

Abstract
The term liquid biopsy is used in contraposition to the traditional “solid” tissue biopsy. In the oncology field it has opened a new plethora of clinical opportunities as tumor-derived material is shedded into the different biofluids from where it can be isolated and analyzed. Common biofluids include blood, urine, saliva, cerebrospinal fluid (CSF), pleural effusion or bile. Starting from these biological specimens several analytes can be isolated, among which we will review the most widely used: circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), circulating tumor RNA (ctRNA), proteins, metabolites, and exosomes. Regarding the nature of the biomarkers it will depend on the analyte, the type of tumor and the clinical application of the liquid biopsy and it includes, somatic point mutations, deletions, amplifications, gene-fusions, DNA-methylated marks, tumor-specific miRNAs, proteins or metabolites. Here we review the characteristics of the analytes and the methodologies used for their isolation. We also describe the applications of the liquid biopsy in the management of patients with cancer, from the early detection of cancers to treatment guidance in patients with advanced tumors. Finally, we also discuss some current limitations and still open questions.
Keywords: circulating biomarkers, circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), personalized medicine, tumor circulome
Introduction
Even before the definition of the term biomarker by a consensus panel at the WHO in Geneva in 2001 as “any substance, structure, or process that can be measured in the body or its products and influence or predict the incidence of outcome or disease” [1], the diagnosis and prognosis of multiple diseases had relied on the identification of different biomarkers in blood tests. Well-known examples are for instance the determination of the levels of different metabolites such as glucose or cholesterol; the presence of enzymes such as transaminases coming from dying hepatocytes; or the levels of monoclonal immunoglobulins, carcinoembryonic antigen (CEA), alfa-fetoprotein (AFP), prostate specific antigen (PSA), cancer antigen 125 (CA 125) or human chorionic gonadotropin (HCG) in the bloodstream [2], [3]. All these biomarkers have been commonly used with variable sensitivity and specificity rates to diagnose diabetes, liver diseases or the presence of myeloma, colon, liver, prostate, ovarian or germ-cell tumors.
However, in the oncology field these biomarkers in biological fluids have shown limited reliability [4]. In addition, during the last decades the large amount of high throughput molecular data obtained together with the progress of pharmacogenomics have demonstrated that it is required to characterize the molecular profile of the tumor for tailoring treatment regimens, the so-called precision medicine, monitoring therapy responses and detecting the emergence of therapy resistance [5]. Importantly, it is also known now that the genetic landscape of tumors is spatially heterogeneous and temporally dynamic [6], [7].
In this sense, the traditional method of tumor sampling by tissue biopsy has several limitations, including the fact that in some cases tumor accessibility is difficult and performing a biopsy may not be possible. In addition, given its invasive nature, tissue biopsies can pose risks to the patient what will also limit longitudinal monitoring. Importantly, tissue biopsies may present sample bias as they provide the tumor picture at a single location missing spatial tumor genetic heterogeneity and impacting in accuracy and sensitivity [6].
During the last years, the effort of multiple groups and the development of different sensitive technologies have allowed the advance of non-invasive methodologies to detect tumor-derived material in biofluids to be used for cancer diagnosis, prognosis, monitoring, and therapy guidance [8], [9], [10], [11], [12], [13].
The term liquid biopsy is therefore used in contraposition to the traditional surgical tumor biopsy and represents the analysis of cancer biomarkers in tumor-derived material isolated typically from the bloodstream “tumor circulome” [13] or other biofluids such as urine, saliva, cerebrospinal fluid (CSF), pleural effusion (PE), or bile of cancer patients [8]. The analytes used for liquid biopsy include circulating tumor cells (CTCs), cell-free DNA (cfDNA), proteins, metabolites, extracellular vesicles, and cell-free RNA [10], [14]. Regarding the nature of the biomarkers it will depend on the analyte, the type of tumor and the clinical application of the liquid biopsy [8] and include, somatic point mutations, deletions, amplifications, gene-fusions, DNA-methylated marks, tumor-specific miRNAs, proteins or metabolites, etc (Figure 1). Based on the analyte and the biomarker, different technologies are applied to identify and characterize them [12]. Noteworthy, the revolution of Next Generation Sequencing (NGS) technologies has enormously boosted the capabilities of nucleic acid-based liquid biopsies [11].
Figure 1:

Biofluids, analytes and biomarkers encompassed in liquid biopsy.
Met-CpG: methylated CpGs (dinucleotide C-G). CCNA: chromosomal copy number aberrations; InDel: insertions and deletions; CTCs: circulating tumor cells; cfDNA: cell-free DNA; cfRNA: cell-free RNA; miRNAs: microRNAs; PE: pleural effusion; CSF: cerebrospinal fluid.
In 2013, the U.S. Food and Drug Administration (FDA) approved the first liquid biopsy test, the CellSearch® CTC enumeration platform (K073338-FDA) intended to monitor advanced metastasic breast, colon, and prostate cancers based on CTCs counts [15]. Three years later, in 2016, the first cfDNA-based liquid biopsy test was approved. A companion diagnostic test to detect epidermal growth factor receptor (EGFR) mutations in the circulating tumor DNA (ctDNA) of patients with non-small cell lung cancer (NSCLC) who may benefit from targeted therapy with erlotinib and osimertinib [16].
However, many limitations remain unsolved before the routine clinical implementation of liquid biopsies in the field of oncology. Besides the optimization and standardization of protocols to isolate the analytes, there are still open questions such as the complete understanding of tumors behavior and shedding dynamics that challenge the full exploitation of liquid biopsy as a new tool for cancer management. In this still growing scenario, and aiming at accelerating the development, validation, and clinical use of liquid biopsy assays, a collaborative consortium between public, industry, academia, and regulatory agencies was established in both Europe (Cancer-ID; https://www.cancer-id.eu/) and the United States (BloodPAC; https://www.bloodpac.org/).
Here, we review the latest advances of liquid biopsy technology describing the methodologies used to isolate the analytes and to identify the biomarkers, with special focus on CTCs and cfDNA. We also describe the applications of the liquid biopsy in the management of patients with cancer, from the early detection of cancers to treatment guidance in patients with advanced tumors. Finally, we also discuss some current limitations and still relevant questions in the field.
Types of analytes in cancer patients
Different types of tumor components can be detected in liquid biofluids of cancer patients. As mentioned above, these tumor analytes include CTCs, cfDNA, proteins, metabolites, extracellular vesicles, and cell-free RNA [10]. However, and common to all of them, the tumor analyte is surrounded by a vast majority of non-cancer analytes shedded by normal cells, hindering the isolation and detection of the tumoral fraction of interest. In this scenario, the development of improved sensitive technologies has boosted the interest on this field allowing the use of liquid biopsies as a minimally-invasive source of neoplastic material for molecular analyses. Each analyte has its own advantages and disadvantages, and will provide different information. In some cases, their mere isolation can be already informative or they can be used as the starting material for biomarker detection. Notably, the existence of a proportion of non-shedding tumors (20–58%) has been reported [17], [18]. Therefore, patients with these tumors would not benefit from liquid biopsy advances, and moreover they would increase the false negative rate. However, these affirmations would need to be revisited after the development of more sensitive techniques, as detection sensitivity could be behind this lack of tumor analytes.
