

Abstract
Excessive consumption of red and processed meat has been associated with a higher risk of developing colorectal cancer. There are many attempts to explain the risk of colorectal cancer associated with the consumption of red and processed meat:
The temperature cooking of meat such as grilling and smoking contribute to the formation of mutagenic compounds including heterocyclic amines and polycyclic aromatic hydrocarbons.
Heme iron in red meat is involved in the formation of N-nitroso compounds and lipid peroxidation products in the digestive tract.
Fatty red meat is involved in the production of secondary bile acids by the bacteria of the gut microbiota.
Many of the products formed are genotoxic and can cause DNA damage and initiate carcinogenesis of colorectal cancer. Various mechanisms contributing to their genotoxic role have been established in human and animal studies. In addition, there is increasing evidence that compounds formed from red and processed meat interact with the gut microbiota in colorectal cancer pathways. Although several early studies in animals and humans suggest a direct causal role of the gut microbiota in the development of colorectal cancer, the links between diet, gut microbiota, and colonic carcinogenesis are largely associations rather than proven causal relationships. Various biological mechanisms, including inflammation and oxidative stress can lead to DNA damage, gut dysbiosis, and therefore increase the risk of colorectal cancer. Dysbiosis of the gut microbiota may increase the risk of colorectal cancer through dietary component promotion of colonic carcinogenesis. In this paper, we review and update current knowledge about the relationships between red meat consumption, gut microbiota, and colorectal cancer.
Keywords: Diet, Red meat, Gut microbiota, Colorectal Cancer, Dysbiosis
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer, diagnosed in both men and women, with an estimated 1.9 million new cases and 935 000 deaths reported in 2020, worldwide [1–3]. However, substantial disparities in both CRC incidence and mortality exist by geographical region, as shown in Tables 1 and 2.
Table 1:
Region-specific incidence rates by sex for cancers of the colon and rectum in 2020.
| Region | Males/ Incidence rate per 100,000 residents | Females/ Incidence rate per 100,000 residents | Males/Incidence rate per 100,000 residents | Females/Incidence rate per 100,000 residents |
|---|---|---|---|---|
| Colon cancer | Colon cancer | Rectum cancer | Rectum cancer | |
| Southern Europe | 25.3 | 16.4 | 14.1 | 7.3 |
| Northern Europe | 23.2 | 18.8 | 15.1 | 8.4 |
| Australia/New Zealand | 22.8 | 20.0 | 13.6 | 7.7 |
| Eastern Europe | 21.1 | 14.0 | 16.9 | 8.9 |
| Western Europe | 20.0 | 15.1 | 13.3 | 6.8 |
| Northern America | 17.4 | 15.0 | 11.0 | 6.6 |
| Eastern Asia | 16.4 | 13.0 | 14.6 | 7.7 |
| Caribbean | 14.1 | 13.8 | 3.9 | 3.4 |
| South America | 12.8 | 10.8 | 7.2 | 5.0 |
| Micronesia/Polynesia | 12.3 | 7.9 | 7.0 | 5.0 |
| Western Asia | 11.7 | 8.7 | 7.9 | 5.2 |
| South-Eastern Asia | 9.5 | 6.3 | 8.5 | 5.3 |
| Central America | 8.7 | 7.0 | 3.0 | 1.9 |
| Southern Africa | 8.7 | 5.9 | 7.2 | 5.0 |
| Melanesia | 6.7 | 3.4 | 5.8 | 3.9 |
| Northern Africa | 5.9 | 5.4 | 4.1 | 3.4 |
| Eastern Africa | 4.2 | 3.5 | 3.5 | 3.1 |
| Western Africa | 4.0 | 2.9 | 3.0 | 2.2 |
| South-Central Asia | 3.4 | 2.2 | 2.8 | 1.9 |
| Middle Africa | 2.9 | 2.3 | 3.6 | 2.8 |
Table 2:
Colorectal cancer new cases and deaths by geographic region (2020).
| Region | New cases | Deaths |
|---|---|---|
| Asia | 957,896 (51.8%) | 461,422 (52.4%) |
| Europe | 499,667 (27.0%) | 242,483 (27.5%) |
| North America | 179,715 (9.7%) | 64,105 (7.3%) |
| Latin America, Caribbean | 128,006 (6.9%) | 64,666 (7.3%) |
| Africa | 61,846 (3.3%) | 40,034 (4.5%) |
| Oceania | 22,332 (1.2%) | 8,066 (0.9%) |
Rectal and colon cancers related deaths are estimated to be in 60% and 71.5% respectively in 2035), making CRC a worldwide public health concern [3–5]. Of major concern, as well, is the increasing incidence of cases and deaths in youth CRC pathophysiology is associated with multiple risk factors including diet, diabetes, obesity, lifestyle, genes, and specific diseases, such as Crohn’s disease, ulcerative colitis, and dysbiosis [6–8]. Epidemiological studies have highlighted specific diets that are likely to be associated with CRC risk. On particular, a red meat enriched diet, low fiber intake, and heavy alcohol intake have been shown to adversely affect the risk of CRC [9,10]. Because of the way they are preserved (combination of salt, nitrate or nitrite), processed meats are exposed to the formation of carcinogens during the high temperature cooking process [11,12]. Such a long-term regimen may promote an increased risk of CRC. Other modifiable risk factors, such as low levels of physical activity, being overweight and smoking may increase CRC risk [13,14]. This evidence suggests that the risk of CRC may be reduced by diet, in addition to health behaviors.
Of 478,040 participants enrolled in the European Prospective Investigation into Cancer and Nutrition (EPIC) trial who were prospectively followed between 1992–1998, 1,329 CRC cases were detected and an association with red and processed meat consumption was observed [15]. Recently, the International Agency for Research on Cancer (IARC) and World Cancer Research Fund-American Institute for Cancer Research (WCRF-AICR) concluded that there is sufficient evidence to support that high consumption of processed meat may increase CRC risk and that evidence of increased risk from red meat consumption is thought to be either putative or probable [16]. Indeed, excessive consumption of red meat has a significant impact on the composition and function of the gut microbiota [17–19]. The human digestive tract is home to no less than 1012 to 1014 microorganisms, including bacteria (the most abundant), viruses, parasites, and non-pathogenic fungi [20,21]. The gut microbiota contains10-fold more cells than the human body and about 150-fold more genes than the human genome [22]. The gut microbiota composition changes with diet, sex, age, race, and lifestyle [21]. After colonization at birth to about 2 years of age, the gut microbiota is unique to each individual before becoming stable, over time [20]. In addition, environmental pollutants such as heterocyclic amines (HCA), polycyclic aromatic hydrocarbons (PAH) that contaminate red meat during cooking process at high temperature and the metabolic by-products (N-nitroso compounds, secondary bile acids, heme, trimethylamine-N-oxide (TMAO)) may interact with the gut microbiota and promote CRC carcinogenesis [17,19,23–25]. Moreover, the microbiome community pattern of the gut is disrupted in patients with CRC compared to healthy individuals [26,27].
In this paper, we review and update current knowledge regarding the interaction between the gut microbiota and CRC, the impact of red meat consumption on the gut microbiota, and the contribution of red meat consumption to the pathogenesis of CRC.
LITERATURE REVIEW
Red meat consumption and colorectal cancer
Numerous epidemiological and scientific studies suggest that the risk of CRC is increased by red and processed meat consumption [21,28,29]. According to WCRF/AICR reports published in 2007 and 2018, excessive consumption of red and processed meat convincingly increases the risk of CRC [30,31]. Red and processed meat was classified as a carcinogen at the same risk level as cigarettes and alcohol by the World Health Organizations International Agency for Research on Cancer in 2015 [32]. Because of its probable carcinogenicity, red and processed meat has been classified as a carcinogen 2A Group [33]. The risk of CRC was found to be increased by 16% for an additional 50 g/day of red and processed meat consumption and by 22% if consumption increased to 100 g/day [34]. Because of its nitrite conservation method, processed meat may present a higher risk per gram of intake than red meat [35,36]. Once produced, processed meat is preserved by methods other than freezing, including curing (the combination of salt, sugar and nitrate or nitrite), drying, smoking, cooking and packaging. According to traditional recipes specific to regions, this list of processed meat and conservation methods is not exhaustive, as there are many other manufacturing and conservation methods worldwide. However, these different conservation methods expose the meat to carcinogenic products (N-nitroso, HCA, HAP) formed during the cooking process at high temperature, especially brining and smoking [11,12]. To date, there are no clearly established biological mechanisms that could explain the role of red and processed meat in the process of CRC carcinogenesis. However, several hypotheses have been formulated and tested by experimental studies to try to explain how red and processed meat could increase the risk of CRC as shown in Figure 1. Experimentally the hypotheses that were tested are: (a) that high-fat meat might promote CRC carcinogenesis by induction of cytotoxic secondary bile acid production; (b) that high temperature meat cooking processes form mutagenic heterocyclic HCA and PAH; (c) that potentially carcinogenic N-nitroso compounds (NOC) are formed exogenously in meat and/or endogenously by nitrosation of amines and amides; (d) that heme iron from red meat may promote carcinogenesis through the formation of NOC and lipid peroxidation products.
Figure 1:
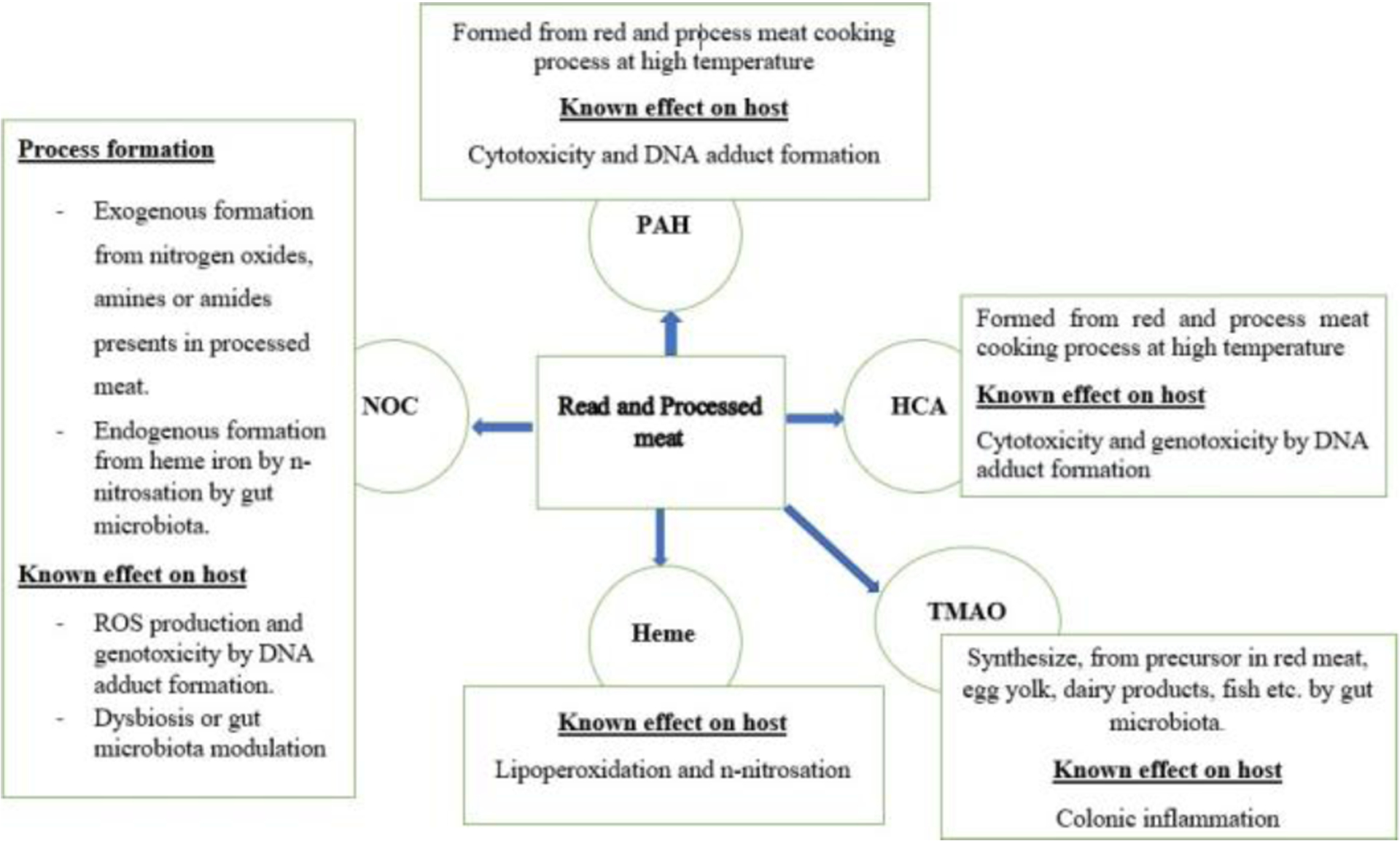
Read meat and processed increases CRC risk. Red and processed meat could promote colon carcinogenesis via different mechanisms such as toxic metabolites production, Lipoperoxidation and n-nitrosation, DNA adduct formation, and colonic inflammation. Red and processed meat can be contaminated by environmental pollutants, including HCA and PAH, during cooking. The International Agency for Research on Cancer (IARC) has classified the HCA and PAH as potential human carcinogens. The intestinal microbiota digests the proteins contained in ingested red meat to generate carcinogenic metabolites such as NOC, TMAO, and heme iron.
