

SUMMARY
CD8+ T cell recruitment to the tumor microenvironment is critical for the success of adoptive cell therapy (ACT). Unfortunately, only a small fraction of transferred cells home to solid tumors. Adhesive ligand-receptor interactions have been implicated in CD8+ T cell homing; however, there is a lack of understanding of how CD8+ T cells interact with tumor vasculature-expressed adhesive ligands under the influence of hemodynamic flow. Here, the capacity of CD8+ T cells to home to melanomas is modeled ex vivo using an engineered microfluidic device that recapitulates the hemodynamic microenvironment of the tumor vasculature. Adoptively transferred CD8+ T cells with enhanced adhesion in flow in vitro and tumor homing in vivo improve tumor control by ACT in combination with immune checkpoint blockade. These results show that engineered microfluidic devices can model the microenvironment of the tumor vasculature to identify subsets of T cells with enhanced tumor infiltrating capabilities, a key limitation in ACT.
Graphical Abstract
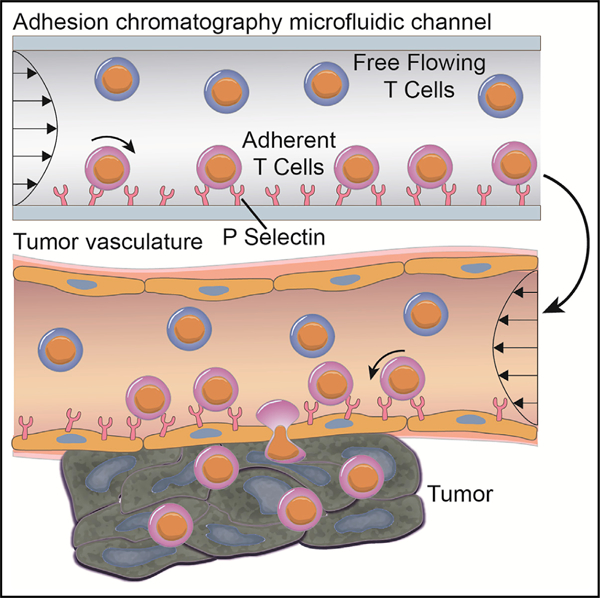
In brief
Camargo et al. engineered a tumor vasculature-mimicking microfluidic device for predicting in vivo tumor homing of CD8+ T cells. Microfluidically enriched CD8+ T cells for P-selectin adhesion in fluid flow in vitro show superior tumor homing in vivo, remodel the tumor microenvironment, and augment the immunotherapeutic benefit of immune checkpoint blockade.
INTRODUCTION
Adoptive cell therapy (ACT) has emerged as a powerful treatment option for patients with metastatic melanoma.1,2 ACT consisting of autologous tumor-infiltrating lymphocytes expanded ex vivo and transferred back into the patient in combination with interleukin (IL)-2 can boost anti-tumor immunity.1–3 This has yielded good clinical responses for the treatment of metastatic melanoma, but at overall low rates.1,2 It is now known that low patient rates of response are due to poor trafficking of transferred cells to relevant tissues.1
In various cancer types, including melanoma, tumor infiltration by CD8+ T cells is correlated with reduction in disease burden and improved survival.2,4–11 In the context of ACT, cultures containing more CD8+ T cells also tend to yield better clinical responses.2,4,5 Unfortunately, only a small fraction of infused T cells make it into either primary or metastatic lesions.1,12,13 This means that infusion of an extremely high number of T cells is required to have any therapeutic effect,12,14 thus pointing to the central role T cell homing to tumors may play in the efficacy of ACT.11,15–17 Unfortunately, methods to predict whether ACT cultures will result in robust tumor homing are lacking, as are methods to enrich cells that exhibit these features associated with favorable responses.
CD8+ T cells traffic to quiescent or inflamed tissues in a highly dynamic process that involves a variety of receptor-ligand inter-actions. The steps involve rolling adhesion that decelerates the cell against the force of blood flow, chemokine-triggered integrin activation, and integrin-mediated firm adhesion leading to transmigration through the endothelial layer.17–19 Due to their role as the initial kickoff step in this adhesion cascade, selectins and their ligands have been correlatively or directly implicated in homing of T cells into melanomas, inflamed skin, and other tumor types.20–26 In experimental animal models of lymphocyte homing, endothelial cells isolated from melanomas are shown to express E- and P-selectin, among other adhesion receptors,27 mirroring human disease.20,28,29 Suggestive of their role in supporting tumor control, melanoma growth is accelerated and CD8+ T cell infiltration is decreased in mice lacking P-, but not E-, selectin or its ligand P-selectin glycoprotein ligand (PSGL)-1.26 Despite the apparent contribution of P-selectin and its ligand(s) in CD8+ T cell homing and disease progression in the context of melanoma, how P-selectin may contribute to the adhesion and homing of CD8+ T cells in ACT has not been explored.
Herein, profiles of endogenous or adoptively transferred murine CD8+ T cells harvested from tumors in an in vivo melanoma model were found to mirror those enriched for their capacity to mediate adhesion to P-selectin in flow in vitro using a cell-adhesion chromatography system30–32 that recapitulates the wall shear stress (WSS) environment of the tumor vasculature.30–32 Both murine and human CD8+ T cells could be enriched for adhesion to P-selectin in flow in vitro, which, when applied to a murine tumor model, resulted in enhanced tumor homing that was associated with improved tumor control of ACT in combination with therapeutic blockade of immune checkpoint programmed cell death (PD-1). These results demonstrate the utility of an engineered microfluidic device in recapitulating the hemodynamic microenvironment of the tumor vasculature in modeling the in vivo homing of lymphocytes relevant to ACT.
RESULTS
P-selectin ligand+ CD8+ T cells preferentially traffic to the TME versus lymphoid tissues
The phenotypes and cellular characteristics of endogenous CD8+ T cells in the tumor microenvironment (TME) versus secondary lymphoid tissues from C57BL/6 mice bearing B16F10 melanomas were evaluated. Endogenous CD8+ T cells infiltrating the TME exhibited more central memory (CD44+CD62L+; CM) and effector (CD44+CD62L−; EFF) phenotypes, while those recovered from the spleens of tumor-bearing animals exhibited a more naive (CD44−CD62L+) phenotype (Figure S1A). P-selectin has been implicated in T cell homing and is expressed in human melanomas,28,29,33 numerous preclinical models,27,28,34 and melanomas that form in the skin of C57BL/6 animals (Figure 1A) at a higher extent compared with naive skin (Figure S2). Furthermore, T cells can be found near P-selectin-expressing vessels via immunohistochemistry (Figure 1B). To this end, the expression of P-selectin ligands by lymphocytes harvested from various tissues of tumor-bearing or naive animals was assessed flow cytometrically by staining with a recombinant P-selectin-Fc chimera (Figure 1C). Recombinant P-selectin-Fc was used to quantify the extent of P-selectin ligand expression by CD8+ T cells instead of antibodies recognizing canonical selectin ligands, because, whereas one such ligand, PSGL-1, is constitutively expressed on lymphocytes,35 it mediates selectin binding only in the context of appropriate glycosylation that cannot be distinguished by antibody-mediated staining of PSGL-1’s protein backbone.35 In so doing, we found a low frequency of P-selectin ligand+ CD8+ T cells in secondary lymphoid tissues such as the spleen and lymph nodes (LNs), including non-tumor-draining (NDLNs) and tumor-draining (TDLNs) LNs (Figure 1D). In contrast, a high frequency of P-selectin ligand+ CD8+ T cells were found in the TME and the skin of tumor-naive animals (Figure 1D). However, overall numbers of P-selectin ligand-expressing CD8+ T cells were higher in tumors compared with the naive skin (Figure 1E).
Figure 1. Endogenous and donor CD8+ T cells recovered from the TME are enriched for P-selectin ligand expression compared with lymphoid tissues.
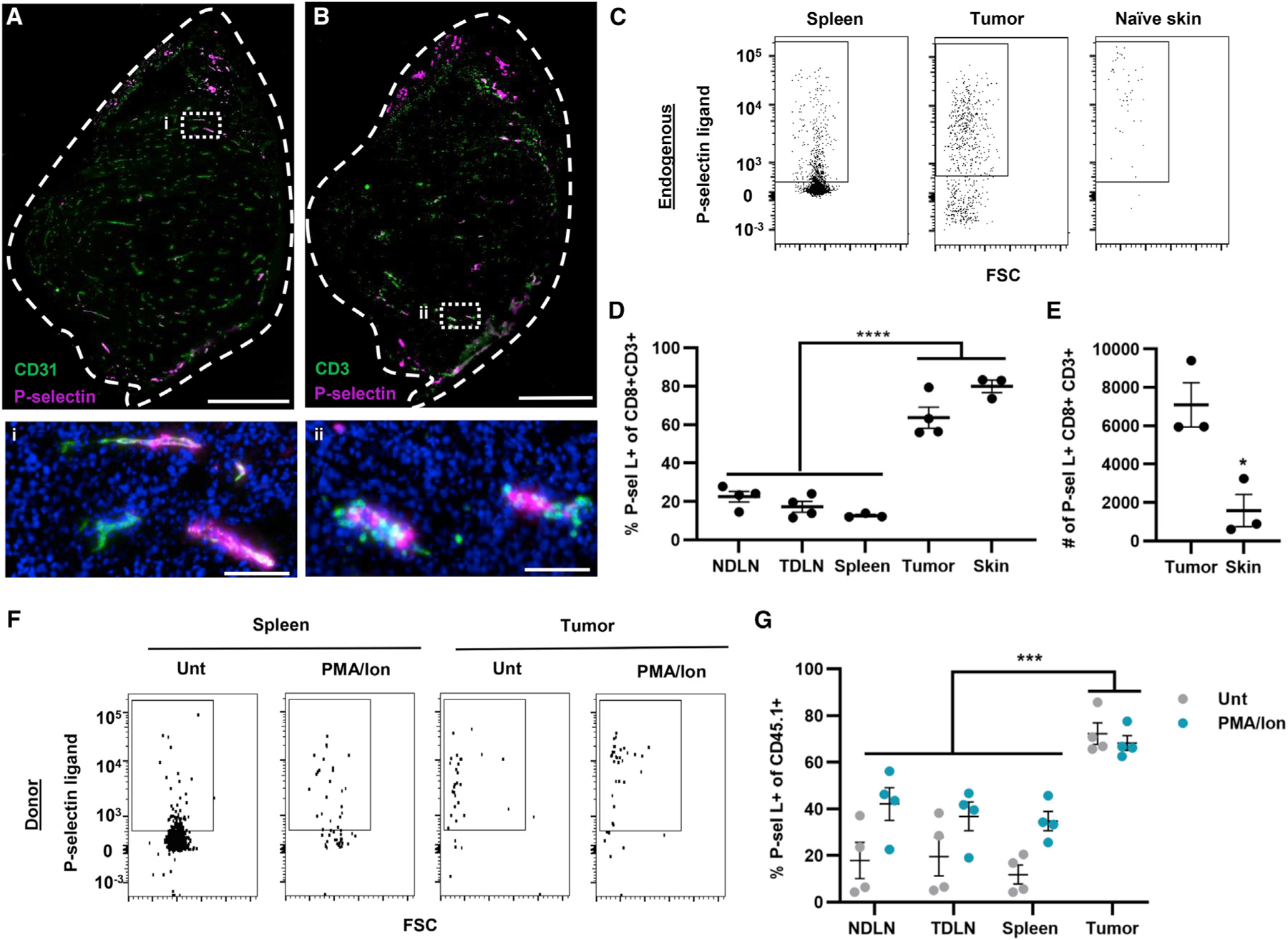
(A and B) Immunohistochemistry staining for P-selectin and CD31 (A) or CD3 (B) in 8-μm-thick sections of B16F10 tumors formed in C57BL/6 mice. Scale bars: top, 400 μm; bottom, 50 μm.
(C) Representative flow cytometry data scatterplots for P-selectin ligand+ expression by CD8+ T cells recovered from the spleen, tumor, and naive skin.
(D) Percentage of CD8+ T cells recovered from the LNs, spleen, tumor, or naive skin expressing P-selectin ligand. Statistical comparisons by one-way ANOVA with Dunnett’s multiple comparisons test.
(E) Total number of P-selectin ligand-expressing (P-sel L+) cells recovered from tumors or naive skin. Statistical comparisons by two-tailed parametric t test.
(F) Representative flow cytometry scatterplots for P-selectin ligand expression by CD45.1+CD8+ T cells recovered from the spleen, tumor, and naive skin 16 h post-adoptive transfer of 106 untreated or PMA/Ion-treated CD45.1+CD8+ T cells into melanoma-bearing mice.
(G) Percentage of transferred CD45.1+CD8+ T cells recovered from various tissues 16 h post-transfer that express P-selectin ligand. Statistical comparisons by two-way ANOVA with Dunnett’s multiple comparisons test. Points represent data from an individual animal. Data in all panels represent the mean ± SEM;
*p < 0.05, ***p < 0.001, ****p < 0.0001. See also Figures S1–S4.
To determine whether differential phenotypes of CD8+ T cells recovered from tumors and skin versus lymphoid tissues reflected differences in activation state, the in vivo homing of CD8+ T cells negatively isolated from spleens of CD45.1 mice left untreated or activated with PMA/Ionomycin (PMA/Ion) (Figure S3) was assessed. As a result, infused populations of cells exhibited either primarily naive or differentiated EFF cells, respectively (Figure S1B). PMA/Ion-treated CD8+ T cells expectedly exhibited reduced L-selectin and increased CD44 expression (Figure S3). They also exhibited increased expression of lymphocyte function-associated antigen (LFA)-1 and P-selectin ligand, as measured by staining with P-selectin-Fc (Figure S3). However, PSGL-1 expression remained unchanged (Figures S3F and S3G). Upon transfer into B16F10 melanoma-bearing CD45.2 animals, PMA/Ion-treated cells trafficked to LNs and the spleen less compared with the tumor (Figure S4). Surprisingly, there was no difference in tumor trafficking between CD8+ T cell treatments (Figure S4). About 60% of donor cells infiltrating the tumor 16 h post-transfer exhibited a CM phenotype, whereas in spleens, donor cells were largely naive, trends seen in both tissues irrespective of the donor cell population (Figure S1C). Mirroring trends in endogenous CD8+ T cells in each tissue, the proportion of donor cells that were P-selectin ligand+ was low versus high in spleens versus tumors (Figures 1F and 1G), irrespective of donor cell population (untreated or PMA/Ion treated). Similarly, trends seen in the spleen with low levels of trafficked donor P-selectin ligand+ CD8+ T cells were recapitulated in LNs (Figure 1G).
