

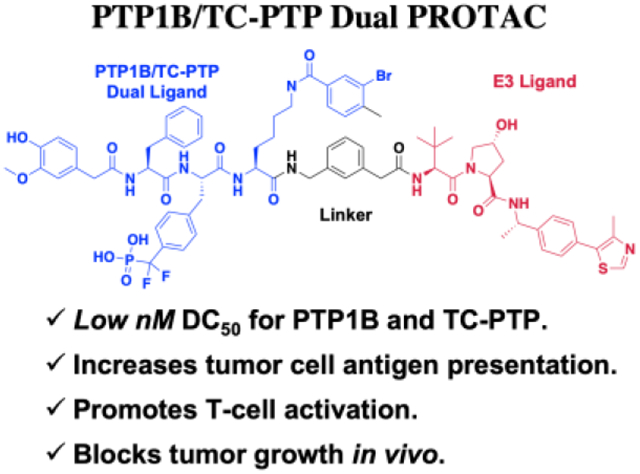
Abstract
Protein tyrosine phosphatase 1B (PTP1B) and T-cell protein tyrosine phosphatase (TC-PTP) play non-redundant negative regulatory roles in T-cell activation, tumor antigen presentation, insulin and leptin signaling, and are potential targets for several therapeutic applications. Here, we report the development of a highly potent and selective small molecule degrader DU-14 for both PTP1B and TC-PTP. DU-14 mediated PTP1B and TC-PTP degradation requires both target protein(s) and VHL E3 ligase engagement and is also ubiquitination- and proteasome-dependent. DU-14 enhances IFN-γ induced JAK1/2-STAT1 pathway activation and promotes MHC-I expression in tumor cells. DU-14 also activates CD8+ T-cells and augments STAT1 and STAT5 phosphorylation. Importantly, DU-14 induces PTP1B and TC-PTP degradation in vivo and suppresses MC38 syngeneic tumor growth. The results indicate that DU-14, as the first PTP1B and TC-PTP dual degrader, merits further development for treating cancer and other indications.
Keywords: PTP1B, TC-PTP, PROTAC degrader, immunotherapy
Graphical Abstract
An inhibitor for protein tyrosine phosphatase 1B (PTP1B) and T-cell protein tyrosine phosphatase (TC-PTP) was discovered and transformed into a first-in-class, potent, and selective dual PROTAC degrader for both targets. The degrader promotes antigen presentation of tumor cells and activates CD8+ T cells. Importantly, the degrader is effective in vivo and suppresses syngeneic tumor growth.
Introduction
Protein tyrosine phosphorylation is controlled by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) and plays a key role in regulating essential cellular functions. Given that numerous human diseases are associated with aberrant protein tyrosine phosphorylation, it is not surprising that PTKs and PTPs are highly sought-after targets for drug discovery. Tremendous success has been achieved in modulating PTKs for therapeutic applications[1], although the PTPs still remain underexplored as a target class[2]. Among members of the PTP family, protein tyrosine phosphatase 1B (PTP1B, also called PTPN1) and T-cell protein tyrosine phosphatase (TC-PTP, also called PTPN2) are the two most closely related PTPs sharing over 72% amino acid sequence identity between their catalytic domains [3,4]. Despite their structural similarity, PTP1B and TC-PTP are known to play non-redundant and synergistic roles in coordinating several important signal pathways.
PTP1B and TC-PTP have long been known to function in concert in regulating both insulin and leptin mediated cellular processes[5]. PTP1B negatively regulates the amplitude of insulin action by dephosphorylating the insulin receptor and insulin receptor substrate 1[6], while TC-PTP catalyzes insulin receptor dephosphorylation to limit the duration of insulin signaling[7]. PTP1B and TC-PTP also attenuate leptin signaling by bringing about the dephosphorylation of JAK2[8] and STAT3[9], respectively. Indeed, mice lacking PTP1B[10] or deficient in TC-PTP [11-15] in peripheral tissues and/or the brain exhibit significantly enhanced insulin sensitivity and are resistant to weight gain, lending support to them as therapeutic targets for Type II diabetes and obesity. Notably, recent studies have shown that the combined intranasal targeting of PTP1B and TC-PTP in the hypothalamus of obese mice can increase energy expenditure and repress feeding to promote weight loss and improve glucose homeostasis[15].
Recent studies also reveal that PTP1B and TC-PTP play non-redundant roles in attenuating IFN-γ signaling[16]. Elimination of PTP1B increases JAK2 phosphorylation and enhances IFN-γ mediated STAT1 activation[8], while TC-PTP removal from tumor cells augments IFN-γ signaling and antigen presentation as a result of increased phosphorylation of JAK1 and its downstream effector STAT1[17,18]. Interestingly, PTP1B and TC-PTP also serve distinct functions as negative regulators of T cell activation [19,20]. To this end, PTP1B deletion in T cells promotes antigen-induced expansion and cytotoxicity of CD8+ T cells against solid tumors through increased JAK2/STAT5 phosphorylation[20]. Genetic ablation of TC-PTP in T cells increases the expansion and survival of CD8+ T cells[21-23] and promotes the activation of CD8+ T cells through amplifying LCK and STAT5 phosphorylation[18,24].
In light of our current understanding of the roles of PTP1B and TC-PTP in cellular signaling, there is compelling rationale for concomitant blockage of PTP1B and TC-PTP to produce synergistic effects for a number of therapeutic applications including Type II diabetes, obesity, and anti-cancer immunotherapies. Several small molecule inhibitors for PTP1B and TC-PTP have been described[25]. Theoretically, co-administration of a PTP1B inhibitor with a TC-PTP-inhibiting agent could be employed to test these therapeutic hypotheses. However, combination therapies are prone to elicit complex pharmacokinetics/pharmacodynamics, unanticipated drug-drug interactions, toxicity and/or patient compliance problems. Known as polypharmacology, the design or use of multi-targeting compounds that act on two or more selected targets have gained considerable interest in drug discovery owing to the increasing appreciation of the complexity of multifactorial human diseases[26,27]. Compared to single-targeting drugs or a combination of multiple drugs, a polypharmacological agent or multi-targeted ligand offers a valuable alternative with several advantages, such as superior therapeutic effects, reduced risk of drug–drug interactions, more predictable pharmacokinetic/pharmacodynamic profiles, and simplification of treatment regimen[26].
