

Abstract
In silico mechanistic modeling approaches have been designed by various stakeholders with the goal of supporting development and approval of generic orally inhaled drug products in the United States. This review summarizes the presentations and panel discussion that comprised a workshop session concentrated on the use of in silico models to predict various outcomes following orally inhaled drug product administration, including the status of such models and how model credibility may be effectively established.
BACKGROUND
Orally inhaled drug products (OIDPs) are commonly used to treat a variety of diseases that include asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis, and influenza, among others. 1 Drug delivery via oral inhalation offers several advantages compared with other dosage forms, including the relatively high epithelial permeability of the lung, its large surface area, relatively small amounts of aqueous fluid at the surface, and the presence of fewer metabolizing enzymes compared with the liver and gastrointestinal (GI) tract. 2 , 3 , 4 , 5 , 6 However, the cost of potentially lifesaving OIDPs used to treat asthma and COPD is often burdensome and may reduce patient compliance. 7 , 8 , 9 , 10 , 11 , 12 Generic drug product competition is one way to reduce associated costs of OIDPs, but the complicated interplay between device, formulation, and patient use factors presents several challenges for the evaluation of generic OIDPs. 11 To facilitate accelerated development and approval of generic OIDPs, new methods are needed that are capable of considering the complexities of orally inhaled drug delivery while reducing the cost of such methods.
Various in silico mechanistic modeling approaches have been used to answer a variety of questions related to OIDP function and performance, which may be useful for facilitating development and approval of generic OIDPs. 13 These in silico methods are used to either predict regional deposition or to predict the local and systemic pharmacokinetics (PK) for the drug of interest. Regional deposition may be predicted using a semi‐empirical method such as the model developed by the National Council on Radiation Protection and Measurements, 14 which captures the impact of aerodynamic particle size distribution (APSD) on deposition. Computational fluid dynamics (CFD) is a physics‐based technique that can predict fluid and particle transport in realistic device and lung geometries and may be used to examine the influence of APSD, plume geometry, spray pattern, and inhalation waveform characteristics on regional deposition. 13 Physiologically based PK (PBPK) modeling is a mechanistic compartmental modeling technique that may be used to predict PK by considering the fate of deposited particles with model components for dissolution, mucociliary clearance, absorption, distribution, metabolism, and excretion. 15 Typically, PBPK is combined with regional deposition predictions from either a semi‐empirical model or a CFD model, although experimental data may also be used as direct inputs to the model.
The use of various modeling approaches to support development and approval of OIDPs was the subject of one of the sessions that were part of a workshop organized by the Center for Research on Complex Generics and the US Food and Drug Administration (FDA) with the title “Regulatory Utility of Mechanistic Modeling to Support Alternative Bioequivalence Approaches” that was held virtually on September 30 through October 1, 2021, and included a symposium on the first day with the title “Mechanistic Modeling of Locally‐Acting Generic Drug Products.” 16 The first part of that symposium was a session with the title “Mechanistic Modeling of Orally Inhaled Generic Drug Products—Speakers and Panel Discussion.” The primary purposes of this session of the workshop were to promote discussion among various stakeholders from the generic drug industry, academia, and FDA regarding the use of in silico models to increase the number of generic OIDPs in the US market, summarize the status of relevant in silico model development, and examine best practices for establishing model credibility. The session was organized into a series of seven presentations with European and American speakers from industry, academia, and FDA, followed by a 45‐min panel discussion with the seven speakers as well as the two session moderators and four additional panelists. The presentations covered a variety of topics, including an overview of complex generic OIDPs, establishment of model credibility, semi‐empirical and CFD regional deposition model case studies, the use OIDP modeling for regulatory purposes, FDA experience with models designed to support generic OIDP approval, and PBPK modeling to show how relationships between in vitro metrics and systemic PK metrics may be identified. This review article summarizes the presentations and panel discussion from this workshop session followed by some conclusions. The content from other sessions and symposiums in the workshop will also be published separately.
PRESENTATIONS
Overview of complex generic OIDPs
Dr. Bryan Newman (FDA) gave a presentation on the challenges and complexities associated with developing a generic OIDP, including the current and recent advancements in FDA's framework for establishing bioequivalence (BE) across a range of dosage forms. Although the characteristic features of respiratory diseases (e.g., their causes, pathological presentation, and affected patient population) can vary considerably, the method of choice for therapy has generally focused on delivering treatments directly to the lungs through patient inhalation. Achieving adequate delivery of the medication to the specifically affected lung regions is often considerably challenging for drug developers. For locally acting OIDPs, adequate regional lung deposition requires aerosolization of appropriately sized drug particles to ensure that off‐target deposition in regions such as the mouth and throat are minimized, which can also minimize the likelihood for adverse effects from systemic exposure.
During the first portion of his talk, Dr. Newman introduced some of the various methods the pharmaceutical industry has used for inhaled drug delivery, including metered dose inhalers (MDIs), dry powder inhalers (DPIs), nebulizer‐driven inhalation solutions and suspensions, and the more recently developed inhalation spray products (i.e., Soft Mist™ inhalers [Boehringer Ingelheim, Ingelheim am Rhein, Germany]). Each of these dosage forms presents unique sources of complexity to consider during product development. The mechanisms for aerosolizing the dose, formulation composition, and nature of the depositing particles can differ between the different dosage forms. Likewise, the administration steps and user interface associated with each dosage form's device constituent affect the patient's interaction with the dosage form during drug delivery and contribute to the level of complexity. Given these differing aspects of complexity for locally acting OIDPs, the FDA has generally used a weight‐of‐evidence approach for establishing BE between a test and reference OIDP, where variations of this approach are detailed in publicly posted product‐specific guidances (PSGs). The recommended BE approach included in an OIDP's weight‐of‐evidence approach can differ, but for a locally acting OIDP, it often includes both in vitro and in vivo BE studies along with formulation sameness and device similarity. The recommended in vitro BE studies provide a sensitive approach for detecting formulation differences that could impact performance, and although the type of in vitro BE study can differ between dosage forms, each is recommended to be conducted across all strengths using three test and reference batches. For the recommended in vivo BE studies, these include PK BE studies either alone or along with comparative clinical endpoint (CCEP) or pharmacodynamic (PD) BE studies. PK BE studies are included to evaluate whether there are differences in systemic exposure between the test and reference OIDP and are also recommended to be conducted across all strengths. Lastly, and depending on the dosage form, CCEP or PD BE studies are included for evaluating the local drug delivery equivalence between a test and reference OIDP. These studies often present the most challenges for a generic applicant to complete given their longer study duration and associated higher costs.
Given the challenges that generic applicants encounter with CCEP or PD BE studies, Dr. Newman concluded his talk with a focus on FDA's current thinking for potential alternative approaches to these studies. In its evaluation of local drug delivery equivalence, the CCEP or PD BE study incorporates all steps from actuation of the OIDP to deposition of the drug particles in the lungs. As such, FDA's current thinking is that through a set of in vitro, in silico, and/or alternative in vivo studies, a body of supporting data can be obtained that can serve as an alternative approach to conducting the CCEP or PD BE study within the context of the weight‐of‐evidence approach. Stemming from results from the FDA's regulatory science initiatives funded through the Generic Drug User Fee Amendments, recommendations for an alternative approach to the CCEP or PD BE study have been included in recent revisions to the PSGs for several solution‐based MDIs. 17 , 18 , 19 , 20 Notably, the types of studies that may serve as alternative BE approaches may depend on the specific dosage form and formulation of the OIDP. As stated previously, these alternative BE approaches can incorporate in silico methods that may be useful in demonstrating how the results from the other alternative BE studies can work together to establish local drug delivery equivalence. Potential examples for in silico approaches that could be incorporated include semi‐empirical regional deposition models, CFD models, and PBPK models. Semi‐empirical models could be used to provide branch‐specific deposition predictions that could be summed together to obtain predictions on regional deposition, whereas CFD models could be used to better predict how an aerosolized dose can be influenced by realistic airway geometries in healthy subjects or patients to impact regional deposition. Inclusion of a PBPK model may offer a way to predict both local and systemic PK and examine how factors such as drug dissolution, absorption, and metabolism may influence local drug concentrations.