CD8+ T cells mediate adhesion to P-selectin-functionalized substrates in hemodynamic flow
Expression of P-selectin ligands by endogenous and donor lymphocytes in the TME raised the hypothesis that CD8+ T cells mediate adhesion to P-selectin in flow, as has been reported for CD4+ T cells.36 As such, the capacity of CD8+ T cells to mediate interactions with inflamed vasculature-expressed adhesive ligands, P-selectin, and ICAM, alone or in combination, under conditions of physiological levels of fluid force was evaluated in vitro. The extent of adhesion of CD8+ T cells to a P-selectin-functionalized substrate in a parallel plate flow chamber assay was measured using videomicroscopy. These experiments were performed under two conditions: static, where after cells were loaded into the channel, the flow was paused for 5 min before restarting, allowing cells within the channel to interact with the substrate to initiate adhesion in the absence of fluid force (Figure 2A), and continuous flow, in which the fluid perfusion was never interrupted (Figure 2B). These experiments were also performed with untreated or PMA/Ion-treated CD8+ T cells to evaluate the effects of cell activation state on adhesion. Under static conditions, untreated CD8+ T cells exhibited adhesion to P-selectin with or without ICAM, but not to ICAM alone (Figure 2C). On the other hand, PMA/Ion-treated CD8+ T cells exhibited adhesion to both ICAM and P-selectin alone, with the combination of the two having the highest extent of adhesion (Figure 2D). However, for both untreated and PMA/Ion-treated cells, the addition of ICAM to P-selectin did not increase the extent of adhesion compared with that observed on P-selectin alone (Figure 2E). The adhesive quality was also assessed, with untreated CD8+ T cells largely exhibiting more rolling adhesion, whereas PMA/Ion-treated CD8+ T cells exhibited more firm adhesion (Figures S5A and S5B). Under continuous flow, similar results were observed, with CD8+ T cells adhering to P-selectin with or without co-presentation of ICAM, but not to ICAM alone, albeit in a shear-stress-sensitive manner (Figure 2F). PMA/Ion-treated CD8+ T cells under continuous flow also exhibited adhesion to ICAM and P-selectin individually, with their combination resulting in the highest extent of adhesion that was WSS dependent (Figure 2G). Co-presentation of P-selectin with ICAM, interactions with which in flow are known to be selectin enabled,36,37 expectedly increased the total extent of adhesion by PMA/Ion-treated but not untreated CD8+ T cells compared with P-selectin alone (Figure 2H). Whereas untreated cells interacted in flow with P-selectin-functionalized substrates largely via rolling adhesion, PMA/Ion-treated CD8+ T cells inter-acted more substantially via firm adhesion, albeit at similar total extents that were WSS dependent (Figures S5C and S5D). Firmer adhesion to the P-selectin substrate by activated CD8+ T cells also resulted in overall lower average velocities of adhesion in flow (Figures S6). Results from experiments under static and continuous flow conditions suggest that both untreated and PMA/Ion-treated CD8+ T cells engage to some extent with P-selectin but not ICAM. Correlation analyses of the number of cells adhering to P-selectin and the frequency of CD8+ T cells expressing P-selectin ligands in perfused populations revealed that, while predictive of the extent of in vitro adhesion to P-selectin under static conditions, adhesion under continuous flow was not predicted by P-selectin ligand expression of perfused CD8+ T cells (Figures 2I–2K). Therefore, P-selectin ligand expression does not predict the extent of adhesion to P-selectin under conditions where adhesion is initiated in the presence of flow.
Figure 2. Adhesion of CD8+ T cells to P-selectin-functionalized substrates is not correlated to P-selectin ligand expression under conditions of continuous flow at physiological levels of wall shear stress.
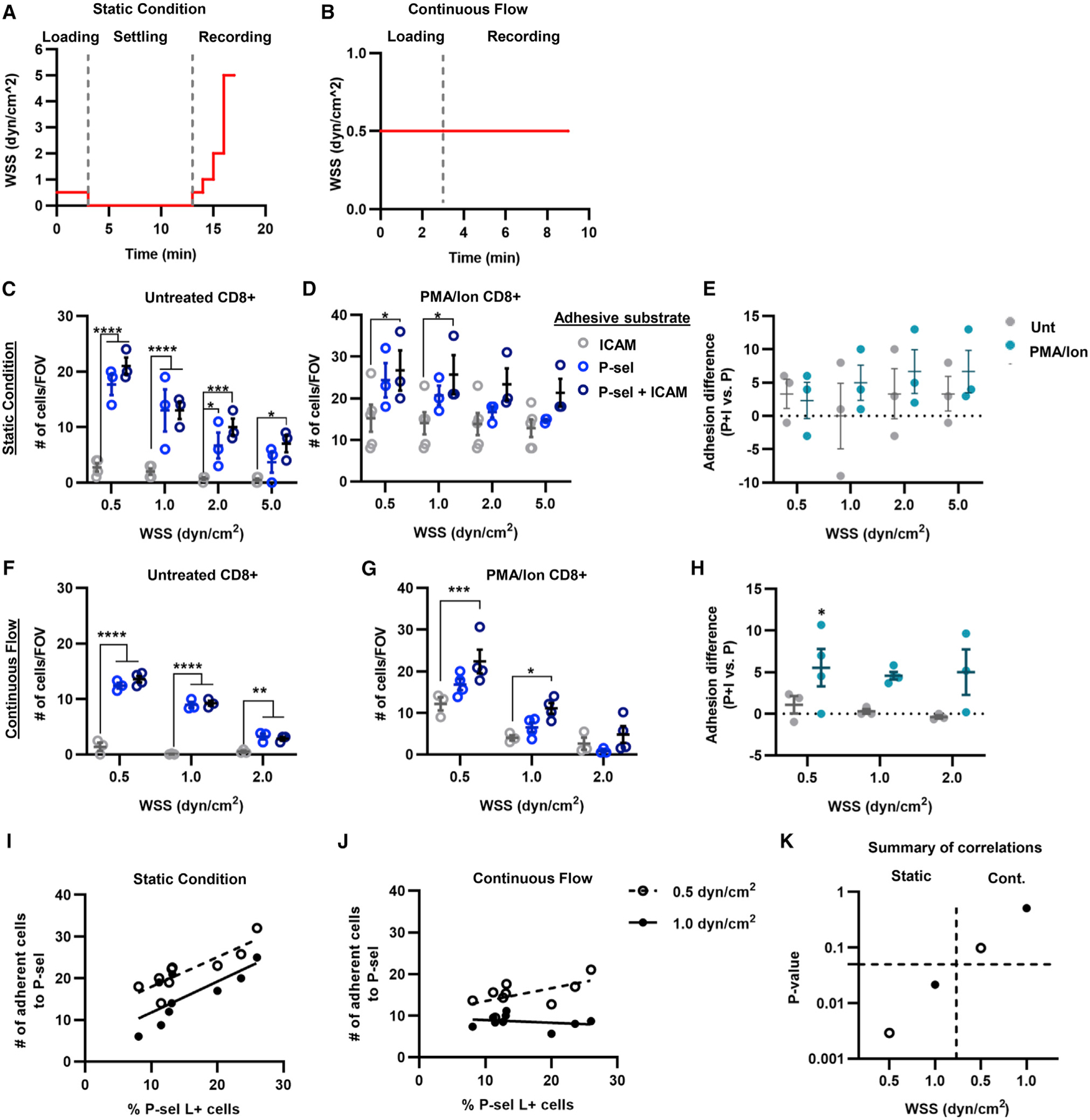
(A and B) Schematics outlining perfusion conditions for adhesion experiments conducted under static (A) and continuous flow (B) conditions.
(C and D) Numbers of untreated (C) or PMA/Ion-treated (D) CD8+ T cells per field view interacting with ICAM, P-selectin, or P-selectin + ICAM functionalized substrate under static conditions.
(E) Differences in total adhesion of perfused CD8+ T cells to P-selectin + ICAM versus P-selectin alone under static conditions.
(F and G) Numbers of untreated (F) or PMA/Ion-treated (G) CD8+ T cells per field view interacting with an ICAM, P-selectin, or P-selectin + ICAM functionalized substrate under continuous flow conditions.
(H) Differences in total adhesion of perfused CD8+ T cells to P-selectin + ICAM versus P-selectin alone under conditions of continuous flow.
(I and J) Correlation analysis between number of adherent cells to P-selectin and percentage of P-selectin ligand+ cells under static (I) or continuous flow (J).
(K) The p values for the correlation analysis between cells adhering to P-selectin under static conditions or continuous flow and the frequency of CD8+ T cells expressing P-selectin ligand; dashed line represents p = 0.05. Data in all panels represent the mean ± SEM of three independently performed experiments. Statistical comparisons performed by two-way ANOVA with Tukey’s multiple comparisons test; *p < 0.05, **p < 0.01. ***p < 0.001, ****p < 0.0001. See also Figures S5 and S6.
Characteristics of CD8+ T cells that interact with P-selectin under shear flow
To evaluate the cellular characteristics of CD8+ T cells that do or do not adhere to P-selectin-functionalized substrates under conditions of physiological levels of shear flow, a cell-adhesion-based chromatography microfluidic system (Figure 3A) that exerts an almost uniform shear stress level across the width of the adhesive substrate was implemented.31,32,38 Calculated based on Stokes flow and experimentally validated (Figure S6B), the channel was designed to allow cells to settle to the bottom of the channel prior to their encounter with the adhesive substrate, allowing uniform substrate contact for all perfused cells. With this system, cells that can mediate adhesion can thus be separated from non-inter-acting cells based on their channel elution time (Figure 3B) and recovered for off-chip analyses.31,32 Loaded cells were verified to interact only with the functionalized portion of the channel and, after recovery by flow-driven unloading, maintained high purity and viability (Figure S7). Demonstrating that cell adhesion in the perfusion channel is P-selectin dependent, pretreatment of cell suspensions for 30 min before perfusion with saturating levels (25 μg/mL) of P-selectin-Fc chimera decreased their adhesion to P-selectin substrates at 0.5 dyn/cm2 (Figure 3C). Furthermore, upon recovery from the channel, untreated and PMA/Ion-treated CD8+ T cells that were reperfused retained the general characteristics of the population from which they originally eluted, e.g., cells that were recovered from the free-flow (FF) fraction exhibited a higher frequency of cells in the FF rather than the adherent (Adh) fraction after reperfusion, and vice versa (Figure 3D). To ensure yields sufficient for statistical comparisons between representative isolated populations, a WSS of 0.5 dyn/cm2 was implemented for all chromatography experiments, given the higher extents of adhesion observed for CD8+ T cells of either treatment type, despite their highly divergent qualities.
Figure 3. Perfusion of CD8+ T cells through an adhesion chromatography channel functionalized with P-selectin under physiological conditions of fluid flow.
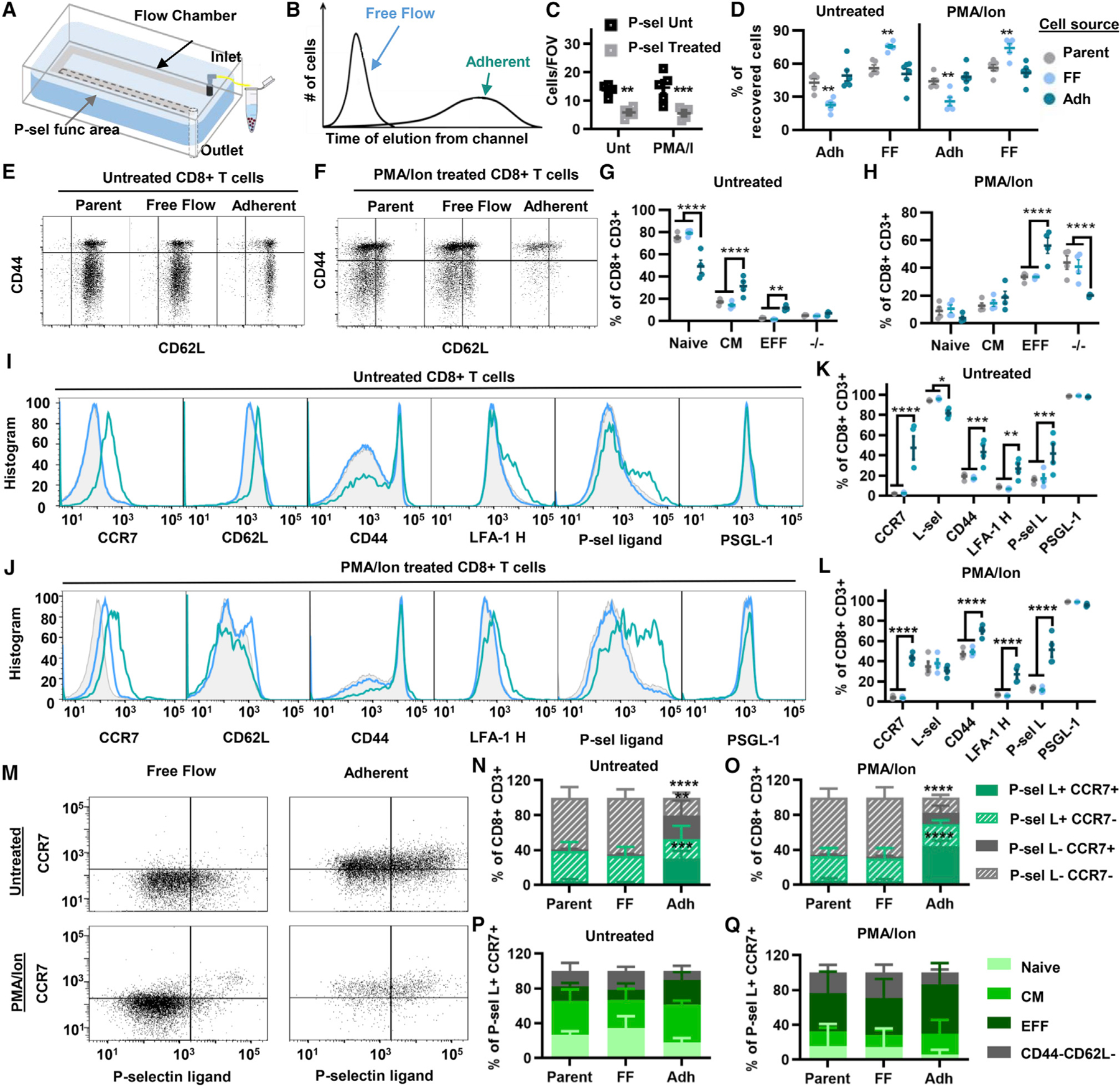
(A) Schematic diagram of hemodynamic microenvironment-mimicking adhesion chromatography microfluidic system.