Historically, circulating tumor-derived protein-based tests have been the gold-standard approach for screening and guidance of different types of cancers. While widely used, their major and always questioned limitation has been the overdiagnosis [19]. Current investigations are focused on the identification of protein signatures instead of single proteins to overcome the lack of specificity [20] and on improving the detection approaches [21]. Tumor derived-extracellular vesicles are lipid-bilayer-enclosed nanovesicles secreted by tumor cells carrying proteins and nucleic acids [22]. They can be found in almost every biofluid, especially in blood. It has been reported that their levels are increased in different types of cancers [23]. Moreover, by analyzing their content researchers have for instance found that exosomal glypican-1 (GPC1) protein distinguished patients with pancreatic cancer with absolute specificity and sensitivity from healthy subjects [23]. Exosomal KRAS and p53 mutations [24] or specific exosomal microRNA signatures [25] were detected in pancreatic cancer patients. The different types of RNA (mRNAs, miRNAs, and lncRNAs) are also released by tumor cells into the bloodstream. However, RNA is a relative unstable molecule and its detection is normally associated with extracellular vesicles. Several examples of exosomal RNA biomarkers in cancer have already been reported [26], [27]. Nevertheless, among tumor liquid biopsy analytes, research on CTCs and cfDNA has experienced an exponential growth in the last decades. Therefore, a detailed revision of these two analytes is presented in the next sections.
Circulating tumor cells (CTCs)
CTCs are a population of cells that have detached from the tumor mass and circulate in the bloodstream. We have to travel one hundred and 50 years ago for the first documented observation of CTCs. It was in 1869 when the Australian physician Thomas R. Ashworth observed cells that closely resembled the primary tumor in the blood of a man who died due to a metastatic cancer [28]. Nevertheless, it was not until the late 90s of the 20th century when the boom of CTC started, after Racila and colleagues developed the later widely used immunomagnetic enrichment-based methodology for CTC detection. Importantly, they also demonstrated that CTCs exist early in disease and pointed out the potential use of these cells for monitoring treatment responses and recurrences [29], [30].
CTCs sources and characteristics
The dissemination of tumor cells into the circulatory system is the first step in the metastatic event [31]. Although the mechanisms implicated in this process are still under investigation, it is known that CTC intravasation can occur through a passive release from the primary tumor or through an active intravasation associated to an epithelial-to-mesenchymal transition (EMT) [32]. Once there, if cells survive and spread, they might establish a separate secondary tumor site in a new host organ [31]. However, metastasis is a highly ineffective event, and most of these cells die in circulation owing to trauma, oxidative stress, and attack by the immune system [33]. Indeed, the presence of CTCs in the blood of a cancer patient is an extremely rare event with one CTC per 1 × 106–107 blood cells depending on the disease state [34] and with a half-life in circulation of less than 2.5 h [35]. CTCs are a highly heterogeneous population, as observed at the genetic, transcriptomic, proteomic, or metabolomic level [36] which could reflect their origin from different clones of the primary tumor [37]. CTCs can be distinguished from the mesenchymal blood cells by the expression of epithelial markers such as the epithelial cell adhesion molecule (EpCam) or proteins of the cytokeratin family (CK8, CK18, and CK19), or even by their epithelial morphology [34]. However, as mentioned above and importantly for the isolation protocols, CTCs may have undergone an EMT transition, losing the epithelial markers and characteristic morphology, and expressing instead known EMT regulators, including transforming growth factor (TGF)-β pathway components, vimentin, N-cadherin and the FOXC1 transcription factor [32], [38]. Furthermore, a percentage of the CTC population acquires stem-cell-like properties, and consistently express stem-cell markers, including ALDH7A1, CD44, and KLF4 [39].
Recent studies have reported that CTCs also exist in multicellular clusters. They are extremely rare compared to single CTCs, but possess a greater metastatic potential [40]. These clusters can be directly derived from the tumor through passive shedding or collective migration [40], [41], or can be de novo formed within the circulation through the aggregation of single CTCs [42]. CTC clusters are comprised of around 2–50 cells with strong cell-cell contacts. In these multicellular aggregates, tumor cells are accompanied by other non-tumor cells including platelets, immune cells, or cancer-associated fibroblasts, suggested to favor CTC clusters survival [32], [43]. Moreover, the proportion of these clusters may be larger than previously anticipated, and increases during cancer metastasis [44]. These CTC clusters have been reported to exhibit enriched expression of keratin 14 [41], plakoglobin [40], and CD44 [42] as well as mesenchymal markers [32].
Isolation of CTCs
In the last decades several strategies have been developed for the isolation of single CTCs from blood. These technologies are based either on the biological or on the physical differences existing between CTCs and non-tumor blood cells. However, CTCs isolation is still challenging mainly due to their extreme rarity and heterogeneity [36]. Thus, and despite the wide number of approaches already developed and tested, big efforts are still ongoing in this field trying to solve the limitations of current technologies. In addition to the isolation method, pre-analytical variables such as the type of blood collection tube, time between sampling, and processing or storage temperature must also be considered as they will impact the downstream analysis [45]. Here, we will give a general overview of the most relevant CTC isolation methods, but specific review manuscripts focused on CTC technologies are recommended for a comprehensive description [14], [46], [47], [48], [49].
Isolation of CTCs based on their biological properties
Methods based on CTCs biological properties rely on specific biomarkers expressed on the cell surface that allow their capture from the blood sample. Positive CTC selection is carried out using tumor-associated cell surface antigens, generally EpCAM and cytokeratins (CK8, CK18, and CK19). However, and as mentioned above, some CTCs loss these epithelial markers and instead express mesenchymal and stem cells markers [32], [38] drawing a blood sample scenario with several CTCs phenotypes. Therefore, a major limitation of the positive-enrichment methods is the bias imposed by the selection marker, which causes a selective capture of specific CTC subpopulations, while losing others. It is worth mentioning as well that EpCAM-based capture has been reported to isolate circulating epithelial cells in patients with benign colon diseases [50], which could therefore result in overdiagnosis. Negative CTC selection relies on the use of antigens expressed in peripheral blood cells but not in CTCs to capture and deplete non-cancer cells from the sample, leaving intact the CTC cells [51]. In this strategy, CD45 antigen is generally used. Although this method results in lower purity compared with the positive enrichment strategy, heterogeneous CTC subpopulations will be isolated together [51], [52]. To apply the immunoaffinity-based principle two different devices are widely used, consisting on selected antibodies bound either to immunomagnetic beads or to microfluidic chips. An additional drawback of these label-capture methodologies is that the binding of CTCs to the surface of the device can complicate CTC recovery and downstream analysis. Currently the CellSearch® system (Menarini Silicon Biosystems) is the only CTC technology approved by the FDA which combines the positive and negative enrichments. It is intended for the enumeration of CTCs in whole blood by an enrichment based on CD45-, EpCAM+, and cytokeratins 8+, 18+, and/or 19+ [15].
