

Abstract
Metalloenzymes catalyze a variety of reactions using a limited number of natural amino acids and metallocofactors. Therefore, the environment beyond the primary coordination sphere must play an important role in both conferring and tuning their phenomenal catalytic properties, enabling active sites with otherwise similar primary coordination environments to perform a diverse array of biological functions. However, since the interactions beyond the primary coordination sphere are numerous and weak, it has been difficult to pinpoint structural features responsible for the tuning of activities of native enzymes. Designing artificial metalloenzymes (ArMs) offers an excellent basis to elucidate the roles of these interactions and to further develop practical biological catalysts. In this review, we highlight how the secondary coordination spheres of ArMs influence metal binding and catalysis, with particular focus on the use of native protein scaffolds as templates for the design of ArMs by either rational design aided by computational modeling, directed evolution, or a combination of both approaches. In describing successes in designing heme, nonheme Fe, and Cu metalloenzymes, heteronuclear metalloenzymes containing heme, and those ArMs containing other metal centers (including those with non-native metal ions and metallocofactors), we have summarized insights gained on how careful controls of the interactions in the secondary coordination sphere, including hydrophobic and hydrogen bonding interactions, allow the generation and tuning of these respective systems to approach, rival, and, in a few cases, exceed those of native enzymes. We have also provided an outlook on the remaining challenges and future directions gain a deeper understanding of the secondary coordination sphere, and in turn guide the design of a broader and more efficient variety of ArMs.
Graphical Abstract
1. INTRODUCTION
Metalloproteins are ubiquitous in nature; a bioinformatic search of three-dimensional protein structures deposited in the Protein Data Bank in 2008 found ~40% to bind metal ions, with estimates ranging between 36 and 59% in different classes.1 A metallomics study in 2010 indicated that microbial metalloproteomes are largely uncharacterized,2 putting the estimate of metalloproteins among all proteins over 50%. Many of these metalloproteins function as metalloenzymes, performing the essential processes to support life as it is known, ranging from metabolism3-14 and DNA synthesis15-18 to the generation of atmospheric O219,20 and nitrogen fixation.21-24 Even more so, the stunning range of chemistry performed in these native systems is done so not only within the limited conditions conducive to life but also using a comparatively narrow range of biologically available ligands and metallocofactors. It is little wonder then that metalloenzymes have captivated chemists and biochemists alike for decades, and tremendous efforts have been undertaken across these fields to understand the intimate details of metalloenzyme functions.
The biologically available ligands for the formation of the primary coordination sphere (PCS) in metalloenzymes include the side chains of nucleophilic amino acid residues such as His, Asp, Glu, Arg, Met, Ser, Tyr, and Cys, the amine and carboxylate protein termini, the carbonyl and amide moieties of the protein backbone, small molecules such as CO, CN−, H2O, O2, S2−, and −OH, and biosynthesized organic cofactors such as protoporphyrins, corrins, pterins, azadithiolate, and homocitrate.25 These PCSs play critical roles in defining the available chemistry of an active site. A primary example is the heme b center, which consists of an iron center equatorially coordinated by the four nitrogen atoms of the protoporphyrin IX macrocycle and axially by one or two amino acids above and/or below the protoporphyrin ring. While undeniably important, abundant evidence from the study of metalloenzymes has shown that the same PCS environment, such as heme b, can display a variety of different properties from electron transfer to reversible O2 binding, dioxygen reduction, and small molecule functionalization.26 As a result, these heme proteins perform a wide range of biological processes, including O2 transport (globins),27-29 cell signaling,30-32 electron transfer,33-35 and enzymatic activities such as catalases,36-40 oxidases,41,42 oxygenases,43-45 peroxidases,46-48 nitric oxide reductases,49-51 and sulfite reductases.52-54 Therefore, features of the protein environment beyond the PCS are essential in not only fine-tuning the activities of metalloenzymes but also conferring new activities in otherwise similar systems.
Unlike the PCS itself, the environment surrounding the PCS consists of many weak and noncoordinating interactions such as hydrogen bonding (H-bonding), electrostatic, and hydrophobic interactions. Some may contribute little, if any, to protein function, while others play major roles that enable many metalloenzymes to catalyze reactions that are currently unparalleled by synthetic catalysts in terms of activity and/or reaction efficiency. Therefore, elucidating the structural features in these secondary coordinate spheres (SCSs) that are responsible for activity not only advances our knowledge about biology but can also result in novel synthetic catalysts or artificial metalloenzymes (ArMs) with new activity or reactivity for many industrial applications, such as producing energy from sustainable sources and synthesizing pharmaceutical drugs.