(B) Schematic diagram of elution times of CD8+ T cells that do (adherent) or do not (free flow) exhibit adhesion to P-selectin.
(C) Number of interacting CD8+ T cells per field of view left untreated or pretreated with P-selectin chimera and perfused through the chromatography channel.
(D) Percentage of recovered untreated or PMA/Ion-treated CD8+ T cells in either the free flow (FF) or the adherent (Adh) fraction of the parent (unsorted) population or reperfused cell subpopulations immediately after perfusion.
(E and F) Gating strategy for fractionated CD8+ T cell subtypes through a P-selectin-functionalized channel. (G and H) Data from (E) and (F), represented as frequency of CD8+ T cells.
(I and J) Representative flow cytometry histograms of different adhesion markers’ expression on parent, FF, and Adh populations. (K and L) Data from (I) and (J), represented as frequency of CD8+ T cells.
(M) Representative flow cytometry plots of CCR7 versus P-selectin ligand expression by free flow or adherent recovered subpopulations from perfused CD8+ T cells left untreated or pretreated with PMA/Ion.
(N and O) Data from (M) represented as frequency of CD8+ T cells.
(P and Q) Frequency of subtypes of P-sel L+CCR7+CD8+ T cells recovered in the different fractions. Points represent results using splenocytes harvested from an individual animal. Data in all panels represent the mean ± SEM. Results represent a minimum of three independently performed experiments. Statistical comparisons performed by two-way ANOVA with Dunnett’s multiple comparisons test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figures S7–S9.
Upon separation of cell suspensions into Adh and FF fractions by perfusion over P-selectin-functionalized channels, fractions enriched from untreated versus PMA/Ion-treated CD8+ T cells were found to exhibit different cellular phenotypes, with cells in the Adh fraction being enriched in CM and EFF cells (Figures 3E–3H). Conversely, levels of naive CD8+ T cells were diminished in the Adh compared with the FF population (Figures 3E–3H). These results match previous flow cytometry analyses of P-selectin ligand−/+ cells, where P-selectin ligand-expressing cells were found to be composed of more CM and EFF subtypes of CD8+ T cells (Figures S3H–S3J).
Evaluation of adhesion molecule expression in the various cell fractions revealed tumor homing molecules such as CD44, LFA-1, and P-selectin ligands to be upregulated in the Adh compared with the FF population of both untreated and PMA/Ion-treated CD8+ T cells (Figures 3I–3L). LN homing receptor C-C motif receptor (CCR) 7 was also enriched in the Adh fractions of cells treated in either manner (Figures 3I–3L). To verify that this signal was not induced by stimulation of CD8+ T cells from P-selectin engagement, expression of CCR7 and other adhesion receptors was evaluated and found not to be upregulated by co-incubation for 1 or 4 h with P-selectin-Fc chimera (Figures S8A and S8B). Furthermore, the effects of mechanical forces on CCR7 expression of cells were tested by perfusing cells through unfunctionalized channels and found to be negligible (Figures S8G and S8H). Together, this suggests that increases in the frequency of CCR7+ cells in the Adh population result from their functional enrichment from the parent population and not a cell signaling response triggered by hemodynamic force resulting from perfusion.
The concurrent versus individual expression of CCR7 and P-selectin ligand among parent and enriched CD8+ T cells of either source (untreated versus PMA/Ion-treated) was evaluated. Within the parent populations of both cell sources, most cells that were P-selectin ligand+ were found to be CCR7 negative (Figures 3M and 3N). However, in the Adh population, the frequency of cells that were double positive for P-selectin ligand and CCR7 was increased (Figures 3M and 3N). Subtype analysis also revealed P-selectin ligand+CCR7+ cells enriched within the different fractionation groups were not of a specific subtype and had no difference across the various fractionation groups (Figures 3P and 3Q). The chromatography channel thus enriches for CD8+ T cells of a more differentiated phenotype that are enriched for co-expression of P-selectin ligand+ and CCR7 from source cells that vary substantially in their initial qualities.
The ex vivo expansion capabilities of CD8+ T cells enriched from a P-selectin-functionalized channel were evaluated by incubating with Dynabeads and IL-2 immediately after perfusion. The cells expanded ~6-fold by day 8 irrespective of fractionation group (Figure S9A), with changes in cell viability resulting from culture that were equivalent between fractionation groups (Figure S9B). Further, CD8+ T cells positive for Ki-67, a proliferation marker,39 were unchanged between different fractionation groups (Figure S14C), as were granzyme-B and PD-1 (Figures S9D and S9E). And despite P-selectin ligand expression being higher preexpansion (day 0) for the Adh fraction compared with other groups, differences were lost with expansion (Figure S14F). Irrespective of fractionation group, cells thus exhibit the same proliferation and differentiation capabilities.
Sorted CD8+ T cell fractions exhibit different in vivo tissue-homing capabilities
The in vivo homing capabilities of cells enriched for adhesion to P-selectin in vitro using the adhesion chromatography system were evaluated by adoptively transferring by intravenous administration FF and Adh fractions of untreated or PMA/Ion-treated CD45.1+CD8+ T cells into B16F10 melanoma-bearing mice (Figure 4A). Sixteen hours post-transfer, cells isolated from various tissues, including the tumor, spleen, TDLNs, and NDLNs, were stained using fluorescent antibodies and flow cytometrically analyzed. Irrespective of pretreatment condition, recovery of total CD45.1 cells in the tumor was highest for animals into which the Adh fractions were transferred compared with animals that received the parent population or FF fraction (Figure 4B). Correspondingly, animals that received the Adh fraction with both cell sources yielded low recoveries in the spleen (Figure 4C). The Adh fraction of untreated CD8+ T cells also had low yields in the NDLNs and TDLNs compared with other groups (Figure 4C). As reflected in the fraction of total CD8+ T cells in each tissue, recipient animals that received donor cells from the Adh fraction exhibited higher frequencies of donor cells in the tumor compared with the parent and FF, irrespective of parent population pretreatment (Figure S10A). In contrast, the frequency of donor cells in lymphoid tissues was lower in mice that received donor cells from Adh fractions compared with other groups, for both untreated and PMA/Ion populations (Figure S10B). When evaluated for their distribution of CD8+ T cell subtypes, donor cells that trafficked to the tumor exhibited no differences in subtype across the fractionation groups, irrespective of activation state (Figure S10C). Donor cells from the Adh fraction recovered from the spleen exhibited a lower frequency of naive cells compared with donors from the FF and parent cells for both pretreatment conditions (Figure S10D). Overall, distributions of CD8+ T cell subtypes appear to be independent of fractionation group in the tumor while dependent on fractionation group in the spleen.
Figure 4. CD8+ T cells recovered using the adhesion chromatography system to enrich for adhesion to P-selectin in flow exhibit enhanced tumor homing compared with cells recovered in the free flow and parent fractions.
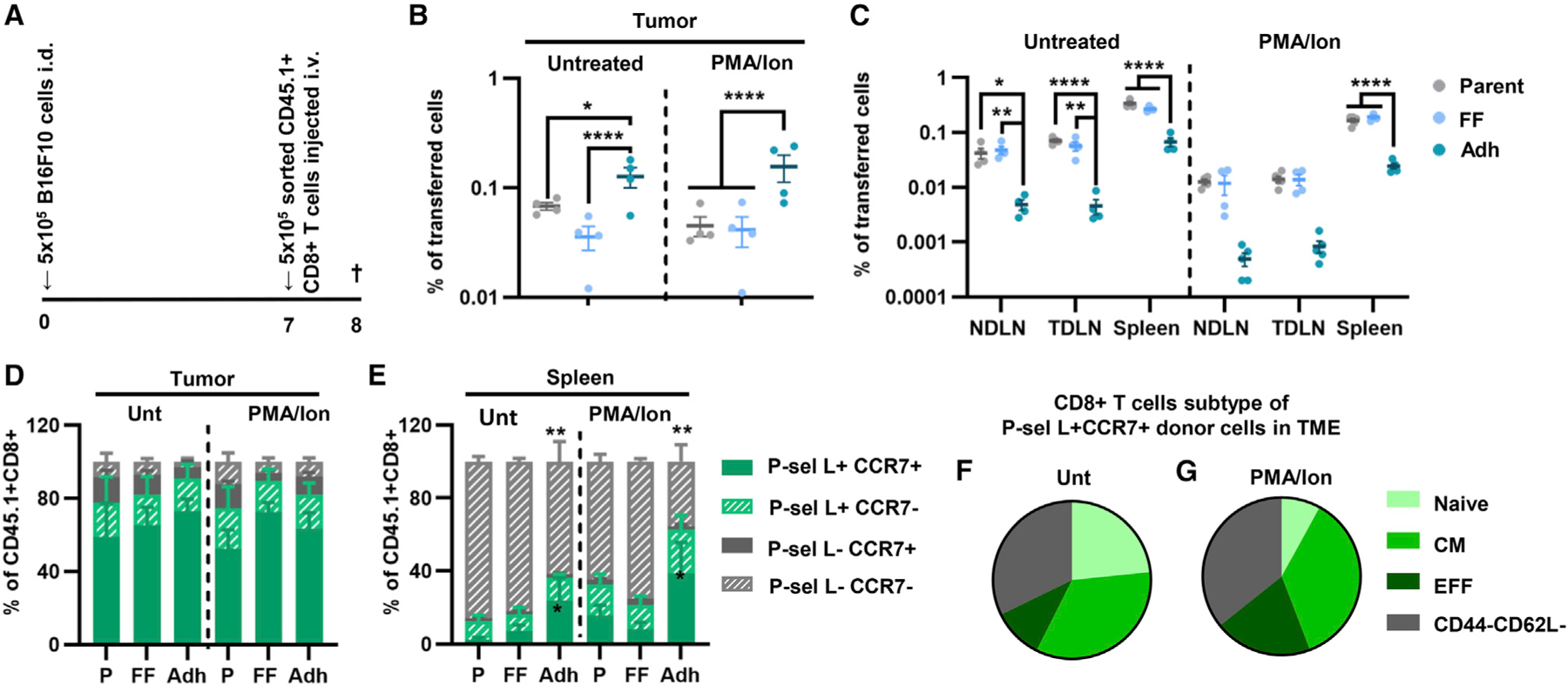
(A) Schematic diagram outlining the experimental design.
(B and C) Percentage of parent untreated and PMA/Ion CD8+ T cells of cells fractionated based on their adhesion to P-selectin in flow recovered in various tissues 16 h after adoptive-transfer into B16F10 melanoma-bearing animals.
(D and E) Frequency of single and co-expression of P-selectin ligand and CCR7 by donor CD8+ T cells recovered in the tumor and the spleen that were left untreated or were PMA/Ion treated.
(F and G) Subtype distribution of donor P-sel L+CCR7+ untreated (F) or PMA/Ion-treated (G) CD8+ T cells recovered from the tumor. Points represent individual results from individual animal. Data in all panels represent the mean ± SEM of three or more independently run experiments; two-way ANOVA with Tukey’s multiple comparisons test; *p < 0.05, **p < 0.01, ****p < 0.0001. See also Figure S10.
When evaluated for their expression of adhesion receptors/ligands, donor cells recovered from the tumor exhibited a high frequency of double positivity for P-selectin ligand and CCR7 expression irrespective of treatment or fractionation group (Figure 4D). This trend was also observed with the P-selectin-functionalized channel where, irrespective of treatment group, the Adh fraction enriched for P-selectin ligand+CCR7+ CD8+ T cells. In contrast, cells recovered from lymphoid tissues varied based on their fractionation group, with the Adh fraction having a higher frequency of trafficked P-selectin ligand+CCR7+ cells and lower P-selectin ligand−CCR7− cells in the spleen and LNs (Figures 4E, S10E, and S10F). Furthermore, the P-selectin ligand+CCR7+ cells that were enriched in the tumor were not composed of a specific subtype (Figures 4F and 4G), suggesting that adhesive quality and not CD8+ T cell subtype might be a better indicator of tumor homing capabilities. These data demonstrate that the in vitro selection for CD8+ T cells that can adhere to P-selectin in physiological shear flow enriches for a subset of cells both with an enhanced tumor homing capability and that mirrors the quality of CD8+ T cells enriched in the tumor compared with lymphoid tissues.
Ex vivo-expanded CD8+ T cells that exhibit higher extents of adhesion to P-selectin in flow in vitro home to greater extents to the TME in vivo
How the capacity to adhere to P-selectin in physiological fluid flow in vitro and home to tumors in vivo varies as CD8+ T cells are expanded was evaluated. Primary mouse CD8+ T cells were incubated with Dynabeads and IL-2 to yield cultures that expanded ~30-fold by day 10 (Figure 5A). Adhesion by CD8+ T cells at various days of expansion was evaluated under static versus continuous flow conditions (Figure S11). Adhesion by untreated CD8+ T cells (day 0) was far less than that of cells at later expansion stages (day 2 and 8) under both static (Figure S11D) and continuous flow conditions (Figure S11H). In addition, cells at day 2 of expansion predominantly mediated rolling adhesion (Figures S11B and S11F), whereas cells at day 8 interacted at high extents via firm adhesion (Figures S11C and S11G). Correspondingly, the frequency of cells expressing P-selectin ligand was low prior to activation and expansion but dramatically increased by days 2 and 8 of expansion (Figure 5B). Using the adhesion chromatography channel, the extent of CD8+ T cell adhesion to a P-selectin-functionalized substrate at a continuous WSS level of 0.5 dyn/cm2 was also found to increase at days 2 and 8 compared with unexpanded (day 0) cells (Figure 5C). Conversely, rolling velocity, which correlates with adhesion extent and cell avisity,32 decreased during expansion (Figure 5C).
Figure 5. Enrichment of P-selectin ligand and CCR7 expression by Dynabead-expanded CD8+ T cells recovered by perfusion through a P-selectin-functionalized channel is correlated with the enrichment of donor cells trafficking to the TME.