Isolation of CTCs based on their physical properties
Methods based on CTCs physical properties rely on the physical differences such as size, density, deformability or electric charge, between CTCs and other blood cells (mainly leukocytes) [47]. CTCs are supposed to be larger in size than normal blood cells [53]. Density of CTCs is similar to nucleate blood cells, lying between plasma and red blood cells after centrifugation. Several reports have determined that CTCs exhibit greater deformability capacity than normal cells [54], [55]. Additionally, electrical features of CTCs have been also applied to discriminate them from non-tumorigenic blood cells using dielectrophoresis [56]. Finally, there are other CTC isolation methods based on their functional features, including their ability to digest cell adhesion matrix, to secrete different proteins or to overexpress the enzyme telomerase [13].
Usefulness, current limitations and important considerations of CTCs
Despite the numerous platforms developed for CTC analysis and the exponential number of publications in this field, the translation into the clinical practice is still limited. To date, several reports have determined that the mere isolation of CTCs and their count number per mL of blood is already a biomarker itself with potential clinical implications. However, only the CellSearch® CTC enumeration platform is nowadays approved by the FDA as a prognostic predictor, providing reliable information on progression-free survival and overall survival, in metastatic breast [44], colon [53], and prostate [56] cancers. Patients with metastatic disease holding < 5 CTCs per 7.5 mL of blood are more likely to have a better clinical response than those with > 5 CTCs per 7.5 mL of blood [57]. Additionally, the clinical validity of CTC counts to select the first-line treatment for metastatic, hormone-receptors positive, breast cancer patients is currently being evaluated (MATABREAST trial; NCT01710605). However, as discussed by Aceto [58], fluctuations in CTC numbers in patients with comparable disease status, overall burden, and metastatic profile have been observed, calling the clinical utility of CTCs presence and abundance into question. Further knowledge regarding the factors influencing CTC shedding, dynamics, and clearance are still needed to have a complete picture and therefore develop more accurate tools for their interpretation.
Nevertheless, CTCs can be as well the analyte for subsequent biomarker analyses. In this regard, CTCs can provide real-time proteomic, (epi) genetic, genomic, or transcriptomic information of the tumor. For example, a test has been developed to detect nuclear androgen-receptor splicing variant 7 (AR-V7) protein expressions in isolated CTCs to guide treatment selection in patients with metastatic castration-resistant prostate cancer [59]. Methylation profiles of tumor suppressor genes in CTCs correlates with metastatic potential and poorer prognosis [60]. Moreover, a microfluidic western blot system to assess the level of eight proteins in individual CTCs derived from estrogen receptor-positive breast cancer patients has been recently developed [61].
Circulating cell-free DNA
Small fragments of DNA circulate freely in the peripheral blood of healthy and diseased individuals. In the case of cancer patients, a fraction of these circulating cell-free DNA (cfDNA) molecules correspond to circulating tumor DNA (ctDNA). If we look back, the presence of soluble DNA in the blood was firstly reported in 1948 by Mandel and Metais [62]. However, it was not until 1977 when serum cfDNA levels were associated to cancer patients and the effect of therapy [63]. We had to wait until 1994 for the discovery of the presence of a KRAS mutation in the cfDNA extracted from plasma of patients with pancreatic cancer [64].
cfDNA sources and characteristics
DNA fragments are released into the bloodstream mainly via cell apoptosis, but also through other type of cell death processes such as cell necrosis, pyroptosis, or autophagy, as well as through active cell secretion [65]. cfDNA can originate from both nuclear and mitochondrial sources [66]. These circulating cfDNA fragments are about 166 base pairs in length, which correspond to the length occupied by a nucleosome. Once there, cfDNA clearance occurs through enzymatic degradation (DNase I, plasma factor VII-activating protease, and factor H), renal excretion, and liver and spleen metabolism [67]. The balance between release and clearance determine cfDNA half-life and although it varies between individuals and conditions, it normally ranges from 16 min to 2.5 h [67], [68]. Epigenetic studies have demonstrated that the circulating nucleosome footprints and the cfDNA methylation patterns of healthy individuals strongly correlate with that of lymphoid and myeloid cells [69], [70], pointing out to the hematopoietic system as the predominant source of cfDNA.
In cancer patients, a proportion of these cfDNA molecules also derive from the primary and secondary tumors [64]. Although it was originally thought that the higher level of cfDNA in the blood of cancer patients might be a cancer biomarker itself, it has been since shown that many other conditions result in similar cfDNA increase. In this regard, important points must be considered: i) concentrations of cfDNA vary enormously between individuals and their physiopathological conditions, being increased not only in advanced cancer patients but also in other scenarios including autoimmune diseases, trauma, exhaustive exercise, or pregnancy; ii) in most early stage cancers, the amount of cfDNA is very low, similar to healthy subjects [71]; iii) the fraction of ctDNA fragments in the total cfDNA is very small, varying from less than 0.01% to over 10% according to tumor burden [72] and tumor metabolism [73]. ctDNA fragments are usually smaller than the cfDNA released by healthy cells. A recent study demonstrated that these different fragmentation profiles could be used not only for cancer screening but also for the determination of the tissue of origin of cancers [74].
Isolation of circulating cfDNA
Although cfDNA can be obtained from different biofluids, to date the majority of studies have been performed with blood as the starting material. This blood is usually collected from peripheral veins; however, it remains unknown whether higher cfDNA yield and ctDNA proportion would change depending on the sampling site. In spite of the exponential focus and advances in this field, we still lack standardized protocols, both for preanalytical sample preparation after blood collection and for plasma cfDNA purification [75]. It should be noted that the variables inherent in these steps may affect the quality of the analyte, and could compromise the interpretation of the final outcome and the comparison between studies.
Several studies have been published aiming at comparing the different methodological options at each single step and recent reviews have put together the obtained information [45], [75], [76] (Table 1). Although both serum and plasma could be used as cfDNA source, plasma is preferred to avoid contamination with genomic DNA derived from white blood cells lysis [77]. Thus, blood should be collected in EDTA anticoagulant tubes and processed within the first 4–6 h after sampling. If quick processing is not possible, specialized cell-stabilizing tubes are available that prevent leukocytes lysis [78]. Evidence suggests that a two-centrifugation steps protocol -first one at low, and the second at high speed- is optimal for plasma recovery and subsequent cfDNA isolation [75]. An optimal cfDNA extraction method should purify all cfDNA fragments avoiding genomic DNA, and minimize the presence of PCR inhibitors. Several methods are commercially available for cfDNA-specific extraction based on either magnetic beads or silica column-based membranes. Some of these kits are manual, whereas others use automated systems that minimize sample handling. Although the variations between the different kits and protocols are unclear, the choice of the extraction method can result in different cfDNA yield [76]. The concentration of cfDNA can be determined using different methodologies ranging from fluorometer approaches to PCR-based assays [75], [76].