ASME V&V 40 for establishing credibility of CFD models
Dr. Brent Craven (FDA) gave a presentation on the credibility assessment of computational modeling and simulation (M&S) of medical devices, which may be applicable to M&S for OIDPs. He first provided an overview of the American Society of Mechanical Engineers (ASME) Verification and Validation (V&V) 40–2018 risk‐informed framework for assessing M&S credibility. 21 The framework is founded on the principle that the credibility of a computational model should be commensurate with the risk incurred by using the model to influence a decision. For example, if M&S results are to be used as a primary source of evidence to inform a decision that has the potential to cause severe patient harm, then the model should be shown to be highly credible. Model credibility is generally established through verification, validation, and uncertainty quantification (VVUQ) activities.
A flow chart illustrating the credibility assessment framework of ASME V&V 40–2018 is illustrated in Figure 1. The first step is to define the question of interest that describes the specific decision that is to be made, which is usually a high‐level decision concerning the medical device that is often outside of the scope of the computational model (e.g., “Is the pulmonary regional deposition of the generic orally inhaled drug product equivalent to the brand‐name product?”). The model context of use (COU) is then defined that describes precisely how the computational model will be used to address the question of interest. Next, a model risk assessment is performed to establish how much risk is incurred by using the model to inform the question of interest. The model risk is generally characterized using a semiquantitative scale, ranging from “low risk” to “high risk.” As shown in Figure 1, a credibility plan is then formulated that describes the specific VVUQ and applicability analyses that will be performed to demonstrate model credibility. ASME V&V 40–2018 introduces a sequence of credibility “factors” that are used to guide the credibility plan. 21 As part of this, credibility goals are established for each activity such that if all goals are achieved, the overall level of model credibility would be commensurate with the model risk. The analyst then performs the modeling and executes the credibility activities. Finally, an adequacy assessment is performed in which the model results and the overall level of model credibility are considered relative to the established model risk. If both the model results and the credibility are acceptable, then the activities are documented, and the results may be used to inform the question of interest. If, however, the model results or the credibility are not acceptable, then there are several options. The model could be improved or additional VVUQ analyses could be performed to increase the credibility. Alternatively, the COU could potentially be revised to change how modeling is used to reduce the model risk. Otherwise, as a last resort, the model may be abandoned and other information (e.g., experiments) used to answer the question of interest. Such may be the case if, for example, revising the model is more costly than acquiring experimental data that may be relied on instead.
FIGURE 1.
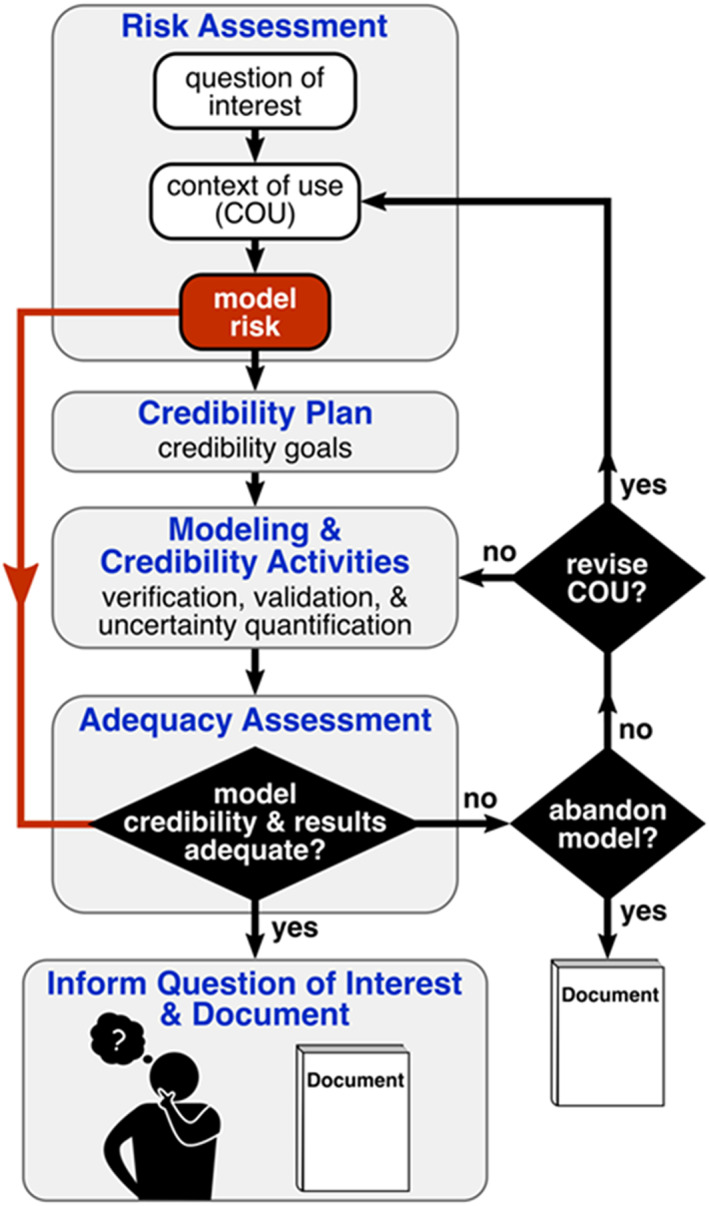
Flow chart illustrating the American Society of Mechanical Engineers Verification and Validation 40–2018 framework for establishing the credibility of computational modeling and simulation of medical devices. 70
In the second part of his talk, Dr. Craven summarized a collaborative regulatory science research project between the Center for Devices and Radiological Health (CDRH) at FDA, academia, and industry titled “A Mock Submission to Initiate a Medical Device Clinical Trial Using Modeling and Simulation” that aims to provide a real‐world, end‐to‐end example of using M&S in a regulatory submission to CDRH. Using a generic inferior vena cava filter that the team designed for the research project, they are using M&S as a primary source of evidence to support the evaluation of two nonclinical bench tests that are recommended for these devices: fatigue resistance and clot trapping. 22 , 23 In each case, they are performing M&S to replicate the nonclinical bench test, acquiring experimental measurements for validation, and demonstrating M&S credibility through VVUQ analyses following the ASME V&V 40–2018 standard. 21 The credibility evidence and the M&S results are being used in a mock regulatory submission to FDA following the CDRH M&S reporting Guidance. 24 A blind and independent regulatory review team at CDRH is evaluating the submission and providing feedback. The overall goal of the project is to inform the future revision of both ASME V&V 40–201821 and the CDRH M&S reporting Guidance. 24 In addition, the team is distributing several CDRH regulatory science tools, 25 including the mock submission documents that will serve as a real‐world, end‐to‐end example of using M&S in a medical device regulatory submission.
Validation of computational predictions of regional lung deposition
Dr. Bo Olsson (Emmace Consulting) talked about validation of generation‐level lung deposition models that use a semi‐empirical modeling approach. Validation of generation‐level models must be indirect because generation‐level in vivo data are lacking. Hence, either in vivo data or in silico predictions must be transformed for them to be comparable. Two validation approaches were discussed. The first of these approaches uses planar scintigraphy, which is the most common type of in vivo data that quantifies regional lung deposition. This technique provides images of radioactivity in the lungs where central (C), intermediary (I), and peripheral (P) regions of interest (ROIs) are defined. However, because each region contains a mixture of airway generations, the terms are slightly misleading. It also prohibits a direct comparison of, for example, calculated deposition in the first eight bifurcations (the tracheobronchial airways) with activity measured in the C ROI. Obviously, the planar image of C contains a significant number of acini. Fortunately, Schroeter et al. 26 published a translation map derived from a two‐dimensional (2D) projection of a three‐dimensional (3D) mathematical generation‐level model of the lung that maps the contribution of each airway generation to the different ROIs. Such a map makes it possible to transform generation‐level deposition to expected activity in the ROIs. Essentially, this transformation of calculated data makes it directly comparable with measured ROI activity but at the expense of downgrading generation‐level deposition to a blurred image. Olsson and Kassinos 27 used this validation approach for the Mimetikos Preludium™ software package (Emmace Consulting, Lund, Sweden) comparing ROI activity in 18 scintigraphic study legs involving nine DPI brands with computed regional lung deposition based on the accompanying in vitro data. The computed oropharyngeal and total lung deposition correlated with high significance (p < 0.0001) to the scintigraphic results with virtually no bias over a wide range. For the regional lung distribution, computed C, P, and P/C results correlated with high significance (p < 0.01) to the corresponding scintigraphic measures but with a bias toward underpredicting C and overpredicting P/C, as shown in Figure 2. During the presentation, it was concluded that mapping of generational‐level deposition to scintigraphic ROI is valid, that the map developed by Schroeter et al. 26 probably can be improved based on new high‐resolution tomographic data, and that direct comparison of generational‐level deposition to scintigraphic ROI is an invalid approach.
FIGURE 2.