To begin to assess the therapeutic potential of PTP1B and TC-PTP dual-targeting, we describe the discovery of an active site-directed ligand that inhibits both PTP1B and TC-PTP with low nM affinity. To further enhance the potency and selectivity of the dual-targeting agent, we leverage the PROteolysis TArgeting Chimeras (PROTAC) approach[28,29] to transform the PTP1B/TC-PTP inhibitor into a series of highly efficacious and selective small molecule degraders for both PTP1B and TC-PTP. The lead dual PROTAC molecule DU-14 degrades both PTP1B and TC-PTP with low nanomolar efficacies in a number of cell lines, increases IFN-γ signaling and antigen presentation in tumor cells, and promotes CD8+ T-cell activation. Importantly, DU-14 exhibits excellent pharmacokinetic properties and blocks tumor growth in a syngeneic mouse model. Collectively, these results provide a proof-of-concept for polypharmacological targeting of both PTP1B and TC-PTP as a therapeutic approach for cancer immunotherapy.
Results and Discussion
Discovery of a potent and selective dual targeting inhibitor for PTP1B and TC-PTP
Given the critical requirement of phosphotyrosine (pTyr) for PTP substrate recognition[30,31], an effective strategy for PTP inhibitor discovery is through a fragment-based approach by tethering appropriate diversity molecules to a nonhydrolyzable pTyr mimetic in order to engage both the active site and nearby distinct peripheral pockets[25,32]. These types of inhibitors are expected to possess enhanced potency and selectivity due to the additivity of free energy of binding and the lack of structural similarity for nonactive site interactions in different PTPs. Among nonhydrolyzable pTyr mimetics that can efficiently engage the PTP active site pocket, phosphonodifluoromethyl phenylalanine (F2Pmp) is widely utilized for PTP inhibitor design [33]. To that end, we have previously developed potent and selective F2Pmp-based competitive inhibitors for PTP1B, TC-PTP, and PTP-Meg2 [34-36]. To identify F2Pmp-based inhibitors targeting both PTP1B and TC-PTP, a stepwise combinatorial synthesis and screening strategy (Scheme 1) was deployed [35,36]. We started from a fluorescein labeled F2Pmp-Lysine dipeptide (DP-01), which contains the active site-directed F2Pmp, a free amine on the side chain of Lysine that can be used for library construction, and a fluorescein tag connected through an Ala-Lys linker. The fluorescein serves as an integral part of all library members, which furnishes a mechanism for the identification of high affinity binders via a competitive homogeneous fluorescence polarization (FP) displacement assay[35,36].
Scheme 1.

Development of PTP1B/TC-PTP dual-inhibitor DI-03 through a combinatorial synthesis and screening strategy.
Through three iterative rounds of fluorescein-tagged library synthesis and affinity-based screening (for details see Supplemental Information and Tables S1, S2, and S3), we identified compound DI-03, which inhibited PTP1B and TC-PTP with IC50 values of 4.7 ± 0.3 and 7.1 ± 0.4 nM, respectively. As expected, DI-03 behaved as a reversible and competitive inhibitor for both PTP1B and TC-PTP with Ki values of 2.4 ± 0.1 and 3.6 ± 0.2 nM, respectively (Figure 1A and B). Importantly, DI-03 displayed 2 to 3 orders of magnitude of selectivity for PTP1B and TC-PTP over a panel of 12 representative PTPs, including PTP Meg2, SHP2, LYP, HePTP, Laforin, STEP, LMW-PTP, Cdc14A, CD45, FAP1, VHR, and PTPα (Figure 1C).
Figure 1.

DI-03 is a potent and selective PTP1B/TC-PTP dual competitive inhibitor. (A) Effect of DI-03 on PTP1B catalyzed pNPP hydrolysis. (B) Effect of DI-03 on TC-PTP catalyzed pNPP hydrolysis. The Lineweaver-Burk plot displayed the characteristic intersecting line pattern, consistent with competitive inhibition. DI-03 concentrations were 0 (●), 5 (■), and 10 nM (▲), respectively. DI-03 inhibit PTP1B and TC-PTP with values of 2.4 ± 0.1 and 3.6 ± 0.2 nM, respectively. (C) Selectivity of DI-03 over a panel of 12 mammalian PTPs.
Design and synthesis of DI-03 based dual PROTAC degraders for PTP1B and TC-PTP
Acquisition of target selectivity is vital in the development and use of a chemical probe for functional interrogation and testing therapeutic hypothesis. Given the conceptual novelty of PTP1B and TC-PTP dual-targeting, we sought to further enhance the selectivity of DI-03 for PTP1B and TC-PTP through application of the PROTAC technology. PROTACs are bifunctional molecules consisting of a ligand for the protein of interest, a linker, and an E3 ligase binding moiety that exploit the cell’s ubiquitin–proteasome machinery to achieve selective target protein degradation[28,29]. Compared to the traditional occupancy-based inhibitors, the event-driven PROTACs exhibit several notable advantages including prolonged efficacy as a result of target elimination, sub-stoichiometric concentrations needed due to the catalytic nature, and higher selectivity, which is dictated by the obligatory ternary complex formation that brings the targe protein into proximity of the E3 ligase for efficient target protein ubiquitination and subsequent proteasome-mediated degradation. Indeed, the observation that PROTAC-mediated target protein degradation selectivity can greatly exceed the binding selectivity of the corresponding ligand for the target protein[37] spurred our interest in pursuing PROTAC molecules that can simultaneously degrade both PTP1B and TC-PTP.
To develop DI-03 based PTP1B and TC-PTP dual PROTAC degraders, we needed to identify a suitable tethering site for linker installment. Although there are no crystal structures of PTP1B or TC-PTP in complex with DI-03 that can be used to guide tethering site selection, the combinatorial synthesis and screening strategy used for PTP1B and TC-PTP dual-inhibitor discovery revealed a potential linker attachment site in DI-03 (Scheme 1). The FP displacement results showed that the fluorescein tagged DP-04 exhibited high binding affinity for both PTP1B and TC-PTP, indicating that modifications on the primary amide moiety (circled in Scheme 1) are tolerable. To further ascertain the suitability of the DI-03 primary amide for linker attachment, we performed molecular docking studies. Since PTP1B and TC-PTP are highly homologous, our docking experiments were solely performed with a previously described PTP1B-ligand co-crystal structure [38]. The docking poses of the cognate ligand agreed to the ligand bound crystal structure (Figure S1), which validated the computational model. As expected, molecular docking results indicated that the DI-03 primary amide was solvent exposed and did not interact with PTP1B (Figure 2A), suggesting that linker attachment at this functionality will not perturb PTP1B binding.
Figure 2.

Molecular docking results suggest the primary amide moiety of DI-03 is solvent exposing and guide the PROTACs design. (A) Binding pose of DI-03 (green stick) to PTP1B shown in transparent surface representation. The solvent-exposing primary amide moiety is circled in red. Hydrogen bonds and ionic bonds are represented by yellow dashes. π-π stacking and cation-π interactions are shown with orange and purple dashes, respectively. Interaction distances are shown in Angstroms. (B) General synthetic route for dual PTP1B and TC-PTP ligand and PROTACs.