Table 1:
Methodological variables in processing and analysis of cfDNA.
| Protocol steps | Variables/Considerations |
|---|---|
| 1. Blood collection [75] |
Specimen type: plasma is preferred to serum to avoid contamination with genomic DNA.Collection tube: it will depend on the time to processing:
|
| 2. Centrifugation for plasma isolation [75] |
Protocol: a two-centrifugation steps protocol is required. First at low (∼2500 g), second at high speed (∼14000 g). Apparently, temperature (room temperature or 4 °C) is not critical. *After the first centrifugation, plasma can be frozen at −80 °C for later processing. |
| 3. cfDNA extraction [75], [76] |
Method: different cfDNA isolation kits (from Qiagen, Promega, Applied Biosystems, Zymo Research, Norgen Biotek, EpiGenTek, among others) are available. Some considerations to take into account:
*If plasma was frozen at −80 °C, thawing must be done slowly in ice. |
| 4. cfDNA quantification [75], [76] |
Method: quantification of cfDNA yield can be performed by:
|
| 5. Biomarker analysis [12], [81] |
Biomarker features: mutation, amplification, fusion, methylation, among others. Approach: according to the biomarker's nature , additional processing steps may be required (for instance, DNA bisulfite treatment for methylation detection).
|
Biomarkers in cfDNA, current limitations and important considerations
As the circulating cfDNA released by tumor cells preserves the characteristics of the cell of origin, the identification of specific genetic and epigenetic tumor alterations in this liquid biopsy analyte has emerged as a promising tool with immense potential in cancer management. As mentioned before, the pool of ctDNA would better reflect the entire tumor epi/genetic picture rather than the traditional tumor biopsy, recapitulating tumor heterogeneity. However, analysis of cfDNA for cancer management still has several drawbacks. We can highlight the low amounts of mutant fragments in the total cfDNA sample which limits the detection success giving rise to false negative results [72]. In addition, it has been recently demonstrated that mutations can derive from clonal hematopoiesis rather than from tumor cells, thus giving rise to false positive results [79]. Indeed, according to a recent publication, a high percentage of cfDNA mutations found in both controls and cancer patients originate from clonal hematopoiesis, highlighting the importance of processing in parallel matched cfDNA and white blood cell DNA [80]. Caution is therefore needed when selecting biomarkers and interpreting cfDNA outcomes. The methodologies used to study the tumor material can be divided into targeted or untargeted strategies. When specific biomarkers are interrogated, digital PCR-based technologies such as droplet digital PCR (ddPCR) and BEAMing (beads, emulsion, amplification, and magnetics) show very high sensitivity, being fast and relatively inexpensive [81]. When a variable number of specific candidate biomarkers are interrogated, NGS-based panels have been designed, such as Tam-Seq (Tagged AMplicon deep sequencing), Safe-Seq (safe sequencing system) or CAPP-Seq (Cancer Personalized Profiling by deep sequencing) (review in ref. [12], [81]). To identify novel biomarkers, NGS technologies need to be applied. They have the limitation of lower sensitivity and higher cfDNA input [81]. Besides the enormous amount of published studies in the field and the huge efforts done by researchers in the last decades, to date only three cfDNA-based tests have been approved by the FDA (Table 2). As mentioned before, the first approved cfDNA-based liquid biopsy was a companion diagnostic test. The Cobas EGFR Mutation test v2 (Roche Molecular System, Inc), relies on real-time PCR detection of 42 mutations in exons 18, 19, 20 and 21 in the EGFR gene including L858R, exon 19 deletions, and T790 M mutation using plasma cfDNA within 4 h. The approved application of this test was to identify among patients with advanced or metastatic non-small cell lung cancer (NSCLC) those with an EGFR mutation, being therefore suitable for EGFR-targeted therapy [16]. Other FDA approved cfDNA-based assay is the Epi proColon test (Epigenomics AG) as a screening test for colorectal cancer (CRC). This test is designed for the detection of methylated SEPT9 in bisulfite converted plasma cfDNA by real-time PCR. The whole process takes around 10 h. Methylation of SEPT9 promoter has been associated with CRCoccurrence [82]. For their straightforward application, the corresponding companies (Roche Molecular System and Epigenomics AG) have developed kits including all the material and instructions needed from sample preparation to interpretations of the results. Whereas a positive result is very informative and useful for patients' management, a negative result in these liquid biopsy tests should be considered inconclusive. Finally, the multiplex PCR and NGS based ClonoSEQ kit, which gained FDA approval in September 2018, is intended to follow disease burden changes over time in response to treatment or during remission in both acute lymphoblastic leukemia or multiple myeloma patients [83]. It assesses and quantifies a panel of immunoglobulin receptor gene sequences and frequently translocated regions of the genome being able to detect a single cancer cell among a million cells [83].
Table 2:
Liquid biopsy FDA-approved tests.
| Kit/test | Company | FDA Status | Alteration detected /technology | Application | Other information |
|---|---|---|---|---|---|
| T Cell Search Circulating Tumor Cell Kit [15] | Menarini Silicon Biosystems | FDA-approved (August 2013) | Enumeration of circulating tumor cells (CTC) of epithelial origin: EpCAM positive enrichment and detection of cytokeratins 8, 18, and/or 19. | Prognosis of metastatic breast, colorectal, or prostate cancer | The sensitivity of this kit will depend on the positivity of CTCs for EpCAM surface marker as well as cytokeratins 8, 18, and/or 19. |
| Epi proColon DNA-methylation blood test [82] | Epigenomics AG | FDA-approved (April 2016) | Detection of methylated cytosine residues in the SEPTIN9 gene in ctDNA by real-time PCR | Screening for colon cancer | The company has developed a kit to assess SEPTIN9 methylation that will be completed in around 32 h. According to the literature, its sensitivity ranges from 69-72%. |
| Cobas EGFR Mutation Test v2 [16] | Roche Diagnostics | FDA-approved (June 2016) | Detection of 42 defined mutations in the epidermal growth factor receptor (EGFR) gene by real-time PCR | Guiding treatment selection in non-small-cell lung carcinoma | This test allows for detection of mutations in cfDNA in less than 4 h (12). 75% of sensitivity and 98% of specificity. |
| ClonoSEQ [83] | Adaptiv Biotechnologies | FDA-approved (Septembre 2018) | Detection of immunoglobulin receptor gene sequences and frequently translocated regions by multiplex PCR and next-generation sequencing (NGS). | Detection of minimal residual disease in acute lymphoblastic leukemia or multiple myeloma | ClonoSeq detects a single cancer cell among a million cells. Data processing will take between 7 and 14 days. |
Applications and current limitations of liquid biopsies for cancer
Despite the great advances in cancer management, this disease is still one of the world's most pressing health care concerns. Current important challenges include early diagnosis, accurate patient stratification and treatment selection, monitoring response to therapy and detection of minimal residual disease, and risk of relapse (Figure 2). To address all these concerns, liquid biopsy-based tools are continuously showing increased potential and, besides of researchers, have attracted the attention of investors [8], [10]. However, hard work is still needed as to date few tests have received the FDA approval or FDA Breakthrough Device Designation (Table 2 and 3).