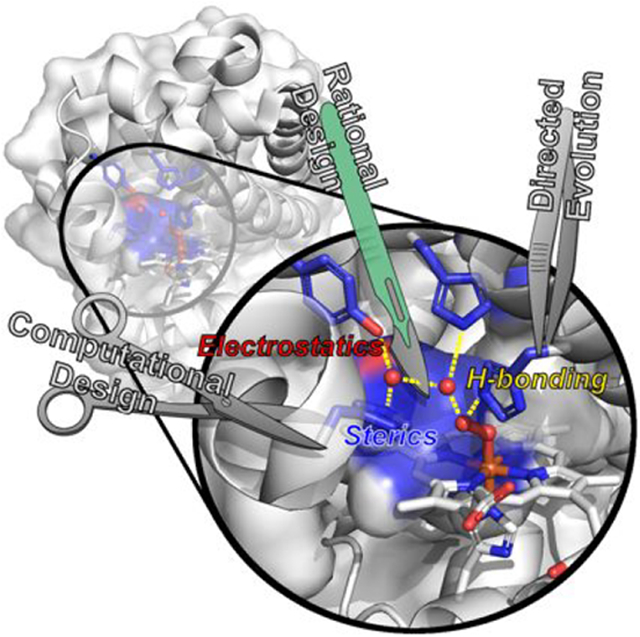
The study of native metalloenzymes is undeniably essential in providing a detailed understanding of their function. Despite major progress in many facets, however, it has been difficult to comprehensively elucidate how these enzymes catalyze so many reactions highly efficiently using structural features beyond the PCS. Such difficulties arise in large part because the PCSs of many native enzymes reside in large and complicated proteins, some of which are membrane proteins, that are not (yet) compatible with heterologous expression. As a result, it is very difficult to carry out site-directed mutagenesis (SDM) to probe the roles of SCS residues and to purify them to homogeneity in high enough yields for traditional studies. In addition, many native metalloenzymes contain multiple metallocofactors that can produce overlapping spectroscopic signals which make it challenging to focus on the PCS of interest.12,55,56 Furthermore, many similar PCSs reside in very different protein scaffolds containing numerous different residues, making it difficult to identify the key SCS residues responsible for the different reactivity. Designing artificial metalloenzymes (ArMs) using small robust protein scaffolds that can be readily constructed, expressed, and purified in simple hosts such as E. coli can overcome the above limitations, because these ArMs contain only the PCS of interest and free of interferences from other cofactors. By placing the same PCS in the same protein scaffold and systematically introducing SCS residues from different enzymes with varying reactivity, we can provide a holistic view of the key structural features responsible for the differences in activity. Therefore, designing ArMs that reproduce the catalytic capacities of native enzymes not only provides the ultimate basis to test what we know about native catalytic mechanisms but also reveals new insights that are not readily obtainable from studying native enzymes, much in spirit with a quote from the late Prof. Richard Feynman, “What I cannot create, I do not understand.”57 Given their small, robust scaffolds, ArMs provide a new, divergent opportunity to combine the benefits of both chemical catalysis and biocatalysis, allowing the power of SCS interactions to be harnessed to enhance catalytic selectivity in new-to-nature reactions.
ArMs can be generally designed using one of the following systems: (1) modifying an existing meta-binding site in a native enzymatic scaffold to promote non-native reactivity, (2) introducing a new metal-binding site to a native protein scaffold without such a site, and (3) designing a metal-binding site in a de novo designed protein scaffold. To achieve this goal, the most common strategies employed include rational design and directed evolution. Rational design involves the purposeful mutation of select residues or regions of a protein scaffold using SDM to incorporate a metal-binding site and/or modulate activity based on careful deliberation over available structural information and often from comparisons with related systems. Such design is often aided by comparing key residues between the native enzymes and ArMs and using the experience and knowledge gained from studying native enzymes, or by computations, to predict and then experimentally verify the roles of these residues. Although immensely successful, this approach is still limited by human intuition and/or the ability of computational programs to capture the structural and functional properties of metalloenzymes, and remains challenging because of highly variable PCSs and weak SCS interactions. Alternatively, directed evolution based on a random library to screen or select for desired activity followed by detailed structural and mechanistic studies of the “winners” can provide an understanding of the roles of residues in not only the SCS but also those much further away in conferring and fine-tuning the activities.58-60 More recently, computational methods using programs that employ large structural databases and machine learning, such as Rosetta61,62 and AlphaFold,63,64 have emerged as powerful tools for the exploratory design of novel ArMs in silico, allowing a much broader scope of protein scaffolds and variants to be surveyed for desired properties, which can then be tested in vitro.