Ingestion of red meat rich in fat and excessive secretion bile acid (BA) have been cited in several animal and human experimental studies as one of the factors promoting the increased risk of CRC. Primary BAs, including cholic acid (CA) and chenodeoxycholic acid (CDCA), are synthesized by the liver following the digestion of dietary lipids in the stomach to emulsify fats. After lipid digestion, most primary BAs are deconjugated and reabsorbed (enterohepatic recycling). Still, a small amount may pass into the colon where they are transformed into secondary BAs (deoxycholic acid and lithocholic acid) by colonic bacteria by enzyme 7α-dehydroxylation as shown in Figure 2. The genus Clostridium is the main 7α-dehydroxylation-producing colonic bacterium [22,26,37]. Mice fed with deoxycholic acid developed significant intestinal inflammation 24 weeks after accumulation of deoxycholic acid in their feces [38]. A 3 to 4-fold higher level of secondary BA has been reported in a study of African-Americans who consumed a high-fat diet compared to native Africans who consumed a low-fat diet, suggesting an elevated risk of CRC in African Americans [39]. A similar study, in 2015, based on a change in the diet of African-Americans, from a high-fat to a low-fat diet, showed a significant decrease in secondary BA secretion and a decrease inflammation of the colonic mucosa [40]. BA with detergent properties can damage the colonic epithelium when presented in high concentrations in the colon. This destruction of colonic cells can lead to inflammation and increased proliferation of stem cells in the colon, leading to a pre-cancerous state [41]. Secondary BA may also promote the growth and multiplication of transformed stem cells in the colon by modulating the Wnt/β-catenin and M3R signaling pathways in cancer cells [42]. Short-term exposure to secondary BAs induce reactive oxygen species (ROS) production in the colon responsible for DNA damage. In contrast, long-term exposure causes inhibition of the tumor suppressor gene (TP53) or activation of the PI3K/Akt signaling pathway in colonic cells contributing to CRC carcinogenesis [43–46]. Through the farnesoid X receptor located on the nuclear membrane of colon cells, deoxycholic acid can mediate CRC carcinogenesis by inhibiting mucosal scarring in an in vivo mouse model [47].
Figure 2:

CRC risk related to the consumption of high-fat red meat. A high-fat diet upregulates the production of primary bile acid in the stomach. Some of this primary bile acid transits into the colon, transforming it into secondary BA by the gut microbiota. Secondary BAs are involved in several mechanisms leading to CRC carcinogenesis, including producing genotoxic substances (aromatic amino acids, hydrogen sulfide) and dysfunctional gut microbiota metabolism.
HCA are formed from products in processed meat (creatinine, amino acids, sugars) when cooked at high temperature [48]. The formation of HCA depends on the type of meat, temperature, time and cooking method. In addition to red and processed meat, poultry and fish also have HCA. The greatest amounts of HCA are produced during high temperature cooking processes such as grilling, frying and barbecuing [49]. The main HCA formed are 2-amino-3,4,8-dimethylimidazo (4,5-f) quinoxaline (DiMeIQx), amino-3-methylimidazo (4,5-f) quinolone (IQ); 2-amino-1-methyl-6-phenylimidazo(4,5-b) pyridine (PhIP); 2-amino-3,8-dimethylimidazo(4,5-f) quinoxaline (MeIQx); and 2-amino-3,4-dimethylimidazo (4,5-f) quinoline (MeIQ). The International Agency for Research on Cancer (IARC) has classified the HCA PhIP, MeIQ, and MeIQx as potential human carcinogens, while IQ is considered a probable human carcinogen [33,50]. In 2015, a Japanese study, of colonoscopies of Japanese women, showed that a high concentration of MeIQ with exposure to PhIP was correlated with a high risk of CRC [51]. A European prospective cohort study of 25,540 participants similarly reported that PhIP was associated with a high risk of colorectal adenoma [52]. Conversely, several other studies have shown that an increase in the co-occurring concentration of the HCA DiMeIQx, MeIQx, and PhIP are positively associated with a significant risk of colorectal adenoma [49,53]. HCA are genotoxic and carcinogenic in animal models. They are involved in the formation of DNA adducts following an N-oxidation reaction catalyzed by the cytochrome P450 enzyme and an O-esterification by N-acetyltransferases [54,55]. PAH are formed from high-temperature domestic cooking and industrial activities as a result of the incompetent combustion of organic materials (coal, crude oil, and gasoline). During cooking, especially red and processed meat, food may be contaminated with PAH over an open flame. With the same action characteristics as HCA, PAH may also increase the risk of CRC in humans. (49) PAH, mainly benzopyrenes or called benzo(α)pyrene (B(α)P), have been suspected to increase the risk of CRC in humans [49]. In experimental studies, particularly in mice, B(α)P were implicated in different mechanisms of CRC carcinogenesis, including DNA adduct formation, induction of oxidative stress, and increased expression of proinflammatory cytokines with dysregulation of the wnt/β-catenin signaling pathway [56,57].
Hughes et al. reported, for the first time in 2001, a significant excretion of NOC in the feces of volunteers who consumed large amounts of red meat [58]. NOC are formed exogenously from nitrogen oxides and amines or amides present in processed meat on the one hand and on the other hand endogenously by decarboxylation of the gut microbiota, followed by n-nitrosation in presence of nitrite [15,49]. These are most often found in certain processed foods such as smoked cheese, smoked fish, cold cuts, ham, sausages. It has been shown that heme iron lead to increased endogenous formation of NOC including N-nitrosothiols, and nitrosylated heme in the gastrointestinal tract during digestion of red meat [59]. These NOC formed can alkylate guanine at the O6 position on DNA resulting in the formation of promutagenic O 6-methylguanine and O 6-carboxymethylguanine lesions, which if not repaired quickly by the DNA repair enzyme O 6-methylguanine-DNA methyltransferase, could lead to genetic mutations and subsequently to the development of CRC [60–62]. Furthermore, in another study, a group of DNA adducts related to NOC and lipid peroxidation were identified as potential markers of red meat digestion. These DNA adducts include methylguanine; 3,N4-etheno-C; guanidinohydantoin; carboxyethyl-T; dimethyl-T; hydroxymethyl-T; tetramethyl-T; 6-carboxymethylguanine; and hydroxyethyl-T [63]. In addition, DNA adducts related to nitroso compounds and lipid peroxidation have been implicated in multiple genetic alterations by causing mutations in key colon cancer genes such as Adenomatous Polyposis Coli (APC), tumor suppressor gene (TP53) and Kirsten rat sarcoma virus (KRAS) [64].
This evidence suggests that NOC, formed from red and processed meat, can promote the development of CRC by inducing mutations in key tumor suppressor genes, including APC and TP53, and oncogenes such as KRAS.
The carcinogenic role of heme iron in red and processed meats is supported by numerous epidemiological and experimental studies [64,65]. Three main mechanisms of heme-induced colorectal carcinogenesis have been described: cytotoxicity of heme by accelerating programmed cell death and epithelial hyperplasia; heme-induced lipid peroxidation and DNA adduct formation and mutation of the APC gene; and heme catalysis of NOC resulting in genetic mutation as explained above [66–69]. Heme is catalyzed by transforming hydroperoxides into ROS [64]. The ROS formed have been suspected of being responsible for the cytotoxic effect of heme, according to a study by Pierre et al. in 2007 [70]. An in vitro study, performed on colonic cells, showed that a heme concentration higher than 100 μM resulted in colonocyte toxicity [71]. Heme increases the membrane permeability of the colonocytes, which subsequently leads to cell lysis.(64) Similarly, in 2012, Ijessennagger et al. showed that mice fed a high heme concentration during the first two days old exhibited acute oxidative stress of their colonocytes, as revealed by the release of Vnn1, a marker of oxidative stress. Cross et al., in 2003, in an evaluation of the effect of heme iron supplementation of a red meat diet, showed a significant increase in the fecal concentration of NOC [72]. Similarly, in 2010, they found a strong correlation between CRC risk and consumption of heme iron, nitrates, and processed meat [73].
The mechanism of trimethylamine–N-oxide (TMAO)-mediated CRC carcinogenesis in humans and animals has been examined in several studies [74]. TMAO is a metabolite of the gut microbiota produced from precursor molecules (choline, L-carnitine, phosphatidylcholine, betaine), which are abundantly present in red meat, egg yolk, dairy products, fish, vegetables, and fruits. The precursors of TMAO are converted to trimethylamine by the gut microbiota, then absorbed in the small intestine, and transported by the portal vein to the liver where they interact with flavin monooxygenase 3 (FMO3) and produce TMAO [26,75].
The inescapable role of the gut microbiota in the generation of TMAO was demonstrated by a study of antibiotics given to human subjects for one week to eliminate the gut microbiota [76]. The elimination of the gut microbiota by antibiotics was positively associated with decreased plasma and urinary TMAO levels compared to untreated controls. The same phenomenon has been reported in animal experiments [77]. In 2014, a study showed a correlation between plasma TMAO levels and CRC in women with low plasma vitamin B12 levels [78]. A similar study, in 2017, revealed that the precursor choline was associated with a 3-fold increased risk of CRC in men with elevated blood levels [79]. Evidence has suggested that TMAO may promote inflammation via various mechanisms, including increased expression of pro-inflammatory genes (cytokine genes, IL6, TNF-α, and chemokine ligands CXCL1, CXCL2) and increased pro-inflammatory effects mediated by H. pylori infection in the stomach [80–84]. The generation of N-nitroso compounds, known as genotoxic agents, is another mechanism of TMAO involved in the carcinogenesis of CRC [85]. It may also be involved in NLRP3 inflammasome activation and ROS production in human colonic cells, preventing their down-regulation [86]. In view of this evidence, CRC carcinogenesis may be activated by TMAO pro-inflammatory roles.