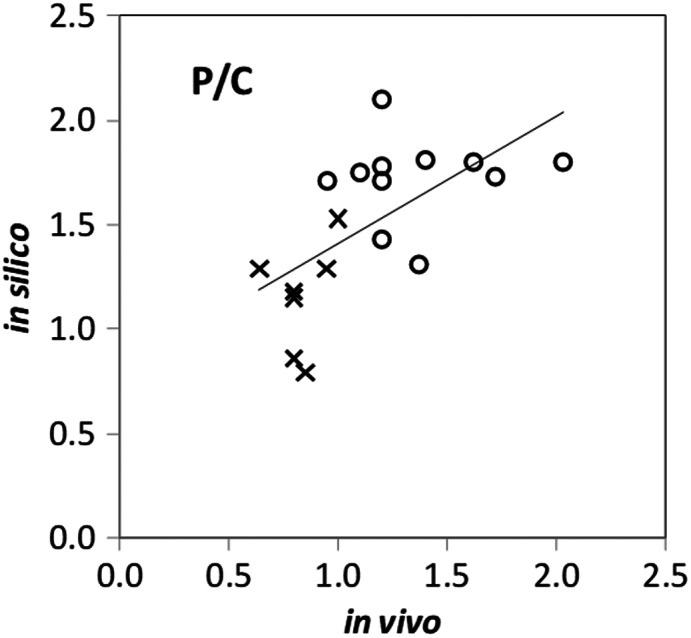
Unweighted linear regression of in silico on in vivo results for the peripheral/central (P/C) ratio. O indicates healthy volunteers, and X indicates asthmatics. Reproduced from Olsson and Kassinos. 27
The second validation technique discussed by Dr. Olsson uses PK data such as plasma concentration–time curves and associated metrics (maximum concentration [C max], area under the plasma concentration‐time curve [AUC], etc.) to indirectly validate regional deposition calculations when different regions in the lung have different rates and/or extents of absorption. To establish this relationship, a mechanistic PBPK model is required together with high‐quality PK data. To explore the use of this technique, PK data were obtained from three two‐way crossover BE studies that were conducted under fasting conditions for three strengths of brand‐name and generic versions of fluticasone propionate (FP) and salmeterol xinafoate inhalation powder (i.e., Advair Diskus [GlaxoSmithKline, Durham, NC, USA] and Wixela Inhub [Mylan Pharmaceuticals Inc., Canonsburg, PA, USA]). 28 Concurrently, in vitro studies that measured APSD and dissolution kinetics in the same product batches were conducted, where it was observed that the measured APSD values, which are directly related to regional deposition values, were very similar for the three strengths of the brand‐name product, whereas the dissolution kinetics differed markedly with the low strength having the fastest dissolution and the high strength having the slowest. 29 This was accompanied with markedly different plasma concentration peak shapes (C max/AUC), with the low strength having the sharpest peak and the high strength having the dullest. 28 Using Mimetikos Preludium, Bäckman and Olsson 29 simulated deposition, dissolution, absorption, and systemic disposition of FP delivered from the brand‐name product based on this study. The results from that study showed that simulated plasma curves matched well with the observed data, as evidenced by the ratios of simulated to observed values for AUC, C max, and C max/AUC for the three strengths, which ranged between 0.85 to 1.09. Based on a sensitivity analysis that demonstrated a pronounced influence of regional distribution on PK output, it was concluded during the presentation that the deposition and dissolution models are valid.
Case study: Predicting regional lung deposition of pharmaceutical aerosols with CFD
Dr. Worth Longest (Virginia Commonwealth University) discussed the use of CFD models to predict regional deposition following OIDP administration. CFD models that consider drug delivery from OIDPs have advanced to the point where they are capable of highly accurate predictions of pharmaceutical aerosol deposition within specific regions of the airways 30 , 31 , 32 , 33 , 34 and throughout the lungs. 35 , 36 , 37 , 38 , 39 , 40 For simulating pharmaceutical aerosol transport and deposition throughout the airways, multiple groups have proposed different simplification techniques to address the myriad of individual bifurcations and structures in the lungs based on the premise that full resolution of aerosol deposition in every individual bifurcation or structure is unnecessary. 38 , 39 , 41 , 42 , 43 , 44 One such approach developed at Virginia Commonwealth University is referred to as a complete‐airway simulation using the stochastic individual pathway (SIP) approach. 42 , 43 , 44 Using this sampling methodology, a fully resolved patient‐specific or characteristic CFD model of the mouth–throat (MT) and upper tracheobronchial (TB) region through approximately the third bifurcation (B3; lobar bronchi) is first developed. 34 Beyond B3, individual 3D pathways are generated into the five lung lobes through the terminal bronchioles (~B15), and at each bifurcation, one branch is continued and the other is not. Additional pathways are stochastically sampled and simulated until the regional deposition values fall within a predefined convergence criterion, for example, 1% change in relative difference. Advanced one‐dimensional, complete‐airway anatomical and ventilation models 45 , 46 , 47 can be used to define the bifurcating geometrical pathway, which is then sampled using the SIP approach and rendered as a 3D structure for CFD simulation. These complete‐airway models can also be applied to define the heterogeneous ventilation distribution map that occurs within the lungs for application to the incomplete SIP pathway outlets. Transport and deposition in the alveolar structure is simulated using a space‐filling, moving‐wall acinar structure 48 or associated correlations from this model. Several recent studies have demonstrated good agreement between complete‐airway SIP model predictions of pharmaceutical aerosol deposition and 2D gamma scintigraphy data for multiple inhaler types. 36 , 37
The use of the complete‐airway SIP approach was described by Dr. Longest, who summarized a previous study that compared the regional pharmaceutical aerosol deposition of a commonly used MDI (FP inhalation metered aerosol, with the brand‐name Flovent HFA [GlaxoSmithKline, Durham, NC, USA]) and DPI (FP inhalation powder, with the brand‐name Flovent Diskus [GlaxoSmithKline, Durham, NC, USA]), both delivering 250 μg of FP. 43 The inhalers were connected to characteristic MT and upper TB airways, and SIP models were passed into each lung lobe, as illustrated in Figure 3a. Correct inhalation waveforms based on patient use instructions were slow‐and‐deep (SD; time‐averaged mean flow of 37 L/min) inhalation with the MDI and quick‐and‐deep (QD; time‐averaged mean flow of 75 L/min) inhalation with the DPI (Figure 3b). Incorrect use of each inhaler was also simulated by assigning the QD profile to the MDI and the SD profile to the DPI. Cascade impaction experiments at different mean flow rates were implemented to determine the associated polydisperse size distribution (PSD) from each inhaler, which was implemented as the initial size distribution at the aerosol injection position within each inhaler mouthpiece. Although this approach is a significant simplification of MDI droplet breakup and evaporation and DPI aggregate breakup, it significantly expedites the CFD calculations with accurate final‐state particle data and has shown good agreement with in vitro 32 , 33 , 42 , 43 , 44 , 49 and in vivo 36 , 37 data in many cases. Further details on the complete‐airway setup, CFD simulations, and assumptions can be found in Longest et al. 43
FIGURE 3.