Figure 2B depicts our strategy to create PROTAC degraders for PTP1B and TC-PTP. The dual ligand (DL) for the two PTPs with a free carboxylic acid at the tethering site was synthesized from the three fragments F1, F2, and F3 (see Supporting Information). The von Hippel–Lindau (VHL) E3 ligase ligands (S,R,S)-AHPC and (S,R,S)-AHPC-Me [39-41] were employed to construct the dual PROTACs. Given the importance of linker length in influencing PROTAC-mediated target protein degradation potency, we synthesized an initial series of bivalent compounds (DU-01 to DU-06) with a linear alkyl chain of various lengths (Table 1). As expected, all of these candidate PROTAC molecules exhibited low nanomolar IC50 values comparable to those of DI-03 when tested against PTP1B and TC-PTP in biochemical phosphatase assays (Table S4), indicating that the linker installment did not affect the ligand binding affinity to the target proteins. We then assessed the effects of these compounds on reducing PTP1B and TC-PTP abundance in HEK293 cells at 1 μM concentration using Western blotting (Table 1, Figure S2A and S2B). Among this series, DU-02 with an n-pentane linker displayed the highest degradation potency. After 16 hours treatment, 1 μM of DU-02 induced the depletion of 81% PTP1B and 83% TC-PTP. Increasing or decreasing the linker length in DU-02 by one additional methylene (DU-01, DU-03) significantly diminished the degradation potency on both PTP1B and TC-PTP. Further increase in linker length generated PROTACs with even lower degradation efficacy. Therefore, the n-pentane chain appeared to represent the optimal linker length for DI-03 based VHL recruiting PTP1B/TC-PCP degraders. Lower degradation efficiency was observed at 0.1 μM compound concentration (Table S5, Figure S2A and S2B), indicating no obvious hook effect[42] in this concentration range.
Table 1.
Structure and degradation results of the initial set of degraders. Degradation assay was conducted in HEK293 cells with 1 μM degrader and 16 hours incubation.
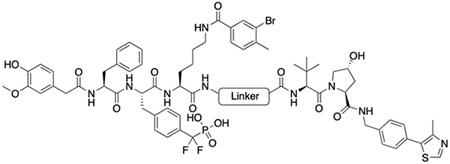
| |||
|---|---|---|---|
| Compound | Linker Structure | % Degradation | |
| PTP1B | TC-PTP | ||
| DMSO | N/A | < 5 | < 5 |
| DI-03 | N/A | < 5 | < 5 |
| DU-01 |
|
53 | 58 |
| DU-02 |
|
81 | 83 |
| DU-03 |
|
61 | 69 |
| DU-04 |
|
58 | 66 |
| DU-05 |
|
38 | 62 |
| DU-06 |
|
29 | 30 |
The chemical nature and flexibility of linkers are also known to affect the formation and stability of the target protein - PROTAC - E3 ligase ternary complex and, thus can influence the degradative efficiency. Accordingly, to further improve the potency of the PTP1B and TC-PTP dual PROTAC degraders, we sought to modify the linker composition while keeping the linker length similar to that of DU-02 (DU-07 to DU-13, Table 2, and Figure S2C and S2D). We first synthesized DU-07 by replacing the n-pentane linker with the corresponding polyethylene glycol to assess the potential influence of linker hydrophobicity on the degradation potency. Treatment of HEK293 cell with 0.2 μM of DU-07 for 16 hours led to the depletion of 45% PTP1B and 47% TC-PTP respectively, which are significantly lower than those induced by DU-02 under the same conditions. We then investigated the effect of linker conformational constraint on PTP1B and TC-PTP degradation by inserting a rigid benzene, piperidine, or piperazine ring into the flexible linear alkyl chain (DU-09 to DU-13). Compared to DU-02, DU-10 with a m-xylene linker exhibited markedly improved degradation potency, while DU-09 with an o-xylene and DU-11 with a p-xylene linker showed lower degradation potencies. On the other hand, PTP1B and TC-PTP were less robustly degraded by DU-12 and DU-13 when compared to DU-02, indicating that the piperidine or piperazine linker is less effective. Finally, we ascertained whether replacement of (S,R,S)-AHPC with (S,R,S)-AHPC-Me, a VHL ligand that was reported to possess higher binding affinity for the E3 ligase [43], in DU-07 and DU-10. To our delight, DU-08 and DU-14 were significantly more potent than their (S,R,S)-AHPC counterparts. In particular, treatment of HEK293 cells for 16 hours with 0.2 μM DU-14 induced 95% and 92% depletion of PTP1B and TC-PTP, respectively, making DU-14 the most potent PTP1B and TC-PTP dual PROTAC degrader in this study. Again, significantly decreased PTP1B and TC-PTP degradation was observed at 0.05 μM degrader concentration (Table S6, Figure S2C and S2D). Indeed, our most potent dual PROTAC DU-14 showed no Hook effect at concentrations up to 6.25 μM (Figure S3).
Table 2.
Structure and degradation results of the second set of degraders. Degradation assay was conducted in HEK293 cells with 0.2 μM degrader and 16 hours incubation.