Red meat consumption and gut microbiota
The interaction between compounds formed from red meat directly or indirectly after its ingestion, including N-nitroso compounds (NOC), heterocyclic amines (HCA), polycyclic aromatic hydrocarbons (PAH), heme iron, bile acids (BA), Trimethylamine-N-oxide (TMAO) and the gut microbiota has been reported in numerous animal and human studies as shown in Table 3. Naturally, the number of NOC-producing bacteria (Escherichia, Pseudomonas, Proteus, Klebsiella, and Neisseria) is low in humans, but may increase with a diet rich in nitrates or nitrites [32,87–89]. Nitrates are found in high concentrations in processed meats and in certain foods, such as vegetables (beets, celery, lettuce, radishes, spinach) [90]. Nitrate ingested through these foods is reduced to nitrite by oral and digestive tract bacteria. The nitrites once formed react with amines, amides and other precursors by nitrosation in the gastrointestinal tract to form NOC compounds. Animal studies have shown that NOC compounds are potent carcinogens in animals [91–93]. In mice, for example, the colon in mice with colitis is enriched with E. coli during an intestinal inflammatory response mediated by nitrate compounds [17]. Previous studies have not been able to provide consistent evidence of an association between exposure to NOC and an increased risk of CRC in humans [94,95]. A study of European populations showed an association between N-nitrosodimethylamine from food sources and increased risk of CRC [94]. Although there have been few studies of NOC compounds and increased risk of CRC, there is strong evidence of an association between red and processed meat consumption and increased risk of CRC [28–32]. Therefore, excessive consumption of processed meat can lead to intestinal dysbiosis and a high risk of CRC carcinogenesis by promoting the multiplication of NOC-producing bacteria [26,96,97].
Table 3:
Interactions between red meat-associated agents and gut microbiota that may increase CRC risk.
| Food ingredients | Derived compounds | Action of derived compounds on gut microbiota | Action of gut microbiota on derived compounds | References |
|---|---|---|---|---|
| NOC | Promotes selective growth of NOC-producing bacteria, creating a state of dysbiosis that is the origin of CRC. | - | (89) | |
| HCA and PAH | Promote colonic inflammation by altering the abundance, composition and metabolic activities of the gut microbiota. | Transform HCA and PAH into less toxic substances. | (23,99,100,193–195) | |
| Read and processed meat | TMAO | - | Involved in the synthesis of TMAO from precursors such as choline, L-carnitine, phosphatidylcholine, and betaine. | (76,196–198) |
| Heme | Increases proteobacteria and Bacteroides abundance; reduces Firmicutes and Deferribacteres; promotes adenoma formation; decreases stool butyrate levels. | Increased heme-induced lipoperoxidation, hyperproliferation ofcolonic tissue. | (18,19,102,199) | |
| Fats | Secondary BA | Increases the ratio of Firmicutes/Bacteroidetes, which is associated with obesity; also promotes the increase of mucin-degrading Actinobacteria; reduces the abundance of Bifidobacterium, Lactobacillus and Akkermansia considered as the good bacteria of the gut microbiota. | Transforms primary bile acids into secondary bile acids which are involved in inflammatory bowel diseases. | (26,27,44,200,201) |
Note: HCA: heterocyclic amines, PAH: polycyclic aromatic hydrocarbons NOC: N-nitroso compounds, Secondary BA: secondary bile acids, TMAO: trimethylamine-N-oxide
It is not known, however, whether or not the combined action of nitrate-induced E. coli multiplication and the formation of NOCs are responsible for promoting inflammation or the development of CRC. Exposure of mouse models to PAHs also causes change in the composition of the gut microbiota after colonic inflammation. In humans, PAHs and HCAs altered the volatile profile of the fecal microbiota and their metabolite activities after high exposure of the gut microbiota over 24 hours to these substances [23,98,99].
Certain bacteria may reduce the risk of CRC associated with HCA consumption in the gut microbiota (e.g., Eulonchus halli) through their beta-glucuronidase and glycerol/diol dehydratase activities that convert HCA to HCA-M1 [99,100].
The production of TMAO from precursors such as choline, L-carnitine, phosphatidylcholine, and betaine is influenced by the gut microbiota in humans [24]. TMAO precursors are biodegraded by gut microbiota to generate pro-inflammatory molecules. The Eubacterium limosum has proved to be very effective in converting TMAO precursors by demethylation of L-carnitine and reducing the amount of TMAO produced in the gut [101]. Mice receiving a diet rich in heme increased the number of bacteria such as Proteobacteria or Bacteroidetes accompanied by a reduction of Firmicutes bacteria and Deferribacteres in their intestine [18,19,102].
Other infectious agents, including viruses bovine origin, thermoresistant, and potentially oncogenic, have also been reported as agents that may be involved in the process of CRC carcinogenesis [103]. Nowadays, only the transmission of heat-resistant bovine viruses during the meat consumption is suspected as an infectious factor involved in the mechanisms of CRC carcinogenesis [104]. These are mainly polyomaviruses, papillomaviruses, and probably the Torque Teno virus (TTV). Once consumed through beef contaminated by environmental substances or by substances generated during the cooking of the meat (HCA, PAH) these viruses could initiate CRC by synergistic interaction with these substances [103]. However, no experimental evidence has shown a direct correlation of these infectious agents with CRC development. This new hypothesis deserves further attention and exploration.
Based on this evidence, the gut microbiota and compounds derived from red meat appear to have close interactions that may influence the composition of the gut microbiota leading to protection against CRC or exposure to CRC risk.
Interaction between gut microbiota and colorectal cancer
In 1975, for the first time, the link between gut microbiota and CRC was established in germ-free compared to conventional rats, with the development of a colorectal tumor after chemical induction [105]. They found that 93% of the conventional rats developed multiple colon tumors and only 20% of the germ-free rats developed colon tumors. After subcutaneous injection of azoxymethane in both groups of rats, the incidence and multiplicity of colonic tumors were increased in germ-free rats compared to conventional rats [106]. Gut microbial dysbiosis in mice was observed during both spontaneous and chemically induced colon tumorigenesis. The diversity of the gut microbiota was reduced in C57BL/6J Apc Min/+ mice compared with wild-type C57BL/6J mice [107]. Since then, several studies found disruption of some microorganisms in the gut microbiota of patients with CRC compared to healthy controls [108]. CRC patients showed a reduction in bacterial diversity and richness compared to healthy individuals [109].
Whole genome sequencing of gut microbial species has allowed researchers to study the microbial communities that colonize colonic tumors as well as non-tumor colonic sites and to characterize individual oncogenic microbiomes [110]. The proliferation of certain bacterial populations including Helicobacter pylori (H. pylori), Escherichia coli (E.coli), Streptococcus bovis (S. bovis), Enterococcus faecalis (E. faecalis), Clostridium septicum (C. septicum), Fusobacterium nucleatum (F. nucleatum), Enterotoxigenic Bacteroides fragilis (ETBF) and Streptococcus gallolyticus (S. gallolyticus) were suspected as factors promoting CRC [108,111]. To better understand the role of the gut microbiota in the carcinogenesis and progression of CRC, hypotheses have been proposed. For example, the driver–passenger model has been proposed to classify commensal bacteria into two different groups, the driver bacteria and the passenger bacteria [112]. Driver bacteria cause DNA damage in colonic cells, that can initiate or cause CRC progression in the first spatial location, then the tumor microenvironment changes to promote infiltration and proliferation of passenger bacteria that may be dominant later in the colonic tumor site. This model attempts to explain that the driver bacteria in initiating CRC, will not always exist as an oncogenic marker in the tumor environment, but will disappear and will be replaced by passenger bacteria in the cancerous tissue. This model can help to clearly understand the discrepancy between results in different studies, clarifying the ambiguous relationship between gut microbiota and CRC.
The other model proposed is the keystone model, which supports the role of a key pathogen in the process of dysbiosis associated with a given disease [113]. This hypothesis is not based on the abundance or level of strength of the microbiota related to the disease, but on its functions that contribute to dysbiosis and its maintenance. For example, Klebsiella pneumonia and Proteus mirabilis could be treated as key pathogens of inflammatory bowel disease, and the role of ETBF in CRC. This model may provide new insights to review the potential role of gut pathogens in the initiation and progression of related disorders.
The potential role of F nucleatum in CRC carcinogenesis has been reported through studies [114]. Analysis of rectal mucosa, feces, and tumor samples from CRC patients showed a high prevalence of bacteria belonging to the genus Fusobacterium compared to healthy subjects or remotely adjacent healthy mucosa in these same CRC patients [115]. F. nucleatum infiltrates the colonic tumor through its adhesin (FadA), selectively binding to E-cadherin and activates the β-catenin signaling pathway, inducing inflammatory responses allowing CRC progression [116]. F. nucleatum also inhibits T cell and natural killer cell activity through another adhesin, fibroblast activation protein 2 (Fap2), which binds to T cell immunoglobulin and the ITIM domain [117]. Fap2 adhesin is used by F. nucleatum to infiltrate the colonic tumor by binding to the carbohydrate moiety D-galactose-β (1–3)-N-acetyl-D-galactosamine (Gal-GalNAc), which is overexpressed in CRC cells [118]. Interleukin IL-17A is highly expressed in CRC patients with F. nucleatum-enriched colonic tumors [119]. A recent study showed that CRC cell metastasis was dependent on the F. nucleatum adhesin Fap2, which induced the secretion of the pro-inflammatory cytokines, IL-8 and CXCL1 [120]. The carcinogenic property of F. nucleatum in Apc Min/+ mice and in human CRC cell lines was indicated by Nuclear factor-kappa B (NF-κB) activation which in turn induced miR21 gene expression promoting inflammatory responses [121]. Another recent study showed that F. nucleatum was significantly increased in patients with early stage CRC and that the presence of F. nucleatum in CRC tissues is associated with a poor prognosis of the disease [122,123,124].
Several studies have reported the link between ETBF and CRC and its use as a potential biomarker in the diagnosis of CRC [125–127]. ETBF secretes a 21 kDa B. fragilis toxin (BFT) that cleaves E-cadherin on colonic epithelial cells resulting in disruption of the colonic barrier [128]. The disrupted colonic barrier causes diarrhea and inflammatory bowel disease [129,130]. A recent study reported that E. faecalis and ETBF copy number were significantly higher in CRC tissue samples compared to the no CRC group [130]. Infection of Apc Min/+ mice with ETBF induced selective activation of Signal transducer and activator of transcription 3 (STAT3) with CRC characterized by Th17 responses. ETBF promotes CRC progression by secreting particles that stimulate colonic epithelial cells to produce exosome-like nanoparticles containing high levels of sphingosine-1-phosphate, CCL20, and prostaglandin E2 (PGE2) that are required for recruitment of Th17 cells into CRC tissues to support their growth and survival [131]. ETBF also plays an important role in mediating inflammatory responses during CRC carcinogenesis and chronic inflammatory bowel disease by activating the NF-κB signaling pathway to recruit immature polymorphonuclear myeloid cells [132,133]. ETBF promoted inflammation and CRC cells multiplication by downregulating exosomal miR-149–3p both in vitro and in vivo [134].
The pilus 3 (pil3) of S. gallolyticus is essential for its attachment to human mucus-producing epithelial cells (135). Interestingly, pil3 binds both to the human mucin 2 (MUC2), which predominates in healthy colonic tissue, and to mucin 5AC (MUC5AC), which is overexpressed in cancerous colonic tissue [135]. It has been postulated that commensal colonization of S. gallolyticus is facilitated by its binding to the mucin MUC2, while it’s binding to MUC5AC gives a growth advantage to bacterial species of the gut microbiota in the tumor microenvironment [136]. This helps explain the higher carriage rate of S. gallolyticus in the presence of colonic tumors. Colonic epithelial cells from CRC patients showed high NF-κB gene expression in the S. gallolyticus-positive group compared with the S. gallolyticus-negative group [137]. NF-κB plays an important role in mediating inflammatory responses during chronic inflammatory bowel disease and CRC carcinogenesis. In another study the same authors showed strong expression of IL-1 and Cyclo-oxygenase-2 (COX-2) genes by colonic cells of S. gallolyticus seropositive CRC patients, both of which are products of NF-κB activity [138].