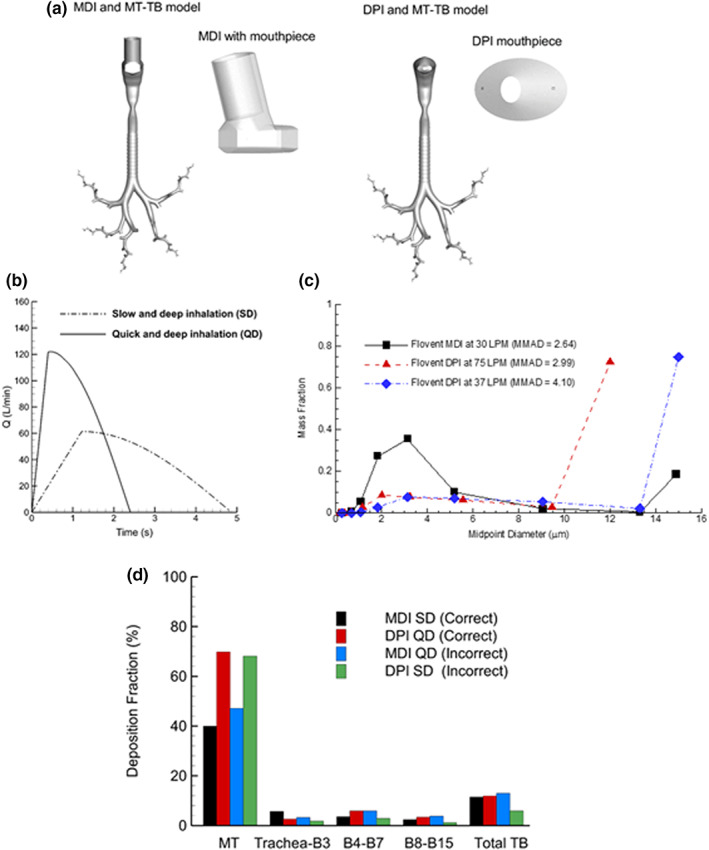
Elements of a computational fluid dynamics–based complete‐airway stochastic individual pathway model for comparison of regional MDI and DPI drug delivery including (a) computational models of the inhaler flow pathways and conducting airways, (b) prescribed SD and QD inhalation profiles, (c) experimentally measured particle size distributions at different tested flow rates, and (d) deposition fractions in specific airway regions as a percentage of aerosolized drug. DPI, dry powder inhaler; MDI, metered dose inhaler; MMAD, mass median aerodynamic diameter; MT, mouth–throat; Q, flow rate; QD, quick and deep; SD, slow and deep; TB, tracheobronchial. Reprinted by permission from Springer Nature: Springer Nature, Pharmaceutical Research, Comparing MDI and DPI aerosol deposition using in vitro experiments and a new stochastic individual path (SIP) model of the conducting airways Longest et al. 43
The impact of breathing profiles on deposition was discussed by Dr. Longest. Based on the complete‐airway model simulations (Figure 3d), MT deposition was not largely impacted by correct versus incorrect inhalation; however, DPI aerosol loss was approximately twofold higher than with the MDI. Within the lung for correct inhalation, the DPI was observed to deliver more drug to the intermediate (B4–B7) and lower (B8–B15) regions. However, this trend reversed when incorrect inhalation was employed (Figure 3d and Table 1). Furthermore, the DPI appeared significantly more sensitive to correct versus incorrect use. These comparisons illustrate how regional deposition modeling can be employed to compare different inhalation profiles and enable a better understanding of the association between regional drug delivery and disease response. 50 , 51 , 52
TABLE 1.
Deposition fractions as a percentage of aerosolized dose with correct inhalation (MDI with SD and DPI with QD).
| Region | MDI | DPI | Relative difference (%) a |
|---|---|---|---|
| MT b | 40.0 | 69.8 | 54 |
| Trachea–B3 | 5.7 | 2.6 | 75 |
| B4–B7 | 1.5 | 1.0 | 40 |
| B8–B15 | 0.9 | 0.6 | 40 |
| Total TB | 8.1 | 4.2 | 63 |
| Alveolar | 51.9 | 26.1 | 66 |
Abbreviations: DPI, dry powder inhaler; MDI, metered dose inhaler; MT, mouth–throat; QD, quick and deep; SD, slow and deep; TB, tracheobronchial.
Relative difference is the absolute value of the difference divided by the average, multiplied by 100%.
Includes deposition from the MDI inhaler.
The presentation was concluded with a discussion on the potential use of CFD modeling for BE assessments of OIDPs. Comparisons can be made between a reference listed drug (RLD) and potential generic drug products using CFD modeling to understand differences in regional deposition on a quantitative basis. For example, Table 1 provides an illustration of relative differences between MDI and DPI drug delivery within each of the sample regions. As expected, these relative differences are high (40%–75%) due to the inherent differences between the two platforms. Dr. Longest suggested that a similar approach could be used in the comparison of an RLD product and a generic product with a relative difference threshold of, for example, 10%. According to Dr. Longest, the exact threshold for different inhalation products would depend on the specific PK, PD, and safety profiles and may occur over a range of, for example, 5% to 20% when tested in a single characteristic adult airway model. These results can serve as a starting point for subsequent dissolution, absorption, and clearance modeling 53 , 54 or as an input for a whole‐body PBPK model. 55 Alternatively, regional deposition can potentially serve as a stand‐alone comparison between an RLD product and potential generic product that is evaluated in parallel with separate methods to evaluate dissolution and epithelial cell absorption.
Modeling to support regulatory needs of OIDPs
Dr. Raja Mohamed (Sandoz) provided his viewpoint on how modeling may be used to support approval of generic OIDPs in the United States. A weight‐of‐evidence approach has been used by the FDA to evaluate the BE of potential generic OIDPs, which includes recommendations for formulation and device sameness and comparative in vitro and PK equivalence as well as comparative PD or CCEP studies. 56 , 57 This weight‐of‐evidence approach is recommended by the FDA because, unlike drug administration from solid oral dosage forms, in vivo PK data measured following OIDP administration capture processes that are downstream of the site of action, and thus an approach that relies only on PK data is not considered by the FDA to be sufficient. 56 Thus, it was concluded during the presentation that the complexity of drug delivery to the site of action following OIDP administration, as illustrated by Hatipoglu et al. 58 and shown in Figure 4, is the primary reason for the selection of a weight‐of‐evidence approach by the FDA. However, the weight‐of‐evidence approach poses many challenges for generic product developers, where large sample sizes may be needed for recommended PD/CCEP BE studies due to expected high variability of the related clinical endpoints and potentially reduced sensitivity to formulation differences, which may require additional time and cost. 59 , 60 Considering these factors, in recent years the FDA has included language describing alternative BE approaches in lieu of recommended CCEP BE studies in several PSGs, including the possible use of PBPK and CFD modeling to support determination of BE for beclomethasone propionate inhalation metered aerosol 17 , 20 and a morphology‐directed Raman spectroscopy approach for mometasone furoate nasal spray and fluticasone furoate nasal spray. 61 , 62
FIGURE 4.
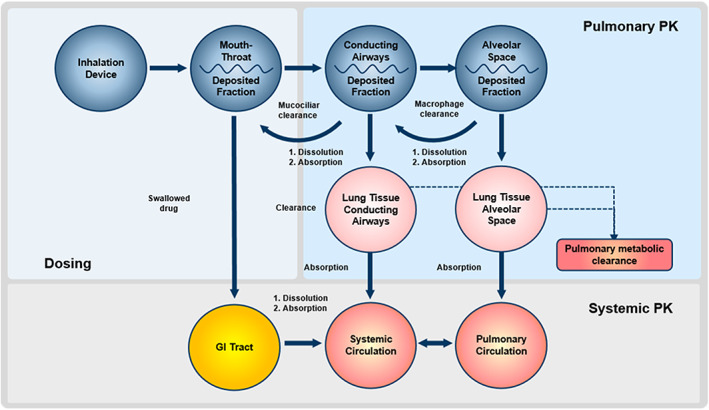
Dosing, pulmonary PK, and systemic PK following orally inhaled drug product administration. GI, gastrointestinal; PK, pharmacokinetics. Reprinted from International Journal of Pharmaceutics, 549, Hatipoglu MK, Hickey AJ, Garcia‐Contreras L, Pharmacokinetics and pharmacodynamics of high doses of inhaled dry powder drugs, 306–316, Copyright 2018, with permission from Elsevier. 58
Following discussion of the weight‐of‐evidence approach, Dr. Mohamed described how modeling may be used as a tool to understand the performance of OIDPs, including the potential impacts from device and formulation differences, as well as the effects of intersubject variability. To be successful, it was proposed that in silico models should focus on linking in vitro data such as those collected from APSD and dissolution studies, among others, with in vivo PK and PD data. Models may be developed using systemic disposition and elimination properties obtained from experimental data available in the literature, along with data collected with relevant in vitro studies that may include solubility, dissolution, APSD, and characterization of powder emission, among others. The overall modeling strategy may be based on separate processes such as deposition and PK or integrated such that all relevant processes are linked, where an integrated model may be capable of simultaneously predicting local and systemic drug concentration values. Model validation is essential for demonstrating the value of the model in the context of any proposed alternative BE approach, where validation may be strengthened by using both registration and manufacturing scale batches to produce comparator data. The presentation was concluded with a few examples of how semi‐empirical and CFD regional deposition models, PBPK modeling, and dissolution modeling have been used to support the development of potential generic OIDPs.