In the present review, we focus on how rational design and directed evolution have provided insight into the roles of the SCS in tuning the functionality of ArMs, with particular emphasis toward systems employing natural protein scaffolds and exhibiting catalytic activity. A more comprehensive perspective on catalysis and electron transfer by de novo metalloenzymes can be found in the review of Pecoraro and co-workers in this issue, and several recent reviews provide detailed coverage of the design and breadth of applications of these scaffolds.65-68 For extensive reviews of the design and catalytic capabilities of ArMs, we refer the reader to many outstanding reviews in this area.69-77 Fe- and Cu-based ArMs constitute a major portion of designed metalloenzymes, and therefore, this review has been broadly organized into sections addressing the fields of heme Fe, nonheme Fe, and Cu followed by multinuclear ArMs, and finally systems containing alternative metal cofactors including V, Cr, Mn, Co, Ni, Zn, Ru, Rh, Pd, and Ir.
2. CATALYSIS BEYOND THE PRIMARY COORDINATION SPHERE BY HEME PROTEINS WITH NON-NATIVE FUNCTIONS
Heme proteins are among the most, if not the most, studied metalloproteins, and their functions range from electron transfer to O2 carrier, catalases, peroxidases, oxidase, and oxygenase. They perform these functions using different types of heme, such as heme a, b, c, d, and f. The different types of the heme have a similar core consisting of Fe and a porphyrin ring. Even the same type of heme (e.g., heme b) can catalyze very different and diverse reactions. Therefore, structural features beyond such PCSs must play important roles in defining and tuning the functions. Designing ArMs to mimic these native heme proteins using much smaller and robust heme proteins, such as myoglobin (Mb), cytochrome c (Cc), and cytochrome c peroxidase (CcP), allows not only testing of what we know about similarities and differences among these proteins but also revelation of new insights.
2.1. Rational Design Using Myoglobin as a Scaffold
Myoglobin (Mb) is a molecular oxygen carrier that natively serves to reversibly bind O2 using a heme b center.78,79 The ready heterologous expression of Mb variants in E. coli. has made this enzyme a popular scaffold for rational designs of heme enzymes with a range of activity far beyond its native function.80-82 Furthermore, the small size, high solubility in water, and ready crystallizability of Mb have also made it accessible to an array of biochemical and biophysical methods to investigate the role of SCSs in catalysis.83-88
Myoglobin belongs to the globin superfamily of proteins, consisting of eight α-helices connected by a short loop region (Figure 1a).89 The endogenous active site of Mb consists of a heme b center (Figure 1b) in which Fe is proximally coordinated by histidine through the Nε position. This PCS is ubiquitous among globins and is presented in some heme peroxidases as well.90-92 Therefore, the differences in activity observed between Mb, other globins, and peroxidases must arise from differences in structure beyond the PCS.
Figure 1.

(a) Crystal structure of WT-sperm whale Mb (swMb) (PDB ID: 5YCE).89 (b) Chemical structure of heme b. (c) Crystal structure of the active site of cytochrome c peroxidase (PDB ID: 1ZBY) with the distal His52 and Arg48 residues shown in yellow.90 (d) Crystal structure of the active site of the F43H/H64L Mb mutant (PDB ID: 1OFK)102 with the two mutated residues shown in yellow. The distances between the Nε atom of the distal His residue and the heme iron in each crystal structure are labeled.
