in the brain, such as schizophrenia [234], traumatic brain injury [235], and multiple sclerosis [236]. The protective effect of the ketogenic diet on neurons may be realized by appropriately improving the autophagic function of neurons since the ketogenic diet can activate Sirt1 and hypoxia-induced factor-1α (HIF-1α), which can synergistically induce the autophagy of neurons [237]. However, the safety of ketone as an alternative fuel remains to be determined since early exposure to ketone resulted in impaired cognitive function in young normal mice [238]. These studies suggested that the functions of ketones may be dependent on the pathological states, which need to be further elucidated.
5.2. Improvement of Glucose Metabolism
Glucose hypometabolism occurs in the brains of AD patients, thus improvement of the glucose uptake and glucose utilization are taken into consideration in the treatment of AD. Liraglutide, a glucagon-like peptide 1 analog that is commonly used by type-2 diabetes patients to improve β cell function and suppress glucagon to restore normoglycemia, was shown to have neuroprotective effects on AD via reducing Aβ production and tau phosphorylation [239]. Recent studies also revealed that liraglutide could improve the neuronal supportive ability of astrocytes via enhancing the astrocytic glycolysis and mitochondrial functions in AD animal models [240, 241]. Based on these observations, liraglutide is in ongoing clinical trials for treating AD. Besides the liraglutide, a first-line antihyperglycemic medication, metformin, was also performed to explore the effects on AD. Studies exerted that metformin could effectively reduce the Aβ load, tau phosphorylation, and neuroinflammation, and promote glucose delivery and uptake in brains of several AD models, suggesting its multi-benefits for combating AD [242]. Nevertheless, it is worth noting that both the liraglutide and metformin have the abilities to reduce Aβ load and tau phosphorylation; however whether these effects are ascribed to the improvements in glucose metabolism remains to be further explored.
5.3. Insulin Application
Disturbance of insulin signaling is a contributor to the energy crisis in the brain of AD patients. Therefore, insulin and antidiabetic drugs become feasible ideas for the treatment of AD. Due to the limitations of hypoglycemia and BBB, intranasal administration is wildly used to investigate the effects of insulin in neurodegenerative disorders [243]. Intranasal insulin was shown biosecurity in the treatment of Down’s syndrome [244], and could effectively alleviate nerve damage and insulin resistance by inhibiting GSK3β in several animal models of cognitive impairments [245-247]. Importantly, the improvements in learning and memory function [248], and reductions in Aβ aggregation and p-Tau levels [249] were also observed in the intranasal insulin-treated animal models of AD. Therefore, several groups have performed clinical trials to investigate the effects of intranasal insulin in AD patients. Kellar D, et al uncovered that intranasal insulin treatment for 12 months significantly reduced changes in white matter hyperintensity volume in deep and frontal regions, and also modulated the inflammation and immune function in MCI patients and AD patients [250, 251]. Similarly, intranasal insulin treatment also improved delayed memory, preserved general cognition, and functional abilities, and retarded the reduction of glucose uptake in AD patients [252]. However, in another randomized clinical trial, although no clinically important adverse events were found to be associated with the treatment, intranasal insulin had no cognitive or functional benefits in AD patients [253]. The paradoxical results remind us that the effects of insulin on AD need more investigation.
CONCLUSION
In this review, the characteristics of the energy crisis in AD were summarized (Fig. 4). Particularly, energy crisis links to the modulations of autophagy, indicating that autophagy may facilitate the rescue of energy crisis in the early stage of AD, and may be out of control in the pathological process of AD, which in turn, leads to the exacerbation of AD. Furthermore, since the occurrence of ferroptosis is partly autophagy-dependent from the current research point of view, it prompts the possibility that ferroptosis may be the outcome of the impaired autophagy induced by the energy crisis. Surely, recent evidence uncovered that energy crisis could inhibit ferroptosis in cancer cells [254, 255], suggesting a certain relation between ferroptosis and energy crisis. However, whether energy crisis induces or represses ferroptosis under the condition of AD pathology merits further investigations. Taken together, despite considerable advances in the experimental phase in many reagents and therapeutic directions, the treatment of AD remains very difficult. Considering that the occurrence of the energy crisis is far earlier than the Aβ deposition, tau phosphorylation, neuroinflammation, and ferroptosis, the energy crisis in the progression of AD should gain more attention in future studies.
Fig. (4).
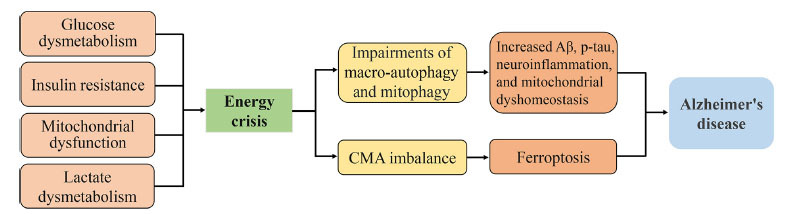
Energy crisis contributes to the progress of AD. The reduced glucose uptake and utilization, insulin resistance, mitochondrial dysfunction, as well as lactate dysmetabolism promote the energy crisis in the brain, which impairs the macro-autophagy and mitophagy, and subsequently causes the productions of Aβ and p-tau, and promotes the neuroinflammation and mitochondrial dysmetabolism; On the other hand, energy crisis may also promote ferroptosis via regulating CMA in neurons, and eventually promotes the progress of AD.
ACKNOWLEDGEMENTS
Declared none.
LIST OF ABBREVIATIONS
- GLUTs
Glucose transporters
- PPAR
Proliferators-activated receptor
- TFEB
Transcription factor EB
- AMPK
AMP-activated protein kinase
- AD
Alzheimer’s disease
- SPs
Senile plaques
- NEP
Neprilysin
- IDE
Insulin-degrading enzyme
AUTHOR CONTRIBUTIONS
D-LH and Y-GF wrote the manuscript and drawn the figures. Y-GF and Z-YW approved and revised the final manuscript. All authors have read and approved the final manuscript.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was financially supported by the Natural Science Foundation of China (Grant No. 92049106), and the Natural Science Foundation of Liaoning Province (Grant Nos. LJKZ0736; 2019JH2/10300003).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Alzheimer’s disease facts and figures. Alzheimers Dement. 2020;2020(Mar):10. doi: 10.1002/alz.12068. [DOI] [PubMed] [Google Scholar]
- 2.Liu P.P., Xie Y., Meng X.Y., Kang J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019;4(1):29. doi: 10.1038/s41392-019-0063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long J.M., Holtzman D.M. Alzheimer disease: An update on pathobiology and treatment strategies. Cell. 2019;179(2):312–339. doi: 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cunnane S., Nugent S., Roy M., Courchesne-Loyer A., Croteau E., Tremblay S., Castellano A., Pifferi F., Bocti C., Paquet N., Begdouri H., Bentourkia M., Turcotte E., Allard M., Barberger-Gateau P., Fulop T., Rapoport S.I. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition. 2011;27(1):3–20. doi: 10.1016/j.nut.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reiman E.M., Caselli R.J., Yun L.S., Chen K., Bandy D., Minoshima S., Thibodeau S.N., Osborne D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N. Engl. J. Med. 1996;334(12):752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- 6.Glick D., Barth S., Macleod K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010;221(1):3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dikic I., Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018;19(6):349–364. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 8.Jack C.R., Jr, Bennett D.A., Blennow K., Carrillo M.C., Dunn B., Haeberlein S.B., Holtzman D.M., Jagust W., Jessen F., Karlawish J., Liu E., Molinuevo J.L., Montine T., Phelps C., Rankin K.P., Rowe C.C., Scheltens P., Siemers E., Snyder H.M., Sperling R., Elliott C., Masliah E., Ryan L., Silverberg N. NIA‐AA Research framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernier G., Nardini E., Hogan R., Flamier A. Alzheimer’s disease: A tale of two diseases? Neural Regen. Res. 2021;16(10):1958–1964. doi: 10.4103/1673-5374.308070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jha N.K., Jha S.K., Kumar D., Kejriwal N., Sharma R., Ambasta R.K., Kumar P. Impact of insulin degrading enzyme and neprilysin in alzheimer’s disease biology: Characterization of putative cognates for therapeutic applications. J. Alzheimers Dis. 2015;48(4):891–917. doi: 10.3233/JAD-150379. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Y., Long Z., Ding Y., Jiang T., Liu J., Li Y., Liu Y., Peng X., Wang K., Feng M., He G. Dihydroartemisinin ameliorates learning and memory in Alzheimer’s disease through promoting autophagosome-lysosome fusion and autolysosomal degradation for Abeta clearance. Front. Aging Neurosci. 2020;12:47. doi: 10.3389/fnagi.2020.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y., Chen Y., Liang Y., Chen H., Ji X., Huang M. Berberine mitigates cognitive decline in an Alzheimer’s Disease Mouse Model by targeting both tau hyperphosphorylation and autophagic clearance. Biomed. Pharmacother. 2020;121:109670. doi: 10.1016/j.biopha.2019.109670. [DOI] [PubMed] [Google Scholar]