ANDA and Pre‐ANDA experience with OIDP modeling
Dr. Ross Walenga (FDA) described abbreviated new drug application (ANDA) and pre‐ANDA interactions with the FDA that have included modeling approaches to support the determination of BE for OIDPs. Various firms have included semi‐empirical and CFD regional deposition, PBPK, and population PK models in pre‐ANDA meeting request packages and ANDA submissions for OIDPs. Although an end‐to‐end approach, including simulation of inhaled aerosols from the device to the point of deposition and subsequent absorption, would be beneficial for supporting pre‐ANDA and ANDA submissions that propose alternative BE approaches, these models usually simulate only one part of the drug delivery process and are intended to work together with other in vitro and/or in vivo studies to consider the whole process. Typically, these models aim to show that local drug delivery to the site of action in the lungs is equivalent via either direct comparison of regional deposition predictions or by establishing a connection between PK metrics and regional deposition. For instances when the goal is to show that PK metrics and regional deposition are related, the model may then be used to compare PK metrics with acceptance criteria carefully selected to ensure BE at the site of action.
Several common areas for improvement were identified in the presentation, and potential solutions to these issues were offered by Dr. Walenga. Models are often submitted to the FDA with inadequate demonstration of model credibility, where verification and validation activities may be absent or insufficient, and validation may only be performed for one drug product and thus may not fully demonstrate the capacity of the model to consider formulation differences. For models that predict regional deposition, it was clarified that comparator data from either gamma scintigraphy or single photon emission computed tomography (SPECT)/computed tomography (CT) in vivo studies are preferred and that the mismatch of 2D in vivo data with 3D predictions may be addressed by using a mapping procedure such as the one described in Schroeter et al. 26 Another common issue is that regional deposition models may not justify the location of the division between the C and P lung regions, where justification based on literature data 50 , 63 should be used to support the division of lung regions. Some regional deposition models have been proposed for products such as solution‐based MDIs or DPIs, but they did not include model components to account for relevant physical processes such as evaporation or agglomeration and deagglomeration. To demonstrate how this may be accomplished, two CFD case studies that included model components to account for evaporation in a solution‐based MDI 64 and agglomeration and deagglomeration in a DPI 65 were presented. Although evaporation may not be a crucial physical process to model for suspension‐based MDIs due to the rapid evaporation of the propellant that is expected to occur prior to exit from the device, agglomeration and deagglomeration may be important processes to consider. Physically, processes such as evaporation and hygroscopic growth may also be potentially considered in semi‐empirical deposition models, as suggested by Youn et al. 66 Several PBPK models provided by industry have attempted to use systemic PK predictions to infer BE at the site of action without providing sufficient evidence to establish this link. Alternatively, it was proposed during the presentation that PBPK may instead be used to understand the impact of dissolution and permeation following drug deposition to better understand if regional deposition predictions may be used alone to evaluate BE at the site of action or if dissolution and/or permeation may be sufficiently slow to create significant differences between regional deposition and regional absorption. Other issues that were identified were contradicting predictions from regional deposition models developed in parallel, insufficiently developed statistical analysis methods, a lack of connection between in vitro data generated by the firm and model input parameters, and a lack of details for the simulation methods. The presentation concluded with an affirmation that modeling may be useful for supporting an alternative BE approach for OIDPs and that the potential issues and remedies that were identified should be considered to improve the chances of success.
Use of mechanistic modeling to determine the sensitivity of in vitro critical quality attributes (CQAs) to regional lung deposition and predict PK for OIDPs
Dr. Clare Butler (Teva) presented two case studies that described the use of Mimetikos Preludium to predict deposition and PK following administration of a DPI. Mimetikos Preludium is a semimechanistic modeling platform for OIDPs that uses a semi‐empirical approach to predict deposition and ordinary differential equations to describe the absorption and elimination processes of inhaled compounds. 67 The software package simulates the absorption of drug deposited in the lung according to specifications of regional deposition, lung and systemic properties, compound and formulation properties, and dosing. Mimetikos Preludium calculates the fraction of drug deposited within specific regions of the lung using regressed cascade impactor data and infers the relative contribution of each lung region (extrathoracic, tracheobronchial, bronchiolar, and alveolar–interstitial) to systemic absorption. Changes in particle size distribution and/or dissolution properties of an inhaled drug can be reflected by altered simulated PK profiles in Mimetikos Preludium. 67
As described in the presentation, a Mimetikos Preludium model was previously generated and validated for tiotropium bromide inhalation powder. The systemic distribution and elimination of tiotropium was simulated in Mimetikos Preludium using compartmental parameters calculated from a PK study of intravenously administered tiotropium. 68 Then, a PK study was conducted using two different dry powder formulations with equivalent 18 μg of tiotropium base per inhalation (i.e., Eq 0.018 mg Base/inh) that were labeled as Lot A and Lot B. An APSD study was conducted, determining similar fine‐particle doses for each formulation, where the results were 3.39 μg and 3.37 μg for Lot A and Lot B, respectively. In vivo PK data showed negligible differences in the resulting C max, time to C max, and AUC from 0 to 72 h (AUC0–72h) values for both formulations. Mimetikos Preludium PK simulations were then conducted to provide predictions, and Table 2 shows the observed and simulated PK results. It was concluded by Dr. Butler that the PK data simulated by Mimetikos Preludium agree well with the observed data based on the comparison of natural logarithms of PK metrics from the observed data and simulated values. The PK profile for a hypothetical formulation with a strength of Eq 0.018 mg Base/inh with a significantly higher fine‐particle dose of 4.45 μg, which was labeled as Lot C, was subsequently simulated using the validated model, and the results are also provided in Table 2. Compared with Lot A and Lot B, the resulting simulated PK profile for Lot C yielded an approximate 36% and 27% increase in C max and AUC0–72h, which highlights the potentially significant effects of particle size differences on PK. Regional lung deposition predictions for Lot C, as shown in Table 3, were also significantly different than predictions based on Lot A and Lot B, with an approximate 15% increase in deposition observed in the alveolar–interstitial region. Taken together, these results support the use of mechanistic modeling to produce accurate PK predictions for OIDPs, determine the effects of differing in vitro particle size distributions on PK outcomes, and demonstrate that the PK outcome is sensitive to changes in regional deposition.
TABLE 2.
PK metrics as calculated using observed in vivo PK data and simulated PK, following administration of three lots of a potential generic version of tiotropium bromide inhalation powder with a strength of Eq 0.018 mg Base/inh, where Lots A, B, and C have measured fine‐particle doses of 3.39 μg, 3.37 μg, and 4.45 μg, respectively.
| C max (pg/ml) | t max (h) | AUC0–72h (pg‐h/ml) | |
|---|---|---|---|
| Observed, Lot A | 22.41 | 0.1 | 99.84 |
| Simulated, Lot A | 27.35 | 0.1 | 102.22 |
| Observed, Lot B | 21.71 | 0.1 | 100.47 |
| Simulated, Lot B | 28.26 | 0.1 | 101.77 |
| Simulated, Lot C | 37.81 | 0.1 | 129.72 |
Abbreviations: AUC0–72h, area under the plasma concentration‐time curve from 0 to 72 h; C max, maximum concentration; PK, pharmacokinetics; t max, time to maximum concentration.
TABLE 3.
Regional deposition predictions for administration of three lots of a potential generic version of tiotropium bromide inhalation powder with a strength of Eq 0.018 mg Base/inh, where Lots A, B, and C have measured fine‐particle doses of 3.39 μg, 3.37 μg, and 4.45 μg, respectively.
| Extrathoracic | Tracheobronchial | Bronchiolar | Alveolar–Interstitial | |
|---|---|---|---|---|
| Lot A | 53.54 | 10.33 | 5.47 | 30.58 |
| Lot B | 56.86 | 12.00 | 5.75 | 25.29 |
| Lot C | 41.32 | 10.54 | 6.25 | 41.40 |
PANEL DISCUSSION
The session was concluded by a panel discussion that was moderated by Dr. Andrzej Przekwas (CFD Research Corporation) and Dr. Robert Lionberger (FDA). The panel included the seven presenters introduced previously in this article as well as Dr. Günther Hochhaus (University of Florida), Dr. Bing Li (FDA), Dr. Markham Luke (FDA), and Dr. Liang Zhao (FDA). Dr. Przekwas began the conversation by noting the significant progress that has been made for regional deposition and PBPK models while noting that there is still a need to link modeling of drug dissolution, clearance, and absorption processes in the airway barrier. Following up on the comment from Dr. Przekwas on advancements for semi‐empirical models, Dr. Olsson described the addition of a GI tract compartment to Mimetikos Preludium and the potential addition of a PBPK model of the GI tract. It was asked if a device compartment may be added to Mimetikos Preludium, but the response was that users are currently encouraged to use in vitro realistic MT APSD data for model inputs from the device and MT rather than to design a new compartment for that purpose. Regarding the relative difficulty of using CFD, Dr. Longest indicated that the state of the art is such that specialists with advanced training are needed to conduct these types of simulations. It was also emphasized that CFD should be paired with in vitro data such as APSD, emitted dose, and spray velocity to be used as model inputs. Regarding the limitations of CFD, access to commercial software packages was named as a potential barrier due to challenges with integrating custom models and with cost, where the use of open‐source software packages such as OpenFOAM are attractive options to overcome these problems. With respect to computational limitations, the burden appears to be less than in the past, where a workstation that costs $30,000–$40,000 may be sufficient. The future of CFD modeling in this area may include integration with dissolution and absorption. Dr. Walenga commented on the use of regional deposition models for regulatory purposes using either semi‐empirical or CFD methods, where the utility of such models may be dictated by characteristics of the compound. If the compound is quickly dissolving and absorbing, regional deposition predictions may be enough to support BE, but if either process is rate‐limiting, then there may be more weight on the results of related in vitro studies such as dissolution. The use of PBPK to better understand the relationship between regional deposition and regional absorption in this scenario may be appropriate, where model parameterization may be supported by in vitro measurements of drug permeability and dissolution.