2.1.1. From Myoglobin to Peroxidase.
In addition to its well-known function as an O2 carrier, Mb is also capable of performing oxidation reactions in the presence of hydrogen peroxide (H2O2).93-95 However, the peroxidase activity of wild-type (WT) Mb is much lower than that of native peroxidases (2.7 M−1s−1 vs 130 M−1s−1).96 Native peroxidases are a large group of enzymes that catalyze several critical redox reactions, including alkene oxidation, sulfoxidation, and dehalogenation. All peroxidases share similar PCS environments to that of ferric Mb, with the fifth coordination site of Fe occupied by a proximal histidine side chain and the sixth coordination position occupied by a water in the resting state. However, peroxidases possess a highly conserved Arg residue in the distal pocket, which is absent in Mb (Figure 1c).97,98 Interestingly, both Mb and peroxidases contain a His residue in the distal pockets (His64 in Mb and His52 in CcP). Comparing the X-ray structures of CcP and Mb reveals that the distal His of Mb is closer to the heme iron than in CcP, which may be responsible for the differences in the interaction of these enzymes with the H2O2 substrate.89,97 By relocating the distal His residue in Mb through a F43H/H64L double mutant, the Watanabe group was able to produce a Mb with catalytic rates for sulfoxidation and epoxidation similar to those of native CcP, without introducing Arg.99 The distal His64 in WT-Mb, which is 4.3 Å from the heme iron, was determined to be a critical residue for the lifetime of the active intermediate (compound I) and, therefore, activity with H2O2. The presence of His64 in WT-Mb presumably results in rapid electron transfer from compound I to His64. Replacement of His64 with a non-oxidizable Leu residue may prevent generation of the protein radical. As the His64 in native Mb is too close to the heme Fe to support the heterolytic cleavage of H2O2, the relocation of the distal His by F43H mutation led to a proper distancing between the Nε atom of His and the heme Fe, which perfectly resembled the corresponding distances in native peroxidases. The appropriate positioning of the distal His in F43H/H64L allows the distal His to function as both a general base to facilitate the H2O2 binding through the deprotonation step and a general acid to aid the heterolytic cleavage of the O─O bond in H2O2 via H-bonding. As another corroboration of the importance of the proper positioning of the distal His in Mb for peroxidase functions, the L29H/H64V mutant exhibited a ~3–6 slower reaction rate with H2O2 relative to the native Mb as the imidazole of His29 is too far away to interact with the H2O2 substrate.100 As another example of mimicking the structural features of the native peroxidase, Wu, et al. found that introduction of two His residues near the active site through the L29H/F43H double mutant produces a SCS resembling the His-Arg pair that is frequently observed in native peroxidases.101 An additional H64A mutation further opened a channel to the heme active center, positively influencing the peroxidase activity.
In addition to enhancing the sulfoxidation and epoxidation reactions in Mb, arene oxidation activity has also been introduced into Mb by mimicking the active site of cytochrome P450 (P450). The differences in reactivity between WT-Mb and P450 may arise from variations in the SCS; for example, d-camphor is capable of tightly binding to the hydrophobic heme pocket of P450 through a hydrogen bonding interaction with the hydroxyl group of Tyr96,103 while the active site of WT-Mb does not afford specific interactions for accommodating organic substrates. Using crystallography, it was found that the distance between carbon atoms C6 and C7 of the electron-rich Trp43 residue and the heme iron in the F43W/H64L Mb mutant was similar to the distance between C5 of d-camphor and the iron in P450cam (Figure 2). The development of the double mutant F43W/H65L Mb resulted in a six-electron oxidative modification of Trp43 in the presence of strong sacrificial oxidant, m-chloroperbenzoic acid (mCPBA), representing the first example of oxidation of an aromatic ring by Mb.104
Figure 2.

(a) Crystal structure of P450cam (left, PDB ID: 2CPP)103 and structural model of F43W/H64L Mb (right). (b) Chemical structures of tryptophan and the six-electron modified tryptophan products.
As conserved residues near the active site of Mb, both His64 and Val68 have been shown to influence the affinity of ligand binding (e.g., O2 and CO).105,107 To investigate the role of Val68 in influencing the rate and enantioselectivity of oxidative reactions, Yang et al. engineered a series of H64D/V68X Mb mutants (X = Ala, Ser, Val, Ile, Leu, and Phe) (Figure 3).106 Along this series, the V68L mutation was observed to activate H2O2 most efficiently, while decreased peroxidase activity was found for both V68I and V68A variants, highlighting that subtle changes in the interactions between residue 68 and the substrate can have profound effects on catalysis. In this case, the restricted rotation of the Ile side chain was found to hinder the association of H2O2 at the heme iron center. While removing this steric constraint via a V68A mutation should hypothetically allow association, structural data indicate that this variant allows for the introduction of a distal water molecule which then blocks substrate entrance, demonstrating the balance required for optimization of catalytic activity.
Figure 3.