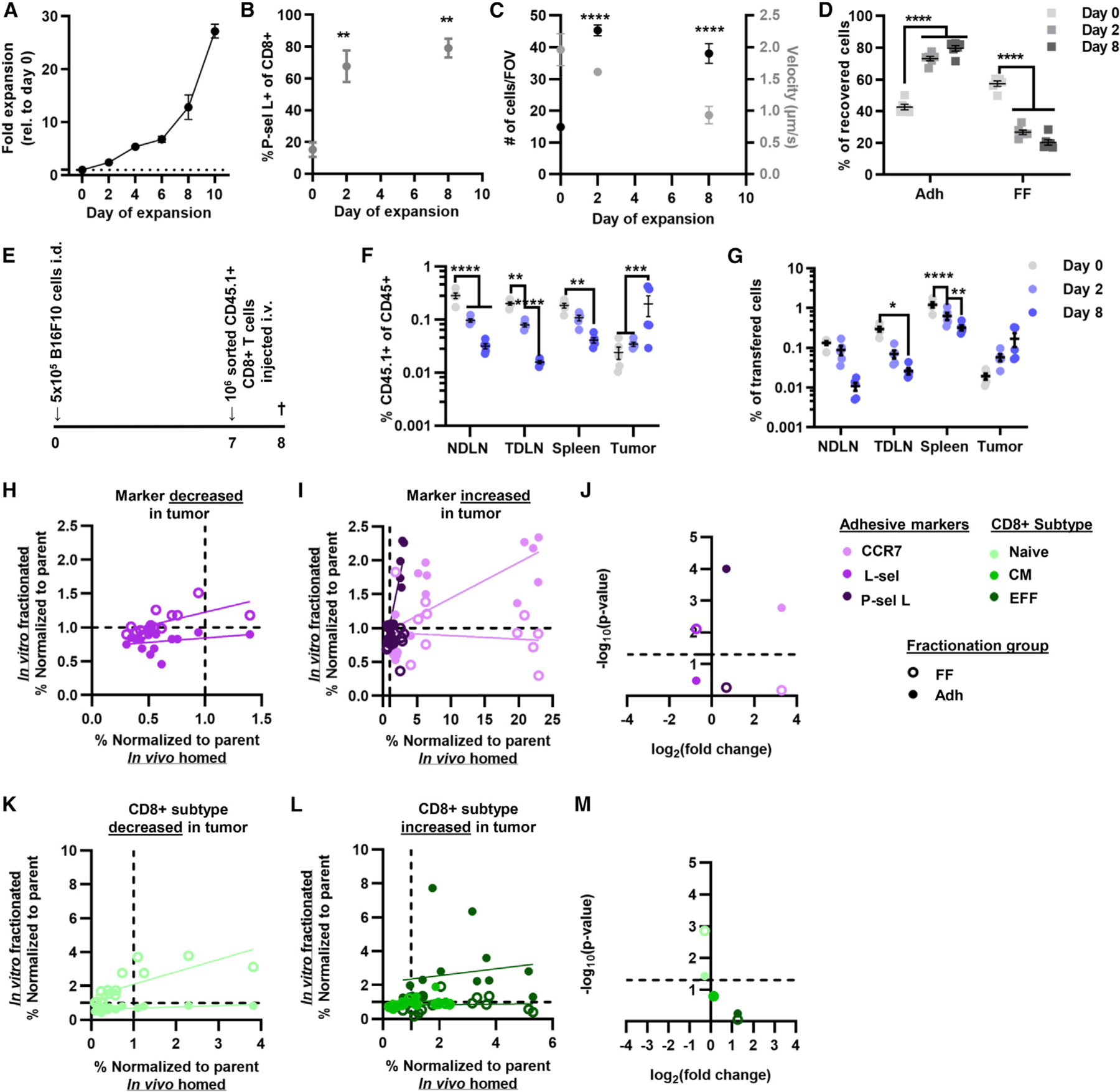
(A) Fold expansion of CD8+ T cells cultured with Dynabeads and IL-2.
(B) Frequency of P-selectin ligand-expressing CD8+ T cells at different days of expansion.
(C) Number and velocity of Dynabead-expanded CD8+ T cells adherent to a P-selectin-functionalized substrate.
(D) Percentage of Dynabead-expanded CD8+ T cells recovered within the Adh and FF fractions by perfusion through the adhesion chromatography system.
(E) Schematic outlining adoptive transfer experimental design.
(F) Frequency of donor CD8+ T cells (CD45.1+CD45.2−) of all CD45+ (CD45.1+CD45.2+) cells into CD45.2 animals bearing B16F10 melanomas.
(G) Frequency of otal CD8+ donor T cells ecovered from analyzed tissues.
(H–M) Correlation analysis of adhesion molecule expression (H and I) and subtype (K and L) enrichment between CD8+ T cells enriched for adhesion to P-selectin in flow and that traffic to the tumors. −log10(p value) versus log2(fold change) of adhesion molecule expression (J) and subtype (M) by CD8+ T cells recovered from the tumor. (F–I, K, and L) Points represent results from one individual animal. (A–D, F, and G) Data represent the mean ± SEM. All data represent results from three or more independently run experiments; one-way ANOVA (B) and two-way ANOVA (C, D, F, and G) with Dunnett’s multiple comparisons test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figures S11–S14.
When CD8+ T cells were fractionated based on adhesion to P-selectin, more CD8+ T cells were recovered in the Adh fractions at days 2 and 8 of expansion relative to unexpanded cells (Figure 5D). Cells recovered in the Adh fractions also expressed higher levels of P-selectin ligand compared with both other groups (Figure S12A). However, after expansion, the fraction of cells recovered in the Adh population that expressed P-selectin ligand was the same as that of the parent population but exceeded that of the FF fraction (Figure S12A). Adh cells at day 2 also expressed higher CCR7 (Figure S12B), and L-selectin was expressed at lower levels compared with FF and parent throughout expansion (Figure S12C). Irrespective of expansion stage, the Adh fractions contained a lower proportion of naive CD8+ T cells compared with the FF fraction and parent population, save at day 8 (Figure S12D). Adh fractions exhibited no distinct differences in the proportion of CM cells (Figure S12E). However, EFF CD8+ T cells were enriched in Adh versus FF fraction from populations that had been expanded, but not relative to the parent population of day 8 cultures (Figure S12F).
The homing of unexpanded and day 2- and 8-expanded CD45.1+CD8+ T cells adoptively transferred into B16F10 melanoma-bearing mice was next evaluated 16 h post-intravenous (i.v.) injection (Figure 5E). With increasing day of expansion, trafficking to lymphoid tissues decreased, as reflected by the lower frequency of donor cells of total leukocytes (CD45+) in these tissues (Figure 5F). Contrastingly, day 8-expanded CD8+ T cells accumulated to the greatest extent in the tumor (Figure 5F). These differences in total infiltration also altered the recovery of CD8+ T cells in various tissues (Figure S13A). A similar trend was seen with respect to the fraction of transferred cells, where at later days of expansion there was decreased accumulation in lymphoid tissues and increase accumulation in the tumor (Figure 5G). Viability of the donor CD8+ T cells was decreased in TME compared with lymphoid tissues, irrespective of the day of expansion (Figure S13B), as a result of the TME being immunosuppressive and rendering T cells functionally impaired.40–42
To evaluate whether there are any adhesive markers or CD8+ T cell subtypes enriched in lymphoid tissues or the TME throughout expansion, the fold-change expression from pre-transfer levels of different markers and subtype was analyzed. The recovered cells in the TME were enriched for CCR7+ cells compared with untransferred on each respective expansion day, except day 8 (Figure S13C). Of recovered cells, the proportion that were L-selectin+ in the TDLNs and spleen was increased relative to the parent population in day 8 cultures (Figure S13D). Likewise, the proportion of cells expressing L-selectin recovered from tumors was diminished compared with their proportion in the donor cell population pretransfer (Figure S13D). Similarly, the proportion of cells recovered from LNs that express P-selectin ligand was decreased in LNs irrespective of expansion day (Figure S13E). In the adoptive transfer model, enrichment of P-selectin ligand-expressing cells in the TME was seen only in unexpanded cells, due to the fact that day 2- and day 8-expanded CD8+ T cells had higher levels of P-selectin ligand expression prior to transfer, resulting in the fold change relative to parent being low (Figure S13E). Correlation analysis was performed to compare the enrichment of CD8+ T cells expressing adhesion markers that distribute in vivo to particular tissues versus those that are collected in the sorted cell fractions (Figures 5H, 5I, S14A, S14B, and S14G–S14J). In the tumor, increased expression of CCR7 and P-selectin ligand correlated with the fold change in expression of these markers in the Adh fraction but not the FF fraction (Figure 5J). On the other hand, expression of L-selectin that is diminished in donor CD8+ T cells recovered from the TME correlated with the fold change in L-selectin expression by cells recovered from the FF fraction (Figure 5J). The fold change in L-selectin expression by donor CD8+ T cells in lymphoid tissues was similarly correlated with the fold change in L-selectin expression by CD8+ T cells recovered from FF fractions (Figures S14C, S14O, and S14P). The fold change in CCR7 expression in donor CD8+ T cells recovered from lymphoid tissues also correlated with the fold change in CCR7 expression by CD8+ T cells in the Adh fraction (Figures S14C, S14O, and S14P). Likewise, the fold change in P-selectin expression by donor cells recovered from the TDLNs and spleen correlated with the fold change in cells recovered in the Adh fraction (Figures S14C and S14P). This suggests that enrichment of cells expressing P-selectin ligand and CCR7 in the chromatography channel was predictive of enrichment in the TME of cells expressing these adhesion receptors, irrespective of expansion time.
When analyzed with respect to differentiation state, naive CD8+ T cells were enriched in LNs upon transfer of cells expanded until day 8 (Figure S13F). In contrast, CM cells were enriched in the tumor only when transferred without expansion (Figure S13G). On the other hand, EFF cells were enriched in the tumor at earlier days of expansion (day 0 and 2) (Figure S13H). Correlation analysis of CD8+ T cell subtype within cultures of various dates of expansion that were found to be enriched in the in vitro fractionated cells versus those that were recovered from various tissues after adoptive transfer in vivo was performed (Figures 5K, 5L, S14D, S14E, and S14K–S14N) to determine if the chromatography channel predicted subtype enrichment in the TME. The diminishments in naive CD8+ T cells in the tumor correlated with the fold change in naive CD8+ T cells in both the FF and the Adh fractions (Figure 5M). Naive CD8+ T cell enrichment in the LNs also correlated with the fold change in naive CD8+ T cells in both Adh and FF fractions (Figures S14F, S14Q, and S14R). EFF CD8+ T cells were correlated with the fold change of EFF cells in the FF but not the Adh fraction (Figures S14F, S14Q, and S14R). Therefore, the fold change relative to parent in naive and, to some extent, EFF CD8+ T cells in the FF and Adh fractions predicts their fold change in the TME and lymphoid tissues. Overall, the enrichment of various sub-types of CD8+ T cells and the in vivo homing behavior of adoptively transferred CD8+ T cells at various states of expansion are predicted by their extent of enrichment for P-selectin adhesion in flow using the chromatography channel.
Tumor-specific CD8+ T cells enriched for adhesion to P-selectin in flow home to and remodel the TME and augment the efficacy of ACT with immune checkpoint blockade
The homing and engraftment of adoptively transferred CD8+ T cells that are tumor specific were evaluated. OT-I CD8+ T cells (CD45.2+) were enriched for adhesion to P-selectin using the adhesion chromatography system, and the FF and Adh fractions were collected and immediately transferred intravenously into B16F10-OVA melanoma-bearing mice (CD45.1) (Figure 6A). At 16 and 64 h post-transfer, cells were isolated from the tumor, spleen, NDLNs, and TDLNs; stimulated in vitro with SIINFEKL for 6 h; antibody stained; and flow-cytometrically analyzed. Consistent with results from analogous experiments using polyclonal CD8+ T cells from wild-type (WT) CD45.1 mice, OT-I CD8+ T cells from the Adh fraction exhibited higher tumor trafficking compared with parent and FF fractions, reflected as the fraction of all CD8+ T cells and the fraction of the total number of transferred cells (Figures 6B and 6C). Trafficking to secondary lymphoid tissues, on the other hand, was lower for cells in the Adh fraction compared with other groups (Figures S15A and S15B). Viability of donor cells in the tumor was the same between fractionation groups at both 16 and 64 h post-transfer (Figure 6D). Similarly, viability of donor cells in lymphoid tissues was the same between fractionation groups at 16 h, but in the spleen at 64 h, FF cells had higher viability compared with other fractions (Figure S15C). At this initial analysis time point, all donor cell groups exhibited the same frequencies of granzyme-B, IFN-γ, TNF-α, and Ki-67 expression in the tumor and lymphoid tissues (Figures 6E, 6F, S15D, and S15E), demonstrating that, despite differences in the extent of total homing to various tissues, donor CD8+ T cells that trafficked to the tumor have the same initial cytotoxic and proliferative capabilities. However, by 64 h post-transfer, TME-infiltrating donor cells from the Adh fraction exhibit increased frequencies of granzyme-B positivity (Figure 6E). In the TDLNs (Figure 6F) but not the NDLNs and spleen (Figures S15D and S15E) at 64 h, FF donor cells exhibited lower Ki-67 expression compared with Adh donor cells, demonstrating that Adh cells proliferate to a greater extent in the TDLN than FF cells. Furthermore, Ki-67 expression of donor cells decreased in the tumor and increased in the TDLNs, indicating this to be a site of donor cell proliferation (Figures 6E and 6F).
Figure 6. CD8+ T cells enriched for adhesion to P-selectin in physiological flow improve tumor control by ACT in combination with immune checkpoint blockade (ICB) aPD-1.