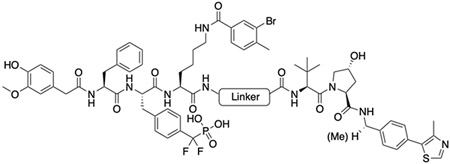
| ||||
|---|---|---|---|---|
| Compound | Linker | E3 Ligase | % Degradation | |
| PTP1B | TC-PTP | |||
| DMSO | N/A | N/A | < 5 | < 5 |
| DI-03 | N/A | N/A | < 5 | < 5 |
| DU-02 |
|
(S,R,S)-AHPC | 61 | 58 |
| DU-07 |
|
(S,R,S)-AHPC | 45 | 47 |
| DU-08 |
|
(S,R,S)-AHPC-Me | 75 | 63 |
| DU-09 |
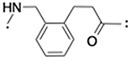
|
(S,R,S)-AHPC | 16 | < 5 |
| DU-10 |
|
(S,R,S)-AHPC | 75 | 70 |
| DU-11 |
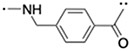
|
(S,R,S)-AHPC | 24 | 37 |
| DU-12 |
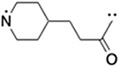
|
(S,R,S)-AHPC | 26 | 32 |
| DU-13 |
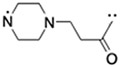
|
(S,R,S)-AHPC | 30 | 14 |
| DU-14 |
|
(S,R,S)-AHPC-Me | 95 | 92 |
DU-14 is a potent, selective, and bona fide PROTAC degrader for PTP1B and TC-PTP
To further establish DU-14 as a genuine dual PROTAC degrader for PTP1B and TC-PTP, we determined its degradation efficacy at a wide range of concentrations in HEK293 and 9 additional cell lines (Figure 3A, 3B, and Figure S4). The DC50 (compound concentration needed to induce target protein degradation by 50%) values for DU-14 mediated PTP1B and TC-PTP degradation were 4.3 and 4.8 nM, respectively, in HEK293 cells after 16 hours treatment. Equipotent low-nanomolar DC50s for PTP1B and TC-PTP degradation were also observed with DU-14 in Jurkat T cells, mouse embryonic fibroblast (MEF) cells, U2OS human bone osteosarcoma epithelial cells, and 6 other cancer cell lines (Figure 3C). Next, we determined the time course of PTP1B and TC-PTP degradation by treating HEK293 cells with 100 nM DU-14 for 1, 3, 6, 8, 16, and 24 hours. We observed a nearly complete loss of PTP1B and TC-PTP within 6 hours, with an estimated t1/2 (the time required to reduce the target protein to half of its initial amount) of 3.1 ± 0.2 and 2.5 ± 0.1 hours for PTP1B or TC-PTP, respectively (Fig. 3D&E). To investigate the binding selectivity of DU-14 for the targeted proteins of interest, we determined that DU-14 inhibits the phosphatase activity of PTP1B and TC-PTP with IC50 values of 24.2 ± 1.7 nM and 30.1 ± 2.3 nM respectively, and exhibits >88-fold selectivity for PTP1B and TC-PTP over a representative list of 12 PTPs, consisting of receptor-like, nonreceptor-like, and dual specific phosphatases (Figure 3F). The biochemical binding selectivity of DU-14 for PTP1B and TC PTP is slightly lower than that of DI-03, as measured by the ability of the compounds to inhibit the phosphatase activity of purified recombinant PTP proteins. This might be due to linker and E3 ligase ligand attachment on DI-03 which may perturb it’s binding to the PTPs. We next evaluated the selectivity of DU-14 as a degrader for PTP1B and TC-PTP. We found that treatment of HEK293 cells with 0.5 μM DU-14 for 16 hours led to complete removal of PTP1B and TC-PTP, whereas none of the other PTP members including SHP2, PTP-Meg2, LYP and PRL2 were affected (Figure 3G). Moreover, no appreciable degradation of PTP-Meg 2 and SHP2 was observed in HEK293 cells treated with up to 6.25 μM DU-14 for 6 hours (Figure S3), indicating that DU-14 exhibits at least 600-fold degradation selectivity for PTP1B and TC-PTP over these two PTPs. Thus, the DU-14 mediated PTP1B and TC-PTP degradation selectivity way exceeded the inhibition/binding selectivity of DI-03 for PTP1B and TC-PTP over PTP-Meg2 (155-fold) and SHP2. We also noted that under the same conditions, DU-14 did not cause any appreciable degradation of other signaling proteins including STAT1, STAT5, AKT, GSK3α and p70S6K (Figure 3G). To further assess the proteome-wide degradation selectivity of DU-14, we performed quantitative mass spectrometry-based proteomic experiments to determine the range of targets degraded by DU-14 in HEK293 cells. As shown in Figure 3H, PTP1B was the only protein whose levels was significantly reduced by 100 nM of DU-14. Unfortunately, TC-PTP was not detected by the mass spectrometry measurements under the given conditions. Taken together, the above results indicate that DU-14 is a potent PTP1B and TC-PTP dual degrader with remarkably high selectivity.
Figure 3.

DU-14 is a potent and selective PTP1B/TC-PTP dual degrader. (A-B) Immunoblots of cell lysates from HEK293 (A) and MC38 (B) cells treated with DU-14 at indicated concentrations for 16 hours showing dose-dependent degradation of PTP1B and TC-PTP achieved by low nanomolar concentration of DU-14. (C) DC50 values of DU-14 in 10 cell lines. Cells were treated with a series concentration of DU-14 for 16 hours then lysed for immunoblot. PTP1B and TC-PTP degradation were quantified by densitometry and normalized to GAPDH. (D-E) Degradation kinetics of DU-14 in the HEK293 cell lines. Cells were treated with 100 nM of DU-14 for 1,3,6,8,16 and 24 hours. PTP1B (●) and TC-PTP (■) protein was examined by immunoblot and normalized to GAPDH to determine the degradation. (F) Biochemical IC50 of DU-14 on a panel of 12 PTPs. (G) Degradation selectivity of DU-14. Immunoblots of cell lysates from HEK293 cells treated with vehicle (DMSO), 0.5 μM DU-14, or 0.5 μM DU-14 along with 2 μM MLN4924 for 16 hours. (H) Proteomic analysis showing specificity of DU-14 for PTP1B degradation by using 4 hours DMSO or 100 nM DU-14 treated HEK293 cells
PROTAC-mediated protein degradation requires the obligatory formation of a functional ternary complex of target protein-PROTAC-E3 ligase in order to enable target protein ubiquitination by the E3 ligase, followed by proteasomal degradation[28]. The PTP1B and TC-PTP inhibition data (Figure 3F) indicate that DU-14 can selectively engage the intended target proteins. To confirm the VHL E3 ligase dependency for DU-14 induced PTP1B and TC-PTP degradation, we prepared cis-DU-14 in which the VHL E3 ligand was replaced with (S,S,S)-AHPC-Me, a epimer of (S,R,S)-AHPC-Me with diminished affinity for VHL[44]. We established that the IC50 values of cis-DU-14 for PTP1B and TC-PTP (25.1 ± 1.6 and 29.7 ± 2.1 nM) are similar to those of DU-14 (Figure 3F). Consistent with the impaired binding of (S,S,S)-AHPC-Me to VHL, cis-DU-14 failed to degrade either PTP1B or TC-PTP in HEK293 cells (Figure 4A). Moreover, addition of the VHL ligand (S,R,S)-AHPC-Me reduced the DU-14 mediated degradation of the two proteins (Figure 4A). In contrast, pretreatment of the cell with Lenalidomide, a ligand for Cereblon (another E3 ligase commonly employed for PROTAC development), had no effect on DU-14-mediated PTP1B and TC-PTP degradation (Figure 4A). These observations suggest that DU-14 induced PTP1B and TC-PTP degradation is VHL-dependent. To demonstrate the ubiquitination- and proteasome-dependency of the DU-14 induced PTP1B and TC-PTP degradation, we determined the effect of MLN-4924, an inhibitor of the E1 ubiquitin-activating enzyme, and the proteasome inhibitor MG-132. As expected, pretreatment of the cells for 30 minutes with these inhibitors markedly reduced the extent of PTP1B and TC-PTP degradation by DU-14, indicating that the E1 ubiquitin-activating enzyme and 26S proteasome were indeed required for the DU-14 induced PTP1B and TC-PTP degradation (Figure 4A). Finally, the DU-14 mediated PTP1B and TC-PTP degradation and proteasome dependency were also verified by immunofluorescent imaging (Figure 4B and 4C). Taken together, our data demonstrated that DU-14 is a genuine PTP1B and TC-PTP dual-PROTAC degrader with high potency and selectivity.