Crystal structures of the (a) V68L (rose brown, PDB ID: 1MLS),105 (b) H64D/V68A (orange, PDB ID: 1LUE),106 and (c) V68I (magenta, PDB ID: 1MLO)105 Mb variants. Different residues at position 68 are shown in cyan. Water molecules are denoted as red spheres. The hydrogen bonds are represented by black dashed lines.
As another route to engineering peroxidase activity, the introduction of stabilizing forces through disulfide bond formation offers a great opportunity to increase protein stability while tuning enzymatic function. For instance, disulfide bonds have been observed in neuroglobin (Ngb)108 and cytoglobin (Cgb)109 and have been shown to regulate ligand binding and migration. Similar disulfide bonds have been incorporated into Mb at different positions through variants containing F46C/M55C and V21C/V66C/F46S mutations (Figure 4).110,111 The presence of the disulfide bond in F46C/M55C Mb was found to increase both protein stability and peroxidase function.110 Meanwhile, the V21C/V66C/F46S Mb variant demonstrated comparable activity to F46S Mb, but with a ~30% reduction in Km, indicating that the presence of the disulfide bond stabilizes a favorable conformation for substrate binding.111
Figure 4.

Crystal structures of the V21C/V66C/F46S Mb variant (PDB ID: 5ZEO).111 The disulfide bond between the Cys21 and Cys66 residues is highlighted in the red dashed circle. The hydrogen bonds involving the distal His64 and the Ser46 residues are shown by black dashed lines.
2.1.2. From Myoglobin to Dehaloperoxidase.
Dehaloperoxidase (DHP) is a heme protein that catalyzes the dehalogenation of halophenols to nontoxic quinone products.112,113 Halophenols are involved in the synthesis of pesticides and even used as pesticides themselves, which may lead to severe damage to the mammalian liver and immune system.82 They can also be readily oxidized to produce carcinogenic radicals capable of modifying DNA bases. Therefore, native peroxidases such as DHP may have important environmental and economic implications for degradation of industrial dyes and halophenols. Despite their potential applications, the use of bioremediation is often hindered by the limited catalytic efficiency of native enzymes.82 Rationally designed artificial enzymes offer an alternative route to generating functional biocatalysts. Mb-based ArM models for DHP have emerged as efficient catalysts for biodegradation. Despite the low sequence homology between DHP and Mb, the two proteins share a remarkably similar protein fold, with the RMSD between the positions of C atoms being 1.8 Å (Figure 5).92
Figure 5.

(a) Overlay of the crystal structures of native DHP (red, PDB ID: 1EW6)92 and WT-Mb (blue, PDB ID: 5YCE).89 (b) Active sites of native DHP and WT-Mb. The distances between the Nε atoms of the distal His residues and the heme irons in DHP, Mb, and the G65T and G65I Mb variants are summarized in the table on the right.
The mechanism of dehalogenation by dehaloperoxidases has also been analyzed using both “peroxidase-like” Mb mutants and “Mb-like” DHP.114 To mimic the distal His-to-Fe distance in DHP, the authors successfully generated G65T and G65I Mb variants in an effort to position the distal His64 farther from the heme iron (Figure 5). As a result, a more than 5-fold increase in kcat for the oxidative dichlorination reaction was observed relative to WT-Mb, highlighting the subtle changes of nonactive site residues can induce changes in the PCS through both the restrictions imposed by the position of the peptide backbone as well as through the hydrogen bond network.
Studies have shown that a novel tyrosine-heme covalent C─O bond cross-link can be formed in the F43Y Mb mutant (Figure 6a),115 which has provided a platform for engineering Mb variants as biosynthetic models of dehaloperoxidases. The F43Y Mb variant itself displays enhanced dehaloperoxidase activity 115-fold higher than WT-Mb and 9-fold higher than native DHP in the presence of 2,4,6-trichlorophenol (TCP) substrate.116 The presence of multiple Tyr and Trp residues in native dye-decolorizing peroxidases (DyPs) has also inspired the engineering of Mb variants with additional Tyr or Trp mutations, building on the F43Y Mb platform. Interestingly, the double mutant F43Y/F138W exhibits ~4.3-fold greater peroxidase activity than native DyP from Vibrio cholerae.117 This enhanced activity presumably arises from the more rapid generation of the oxidizing species and protein radicals.
Figure 6.