Figure 2:
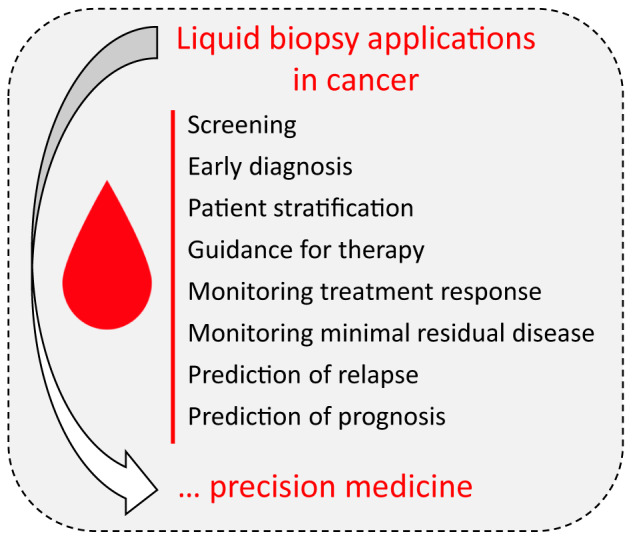
Summary of liquid biopsy applications for cancer management.
Table 3:
Selected liquid biopsy tests within the FDA's Breakthrough Devices Program.
| Kit/test | Company | Technology/application | FDA Status |
|---|---|---|---|
| Foundation One Liquid test | Roche Foundation Medicine | NGS test to detect clinically relevant indels, substitutions, CNV and selected genetic rearrangements in 70 oncogenes for companion diagnostic | FDA granted Breakthrough Device Designation (April 2018) |
| Multicancer early detection test | Grail | NGS blood test analyzing ctDNA methylation patterns for detecting multiple cancer types | FDA granted Breakthrough Device Designation (May 2019) |
| Guardant 360 | Guardant Health | ctDNA test of mutations (73 genes), amplifications (18 genes), fusions (6 genes), indels (23 genes), to guide treatment selection in non-small-cell lung carcinoma | FDA granted Breakthrough Device Designation (May 2019) |
| Resolution HRD | Resolution Bioscience | NGS assay to detect sequence variations in genes associated with homologous recombination deficiency (HRD). Companion diagnostic for prostate cancer | FDA granted Breakthrough Device Designation (May 2019) |
| Cancer SEEK | Thrive Earlier Detection | Multianalyte test that combines multiplexed PCR detection of mutations in ctDNA at 1,933 loci with measurements of validated protein biomarkers to diagnose eight common cancer types | FDA granted Breakthrough Device Designation (May 2019) |
| ExoDx Prostate IntelliScore (EPI) test | Bio-Techne | Urine exosome-based genomic test for the diagnosis of prostate cancer | FDA granted Breakthrough Device Designation (June 2019) |
| Ivy Gene CORE Test; IvyGene DX Liver Test | Laboratory for Advanced Medicine (LAM) | Hyper-methylated ctDNA from multiple gene targets. To confirm the presence of breast, colon, liver, and lung cancers as early as stage 1 | FDA granted Breakthrough Device Designation (September 2019) |
| [88], [89], [90] |
Evidence show that the efficacy of any anti-tumoral therapy including conventional chemotherapy, targeted therapy, or immune checkpoint inhibitors significantly improves when the tumor burden is low, making urgent the implementation of early detection methods. In this regard, liquid biopsy appears as a promising technique for cancer screening and early diagnosis. However, some limitations need to be solved before its incorporation in the clinical practice [11]. As mentioned above, one of the major obstacles in liquid biopsy-based early diagnosis is that biofluids of these patients harbour almost undetectable tumor analytes. In addition, the identification of the organ of origin, the high rate of false positive (overdiagnosis) and false negative (underdiagnosis) results due to both technical (biomarkers selection, sample processing, limited sensitivity), and biological (non-shedding tumors or clonal hematopoiesis of indeterminate potential) factors must be upgraded [11], [17], [79]. In this sense, the use of multiple types of biomarkers combining different analytes appears as a promising alternative to increase sensitivity and specificity [9], [84]. For instance, the CancerSEEK blood test is able to detect eight common types of cancer by assessing eight protein biomarkers and tumor-specific mutations in circulating cfDNA found in blood samples [84].
Once cancer diagnosis has been confirmed patient stratification and treatment decisions will depend on an accurate molecular profiling of the tumor. In this regard, besides being minimally-invasive, liquid biopsies have the advantage over tissue biopsy of recapitulating a more complete tumoral picture, theoretically allowing a more precise analysis. In this scenario, efforts are being made to identify actionable mutations or gene expression patterns in liquid biopsy analytes that could guide therapy. For example, as mentioned before, the FDA approved Cobas EGFR Mutation test v2 (Roche Molecular System, Inc) aims at distinguishing NSCLC patients that will benefit from EGFR-targeted therapy [16] and the Guardan360 (Guardant Health) examines a 73-gene panel in cfDNA to help NSCLC treatment selection. Similarly, ongoing research suggests that exosomal PD-L1 levels may constitute a predictor for anti-PD-1 therapy [85].
Monitoring treatment response and early detection of recurrence is essential to gain time for a quick second shot against cancer. Liquid biopsies as well hold promise in this context, with the great advantage of easily allowing longitudinal sample collection for a precise and continued follow-up. Tumor-derived analytes should decrease after complete surgical resection or during the evolution of a curative treatment. If specific tumor features or CTCs have been identified in the diagnostic phase, they could be followed-up as a warning alarm. In agreement, it has been reported that persistent detection of tumor analytes predicts high risk of relapse in different types of tumors [72], [86], [87]. Remarkably, the FDA-approved ClonoSEQ test is intended to detect minimal residual disease in acute lymphoblastic leukemia or multiple myeloma patients after completion of initial therapy [83].
Concluding remarks and open questions
In the last decades, liquid biopsy has emerged as a promising minimally-invasive tool for cancer management. Behind the term “liquid biopsy” a wide bulk of concepts are included since it encompasses different biofluids, analytes, biomarkers, technologies, and applications. Researchers are progressing at giant steps and are generating new knowledge that is forming the basis for the development of liquid biopsy tests. However, the lack of homogeneity in the protocols and the competitiveness in the field, makes it difficult to conclude with firmness whether we are about to implement those liquid biopsy assays in a real-world clinical setting. Moreover, relevant and unsolved questions still remain open. After a quick look at the literature, several protocols with a number of different variables are found when one tries to elucidate how to isolate a liquid biopsy analyte. Would it be possible to develop a unique standard isolation protocol for each component of the tumor circulome? This would help regarding the standardization and interpretation of results. The majority of the diagnostic studies have been performed comparing cancer patients versus healthy controls, but in a true clinicalpractice setting screenings would be done in patients with different non-tumoral pathological conditions. Will tests maintain their calculated specificity? Moreover, and as mentioned, the existence of non-shedding tumors has been described [17], [18]. Do these tumors really exist and in which percentage? If yes, they may increase the number of underdiagnosed cases and complicate the interpretation of tests. Where should we establish the sensitivity threshold of a liquid biopsy test to be considered useful? Regarding overdiagnosis, and before considering a test valid, which should be the size and the characteristics of the individuals tested as controls? In spite of these opened questions, the studies reviewed here underline the potential of liquid biopsy for cancer precision medicine. With researchers all over the world focused on accelerate liquid biopsy knowledge and applications, soon these questions will be solved and the translation to the clinical practice of these minimally-invasive technologies will revolutionize cancer patient management.