Figure 4.

DU-14 is a bona fide PTP1B/TC-PTP dual PROTAC. (A) Mechanistic investigation of PTP1B/TC-PTP degradation induced by DU-14 in HEK293 cells. Cells were pre-treated with indicated concentration of MLN4924 (prevents ubiquitination), MG132 (blocks proteasome activity), Lenalidomide (prevents CRBN binding) or (S,R,S)AHPC-Me (prevents VHL binding) followed by 4 h treatment with DU-14 at 100 nM showing DU-14 mediated PTP1B/TC-PTP degradation depends on the ubiquitination-proteasome pathway. Cells were also treated with 100 nM cis-DU-14 (inactivated degrader) for 4 h and no degradation observed. (B-C) Immunofluorescences of PTP1B (Green) and TC-PTP (Red) with U2OS cells treated with DMSO, and 1 μM DU-14 for 3 and 24 hours showing DU-14 degrades cytoplasm localized PTP1B and both nucleus and cytoplasm localized TC-PTP. 20 μM MG132 was used along with DU-14 to block PTP1B and TC-PTP degradation.
DU-14 amplifies INF-γ signaling and promotes T cell activation
We next proceeded to ascertain target engagement by DU-14 inside the cell. As negative regulators of IFN-γ signaling, PTP1B and TC-PTP dephosphorylate JAK2 at Y1007/Y1008[8] and JAK1 at Y1034/Y1035[17,45], respectively. In addition, TC-PTP can also directly dephosphorylate STAT family members, including STAT1[46] and STAT3[9] in the nucleus to comprehensively attenuate IFN signaling. As expected, treatment of MEF cells with 500 nM DU-14 for 16 hours let to the complete removal of both PTP1B and TC-PTP and augmented IFN-γ stimulated JAK2 Y1007/Y1008 and JAK1 Y1034/Y1035 phosphorylation (Figure 5A). To demonstrate that the increase in pJAK1/Y1034/Y1035 and pJAK2/Y1007/Y1008 was indeed caused by PTP1B/TC-PTP degradation, we utilized MEF cells that either lack PTP1B or TC-PTP as negative controls. As shown in Figure 5A, DU-14 treatment of TC-PTP−/− MEF cells further elevated IFN-γ mediated PTP1B substrate JAK2/Y1007/Y1008 phosphorylation with no effect on the level of TC-PTP substrate pJAK1/Y1034/Y1035. In contrast, addition of DU-14 to PTP1B−/− MEF cells further increased the IFN-γ mediated TC-PTP substrate pJAK1/Y1034/Y1035 level with no alteration of PTP1B substrate JAK2/Y1007/Y1008 phosphorylation. These results indicate that DU-14 can block both PTP1B and TC-PTP catalyzed substrates dephosphorylation.
Figure 5.

DU-14 efficiently amplifies cellular IFN-γ signaling by degrading PTP1B and TC-PTP. (A) Immunoblots of whole cell lysates from wild-type, PTP1B deleted or TC-PTP deleted MEF cells that treated with 0.5 μM DU-14 for 16 hours and stimulated with 20 ng/ml mouse IFN-γ for 15 mins. Deletion of TC-PTP or PTP1B abolished the DU-14 induced phosphorylation level increase of the TC-PTP substrate JAK1 or the PTP1B substrate JAK2. (B) Immunoblots of whole cell lysates from MC38 cells that treated with DU-14 for 16 hours at indicated concentration and stimulated with 20 ng/ml mouse IFN-γ for 15 mins showing DU-14 dose-dependently elevates IFN-γ mediated STAT1 activation (C) U2OS cells were treated with DMSO or 0.2 μM DU-14 for 16 hours and stimulated with 20 ng/ml IFN-γ for 30 minutes. Immunofluorescence showing DU-14 dramatically enhanced IFN-γ mediated STAT1 phosphorylation (Green) and nucleus translocation. DAPI was used to stain cell nucleus. (D) MC38 cells were treated with DMSO (Blue Peak) or 500 nM DU-14 (Red Peak) for 16 hours for PTP1B and TC-PTP degradation then stimulated with 20 ng/ml mouse IFN-γ for 48 hours to induce MHC-I expression. Mouse MHC-I complex was stained with mouse H2K(b)/H2D(b) antibody and measured by flow cytometry. DU-14 treated MC38 cells exhibited elevated expression of MHC-I.
IFN-γ signaling enhances tumor cell recognition and elimination by recruiting cytotoxic T lymphocytes to tumor cells[47]. This is achieved by the INF-γ induced JAK1/2-STAT1 pathway activation, resulting in the upregulation of interferon response genes, including genes for T cell chemoattractants, such as CXCL9 and antigen presentation, such as MHC-I complex components[48]. Importantly, previous studies have shown that the deletion of TC-PTP in tumor cells can enhance IFN-γ induced JAK/STAT1 signaling to facilitate antigen presentation, T cell recruitment, and anti-tumor immunity[17,18]. To further investigate the impact of DU-14 on IFN-γ signaling in tumor cells, we measured pSTAT1/Y701 as a readout for pathway activation. As shown in Figure 5B, treatment of MC38 cells with DU-14 led to a dose dependent degradation of PTP1B/TC-PTP and increase in pSTAT1/Y701. Immunofluorescent imaging also showed enhanced IFN-γ induced STAT1 phosphorylation (Green) and nuclear translocation in U2OS cells upon DU-14 treatment, which is required for the initiation of STAT1 mediated transcription (Figure 5C). A positive correlation between the DU-14 induced increase in pSTAT1/Y701 and the extent of PTP1B/TC-PTP degradation was also observed in a number of different cell lines including HEK293, Jurkat, MiaPaca2, H116, H358, HepG2, U2OS, MC38, MEF and B16F10 (Figure S4). Consistent with the enhanced upstream INF-γ signaling activation, we observed over 80% higher cell surface MHC-I expression in MC38 cells in the presence of DU-14 as result of IFN-γ stimulation (Figure 5D). Finally, we have compared the efficacy of PTP1B and TC-PTP dual degradation to dual inhibition. As shown in Figure S5, DU-14 treatment amplified IFN-γ induced pSTAT1/Y701 with significantly higher efficiency (EC50 of 76.2 ± 12.3 nM) than that effected by the DI-03 (EC50 > 2.5 μM). The results further confirmed that bifunctional PROTAC molecules possess improved biological activity compared to their corresponding ligands. Collectively, we demonstrated that DU-14 efficiently amplified IFN-γ signaling through inhibition of PTP1B and TC-PTP catalyzed tyrosine dephosphorylation of JAK2/Y1007/Y1008, JAK1/Y1034/Y1035 and/or STAT1/Y701, leading to increased cell surface MHC-I expression, which is required for efficient T-cell mediated killing of tumor cells.