E. coli, is an anaerobic gram-negative commensal bacterium, commonly found in the intestinal microenvironment. Some groups of E. coli called pathotypes belonging mainly to the B2 and D phylogroups have been identified as potentially oncogenic and pro-inflammatory [139]. Several studies have linked these pathogenic E. coli groups to CRC risk. Enrichment in B2 and D phylogroups was reported in CRC patients compared to control subjects (without CRC) [140]. Indeed, these phylogroups were identified in 90% and 93% of adenoma and carcinoma patients, respectively, while only 3% of colon biopsies from asymptomatic controls were positive for these phylogroups. A similar study showed that among 21 CRC patients 70% had E. coli enriched colorectal tissue, compared with 42% of 24 control biopsies without CRC [141]. In addition, many other studies have confirmed the enrichment of tumor tissues with E. coli in CRC patients compared to healthy subjects [142–144]. Therefore, a link between these pathogenic E. coli groups and CRC risk has been proposed through several studies. Nevertheless, the mechanism involved is very poorly understood to date. But according to studies, pathogenic E. coli strains producing cyclomodulins, and toxins are responsible for induction of DNA damage and cell cycle disruption in eukaryotes [131,139,145]. A high prevalence of cyclomodulin and toxin-producing E. coli was observed in CRC patients compared to healthy subjects [139,143]. These genotoxic toxins include the polyketide synthase (pks) pathogenicity responsible for colibactin expression, cytolethal distention toxin (CDT), cytotoxic necrotizing factor (CNF), cycle-inhibiting factor (CIF), and afimbriale adhesin (afa) [144,146,147]. The pks genomic island codes for the polyketide-peptide genotoxin, colibactin [148–150]. Culture of mammalian epithelial cells exposed to pks+ E. coli to show transient DNA damage [149]. According to experimental studies, colibactin could promote tumor growth by forming cross-links with DNA in cellulo as an alkylating agent, and DNA double-strand breaks or by promoting the emergence of senescent cells with an irreversible cell cycle arrest [151,152]. In AOM/IL10−/− or ApcMin/+ mouse models with chemo-induced tumor, infection with colibactin-producing E. coli strains induced an acceleration of tumor development compared to uninfected control mice or mice infected with a non-colibactin-producing mutant of these strains, or animals infected and treated with molecules inhibiting colibactin synthesis [139,142,144,153]. CNF toxin binds to the tight junctions of colonic epithelial cells which internalize it by endocytosis and promotes cell proliferation by encouraging entry into the cell cycle and the G1/S transition for cell survival by inducing, in particular, the expression of the anti-apoptotic proteins Bcl-2 (B-cell lymphoma 2) and Bcl-xl (B-cell lymphoma-extra-large) [154]. CIF promotes actin cytoskeleton rearrangement and induces G2/M cell cycle arrest characterized by inactive phosphorylation of cyclin-1-dependent kinase, an essential player in cell cycle regulation [155]. CNF-1 also induces transient activation of COX-2 and Rho GTPases characterized by alterations in the cytoskeleton and thus affects the cell cycle [156,157]. The CDT toxin secreted by pathogenic E. coli strains is known to have DNAase activity resulting in DNA double strand breaks, cell division arrest and inhibition of cell apoptosis [157].
Like E. coli, E. faecalis is part of the facultative anaerobic Gram-positive commensal flora and does not appear to be offensive to humans. However, studies have shown enrichment of CRC patients’ fecal samples and tumor tissues by E. faecalis compared to those healthy individuals [158,159]. A recent study found that E. faecalis species was significantly lower in obese patients than in non-obese patients with CRC [160]. Similarly, the abundance of E. faecalis was relatively higher in obese subjects than in non-obese subjects. This study demonstrated that a reduced presence of E. faecalis may be associated with obesity-related CRC carcinogenesis. Another recent study conducted on 256 fresh frozen CRC tissues detected E. faecalis in 193 of the 256 CRC tissues [161]. E. faecalis bacteremia was observed in an 86-year-old white male during a secondary gastrointestinal hemorrhage with confirmation of colorectal adenocarcinoma by colonoscopy [162]. E. faecalis was able to promote and maintain colitis in Il10−/− or Il10 gene deficient mice with induction of rectal dysplasia and carcinoma [163]. Intestinal epithelial cells from wild-type mice expressed the cytokine TGF-β upon infection with colitogenic E. faecalis, thereby activating Smad signaling. Interestingly, these mice lost toll-like receptor (TLR2) expression with NF-κB-dependent pro-inflammatory gene inhibition, in contrast to Il10−/− mice that failed to inhibit TLR2 receptor-mediated pro-inflammatory gene expression in intestinal epithelial cells upon colonization by E. faecalis. (163) Blood isolates of E. faecalis have also been associated with the production of extracellular superoxide (O2−) and hydrogen peroxide (H2O2) in the intestine [164,165]. These extracellular free radicals induced DNA damage in the studies [165]. Similarly, E. faecalis is able to induce DNA damage in vivo in colonic cells in rats. Extracellular infection of mammalian cells by E. faecalis can result in the production of superoxide (O2−) leading to overexpression of COX-2 in macrophages and promotes chromosomal instability in primary colonic epithelial cells [166]. Similarly E. faecalis is able to polarize colonic macrophages into an M1 phenotype, which in turn induces aneuploidy and chromosomal instability in colonic epithelial cells commonly found in cancers [167]. These data may explain the mechanisms by which E. faecalis exerts its impact on CRC.
McCOY WC et al. first established the link between S. bovis and CRC in 1951 [168]. Subsequently, around 1977, the S. bovis strain was isolated from stool samples of 35 out of 63 CRC patients compared to 11 out of 105 control individuals without CRC [169]. Subsequent studies have confirmed the link between S. bovis and CRC [170,171]. According to an in vitro study carried out in 2004, infection of colonic cells by S. bovis induced an overexpression of pro-inflammatory mediators, notably IL-8, COX-2 and PGE2 [172]. Other animal studies haveconfirmedthecarcinogenicpropertiesofS.bovis.Azoxymethane-treated rats confirmed the release of pro-inflammatory mediators after infection with S. bovis, which explains the increase in the number of aberrant crypts. Interestingly of the azoxymethane-treated rats, three of the six treated rats developed polyps during S. bovis infection, whereas no polyps were detected in the uninfected azoxymethane treated rats. Hyperproliferative crypts were also detected in azoxymethane-treated rats with S. bovis infection, demonstrating the involvement of this bacterium in CRC carcinogenesis [173]. Through samples of human origin including stool, tumor and non-tumor CRC tissues, studies have shown an enrichment of this bacterium in these samples in CRC patients compared to control subjects without CRC [138]. Studies have also reported significant overexpression of mRNAs that encode the proinflammatory mediators IL-1β, COX-2, and IL-8 in S. bovis-infiltrated tissues compared with non-infiltrated tissues. Similarly the expression of these mRNAs was elevated in tumor tissues compared to non-tumor tissues. The pro-inflammatory profile of S. bovis may increase the risk of CRC development and progression.
H. pylori is a gram-negative bacterium that preferentially infects and colonizes gastric tissue in humans. Although most infected individuals remain asymptomatic, H. pylori can induce chronic inflammatory responses increasing the risk of gastric ulcer, and adenocarcinoma of the stomach [174]. Despite gastric colonization by H. pylori, it has been shown that its toxicity can extend outside the stomach. The link between H. pylori infection and CRC remains controversial with studies showing a strong association with a high prevalence of H. pylori infection in patients with colonic adenomas and carcinomas [175,176]. While other studies have shown no association [177,178]. Many recent studies have reported a significant association between H. pylori infection and an increased occurrence of CRC [179–181]. Similarly Yan et al. showed a positive association between H. pylori infection and CRC. Despite the ambiguity between some studies on the link between H. pylori infection and CRC, others have attempted to explain the molecular mechanism underlying the potential association between H. pylori infection and CRC with some supporting hypotheses such as toxin release, dysbiosis and chronic inflammation. For example, significant gastrin secretion mediated by H. pylori infection was associated with increased expression of COX-2 and anti-apoptotic B-cell lymphoma 2 (BCL2) protein compared to pro-apoptotic BCL2 Associated X(BAX) protein resulting in decreased apoptosis in CRC [182]. High gastrin production disrupts gastric acid production and the gastric barrier, leading to an imbalance in the intestinal microbiota [183]. This dysbiosis could facilitate the colonization and multiplication of oncogenic bacteria associated with CRC such as B. fragilis and E. faecalis. Other mechanisms of CRC carcinogenesis mediated by H. pylori infection have been proposed. These include the production of ROS and reactive nitrogen species (RNS) that can lead to DNA damage, which could promote CRC carcinogenesis [184]. In addition, H. pylori strains toxicities’ vary from patients. For example, strains that carry the virulence factor cytotoxin-associated gene A (CagA) are more toxic than those without, and patients with these strains have an increased risk of developing gastric cancer and CRC compared to those without [185]. The VacA protein secreted by these H. pylori strains can induce production of pro-inflammatory mediators such as TNF-α, IFN-γ, IL-1β, IL-6 and IL-8 by infected cells [186,187]. The toxins secreted by H. pylori give it a pro-inflammatory property that can promote gastric and CRC carcinogenesis.
C. septicum is a Gram-positive, anaerobic, spore-forming bacillus, which is not normally present in the intestinal flora, but can cause direct and spontaneous infections in the gut.
This bacterium produces alpha-toxin, a virulence factor which is both lethal and hemolytic in mice [188]. Several studies have reported a probable association between C. septicum infection and CRC [189–191]. This association may be due to the hypoxic and acidic tumor environment that favors germination of C. septicum spores [190]. Eighty percent (80%) of patients infected with C. septicum were associated with malignancy. The alpha-toxin-producing C. septicum group is associated with the release of TNF-α after activation of the mitogen-activated protein kinase (MAPK) pathway, which has been shown to be dysregulated in cancers [192–201]. This pro-inflammatory property of C. septicum may promote carcinogenesis. However, despite available data, no direct link between C. septicum and CRC has been defined to date.
DISCUSSION AND CONCLUSION
This review examined current knowledge about risk factors of CRC carcinogenesis. Red and processed meat consumption and its interaction with the gut microbiota are found to be major associated factors. The CRC-associated gut microbiota is made of pro-inflammatory or pro-carcinogenic bacteria and opportunistic pathogenic bacteria that enrich the tumor microenvironment by promoting disease progression. Bacteria such as E. coli, S. gallolyticus, and F. nucleatum are frequently initiators of colonic carcinogenesis through virulence factors and responsible for CRC progression after their infiltration into the tumor microenvironment. Animal and human experimental studies also strongly support the evidence of diet being a major risk factor for CRC. More longitudinal clinical studies are needed to confirm and better understand the mechanisms underlying the diet-mediated disruption of gut microbiota in humans and establish the direct cause and impact that dysbiosis has on the initiation and progression of CRC.
ACKNOWLEDGMENTS
The authors thank Harvard University, Boston University, Northwestern University, and University of New Mexico (HBNU) Consortium, Global Health, Fogarty International Center and the NIH for their training support (D43 TW010543). We also thank the <<Centre de Recherche et de Formation sur les Pathologies Moléculaires, CREFPAM (Molecular Pathologies Research and Training Center) >> at the Faculty of Medicine and Odontostomatology of the Université des Sciences, Techniques et Technologies de Bamako and University Hospital Center of Point G, Bamako for logistical support.
FUNDING
Research reported was supported by the Institute of Global Health (IGH) Catalyzer of Northwestern University (NU) and the National Institutes of Health (D43CA260658, D43TW010350 and R21AI148033).