- 13.Uddin M.S., Kabir M.T., Niaz K., Jeandet P., Clément C., Mathew B., Rauf A., Rengasamy K.R.R., Sobarzo-Sánchez E., Ashraf G.M., Aleya L. Molecular insight into the therapeutic promise of flavonoids against Alzheimer’s disease. Molecules. 2020;25(6):1267. doi: 10.3390/molecules25061267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamano T., Shirafuji N., Yen S.H., Yoshida H., Kanaan N.M., Hayashi K., Ikawa M., Yamamura O., Fujita Y., Kuriyama M., Nakamoto Y. Rho-kinase ROCK inhibitors reduce oligomeric tau protein. Neurobiol. Aging. 2020;89:41–54. doi: 10.1016/j.neurobiolaging.2019.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madadi S., Schwarzenbach H., Saidijam M., Mahjub R., Soleimani M. Potential microRNA-related targets in clearance pathways of amyloid-β: Novel therapeutic approach for the treatment of Alzheimer’s disease. Cell Biosci. 2019;9(1):91. doi: 10.1186/s13578-019-0354-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang P., Wang Z.Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2017;35:265–290. doi: 10.1016/j.arr.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 17.Liu J.L., Fan Y.G., Yang Z.S., Wang Z.Y., Guo C. Iron and Alzheimer’s disease: From pathogenesis to therapeutic implications. Front. Neurosci. 2018;12:632. doi: 10.3389/fnins.2018.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y.H., Wang D.W., Xu S.F., Zhang S., Fan Y.G., Yang Y.Y., Guo S.Q., Wang S., Guo T., Wang Z.Y., Guo C. α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 2018;14:535–548. doi: 10.1016/j.redox.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daulatzai M.A. Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J. Neurosci. Res. 2017;95(4):943–972. doi: 10.1002/jnr.23777. [DOI] [PubMed] [Google Scholar]
- 20.Sadowski M., Pankiewicz J., Scholtzova H., Ji Y., Quartermain D., Jensen C.H., Duff K., Nixon R.A., Gruen R.J., Wisniewski T. Amyloid-beta deposition is associated with decreased hippocampal glucose metabolism and spatial memory impairment in APP/PS1 mice. J. Neuropathol. Exp. Neurol. 2004;63(5):418–428. doi: 10.1093/jnen/63.5.418. [DOI] [PubMed] [Google Scholar]
- 21.Bélanger M., Allaman I., Magistretti P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14(6):724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 22.Maqbool M., Mobashir M., Hoda N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016;107:63–81. doi: 10.1016/j.ejmech.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 23.Arnold S.E., Arvanitakis Z., Macauley-Rambach S.L., Koenig A.M., Wang H.Y., Ahima R.S., Craft S., Gandy S., Buettner C., Stoeckel L.E., Holtzman D.M., Nathan D.M. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018;14(3):168–181. doi: 10.1038/nrneurol.2017.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alberini C.M., Cruz E., Descalzi G., Bessières B., Gao V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia. 2018;66(6):1244–1262. doi: 10.1002/glia.23250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magistretti P.J., Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015;86(4):883–901. doi: 10.1016/j.neuron.2015.03.035. [DOI] [PubMed] [Google Scholar]
- 26.Vilchez D., Ros S., Cifuentes D., Pujadas L., Vallès J., García-Fojeda B., Criado-García O., Fernández-Sánchez E., Medraño-Fernández I., Domínguez J., García-Rocha M., Soriano E., Rodríguez de Córdoba S., Guinovart J.J. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 2007;10(11):1407–1413. doi: 10.1038/nn1998. [DOI] [PubMed] [Google Scholar]
- 27.Carpenter K.L.H., Jalloh I., Hutchinson P.J. Glycolysis and the significance of lactate in traumatic brain injury. Front. Neurosci. 2015;9:112. doi: 10.3389/fnins.2015.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pellerin L., Magistretti P.J. Sweet Sixteen for ANLS. J. Cereb. Blood Flow Metab. 2012;32(7):1152–1166. doi: 10.1038/jcbfm.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi S. Metabolic compartmentalization between astroglia and neurons in physiological and pathophysiological conditions of the neurovascular unit. Neuropathology. 2020;40(2):121–137. doi: 10.1111/neup.12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morland C., Lauritzen K.H., Puchades M., Holm-Hansen S., Andersson K., Gjedde A., Attramadal H., Storm-Mathisen J., Bergersen L.H. The lactate receptor, G-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1: Expression and action in brain. J. Neurosci. Res. 2015;93(7):1045–1055. doi: 10.1002/jnr.23593. [DOI] [PubMed] [Google Scholar]
- 31.Brooks G.A. The science and translation of lactate shuttle theory. Cell Metab. 2018;27(4):757–785. doi: 10.1016/j.cmet.2018.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Bouzier-Sore A.K., Voisin P., Bouchaud V., Bezancon E., Franconi J.M., Pellerin L. Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: A comparative NMR study. Eur. J. Neurosci. 2006;24(6):1687–1694. doi: 10.1111/j.1460-9568.2006.05056.x. [DOI] [PubMed] [Google Scholar]
- 33.Mayorga-Weber G., Rivera F.J., Castro M.A. Neuron‐glia (mis)interactions in brain energy metabolism during aging. J. Neurosci. Res. 2022;100(3):835–854. doi: 10.1002/jnr.25015. [DOI] [PubMed] [Google Scholar]
- 34.Chuquet J., Quilichini P., Nimchinsky E.A., Buzsáki G. Predominant enhancement of glucose uptake in astrocytes versus neurons during activation of the somatosensory cortex. J. Neurosci. 2010;30(45):15298–15303. doi: 10.1523/JNEUROSCI.0762-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magistretti P.J., Allaman I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018;19(4):235–249. doi: 10.1038/nrn.2018.19. [DOI] [PubMed] [Google Scholar]
- 36.Supplie L.M., Düking T., Campbell G., Diaz F., Moraes C.T., Götz M., Hamprecht B., Boretius S., Mahad D., Nave K.A. Respiration-deficient astrocytes survive as glycolytic cells in vivo. J. Neurosci. 2017;37(16):4231–4242. doi: 10.1523/JNEUROSCI.0756-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong X., Zhang Q., Yu X., Wang D., Ma J., Ma J., Shi S.H. Metabolic lactate production coordinates vasculature development and progenitor behavior in the developing mouse neocortex. Nat. Neurosci. 2022;25(7):865–875. doi: 10.1038/s41593-022-01093-7. [DOI] [PubMed] [Google Scholar]
- 38.Beard E., Lengacher S., Dias S., Magistretti P.J., Finsterwald C. Astrocytes as key regulators of brain energy metabolism: New therapeutic perspectives. Front. Physiol. 2022;12:825816. doi: 10.3389/fphys.2021.825816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henn R.E., Noureldein M.H., Elzinga S.E., Kim B., Savelieff M.G., Feldman E.L. Glial-neuron crosstalk in health and disease: A focus on metabolism, obesity, and cognitive impairment. Neurobiol. Dis. 2022;170:105766. doi: 10.1016/j.nbd.2022.105766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lauro C., Limatola C. Metabolic reprograming of microglia in the regulation of the innate inflammatory response. Front. Immunol. 2020;11:493. doi: 10.3389/fimmu.2020.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baik S.H., Kang S., Lee W., Choi H., Chung S., Kim J.I., Mook-Jung I. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 2019;30(3):493–507.e6. doi: 10.1016/j.cmet.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 42.Li Y., Lu B., Sheng L., Zhu Z., Sun H., Zhou Y., Yang Y., Xue D., Chen W., Tian X., Du Y., Yan M., Zhu W., Xing F., Li K., Lin S., Qiu P., Su X., Huang Y., Yan G., Yin W. Hexokinase 2-dependent hyperglycolysis driving microglial activation contributes to ischemic brain injury. J. Neurochem. 2018;144(2):186–200. doi: 10.1111/jnc.14267. [DOI] [PubMed] [Google Scholar]
- 43.Monsorno K., Buckinx A., Paolicelli R.C. Microglial metabolic flexibility: Emerging roles for lactate. Trends Endocrinol. Metab. 2022;33(3):186–195. doi: 10.1016/j.tem.2021.12.001. [DOI] [PubMed] [Google Scholar]
- 44.Manceau R., Majeur D., Alquier T. Neuronal control of peripheral nutrient partitioning. Diabetologia. 2020;63(4):673–682. doi: 10.1007/s00125-020-05104-9. [DOI] [PubMed] [Google Scholar]
- 45.Morita-Takemura S., Wanaka A. Blood-to-brain communication in the hypothalamus for energy intake regulation. Neurochem. Int. 2019;128:135–142. doi: 10.1016/j.neuint.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 46.Shao X., Tang Y., Long H., Gu H., Zhang J., Deng P., Zhao Y., Cen X. HMG-CoA synthase 2 drives brain metabolic reprogramming in cocaine exposure. Neuropharmacology. 2019;148:377–393. doi: 10.1016/j.neuropharm.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 47.Puchalska P., Crawford P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017;25(2):262–284. doi: 10.1016/j.cmet.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grabacka M., Pierzchalska M., Dean M., Reiss K. Regulation of ketone body metabolism and the role of pparalpha. Int. J. Mol. Sci. 2016;17(12):2093. doi: 10.3390/ijms17122093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Güemes M., Hussain K. Hyperinsulinemic Hypoglycemia. Pediatr. Clin. North Am. 2015;62(4):1017–1036. doi: 10.1016/j.pcl.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 50.Voronina P.P., Adamovich K.V., Adamovich T.V., Dubouskaya T.G., Hrynevich S.V., Waseem T.V., Fedorovich S.V. High concentration of ketone body beta-hydroxybutyrate modifies synaptic vesicle cycle and depolarizes plasma membrane of rat brain synaptosomes. J. Mol. Neurosci. 2020;70(1):112–119. doi: 10.1007/s12031-019-01406-9. [DOI] [PubMed] [Google Scholar]
- 51.Suissa L, Flachon V, Guigonis JM. Urinary ketone body loss leads to degeneration of brain white matter in elderly SLC5A8- deficient mice. J. Cereb. Blood Flow Metab. 2019. p. 271678X19873662. [DOI] [PMC free article] [PubMed]