(a) Crystal structure of F43Y Mb (PDB ID: 4QAU)115 showing the Tyr-heme covalent C─O bond and two distal water molecules forming H-bonding interactions (dotted lines).115 (b) Overlay of the crystal structures of ferric F43Y/H64D Mb (blue, PDB ID: 5ZZF)118 and the heme active site of native chloroperoxidase (orange, PDB ID: 1CPO),119 showing the H-bonding network. (c) Crystal structure of ferric F43Y/H64D Mb in complex with TCP (PDB ID: 5ZZG),118 showing the conformation of Tyr43 and TCP and the H-bonding interactions in the heme center. The distance between the Cl4 atom and the heme Fe (3.91 Å) is indicated. Reproduced with permission from ref 118. Copyright 2018 American Chemical Society.
In 2018 Yin et al. combined structural features from both chloroperoxidase (distal Glu) and DHP (distal Tyr) by generating a F43Y/H64D Mb mutant (Figure 6b), which resulted in a 1000-fold increase in catalytic efficiency relative to native DHP from A. ornata.118 The crystal structure (Figure 6c) of this mutant in complex with TCP revealed that Asp64 adapts an open conformation when substrate is bound, highlighting the crucial role of this residue in modulating the substrate binding.
Beyond dehalogenation, the F43Y Mb platform has been employed in the biosynthesis of indigo from indole. While it has been shown that replacement of the distal His64 in Mb with Asp would favor the H2O2-dependent oxidation reaction,120 the double mutant F43Y/H64D exhibits the highest efficiency toward the oxidation of indole to indigo reported to-date.121 More recent work employing a triple mutant based on the F43Y platform has achieved comparable catalytic efficiency to the most efficient native horseradish peroxidase (HRP), where the combination of T67R and F138W mutations is used to mimic the His-Arg pair and conserved Trp residues of native peroxidases.122 The scope of engineered Mb-based catalysts has also been expanded to biodegradation applications; for example, the F43H/H64A Mb mutant successfully catalyzed the biodegradation of malachite green (MG) with even higher efficiency than natural enzymes such as dye-decolorizing peroxidase and laccase.123 Molecular modeling indicates that these active site mutations favor the binding of MG in the heme distal pocket.
2.1.3. From Myoglobin to Carbene Transferase.
Beyond peroxidase and dehaloperoxidase activity, Mb has also been redesigned to promote carbene transfer reactions.124-128 Carbenes are formally neutral divalent carbon species whose activity can be attributed to an incomplete octet electronic configuration.129 Recent protein engineering efforts have revealed that carbene transfer can be catalyzed in heme proteins, including Mb, via an iron porphyrin carbene intermediate, providing a whole new pathway for the biosynthesis of important molecules (Scheme 1).130 In 2015, Bordeaux et al. discovered that WT-swMb exhibits cyclopropanation activity without asymmetric induction.124 Inspired by these results, further Mb-based catalysts have since been developed with both higher activity and selectivity toward cyclopropanation, as well as expanded the substrate scope, including olefin cyclopropanation,124 carbene NH insertion,131 and carbene SH insertion reactions.125
Scheme 1. Carbene Transfer Reactions Catalyzed by Evolved Hemoproteinsa.

a Reprinted with permission from ref 130. Copyright 2021 American Chemical Society.
One of the first efforts to improve the cyclopropanation activity of Mb involved varying the steric bulk properties at the active site residues Phe43, His64, and Val68 of Mb, resulting in the development of the H64V/V68AMb variant, which exhibits high activity and selectivity toward the cyclopropanation of styrene with ethyl diazoacetate (EDA) (Figure 7).124 Based on the available crystal structures of related Mb- and Fe-(porphyrin)/carbene complexes, it has been proposed that the H64V mutation aids in promoting substrate access to heme, while the V68A mutation expands the size of the distal cavity and, thus, has a dramatic effect on enantioselectivity.
Figure 7.

(a) Top view of the crystal structure of WT-swMb (PDB ID: 5YCE) with the key residues in the distal heme pocket shown in magenta.89 (b) Pocket cavity space (mesh) above the heme cofactor in H64V/V68A Mb (left) and WT-swMb (right). The pocket volumes were calculated to correspond to 243 and 125 Å3, respectively. Reproduced with permission from ref 135. Copyright 2019 American Chemical Society. (c) Structural model of the carbene-bound transition state (TSO/ins) of H64G/V68A Mb. The benzofuran substrate is shown in cyan. Reproduced with permission from ref 133. Copyright 2019 John Wiley & Sons.