ABBREVIATIONS
- AFA
Afimbriale adhesin
- APC
Adenomatous Polyposis Coli
- AKT
Protein Kinase B
- BA
Bile Acid
- B(α)P
Benzo(Α)Pyrene
- BAX
BCL2 Associated X
- BCL-2
B-Cell Lymphoma 2
- Bcl-xl
B-Cell Lymphoma- Extra-Large
- CA
Cholic Acid
- CagA
Cytotoxin-Associated Gene A
- CDCA
Chenodeoxycholic Acid
- CDT
Cytolethal Distention Toxin
- CIF
Cycle-Inhibiting Factor
- CNF
Cytotoxic Necrotizing Factor
- CXCL1
Chemokine (C-X-C Motif) Ligand 1
- CXCL2
Chemokine (C-X-C Motif) Ligand 2
- CCL20
C-C Motif Chemokine Ligand 20
- COX-2
Cyclo-Oxygenase-2
- CRC
Colorectal Cancer
- DiMeIQx
2-Amino-3,4,8-Dimethylimidazo (4,5-F) Quinoxaline
- DNA
Deoxyribonucleic Acid
- EPIC
The European Prospective Investigation into Cancer and Nutrition
- ETBF
Enterotoxigenic Bacteroides Fragilis
- FadA
Fusobacterium Adhesin A
- FMO3
Flavin Monooxygenase 3
- Gal-GalNAc
D-Galactose-Β (1–3)-N-Acetyl-D-Galactosamine
- HCA
Heterocyclic Amines
- IARC
International Agency for Research on Cancer
- IFN-γ
Interferon Gamma
- IL-1β
Interleukin 1 Beta
- IL-6
Interleukin 6
- IL-8
Interleukin 8
- IL 17
Interleukin-17
- KRAS
Kirsten Rat Sarcoma Virus
- M3R
Muscarinic Receptor
- MAPK
Mitogen-Activated Protein Kinase
- MIR21
MicroRNA21
- MUC2
Mucin 2
- MUC5AC
Mucin 5AC
- NF-κB
Nuclear Factor Kappa
- NLPP3
NOD-Like Receptor Family Pyrin Domain Containing 3
- NOC
N-Nitroso Compounds
- PAH
Polycyclic Aromatic Hydrocarbons
- PI3K
Phosphoinositide 3-Kinases
- PGE2
Prostaglandin E2
- PKS
Polyketide Synthase
- ROS
Reactive Oxygen Species
- RNS
Reactive Nitrogen Species
- TH17
T Helper 17 Cells
- TMAO
Trimethylamine-N-oxide
- TGF-β
Transforming Growth Factor Beta
- TLR2
Toll-Like Receptor 2
- TNF-α
Tumor Necrosis Factorα
- TP53
Tumor Suppressor Gene
Footnotes
COMPETING INTERESTS
The authors declare that they have no competing interests.
Ethics approval and consent to participate
REFERENCES
- 1.Hammerling U, Bergman Laurila J, Grafström R, Ilbäck N-G. Consumption of red/processed meat and colorectal carcinoma: Possible mechanisms underlying the significant association. Crit Rev Food Sci Nutr. 2016; 56:614–634. [DOI] [PubMed] [Google Scholar]
- 2.Liu W, Zhang R, Shu R, Yu J, Li H, Long H, et al. Study of the relationship between microbiome and colorectal cancer susceptibility using 16srrna sequencing. BioMed Res Int. 2020; 7828392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021; 71:209–249. [DOI] [PubMed] [Google Scholar]
- 4.Sawicki T, Ruszkowska M, Danielewicz A, Niedźwiedzka E, Arłukowicz T, Przybyłowicz KE. A review of colorectal cancer in terms of epidemiology, risk factors, development, symptoms and diagnosis. Cancers. 2021; 13:2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loomans-Kropp HA, Umar A. Increasing incidence of colorectal cancer in young adults. J Cancer Epidemiol. 2019; 2019:9841295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montalban-Arques A, Scharl M. Intestinal microbiota and colorectal carcinoma: Implications for pathogenesis, diagnosis, and therapy. EBioMedicine. 2019; 48:648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costea T, Hudiță A, Ciolac O-A, Gălățeanu B, Ginghină O, Costache M, et al. Chemoprevention of colorectal cancer by dietary compounds. Int J Mol Sci. 2018; 19:3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ocvirk S, Wilson AS, Appolonia CN, Thomas TK, O’Keefe SJD. Fiber, fat, and colorectal cancer: new insight into modifiable dietary risk factors. Curr Gastroenterol Rep. 2019; 21:62. [DOI] [PubMed] [Google Scholar]
- 9.Alexander DD, Weed DL, Miller PE, Mohamed MA. Red meat and colorectal cancer: A quantitative update on the state of the epidemiologic science. J am coll nutr. 2015; 34:521–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruno-Barcena JM, Azcarate-Peril MA. Galacto-oligosaccharides and colorectal cancer: Feeding our intestinal probiome. J Funct Foods. 2015; 12:92–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santarelli RL, Pierre F, Corpet DE. Processed meat and colorectal cancer: A review of epidemiologic and experimental evidence. Nutr Cancer. 2008; 60:131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joosen AMCP Kuhnle GGC, Aspinall SM Barrow TM, Lecommandeur E, Azqueta A, et al. Effect of processed and red meat on endogenous nitrosation and DNA damage. Carcinogenesis. 2009; 30:1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bardou M, Barkun AN, Martel M. Obesity and colorectal cancer. Gut. 2013; 62:933–947. [DOI] [PubMed] [Google Scholar]
- 14.Blomain ES, Waldman SA. Does obesity promote the development of colorectal cancer? Expert Rev Anticancer Ther. 2016; 16:465–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aykan NF. Red meat and colorectal cancer. Oncol Rev. 2015; 9:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carr PR, Walter V, Brenner H, Hoffmeister M. Meat subtypes and their association with colorectal cancer: Systematic review and meta-analysis. Int J Cancer. 2016; 138:293–302. [DOI] [PubMed] [Google Scholar]
- 17.Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013; 339:708–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Constante M, Fragoso G, Calvé A, Samba-Mondonga M, Santos MM. Dietary heme induces gut dysbiosis, aggravates colitis, and potentiates the development of adenomas in mice. Front Microbiol. 2017; 8:1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.IJssennagger N, Derrien M, van Doorn GM, Rijnierse A, van den Bogert B, Müller M, et al. Dietary heme alters microbiota and mucosa of mouse colon without functional changes in host-microbe cross-talk. PloS One. 2012; 7:e49868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sekirov I, Russell SL, Antunes LCM, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010; 90:859–904. [DOI] [PubMed] [Google Scholar]
- 21.Yang J, Yu J. The association of diet, gut microbiota and colorectal cancer: What we eat may imply what we get. Protein Cell. 2018; 9:474–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verspreet J, Damen B, Broekaert WF, Verbeke K, Delcour JA, Courtin CM. A critical look at prebiotics within the dietary fiber concept. Annu Rev Food Sci Technol. 2016; 7:167–190. [DOI] [PubMed] [Google Scholar]
- 23.Defois C, Ratel J, Garrait G, Denis S, Le Goff O, Talvas J, et al. Food chemicals disrupt human gut microbiota activity and impact intestinal homeostasis as revealed by in vitro systems. Sci Rep. 2018; 8:11006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Bergeron N, Levison BS, Li XS, Chiu S, Jia X, et al. Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine N-oxide metabolism and renal excretion in healthy men and women. Eur Heart J. 2019; 40:583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ijssennagger N, Rijnierse A, de Wit NJW, Boekschoten MV, Dekker J, Schonewille A, et al. Dietary heme induces acute oxidative stress, but delayed cytotoxicity and compensatory hyperproliferation in mouse colon. Carcinogenesis. 2013; 34:1628–1635. [DOI] [PubMed] [Google Scholar]
- 26.Zou S, Fang L, Lee M-H. Dysbiosis of gut microbiota in promoting the development of colorectal cancer. Gastroenterol Rep. 2018; 6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert M-A, Quervain E, et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut. 2013; 62:531–539. [DOI] [PubMed] [Google Scholar]
- 28.Silvester KR, Cummings JH. Does digestibility of meat protein help explain large bowel cancer risk? Nutr Cancer. 1995; 24:279–288. [DOI] [PubMed] [Google Scholar]
- 29.Burkitt DP. Epidemiology of cancer of the colon and rectum. Cancer. 1971; 28:3–13. [DOI] [PubMed] [Google Scholar]
- 30.Clinton SK, Giovannucci EL, Hursting SD. The world cancer research fund/american institute for cancer research third expert report on diet, nutrition, physical activity, and cancer: impact and future directions. J Nutr. 2020; 150:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wiseman M The second world cancer research fund/american institute for cancer research expert report. Food, nutrition, physical activity, and the prevention of cancer: a global perspective: nutrition society and bapen medical symposium on ‘nutrition support in cancer therapy.’ Proc Nutr Soc. 2008; 67:253–256. [DOI] [PubMed] [Google Scholar]
- 32.Loke YL, Chew MT, Ngeow YF, Lim WWD, Peh SC. Colon carcinogenesis: The interplay between diet and gut microbiota. Front cell infect microbiol. 2020; 10:603086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouvard V, Loomis D, Guyton KZ, Grosse Y, Ghissassi FE, Benbrahim-Tallaa L, et al. International agency for research on cancer monograph working group. Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 2015; 16:1599–1600. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Z, Feng Q, Yin Z, Shuang J, Bai B, Yu P, et al. Red and processed meat consumption and colorectal cancer risk: a systematic review and meta-analysis. Oncotarget. 2017; 8:83306–83314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cross AJ, Leitzmann MF, Gail MH, Hollenbeck AR, Schatzkin A, Sinha R. A prospective study of red and processed meat intake in relation to cancer risk. PLoS Med. 2007; 4:e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Norat T, Bingham S, Ferrari P, Slimani N, Jenab M, Mazuir M, et al. Meat, fish, and colorectal cancer risk: the european prospective investigation into cancer and nutrition. J Natl Cancer Inst. 2005; 97:906–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ocvirk S, O’Keefe SJ. Influence of bile acids on colorectal cancer risk: potential mechanisms mediated by diet - gut microbiota interactions. Curr Nutr Rep.2017; 6:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu M, Cen M, Shen Y, Zhu Y, Cheng F, Tang L, et al. Deoxycholic acid-induced gut dysbiosis disrupts bile acid enterohepatic circulation and promotes intestinal inflammation. Dig Dis Sci. 2021; 66:568–576. [DOI] [PubMed] [Google Scholar]
- 39.Ou J, DeLany JP, Zhang M, Sharma S, O’Keefe SJD. Association between low colonic short-chain fatty acids and high bile acids in high colon cancer risk populations. Nutr Cancer.2012; 64:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Keefe SJD, Li JV, Lahti L, Ou J, Carbonero F, Mohammed K, et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat Commun. 2015; 6:6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen TT, Ung TT, Kim NH, Jung YD. Role of bile acids in colon carcinogenesis. World J Clin Cases. 2018; 6:577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farhana L, Nangia-Makker P, Arbit E, Shango K, Sarkar S, Mahmud H, et al. Bile acid: a potential inducer of colon cancer stem cells. Stem Cell Res Ther. 2016; 7:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol. 2011; 85:863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolf PG, Byrd DA, Cares K, Dai H, Odoms-Young A, Gaskins HR, et al. Bile acids, gut microbes, and the neighborhood food environment-a potential driver of colorectal cancer health disparities. MSystems. 7:e01174–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ignacio Barrasa J, Olmo N, Pérez-Ramos P, Santiago-Gómez A, Lecona E, Turnay J, et al. Deoxycholic and chenodeoxycholic bile acids induce apoptosis via oxidative stress in human colon adenocarcinoma cells. Apoptosis Int J Program Cell Death. 2011; 16:1054–1067. [DOI] [PubMed] [Google Scholar]