- 52.Grillo C.A., Piroli G.G., Hendry R.M., Reagan L.P. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res. 2009;1296:35–45. doi: 10.1016/j.brainres.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fernandez A.M., Hernandez-Garzón E., Perez-Domper P., Perez-Alvarez A., Mederos S., Matsui T., Santi A., Trueba-Saiz A., García-Guerra L., Pose-Utrilla J., Fielitz J., Olson E.N., Fernandez de la Rosa R., Garcia Garcia L., Pozo M.A., Iglesias T., Araque A., Soya H., Perea G., Martin E.D., Torres Aleman I. Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes. 2017;66(1):64–74. doi: 10.2337/db16-0861. [DOI] [PubMed] [Google Scholar]
- 54.Bromander S., Anckarsäter R., Ahrén B., Kristiansson M., Blennow K., Holmäng A., Zetterberg H., Anckarsäter H., Wass C.E. Cerebrospinal fluid insulin during non-neurological surgery. J. Neural Transm. (Vienna) 2010;117(10):1167–1170. doi: 10.1007/s00702-010-0456-x. [DOI] [PubMed] [Google Scholar]
- 55.Didier S., Sauvé F., Domise M., Buée L., Marinangeli C., Vingtdeux V. AMP-activated protein kinase controls immediate early genes expression following synaptic activation through the PKA/CREB pathway. Int. J. Mol. Sci. 2018;19(12):3716. doi: 10.3390/ijms19123716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uehara T., Yamasaki T., Okamoto T., Koike T., Kan S., Miyauchi S., Kira J., Tobimatsu S. Efficiency of a “small-world” brain network depends on consciousness level: A resting-state FMRI study. Cereb. Cortex. 2014;24(6):1529–1539. doi: 10.1093/cercor/bht004. [DOI] [PubMed] [Google Scholar]
- 57.Tomasi D., Wang G.J., Volkow N.D. Energetic cost of brain functional connectivity. Proc. Natl. Acad. Sci. USA. 2013;110(33):13642–13647. doi: 10.1073/pnas.1303346110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kealy J., Bennett R., Woods B., Lowry J.P. Real-time changes in hippocampal energy demands during a spatial working memory task. Behav. Brain Res. 2017;326:59–68. doi: 10.1016/j.bbr.2017.02.034. [DOI] [PubMed] [Google Scholar]
- 59.Minoshima S., Foster N.L., Sima A.A.F., Frey K.A., Albin R.L., Kuhl D.E. Alzheimer’s disease versus dementia with Lewy bodies: Cerebral metabolic distinction with autopsy confirmation. Ann. Neurol. 2001;50(3):358–365. doi: 10.1002/ana.1133. [DOI] [PubMed] [Google Scholar]
- 60.Barros L.F., Bolaños J.P., Bonvento G., Bouzier-Sore A.K., Brown A., Hirrlinger J., Kasparov S., Kirchhoff F., Murphy A.N., Pellerin L., Robinson M.B., Weber B. Current technical approaches to brain energy metabolism. Glia. 2018;66(6):1138–1159. doi: 10.1002/glia.23248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meng X., Liu J., Fan X., Bian C., Wei Q., Wang Z., Liu W., Jiao Z. Multi-modal neuroimaging neural network-based feature detection for diagnosis of Alzheimer’s disease. Front. Aging Neurosci. 2022;14:911220. doi: 10.3389/fnagi.2022.911220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Engel M.G., Smith J., Mao K., Quipildor G.F., Cui M.H., Gulinello M., Branch C.A., Gandy S.E., Huffman D.M. Evidence for preserved insulin responsiveness in the aging rat brain. Geroscience. 2022 doi: 10.1007/s11357-022-00618-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu Y.Q., Wang Y.N., Zhang L.J., Liu L.Q., Pan Y.C., Su T., Liao X.L., Shu H.Y., Kang M., Ying P., Xu S.H., Shao Y. Regional homogeneity in patients with mild cognitive impairment: A resting-state functional magnetic resonance imaging study. Front. Aging Neurosci. 2022;14:877281. doi: 10.3389/fnagi.2022.877281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sheline Y.I., Morris J.C., Snyder A.Z., Price J.L., Yan Z., D’Angelo G., Liu C., Dixit S., Benzinger T., Fagan A., Goate A., Mintun M.A. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Aβ42. J. Neurosci. 2010;30(50):17035–17040. doi: 10.1523/JNEUROSCI.3987-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heise V., Filippini N., Trachtenberg A.J., Suri S., Ebmeier K.P., Mackay C.E. Apolipoprotein E genotype, gender and age modulate connectivity of the hippocampus in healthy adults. Neuroimage. 2014;98:23–30. doi: 10.1016/j.neuroimage.2014.04.081. [DOI] [PubMed] [Google Scholar]
- 66.Song T., Song X., Zhu C., Patrick R., Skurla M., Santangelo I., Green M., Harper D., Ren B., Forester B.P., Öngür D., Du F. Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer’s disease: A meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res. Rev. 2021;72:101503. doi: 10.1016/j.arr.2021.101503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bonvento G., Valette J., Flament J., Mochel F., Brouillet E. Imaging and spectroscopic approaches to probe brain energy metabolism dysregulation in neurodegenerative diseases. J. Cereb. Blood Flow Metab. 2017;37(6):1927–1943. doi: 10.1177/0271678X17697989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Q., Abrigo J., Liu W., Han E.Y., Yeung D.K.W., Shi L., Au L.W.C., Deng M., Chen S., Leung E.Y.L., Ho C.L., Mok V.C.T., Chu W.C.W. Lower posterior cingulate n-acetylaspartate to creatine level in early detection of biologically defined Alzheimer’s disease. Brain Sci. 2022;12(6):722. doi: 10.3390/brainsci12060722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rijpma A., van der Graaf M., Meulenbroek O., Olde Rikkert M.G.M., Heerschap A. Altered brain high-energy phosphate metabolism in mild Alzheimer’s disease: A 3-dimensional 31P MR spectroscopic imaging study. Neuroimage Clin. 2018;18:254–261. doi: 10.1016/j.nicl.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Das N., Ren J., Spence J.S., Rackley A., Chapman S.B. Relationship of parieto-occipital brain energy phosphate metabolism and cognition using (31)P MRS at 7-tesla in amnestic mild cognitive impairment. Front. Aging Neurosci. 2020;12:222. doi: 10.3389/fnagi.2020.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mandal P.K., Guha Roy R., Samkaria A., Maroon J.C., Arora Y. In vivo (13)C magnetic resonance spectroscopy for assessing brain biochemistry in health and disease. Neurochem. Res. 2022;47(5):1183–1201. doi: 10.1007/s11064-022-03538-8. [DOI] [PubMed] [Google Scholar]
- 72.Minoshima S., Cross D., Thientunyakit T., Foster N.L., Drzezga A. (18)F-FDG PET imaging in neurodegenerative dementing disorders: Insights into subtype classification, emerging disease categories, and mixed dementia with copathologies. J. Nucl. Med. 2022;63(Suppl. 1):2S–12S. doi: 10.2967/jnumed.121.263194. [DOI] [PubMed] [Google Scholar]
- 73.Silverman D.H.S., Small G.W., Chang C.Y., Lu C.S., de Aburto M.A.K., Chen W., Czernin J., Rapoport S.I., Pietrini P., Alexander G.E., Schapiro M.B., Jagust W.J., Hoffman J.M., Welsh-Bohmer K.A., Alavi A., Clark C.M., Salmon E., de Leon M.J., Mielke R., Cummings J.L., Kowell A.P., Gambhir S.S., Hoh C.K., Phelps M.E. Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome. JAMA. 2001;286(17):2120–2127. doi: 10.1001/jama.286.17.2120. [DOI] [PubMed] [Google Scholar]
- 74.Marcus C., Mena E., Subramaniam R.M. Brain PET in the diagnosis of Alzheimer’s disease. Clin. Nucl. Med. 2014;39(10):e413–e426. doi: 10.1097/RLU.0000000000000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pezzoli S., Manca R., Cagnin A., Venneri A. Alzheimer’s Disease Neuroimaging I. A multimodal neuroimaging and neuropsychological study of visual hallucinations in Alzheimer’s disease. J. Alzheimers Dis. 2022:1–17. doi: 10.3233/JAD-215107. [DOI] [PubMed] [Google Scholar]
- 76.Perovnik M., Tomše P., Jamšek J., Emeršič A., Tang C., Eidelberg D., Trošt M. Identification and validation of Alzheimer’s disease-related metabolic brain pattern in biomarker confirmed Alzheimer’s dementia patients. Sci. Rep. 2022;12(1):11752. doi: 10.1038/s41598-022-15667-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fernandes M., Manfredi N., Aluisantonio L., Franchini F., Chiaravalloti A., Izzi F., Di Santo S., Schillaci O., Mercuri N.B., Placidi F., Liguori C. Cognitive functioning, cerebrospinal fluid Alzheimer’s disease biomarkers and cerebral glucose metabolism in late‐onset epilepsy of unknown aetiology: A prospective study. Eur. J. Neurosci. 2022. p. ejn.15734. [DOI] [PubMed]
- 78.Croteau E., Castellano C.A., Fortier M., Bocti C., Fulop T., Paquet N., Cunnane S.C. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 2018;107:18–26. doi: 10.1016/j.exger.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 79.Ossenkoppele R., Tolboom N., Foster-Dingley J.C., Adriaanse S.F., Boellaard R., Yaqub M., Windhorst A.D., Barkhof F., Lammertsma A.A., Scheltens P., van der Flier W.M., van Berckel B.N.M. Longitudinal imaging of Alzheimer pathology using [11C]PIB, [18F]FDDNP and [18F]FDG PET. Eur. J. Nucl. Med. Mol. Imaging. 2012;39(6):990–1000. doi: 10.1007/s00259-012-2102-3. [DOI] [PubMed] [Google Scholar]
- 80.Vercruysse P., Vieau D., Blum D., Petersén Å., Dupuis L. Hypothalamic Alterations in neurodegenerative diseases and their relation to abnormal energy metabolism. Front. Mol. Neurosci. 2018;11:2. doi: 10.3389/fnmol.2018.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ishii M., Iadecola C. Metabolic and non-cognitive manifestations of Alzheimer’s disease: The hypothalamus as both culprit and target of pathology. Cell Metab. 2015;22(5):761–776. doi: 10.1016/j.cmet.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Niwa K., Kazama K., Younkin S.G., Carlson G.A., Iadecola C. Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol. Dis. 2002;9(1):61–68. doi: 10.1006/nbdi.2001.0460. [DOI] [PubMed] [Google Scholar]
- 83.Lowe M.T.J., Faull R.L.M., Christie D.L., Waldvogel H.J. Distribution of the creatine transporter throughout the human brain reveals a spectrum of creatine transporter immunoreactivity. J. Comp. Neurol. 2015;523(5):699–725. doi: 10.1002/cne.23667. [DOI] [PubMed] [Google Scholar]
- 84.Leveugle B., Spik G., Perl D.P., Bouras C., Fillit H.M., Hof P.R. The iron-binding protein lactotransferrin is present in pathologic lesions in a variety of neurodegenerative disorders: A comparative immunohistochemical analysis. Brain Res. 1994;650(1):20–31. doi: 10.1016/0006-8993(94)90202-X. [DOI] [PubMed] [Google Scholar]