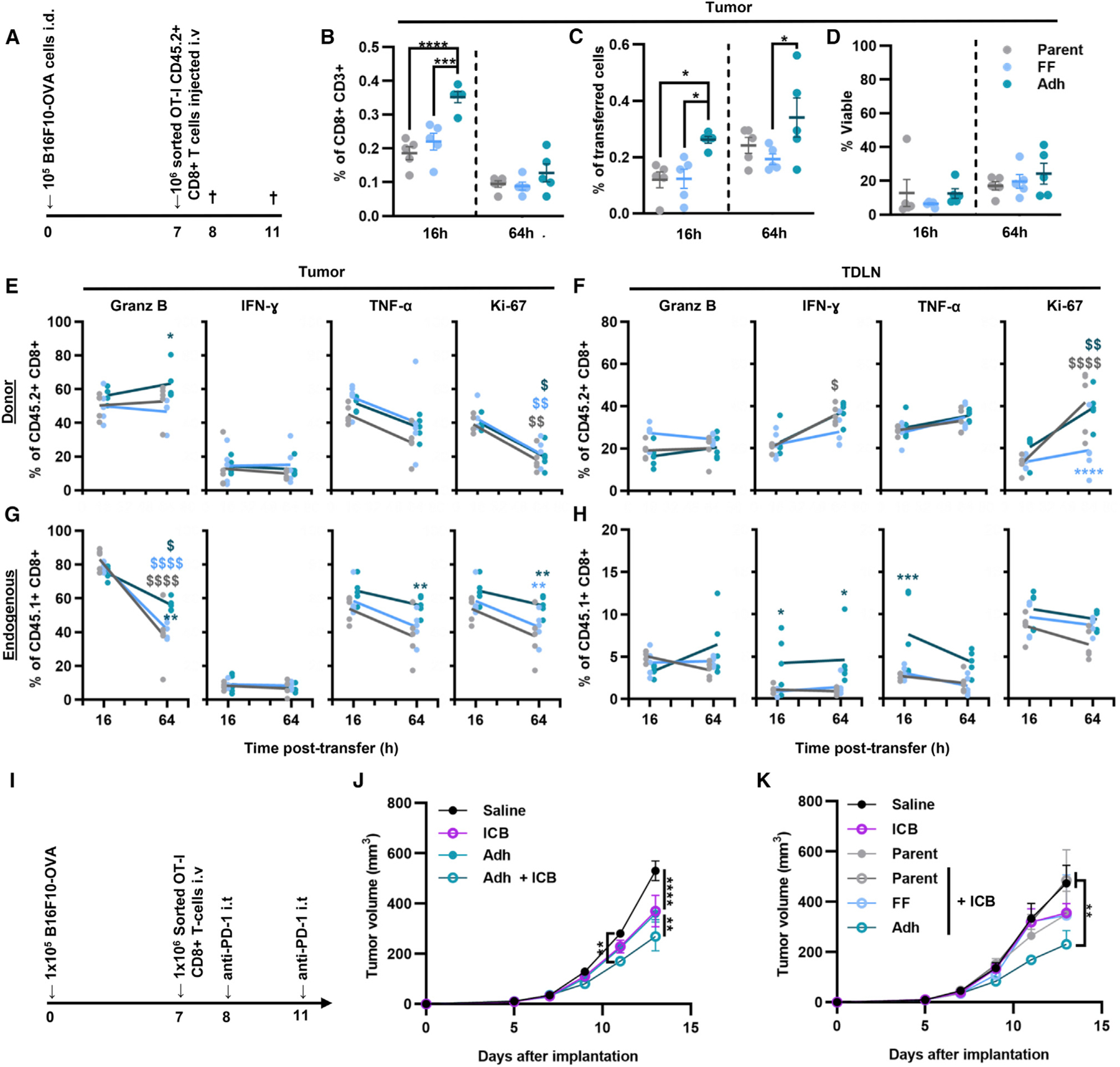
(A) Schematic outlining the experimental design.
(B) Frequency of donor CD8+ T cells in all CD8+ T cells recovered in the tumor.
(C) Donor CD8+ T cells recovered in the tumor as the frequency of the total number of cells transferred into the B16F10-OVA tumor-bearing mice.
(D) Viability of donor CD8+ T cells in the tumor.
(E–H) Percentage of cytokine-producing or Ki-67+ cells of donor CD8+ T cells in the tumor (E) or TDLNs (F). Percentage of cytokine-producing or Ki-67+ cells of endogenous CD8+ T cells in the tumor (G) or TDLNs (H).
(I) cSchematic outlining the experimental design.
(J) B16F10-OVA tumor growth after treatment with aPD-1 alone or adoptive transfer (AT) of Adh OT-1 CD8+ T cells with and without aPD-1.
(K) B16F10-OVA tumor growth after therapy with aPD-1 alone or in combination AT of parent or sorted OT-1 CD8+ T cells (FF and Adh). All graphs represent mean ± SEM (n = 5); statistics were performed by two-way ANOVA with Tukey’s multiple comparisons test. (E–H) * indicates significance of comparison to parent population, $ indicates significance of comparison between time points; *p < 0.05, **p < 0.01. ***p < 0.001, ****p < 0.0001. See also Figure S15.
When endogenous CD8+ T cells were evaluated, no differences in activation marker expression by CD8+ T cells infiltrating the TME were observed between mice that received different donor cells at 16 h (Figure 6G). Endogenous CD8+ T cells recovered from the TDLNs of mice into which donor CD8+ T cells were transferred from Adh fractions, however, exhibited a higher frequency of IFN-γ and TNF-α production, an effect that was sustained for IFN-γ at 64 h (Figure 6H). Given the TDLNs’ role in facilitating priming of lymphocytes in response to tumor-derived antigen,43–46 this is suggestive of an anti-tumor CD8+ T cell response being locally elicited. Consistent with this, despite no differences at the initial 16 h time point, by 64 h post-transfer, frequencies of host CD8+ T cells producing granzyme-B and TNF-α infiltrating the tumor were increased for animals that received donor cells from Adh fractions (Figure 6G). However, the overall frequency of granzyme-B-producing cells decreased between 16 and 64 h (Figure 6G). Increased homing to tumors by tumor-specific donor CD8+ T cells enabled by enrichment for adhesion to P-selectin in flow drives faster initial tumor killing. Moreover, this homing drives further priming and expansion within TDLNs to facilitate anti-tumor CD8+ T cell immunity.
The effects of tumor homing differences on the therapeutic potency of donor tumor-specific CD8+ T cells were evaluated by intravenously infusing OT-I CD8+ T cells fractionated in the adhesion chromatography system into B16F10-OVA tumor-bearing animals. The benefit of ACT in combination with blockade of PD-1, whose efficacy in melanoma is highly correlated with CD8+ T cell tumor infiltration,47,48 was evaluated (Figure 6I) with or without concurrent anti-PD-1 (aPD-1) treatment, with effects compared with aPD-1 alone or saline control. The combination therapy slowed tumor growth by day 11, and by day 13 both aPD-1 and ACT using Adh donor cells slowed tumor growth as a monotherapy, albeit at more modest extents compared with the two used in combination (Figure 6J). When donor cell quality synergies with aPD-1 were evaluated, however, ACT with parent or FF donor cells did not improve the effects of aPD-1 (Figure 6K). Therefore, cells enriched for their in vitro adhesion to P-selectin in flow with increased tumor homing in vivo improve the therapeutic synergies of ACT with aPD-1.
Enrichment of ex vivo-expanded human CD8+ T cells for in vitro adhesion to P-selectin in flow
The relevance of the adhesion chromatography system to the analysis of human CD8+ T cells expanded using a clinically relevant expansion protocol was evaluated. Human CD8+ T cells were isolated from healthy donor peripheral blood mononuclear cells and incubated with Dynabeads and IL-2. Analysis of expanded cells (Figure 7A) sorted through a P-selectin-functionalized channel revealed that CD8+ T cell recovery within Adh fractions increased upon expansion (Figure 7B). Adh fractions were also enriched for cells with P-selectin ligand expression, although the extent of enrichment was dependent on expansion day (Figures 7C–7E). Although no difference in CCR7 expression between sorted samples was found (Figures S16A and S16D), in contrast to results found with murine CD8+ T cells, L-selectin expression was higher in cells recovered in FF cells on days 5 and 7 of expansion (Figure S16E), in agreement with results from murine studies. Sialyl Lewis a/x (sLea/x), a tetrasaccharide carbohydrate P-selectin ligand that plays an important role in selectin-mediated adhesion in flow,35 was measured on fractionated groups and was found to be highly expressed by cells recovered in the Adh versus FF fraction (Figure S16F). Overall, human CD8+ T cells fractionated for their capacity to interact with P-selectin in flow appear enriched for high expression of tumor homing molecules such as P-selectin ligand and sLea/x, while CD8+ T cells that do not interact with P-selectin are more frequently L-selectin positive. The adhesion chromatography system is thus amenable for analysis of human biospecimens and, in line with results with a preclinical mouse tumor immunotherapy model, enriches for cells with high expression of tumor homing ligands.
Figure 7. Human CD8+ T cells exhibit differential adhesion to P-selectin in physiological levels of fluid flow during expansion.
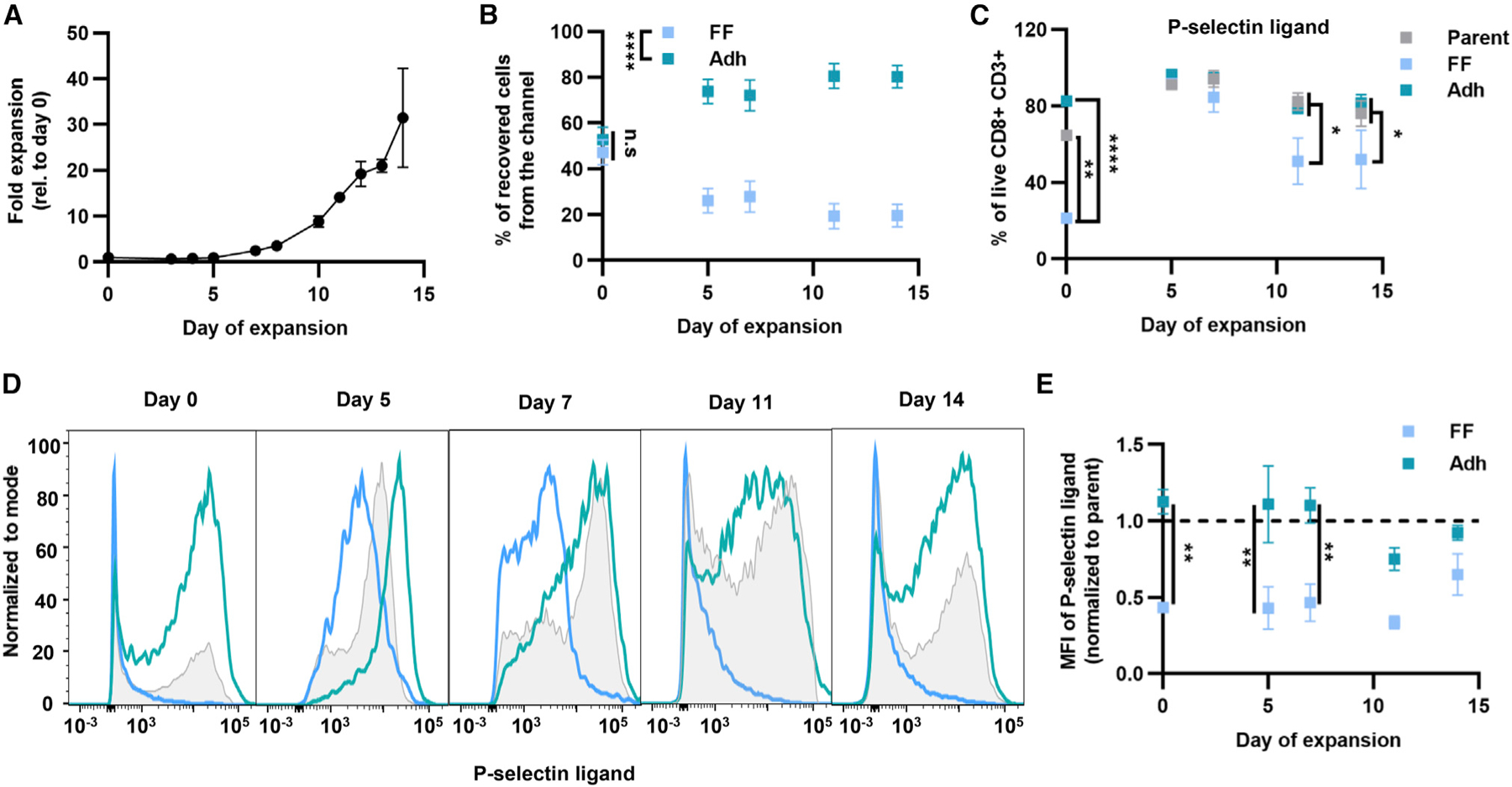
(A) Expansion of human CD8+ T cells from healthy donors cultured with Dynabeads and IL-2.
(B) Percentage of CD8+ T cells recovered in Adh and FF fractions using P-selectin-functionalized channel.
(C) Frequency of P-selectin ligand+ cells of live CD8+ T cells of parent populations or those recovered in FF or Adh fractions after perfusion through a P-selectin-functionalized channel at different days of expansion.
(D) Histograms of P-selectin ligand expression at different days of expansion of human CD8+ T cells.
(E) Mean fluorescence intensity of P-selectin ligand+ cells normalized to parent cell mean fluorescence. For all graphs the data represent the mean ± SEM of three or more independently run experiments; two-way ANOVA with Bonferroni’s multiple comparisons test; *p < 0.05, **p < 0.01, ****p < 0.0001. See also Figure S16.
DISCUSSION
ACT has emerged as a promising therapy for metastatic melanoma, but this treatment has low rates of response due in part to poor cell trafficking to diseased tissues.1,2 Understanding the mechanisms underlying CD8+ T cell infiltration to the TME holds promise for improving the clinical outcomes of ACT. In this study, an engineered microfluidic device was implemented to characterize what adhesion and chemokine receptors, as well as differentiation states, are associated with enhanced adhesion by CD8+ T cells to P-selectin in physiological flow. Biodistribution analysis of adoptively transferred cells into a preclinical B16F10 melanoma tumor model revealed the predictive benefit of cell adhesion to P-selectin in in vitro and in vivo tumor homing, which led to superior therapeutic effects in potentiating combination immunotherapy with aPD-1.
Preferential trafficking of P-selectin ligand+ CD8+ T cells to tumors seen herein is consistent with previous studies demonstrating that the expression of P-selectin and its ligands plays an important role in CD8+ T cell homing to tumors and in disease progression.21,26 Results from in vitro flow-based perfusion experiments suggest that physiological force from fluid flow influences the mechanisms of CD8+ T cell adhesion to endothelial-expressed adhesive ligands. Furthermore, P-selectin ligand expression correlates with the extent of cell adhesion under conditions under which adhesion is initiated in the absence but not in the presence of flow. This is consistent with the potential for a greater number of low-affinity receptor-ligand interactions to occur under static conditions,49 which presumably are not able to form under continuously applied shear stress. Therefore, continuous perfusion could be considered a more physiologically relevant method to investigate mechanisms underlying CD8+ T cell adhesion in vivo relevant to cellular homing during ACT.