Acknowledgements
Work in authors' laboratory was supported by: CIBERehd; Grant numbers PI19/00613 and PI16/01126 from Instituto de Salud Carlos III (ISCIII) co-financed by “Fondo Europeo de Desarrollo Regional” (FEDER) “Una manera de hacer Europa”; MINECO/AEI/FEDER,UE [SAF2016-75972-R]; Grant numbers 2018-055 and 2017-058 from the Department of Health of the Gobierno de Navarra; HEPACARE Project from Fundación La Caixa; AECC post-doctoral fellowship (MA) from the AECC Scientific Foundation (POSTD18014AREC); Fundación Eugenio Rodríguez Pascual; Fundación Echebano; Fundación Mario Losantos; Fundación M Torres and Mr. Eduardo Ávila and Mr. Sergio Durá contribution.
Footnotes
Research funding: Work in authors' laboratory was supported by: CIBERehd; Grant numbers PI19/00613 and PI16/01126 from Instituto de Salud Carlos III (ISCIII) co-financed by “Fondo Europeo de Desarrollo Regional” (FEDER) “Una manera de hacer Europa”; MINECO/AEI/FEDER,UE [SAF2016-75972-R]; Grant numbers 2018-055 and 2017-058 from the Departamento de Salud del Gobierno de Navarra; HEPACARE Project from Fundación La Caixa; AECC post-doctoral fellowship (MA) from the AECC Scientific Foundation (POSTD18014AREC); Fundación Eugenio Rodríguez Pascual; Fundación Echebano; Fundación Mario Losantos; Fundación M Torres and Mr. Eduardo Ávila and Mr. Sergio Durá contribution.
Author contributions: All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
Competing interests: Authors state no conflict of interest.
Article Note: A translation of this article can be found here: https://doi.org/10.1515/almed-2020-0038
Contributor Information
María Arechederra, Email: macalderon@unav.es.
Carmen Berasain, Email: cberasain@unav.es.
References
- 1.Programme UNE , Organisation IL , Safety WHO& IP on C . Biomarkers in risk assessment: validity and validation. Geneva: World Health Organization; 2001. [Google Scholar]
- 2.Henry LN, Hayes DF. Cancer biomarkers. Mol Oncol. 2012;6:140–6. doi: 10.1016/j.molonc.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar M, Sarin SK. Curr Trends Sci. 2009. Biomarkers of diseases in medicine; pp. 403–17. Platinum Jubilee Special Issue. [Google Scholar]
- 4.Duffy MJ. Tumor markers in clinical practice: a review focusing on common solid cancers. Med Princ Pract. 2013;22:4–11. doi: 10.1159/000338393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel JN. Cancer pharmacogenomics, challenges in implementation, and patient-focused perspectives. Pharmacogenomics Personalized Med. 2016;9:65–77. doi: 10.2147/pgpm.s62918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/nejmoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613–28. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 8.Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017;14:531–48. doi: 10.1038/nrclinonc.2017.14. [DOI] [PubMed] [Google Scholar]
- 9.Marrugo-Ramírez J, Mir M, Samitier J. Blood-based cancer biomarkers in liquid biopsy: a promising non-invasive alternative to tissue biopsy. Int J Mol Sci. 2018;19:2877. doi: 10.3390/ijms19102877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mattox AK, Bettegowda C, Zhou S, Papadopoulos N, Kinzler KW, Vogelstein B. Applications of liquid biopsies for cancer. Sci Transl Med. 2019;11:eaay1984. doi: 10.1126/scitranslmed.aay1984. [DOI] [PubMed] [Google Scholar]
- 11.Chen M, Zhao H. Next-generation sequencing in liquid biopsy: cancer screening and early detection. Hum Genom. 2019;13:34. doi: 10.1186/s40246-019-0220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmirotta R, Lovero D, Cafforio P, Felici C, Mannavola F, Pellè E, et al. Liquid biopsy of cancer: a multimodal diagnostic tool in clinical oncology. Ther Adv Med Oncol. 2018;10 doi: 10.1177/1758835918794630. 1758835918794630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rubis G, Krishnan S, Bebawy M. Liquid biopsies in cancer diagnosis, monitoring, and prognosis. Trends Pharmacol Sci. 2019;40:172–86. doi: 10.1016/j.tips.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Schaffner F, Merlin JL, Von Bubnoff N, editors. Tumor liquid biopsies. vol 215 Cham: Springer; 2020. [Google Scholar]
- 15.Wit S. de, Van, editors; Dalum G, Terstappen LWMM. Detection of circulating tumor cells. Scientifica. 2014;2014:1–11. doi: 10.1155/2014/819362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwapisz D. The first liquid biopsy test approved. Is it a new era of mutation testing for non-small cell lung cancer? Ann Transl Med. 2017;5:46. doi: 10.21037/atm.2017.01.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stetson D, Ahmed A, Xu X, Nuttall BR, Lubinski TJ, Johnson JH, et al. Orthogonal comparison of four plasma NGS tests with tumor suggests technical factors are a major source of assay discordance. JCO Precis Oncol. 2019;3:1–9. doi: 10.1200/po.18.00191. [DOI] [PubMed] [Google Scholar]
- 18.Abbosh C, Birkbak NJ, Swanton C. Early stage NSCLC — challenges to implementing ctDNA-based screening and MRD detection. Nat Rev Clin Oncol. 2018;15:577–86. doi: 10.1038/s41571-018-0058-3. [DOI] [PubMed] [Google Scholar]
- 19.Pinsky PF, Prorok PC, Kramer BS. Prostate cancer screening — a perspective on the current state of the evidence. N Engl J Med. 2017;376:1285–9. doi: 10.1056/nejmsb1616281. [DOI] [PubMed] [Google Scholar]
- 20.Bhardwaj M, Gies A, Werner S, Schrotz-King P, Brenner H. Blood-based protein signatures for early detection of colorectal cancer: a systematic review. Clin Transl Gastroenterol. 2017;8:e128. doi: 10.1038/ctg.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhardwaj M, Gies A, Weigl K, Tikk K, Benner A, Schrotz-King P, et al. Evaluation and validation of plasma proteins using two different protein detection methods for early detection of colorectal cancer. Cancers. 2019;11:1426. doi: 10.3390/cancers11101426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Torrano V, Royo F, Peinado H, Loizaga-Iriarte A, Unda M, Falcón-Perez JM, et al. Vesicle-MaNiA: extracellular vesicles in liquid biopsy and cancer. Curr Opin Pharmacol. 2016;29:47–53. doi: 10.1016/j.coph.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177–82. doi: 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. 2014;289:3869–75. doi: 10.1074/jbc.c113.532267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madhavan B, Yue S, Galli U, Rana S, Gross W, Müller M, et al. Combined evaluation of a panel of protein and miRNA serum-exosome biomarkers for pancreatic cancer diagnosis increases sensitivity and specificity. Int J Canc. 2014;136:2616–27. doi: 10.1002/ijc.29324. [DOI] [PubMed] [Google Scholar]