- 85.Peng W., Tan C., Mo L., Jiang J., Zhou W., Du J., Zhou X., Liu X., Chen L. Glucose transporter 3 in neuronal glucose metabolism: Health and diseases. Metabolism. 2021;123:154869. doi: 10.1016/j.metabol.2021.154869. [DOI] [PubMed] [Google Scholar]
- 86.Li Z., Zhang Y., Zheng Y., Liu W., Zhang X., Li W., Zhang D., Cai Q., Wang S., Meng X., Huang G. Intranasal 15d-PGJ2 ameliorates brain glucose hypometabolism via PPARγ-dependent activation of PGC-1α/GLUT4 signalling in APP/PS1 transgenic mice. Neuropharmacology. 2021;196:108685. doi: 10.1016/j.neuropharm.2021.108685. [DOI] [PubMed] [Google Scholar]
- 87.Ding F., Yao J., Rettberg J.R., Chen S., Brinton R.D. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: Implication for bioenergetic intervention. PLoS One. 2013;8(11):e79977. doi: 10.1371/journal.pone.0079977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ahn K.C., Learman C.R., Dunbar G.L., Maiti P., Jang W.C., Cha H.C., Song M.S. Characterization of impaired cerebrovascular structure in APP/PS1 mouse brains. Neuroscience. 2018;385:246–254. doi: 10.1016/j.neuroscience.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 89.Choi H., Choi Y., Lee E.J., Kim H., Lee Y., Kwon S., Hwang D.W., Lee D.S. Hippocampal glucose uptake as a surrogate of metabolic change of microglia in Alzheimer’s disease. J. Neuroinflammation. 2021;18(1):190. doi: 10.1186/s12974-021-02244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McIntosh A., Mela V., Harty C., Minogue A.M., Costello D.A., Kerskens C., Lynch M.A. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. 2019;29(5):606–621. doi: 10.1111/bpa.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Holland R., McIntosh A.L., Finucane O.M., Mela V., Rubio-Araiz A., Timmons G., McCarthy S.A., Gun’ko Y.K., Lynch M.A. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav. Immun. 2018;68:183–196. doi: 10.1016/j.bbi.2017.10.017. [DOI] [PubMed] [Google Scholar]
- 92.Andersson A.K., Rönnbäck L., Hansson E. Lactate induces tumour necrosis factor-α, interleukin-6 and interleukin-1β release in microglial- and astroglial-enriched primary cultures. J. Neurochem. 2005;93(5):1327–1333. doi: 10.1111/j.1471-4159.2005.03132.x. [DOI] [PubMed] [Google Scholar]
- 93.Pan R.Y., He L., Zhang J., Liu X., Liao Y., Gao J., Liao Y., Yan Y., Li Q., Zhou X., Cheng J., Xing Q., Guan F., Zhang J., Sun L., Yuan Z. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 2022;34(4):634–648.e6. doi: 10.1016/j.cmet.2022.02.013. [DOI] [PubMed] [Google Scholar]
- 94.Doorduijn A.S., van de Rest O., van der Flier W.M., Visser M. de van der Schueren, M.A.E. Energy and protein intake of Alzheimer’s disease patients compared to cognitively normal controls: Systematic review. J. Am. Med. Dir. Assoc. 2019;20(1):14–21. doi: 10.1016/j.jamda.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 95.Vogelsang P., Giil L.M., Lund A., Vedeler C.A., Parkar A.P., Nordrehaug J.E., Kristoffersen E.K. Reduced glucose transporter-1 in brain derived circulating endothelial cells in mild Alzheimer’s disease patients. Brain Res. 2018;1678:304–309. doi: 10.1016/j.brainres.2017.10.035. [DOI] [PubMed] [Google Scholar]
- 96.Winkler E.A., Nishida Y., Sagare A.P., Rege S.V., Bell R.D., Perlmutter D., Sengillo J.D., Hillman S., Kong P., Nelson A.R., Sullivan J.S., Zhao Z., Meiselman H.J., Wenby R.B., Soto J., Abel E.D., Makshanoff J., Zuniga E., De Vivo D.C., Zlokovic B.V. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015;18(4):521–530. doi: 10.1038/nn.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee Y.J., Kim J.E., Hwang I.S., Kwak M.H., Lee J.H., Jung Y.J. an, B.S.; Kwon, H.S.; Kim, B.C.; Kim, S.J.; Kim, J.M.; Hwang, D.Y. Alzheimer’s phenotypes induced by overexpression of human presenilin 2 mutant proteins stimulate significant changes in key factors of glucose metabolism. Mol. Med. Rep. 2013;7(5):1571–1578. doi: 10.3892/mmr.2013.1404. [DOI] [PubMed] [Google Scholar]
- 98.Szablewski L. Glucose transporters in brain: In health and in Alzheimer’s disease. J. Alzheimers Dis. 2016;55(4):1307–1320. doi: 10.3233/JAD-160841. [DOI] [PubMed] [Google Scholar]
- 99.Pearson-Leary J., McNay E.C. Novel roles for the insulin-regulated glucose transporter-4 in hippocampally dependent memory. J. Neurosci. 2016;36(47):11851–11864. doi: 10.1523/JNEUROSCI.1700-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.De Felice F.G., Ferreira S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes. 2014;63(7):2262–2272. doi: 10.2337/db13-1954. [DOI] [PubMed] [Google Scholar]
- 101.Duarte AI, Santos MS, Oliveira CR, Moreira PI. Brain insulin signalling, glucose metabolism and females' reproductive aging: A dangerous triad in Alzheimer's disease. Neuropharmacology. 2018;136(Pt B):223–42. doi: 10.1016/j.neuropharm.2018.01.044. [DOI] [PubMed] [Google Scholar]
- 102.van der Harg J.M., Eggels L., Bangel F.N., Ruigrok S.R., Zwart R., Hoozemans J.J.M., la Fleur S.E., Scheper W. Insulin deficiency results in reversible protein kinase A activation and tau phosphorylation. Neurobiol. Dis. 2017;103:163–173. doi: 10.1016/j.nbd.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 103.Gratuze M., Joly-Amado A., Vieau D., Buée L., Blum D. Mutual relationship between tau and central insulin signalling: Consequences for AD and tauopathies? Neuroendocrinology. 2018;107(2):181–195. doi: 10.1159/000487641. [DOI] [PubMed] [Google Scholar]
- 104.Leboucher A., Ahmed T., Caron E., Tailleux A., Raison S., Joly-Amado A., Marciniak E., Carvalho K., Hamdane M., Bantubungi K., Lancel S., Eddarkaoui S., Caillierez R., Vallez E., Staels B., Vieau D., Balschun D., Buee L., Blum D. Brain insulin response and peripheral metabolic changes in a Tau transgenic mouse model. Neurobiol. Dis. 2019;125:14–22. doi: 10.1016/j.nbd.2019.01.008. [DOI] [PubMed] [Google Scholar]
- 105.Zhang S., Chai R., Yang Y.Y., Guo S.Q., Wang S., Guo T., Xu S.F., Zhang Y.H., Wang Z.Y., Guo C. Chronic diabetic states worsen Alzheimer neuropathology and cognitive deficits accompanying disruption of calcium signaling in leptin-deficient APP/PS1 mice. Oncotarget. 2017;8(27):43617–43634. doi: 10.18632/oncotarget.17116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rahman S.O., Panda B.P., Parvez S., Kaundal M., Hussain S., Akhtar M., Najmi A.K. Neuroprotective role of astaxanthin in hippocampal insulin resistance induced by Aβ peptides in animal model of Alzheimer’s disease. Biomed. Pharmacother. 2019;110:47–58. doi: 10.1016/j.biopha.2018.11.043. [DOI] [PubMed] [Google Scholar]
- 107.Najem D., Bamji-Mirza M., Yang Z., Zhang W. Abeta-induced insulin resistance and the effects of insulin on the cholesterol synthesis pathway and Abeta secretion in neural cells. Neurosci. Bull. 2016;32(3):227–238. doi: 10.1007/s12264-016-0034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhou Y., Xu B. New insights into anti-diabetes effects and molecular mechanisms of dietary saponins. Crit. Rev. Food Sci. Nutr. 2022:1–26. doi: 10.1080/10408398.2022.2101425. [DOI] [PubMed] [Google Scholar]
- 109.Zhou K., Chen Q., Chen J., Liang D., Feng W., Liu M., Wang Q., Wang R., Ouyang Q., Quan C., Chen S. Spatiotemporal regulation of insulin signaling by liquid–liquid phase separation. Cell Discov. 2022;8(1):64. doi: 10.1038/s41421-022-00430-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Akhtar A., Sah S.P. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020;135:104707. doi: 10.1016/j.neuint.2020.104707. [DOI] [PubMed] [Google Scholar]
- 111.Long H-Z., Cheng Y., Zhou Z-W., Luo H-Y., Wen D-D., Gao L-C. Pi3k/Akt signal pathway: a target of natural products in the prevention and treatment of Alzheimer’s disease and Parkinson’s disease. Front. Pharmacol. 2021;12:648636. doi: 10.3389/fphar.2021.648636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Das A., Bhattacharya B., Roy S. Decrypting a path based approach for identifying the interplay between PI3K and GSK3 signaling cascade from the perspective of cancer. Genes Dis. 2022;9(4):868–888. doi: 10.1016/j.gendis.2021.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rapaka D., Bitra V.R., Challa S.R., Adiukwu P.C. mTOR signaling as a molecular target for the alleviation of Alzheimer’s disease pathogenesis. Neurochem. Int. 2022;155:105311. doi: 10.1016/j.neuint.2022.105311. [DOI] [PubMed] [Google Scholar]
- 114.Mulder F.V.M., Peeters E.F.H.I., Westerink J., Zwartkruis F.J.T., de Ranitz-Greven W.L. The long-term effect of mTOR inhibition on lipid and glucose metabolism in tuberous sclerosis complex: Data from the Dutch TSC registry. Orphanet J. Rare Dis. 2022;17(1):252. doi: 10.1186/s13023-022-02385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Karwi Q.G., Lopaschuk G.D. Branched-chain amino acid metabolism in the failing heart. Cardiovasc. Drugs Ther. 2022 doi: 10.1007/s10557-022-07320-4. [DOI] [PubMed] [Google Scholar]