Beyond attenuating JAK/STAT signaling in tumor cells, PTP1B and TC-PTP also have fundamental roles in T cells and the deletion of either TC-PTP or PTP1B in T cells can markedly enhance anti-tumor immunity[20,22,24]. TC-PTP attenuates T cell receptor (TCR) signaling by dephosphorylating and inactivating the Src family kinase LCK[19]. TC-PTP also attenuates JAK/STAT1/5 signaling in response to cytokines such as IFNs and IL-2[19,22,24] that are required for the activation, clonal expansion and differentiation of T cells. The deletion of TC-PTP in T cells enhances immunosurveillance and inhibits the growth of syngeneic tumors in mice and the anti-tumor efficacy of adoptively transferred T cells[24]. PTP1B also negatively regulates IL-2-induced JAK/STAT5 signaling in T cells, and its deletion or inhibition in vivo can enhance the anti-tumor activity of T cells[20]. Accordingly, we sought to assess the impact of targeting PTP1B and TC-PTP with DU-14 on JAK/STAT signaling and the activation of T cells after crosslinking the TCR. The effects of DU-14 were compared with the genetic deletion of TC-PTP (encoded by Ptpn2) in T cells (Lck-Cre; Ptpn2fl/fl). First, we assessed the impact of DU-14 on TC-PTP and PTP1B protein levels by flow cytometry using validated antibodies[19,20,24]. After 48 hr of treatment TC-PTP and PTP1B were effectively degraded in T cells (Figure 6A). The degradation of TC-PTP and PTP1B was accompanied by a more than 3-fold increase in STAT1 Y701 phosphorylation and 2-fold increase in STAT5 Y694 phosphorylation detected after crosslinking the TCR with α-CD3/α-CD28 to activate T cells (Figure 6B). Importantly, the promotion of pSTAT1/Y701 and pSTAT5/Y694 by DU-14 treatment exceeded that achieved by the genetic deletion of TC-PTP, consistent with DU-14 targeting both TC-PTP and PTP1B to enhance signaling. Furthermore, DU-14 treatment also enhanced the TCR-induced activation of T cells, as assessed by monitoring for cell size and the expression of cell surface activation markers, including CD44, CD25 (IL-2 receptor α) and CD69 (Figure 6C). In this instance, DU-14 only moderately, albeit not significantly, increased T cell activation beyond that achieved by the deletion of TC-PTP. This was not necessarily a surprise, since TC-PTP but not PTP1B, attenuates TCR signaling in naive CD8+ T cells[19,20]. Nonetheless, these results demonstrate that the combinatorial targeting of PTP1B and TC-PTP with DU-14 can enhance JAK/STAT signaling and the activation of CD8+ T cells that are instrumental in anti-viral and anti-tumor immunity.
Figure 6.

DU-14 induces PTP1B and TC-PTP degradation in CD8+ naïve T cells, enhances STAT1 and STAT5 phosphorylation, and promotes CD8+ T cell activation. (A) Purified CD8+ naïve T cells from control (Ptpn2fl/fl) and Lck-Cre; Ptpn2fl/fl mice (n=3/genotype/condition) were incubated for 48 h with IL-7 in the presence or absence of DU-14 as indicated and TC-PTP and PTP1B protein levels monitored by flow cytometry. (B-C) Vehicle-treated control, TCPTP-deficient Lck-Cre; Ptpn2fl/fl and DU-14-treated control naïve CD8+ T cells were stimulated with plate-bound α-CD3/α-CD28 for 48 h to promote T cell activation. (B) Basal STAT-1 (p(Y701) STAT-1) and STAT-5 (p(Y694) STAT-5) phosphorylation were assessed by flow cytometry. (C) Cell size (FSC-A) and the T cell activation markers CD25, CD69 and CD44 (MFI; mean fluorescence intensities) were measured by flow cytometry. (● Ptpn2fl/fl + Vehicle, ■ Lck-Cre; Ptpn2fl/fl + Vehicle, ▲ Ptpn2fl/fl + DU-14).
DU-14 suppresses syngeneic tumor growth in immunocompetent mice
Previous studies have shown that the genetic deletion of PTP1B or TC-PTP promotes anti-tumor immunity in both cell-autonomous and non-autonomous manners [17,18,20,22,24]. To explore the therapeutic potential of dual targeting of PTP1B and TC-PTP, we investigated whether systemic administration of DU-14 could repress the growth of syngeneic tumors in mice. Pharmacokinetics (PK) analyses of DU-14 in C57BL/6 mice showed that a single intraperitoneal injection of DU-14 at 25 and 50 mg/kg achieved a peak plasma concentration (Cmax) of 1.6 ± 0.3 and 4.3 ± 0.3 μM with half-lives of 3.0 ± 1.1 and 3.3 ± 0.8 hours, respectively (Figure 7A). At both doses the plasma concentration of DU-14 was maintained above its cellular EC50 for STAT1 activation (34.2 ± 2.2 nM in MC38 cells, Figure S2) for at least 12 hours. Based on the PK profiles, we assessed the anti-tumor activity of DU-14 in C57BL/6 mice bearing MC38 colorectal tumors in the flanks. Mice bearing established tumors (200 mm3) were injected once daily with either saline or 25 or 50 mg/kg DU-14. Compared with the control group, 25 mg/kg DU-14 treatment effectively repressed MC38 tumor growth while 50 mg/kg DU-14 resulted in static tumor growth (Figure 7B). No obvious toxicity was observed based on body weight measurement (Figure 7C). Importantly, nearly complete PTP1B and TC-PTP degradation was observed in whole tumor homogenates after treatment with DU-14 treatment (Figure 7D). Immunohistochemical analyses revealed that the repression of tumor growth was accompanied by a marked increase in CD8+ T cell infiltrates (brown α-CD8α staining) in MC38 tumors (Figure 7E and 7F). These results are consistent with our cell-based studies, which point towards the combined targeting of PTP1B and TC-PTP with DU-14 in tumor cells and T cells promoting both tumor antigen presentation and T cell activation. Together, these data suggest that DU-14 is a promising lead for further development and that dual degradation of PTP1B and TC-PTP could represent a novel and effective immunotherapy strategy.
Figure 7.