- 46.Raufman J-P, Shant J, Guo CY, Roy S, Cheng K. Deoxycholyltaurine rescues human colon cancer cells from apoptosis by activating EGFR-dependent PI3K/Akt signaling. J Cell Physiol. 2008; 215:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mroz MS, Lajczak NK, Goggins BJ, Keely S, Keely SJ. The bile acids, deoxycholic acid and ursodeoxycholic acid, regulate colonic epithelial wound healing. Am J Physiol Gastrointest Liver Physiol.2018; 314:G378–G387. [DOI] [PubMed] [Google Scholar]
- 48.Helmus DS, Thompson CL, Zelenskiy S, Tucker TC, Li L. Red meat-derived heterocyclic amines increase risk of colon cancer: A population-based case-control study. Nutr Cancer. 2013; 65:1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiavarini M, Bertarelli G, Minelli L, Fabiani R. Dietary intake of meat cooking-related mutagens (hcas) and risk of colorectal adenoma and cancer: A systematic review and meta-analysis. Nutrients. 2017; 9:E514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Domingo JL, Nadal M. Carcinogenicity of consumption of red and processed meat: What about environmental contaminants? Environ Res. 2016; 145:109–115. [DOI] [PubMed] [Google Scholar]
- 51.Budhathoki S, Iwasaki M, Yamaji T, Sasazuki S, Takachi R, Sakamoto H,et al. Dietary heterocyclic amine intake, nat2 genetic polymorphism, and colorectal adenoma risk: the colorectal adenoma study in tokyo. Cancer Epidemiol Biomarkers Prev. 2015; 24:613–620. [DOI] [PubMed] [Google Scholar]
- 52.Rohrmann S, Hermann S, Linseisen J. Heterocyclic aromatic amine intake increases colorectal adenoma risk: findings from a prospective European cohort study. Am J Clin Nutr. 2009; 89:1418–1424. [DOI] [PubMed] [Google Scholar]
- 53.Barbir A, Linseisen J, Hermann S, Kaaks R, Teucher B, Eichholzer M, et al. Effects of phenotypes in heterocyclic aromatic amine (HCA) metabolism-related genes on the association of HCA intake with the risk of colorectal adenomas. Cancer Causes Control. 2012; 23:1429–1442. [DOI] [PubMed] [Google Scholar]
- 54.Sugimura T, Wakabayashi K, Nakagama H, Nagao M. Heterocyclic amines: Mutagens/carcinogens produced during cooking of meat and fish. Cancer Sci. 2004; 95:290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turesky RJ. Formation and biochemistry of carcinogenic heterocyclic aromatic amines in cooked meats. Toxicol Lett. 2007; 168:219–227. [DOI] [PubMed] [Google Scholar]
- 56.Ajayi BO, Adedara IA, Farombi EO. Benzo(a)pyrene induces oxidative stress, pro-inflammatory cytokines, expression of nuclear factor-kappa B and deregulation of wnt/beta-catenin signaling in colons of BALB/c mice. Food Chem Toxicol Int J Publ Br Ind Biol Res Assoc. 2016; 95:42–51. [DOI] [PubMed] [Google Scholar]
- 57.Diggs DL, Myers JN, Banks LD, Niaz MS, Hood DB, Roberts LJ, et al. Influence of dietary fat type on benzo(a)pyrene [B(a)P] biotransformation in a B(a)P-induced mouse model of colon cancer. J Nutr Biochem.2013; 24:2051–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hughes R, Cross AJ, Pollock JR, Bingham S. Dose-dependent effect of dietary meat on endogenous colonic N-nitrosation. Carcinogenesis. 2001; 22:199–202. [DOI] [PubMed] [Google Scholar]
- 59.Steinberg P Red meat-derived nitroso compounds, lipid peroxidation products and colorectal cancer. Foods Basel Switz.2019; 8:E252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu L, Qu Y-H, Chu X-D, Wang R, Nelson HH, Gao Y-T, et al. Urinary levels of N-nitroso compounds in relation to risk of gastric cancer: findings from the shanghai cohort study. PloS One. 2015; 10:e0117326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fahrer J, Kaina B. O6-methylguanine-DNA methyltransferase in the defense against N-nitroso compounds and colorectal cancer. Carcinogenesis. 2013; 34:2435–2442. [DOI] [PubMed] [Google Scholar]
- 62.Le Leu RK, Winter JM, Christophersen CT, Young GP, Humphreys KJ, Hu Y, et al. Butyrylated starch intake can prevent red meat-induced O6-methyl-2-deoxyguanosine adducts in human rectal tissue: A randomised clinical trial. Br J Nutr. 2015; 114:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hemeryck LY, Rombouts C, De Paepe E, Vanhaecke L. DNA adduct profiling of in vitro colonic meat digests to map red vs. white meat genotoxicity. Food Chem Toxicol. 2018; 115:73–87. [DOI] [PubMed] [Google Scholar]
- 64.Gamage SMK, Dissabandara L, Lam AK-Y, Gopalan V. The role of heme iron molecules derived from red and processed meat in the pathogenesis of colorectal carcinoma. Crit Rev Oncol Hematol. 2018; 126:121–128. [DOI] [PubMed] [Google Scholar]
- 65.Bastide NM, Chenni F, Audebert M, Santarelli RL, Taché S, Naud N, et al. A central role for heme iron in colon carcinogenesis associated with red meat intake. Cancer Res. 2015; 75:870–879. [DOI] [PubMed] [Google Scholar]
- 66.Kuhnle GGC, Story GW, Reda T, Mani AR, Moore KP, Lunn JC, et al. Diet-induced endogenous formation of nitroso compounds in the GI tract. Free Radic Biol Med. 2007; 43:1040–1047. [DOI] [PubMed] [Google Scholar]
- 67.Pierre FHF, Santarelli RL, Allam O, Tache S, Naud N, Gueraud F, et al. Freeze-dried ham promotes azoxymethane-induced mucin-depleted foci and aberrant crypt foci in rat colon. Nutr Cancer. 2010; 62:567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol. 2014; 5:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chenni FZ, Taché S, Naud N, Guéraud F, Hobbs DA, Kunhle GGC, et al. Heme-induced biomarkers associated with red meat promotion of colon cancer are not modulated by the intake of nitrite. Nutr Cancer. 2013; 65:227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pierre F, Tache S, Guéraud F, Rerole AL, Jourdan M-L, Petit C. Apc mutation induces resistance of colonic cells to lipoperoxide-triggered apoptosis induced by faecal water from haem-fed rats. Carcinogenesis. 2007; 28:321–327. [DOI] [PubMed] [Google Scholar]
- 71.Glei M, Klenow S, Sauer J, Wegewitz U, Richter K, Pool-Zobel BL. Hemoglobin and hemin induce DNA damage in human colon tumor cells HT29 clone 19A and in primary human colonocytes. Mutat Res. 2006; 594:162–171. [DOI] [PubMed] [Google Scholar]
- 72.Cross AJ, Pollock JRA, Bingham SA. Haem, not protein or inorganic iron, is responsible for endogenous intestinal N-nitrosation arising from red meat. Cancer Res. 2003; 63:2358–2360. [PubMed] [Google Scholar]
- 73.Cross AJ, Ferrucci LM, Risch A, Graubard BI, Ward MH, Park Y, et al. A large prospective study of meat consumption and colorectal cancer risk: an investigation of potential mechanisms underlying this association. Cancer Res. 2010; 70:2406–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chan CWH, Law BMH, Waye MMY, Chan JYW, So WKW, Chow KM. Trimethylamine-n-oxide as one hypothetical link for the relationship between intestinal microbiota and cancer - where we are and where shall we go? J Cancer. 2019; 10:5874–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Subramaniam S, Fletcher C. Trimethylamine N-oxide: breathe new life. Br J Pharmacol. 2018; 175:1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013; 19:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011; 472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bae S, Ulrich CM, Neuhouser ML, Malysheva O, Bailey LB, Xiao L, et al. Plasma choline metabolites and colorectal cancer risk in the women’s health initiative observational study. Cancer Res. 2014; 74:7442–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guertin KA, Li XS, Graubard BI, Albanes D, Weinstein SJ, Goedert JJ, et al. Serum trimethylamine n-oxide, carnitine, choline and betaine in relation to colorectal cancer risk in the alpha tocopherol and beta carotene study. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. 2017; 26:945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen M-L, Zhu X-H, Ran L, Lang H-D, Yi L, Mi M-T. Trimethylamine-n-oxide induces vascular inflammation by activating the nlrp3 inflammasome through the sirt3-sod2-mtros signaling pathway. J Am Heart Assoc. 2017; 6:e006347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu D, Cao M, Peng J, Li N, Yi S, Song L, et al. The effect of trimethylamine N-oxide on Helicobacter pylori-induced changes of immunoinflammatory genes expression in gastric epithelial cells. Int Immunopharmacol. 2017; 43:172–178. [DOI] [PubMed] [Google Scholar]
- 82.Huang C-F, Chen L, Li Y-C, Wu L, Yu G-T, Zhang W-F, et al. NLRP3 inflammasome activation promotes inflammation-induced carcinogenesis in head and neck squamous cell carcinoma. J Exp Clin Cancer Res CR. 2017; 36:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis. 2011; 70 Suppl 1:i104–108. [DOI] [PubMed] [Google Scholar]
- 84.Lee HJ, Song I-C, Yun H-J, Jo D-Y, Kim S. CXC chemokines and chemokine receptors in gastric cancer: From basic findings towards therapeutic targeting. World J Gastroenterol WJG. 2014; 20:1681–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oellgaard J, Winther SA, Hansen TS, Rossing P, von Scholten BJ. Trimethylamine n-oxide (tmao) as a new potential therapeutic target for insulin resistance and cancer. Curr Pharm Des. 2017; 23:3699–3712. [DOI] [PubMed] [Google Scholar]
- 86.Yue C, Yang X, Li J, Chen X, Zhao X, Chen Y, et al. Trimethylamine N-oxide prime NLRP3 inflammasome via inhibiting ATG16L1-induced autophagy in colonic epithelial cells. Biochem Biophys Res Commun. 2017; 490:541–551. [DOI] [PubMed] [Google Scholar]
- 87.Yoon K, Kim N. The effect of microbiota on colon carcinogenesis. J Cancer Prev. 2018; 23:117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Engemann A, Focke C, Humpf H-U. Intestinal formation of N-nitroso compounds in the pig cecum model. J Agric Food Chem. 2013; 61:998–1005. [DOI] [PubMed] [Google Scholar]
- 89.Kobayashi J Effect of diet and gut environment on the gastrointestinal formation of N-nitroso compounds: A review. Nitric Oxide Biol Chem. 2018; 73:66–73. [DOI] [PubMed] [Google Scholar]
- 90.Gangolli SD, van den Brandt PA, Feron VJ, Janzowsky C, Koeman JH, Speijers GJ, et al. Nitrate, nitrite and N-nitroso compounds. Eur J Pharmacol. 1994; 292:1–38. [DOI] [PubMed] [Google Scholar]
- 91.Povey AC, Badawi AF, Cooper DP, Hall CN, Harrison KL, Jackson PE, et al. DNA alkylation and repair in the large bowel: Animal and human studies. J Nutr. 2002; 132:3518S–3521S. [DOI] [PubMed] [Google Scholar]
- 92.DellaValle CT, Xiao Q, Yang G, Shu XO, Aschebrook-Kilfoy B, Zheng W, et al. Dietary nitrate and nitrite intake and risk of colorectal cancer in the shanghai women’s health study. Int J Cancer J Int Cancer. 2014; 134:2917–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bogovski P, Bogovski S. Animal species in which N-nitroso compounds induce cancer. Int J Cancer. 1981; 27:471–474. [DOI] [PubMed] [Google Scholar]
- 94.Loh YH, Jakszyn P, Luben RN, Mulligan AA, Mitrou PN, Khaw K-T. N-nitroso compounds and cancer incidence: The European prospective investigation into cancer and nutrition (epic)-norfolk study. Am J Clin Nutr. 2011; 93:1053–1061. [DOI] [PubMed] [Google Scholar]
- 95.IARC monographs on the evaluation of carcinogenic risks to humans. Ingested nitrate and nitrite, and cyanobacterial peptide toxins. Iarc Monogr Eval Carcinog Risks Hum.2010; 94:v–412. [PMC free article] [PubMed] [Google Scholar]