- 26.Re MD, Biasco E, Crucitta S, Derosa L, Rofi E, Orlandini C, et al. The detection of androgen receptor splice variant 7 in plasma-derived exosomal RNA strongly predicts resistance to hormonal therapy in metastatic prostate cancer patients. Eur Urol. 2016;71:680–7. doi: 10.1016/j.eururo.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 27.Li Q, Shao Y, Zhang X, Zheng T, Miao M, Qin L, et al. Plasma long noncoding RNA protected by exosomes as a potential stable biomarker for gastric cancer. Tumor Biol. 2014;36:2007–12. doi: 10.1007/s13277-014-2807-y. [DOI] [PubMed] [Google Scholar]
- 28.Ashworth T.R. A case of cancer in which cells similar to those in the Tumours were seen in the blood after death. Med J Aust. 1869;14:146–7. [Google Scholar]
- 29.Racila E, Euhus D, Weiss AJ, Rao C, McConnell J, Terstappen LWMM, et al. Detection and characterization of carcinoma cells in the blood. Proc Natl Acad Sci Unit States Am. 1998;95:4589–94. doi: 10.1073/pnas.95.8.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krishnan SR, Luk F, Brown RD, Suen H, Kwan Y, Bebawy M. Isolation of human CD138(+) microparticles from the plasma of patients with multiple myeloma. Neoplasia. 2016;18:25–32. doi: 10.1016/j.neo.2015.11.011. New York N Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Canc. 2002;2:563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 32.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–4. doi: 10.1126/science.1228522. New York N Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang WC, Zhang XF, Peng J, Li XF, Wang AL, Bie YQ, et al. Survival mechanisms and influence factors of circulating tumor cells. BioMed Res Int. 2018;2018:6304701. doi: 10.1155/2018/6304701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Canc Res. 2004;10:6897–904. doi: 10.1158/1078-0432.ccr-04-0378. [DOI] [PubMed] [Google Scholar]
- 35.Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Canc Res. 2004;10:8152–62. doi: 10.1158/1078-0432.ccr-04-1110. [DOI] [PubMed] [Google Scholar]
- 36.Micalizzi DS, Maheswaran S, Haber DA. A conduit to metastasis: circulating tumor cell biology. Gene Dev. 2017;31:1827–40. doi: 10.1101/gad.305805.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fehm T, Sagalowsky A, Clifford E, Beitsch P, Saboorian H, Euhus D, et al. Cytogenetic evidence that circulating epithelial cells in patients with carcinoma are malignant. Clin Cancer Res. 2002;8:2073–84. Official J Am Assoc Cancer Res. [PubMed] [Google Scholar]
- 38.Armstrong A, Marengo M, Oltean S, Kemeny G, Bitting R, Turnbull J, et al. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Canc Res. 2011;9:997–1007. doi: 10.1158/1541-7786.mcr-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyamoto D, Zheng Y, Wittner B, Lee R, Zhu H, Broderick K, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349:1351–6. doi: 10.1126/science.aab0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110–22. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci Unit States Am. 2016;113:E854–63. doi: 10.1073/pnas.1508541113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X, Taftaf R, Kawaguchi M, Chang Y-F, Chen W, Entenberg D, et al. Homophilic CD44 interactions mediate tumor cell aggregation and polyclonal metastasis in patient-derived breast cancer models. Canc Discov. 2019;9:96–113. doi: 10.1158/2159-8290.cd-18-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aceto N, Toner M, Maheswaran S, Haber DA. En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Canc. 2015;1:44–52. doi: 10.1016/j.trecan.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 44.Suo Y, Xie C, Zhu X, Fan Z, Yang Z, He H, et al. Proportion of circulating tumor cell clusters increases during cancer metastasis. Cytometry Part J Int Soc Anal Cytol. 2016;91:250–3. doi: 10.1002/cyto.a.23037. [DOI] [PubMed] [Google Scholar]
- 45.Geeurickx E, Hendrix A. Targets, pitfalls and reference materials for liquid biopsy tests in cancer diagnostics. Mol Aspect Med. 2019;8:100828. doi: 10.1016/j.mam.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 46.Yap K, Cohen EN, Reuben JM, Khoury JD. Circulating tumor cells: state-of-the-art update on technologies and clinical applications. Curr Hematol Malig Rep. 2019;14:353–7. doi: 10.1007/s11899-019-00531-x. [DOI] [PubMed] [Google Scholar]
- 47.Ferreira MM, Ramani VC, Jeffrey SS. Circulating tumor cell technologies. Mol Oncol. 2016;10:374–94. doi: 10.1016/j.molonc.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bankó P, Lee SY, Nagygyörgy V, Zrínyi M, Chae CH, Cho DH, et al. Technologies for circulating tumor cell separation from whole blood. J Hematol Oncol. 2019;12:48. doi: 10.1186/s13045-019-0735-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kolinsky MP, Stoecklein N, Lambros M, Gil V, Rodrigues DN, Carreira S, et al. Recent results in cancer research. Recent Results Cancer Res Fortschritte Der Krebsforschung Progres Dans Les Recherches Sur Le Cancer. 2019;215:57–76. doi: 10.1007/978-3-030-26439-0_3. [DOI] [PubMed] [Google Scholar]
- 50.Pantel K, Denève E, Nocca D, Coffy A, Vendrell JP, Maudelonde T, et al. Circulating epithelial cells in patients with benign colon diseases. Clin Chem. 2011;58:936–40. doi: 10.1373/clinchem.2011.175570. [DOI] [PubMed] [Google Scholar]
- 51.Lara O, Tong X, Zborowski M, Chalmers JJ. Enrichment of rare cancer cells through depletion of normal cells using density and flow-through, immunomagnetic cell separation. Exp Hematol. 2004;32:891–904. doi: 10.1016/j.exphem.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 52.Yang L, Lang JC, Balasubramanian P, Jatana KR, Schuller D, Agrawal A, et al. Optimization of an enrichment process for circulating tumor cells from the blood of head and neck cancer patients through depletion of normal cells. Biotechnol Bioeng. 2009;102:521–34. doi: 10.1002/bit.22066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harouaka RA, Nisic M, Zheng SY. Circulating tumor cell enrichment based on physical properties. J Lab Autom. 2013;18:455–68. doi: 10.1177/2211068213494391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cross SE, Jin YS, Rao J, Gimzewski JK. Nanomechanical analysis of cells from cancer patients. Nat Nanotechnol. 2007;2:780–3. doi: 10.1038/nnano.2007.388. [DOI] [PubMed] [Google Scholar]
- 55.Gossett DR, Tse HTK, Lee SA, Ying Y, Lindgren AG, Yang OO, et al. Hydrodynamic stretching of single cells for large population mechanical phenotyping. Proc Natl Acad Sci USA. 2012;109:7630–5. doi: 10.1073/pnas.1200107109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen NV, Jen CP. Impedance detection integrated with dielectrophoresis enrichment platform for lung circulating tumor cells in a microfluidic channel. Biosens Bioelectron. 2018;121:10–8. doi: 10.1016/j.bios.2018.08.059. [DOI] [PubMed] [Google Scholar]