The reaction scope of the H64V/V68A Mb mutant has also been explored beyond cyclopropanation, demonstrating activity toward carbene N-H insertion with arylamine substrates.131 For the H64V/V68A Mb mutant, the yield for N-methylaniline was relatively low. An improved activity could be achieved through the L29A, H64V, and L29A/H64V variants. The beneficial effects of the L29A mutation may arise from a decrease in steric bulk directly above the heme iron site, allowing the amine substrate to be accommodated. In addition, H64V/V68A Mb has also exhibited promising reactivity toward C-H functionalization of indole at the C3 position.132 This enhanced activity (relative to WT-Mb and H64V Mb) is mainly attributed to the V68A mutation, which enlarges the active site near the heme group to facilitate attack of the heme-bound carbene by indole. The presence of an N1 or C2 substituent on indole resulted in significantly diminished substrate conversions, a limitation that was partially overcome in L29F/H64V Mb. This increase in activity was hypothesized to arise from Phe29 helping to orient the indole substrate into a favorable position via π-stacking interactions.
Along a similar vein, Tyagi et al. identified the L29A/H64V mutant as a promising catalyst for carbene S-H insertion by examining a panel of Mb variants with mutations at residues lining the active site, specifically 29, 43, 64, 68, and 107.125 The greatest enhancement for a single mutation was observed in the L29A Mb variant, which may arise from its role in facilitating the nucleophilic attack of thiol or amine to the heme-bound carbene intermediate by expanding the distal cavity above the heme. Further examination of mutations at these positions has also revealed the F43V/V68F Mb variant, which catalyzes the olefination of aldehydes with excellent efficiency and E diastereoselectivity.128
In 2019, Vargas et al. expanded the substrate scope of Mb to accommodate the larger benzofuran substrate and, in doing so, established the important role of residue 64 in H64X/V68A variants in controlling access of the olefin substrate.133 By decreasing side chain steric bulk, progressively improved catalytic activity could be achieved along the series His≪Val < Ala < Gly. This “gating” effect has also been manifested in more recent work, where a decreasing size of the residue 64 side chain enhanced the cyclopropanation activity in the presence of a diazoketone carbene donor by removing the steric hindrance.134
Taken together, these experimental results illustrate how the catalytic activity and selectivity toward different amine substrates can be modulated by modification of the Mb active site. Although the engineered Mb variants represent a superb platform for carbene transfer catalysis, the precise structural features that govern protein-mediated stereocontrols are yet to be fully elucidated. This problem was not resolved until a crystallographic and computational (DFT) analysis of the H64V/V68A mutant (Figure 7) revealed that stereocontrol of the cyclopropanation reaction by active site mutagenesis is achieved by favoring a specific conformation of the heme-bound carbene and control geometry of the attack of the olefin through steric obstruction, van der Waals forces, and π−π interactions.135
2.1.4. From Mb to Nitrite Reductase (NiR).
To study the Tyr-Cys cross-linking phenomenon, which is ubiquitous in a variety of metalloenzymes, an unnatural amino acid 2-amino-3-(4-hydroxy-3-(methylthio)phenyl)propanoic acid (MtTyr) was incorporated into a functional model of T. nitratireducens cytochrome c nitrite reductase (TvNiR) engineered into swMb by Zhou, et al. The enzymatic activity was determined based on the reduction of hydroxylamine to ammonium by models both containing and lacking MtTyr (MtTyrMb and TyrMb, respectively) as mimics for the presence/absence of the Tyr-Cys cross-link. A 4-fold increase in kcat was found for the MtTyrMb variant, supporting the hypothesis that thioester modification on Tyr helps to enhance NiR activity.136
2.2. Rational Design Using Cytochrome c Peroxidase as a Scaffold
Peroxidases are a large family of enzymes that function to catalyze the reduction of H2O2 in the presence of various substrates. Notably, cytochrome c peroxidase (CcP) and ascorbate peroxidase (APX), two iconic members of this family, do not share any common reducing substrates despite having numerous structural similarities. Since the structures of CcP and APX mainly differ in the ascorbate binding site, Meharenna et al. engineered an ascorbate-binding site into CcP to repurpose the function of CcP toward the non-native substrate (Figure 8a).137 This hybrid protein was generated by replacing native residues 30–42 in CcP with the ascorbate binding site residues 27–32 in APX. In addition, a conserved Arg residue in APX, which is thought to facilitate ascorbate binding via H-bonding, was anchored at position 184 in CcP. The resulting hybridized mutant was found to catalyze the peroxidation of ascorbate at a rate of about 12 min−1.