Model credibility and its use for OIDP modeling was discussed. Dr. Craven addressed a question from Dr. Przekwas regarding the potential need for uncertainty quantification with experimental data in much the same way that he had described for modeling. Dr. Craven responded by agreeing that certainly experimentalists should be quantifying uncertainty, although he noted that there are some differences compared with modeling. In most experiments, the influence of any variability is inherently accounted for by performing repeat tests and by using multiple test samples. Deterministic computational models, however, do not account for this variability if they simply use nominal geometry and test conditions. If such variability is important, computational modeling can use uncertainty quantification methods to propagate various sources of uncertainty through the model to quantify their influence on the results. Considering a question concerning the potential for a mock submission of an OIDP model such as the mock submission for medical devices that was described earlier in the session, Dr. Luke commented that there are significant differences between medical devices and OIDPs, such as existing standards for medical devices and the additional complexity of OIDPs, that may make such a mock submission for an OIDP model challenging, although he indicated that such an exercise may still have some educational value.
Concerning the PBPK modeling described as part of Dr. Mohamed's presentation, he elaborated that it is useful to conduct in vivo PK studies that are designed to support PBPK model development and validation and that with the model it may be possible to extrapolate to a larger study size. With respect to the use of CFD to support an ANDA submission, it was posited that CFD modeling may be useful in scenarios where experimental data are not available to provide missing information where needed. Dr. Butler commented on PBPK modeling for highly soluble and poorly soluble drugs, that to obtain a favorable validation there is a need for accurate and reliable in vitro data to be used for model inputs such as APSD data. Dr. Olsson added that for a poorly soluble drug such as FP, the modeling study he described 29 was supported by in vitro 29 and in vivo 28 data collected from the same batch of the same product, all with the same age, with the implication that doing so may improve model validation.
The relative value of different dissolution tests to provide input PBPK data was discussed by Dr. Olsson, who indicated his preference for US Pharmacopeia Apparatus 2 (i.e., paddle) dissolution testing using aerosols that have been sampled from an impactor, where the aerosol sampling method is important. There may be value to dissolution testing with a membrane‐based method for poorly soluble compounds, but for drugs with high solubility, this type of test may measure diffusion rather than dissolution. Dr. Hochhaus asked about the ideal dissolution media and whether water solubility or solubility in biorelevant media may be best for defining a PBPK parameter. Dr. Olsson replied that some type of solubility enhancer is needed for dissolution testing and that the parameterization of the output is important, where the apparent volume median diameter and apparent PSD are useful characterizations of the initial surface area available for dissolution and over time, which are useful for both in vitro and in vivo conditions. With respect to solubility inputs for Mimetikos Preludium, solubility in phosphate‐buffered saline is used along with another parameter that describes the free fraction in the epithelial lining fluid that can act as a reservoir for biphasic systems. Dr. Li expressed that for BE of OIDPs the role of dissolution testing is unclear because for many products dissolution is rapid and asked the panel what challenges must be overcome to use dissolution to establish BE or if there is a role for dissolution to be purely used to support modeling. Dr. Luke answered that, as discussed by Dr. Butler, the role of dissolution would likely be product specific. Dr. Hochhaus referred to recently published work 69 where dissolution testing of three different formulations was valuable for understanding the difference in the behavior of these formulations, even though the dissolution results may not be useful for PBPK model inputs. Dr. Newman expressed that when a dissolution study is designed, it may be optimized to show product differences or it may be constructed to provide biorelevant results to be used for model inputs, and that the degree of solubility may determine the relative weight of a stand‐alone dissolution study within an alternative BE approach for OIDPs.
Dr. Przekwas asked the panel if there are any efforts funded by the FDA to further develop in vivo methods for measuring regional deposition, such as gamma scintigraphy, to improve resolution of these data to be used for regional deposition model validation. In response, Dr. Walenga suggested that SPECT/CT data may be useful due to the ability of the technique to record 3D information and also that the mapping procedure discussed earlier in the session and outlined by Schroeter et al. 26 may be used to provide direct comparisons between modeling predictions and 2D data collected using gamma scintigraphy. Dr. Przekwas emphasized the need for new data collected using improved techniques, and Dr. Luke added that it is important to collect data for new products on the market, such as the products with novel inhalation device design or delivery mechanisms.
A final topic for discussion was introduced by Dr. Butler, who asked about the future of FDA recommendations for in silico models and at what point in time will PSGs include specific information for how to use these models for regulatory submissions. Dr. Walenga replied that pre‐ANDA meetings and ANDA submissions as well as research in this area has proved to be instructive and that the understanding at FDA for how to best use these models is still evolving. Dr. Li added that a recommendation to use M&S is currently present in the PSGs for beclomethasone dipropionate solution‐based MDI products 17 , 20 and that M&S was also used recently to support approval of an OIDP where there was in issue with imputed data in the submitted bronchoprovocation PD BE study. Also, Dr. Newman explained that recommendations for M&S are also present in PSGs for ipratropium bromide 18 and ciclesonide 19 solution‐based MDI products. It was clarified that Dr. Butler was interested in knowing when PSGs that include M&S recommendations for suspension‐based MDIs and DPIs may be available, and Dr. Newman replied that for those more complex products it may take more time, but that there are efforts in place to develop understanding for those. Dr. Przekwas then asked about how determination of BE may differ for different drug types such as corticosteroids, long‐acting β2 adrenergic agonists, and short‐acting β2 adrenergic agonists that may all target different lung regions. Dr. Newman replied that BE recommendations for alternative BE approaches until now have focused on techniques that ensure equivalent delivery to the site of action and have not considered the mechanism of action of the drug, although differences in drug type would manifest in parameter selection for PBPK models. Dr. Zhao commented that the overall momentum toward developing recommendations for alternative BE approaches for OIDPs that include in silico is positive and that the next steps include the establishment of best practices for relevant models and that advances intended for generic drug development and approval may be useful for new drug development as well. In response to the question from Dr. Przekwas on how BE recommendations may differ for different drug types, Dr. Luke clarified that for fixed‐combination drug products as defined by 21 Code of Federal Regulations (CFR) 300.50 71 with more than one active ingredient, the expectation for each active ingredient would be clarified in individual PSGs. Dr. Lionberger concluded the panel discussion by providing support for the enhancements to OIDP modeling capabilities in recent years and indicated that the next step will be to integrate these new modeling strategies into internal FDA processes and into product development processes within industry.
CONCLUSIONS
Increased generic competition is needed for OIDPs to reduce associated costs to the patient and issues with patient compliance 7 , 8 , 9 , 10 , 11 , 12 for these valuable medicines. 1 In silico modeling has been proposed along with other in vivo and in vitro techniques as a means of supporting generic OIDP development and approval by potentially decreasing the need for large clinical studies and thereby helping to reduce development timelines and costs for members of industry. The purpose of this workshop session was to bring various stakeholders together to present the status of modeling for OIDPs and to discuss how to best establish model credibility. The promise of these modeling techniques was evident in several of the presentations in this session, which provided case studies for semi‐empirical and CFD regional deposition as well as PBPK models that all showed good agreement with in vivo data. A variety of current and future challenges were identified for these models with respect to their potential for supporting alternative BE approaches for OIDPs. One recurring theme was the need for quality in vitro and in vivo data to support model validation and parameterization, and it was emphasized that these data are in general preferably collected in parallel with model development rather than obtained retrospectively from other studies because data collected in parallel are more likely to accurately represent the simulated case. Especially, there is a need for new in vivo regional deposition data that can precisely capture regional differences considering the current lack of recent data from literature. Although there were several examples of model validation given in the presentations, there is a need to better understand how to apply a framework such as ASME V&V 40–2018 to assess the credibility of OIDP models intended to support approval of OIDPs. Altogether, there was a consensus that the next step for modeling to support development and approval of OIDPs will be to better understand how to best integrate these models into decision‐making processes across a variety of OIDPs that differ in device principles and active ingredients.