- 116.Du X., Di Malta C., Fang Z., Shen T., Niu X., Chen M., Jin B., Yu H., Lei L., Gao W., Song Y., Wang Z., Xu C., Cao Z., Liu G., Li X. Nuciferine protects against high-fat diet-induced hepatic steatosis and insulin resistance via activating TFEB-mediated autophagy–lysosomal pathway. Acta Pharm. Sin. B. 2022;12(6):2869–2886. doi: 10.1016/j.apsb.2021.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Moloney A.M., Griffin R.J., Timmons S., O’Connor R., Ravid R., O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging. 2010;31(2):224–243. doi: 10.1016/j.neurobiolaging.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 118.Bosco D., Fava A., Plastino M., Montalcini T., Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J. Cell. Mol. Med. 2011;15(9):1807–1821. doi: 10.1111/j.1582-4934.2011.01318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lanzillotta C., Di Domenico F., Perluigi M., Butterfield D.A. Targeting mitochondria in Alzheimer disease: Rationale and perspectives. CNS Drugs. 2019;33(10):957–969. doi: 10.1007/s40263-019-00658-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yu H., Lin X., Wang D., Zhang Z., Guo Y., Ren X., Xu B., Yuan J., Liu J., Spencer P.S., Wang J.Z., Yang X. Mitochondrial molecular abnormalities revealed by proteomic analysis of hippocampal organelles of mice triple transgenic for Alzheimer disease. Front. Mol. Neurosci. 2018;11:74. doi: 10.3389/fnmol.2018.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tang J., Oliveros A., Jang M.H. Dysfunctional mitochondrial bioenergetics and synaptic degeneration in Alzheimer disease. Int. Neurourol. J. 2019;23(Suppl. 1):S5–S10. doi: 10.5213/inj.1938036.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mastroeni D., McKee A., Grover A., Rogers J., Coleman P.D. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS One. 2009;4(8):e6617. doi: 10.1371/journal.pone.0006617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Armand-Ugon M., Ansoleaga B., Berjaoui S., Ferrer I. Reduced mitochondrial activity is early and steady in the entorhinal cortex but it is mainly unmodified in the frontal cortex in Alzheimer’s disease. Curr. Alzheimer Res. 2017;14(12):1327–1334. doi: 10.2174/1567205014666170505095921. [DOI] [PubMed] [Google Scholar]
- 124.Bosetti F., Brizzi F., Barogi S., Mancuso M., Siciliano G., Tendi E.A., Murri L., Rapoport S.I., Solaini G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging. 2002;23(3):371–376. doi: 10.1016/S0197-4580(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 125.Brown A.M., Gordon D., Lee H., Vrièze F.W-D., Cellini E., Bagnoli S., Nacmias B., Sorbi S., Hardy J., Blass J.P. Testing for linkage and association across the dihydrolipoyl dehydrogenase gene region with Alzheimer’s disease in three sample populations. Neurochem. Res. 2007;32(4-5):857–869. doi: 10.1007/s11064-006-9235-3. [DOI] [PubMed] [Google Scholar]
- 126.Ahmad W., Ebert P.R. 5-Methoxyindole-2-carboxylic acid (MICA) suppresses Aβ-mediated pathology in C. elegans. Exp. Gerontol. 2018;108:215–225. doi: 10.1016/j.exger.2018.04.021. [DOI] [PubMed] [Google Scholar]
- 127.Ahmad W. Dihydrolipoamide dehydrogenase suppression induces human tau phosphorylation by increasing whole body glucose levels in a C. elegans model of Alzheimer’s Disease. Exp. Brain Res. 2018;236(11):2857–2866. doi: 10.1007/s00221-018-5341-0. [DOI] [PubMed] [Google Scholar]
- 128.Butterfield D.A., Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019;20(3):148–160. doi: 10.1038/s41583-019-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ahmad W., Ijaz B., Shabbiri K., Ahmed F., Rehman S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/RNS generation. J. Biomed. Sci. 2017;24(1):76. doi: 10.1186/s12929-017-0379-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fan Y.G., Guo T., Han X.R., Liu J.L., Cai Y.T., Xue H., Huang X.S., Li Y.C., Wang Z.Y., Guo C. Paricalcitol accelerates BACE1 lysosomal degradation and inhibits calpain-1 dependent neuronal loss in APP/PS1 transgenic mice. EBioMedicine. 2019;45:393–407. doi: 10.1016/j.ebiom.2019.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rashid H.O., Yadav R.K., Kim H.R., Chae H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy. 2015;11(11):1956–1977. doi: 10.1080/15548627.2015.1091141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Poirier Y., Grimm A., Schmitt K., Eckert A. Link between the unfolded protein response and dysregulation of mitochondrial bioenergetics in Alzheimer’s disease. Cell. Mol. Life Sci. 2019;76(7):1419–1431. doi: 10.1007/s00018-019-03009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Coggan J.S., Keller D., Calì C., Lehväslaiho H., Markram H., Schürmann F., Magistretti P.J. Norepinephrine stimulates glycogenolysis in astrocytes to fuel neurons with lactate. PLOS Comput. Biol. 2018;14(8):e1006392. doi: 10.1371/journal.pcbi.1006392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vohra R., Kolko M. Lactate: More Than merely a metabolic waste product in the inner retina. Mol. Neurobiol. 2020;57(4):2021–2037. doi: 10.1007/s12035-019-01863-8. [DOI] [PubMed] [Google Scholar]
- 135.e, L.; Swerdlow, R.H. Lactate's effect on human neuroblastoma cell bioenergetic fluxes. Biochem. Pharmacol. 2016;99:88–100. doi: 10.1016/j.bcp.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Muraleedharan R., Gawali M.V., Tiwari D., Sukumaran A., Oatman N., Anderson J., Nardini D., Bhuiyan M.A.N., Tkáč I., Ward A.L., Kundu M., Waclaw R., Chow L.M., Gross C., Rao R., Schirmeier S., Dasgupta B. AMPK-regulated astrocytic lactate shuttle plays a non-cell-autonomous role in neuronal survival. Cell Rep. 2020;32(9):108092. doi: 10.1016/j.celrep.2020.108092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Barone E., Di Domenico F., Perluigi M., Butterfield D.A. The interplay among oxidative stress, brain insulin resistance and AMPK dysfunction contribute to neurodegeneration in type 2 diabetes and Alzheimer disease. Free Radic. Biol. Med. 2021;176:16–33. doi: 10.1016/j.freeradbiomed.2021.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lu W., Huang J., Sun S., Huang S., Gan S., Xu J., Yang M., Xu S., Jiang X. Changes in lactate content and monocarboxylate transporter 2 expression in Aβ25-35-treated rat model of Alzheimer’s disease. Neurol. Sci. 2015;36(6):871–876. doi: 10.1007/s10072-015-2087-3. [DOI] [PubMed] [Google Scholar]
- 139.Liguori C., Stefani A., Sancesario G., Sancesario G.M., Marciani M.G., Pierantozzi M. CSF lactate levels, τ proteins, cognitive decline: A dynamic relationship in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. 2015;86(6):655–659. doi: 10.1136/jnnp-2014-308577. [DOI] [PubMed] [Google Scholar]
- 140.Liguori C., Chiaravalloti A., Sancesario G., Stefani A., Sancesario G.M., Mercuri N.B., Schillaci O., Pierantozzi M. Cerebrospinal fluid lactate levels and brain [18F]FDG PET hypometabolism within the default mode network in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging. 2016;43(11):2040–2049. doi: 10.1007/s00259-016-3417-2. [DOI] [PubMed] [Google Scholar]
- 141.Wang Y., Li J., Wang M.Y., Pan Z.Y., Li Z.Q., Wang Z.F. Chronic microglial inflammation promotes neuronal lactate supply but impairs its utilization in primary rat astrocyte-neuron co-cultures. Biochem. Biophys. Res. Commun. 2022;607:28–35. doi: 10.1016/j.bbrc.2022.03.122. [DOI] [PubMed] [Google Scholar]
- 142.Chen S., Guo D., Lei B., Bi J., Yang H. Biglycan protects human neuroblastoma cells from nitric oxide-induced death by inhibiting AMPK-mTOR mediated autophagy and intracellular ROS level. Biotechnol. Lett. 2020;42(4):657–668. doi: 10.1007/s10529-020-02818-z. [DOI] [PubMed] [Google Scholar]
- 143.Pichiah P.B.T., Sankarganesh D., Arunachalam S., Achiraman S. Adipose-derived molecules-untouched horizons in Alzheimer’s disease biology. Front. Aging Neurosci. 2020;12:17. doi: 10.3389/fnagi.2020.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chauhan A.K., Mallick B.N. Association between autophagy and rapid eye movement sleep loss-associated neurodegenerative and patho-physio-behavioral changes. Sleep Med. 2019;63:29–37. doi: 10.1016/j.sleep.2019.04.019. [DOI] [PubMed] [Google Scholar]
- 145.Saito T., Kuma A., Sugiura Y., Ichimura Y., Obata M., Kitamura H., Okuda S., Lee H.C., Ikeda K., Kanegae Y., Saito I., Auwerx J., Motohashi H., Suematsu M., Soga T., Yokomizo T., Waguri S., Mizushima N., Komatsu M. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 2019;10(1):1567. doi: 10.1038/s41467-019-08829-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Harris M., El Hindy M., Usmari-Moraes M., Hudd F., Shafei M., Dong M., Hezwani M., Clark P., House M., Forshaw T., Kehoe P., Conway M.E. BCAT-induced autophagy regulates Aβ load through an interdependence of redox state and PKC phosphorylation-implications in Alzheimer’s disease. Free Radic. Biol. Med. 2020;152:755–766. doi: 10.1016/j.freeradbiomed.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 147.Füllgrabe J., Ghislat G., Cho D.H., Rubinsztein D.C. Transcriptional regulation of mammalian autophagy at a glance. J. Cell Sci. 2016;129(16):3059–3066. doi: 10.1242/jcs.188920. [DOI] [PubMed] [Google Scholar]
- 148.Schmukler E., Pinkas-Kramarski R. Autophagy induction in the treatment of Alzheimer’s disease. Drug Dev. Res. 2020;81(2):184–193. doi: 10.1002/ddr.21605. [DOI] [PubMed] [Google Scholar]
- 149.Hwang J.Y., Yan J., Zukin R.S. Autophagy and synaptic plasticity: Epigenetic regulation. Curr. Opin. Neurobiol. 2019;59:207–212. doi: 10.1016/j.conb.2019.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ruan H.B., Ma Y., Torres S., Zhang B., Feriod C., Heck R.M., Qian K., Fu M., Li X., Nathanson M.H., Bennett A.M., Nie Y., Ehrlich B.E., Yang X. Calcium-dependent O-GlcNAc signaling drives liver autophagy in adaptation to starvation. Genes Dev. 2017;31(16):1655–1665. doi: 10.1101/gad.305441.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yue J., Wang X., Feng B., Hu L., Yang L., Lu L., Zhang K., Wang Y., Liu S. Activation of G-protein-coupled receptor 30 protects neurons against excitotoxicity through inhibiting excessive autophagy induced by glutamate. ACS Chem. Neurosci. 2019;10(10):4227–4236. doi: 10.1021/acschemneuro.9b00287. [DOI] [PubMed] [Google Scholar]