A matter of controversy in the ACT field has been which differentiation state of CD8+ T cells contributes the most to immediate tumor killing versus long-term control of tumor growth.1,3 Numerous studies have shown that less differentiated CD8+ T cells, such naive, stem-cell memory (SCM), and CM CD8+ T cells, have enhanced anti-tumor activity compared with effector memory (EM) and EFF CD8+ T cells.3,14,50–52 This inverse relationship between T cell differentiation and treatment efficacy is believed to result from less differentiated cells having enhanced self-renewal and multipotent capabilities. However, less is known about the differences in cell adhesion and homing behaviors between CD8+ T cell subtypes and how this controls their trafficking and resulting functions. What subtypes of CD8+ T cells have enhanced cell adhesion to a tumor-like substrate was assessed here using an adhesion chromatography system that exposed cells to endothelial-presented adhesion receptors under physiological levels of shear flow. This approach offers an advantage over in vivo analysis methods in which the influences of cytokine stimulation or antigen presentation within the TME can lead to CD8+ T cell activation and differentiation, confounding the effects of adhesion and migration processes alone. This in vitro method instead allows the assay of CD8+ T cell adhesion to be done in a controlled manner, explored here in the context of engagement to P-selectin under the influence of physiological levels of fluid flow. For non-activated CD8+ T cells, an enrichment of CM and EFF CD8+ T cells was found in Adh fractions, whereas only EFF cells are enriched in PMA/Ion-treated CD8+ T cells. Biodistribution analysis of the channel-sorted CD8+ T cells revealed that cells recovered in the Adh fraction trafficked better to the tumor and that the cells in the tumor exhibited higher proportions of cells that expressed both P-selectin ligand and CCR7. These results support the interpretation that the in vitro chromatography channel can be used to predict homing and engraftment behavior of CD8+ T cells in vivo.
When expanding murine CD8+ T cells ex vivo, P-selectin ligand expression was found to increase throughout expansion, leading to an increase in the number of CD8+ T cells mediating adhesion to P-selectin in flow and reducing their velocities of rolling adhesion. Using a P-selectin-functionalized channel to fractionate CD8+ T cells at different days of expansion revealed that P-selectin ligand+ cells were only enriched prior to expansion, signifying that sorted cells would have the greatest therapeutic benefit when starting from less differentiated cell sources. Less differentiated CD8+ T cells that are known to exhibit very potent survival and self-renewal capabilities,53 however, tend to traffic more to LNs rather than tumors.19,54 Using this system to sort less differentiated CD8+ T cell subtypes with greater tumor rather than LN homing could improve their therapeutic effects, an inter-pretation supported by tumor therapy experiments in combination with aPD-1.
When expanded cells were transferred into B16F10 melanoma-bearing mice, later days of expansion resulted in reduced LN homing, with subtle increases in tumor homing, results that closely match the in vitro data where later days of expansion resulted in a reduction of recovered cells in the FF fraction and increased recovery in the Adh fraction. Furthermore, the enrichment of P-selectin ligand and CCR7 expression by cells in the Adh fraction correlated with the enrichment of cells expressing these markers in the tumor. These studies suggest that the chromatography system can be employed with ex vivo expansion protocols to predict homing of CD8+ T cells throughout expansion.
Immune checkpoint blockade is most effective in patients with tumors that are highly infiltrated by CD8+ T cells.47,48 Blockade of PD-1 can restore the activation and cytotoxic capabilities of T cells to result in tumor control.55,56 Many studies have demonstrated that aPD-1 treatment improves the potency of ACT with a high dose of transferred T cells.57–61 ACT comprising cells enriched for adhesion to P-selectin in flow improved the effects of aPD-1 to reduce tumor growth in the B16F10 tumor model. This could be interpreted as ACT with cells with greater adhesion propensity transforming scarcely infiltrated, immunologically “cold” B16F10 tumors into “warm” tumors, such that aPD-1 anti-tumor effects are enhanced, thereby leading to improved therapeutic efficacy.
In conclusion, the capacity of CD8+ T cells to home and engraft within tumors versus lymphoid tissues can be modeled ex vivo using an engineered microfluidic device that recapitulates the hemodynamic microenvironment of the vasculature. Adhesion-based sorting of CD8+ T cells prior to transfer increases tumor homing and improves the therapeutic effects of ACT. Knowing what sub-population of CD8+ T cells homes better to the tumor, as well as determining the minimal timeline to produce cells enriched for this homing behavior, can enable dose sparing for ACT, thus minimizing undesirable side effects. An advantage of this method is its amenability not only to preclinical studies but also to investigations using human biospecimens. This approach, therefore, holds promise in improving the delivery limitations of ACT to increase treatment safety and patient response rates.
Limitations of the study
The main limitation is the in vitro adhesion chromatography system not fully recapitulating all aspects of the microenvironment of the tumor vasculature. Features of the in vivo environment not modeled by the engineered microfluidic system include non-uniform distributions of adhesive ligands presented throughout the vessel,62 expression of multiple adhesion ligands by vessel-lining cells,29,33,63–65 the presence of endothelial cell-presented chemokines,66 variable WSS levels among different tumor vessels,67 pulsatile flow,68,69 and shape of the vessel cross section. On the other hand, the exact cause of CD8+ T cell homing to the TME and its regulation by biophysical force that results from fluid flow cannot be fully interrogated using an in vivo model in isolation. Correlation analyses between in vitro adhesion and in vivo homing motivate future studies using knockout animal models. And although the in vivo studies presented herein used one melanoma model, establishing how adhesion to P-selectin contributes to homing of CD8+ T cells to other melanomas or tumor types where P-selectin has been shown to be expressed, such as ovarian, lung, and breast cancer,28 would broaden the applicability of the findings.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Susan Thomas (susan.thomas@gatech.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
B16F10 and B16F10-OVA cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, and 1% penicillin/streptomycin/amphotericin B. Cells were passaged at ~80% confluence, and maintained at 37°C with 5% CO2 in a standard incubator.
Animal tumor models
C57Bl/6 or B6 CD45.1 female mice were purchased at six weeks of age from the Jackson Laboratory. All protocols were approved by the Institutional Animal Care and Use Committee (IACUC). For tumor-bearing cohorts, 0.1–0.5 × 106 melanoma cells were implanted intradermally in 6- to 8-week-old mice. To evaluate therapeutic effects, tumor size was measured with calipers in three dimensions and reported as an ellipsoidal volume.
METHOD DETAILS
Study design
This study’s objective was to implement a microfluidic system to predict the in vivo homing by CD8+ T-cells by evaluating their adhesion in vitro in a tumor vasculature-like microenvironment that incorporates the effects of hemodynamic flow. Adhesion molecule expression by adherent and non-adherent cells as well as endogenous and adoptively transferred cells that home to the tumor versus lymphoid tissues including the spleen and lymph nodes was evaluated. Cells enriched for their capacity to mediate adhesion in vitro were adoptively transferred into tumor-bearing murine hosts and the resulting cellular biodistribution profiles were compared. The therapeutic effects of adherent versus non-adherent cells as ACT in a tumor immunotherapy model in combination with aPD-1 were evaluated. Sample sizes were chosen based on previously published studies. For animal studies, mice were randomized into various groups before treatment, with one cage having one mouse per group. The endpoint for survival studies was set at the humane endpoint (tumors reaching 1.5 cm in any direction or ulcerating tumors). Experiments were not done in a blinded fashion.
Immunohistochemistry and imaging
B16F10 tumors 7d post intradermal implantation in the dorsal skin with 0.5 × 106 B16F10 cells were surgically excised, embedded in optimum cutting temperature embedding medium, and stored at −80°C. A cryostat was used to slice 8 μm thick tissue sections that were mounted onto histological slides and stored at −20°C. Sections were 2% PFA fixed for 20 min at room temperature, blocked with 10% donkey serum diluted in Dulbecco’s Phosphate Buffered Saline (D-PBS) with calcium and magnesium for 1h at room tempertature, and incubated overnight at 4°C with the following primary antibody: goat anti-mouse CD62P (1:13, R&D Systems, AF737), and rat anti-mouse CD3 (1:50, Invitrogen) or rat anti-mouse CD31 (1:50, Invitrogen). The following day, the slides were incubated for 1h at room temperature with the following secondary antibody: donkey anti-goat Alexa Flour 555 (1:200, Invitrogen) and donkey anti-rat Alexa Flour 647 (1:1000, Invitrogen). In between each staining step, slides were washed three times with gentle agitation in 0.1% Tween 20 diluted in D-PBS with calcium and magnesium. Washed slides were mounted using Vectashied mounting medium with DAPI and microscopic images were taken using a Zeiss AxioObserver Z1 fluorescent microscope with a 10x magnification objective.
Murine CD8+ T cell isolation
C57Bl/6, B6 CD45.1, or OT-I (purchased from Charles River Laboratories and bred in-house) animals were euthanized, and the spleens were harvested and disrupted with 18G needles followed by washing with B-PBS. Cells were passed through a sterile 70-μm cell strainer, washed, and incubated with red blood cell lysing buffer (Sigma-Aldrich) for 5 min at room temperature, quenched with D-PBS, washed, and resuspended for counting. Cells were resuspended at 108 cells/ml buffer (Biolegend, MojoSort Buffer), and then incubated with a biotin-antibody cocktail for 15 min, followed by streptavidin nanobeads for another 15 min (Biolegend, MojoSort Mouse CD8a Selection Kit). Buffer was added to the mixture and placed in a magnet (STEMCELL Technologies), and the supernatant was collected. Cells were then counted and resuspended in either cell media for expansion or activation experiments, saline for adoptive transfer experiments, or 0.1% BSA for perfusion experiments. Cells were maintained in sterile conditions before adoptive transfer. Pre-transfer, purity, viability, and CD8+ T-cell subtypes were confirmed via flow cytometry on a customized BD LSRFortessa flow cytometer.
Human CD8+ T cell isolation
Human peripheral blood (buffy coats) from de-identified healthy donors were purchased (Oklahoma Blood Institute). Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood via centrifugation with Lymphocyte Separation Media (Corning, 25–072-CV). Following PBMC isolation, CD8+ T-cells were isolated using a negative isolation kit from Stem Cell (17953). Cells were cryoed down in HI-FBS with 10% DMSO until further use.
CD8+ T cell activation with PMA/Ion
Isolated CD8+ T-cells were suspended in either cell media (RPMI 1640 with 10% fetal bovine) or cell media supplemented with 20 ng/mL PMA (Sigma-Aldrich) plus 1 μg/ml Ionomycin (Invitrogen, ThermoFisher). Cells were incubated for 4 h at 37°C with 5% CO2 in a standard incubator.
CD8+ T cell expansion
Isolated CD8+ T-cells were suspended at a concentration of 106 cells/mL in culture medium (RPMI 1640-containing L-glutamine, 1% penicillin/streptomycin/amphotericin B, 1% HEPES, 1% non-essential amino acids, 1% sodium pyruvate, 0.05mM 2-mercaptoethanol, and 10% fetal bovine serum). Cells were mixed with Dynabeads (Gibco, ThermoFisher) at a bead-to-cell ratio of 1:1 and 100 U/ml rIL-2 (R&D Systems). Cells were incubated at 37°C with 5% CO2 in a standard incubator. Cells were examined daily, and after day 3, cells were split daily. Beads were removed on day 3 (for human cells) or day 5 (for murine cells), and then maintain in media with 100 U/ml rIL-2.
Adoptive transfer
After activation or expansion, cells were suspended at a concentration of 108 in buffer (D-PBS plus 1mM CaCl2) and mixed with dead cell removal (Annexin V) cocktail, biotin selection cocktail, and RapidSpheres (STEMCELL Technologies). Buffer was added to the mixture and placed in a magnet; the supernatant was collected. Cells were then counted and resuspended at the desired concentration. CD8+ T-cells were suspended in sterile saline at a concentration of 106 cells per 200 μL of sterile saline. After mice were anesthetized, the hair over the neck of mice was removed using depilatory cream and cleaned using warm water, then suspended cells were injected intravenously via the jugular vein.
In vivo biodistribution analysis
At 16 h post-adoptive transfer, mice were euthanized, and tumor-draining lymph nodes, non-draining lymph nodes, spleens, and tumors were collected. Cells were analyzed via flow cytometry, and transferred cells were identified by staining for CD45.1 (donor mice) and CD45.2 (host mice). The percent recovered cells was defined as the number of cells in a given tissue divided by the known number of cells injected into the animal.
Treatment of B16F10-OVA melanoma-bearing mice
B16F10-OVA cells (0:1 × 106) were implanted intradermally on day 0. After 7 days mice were treated with 1 million OT-I CD8+ T-cells (sorted on chromatography channel or unsorted) i.v. On day 8 and 11, mice were i.t injected with 150 μ g of anti-mouse PD-1 (clone RMP1–14; BioXCell) in 30 μ l of saline.
Flow cytometry
Harvested LNs were incubated with 1 mg/mL of collagenase D (Sigma-Aldrich) in D-PBS with calcium and magnesium for 1 h at 37°C, passed through a 70-μm cell strainer, washed, and resuspended in a 96-well plate for staining. Spleens were disrupted using 18G needles, passed through a 70-μm strainer, washed and incubated with red blood cell lysing buffer (Sigma-Aldrich) for 5 min at room temperature, diluted with D-PBS, washed, and resuspended. Tumors were incubated with 1 mg/mL of collagenase D (Sigma-Aldrich) in D-PBS with calcium and magnesium for 4 h at 37°C, passed through a 70-μm cell strainer, washed, and resuspended. All antibodies for flow cytometry were from Biolegend unless otherwise stated. Cells were blocked with anti-mouse CD16/CD32 (clone, 2.4G2) (Tonbo Biosciences) for 5 min on ice, washed, and stained with fixable viability dye Zombie Aqua for 30 min at room temperature, and then washed. Cells were then incubated with 10 μg/ml P-selectin plus 10 μg/ml of FITC anti-IgG (Fc specific) diluted in D-PBS for 30 min on iced and then washed. Antibody cocktails were prepared in flow cytometry buffer (0.1% bovine serum albumin in D-PBS) following manufacturer concentration or preliminary titrations. Cells were incubated with an antibody cocktail for 30 min on ice and then washed. Cells were fixed with 4% paraformaldehyde (VWR International Inc). For cytokine staining, cells were suspended in IC Fixation Buffer (eBioscience, Thermo Fisher Scientific Inc.) for 60 min on ice in the dark. Cells were then incubated with the antibody cocktail in IC Permeabilization buffer for 60 min at room temperature in the dark. Cells were then washed and resuspended in buffer, and kept at 4°C until analyzed with customized BD LSRFortessa flow cytometer (BD Biosciences). Compensation was performed using AbC, ArC, or UltraComp compensation beads (ThermoFisher).