- 57.Toss A, Mu Z, Fernandez S, Cristofanilli M. CTC enumeration and characterization: moving toward personalized medicine. Ann Transl Med. 2014;2:108. doi: 10.3978/j.issn.2305-5839.2014.09.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aceto N. Fluctuating numbers of circulating tumor cells in cancer patients and the meaning of zero counts. Oncotarget. 2019;10:2658–9. doi: 10.18632/oncotarget.26850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol. 2016;2:1441. doi: 10.1001/jamaoncol.2016.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chimonidou M, Strati A, Tzitzira A, Sotiropoulou G, Malamos N, Georgoulias V, et al. DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clin Chem. 2011;57:1169–77. doi: 10.1373/clinchem.2011.165902. [DOI] [PubMed] [Google Scholar]
- 61.Sinkala E, Sollier-Christen E, Renier C, Rosàs-Canyelles E, Che J, Heirich K, et al. Profiling protein expression in circulating tumour cells using microfluidic western blotting. Nat Commun. 2017;8:14622. doi: 10.1038/ncomms14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mandel P, Metais P. Comptes rendus des seances de la Societe de biologie et de ses filiales. Journal de la Société de Biologie. 1948;142:241–3. [PubMed] [Google Scholar]
- 63.Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977;37:646–50. [PubMed] [Google Scholar]
- 64.Sorenson GD, Pribish DM, Valone FH, Memoli VA, Bzik DJ, Yao SL. Soluble normal and mutated DNA sequences from single-copy genes in human blood. Cancer Epidemiology Biomarkers Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. 1994;3:67–71. [PubMed] [Google Scholar]
- 65.Stroun M, Maurice P, Vasioukhin V, Lyautey J, Lederrey C, Lefort F, et al. The origin and mechanism of circulating DNA. Ann N Y Acad Sci. 2006;906:161–8. doi: 10.1111/j.1749-6632.2000.tb06608.x. [DOI] [PubMed] [Google Scholar]
- 66.Yu M. Circulating cell-free mitochondrial DNA as a novel cancer biomarker: opportunities and challenges. Mitochondrial DNA. 2012;23:329–32. doi: 10.3109/19401736.2012.696625. [DOI] [PubMed] [Google Scholar]
- 67.Kustanovich A, Schwartz R, Peretz T, Grinshpun A. Life and death of circulating cell-free DNA. Canc Biol Ther. 2019;20:1057–67. doi: 10.1080/15384047.2019.1598759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu SCY, Lee SWY, Jiang P, Leung TY, Chan KCA, Chiu RWK, et al. High-resolution profiling of fetal DNA clearance from maternal plasma by massively parallel sequencing. Clin Chem. 2013;59:1228–37. doi: 10.1373/clinchem.2013.203679. [DOI] [PubMed] [Google Scholar]
- 69.Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell. 2016;164:57–68. doi: 10.1016/j.cell.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun. 2018;9:5068. doi: 10.1038/s41467-018-07466-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bettegowda C, Sausen M, Leary R, Kinde I, Agrawal N, Bartlett B, et al. Detection of circulating tumor DNA in early and late stage human malignancies. Neuro Oncol. 2014;16:iii7. doi: 10.1093/neuonc/nou206.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–90. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morbelli S, Alama A, Ferrarazzo G, Coco S, Genova C, Rijavec E, et al. Circulating tumor DNA reflects tumor metabolism rather than tumor burden in chemotherapy-naive patients with advanced non-small cell lung cancer: (18)F-FDG PET/CT study. J Nucl MedOff Publ Soc Nucl Med. 2017;58:1764–9. doi: 10.2967/jnumed.117.193201. [DOI] [PubMed] [Google Scholar]
- 74.Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570:385–9. doi: 10.1038/s41586-019-1272-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trigg RM, Martinson LJ, Parpart-Li S, Shaw JA. Factors that influence quality and yield of circulating-free DNA: a systematic review of the methodology literature. Heliyon. 2018;4:e00699. doi: 10.1016/j.heliyon.2018.e00699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Markus H, Contente-Cuomo T, Farooq M, Liang WS, Borad MJ, Sivakumar S, et al. Evaluation of pre-analytical factors affecting plasma DNA analysis. Sci Rep. 2018;8:7375. doi: 10.1038/s41598-018-25810-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chan KCA, Yeung SW, Lui WB, Rainer TH, Lo YMD. Effects of preanalytical factors on the molecular size of cell-free DNA in blood. Clin Chem. 2005;51:781–4. doi: 10.1373/clinchem.2004.046219. [DOI] [PubMed] [Google Scholar]
- 78.Sorber L, Zwaenepoel K, Jacobs J, Winne KD, Casteren KV, Augustus E, et al. Specialized blood collection tubes for liquid biopsy: improving the pre-analytical conditions. Mol Diagn Ther. 2019;24:113–24. doi: 10.1007/s40291-019-00442-w. [DOI] [PubMed] [Google Scholar]
- 79.Hu Y, Ulrich B, Supplee J, Kuang Y, Lizotte PH, Feeney N, et al. False positive plasma genotyping due to clonal hematopoiesis. Clin Canc Res. 2018;24:0143. doi: 10.1158/1078-0432.ccr-18-0143. [DOI] [PubMed] [Google Scholar]
- 80.Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med. 2019;25:1928–37. doi: 10.1038/s41591-019-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Elazezy M, Joosse SA. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput Struct Biotechnol J. 2018;16:370–8. doi: 10.1016/j.csbj.2018.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lamb YN, Dhillon S. Epi proColon® 2.0 CE: a blood-based screening test for colorectal cancer. Mol Diagn Ther. 2017;21:225–32. doi: 10.1007/s40291-017-0259-y. [DOI] [PubMed] [Google Scholar]
- 83.ClonoSEQ cleared for residual cancer testing. Canc Discov. 2018;8:OF6. doi: 10.1158/2159-8290.cd-nb2018-136. [DOI] [PubMed] [Google Scholar]
- 84.Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–30. doi: 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–6. doi: 10.1038/s41586-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:346ra92. doi: 10.1126/scitranslmed.aaf6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Olsson E, Winter C, George A, Chen Y, Howlin J, Tang M-HE, et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol Med. 2015;7:1034–47. doi: 10.15252/emmm.201404913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.BioTech . FDA grants breakthrough device designation to BioTeche's ExoDx Prostate IntelliScore (EPI) test. 2017. [Google Scholar]
- 89.Outlook PT . FDA grants Breakthrough device designation to LAM's novel liver cancer detection test. 2010. [Google Scholar]
- 90.Sheridan C. Investors keep the faith in cancer liquid biopsies. Nat Biotechnol. 2019;37:972–4. doi: 10.1038/d41587-019-00022-7. [DOI] [PubMed] [Google Scholar]