- 152.Loos B., Klionsky D.J., Wong E. Augmenting brain metabolism to increase macro- and chaperone-mediated autophagy for decreasing neuronal proteotoxicity and aging. Prog. Neurobiol. 2017;156:90–106. doi: 10.1016/j.pneurobio.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 153.Rojas-Morales P., Tapia E., Pedraza-Chaverri J. β-Hydroxybutyrate: A signaling metabolite in starvation response? Cell. Signal. 2016;28(8):917–923. doi: 10.1016/j.cellsig.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 154.Onyango A.N. Cellular stresses and stress responses in the pathogenesis of insulin resistance. Oxid. Med. Cell. Longev. 2018;2018:1–27. doi: 10.1155/2018/4321714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Belgrad J., De Pace R., Fields R.D. Autophagy in myelinating glia. J. Neurosci. 2020;40(2):256–266. doi: 10.1523/JNEUROSCI.1066-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.He B., Perez S.E., Lee S.H., Ginsberg S.D., Malek-Ahmadi M., Mufson E.J. Expression profiling of precuneus layer III cathepsin D‐immunopositive pyramidal neurons in mild cognitive impairment and Alzheimer’s disease: Evidence for neuronal signaling vulnerability. J. Comp. Neurol. 2020;528(16):2748–2766. doi: 10.1002/cne.24929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lee J.H., Yu W.H., Kumar A., Lee S., Mohan P.S., Peterhoff C.M., Wolfe D.M., Martinez-Vicente M., Massey A.C., Sovak G., Uchiyama Y., Westaway D., Cuervo A.M., Nixon R.A. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Barbero-Camps E., Roca-Agujetas V., Bartolessis I., de Dios C., Fernández-Checa J.C., Marí M., Morales A., Hartmann T., Colell A. Cholesterol impairs autophagy-mediated clearance of amyloid beta while promoting its secretion. Autophagy. 2018;14(7):1129–1154. doi: 10.1080/15548627.2018.1438807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chan H.H., Koh R.Y., Lim C.L., Leong C.O. Receptor-interacting protein kinase 1 (RIPK1) as a potential therapeutic target: An overview of its possible role in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2019;16(10):907–918. doi: 10.2174/1567205016666191023102422. [DOI] [PubMed] [Google Scholar]
- 160.Dou J., Su P., Xu C., Wen Z., Mao Z., Li W. Targeting Hsc70-based autophagy to eliminate amyloid β oligomers. Biochem. Biophys. Res. Commun. 2020;524(4):923–928. doi: 10.1016/j.bbrc.2020.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Caballero B., Wang Y., Diaz A., Tasset I., Juste Y.R., Stiller B., Mandelkow E.M., Mandelkow E., Cuervo A.M. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell. 2018;17(1):e12692. doi: 10.1111/acel.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Silva P.N., Furuya T.K., Braga I.L., Rasmussen L.T., Labio R.W., Bertolucci P.H., Chen E.S., Turecki G., Mechawar N., Payão S.L., Mill J., Smith M.C. Analysis of HSPA8 and HSPA9 mRNA expression and promoter methylation in the brain and blood of Alzheimer’s disease patients. J. Alzheimers Dis. 2013;38(1):165–170. doi: 10.3233/JAD-130428. [DOI] [PubMed] [Google Scholar]
- 163.Yang X., Tohda C. Heat shock cognate 70 inhibitor, VER-155008, reduces memory deficits and axonal degeneration in a mouse model of Alzheimer’s disease. Front. Pharmacol. 2018;9:48. doi: 10.3389/fphar.2018.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gorantla N.V., Chinnathambi S. Tau protein squired by molecular chaperones during Alzheimer’s disease. J. Mol. Neurosci. 2018;66(3):356–368. doi: 10.1007/s12031-018-1174-3. [DOI] [PubMed] [Google Scholar]
- 165.Shimura H., Schwartz D., Gygi S.P., Kosik K.S. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 2004;279(6):4869–4876. doi: 10.1074/jbc.M305838200. [DOI] [PubMed] [Google Scholar]
- 166.Woo J.A., Liu T., Zhao X., Trotter C., Yrigoin K., Cazzaro S., Narvaez E.D., Khan H., Witas R., Bukhari A., Makati K., Wang X., Dickey C., Kang D.E. Enhanced tau pathology via RanBP9 and Hsp90/Hsc70 chaperone complexes. Hum. Mol. Genet. 2017;26(20):3973–3988. doi: 10.1093/hmg/ddx284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Itoh K., Nakamura K., Iijima M., Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013;23(2):64–71. doi: 10.1016/j.tcb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Barnett A., Brewer G.J. Autophagy in aging and Alzheimer’s disease: Pathologic or protective? J. Alzheimers Dis. 2011;25(3):385–394. doi: 10.3233/JAD-2011-101989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Geisler S., Holmström K.M., Skujat D., Fiesel F.C., Rothfuss O.C., Kahle P.J., Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010;12(2):119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 170.Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 171.Mary A., Eysert F., Checler F., Chami M. Mitophagy in Alzheimer’s disease: Molecular defects and therapeutic approaches. Mol. Psychiatry. 2022 doi: 10.1038/s41380-022-01631-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hu Y., Chen H., Zhang L., Lin X., Li X., Zhuang H., Fan H., Meng T., He Z., Huang H., Gong Q., Zhu D., Xu Y., He P., Li L., Feng D. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy. 2021;17(5):1142–1156. doi: 10.1080/15548627.2020.1749490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Manczak M., Kandimalla R., Yin X., Reddy P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018;27(8):1332–1342. doi: 10.1093/hmg/ddy042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Brewer G.J., Herrera R.A., Philipp S., Sosna J., Reyes-Ruiz J.M., Glabe C.G. Age-related intraneuronal aggregation of amyloid-beta in endosomes, mitochondria, autophagosomes, and lysosomes. J. Alzheimers Dis. 2020;73(1):229–246. doi: 10.3233/JAD-190835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nobili A., Krashia P., D’Amelio M. Cisd2: A promising new target in Alzheimer’s disease †. J. Pathol. 2020;251(2):113–116. doi: 10.1002/path.5436. [DOI] [PubMed] [Google Scholar]
- 176.Cummins N., Tweedie A., Zuryn S., Bertran-Gonzalez J., Götz J. Disease‐associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019;38(3):e99360. doi: 10.15252/embj.201899360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hu Y., Li X.C., Wang Z., Luo Y., Zhang X., Liu X.P., Feng Q., Wang Q., Yue Z., Chen Z., Ye K., Wang J.Z., Liu G.P. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget. 2016;7(14):17356–17368. doi: 10.18632/oncotarget.7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kuang H., Tan C.Y., Tian H.Z., Liu L.H., Yang M.W., Hong F.F., Yang S.L. Exploring the bi‐directional relationship between autophagy and Alzheimer’s disease. CNS Neurosci. Ther. 2020;26(2):155–166. doi: 10.1111/cns.13216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cavieres V.A., González A., Muñoz V.C., Yefi C.P., Bustamante H.A., Barraza R.R., Tapia-Rojas C., Otth C., Barrera M.J., González C., Mardones G.A., Inestrosa N.C., Burgos P.V. Tetrahydrohyperforin inhibits the proteolytic processing of amyloid precursor protein and enhances its degradation by Atg5-dependent autophagy. PLoS One. 2015;10(8):e0136313. doi: 10.1371/journal.pone.0136313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Feng T., Tammineni P., Agrawal C., Jeong Y.Y., Cai Q. Autophagy-mediated regulation of BACE1 protein trafficking and degradation. J. Biol. Chem. 2017;292(5):1679–1690. doi: 10.1074/jbc.M116.766584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wu H., Lu M.H., Wang W., Zhang M.Y., Zhu Q.Q., Xia Y.Y., Xu R.X., Yang Y., Chen L.H., Ma Q.H. Lamotrigine reduces beta-site AbetaPP-cleaving enzyme 1 protein levels through induction of autophagy. J. Alzheimers Dis. 2015;46(4):863–876. doi: 10.3233/JAD-143162. [DOI] [PubMed] [Google Scholar]
- 182.Nilsson P., Loganathan K., Sekiguchi M., Matsuba Y., Hui K., Tsubuki S., Tanaka M., Iwata N., Saito T., Saido T.C. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5(1):61–69. doi: 10.1016/j.celrep.2013.08.042. [DOI] [PubMed] [Google Scholar]
- 183.Jiang S., Zhao Y., Zhang T., Lan J., Yang J., Yuan L., Zhang Q., Pan K., Zhang K. Galantamine inhibits β-amyloid-induced cytostatic autophagy in PC12 cells through decreasing ROS production. Cell Prolif. 2018;51(3):e12427. doi: 10.1111/cpr.12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Benito-Cuesta I., Ordonez-Gutierrez L., Wandosell F. AMPK activation does not enhance autophagy in neurons in contrast to mTORc1 inhibition: Different impact on beta-amyloid clearance. Autophagy. 2020;•••:1–16. doi: 10.1080/15548627.2020.1728095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Vartak R.S., Rodin A., Oddo S. Differential activation of the mTOR/autophagy pathway predicts cognitive performance in APP/PS1 mice. Neurobiol. Aging. 2019;83:105–113. doi: 10.1016/j.neurobiolaging.2019.08.018. [DOI] [PubMed] [Google Scholar]
- 186.Han K., Kim S.H., Choi M. Computational modeling of the effects of autophagy on amyloid-β peptide levels. Theor. Biol. Med. Model. 2020;17(1):2. doi: 10.1186/s12976-020-00119-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Chen J.L., Luo C., Pu D., Zhang G.Q., Zhao Y.X., Sun Y., Zhao K.X., Liao Z.Y., Lv A.K., Zhu S.Y., Zhou J., Xiao Q. Metformin attenuates diabetes-induced tau hyperphosphorylation in vitro and in vivo by enhancing autophagic clearance. Exp. Neurol. 2019;311:44–56. doi: 10.1016/j.expneurol.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 188.Li X., Lu J., Xu Y., Wang J., Qiu X., Fan L., Li B., Liu W., Mao F., Zhu J., Shen X., Li J. Discovery of nitazoxanide-based derivatives as autophagy activators for the treatment of Alzheimer’s disease. Acta Pharm. Sin. B. 2020;10(4):646–666. doi: 10.1016/j.apsb.2019.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ejlerskov P., Rubinsztein D.C., Pocock R. IFNB/interferon-β regulates autophagy via a MIR1 -TBC1D15-RAB7 pathway. Autophagy. 2020;16(4):767–769. doi: 10.1080/15548627.2020.1718384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Reddy P.H., Oliver D.M.A. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells. 2019;8(5):488. doi: 10.3390/cells8050488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Menzies F.M., Fleming A., Caricasole A., Bento C.F., Andrews S.P., Ashkenazi A., Füllgrabe J., Jackson A., Jimenez Sanchez M., Karabiyik C., Licitra F., Lopez Ramirez A., Pavel M., Puri C., Renna M., Ricketts T., Schlotawa L., Vicinanza M., Won H., Zhu Y., Skidmore J., Rubinsztein D.C. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93(5):1015–1034. doi: 10.1016/j.neuron.2017.01.022. [DOI] [PubMed] [Google Scholar]