Ex vivo SIINFEKL stimulation
After cell isolation, LNs, spleen, and tumor samples were plated in a sterile 96-well U-bottom plate. SIINFEKL peptide (1 μg/ml) in 100 μL of IMDM (Iscove’s modified Dulbecco’s medium) with 10% heat-inactivated fetal bovine serum and 0.05 mM β-mercaptoethanol (Sigma-Aldrich) was added to each sample and then incubated for a total of 6 h at 37°C with 5% CO2. Three hours into the incubation period, brefeldin A (50 μg/mL) was added to each sample. Cells were then stained for flow cytometry as described above.
Flow-based cell adhesion experiments
A 0.01-inch thick silicone gasket with a 2.5 mm wide rectangular opening was assembled between an acrylic disk with an inlet and outlet ports (GlycoTech Corporation) and a functionalized polystyrene dish via vacuum suction. The chamber and inlet and outlet tubing were filled with 0.1% BSA in D-PBS perfusion medium, taking care that no bubbles formed at connections. The outlet line was connected to a syringe pump (PhD Ultra Harvard Apparatus), and the inlet line was connected to a reservoir. The chamber was placed on an Eclipse Texas Instrument (TI) optical microscope (Nikon), and medium was perfused through the chamber at the desired flow rate via syringe withdrawal. A suspension of 5 × 105 CD8+ T-cells per ml were added to the inlet reservoir. For continuous flow experiments, cells were perfused at a flow rate to attain desired shear stress. For static condition experiments, cells were perfused into the chamber for 3 min, flow was stopped for 10 min to allow cells to settle. Flow was then restarted at the desired flow rate, and after 1 min, the flow rate was continuously increased. Image recording was done using Nikon NIS-Elements software. For continuous flow, six evenly spaced positions within the functionalized region of the dish were imaged for 30 s each, followed by checking non-specific adhesion on the non-functionalized region. For static conditions, one position was imaged for the entire experiment; as the flow rate increased, we can visualize the number of cells detaching from the substrate; at the end, the non-functionalized control was checked. For all experiments, the exposure time was 0.281 μs, the frame rate was 25 frames per second, and the objective was 10x.
Substrate functionalization
In experiments utilizing the vacuum-sealed gasket, a 1.07 × 0.25 cm rectangle in the center of a 35 mm non-tissue culture treated, round, polystyrene dishes were coated with anti-IgG (Fc specific) (Sigma-Aldrich) diluted in D-PBS without calcium and magnesium, at the concentration corresponding to the total P-selectin and ICAM-1 concentration in each condition. The anti-IgG solution was incubated overnight at 4°C, and then washed with D-PBS, blocked with 1% BSA in D-PBS at room temperature for 1h, then washed, incubated at room temperature for 2 h with 10 μg/ml P-selectin (R&D Systems), 5 μg/ml ICAM-1 (R&D systems), or the combination of both dilutes in D-PBS with calcium and magnesium. The non-functionalized region, a 0.53 × 0.25 cm rectangle immediately proximal to the functionalized rectangle, was blocked with 1% BSA in D-PBS. All dishes were stored at 4°C with D-PBS until use in same-day experiments.
Chromatography channel fabrication
The microfluidic channels were made using 100 μm thick double-sided adhesive tape (3M). U-shaped channel of two 2 cm wide by 14 cm long sections connected by a 2 cm wide 1.5 cm long section was cut using a crafted cutter (Silhouette America). On one side, the adhesive tape was attached to PDMS (Ellsworth Adhesives). An inlet hole was made with a biopsy punch before attaching the other side of the adhesive tape to a non-tissue culture-treated polystyrene plate with a drilled outlet hole. PDMS was pre-made by mixing PDMS base with curing agent at a ratio of 9:1 and curing for 4h at 90°C.
Chromatography channel functionalization
The chromatography channel was functionalized by incubating 25 μg/mL anti-IgG (Fc-specific) (R&D Systems) in D-PBS without calcium and magnesium overnight at 4°C, and then washed with D-PBS, blocked with 1% BSA in D-PBS at room temperature for 1 h, then washes again. Then 25 μg/ml of P-selectin diluted in D-PBS with calcium, and magnesium was placed in the functionalized portion of the channel for 2 h at room temperature. Finally, the entire device was blocked with 1% BSA in D-PBS at room temperature for 1 h.
Chromatography channel experiments
An inlet syringe connected to tubing was filled with perfusion media (0.1% BSA in D-PBS), and was connected to a syringe pump (PhD Ultra Harvard Apparatus). The syringe pump was used to withdraw a cell pulse of 1 mL at a concentration of 2.5 × 106 cells/ml into inlet tubing at a rate of 0.4 mL/min. The tubing containing the cell pulse was inserted into the inlet hole of the channel, and a 5 mL test tube was connected to the bottom of the outlet hole as the cell collection reservoir. The channel was placed on an Eclipse Ti optical microscope (Nikon), and medium was perfused through the channel at the desired flow rate; perfusion was then stopped after the free flow cells elution time had been reached. Then, the syringe and inlet tubing was replaced with a new syringe and tubing containing only perfusion media to eject the adherent cell in the channel out of the channel and into a second collection tube. The number of cells in the sorted fraction was counted using a hemocytometer, and then adoptively transferred into mice or analyzed via flow cytometry.
Ex vivo P-selectin stimulation
Isolated CD8+ T-cells were suspended in either cell media (RPMI 1640 with 10% fetal bovine) and incubated with either 1, 2.5, 10, and 20 μg/ml of P-selectin or with 20 ng/mL PMA (Sigma-Aldrich) plus 1 μg/ml Ionomycin (Invitrogen, ThermoFisher) for 1h or 4h. Cells were incubated for 4h at 37°C with 5% CO2 in a standard incubator. After incubation time, cells were analyzed via flow cytometry.
QUANTIFICATION AND STATISTICAL ANALYSIS
Flow cytometry analysis
Flow cytometry data were analyzed using FlowJo software version 10, data is presented in this paper using GraphPad Prism.
Quantification of cell adhesive behavior
Videos were manually post-processed by counting the number of rolling and firmly adherent cells per 30-s video field of view. Firmly adherent cell was defined as a cell that did not move more than 1 radius in length throughout the experiment. Cell velocities were measured for 10–15 rolling cells per FOV with each experiment (with 3 replicas) using a manual particle tracking plugin in ImageJ. Velocity was determined by dividing a cell’s total translational distance over the functionalized area by the total time it took to travel that same distance.
Setting distance analysis
The mean distance of the cells from the bottom of the channel at any given time was experimentally determined and calculated as previously described.30 In short, a cell pulse followed by perfusion media was perfused into an unfunctionalized channel, and 5 min videos were taken with the focal point set at the bottom of the channel at 9 different positions across the channel. The videos were then analyzed using OpenCV-based Traffic Flow Analyzer cell tracking software to detect cell edges and the cell x and y positions of each detected cell. Based on the total tracked distance and video frames per second, cell velocity was calculated using these data. The metric (the vertical location of the cell from the bottom of the channel divided by an effective cell radius) was calculated from Equation 1, and the mean x and y radius were measured during cell tracking.
| (Equation 1) |
Statistics
Data are represented as means accompanied by SEM, and statistics were calculated using Prism 9 software (GraphPad Software). ****p < 0.0001, ***p < 0.001,**p < 0.01, and *p < 0.05 by unpaired t-test or two-way analysis of variance (ANOVA) followers by post hoc test for multiple comparisons.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Goat anti-human IgG (Fc Specific) | Sigma-Aldrich | Cat#I2136 |
| Goat anti-mouse CD62P | R&D Systems | Cat#AF737 |
| Donkey anti-goat Alexa Fluor 555 | Invitrogen | Cat#A21432; RRID:AB_2535853 |
| Rat anti-mouse CD3 | Invitrogen | Cat#14–0032-82; RRID:AB_467053 |
| Rat anti-mouse CD31 | Invitrogen | Cat#14–0311-82; RRID:AB_467201 |
| Donkey anti-rat Alexa Flour 647 | Invitrogen | Cat#A78947; RRID:AB_2910635 |
| InVivoPlus anti-mouse PD-1 (CD279) | BioXCell | Cat#BP0146; RRID:AB_10949053 |
| FITC anti-IgG (Fc specific) | Sigma-Aldrich | Cat#F9512 |
| APC anti-mouse CD44 | Biolegend | Cat#103012; RRID:AB_312963 |
| BV421 anti-mouse CD62L | Biolegend | Cat#104436; RRID:AB_2562560 |
| APC/Cyanine7 anti-mouse CD8a | Biolegend | Cat#100714; RRID:AB_312753 |
| BV785 anti-mouse CCR7 | Biolegend | Cat#145526; RRID:AB_2566799 |
| PE/Cyanine anti-mouse LFA-1 | Biolegend | Cat#141012; RRID:AB_2564308 |
| PE anti-mouse PSGL-1 | BD Biosciences | Cat#555306; RRID:AB_395719 |
| PerCP anti-mouse CD3 | Biolegend | Cat#100326; RRID:AB_893317 |
| PE anti-mouse CD45.1 | Biolegend | Cat#110708; RRID:AB_313497 |
| BV711 anti-mouse CD45.2 | Biolegend | Cat#109847; RRID:AB_2616859 |
| AF700 anti-mouse SCA-1 | Biolegend | Cat#108142; RRID:AB_2565959 |
| BV605 anti-mouse PD-1 | Biolegend | Cat#135220; RRID:AB_2562616 |
| AF700 anti-mouse Ki-67 | Biolegend | Cat#652420; RRID:AB_2564285 |
| FITC anti-mouse Granzyme B | Biolegend | Cat#372206; RRID:AB_2687030 |
| PE anti-mouse IFN-ɣ | Biolegend | Cat#505808; RRID:AB_315402 |
| PE-Cy7 anti-mouse TNF-α | Biolegend | Cat#506324; RRID: AB_2256076 |
| APC-Cy7 anti-Human CD8 | BD Biosciences | Cat#557834; RRID:AB_396892 |
| APC anti-human CD62L | Biolegend | Cat#304809; RRID:AB_314469 |
| PE anti-human HECA-452 (sLea/x) | Biolegend | Cat#321312; RRID:AB_2565589 |
| BV605 anti-human CCR7 | Biolegend | Cat#353223; RRID:AB_11124325 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Recombinant Mouse P-selectin Fc Chimera | R&D Systems | Cat#737-PS-200 |
| Recombinant Mouse ICAM Fc Chimera | R&D Systems | Cat#796-IC-050 |
| Recombinant Human P-selectin Fc Chimera | R&D Systems | Cat#137-PS-050 |
| Polydimethylsiloxane (PDMS), Dow Corning Sylgard 184 | Ellsworth Adhesives | Cat#184 SIL ELAST KIT 3.9KG, SKU 2065622 |
| Phorbol 12-myristate 13-acetate | Sigma-Aldrich | Cat#P8139–1MG |
| Ionomycin, Calcium Salt | Invitrogen | Cat#I24222 |
| Recombinant-IL-2 | Bio-techne | Cat#402-ML-020 |
| SIINFEKL | vivitide | Cat#257–264 |
|
| ||
| Critical commercial assays | ||
|
| ||
| MojoSort Mouse CD8a Selection Kit | Biolegend | Cat#480035 |
| EasySep Human CD8+ T cell Enrichment Kit | STEMCELL | Cat#19053 |
| Dynabeads Mouse T-Activator CD3/CD28 | Gibco | Cat#11453D |
| Dynabeads Human T-Activator CD3/CD28 | Gibco | Cat#11132D |
| EasySep Dead Cell Removal (Annexin V) Kit | STEMCELL | Cat#17899 |
| eBioscience Foxp3/Transcription Factor Staining Buffer Set | ThermoFisher | Cat#00–5523-00 |
| ZombieAqua Fixable Viability Kit | Biolegend | Cat#423102 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| B16F10 | Obtained from Swartz Laboratory | N/A |
| B16F10-OVA | Obtained from Swartz Laboratory | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| 6 week old female C57BL/6J | Jackson Labs | Strain#000664 |
| 6 week old female B6 CD45.1 | Jackson Labs | Strain#002014 |
| OT-I Mouse | Charles River | Strain Code 642 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo | Treestar, Inc | N/A |
| ImageJ | National Institutes of Health | N/A |
| Manual Tracking ImageJ plugin | National Institutes of Health | N/A |
| Prism 9 | GraphPad | N/A |
|
| ||
| Other | ||
|
| ||
| Culture Dishes, Nontreated Sterile | Corning Life Sciences | Cat#431272 |
| Double sided adhesive 96042 | 3M | 3M ID 700063936593 |
| Non-silicone secondary release | 3M | 3M ID 70000118912 |
Highlights.
An engineered microfluidic device models CD8+ T cell tumor homing in vivo
CD8+ T cell adhesion to P-selectin in fluid flow in vitro predicts tumor homing
CD8+ T cells enriched for adhesion to P-selectin better home to and remodel the TME
Transfer of CD8+ T cells enriched for adhesion in vitro improved aPD-1 tumor control
Acknowledgments
We thank Claire McClain for technical assistance. This work was supported by US National Institutes of Health grants R01CA207619 (S.N.T.), U01CA214354 (S.N.T.), and T32EB006343 (L.F.S.). C.P.C. and M.P.M. were supported by National Science Foundation Graduate Research Fellowships (DGE-2039655). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112175.
DECLARATION OF INTERESTS
C.P.C. and S.N.T. have a provisional patent filed associated with this work.