FUNDING INFORMATION
No funding was received for this work.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
ACKNOWLEDGMENTS
The article reflects the views of the authors and should not be construed to represent the US Food and Drug Administration's views or policies.
Walenga RL, Butler C, Craven BA, et al. Mechanistic modeling of generic orally inhaled drug products: A workshop summary report. CPT Pharmacometrics Syst Pharmacol. 2023;12:560‐574. doi: 10.1002/psp4.12889
REFERENCES
- 1. Anderson S, Atkins P, Bäckman P, et al. Inhaled medicines: past, present, and future. Pharmacol Rev. 2022;74:48‐118. [DOI] [PubMed] [Google Scholar]
- 2. Ji CM, Cardoso WV, Gebremichael A, et al. Pulmonary cytochrome P‐450 monooxygenase system and Clara cell differentiation in rats. Am J Physiol Lung Cell Mol Physiol. 1995;269:394‐402. [DOI] [PubMed] [Google Scholar]
- 3. Keith IM, Olson EB, Wilson NM, et al. Immunological identification and effects of 3‐methylcholanthrene and phenobarbital on rat pulmonary cytochrome P‐450. Cancer Res. 1987;47:1878‐1882. [PubMed] [Google Scholar]
- 4. Patton JS. Mechanisms of macromolecule absorption by the lungs. Adv Drug Deliv Rev. 1996;19:3‐36. [Google Scholar]
- 5. Patton JS, Byron PR. Inhaling medicines: delivering drugs to the body through the lungs. Nat Rev Drug Discov. 2007;6:67‐74. [DOI] [PubMed] [Google Scholar]
- 6. Tronde A, Nordén B, Marchner H, Wendel AK, Lennernäs H, Bengtsson UH. Pulmonary absorption rate and bioavailability of drugs in vivo in rats: structure absorption relationships and physicochemical profiling of drugs. J Pharm Sci. 2003;92:1216‐1233. [DOI] [PubMed] [Google Scholar]
- 7. Carrier E, Cunningham P. Medical cost burdens among nonelderly adults with asthma. Am J Manag Care. 2014;20:925‐932. [PubMed] [Google Scholar]
- 8. Castaldi PJ, Rogers WH, Safran DG, WIlson IB. Inhaler costs and medication nonadherence among seniors with chronic pulmonary disease. Chest. 2010;138:614‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DiMatteo MR. Variations in patients' adherence to medical recommendations: a quantitative review of 50 years of research. Med Care. 2004;42:200‐209. [DOI] [PubMed] [Google Scholar]
- 10. Neuman P, Strollo MK, Guterman S, et al. Medicare prescription drug benefit progress report: findings from a 2006 national survey of seniors: the first in‐depth look at the characteristics and experiences of seniors since the implementation of the Medicare drug benefit. Health Aff. (Millwood). 2007;26:w630‐w643. [DOI] [PubMed] [Google Scholar]
- 11. Newman B, Witzmann K. Addressing the regulatory and scientific challenges with generic orally inhaled drug products. Pharmaceut Med. 2020;34:93‐102. [DOI] [PubMed] [Google Scholar]
- 12. Piette JD, Heisler M, Wagner TH. Cost‐related medication underuse among chronically ill adults: the treatments people forgo, how often, and who is at risk. Am J Public Health. 2004;94:1782‐1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walenga RL, Babiskin AH, Zhao L. In silico methods for development of generic drug–device combination orally inhaled drug products. CPT Pharmacometrics Syst Pharmacol. 2019;8:359‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Swift DL. Inspiratory inertial deposition of aerosols in human nasal airway replicate casts: implication for the proposed NCRP lung model. Radiat Prot Dosimetry. 1991;38:29‐34. [Google Scholar]
- 15. Jones HM, Rowland‐Yeo K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst Pharmacol. 2013;2:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Regulatory utility of mechanistic modeling to support alternative bioequivalence approaches. 2022. https://complexgenerics.org/PBPK2021/. Accessed May 23, 2022. [DOI] [PMC free article] [PubMed]
- 17. U.S. Food and Drug Administration . Draft product‐specific guidance on beclomethasone dipropionate. 2020. https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020911.pdf. Accessed May 3, 2022.
- 18. U.S. Food and Drug Administration . Draft product‐specific guidance on ipratropium bromide. 2021. https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021527.pdf. Accessed May 3, 2022.
- 19. U.S. Food and Drug Administration . Draft product‐specific guidance on ciclesonide. 2021. https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_021658.pdf. Accessed May 3, 2022.
- 20. U.S. Food and Drug Administration . Draft product‐specific guidance on beclomethasone dipropionate. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/psg/Beclomethasone%20dipropionate%20Inhalation%20Aerosol%20Metered%20NDA%20207921%20PSG%20Page%20RC%20May%202019.pdf. Accessed May 3, 2022.
- 21. American Society of Mechanical Engineers . Verification and Validation in Computational Modeling of Medical Devices. ASME; 2018. [Google Scholar]
- 22. International Organization for Standardization . ISO 25539‐3:2011: Cardiovascular Implants — Endovascular Devices — Part 3: Vena Cava Filters. International Organization for Standardization; 2011. [Google Scholar]
- 23. US Food and Drug Administration . Guidance for industry and FDA staff: Guidance for cardiovascular intravascular filter 510(k) submissions. 1999. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/guidance‐cardiovascular‐intravascular‐filter‐510k‐submissions‐guidance‐industry‐and‐fda‐staff. Accessed May 3, 2022.
- 24. US Food and Drug Administration . Guidance for industry and FDA staff: Reporting of computational modeling studies in medical device submissions. 2016. https://www.fda.gov/media/87586/download. Accessed May 3, 2022.
- 25. US Food and Drug Administration . Catalog of regulatory science tools to help assess new medical devices. 2021. https://www.fda.gov/medical‐devices/science‐and‐research‐medical‐devices/catalog‐regulatory‐science‐tools‐help‐assess‐new‐medical‐devices. Accessed March 16, 2022.