- 192.Kang S., Son S.M., Baik S.H., Yang J., Mook-Jung I. Autophagy-mediated secretory pathway is responsible for both normal and pathological tau in neurons. J. Alzheimers Dis. 2019;70(3):667–680. doi: 10.3233/JAD-190180. [DOI] [PubMed] [Google Scholar]
- 193.Berger Z., Ravikumar B., Menzies F.M., Oroz L.G., Underwood B.R., Pangalos M.N., Schmitt I., Wullner U., Evert B.O., O’Kane C.J., Rubinsztein D.C. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006;15(3):433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 194.Wang Y., Martinez-Vicente M., Krüger U., Kaushik S., Wong E., Mandelkow E.M., Cuervo A.M., Mandelkow E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009;18(21):4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Schaeffer V., Lavenir I., Ozcelik S., Tolnay M., Winkler D.T., Goedert M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain. 2012;135(7):2169–2177. doi: 10.1093/brain/aws143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ferreon J., Jain A., Choi K.J., Tsoi P., MacKenzie K., Jung S., Ferreon A. Acetylation disfavors tau phase separation. Int. J. Mol. Sci. 2018;19(5):1360. doi: 10.3390/ijms19051360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Aubry S., Shin W., Crary J.F., Lefort R., Qureshi Y.H., Lefebvre C., Califano A., Shelanski M.L. Assembly and interrogation of Alzheimer’s disease genetic networks reveal novel regulators of progression. PLoS One. 2015;10(3):e0120352. doi: 10.1371/journal.pone.0120352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Esteves A.R., Palma A.M., Gomes R., Santos D., Silva D.F., Cardoso S.M. Acetylation as a major determinant to microtubule-dependent autophagy: Relevance to Alzheimer’s and Parkinson disease pathology. Biochim. Biophys. Acta Mol. Basis Dis. 2019;1865(8):2008–2023. doi: 10.1016/j.bbadis.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 199.Chen X., Li Y., Wang C., Tang Y., Mok S.A., Tsai R.M., Rojas J.C., Karydas A., Miller B.L., Boxer A.L., Gestwicki J.E., Arkin M., Cuervo A.M., Gan L. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol. Neurodegener. 2020;15(1):2. doi: 10.1186/s13024-019-0354-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kirilyuk A., Shimoji M., Catania J., Sahu G., Pattabiraman N., Giordano A., Albanese C., Mocchetti I., Toretsky J.A., Uversky V.N., Avantaggiati M.L. An intrinsically disordered region of the acetyltransferase p300 with similarity to prion-like domains plays a role in aggregation. PLoS One. 2012;7(11):e48243. doi: 10.1371/journal.pone.0048243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wen M., Ding L., Zhang L., Zhang T., Teruyoshi Y., Wang Y., Xue C. Eicosapentaenoic acid-enriched phosphatidylcholine mitigated abeta1-42-induced neurotoxicity via autophagy-inflammasome pathway. J. Agric. Food Chem. 2019;67(49):13767–13774. doi: 10.1021/acs.jafc.9b05947. [DOI] [PubMed] [Google Scholar]
- 202.Gabbouj S., Ryhänen S., Marttinen M., Wittrahm R., Takalo M., Kemppainen S., Martiskainen H., Tanila H., Haapasalo A., Hiltunen M., Natunen T. Altered insulin signaling in Alzheimer’s disease brain-special emphasis on PI3K-AKT Pathway. Front. Neurosci. 2019;13:629. doi: 10.3389/fnins.2019.00629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zhang Y., Dong Z., Song W. NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct. Target. Ther. 2020;5(1):37. doi: 10.1038/s41392-020-0145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ali M., Gupta M., Wani A., Sharma A., Abdullaha M., Kour D., Choudhary S., Bharate S.B., Singh G., Kumar A. IIIM-941, a stilbene derivative inhibits NLRP3 inflammasome activation by inducing autophagy. Front. Pharmacol. 2021;12:695712. doi: 10.3389/fphar.2021.695712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zhou W., Xiao D., Zhao Y., Tan B., Long Z., Yu L., He G. Enhanced autolysosomal function ameliorates the inflammatory response mediated by the NLRP3 Inflammasome in Alzheimer’s disease. Front. Aging Neurosci. 2021;13:629891. doi: 10.3389/fnagi.2021.629891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gali C.C., Fanaee-Danesh E., Zandl-Lang M., Albrecher N.M., Tam-Amersdorfer C., Stracke A., Sachdev V., Reichmann F., Sun Y., Avdili A., Reiter M., Kratky D., Holzer P., Lass A., Kandimalla K.K., Panzenboeck U. Amyloid-beta impairs insulin signaling by accelerating autophagy-lysosomal degradation of LRP-1 and IR-β in blood-brain barrier endothelial cells in vitro and in 3XTg-AD mice. Mol. Cell. Neurosci. 2019;99:103390. doi: 10.1016/j.mcn.2019.103390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Puigoriol-Illamola D., Griñán-Ferré C., Vasilopoulou F., Leiva R., Vázquez S., Pallàs M. 11beta-HSD1 inhibition by RL-118 promotes autophagy and correlates with reduced oxidative stress and inflammation, enhancing cognitive performance in SAMP8 mouse model. Mol. Neurobiol. 2018;55(12):8904–8915. doi: 10.1007/s12035-018-1026-8. [DOI] [PubMed] [Google Scholar]
- 208.Hsieh C.F., Liu C.K., Lee C.T., Yu L.E., Wang J.Y. Acute glucose fluctuation impacts microglial activity, leading to inflammatory activation or self-degradation. Sci. Rep. 2019;9(1):840. doi: 10.1038/s41598-018-37215-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lane D.J.R., Ayton S., Bush A.I. Iron and Alzheimer’s disease: An update on emerging mechanisms. J. Alzheimers Dis. 2018;64(s1):S379–S395. doi: 10.3233/JAD-179944. [DOI] [PubMed] [Google Scholar]
- 210.Lee Y.S., Kalimuthu K., Seok Park Y., Makala H., Watkins S.C., Choudry M.H.A., Bartlett D.L., Tae Kwon Y., Lee Y.J. Ferroptotic agent‐induced endoplasmic reticulum stress response plays a pivotal role in the autophagic process outcome. J. Cell. Physiol. 2020;235(10):6767–6778. doi: 10.1002/jcp.29571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wang T., Xu S.F., Fan Y.G., Li L.B., Guo C. Iron pathophysiology in Alzheimer’s diseases. Adv. Exp. Med. Biol. 2019;1173:67–104. doi: 10.1007/978-981-13-9589-5_5. [DOI] [PubMed] [Google Scholar]
- 212.Conway O., Akpinar H.A., Rogov V.V., Kirkin V. Selective autophagy receptors in neuronal health and disease. J. Mol. Biol. 2020;432(8):2483–2509. doi: 10.1016/j.jmb.2019.10.013. [DOI] [PubMed] [Google Scholar]
- 213.Xian-hui D., Wei-juan G., Tie-mei S., Hong-lin X., Jiang-tao B., Jing-yi Z., Xi-qing C. Age-related changes of brain iron load changes in the frontal cortex in APPswe/PS1ΔE9 transgenic mouse model of Alzheimer’s disease. J. Trace Elem. Med. Biol. 2015;30:118–123. doi: 10.1016/j.jtemb.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 214.Wu J., Tuo Q., Lei P. Ferroptosis, a recent defined form of critical cell death in neurological disorders. J. Mol. Neurosci. 2018;66(2):197–206. doi: 10.1007/s12031-018-1155-6. [DOI] [PubMed] [Google Scholar]
- 215.Quiles del Rey M., Mancias J.D. NCOA4-mediated ferritinophagy: A potential link to neurodegeneration. Front. Neurosci. 2019;13:238. doi: 10.3389/fnins.2019.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Fujii J., Homma T., Kobayashi S. Ferroptosis caused by cysteine insufficiency and oxidative insult. Free Radic. Res. 2019:1–12. doi: 10.1080/10715762.2019.1666983. [DOI] [PubMed] [Google Scholar]
- 217.Yan N., Zhang J. Iron metabolism, ferroptosis, and the links with Alzheimer’s disease. Front. Neurosci. 2020;13:1443. doi: 10.3389/fnins.2019.01443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Chen Y., Li N., Wang H., Wang N., Peng H., Wang J., Li Y., Liu M., Li H., Zhang Y., Wang Z. Amentoflavone suppresses cell proliferation and induces cell death through triggering autophagy-dependent ferroptosis in human glioma. Life Sci. 2020;247:117425. doi: 10.1016/j.lfs.2020.117425. [DOI] [PubMed] [Google Scholar]
- 219.Nirmala J.G., Lopus M. Cell death mechanisms in eukaryotes. Cell Biol. Toxicol. 2020;36(3):145–640. doi: 10.1007/s10565-019-09496-2. [DOI] [PubMed] [Google Scholar]
- 220.Song X., Zhu S., Chen P., Hou W., Wen Q., Liu J., Xie Y., Liu J., Klionsky D.J., Kroemer G., Lotze M.T., Zeh H.J., Kang R., Tang D. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(-) activity. Curr. Biol. 2018;28(15):2388–2399.e5. doi: 10.1016/j.cub.2018.05.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kang R., Zhu S., Zeh H.J., Klionsky D.J., Tang D. BECN1 is a new driver of ferroptosis. Autophagy. 2018;14(12):2173–2175. doi: 10.1080/15548627.2018.1513758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Liu J., Yang M., Kang R., Klionsky D.J., Tang D. Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy. 2019;15(11):2033–2035. doi: 10.1080/15548627.2019.1659623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Wu Z., Geng Y., Lu X., Shi Y., Wu G., Zhang M., Shan B., Pan H., Yuan J. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl. Acad. Sci. USA. 2019;116(8):2996–3005. doi: 10.1073/pnas.1819728116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Devos D., Cabantchik Z.I., Moreau C., Danel V., Mahoney-Sanchez L., Bouchaoui H., Gouel F., Rolland A.S., Duce J.A., Devedjian J.C. Conservative iron chelation for neurodegenerative diseases such as Parkinson’s disease and amyotrophic lateral sclerosis. J. Neural Transm. (Vienna) 2020;127(2):189–203. doi: 10.1007/s00702-019-02138-1. [DOI] [PubMed] [Google Scholar]