- 96.Sobhani I, Amiot A, Le Baleur Y, Levy M, Auriault M-L, Van Nhieu JT, et al. Microbial dysbiosis and colon carcinogenesis: could colon cancer be considered a bacteria-related disease? Ther Adv Gastroenterol. 2013; 6:215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jahani-Sherafat S, Alebouyeh M, Moghim S, Ahmadi Amoli H, Ghasemian-Safaei H. Role of gut microbiota in the pathogenesis of colorectal cancer: A review article. Gastroenterol Hepatol Bed Bench. 2018; 11:101–109. [PMC free article] [PubMed] [Google Scholar]
- 98.Defois C, Ratel J, Denis S, Batut B, Beugnot R, Peyretaillade E, et al. Environmental pollutant benzo[a]pyrene impacts the volatile metabolome and transcriptome of the human gut microbiota. Front Microbiol. 2017; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang J, Lacroix C, Wortmann E, Ruscheweyh H-J, Sunagawa S, Sturla SJ, et al. Gut microbial beta-glucuronidase and glycerol/diol dehydratase activity contribute to dietary heterocyclic amine biotransformation. BMC Microbiol.2019; 19:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fekry MI, Engels C, Zhang J, Schwab C, Lacroix C, Sturla SJ, et al. The strict anaerobic gut microbe Eubacterium hallii transforms the carcinogenic dietary heterocyclic amine 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP). Environ Microbiol Rep.2016; 8:201–209. [DOI] [PubMed] [Google Scholar]
- 101.Kountz DJ, Behrman EJ, Zhang L, Krzycki JA. MtcB, a member of the MttB superfamily from the human gut acetogen eubacterium limosum, is a cobalamin-dependent carnitine demethylase. J Biol Chem. 2020; 295:11971–11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Martin OCB, Olier M, Ellero-Simatos S, Naud N, Dupuy J, Huc L, et al. Haem iron reshapes colonic luminal environment: Impact on mucosal homeostasis and microbiome through aldehyde formation. Microbiome. 2019; 7:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alisson-Silva F, Kawanishi K, Varki A. Human risk of diseases associated with red meat intake: Analysis of current theories and proposed role for metabolic incorporation of a non-human sialic acid. Mol Aspects Med. 2016; 51:16–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zur Hausen H Red meat consumption and cancer: Reasons to suspect involvement of bovine infectious factors in colorectal cancer. Int J Cancer.2012; 130:2475–2483. [DOI] [PubMed] [Google Scholar]
- 105.Weisburger JH, Reddy BS, Narisawa T, Wynder EL. Germ-free status and colon tumor induction by N-methyl-N’-nitro-N-nitrosoguanidine. Proc Soc Exp Biol Med Soc Exp Biol Med NYN.1975; 148:1119–1121. [DOI] [PubMed] [Google Scholar]
- 106.Reddy BS, Narisawa T, Weisburger JH. Colon carcinogenesis in germ-free rats with intrarectal 1,2-dimethylhydrazine and subcutaneous azoxymethane. Cancer Res.1976; 36:2874–2876. [PubMed] [Google Scholar]
- 107.Son JS, Khair S, Pettet DW, Ouyang N, Tian X, Zhang Y, et al. Altered interactions between the gut microbiome and colonic mucosa precede polyposis in apcmin/+ mice. PLoS ONE. 2015; 10:e0127985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mendonça LABM, dos Santos Ferreira R, de Cássia Avellaneda Guimarães R, de Castro AP, Franco OL, Matias R, et al. The complex puzzle of interactions among functional food, gut microbiota, and colorectal cancer. Front Oncol. 2018; 8:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PloS One. 2012; 7:e39743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yu J, Feng Q, Wong SH, Zhang D, Liang QY, Qin Y, et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017; 66:70–78. [DOI] [PubMed] [Google Scholar]
- 111.Gagnière J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E, et al. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol. 2016; 22:501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012; 10:575–582. [DOI] [PubMed] [Google Scholar]
- 113.Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012; 10:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cheng Y, Ling Z, Li L. The intestinal microbiota and colorectal cancer. Front Immunol. 2020; 11:615056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of fusobacterium with colorectal carcinoma. Genome Res. 2012; 22:292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its fada adhesin. Cell Host Microbe. 2013; 14:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015; 42:344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Abed J, Emgård JEM, Zamir G, Faroja M, Almogy G, et al. Fap2 mediates fusobacterium nucleatum colorectal adenocarcinoma enrichment by binding to tumor-expressed Gal-GalNAc. Cell Host Microbe.2016; 20:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ye X, Wang R, Bhattacharya R, Boulbes DR, Fan F, Xia L, et al. Fusobacterium nucleatum subspecies animalis influences proinflammatory cytokine expression and monocyte activation in human colorectal tumors. Cancer Prev Res Phila Pa. 2017; 10:398–409. [DOI] [PubMed] [Google Scholar]
- 120.Casasanta MA, Yoo CC, Udayasuryan B, Sanders BE, Umaña A, Zhang Y, et al. Fusobacterium nucleatum host-cell binding and invasion induces IL-8 and CXCL1 secretion that drives colorectal cancer cell migration. Sci Signal. 2020; 13:eaba9157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor-κb, and up-regulating expression of microrna-21. Gastroenterology. 2017; 152:851–866.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu X, Cheng Y, Shao L, Ling Z. Alterations of the predominant fecal microbiota and disruption of the gut mucosal barrier in patients with early-stage colorectal cancer. BioMed Res Int. 2020; 2020:2948282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut.2016; 65:1973–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wei Z, Cao S, Liu S, Yao Z, Sun T, Li Y, et al. Could gut microbiota serve as prognostic biomarker associated with colorectal cancer patients’ survival? A pilot study on relevant mechanism. Oncotarget. 2016; 7:46158–46172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Haghi F, Goli E, Mirzaei B, Zeighami H. The association between fecal enterotoxigenic B. fragilis with colorectal cancer. BMC Cancer. 2019; 19:879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Keenan JI, Aitchison A, Purcell RV, Greenlees R, Pearson JF, Frizelle FA. Screening for enterotoxigenic bacteroides fragilis in stool samples. Anaerobe.2016; 40:50–53. [DOI] [PubMed] [Google Scholar]
- 127.Zamani S, Taslimi R, Sarabi A, Jasemi S, Sechi LA, Feizabadi MM. Enterotoxigenic bacteroides fragilis: A possible etiological candidate for bacterially-induced colorectal precancerous and cancerous lesions. Front Cell Infect Microbiol. 2019; 9:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wu S, Rhee K-J, Zhang M, Franco A, Sears CL. Bacteroides fragilis toxin stimulates intestinal epithelial cell shedding and gamma-secretase-dependent E-cadherin cleavage. J Cell Sci.2007; 120:1944–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chung L, Orberg ET, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. 2018; 23:203–214.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Khodaverdi N, Zeighami H, Jalilvand A, Haghi F, Hesami N. High frequency of enterotoxigenic bacteroides fragilis and enterococcus faecalis in the paraffin-embedded tissues of iranian colorectal cancer patients. BMC Cancer. 2021; 21:1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Deng Z, Mu J, Tseng M, Wattenberg B, Zhuang X, Egilmez NK, et al. Enterobacteria-secreted particles induce production of exosome-like S1P-containing particles by intestinal epithelium to drive Th17-mediated tumorigenesis. Nat Commun. 2015; 6:6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017; 2:17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chung L, Thiele Orberg E, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. 2018; 23:203–214.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cao Y, Wang Z, Yan Y, Ji L, He J, Xuan B, et al. Enterotoxigenic bacteroidesfragilis promotes intestinal inflammation and malignancy by inhibiting exosome-packaged mir-149–3p. Gastroenterology. 2021; 161:1552–1566.e12. [DOI] [PubMed] [Google Scholar]
- 135.Martins M, Aymeric L, du Merle L, Danne C, Robbe-Masselot C, Trieu-Cuot P. Streptococcus gallolyticus pil3 pilus is required for adhesion to colonic mucus and for colonization of mouse distal colon. J Infect Dis. 2015; 212:1646–1655. [DOI] [PubMed] [Google Scholar]
- 136.Martins M, Porrini C, du Merle L, Danne C, Robbe-Masselot C, Trieu-Cuot P, et al. The Pil3 pilus of Streptococcus gallolyticus binds to intestinal mucins and to fibrinogen. Gut Microbes. 2016; 7:526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Abdulamir AS, Hafidh RR, Mahdi LK, Al-jeboori T, Abubaker F. Investigation into the controversial association of Streptococcus gallolyticus with colorectal cancer and adenoma. BMC Cancer. 2009; 9:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Abdulamir AS, Hafidh RR, Bakar FA. Molecular detection, quantification, and isolation of Streptococcus gallolyticus bacteria colonizing colorectal tumors: inflammation-driven potential of carcinogenesis via IL-1, COX-2, and IL-8. Mol Cancer. 2010; 9:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Raisch J, Buc E, Bonnet M, Sauvanet P, Vazeille E, de Vallée A, et al. Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World J Gastroenterol. 2014; 20:6560–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, et al. Association between intraepithelial escherichia coli and colorectal cancer. Gastroenterology. 1998; 115:281–286. [DOI] [PubMed] [Google Scholar]
- 141.Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M. Enhanced escherichia coli adherence and invasion in crohn’s disease and colon cancer. Gastroenterology. 2004; 127:80–93. [DOI] [PubMed] [Google Scholar]
- 142.Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, et al. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res Off J Am Assoc Cancer Res. 2014; 20:859–867. [DOI] [PubMed] [Google Scholar]
- 143.Prorok-Hamon M, Friswell MK, Alswied A, Roberts CL, Song F, Flanagan PK, et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut. 2014; 63:761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan T-J, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012; 338:120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Magdy A, Elhadidy M, Abd Ellatif ME, El Nakeeb A, Abdallah E, Thabet W, et al. Enteropathogenic escherichia coli (epec): Does it have a role in colorectal tumourigenesis? A prospective cohort study. Int J Surg Lond Engl. 2015; 18:169–173. [DOI] [PubMed] [Google Scholar]
- 146.Wilson MR, Jiang Y, Villalta PW, Stornetta A, Boudreau PD, Carrá A, et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science. 2019; 363:eaar7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bossuet-Greif N, Vignard J, Taieb F, Mirey G, Dubois D, Petit C, et al. The colibactin genotoxin generates dna interstrand cross-links in infected cells. mBio. 2018; 9:e02393–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kawanishi M, Shimohara C, Oda Y, Hisatomi Y, Tsunematsu Y, Sato M, et al. Genotyping of a gene cluster for production of colibactin and in vitro genotoxicity analysis of escherichia coli strains obtained from the japan collection of microorganisms. Genes Environ Off J Jpn Environ Mutagen Soc. 2020; 42:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède J-P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A. 2010; 107:11537–11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Arthur JC, Gharaibeh RZ, Mühlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, et al. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat Commun. 2014; 5:4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes. 2014; 5:675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. 2014; 63:1932–1942. [DOI] [PubMed] [Google Scholar]