Targeting PTP1B and TC-PTP with DU-14 represses MC38 tumor in mice. (A) Mice blood concentration of DU-14 overtime after single dose administration of 25 (■) or 50 (●) mg/kg DU-14 i.p. injection. (B) MC38 tumor growth after daily treatment with saline (●) versus 25 (■) or 50 mg/kg (▲) DU-14 (n=8) (C) Body weights of MC38 tumor bearing mice (n=4) treated with DU-14 for 10 days. (D) PTP1B and TC-PTP immunoblots of MC38 whole tumor homogenates (n=4) from mice treated with saline versus 25 or 50 mg/kg DU-14. (E-F) Immunohistochemistry (IHC) analysis of MC38 tumors staining for CD8+ T cells. Representative images and the quantified results from 6 sections are shown.
Conclusion
Immunotherapies aimed at boosting the ability of T cells to locate, infiltrate, and destroy tumors have revolutionized cancer treatment[49]. However, only a small population of patients have benefited from current options, highlighting the need to identify new approaches to improve cancer immunotherapy. Recent studies reveal that PTP1B and TC-PTP play non-redundant negative regulatory roles in T-cell activation as well as in tumor cell antigen presentation[17-20,22,24]. Consequently, targeting both enzymes concurrently may produce synergistic effects on upregulating T-cell mediated anti-tumor immunity. Unfortunately, PTP1B and TC-PTP are among the most difficult-to-drug intracellular targets due to difficulties in developing inhibitors with sufficient selectivity and robust in vivo activity[25]. Here we applied fragment-based methods to discover a highly potent (low nM) and selective (>150 fold over 12 PTPs) active site directed inhibitor DI-03 for PTP1B and TC-PTP. To further enhance the potency and selectivity of the dual-targeting agent, we utilized the PROTAC strategy to transform DI-03 into a highly efficacious and selective PTP1B and TC-PTP dual degrader DU-14. Despite having an unmasked F2Pmp, DU-14 promotes efficient PTP1B and TC-PTP degradation with low nanomolar potencies in a target(s), E3 ligase, ubiquitination and proteasome dependent manner. Notably, DU-14 enhances IFN-γ mediated JAK1/2-STAT1 signaling and antigen presentation in tumor cells. DU-14 also upregulates TCR and IL-2 signaling and enhances CD8+ T cell activation. Importantly, DU-14 exhibits excellent pharmacokinetic properties and inhibits the growth of syngeneic tumors in immunocompetent mice; this is accompanied by the increased infiltration of CD8+ T cells. Given the high molecular weight and negative changes associated with DU-14, it is remarkable that it exhibits robust in vivo efficacy. Although DU-14 is in non-compliance with the Lipinski rule of five[50], it has been increasingly recognized the existence of bioavailable drugs that exceed the proposed constraints[51]. To this end, we note that the molecular attributes of DU-14 are similar to the published PROTACs with biological activities[52] and to those currently in clinical trials[53]. Similar F2Pmp containing PTP-Meg 2 inhibitor 7[36] also possesses highly efficacious cellular activity and is capable of augmenting insulin signaling and improving insulin sensitivity and glucose homeostasis in diet-induced obese mice. Finally, the catalytic feature of the PROTAC mediated degradation may also boost the biological activity of degraders with less than ideal cell permeability. Collectively, our study highlights the translational potential of systemic and concomitant targeting of both PTP1B and TC-PTP as a novel and promising therapeutic approach for cancer immunotherapy. DU-14 is the first small molecule degrader developed from a nonhydrolyzable pTyr mimetic that specifically and efficiently induces PTP1B and TC-PTP degradation in both cells and whole animals. We anticipate that the PROTAC approach will help resuscitate drug discovery efforts targeting the active sites of PTPs.
Supplementary Material
Acknowledgements
This work was supported in part by NIH RO1CA069202 and the Robert C. and Charlotte Anderson Chair Endowment (to Z.-Y.Z.), NIH 3RF1AG064250 (to W.A.T.), the National Health and Medical Research Council of Australia (to T.T.) and Cancer Council Victoria (to F.W.). The authors gratefully acknowledge the support of NIH P30 CA023168 for use of the Flow Cytometry Facility.
References
- [1].Attwood MM, Fabbro D, Sokolov AV, Knapp S, Schiöth HB, Nat Rev Drug Discov 2021, 20, 839–861. [DOI] [PubMed] [Google Scholar]
- [2].Krabill AD, Zhang Z-Y, Biochemical Society Transactions 2021, 49, 1723–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Møller NPH, Mol. Cell. Biol 2001, 21, 7117–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tiganis T, FEBS Journal 2013, 280, 445–458. [DOI] [PubMed] [Google Scholar]
- [5].Zhang Z-Y, Dodd GT, Tiganis T, Trends in Pharmacological Sciences 2015, 36, 661–674. [DOI] [PubMed] [Google Scholar]
- [6].Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D, Molecular Cell 2000, 6, 1401–1412. [DOI] [PubMed] [Google Scholar]
- [7].Galic S, Hauser C, Kahn BB, Haj FG, Neel BG, Tonks NK, Tiganis T, MCB 2005, 25, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, Parisien J-P, Salmeen A, Barford D, Tonks NK, Journal of Biological Chemistry 2001, 276, 47771–47774. [DOI] [PubMed] [Google Scholar]
- [9].Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ, Simonds S, Wiede F, Reichenbach A, Hauser C, Sims NA, Bence KK, Zhang S, Zhang Z-Y, Kahn BB, Neel BG, Andrews ZB, Cowley MA, Tiganis T, Cell Metabolism 2011, 14, 684–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Elchebly M, Science 1999, 283, 1544–1548. [DOI] [PubMed] [Google Scholar]
- [11].Fukushima A, Loh K, Galic S, Fam B, Shields B, Wiede F, Tremblay ML, Watt MJ, Andrikopoulos S, Tiganis T, Diabetes 2010, 59, 1906–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dodd GT, Andrews ZB, Simonds SE, Michael NJ, DeVeer M, Brüning JC, Spanswick D, Cowley MA, Tiganis T, Cell Metabolism 2017, 26, 375–393.e7. [DOI] [PubMed] [Google Scholar]