- 225.Yan N., Zhang J.J. The emerging roles of ferroptosis in vascular cognitive impairment. Front. Neurosci. 2019;13:811. doi: 10.3389/fnins.2019.00811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Ashraf A., Jeandriens J., Parkes H.G., So P.W. Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: Evidence of ferroptosis. Redox Biol. 2020;32:101494. doi: 10.1016/j.redox.2020.101494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Li L.B., Chai R., Zhang S., Xu S.F., Zhang Y.H., Li H.L., Fan Y.G., Guo C. Iron exposure and the cellular mechanisms linked to neuron degeneration in adult mice. Cells. 2019;8(2):198. doi: 10.3390/cells8020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Weiland A., Wang Y., Wu W., Lan X., Han X., Li Q., Wang J. Ferroptosis and its role in diverse brain diseases. Mol. Neurobiol. 2019;56(7):4880–4893. doi: 10.1007/s12035-018-1403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Peña-Bautista C., Vento M., Baquero M., Cháfer-Pericás C. Lipid peroxidation in neurodegeneration. Clin. Chim. Acta. 2019;497:178–188. doi: 10.1016/j.cca.2019.07.037. [DOI] [PubMed] [Google Scholar]
- 230.Li C., Zhang Y., Liu J., Kang R., Klionsky D.J., Tang D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2020:1–13. doi: 10.1080/15548627.2020.1739447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Croteau E., Castellano C.A., Richard M.A., Fortier M., Nugent S., Lepage M., Duchesne S., Whittingstall K., Turcotte É.E., Bocti C., Fülöp T., Cunnane S.C. Ketogenic medium chain triglycerides increase brain energy metabolism in Alzheimer’s disease. J. Alzheimers Dis. 2018;64(2):551–561. doi: 10.3233/JAD-180202. [DOI] [PubMed] [Google Scholar]
- 232.Fortier M., Castellano C.A., Croteau E., Langlois F., Bocti C., St-Pierre V., Vandenberghe C., Bernier M., Roy M., Descoteaux M., Whittingstall K., Lepage M., Turcotte É.E., Fulop T., Cunnane S.C. A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement. 2019;15(5):625–634. doi: 10.1016/j.jalz.2018.12.017. [DOI] [PubMed] [Google Scholar]
- 233.Mujica-Parodi L.R., Amgalan A., Sultan S.F., Antal B., Sun X., Skiena S., Lithen A., Adra N., Ratai E.M., Weistuch C., Govindarajan S.T., Strey H.H., Dill K.A., Stufflebeam S.M., Veech R.L., Clarke K. Diet modulates brain network stability, a biomarker for brain aging, in young adults. Proc. Natl. Acad. Sci. USA. 2020;117(11):6170–6177. doi: 10.1073/pnas.1913042117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kraeuter A.K., Mashavave T., Suvarna A., van den Buuse M., Sarnyai Z. Effects of beta-hydroxybutyrate administration on MK-801-induced schizophrenia-like behaviour in mice. Psychopharmacology (Berl.) 2020;237(5):1397–1405. doi: 10.1007/s00213-020-05467-2. [DOI] [PubMed] [Google Scholar]
- 235.Lee D.C., Vali K., Baldwin S.R., Divino J.N., Feliciano J.L., Fequiere J.R., Fernandez M.A., Frageau J.C., Longo F.K., Madhoun S.S., Mingione V.P., O’Toole T.R., Ruiz M.G., Tanner G.R. Dietary supplementation with the ketogenic diet metabolite beta-hydroxybutyrate ameliorates post-TBI aggression in young-adult male drosophila. Front. Neurosci. 2019;13:1140. doi: 10.3389/fnins.2019.01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Bahr L.S., Bock M., Liebscher D., Bellmann-Strobl J., Franz L., Prüß A., Schumann D., Piper S.K., Kessler C.S., Steckhan N., Michalsen A., Paul F., Mähler A. Ketogenic diet and fasting diet as Nutritional Approaches in Multiple Sclerosis (NAMS): Protocol of a randomized controlled study. Trials. 2020;21(1):3. doi: 10.1186/s13063-019-3928-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.McCarty M.F., DiNicolantonio J.J., O’Keefe J.H. Ketosis may promote brain macroautophagy by activating Sirt1 and hypoxia-inducible factor-1. Med. Hypotheses. 2015;85(5):631–639. doi: 10.1016/j.mehy.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 238.Miles K.N., Skelton M.R. Male mice placed on a ketogenic diet from postnatal day (P) 21 through adulthood have reduced growth, are hypoactive, show increased freezing in a conditioned fear paradigm, and have spatial learning deficits. Brain Res. 2020;1734:146697. doi: 10.1016/j.brainres.2020.146697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Vargas-Soria M., Carranza-Naval M.J., del Marco A., Garcia-Alloza M. Role of liraglutide in Alzheimer’s disease pathology. Alzheimers Res. Ther. 2021;13(1):112. doi: 10.1186/s13195-021-00853-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Xie Y., Zheng J., Li S., Li H., Zhou Y., Zheng W., Zhang M., Liu L., Chen Z. GLP-1 improves the neuronal supportive ability of astrocytes in Alzheimer’s disease by regulating mitochondrial dysfunction via the cAMP/PKA pathway. Biochem. Pharmacol. 2021;188:114578. doi: 10.1016/j.bcp.2021.114578. [DOI] [PubMed] [Google Scholar]
- 241.Zheng J., Xie Y., Ren L., Qi L., Wu L., Pan X., Zhou J., Chen Z., Liu L. GLP-1 improves the supportive ability of astrocytes to neurons by promoting aerobic glycolysis in Alzheimer’s disease. Mol. Metab. 2021;47:101180. doi: 10.1016/j.molmet.2021.101180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Poor S.R., Ettcheto M., Cano A., Sanchez-Lopez E., Manzine P.R., Olloquequi J., Camins A., Javan M. Metformin a potential pharmacological strategy in late onset Alzheimer’s disease treatment. Pharmaceuticals (Basel) 2021;14(9):890. doi: 10.3390/ph14090890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Femminella G.D., Bencivenga L., Petraglia L., Visaggi L., Gioia L., Grieco F.V., de Lucia C., Komici K., Corbi G., Edison P., Rengo G., Ferrara N. Antidiabetic drugs in Alzheimer’s disease: Mechanisms of action and future perspectives. J. Diabetes Res. 2017;2017:1–7. doi: 10.1155/2017/7420796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Rosenbloom M., Barclay T., Johnsen J., Erickson L., Svitak A., Pyle M., Frey W., Hanson L.R. Double-blind placebo-controlled pilot investigation of the safety of a single dose of rapid-acting intranasal insulin in Down Syndrome. Drugs R D. 2020;20(1):11–15. doi: 10.1007/s40268-020-00296-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Lv H., Tang L., Guo C., Jiang Y., Gao C., Wang Y., Jian C. Intranasal insulin administration may be highly effective in improving cognitive function in mice with cognitive dysfunction by reversing brain insulin resistance. Cogn. Neurodynamics. 2020;14(3):323–338. doi: 10.1007/s11571-020-09571-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Yu J-M., Jiang G-H., Zhu Y., Huang Y., Yang J., Tu R., Zhang X., He W-W., Hou C-Y., Wang X-M. Intranasal insulin ameliorates neurological impairment after intracerebral hemorrhage in mice. Neural Regen. Res. 2022;17(1):210–216. doi: 10.4103/1673-5374.314320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Li X., Run X., Wei Z., Zeng K., Liang Z., Huang F., Ke D., Wang Q., Wang J.Z., Liu R., Zhang B., Wang X. Intranasal insulin prevents anesthesia-induced cognitive impairments in aged mice. Curr. Alzheimer Res. 2018;16(1):8–18. doi: 10.2174/1567205015666181031145045. [DOI] [PubMed] [Google Scholar]
- 248.Bazrgar M., Khodabakhsh P., Dargahi L., Mohagheghi F., Ahmadiani A. MicroRNA modulation is a potential molecular mechanism for neuroprotective effects of intranasal insulin administration in amyloid βeta oligomer induced Alzheimer’s like rat model. Exp. Gerontol. 2022;164:111812. doi: 10.1016/j.exger.2022.111812. [DOI] [PubMed] [Google Scholar]
- 249.Barone E., Tramutola A., Triani F., Calcagnini S., Di Domenico F., Ripoli C., Gaetani S., Grassi C., Butterfield D.A., Cassano T., Perluigi M. Biliverdin reductase-a mediates the beneficial effects of intranasal insulin in Alzheimer disease. Mol. Neurobiol. 2019;56(4):2922–2943. doi: 10.1007/s12035-018-1231-5. [DOI] [PubMed] [Google Scholar]
- 250.Kellar D., Lockhart S.N., Aisen P., Raman R., Rissman R.A., Brewer J., Craft S. Intranasal insulin reduces white matter hyperintensity progression in association with improvements in cognition and CSF biomarker profiles in mild cognitive impairment and Alzheimer’s disease. J. Prev. Alzheimers Dis. 2021;8(3):240–248. doi: 10.14283/jpad.2021.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kellar D., Register T., Lockhart S.N., Aisen P., Raman R., Rissman R.A., Brewer J., Craft S. Intranasal insulin modulates cerebrospinal fluid markers of neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A randomized trial. Sci. Rep. 2022;12(1):1346. doi: 10.1038/s41598-022-05165-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Craft S., Baker L.D., Montine T.J., Minoshima S., Watson G.S., Claxton A., Arbuckle M., Callaghan M., Tsai E., Plymate S.R., Green P.S., Leverenz J., Cross D., Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012;69(1):29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Craft S., Raman R., Chow T.W., Rafii M.S., Sun C.K., Rissman R.A., Donohue M.C., Brewer J.B., Jenkins C., Harless K., Gessert D., Aisen P.S. Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer disease dementia: A randomized clinical trial. JAMA Neurol. 2020;77(9):1099–1109. doi: 10.1001/jamaneurol.2020.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lee H., Zandkarimi F., Zhang Y., Meena J.K., Kim J., Zhuang L., Tyagi S., Ma L., Westbrook T.F., Steinberg G.R., Nakada D., Stockwell B.R., Gan B. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020;22(2):225–234. doi: 10.1038/s41556-020-0461-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Lee H., Zhuang L., Gan B. Energy stress inhibits ferroptosis via AMPK. Mol. Cell. Oncol. 2020;7(4):1761242. doi: 10.1080/23723556.2020.1761242. [DOI] [PMC free article] [PubMed] [Google Scholar]