- 26. Schroeter JD, Pritchard JN, Hwang D, Martonen TB. Airway identification within planar gamma camera images using computer models of lung morphology. Pharm Res. 2005;22:1692‐1699. [DOI] [PubMed] [Google Scholar]
- 27. Olsson B, Kassinos SC. On the validation of generational lung deposition computer models using planar scintigraphic images: the case of Mimetikos Preludium. J Aerosol Med Pulm Drug Deliv. 2021;34:115‐123. [DOI] [PubMed] [Google Scholar]
- 28. Haughie S, Allan R, Wood N, Ward J. Equivalent systemic exposure to fluticasone propionate/salmeterol following single inhaled doses from Advair Diskus and Wixela Inhub: results of three pharmacokinetic bioequivalence studies. J Aerosol Med Pulm Drug Deliv. 2020;33:34‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bäckman P, Olsson B. Pulmonary drug dissolution, regional retention & systemic absorption: understanding their interactions through mechanistic modeling. In: Byron PR, ed. Respiratory Drug Delivery. Davis Healthcare International Publishing; 2020. [Google Scholar]
- 30. Zhao J, Haghnegahdar A, Feng Y, et al. Prediction of the carrier shape effect on particle transport, interaction and deposition in two dry powder inhalers and a mouth‐to‐G13 human respiratory system: a CFD‐DEM study. J Aerosol Sci. 2022;160:105899. [Google Scholar]
- 31. Zhao J, Feng Y, Fromen CA. Glottis motion effects on the particle transport and deposition in a subject‐specific mouth‐to‐trachea model: a CFPD study. Comput Biol Med. 2020;116:103532. [DOI] [PubMed] [Google Scholar]
- 32. Walenga RL, Tian G, Longest PW. Development of characteristic upper tracheobronchial airway models for testing pharmaceutical aerosol delivery. J Biomech Eng. 2013;135:091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Longest PW, Hindle M. Evaluation of the Respimat soft mist inhaler using a concurrent CFD and in vitro approach. J Aerosol Med Pulm Drug Deliv. 2009;22:99‐112. [DOI] [PubMed] [Google Scholar]
- 34. Longest PW, Holbrook LT. In silico models of aerosol delivery to the respiratory tract—development and applications. Adv Drug Deliv Rev. 2012;64:296‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kleinstreuer C, Feng Y, Childress E. Drug‐targeting methodologies with applications: a review. World J Clin Cases. 2014;2:742‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Longest PW, Tian G, Khajeh‐Hosseini‐Dalasm N, Hindle M. Validating whole‐airway CFD predictions of DPI aerosol deposition at multiple flow rates. J Aerosol Med Pulm Drug Deliv. 2016;29:461‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tian G, Hindle M, Lee S, Longest PW. Validating CFD predictions of pharmaceutical aerosol deposition with in vivo data. Pharm Res. 2015;32:3170‐3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kolanjiyil AV, Kleinstreuer C. Computationally efficient analysis of particle transport and deposition in a human whole‐lung‐airway model. Part I: theory and model validation. Comput Biol Med. 2016;79:193‐204. [DOI] [PubMed] [Google Scholar]
- 39. Kolanjiyil AV, Kleinstreuer C. Computational analysis of aerosol‐dynamics in a human whole‐lung airway model. J Aerosol Sci. 2017;114:301‐316. [Google Scholar]
- 40. Longest PW, Bass K, Dutta R, et al. Use of computational fluid dynamics deposition modeling in respiratory drug delivery. Expert Opin Drug Deliv. 2019;16:7‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kolanjiyil AV, Kleinstreuer C, Sadikot RT. Computationally efficient analysis of particle transport and deposition in a human whole‐lung‐airway model. Part II: dry powder inhaler application. Comput Biol Med. 2017;84:247‐253. [DOI] [PubMed] [Google Scholar]
- 42. Longest PW, Tian G, Delvadia R, Hindle M. Development of a stochastic individual path (SIP) model for predicting the deposition of pharmaceutical aerosols: effects of turbulence, polydisperse aerosol size, and evaluation of multiple lung lobes. Aerosol Sci Tech. 2012;46:1271‐1285. [Google Scholar]
- 43. Longest PW, Tian G, Walenga RL, Hindle M. Comparing MDI and DPI aerosol deposition using in vitro experiments and a new stochastic individual path (SIP) model of the conducting airways. Pharm Res. 2012;29:1670‐1688. [DOI] [PubMed] [Google Scholar]
- 44. Tian G, Longest PW, Su G, Walenga RL, Hindle M. Development of a stochastic individual path (SIP) model for predicting the tracheobronchial deposition of pharmaceutical aerosols: effects of transient inhalation and sampling the airways. J Aerosol Sci. 2011;42:781‐799. [Google Scholar]
- 45. Nousias S, Zacharaki EI, Moustakas K. AVATREE: an open‐source computational modelling framework modelling anatomically valid airway TREE conformations. PLoS One. 2020;15:e0230259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tawhai MH, Hunter P, Tschirren J, Reinhardt J, McLennan G, Hoffman EA. CT‐based geometry analysis and finite element models of the human and ovine bronchial tree. J Appl Physiol. 2004;97:2310‐2321. [DOI] [PubMed] [Google Scholar]
- 47. Tawhai MH, Pullan AJ, Hunter PJ. Generation of an anatomically based three‐dimensional model of the conducting airways. Ann Biomed Eng. 2000;28:793‐802. [DOI] [PubMed] [Google Scholar]
- 48. Khajeh‐Hosseini‐Dalasm N, Longest PW. Deposition of particles in the alveolar airways: inhalation and breath‐hold with pharmaceutical aerosols. J Aerosol Sci. 2015;79:15‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Delvadia RR, Longest PW, Hindle M, Byron PR. In vitro tests for aerosol deposition III: effect of inhaler insertion angle on aerosol deposition. J Aerosol Med Pulm Drug Deliv. 2013;26:145‐156. [DOI] [PubMed] [Google Scholar]
- 50. Usmani OS, Barnes PJ. Assessing and treating small airways disease in asthma and chronic obstructive pulmonary disease. Ann Med. 2012;44:146‐156. [DOI] [PubMed] [Google Scholar]
- 51. Van den Berge M, Ten Hacken NHT, Van der Wiel E, Postma DS. Treatment of the bronchial tree from beginning to end: targeting small airway inflammation in asthma. Allergy. 2013;68:16‐26. [DOI] [PubMed] [Google Scholar]
- 52. Walenga RL, Longest PW. Current inhalers deliver very small doses to the lower tracheobronchial airways: assessment of healthy and constricted lungs. J Pharm Sci. 2016;105:147‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Longest PW, Hindle M. Small airway absorption and microdosimetry of inhaled corticosteroid particles after deposition. Pharm Res. 2017;34:2049‐2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rygg A, Hindle M, Longest PW. Absorption and clearance of pharmaceutical aerosols in the human nose: effects of nasal spray suspension particle size and properties. Pharm Res. 2016;33:909‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Weber B, Hochhaus G. A pharmacokinetic simulation tool for inhaled corticosteroids. AAPS J. 2013;15:159‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Adams WP, Ahrens RC, Chen ML, et al. Demonstrating bioequivalence of locally acting orally inhaled drug products (OIPs): workshop summary report. J Aerosol Med Pulm Drug Deliv. 2010;23:1‐29. [DOI] [PubMed] [Google Scholar]
- 57. Lee SL, Adams WP, Li BV, et al. In vitro considerations to support bioequivalence of locally acting drugs in dry powder inhalers for lung diseases. AAPS J. 2009;11:414‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hatipoglu MK, Hickey AJ, Garcia‐Contreras L. Pharmacokinetics and pharmacodynamics of high doses of inhaled dry powder drugs. Int J Pharm. 2018;549:306‐316. [DOI] [PubMed] [Google Scholar]
- 59. Longphre MV, Getz EB, Fuller R. Clinical bioequivalence of OT329 SOLIS and ADVAIR DISKUS in adults with asthma. Ann Am Thorac Soc. 2017;14:182‐189. [DOI] [PubMed] [Google Scholar]
- 60. Ng D, Kerwin EM, White MV, et al. Clinical bioequivalence of Wixela Inhub and Advair Diskus in adults with asthma. J Aerosol Med Pulm Drug Deliv. 2020;33:99‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. U.S. Food and Drug Administration . Draft Product‐Specific Guidance on Mometasone Furoate. 2020. https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020762.pdf. Accessed May 3, 2022.
- 62. U.S. Food and Drug Administration . Draft Product‐Specific Guidance on Fluticasone Furoate. 2020. https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022051.pdf. Accessed May 3, 2022.
- 63. Yeh HC, Schum GM. Models of human lung airways and their application to inhaled particle deposition. Bull Math Biol. 1980;42:461‐480. [DOI] [PubMed] [Google Scholar]
- 64. Walenga RL, Conti DS, Oguntimein O, et al. Droplet evaporation from a solution‐based metered dose inhaler: a computational approach. AAPS PharmSci. 2018;360(1):M1130‐05‐037. [Google Scholar]
- 65. Tong ZB, Yang RY, Yu AB. CFD‐DEM study of the aerosolisation mechanism of carrier‐based formulations with high drug loadings. Powder Technol. 2017;314:620‐626. [Google Scholar]
- 66. Youn JS, Csavina J, Rine KP, et al. Hygroscopic properties and respiratory system deposition behavior of particulate matter emitted by mining and smelting operations. Environ Sci Technol. 2016;50:11706‐11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Olsson B, Bäckman P. Mimetikos Preludium™: a new pharma‐friendly aerosol drug deposition calculator. Respiratory Drug Delivery. 2018;1:103‐112. [Google Scholar]
- 68. Türck D, Weber W, Sigmund R, et al. Pharmacokinetics of intravenous, single‐dose tiotropium in subjects with different degrees of renal impairment. J Clin Pharmacol. 2004;44:163‐172. [DOI] [PubMed] [Google Scholar]
- 69. Hochhaus G, Chen MJ, Kurumaddali A, et al. Can pharmacokinetic studies assess the pulmonary fate of dry powder inhaler formulations of fluticasone propionate? AAPS J. 2021;23:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Craven BA, Aycock KI. Flow chart illustrating the ASME V&V 40 credibility assessment framework for computational modeling and simulation of medical devices. 2022. 10.6084/m9.figshare.19448282.v1. Accessed May 3, 2022. [DOI]
- 71. US Department of Health and Human Services . Fixed‐combination prescription drugs for humans. 21 CFR 300.50. Revised January 5, 1999. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=300.50. Accessed November 16, 2022.