- [13].Dodd GT, Decherf S, Loh K, Simonds SE, Wiede F, Balland E, Merry TL, Münzberg H, Zhang Z-Y, Kahn BB, Neel BG, Bence KK, Andrews ZB, Cowley MA, Tiganis T, Cell 2015, 160, 88–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dodd GT, Lee-Young RS, Brüning JC, Tiganis T, Diabetes 2018, 67, 1246–1257. [DOI] [PubMed] [Google Scholar]
- [15].Dodd GT, Xirouchaki CE, Eramo M, Mitchell CA, Andrews ZB, Henry BA, Cowley MA, Tiganis T, Cell Reports 2019, 28, 2905–2922.e5. [DOI] [PubMed] [Google Scholar]
- [16].Heinonen KM, Bourdeau A, Doody KM, Tremblay ML, Proceedings of the National Academy of Sciences 2009, 106, 9368–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, Weiss SA, Lo J, Fisher DE, Miao D, Van Allen E, Root DE, Sharpe AH, Doench JG, Haining WN, Nature 2017, 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Goh PK, Wiede F, Zeissig MN, Britt KL, Liang S, Molloy T, Goode N, Xu R, Loi S, Muller M, Humbert PO, McLean C, Tiganis T, Sci. Adv. 2022, 8, eabk3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wiede F, Shields BJ, Chew SH, Kyparissoudis K, van Vliet C, Galic S, Tremblay ML, Russell SM, Godfrey DI, Tiganis T, J. Clin. Invest 2011, 121, 4758–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wiede F, Lu K-H, Du X, Zeissig MN, Xu R, Goh PK, Xirouchaki CE, Hogarth SJ, Greatorex S, Sek K, Daly RJ, Beavis PA, Darcy PK, Tonks NK, Tiganis T, Cancer Discovery 2022, 12, 752–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Flosbach M, Oberle SG, Scherer S, Zecha J, von Hoesslin M, Wiede F, Chennupati V, Cullen JG, List M, Pauling JK, Baumbach J, Kuster B, Tiganis T, Zehn D, Cell Reports 2020, 32, 107957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].LaFleur MW, Nguyen TH, Coxe MA, Miller BC, Yates KB, Gillis JE, Sen DR, Gaudiano EF, Al Abosy R, Freeman GJ, Haining WN, Sharpe AH, Nat Immunol 2019, 20, 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wiede F, La Gruta NL, Tiganis T, Nat Commun 2014, 5, 3073. [DOI] [PubMed] [Google Scholar]
- [24].Wiede F, Lu K, Du X, Liang S, Hochheiser K, Dodd GT, Goh PK, Kearney C, Meyran D, Beavis PA, Henderson MA, Park SL, Waithman J, Zhang S, Zhang Z, Oliaro J, Gebhardt T, Darcy PK, Tiganis T, EMBO J 2020, 39, DOI 10.15252/embj.2019103637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang Z-Y, Acc. Chem. Res 2017, 50, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Anighoro A, Bajorath J, Rastelli G, J. Med. Chem 2014, 57, 7874–7887. [DOI] [PubMed] [Google Scholar]
- [27].Proschak E, Stark H, Merk D, J. Med. Chem 2019, 62, 420–444. [DOI] [PubMed] [Google Scholar]
- [28].Neklesa TK, Winkler JD, Crews CM, Pharmacology & Therapeutics 2017, 174, 138–144. [DOI] [PubMed] [Google Scholar]
- [29].Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, Toure M, Dong H, Qian Y, Wang J, Crew AP, Hines J, Crews CM, Cell Chemical Biology 2018, 25, 67–77.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang ZY, Maclean D, Thiemesefler AM, Roeske RW, Dixon JE, Analytical Biochemistry 1993, 211, 7–15. [DOI] [PubMed] [Google Scholar]
- [31].Jia Z, Barford D, Flint AJ, Tonks NK, Science 1995, 268, 1754–1758. [DOI] [PubMed] [Google Scholar]
- [32].Puius YA, Zhao Y, Sullivan M, Lawrence DS, Almo SC, Zhang Z-Y, Proc. Natl. Acad. Sci. U.S.A 1997, 94, 13420–13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Burke T, CTMC 2006, 6, 1465–1471. [DOI] [PubMed] [Google Scholar]
- [34].Shen K, Keng Y-F, Wu L, Guo X-L, Lawrence DS, Zhang Z-Y, Journal of Biological Chemistry 2001, 276, 47311–47319. [DOI] [PubMed] [Google Scholar]
- [35].Zhang S, Chen L, Luo Y, Gunawan A, Lawrence DS, Zhang Z-Y, J. Am. Chem. Soc 2009, 131, 13072–13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang S, Liu S, Tao R, Wei D, Chen L, Shen W, Yu Z-H, Wang L, Jones DR, Dong XC, Zhang Z-Y, J. Am. Chem. Soc 2012, 134, 18116–18124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gopalsamy A, J. Med. Chem 2022, 65, 8113–8126. [DOI] [PubMed] [Google Scholar]
- [38].Sun J-P, Fedorov AA, Lee S-Y, Guo X-L, Shen K, Lawrence DS, Almo SC, Zhang Z-Y, Journal of Biological Chemistry 2003, 278, 12406–12414. [DOI] [PubMed] [Google Scholar]
- [39].Yang K, Wu H, Zhang Z, Leisten ED, Nie X, Liu B, Wen Z, Zhang J, Cunningham MD, Tang W, ACS Med. Chem. Lett 2020, 11, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang C-Y, Wang M, Zhao L, Xu T, Chinnaswamy K, Delproposto J, Stuckey J, Wang S, J. Med. Chem 2019, 62, 941–964. [DOI] [PubMed] [Google Scholar]
- [41].Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM, ACS Cent. Sci 2020, 6, 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kostic M, Jones LH, Trends in Pharmacological Sciences 2020, 41, 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AMK, Wang J, Chen X, Dong H, Siu K, Winkler JD, Crew AP, Crews CM, Coleman KG, Proc Natl Acad Sci USA 2016, 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Soares P, Gadd MS, Frost J, Galdeano C, Ellis L, Epemolu O, Rocha S, Read KD, Ciulli A, J. Med. Chem 2018, 61, 599–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ, Current Biology 2002, 12, 446–453. [DOI] [PubMed] [Google Scholar]
- [46].ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, Shuai K, Molecular and Cellular Biology 2002, 22, 5662–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ivashkiv LB, Nat Rev Immunol 2018, 18, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hu X, Ivashkiv LB, Immunity 2009, 31, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Waldman AD, Fritz JM, Lenardo MJ, Nat Rev Immunol 2020, 20, 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Advanced Drug Delivery Reviews 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- [51].O’ Donovan DH, De Fusco C, Kuhnke L, Reichel A, J. Med. Chem 2023, 66, 2347–2360. [DOI] [PubMed] [Google Scholar]
- [52].Edmondson SD, Yang B, Fallan C, Bioorganic & Medicinal Chemistry Letters 2019, 29, 1555–1564. [DOI] [PubMed] [Google Scholar]
- [53].O’Brien Laramy MN, Luthra S, Brown MF, Bartlett DW, Nat Rev Drug Discov 2023, DOI 10.1038/s41573-023-00652-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.