- 153.Cougnoux A, Delmas J, Gibold L, Faïs T, Romagnoli C, Robin F, et al. Small-molecule inhibitors prevent the genotoxic and protumoural effects induced by colibactin-producing bacteria. Gut. 2016; 65:278–285. [DOI] [PubMed] [Google Scholar]
- 154.Miraglia AG, Travaglione S, Meschini S, Falzano L, Matarrese P, Quaranta MG, et al. Cytotoxic necrotizing factor 1 prevents apoptosis via the Akt/IkappaB kinase pathway: role of nuclear factor-kappaB and Bcl-2. Mol Biol Cell. 2007; 18:2735–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Marchès O, Ledger TN, Boury M, Ohara M, Tu X, Goffaux F, et al. Enteropathogenic and enterohaemorrhagic escherichia coli deliver a novel effector called cif, which blocks cell cycle g2/m transition. Mol Microbiol. 2003; 50:1553–1567. [DOI] [PubMed] [Google Scholar]
- 156.Flatau G, Lemichez E, Gauthier M, Chardin P, Paris S, Fiorentini C, et al. Toxin-induced activation of the g protein p21 rho by deamidation of glutamine. Nature. 1997; 387:729–733. [DOI] [PubMed] [Google Scholar]
- 157.Taieb F, Petit C, Nougayrède J-P, Oswald E. The enterobacterial genotoxins: Cytolethal distending toxin and colibactin. EcoSal Plus. 2016; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhou Y, He H, Xu H, Li Y, Li Z, Du Y, et al. Association of oncogenic bacteria with colorectal cancer in South China. Oncotarget. 2016; 7:80794–80802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Balamurugan R, Rajendiran E, George S, Samuel GV, Ramakrishna BS. Real-time polymerase chain reaction quantification of specific butyrate-producing bacteria, desulfovibrio and Enterococcus faecalis in the feces of patients with colorectal cancer. J Gastroenterol Hepatol. 2008; 23:1298–1303. [DOI] [PubMed] [Google Scholar]
- 160.Shoji M, Sasaki Y, Abe Y, Nishise S, Yaoita T, Yagi M, et al. Characteristics of the gut microbiome profile in obese patients with colorectal cancer. JGH Open Open Access J Gastroenterol Hepatol. 2021; 5:498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mima K, Sakamoto Y, Kosumi K, Ogata Y, Miyake K, Hiyoshi Y, et al. Mucosal cancer-associated microbes and anastomotic leakage after resection of colorectal carcinoma. Surg Oncol. 2020; 32:63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Amarnani R, Rapose A. Colon cancer and enterococcus bacteremia co-affection: A dangerous alliance. J Infect Public Health. 2017; 10:681–684. [DOI] [PubMed] [Google Scholar]
- 163.Balish E, Warner T. Enterococcus faecalis induces inflammatory bowel disease in interleukin-10 knockout mice. Am J Pathol. 2002; 160:2253–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Huycke MM, Joyce W, Wack MF. Augmented production of extracellular superoxide by blood isolates of Enterococcus faecalis. J Infect Dis. 1996; 173:743–746. [DOI] [PubMed] [Google Scholar]
- 165.Huycke MM, Abrams V, Moore DR. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis. 2002; 23:529–536. [DOI] [PubMed] [Google Scholar]
- 166.Wang X, Huycke MM. Extracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology. 2007; 132:551–561. [DOI] [PubMed] [Google Scholar]
- 167.Wang X, Yang Y, Huycke MM. Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut. 2015; 64:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.McCOY WC, Mason JM. Enterococcal endocarditis associated with carcinoma of the sigmoid; report of a case. J Med Assoc State Ala. 1951; 21:162–166. [PubMed] [Google Scholar]
- 169.Klein RS, Recco RA, Catalano MT, Edberg SC, Casey JI, Steigbigel NH. Association of Streptococcus bovis with carcinoma of the colon. N Engl J Med. 1977; 297:800–802. [DOI] [PubMed] [Google Scholar]
- 170.Boleij A, van Gelder MMHJ, Swinkels DW, Tjalsma H. Clinical Importance of Streptococcus gallolyticus infection among colorectal cancer patients: systematic review and meta-analysis. Clin Infect Dis Off Publ Infect Dis Soc Am. 2011; 53:870–878. [DOI] [PubMed] [Google Scholar]
- 171.Corredoira-Sánchez J, García-Garrote F, Rabuñal R, López-Roses L, García-País MJ, Castro E, et al. Association between bacteremia due to Streptococcus gallolyticus subsp. gallolyticus (Streptococcus bovis I) and colorectal neoplasia: A case-control study. Clin Infect Dis Off Publ Infect Dis Soc Am. 2012; 55:491–496. [DOI] [PubMed] [Google Scholar]
- 172.Biarc J, Nguyen IS, Pini A, Gossé F, Richert S, Thiersé D, et al. Carcinogenic properties of proteins with pro-inflammatory activity from streptococcus infantarius (formerly s.bovis). Carcinogenesis. 2004; 25:1477–1484. [DOI] [PubMed] [Google Scholar]
- 173.Ellmerich S, Schöller M, Duranton B, Gossé F, Galluser M, Klein JP, et al. Promotion of intestinal carcinogenesis by Streptococcus bovis. Carcinogenesis. 2000; 21:753–756. [DOI] [PubMed] [Google Scholar]
- 174.Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002; 347:1175–1186. [DOI] [PubMed] [Google Scholar]
- 175.Hong SN, Lee SM, Kim JH, Lee TY, Kim JH, Choe WH, et al. Helicobacter pylori infection increases the risk of colorectal adenomas: cross-sectional study and meta-analysis. Dig Dis Sci. 2012; 57:2184–2194. [DOI] [PubMed] [Google Scholar]
- 176.Inoue I, Mukoubayashi C, Yoshimura N, Niwa T, Deguchi H, Watanabe M, et al. Elevated risk of colorectal adenoma with Helicobacter pylori-related chronic gastritis: A population-based case-control study. Int J Cancer. 2011; 129:2704–2711. [DOI] [PubMed] [Google Scholar]
- 177.Siddheshwar RK, Muhammad KB, Gray JC, Kelly SB. Seroprevalence of Helicobacter pylori in patients with colorectal polyps and colorectal carcinoma. Am J Gastroenterol. 2001; 96:84–88. [DOI] [PubMed] [Google Scholar]
- 178.Bae RC, Jeon SW, Cho HJ, Jung MK, Kweon YO, Kim SK. Gastric dysplasia may be an independent risk factor of an advanced colorectal neoplasm. World J Gastroenterol. 2009; 15:5722–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yan Y, Chen Y-N, Zhao Q, Chen C, Lin C-J, Jin Y, et al. Helicobacter pylori infection with intestinal metaplasia: An independent risk factor for colorectal adenomas. World J Gastroenterol. 2017; 23:1443–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kim TJ, Kim ER, Chang DK, Kim Y-H, Baek S-Y, Kim K, et al. Helicobacter pylori infection is an independent risk factor of early and advanced colorectal neoplasm. Helicobacter. 2017; 22: [DOI] [PubMed] [Google Scholar]
- 181.Nam JH, Hong CW, Kim BC, Shin A, Ryu KH, Park BJ, et al. Helicobacter pylori infection is an independent risk factor for colonic adenomatous neoplasms. Cancer Causes Control CCC. 2017; 28:107–115. [DOI] [PubMed] [Google Scholar]
- 182.Hartwich A, Konturek SJ, Pierzchalski P, Zuchowicz M, Labza H, Konturek PC, et al. Helicobacter pylori infection, gastrin, cyclooxygenase-2, and apoptosis in colorectal cancer. Int J Colorectal Dis. 2001; 16:202–210. [DOI] [PubMed] [Google Scholar]
- 183.Tatishchev SF, Vanbeek C, Wang HL. Helicobacter pylori infection and colorectal carcinoma: is there a causal association? J Gastrointest Oncol. 2012; 3:380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Handa O, Naito Y, Yoshikawa T. Helicobacter pylori: A ROS-inducing bacterial species in the stomach. Inflamm Res off J Eur Histamine Res Soc Al. 2010; 59:997–1003. [DOI] [PubMed] [Google Scholar]
- 185.Shmuely H, Passaro D, Figer A, Niv Y, Pitlik S, Samra Z, et al. Relationship between helicobacter pylori caga status and colorectal cancer. Am J Gastroenterol. 2001. 96:3406–3410. [DOI] [PubMed] [Google Scholar]
- 186.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007; 117:60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wessler S, Krisch LM, Elmer DP, Aberger F. From inflammation to gastric cancer - the importance of hedgehog/gli signaling in helicobacter pylori-induced chronic inflammatory and neoplastic diseases. Cell Commun Signal CCS. 2017; 15:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ballard J, Bryant A, Stevens D, Tweten RK. Purification and characterization of the lethal toxin (alpha-toxin) of clostridium septicum. Infect immune. 1992; 60:784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mirza NN, McCloud JM, Cheetham MJ. Clostridium septicum sepsis and colorectal cancer - a reminder. World J Surg Oncol. 2009; 7:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chew SS, Lubowski DZ. Clostridium septicum and malignancy. ANZ J Surg. 2001; 71:647–649. [DOI] [PubMed] [Google Scholar]
- 191.Corredoira J, Grau I, Garcia-Rodriguez JF, García-País MJ, Rabuñal R, Ardanuy C, et al. Colorectal neoplasm in cases of Clostridium septicum and Streptococcus gallolyticus subsp. gallolyticus bacteraemia. Eur J Intern Med. 2017; 41:68–73. [DOI] [PubMed] [Google Scholar]
- 192.Chakravorty A, Awad MM, Cheung JK, Hiscox TJ, Lyras D, Rood JI. The pore-forming α-toxin from clostridium septicum activates the MAPK pathway in a Ras-c-Raf-dependent and independent manner. Toxins. 2015; 7:516–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ribière C, Peyret P, Parisot N, Darcha C, Déchelotte PJ, Barnich N, et al. Oral exposure to environmental pollutant benzo[a]pyrene impacts the intestinal epithelium and induces gut microbial shifts in murine model. Sci Rep. 2016; 6:31027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Van de Wiele T, Vanhaecke L, Boeckaert C, Peru K, Headley J, Verstraete W, et al. Human colon microbiota transform polycyclic aromatic hydrocarbons to estrogenic metabolites. Environ Health Perspect. 2005; 113:6–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Khalil A, Villard P-H, Dao MA, Burcelin R, Champion S, Fouchier F, et al. Polycyclic aromatic hydrocarbons potentiate high-fat diet effects on intestinal inflammation. Toxicol Lett. 2010; 196:161–167. [DOI] [PubMed] [Google Scholar]
- 196.Tang WHW, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013; 368:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Romano KA, Vivas EI, Amador-Noguez D, Rey FE. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio. 2015; 6:e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Cho CE, Taesuwan S, Malysheva OV, Bender E, Tulchinsky NF, Yan J, et al. Trimethylamine-N-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: A randomized controlled trial. Mol Nutr Food Res.2017; 61: [DOI] [PubMed] [Google Scholar]
- 199.Ijssennagger N, Belzer C, Hooiveld GJ, Dekker J, van Mil SWC, Müller M, et al. Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc Natl Acad Sci U S A. 2015; 112:10038–10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Cao H, Xu M, Dong W, Deng B, Wang S, Zhang Y, et al. Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int J Cancer. 2017. 140:2545–2556. [DOI] [PubMed] [Google Scholar]
- 201.Islam KBMS, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011; 141:1773–1781. [DOI] [PubMed] [Google Scholar]