REFERENCES
- 1.Wu R, Forget M-A, Chacon J, Bernatchez C, Haymaker C, Chen JQ, Hwu P, and Radvanyi LG (2012). Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: current status and future outlook. Cancer J 18, 160–175. 10.1097/PPO.0b013e31824d4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernatchez C, Radvanyi LG, and Hwu P (2012). Advances in the treatment of metastatic melanoma: adoptive T cell therapy. Semin. Oncol 39, 215–226. 10.1053/j.seminoncol.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Restifo NP, Dudley ME, and Rosenberg SA (2012). Adoptive immunotherapy for cancer : harnessing the T cell response. Nat. Rev. Immunol 12, 269–281. 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prieto PA, Durflinger KH, Wunderlich JR, Rosenberg SA, and Dudley ME (2010). Enrichment of CD8+ cells from melanoma tumor-infiltrating lymphocyte cultures reveals tumor reactivity for use in adoptive cell therapy. J. Immunother 33, 547–556. 10.1097/CJI.0b013e3181d367bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, Wu R, Lizee G, Mahoney S, Alvarado G, et al. (2012). Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res 18, 6758–6770. 10.1158/1078-0432.CCR-12-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pagé s F, Galon J, Dieu-Nosjean MC, Tartour E, Sauté s-Fridman C, and Fridman WH (2010). Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 29, 1093–1102. 10.1038/onc.2009.416. [DOI] [PubMed] [Google Scholar]
- 7.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, et al. (2005). Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 102, 18538–18543. 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackensen A, Ferradini L, Carcelain G, Triebel F, Faure F, Viel S, and Hercend T (1993). Evidence for in situ amplification of cytotoxic T-lymphocytes with antitumor activity in a human regressive melanoma. Cancer Res 53, 3569–3573. [PubMed] [Google Scholar]
- 9.Kim ST, Jeong H, Woo OH, Seo JH, Kim A, Lee ES, Shin SW, Kim YH, Kim JS, and Park KH (2013). Tumor-infiltrating lymphocytes, tumor characteristics, and recurrence in patients with early breast cancer. Am. J. Clin. Oncol 36, 224–231. 10.1097/COC.0b013e3182467d90. [DOI] [PubMed] [Google Scholar]
- 10.Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, and Ohtani H (1998). CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 58, 3491–3494. [PubMed] [Google Scholar]
- 11.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagé s C, Tosolini M, Camus M, Berger A, Wind P, et al. (2006). Type, density and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964. 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 12.Peske JD, Woods AB, and Engelhard VH (2015). Control of CD8 T-cell infiltration into tumors by vasculature and microenvironment. Adv. Cancer Res 128, 263–307. 10.1016/bs.acr.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernhard H, Neudorfer J, Gebhard K, Conrad H, Hermann C, Nährig J, Fend F, Weber W, Busch DH, and Peschel C (2008). Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunol. Immunother 57, 271–280. 10.1007/s00262-007-0355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, Borman ZA, Kerkar SP, Scott CD, Finkelstein SE, et al. (2011). Determinants of successful CD8 + T-cell adoptive immunotherapy for large established tumors in mice. Clin. Cancer Res 17, 5343–5352. 10.1158/1078-0432.CCR-11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanitis E, Dangaj D, Irving M, and Coukos G (2017). Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol 28, xii18–xii32. 10.1093/annonc/mdx238. [DOI] [PubMed] [Google Scholar]
- 16.Kmiecik J, Poli A, Brons NHC, Waha A, Eide GE, Enger PØ, Zimmer J, and Chekenya M (2013). Elevated CD3+and CD8+tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol 264, 71–83. 10.1016/j.jneuroim.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 17.Sackstein R, Schatton T, and Barthel SR (2017). T-lymphocyte homing: an underappreciated yet critical hurdle for successful cancer immunotherapy. Lab. Invest 97, 669–697. 10.1038/labinvest.2017.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nolz JC, Starbeck-Miller GR, and Harty JT (2011). Naive, effector and memory CD8 T-cell trafficking: parallels and distinctions. Immunotherapy 3, 1223–1233. 10.2217/imt.11.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weninger W, Crowley MA, Manjunath N, and von Andrian UH (2001). Migratory properties of naive, effector, and memory CD8+ T cells. J. Exp. Med 194, 953–966. 10.1084/jem.194.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Afanasiev OK, Nagase K, Simonson W, Vandeven N, Blom A, Koelle DM, Clark R, and Nghiem P (2013). Vascular E-selectin expression correlates with CD8 lymphocyte infiltration and improved outcome in Merkel cell carcinoma. J. Invest. Dermatol 133, 2065–2073. 10.1038/jid.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stark FC, Gurnani K, Sad S, and Krishnan L (2012). Lack of functional selectin ligand interactions compromises long term tumor protection by CD8 + T cells. PLoS One 7, e32211. 10.1371/journal.pone.0032211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher DT, Chen Q, Skitzki JJ, Muhitch JB, Zhou L, Appenheimer MM, Vardam TD, Weis EL, Passanese J, Wang WC, et al. (2011). IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J. Clin. Invest 121, 3846–3859. 10.1172/JCI44952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams DH, Yannelli JR, Newman W, Lawley T, Ades E, Rosenberg S a, and Shaw, S. (1997). Adhesion of tumour-infiltrating lymphocytes to endothelium: a phenotypic and functional analysis. Br. J. Cancer 75, 1421–1431. 10.1038/bjc.1997.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirata T, Furie BC, and Furie B (2002). P-E-and L-selectin mediate migration of activated CD8+ T lymphocytes into inflamed skin. J. Immunol 169, 4307–4313. 10.4049/jimmunol.169.8.4307. [DOI] [PubMed] [Google Scholar]
- 25.Tietz W, Allemand Y, Borges E, von Laer D, Hallmann R, Vestweber D, and Hamann A (1998). CD4+ T cells migrate into inflamed skin only if they express ligands for E- and P-selectin. J. Immunol 161, 963–970. [PubMed] [Google Scholar]
- 26.Yamaoka T, Fujimoto M, Ogawa F, Yoshizaki A, Bae SJ, Muroi E, Komura K, Iwata Y, Akiyama Y, Yanaba K, et al. (2011). The roles of P- and E-selectins and P-selectin glycoprotein ligand-1 in primary and metastatic mouse melanomas. J. Dermatol. Sci 64, 99–107. 10.1016/j.jdermsci.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 27.Woods AN, Wilson AL, Srivinisan N, Zeng J, Dutta AB, Peske JD, Tewalt EF, Gregg RK, Ferguson AR, and Engelhard VH (2017). Differential expression of homing receptor ligands on tumor-associated vasculature that control CD8 effector T-cell entry. Cancer Immunol. Res 5, 1062–1073. 10.1158/2326-6066.CIR-17-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shamay Y, Elkabets M, Li H, Shah J, Brook S, Wang F, Adler K, Baut E, Scaltriti M, Jena PV, et al. (2016). P-selectin is a nanotherapeutic delivery target in the tumor microenvironment. Sci. Transl. Med 8, 345ra87. 10.1126/scitranslmed.aaf7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weishaupt C, Munoz KN, Buzney E, Kupper TS, and Fuhlbrigge RC (2007). T-cell distribution and adhesion receptor expression in metastatic melanoma. Clin. Cancer Res 13, 2549–2556. 10.1158/1078-0432.CCR-06-2450. [DOI] [PubMed] [Google Scholar]
- 30.Oh J, Edwards EE, McClatchey PM, and Thomas SN (2015). Analytical cell adhesion chromatography reveals impaired persistence of metastatic cell rolling adhesion to P-selectin. J. Cell Sci 128, 3731–3743. 10.1242/jcs.166439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Birmingham KG, O’Melia MJ, Ban D, Mouw J, Edwards EE, Marcus AI, Mcdonald J, and Thomas SN (2019). Analyzing mechanisms of metastatic cancer cell adhesive phenotype leveraging preparative adhesion chromatography microfluidic. Adv. Biosyst 3, e1800328. 10.1002/adbi.201800328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birmingham KG, Robinson IE, Edwards EE, and Thomas SN (2020). Photoconversion and chromatographic microfluidic system reveals differential cellular phenotypes of adhesion velocity versus persistence in shear flow. Lab Chip 20, 806–822. 10.1039/c9lc00923j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nooijen PT, Westphal JR, Eggermont AM, Schalkwijk C, Max R, de Waal RM, and Ruiter DJ (1998). Endothelial P-selectin expression is reduced in advanced primary melanoma and melanoma metastasis. Am. J. Pathol 152, 679–682. [PMC free article] [PubMed] [Google Scholar]
- 34.Läubli H, and Borsig L (2010). Selectins as mediators of lung metastasis. Cancer Microenviron 3, 97–105. 10.1007/s12307-0100043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tinoco R, Otero DC, Takahashi AA, and Bradley LM (2017). PSGL-1: a new player in the immune checkpoint landscape. Trends Immunol 38, 323–335. 10.1016/j.it.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atarashi K, Hirata T, Matsumoto M, Kanemitsu N, and Miyasaka M (2005). Rolling of Th1 cells via P-selectin glycoprotein ligand-1 stimulates LFA-1-mediated cell binding to ICAM-1. J. Immunol 174, 1424–1432. 10.4049/jimmunol.174.3.1424. [DOI] [PubMed] [Google Scholar]
- 37.Edwards EE, and Thomas SN (2017). P-Selectin and ICAM-1 synergy in mediating THP-1 monocyte adhesion in hemodynamic flow is length dependent. Integr. Biol 9, 313–327. 10.1039/c7ib00020k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong AK, LLanos P, Boroda N, Rosenberg SR, and Rabbany SY (2016). A parallel-plate flow chamber for mechanical characterization of endothelial cells exposed to laminar shear stress. Cell. Mol. Bioeng 9, 127–138. 10.1007/s12195-015-0424-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soares A, Govender L, Hughes J, Mavakla W, de Kock M, Barnard C, Pienaar B, Janse van Rensburg E, Jacobs G, Khomba G, et al. (2010). Novel application of Ki67 to quantify antigen-specific in vitro lymphoproliferation. J. Immunol. Methods 362, 43–50. 10.1016/j.jim.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horton BL, Williams JB, Cabanov A, Spranger S, and Gajewski TF (2018). Intratumoral CD8+ T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol. Res 6, 14–24. 10.1158/2326-6066.CIR-17-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horton BL, and Gajewski TF (2018). Back from the dead: TIL apoptosis in cancer immune evasion. Br. J. Cancer 118, 309–311. 10.1038/bjc.2017.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu J, Powis De Tenbossche CG, Cané S, Colau D, Van Baren N, Lurquin C, Schmitt-Verhulst AM, Liljeströ m P, Uyttenhove C, and Van Den Eynde BJ (2017). Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat. Commun 8, 1404–1415. 10.1038/s41467-017-00784-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’melia MJ, Rohner NA, Manspeaker MP, Francis DM, Kissick HT, and Thomas SN (2020). Quality of CD8 + T cell immunity evoked in lymph nodes is compartmentalized by route of antigen transport and functional in tumor context. Sci. Adv 6, eabd7134. 10.1126/sciadv.abd7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, and Carbone FR (2006). Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 25, 153–162. 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 45.O’Melia MJ, Manspeaker MP, and Thomas SN (2021). Tumor-draining lymph nodes are survival niches that support T cell priming against lymphatic transported tumor antigen and effects of immune checkpoint blockade in TNBC. Cancer Immunol. Immunother 70, 2179–2195. 10.1007/s00262-020-02792-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF (2016). Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 30, 324–336. 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kümpers C, Jokic M, Haase O, Offermann A, Vogel W, Grätz V, Langan EA, Perner S, and Terheyden P (2019). Immune cell infiltration of the primary tumor, not PD-L1 status, is associated with improved response to checkpoint inhibition in metastatic melanoma. Front. Med 6, 27. 10.3389/fmed.2019.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McEver RP, and Zhu C (2010). Rolling cell adhesion. Annu. Rev. Cell Dev. Biol 26, 363–396. 10.1146/annurev.cellbio.042308.113238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klebanoff C. a Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony P. a, Palmer DC, et al. (2004). IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc. Natl. Acad. Sci. USA 101, 1969–1974. 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klebanoff CA, Gattinoni L, Torabi-parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, et al. (2005). Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 102, 9571–9576. 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, and Restifo NP (2005). Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Invest 115, 1616–1626. 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gattinoni L, Klebanoff CA, and Restifo NP (2012). Paths to stemness: building the ultimate antitumour T cell. Nat. Rev. Cancer 12, 671–684. 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mueller SN, Gebhardt T, Carbone FR, and Heath WR (2013). Memory T cell subsets , migration patterns , and tissue residence. Annu. Rev. Immunol 31, 137–161. 10.1146/annurev-immunol-032712-095954. [DOI] [PubMed] [Google Scholar]
- 55.Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264. 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharma P, and Allison JP (2015). The future of immune checkpoint therapy. Science 348, 56–61. 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 57.Elia AR, Grioni M, Basso V, Curnis F, Freschi M, Corti A, Mondino A, and Bellone M (2018). Targeting tumor vasculature with TNF leads effector t cells to the tumor and enhances therapeutic efficacy of immune checkpoint blockers in combination with adoptive cell therapy. Clin. Cancer Res 24, 2171–2181. 10.1158/1078-0432.CCR-17-2210. [DOI] [PubMed] [Google Scholar]
- 58.John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH, and Darcy PK (2013). Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res 19, 5636–5646. 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 59.Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, Song M, Miele MM, Li Z, Wang P, et al. (2018). Targeted delivery of a PD-1-blocking scFV by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol 36, 847–856. 10.1038/nbt.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, and Adusumilli PS (2016). Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest 126, 3130–3144. 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li S, Siriwon N, Zhang X, Yang S, Jin T, He F, Kim YJ, Mac J, Lu Z, Wang S, et al. (2017). Enhanced cancer immunotherapy by chimeric antigen receptor–modified T cells engineered to secrete checkpoint inhibitors. Clin. Cancer Res 23, 6982–6992. 10.1158/1078-0432.CCR-17-0867. [DOI] [PubMed] [Google Scholar]
- 62.Chong BF, Murphy J-E, Kupper TS, and Fuhlbrigge RC (2004). E-selectin, thymus- and activation-regulated chemokine/CCL17, and intercellular adhesion molecule-1 are constitutively coexpressed in dermal microvessels: a foundation for a cutaneous immunosurveillance system. J. Immunol 172, 1575–1581. 10.4049/jimmunol.172.3.1575. [DOI] [PubMed] [Google Scholar]
- 63.Piali L, Fichtel A, Terpe H-J, Imhof BA, and Gisler RH (1995). Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J. Exp. Med 181, 811–816. 10.1084/jem.181.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hendry SA, Farnsworth RH, Solomon B, Achen MG, Stacker SA, and Fox SB (2016). The role of the tumor vasculature in the host immune response: implications for therapeutic strategies targeting the tumor microenvironment. Front. Immunol 7, 621. 10.3389/fimmu.2016.00621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Griffioen AW, Damen CA, Blijham GH, and Groenewegen G (1996). Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood 88, 667–673. [PubMed] [Google Scholar]
- 66.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, Mckee M, and Gajewski TF (2009). Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 69, 3077–3085. 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamoun WS, Chae SS, Lacorre DA, Tyrrell JA, Mitre M, Gillissen MA, Fukumura D, Jain RK, and Munn LL (2010). Simultaneous measurement of RBC velocity, flux, hematocrit and shear rate in vascular networks. Nat. Methods 7, 655–660. 10.1038/nmeth.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Siemann DW (2011). The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat Rev 37, 63–74. 10.1016/j.ctrv.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Topper JN, and Gimbrone MA Jr. (1999). Blood flow and vascular gene expression: fluid shear stress as a modulator of endothelial phenotype. Mol. Med. Today 5, 40–46. 10.1172/JCI83083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.