Figure 8.

(a) Overlay of the crystal structures of WT-CcP (red) and APX (green). Reproduced with permission from ref 137. Copyright 2008 American Chemical Society. The ascorbate binding site and the corresponding loop in CcP are highlighted in the black dashed square. (b) Crystal structure of yeast CcP (PDB ID: 2CCP).139 Three residues that are mutated to Ala are highlighted in magenta.
Peroxidases are natural platforms for engineering peroxygenation reactions since they react efficiently with hydrogen peroxide. The low peroxygenase activity of CcP may be attributed to the low affinity of the heme pocket for apolar substrates. In 2013, Erman et al. employed the CcP platform to build specific peroxygenation activity into CcP to mimic P450 monooxygenases.138 In particular, a triple CcP variant, R48A/W51A/H52A (Figure 8b), was designed to replace the H-bonding residues in the distal heme pocket with residues containing apolar side chains in order to promote binding of the organic substrate 1-methoxynaphthalene. The naphthalene hydroxylation activity of this engineered CcP mutant was 34-fold greater than that ofWT-CcP and even comparable to that of WT-P450.
In many cases, the catalytic potentials of native enzymes originate from key transition metal centers in the active site. However, redox active amino acids, such as Trp and Tyr, can also play a pivotal role in catalysis, and the surrounding microenvironments of these residues are essential in tuning their redox potential.140,141 This is true for lignin peroxidase (LiP), a fungal enzyme that plays a key role in the ligninolytic cycles for the degradation of the complex polymer lignin.142 The surface Trp171 residue of LiP is proposed to be the site of lignin oxidation, where the surrounding negatively charged residues (Asp264, Asp165, Glu168, and Glu250) provide an acidic microenvironment that enhances the oxidation potential of Trp•+.143,144 Taking inspiration from LiP, Smith, Ivancich, and co-workers redesigned Coprinus cinereus peroxidase (CiP) by incorporating a surface Trp residue (D179W) along with two negatively charged neighboring residues (R258E and R272D) to impart activity toward veratryl alcohol.145 More recently, Field et al. created a similar series of CcP variants (A193W, Y229W, and A193W/Y229W) with Trp residues near the protein surface to mimic the function of LiPs.146 The double mutant A193W/Y229W was found to exhibit the highest oxidizing activity, arising from an efficient electron hopping pathway that acted to accelerate oxidation of the aromatic amino acid.
2.3. Rational Design Using Cytochrome c as a Scaffold
Cytochrome c (Cc) is an electron transfer protein that contains a six-coordinated Fe heme center with axial ligation by one Met and one His (Figure 9a).147 The electron provided by Cc can be utilized by other enzymes, such as cytochrome c oxidase (CcO), for their functions. Unlike typical peroxidases that harness pentacoordinate heme iron with a vacant distal coordination site for substrate binding, the occupation of the distal coordination site by Met80 in Cc seems to preclude peroxidase activity. However, Cc-catalyzed peroxidase reactions still occur despite an initial lag phase that corresponds to the in situ conversion of the six-coordinate precatalyst to an active form, which involves permanent displacement of the Met80 ligand from the heme iron.148 Therefore, the intrinsic peroxidase activity of Cc is suppressed by the protein matrix in its native Met80-bound state when compared to other peroxidases such as CcP and HRP. While the proximal His18 ligation to heme iron in Cc is very robust, the 70–85 Ω loop (also known as Ω-loop D) is quite flexible,149,150 contributing to a relatively weak Met80-FeIII bond.151 Consequently, displacing the Met80 ligand by mutating it to a noncoordinating residue and/or perturbing the unstable Ω-loop D provides a route to converting Cc into peroxidase-like metalloenzymes. Moreover, the remarkable stability and compact globular protein structure of Cc, as well as the covalent bound heme group, make it an excellent structural framework for reengineering.
Figure 9.

(a) Crystal structure of yeast iso-1 Cc (top, PDB ID: 2YCC).147 The flexible coordination loop, Ω-loop D, is colored in red. At the bottom is the multiple sequence alignment of the distal heme region of some classical peroxidases which is compared to the sequence of the distal heme region of yeast iso-1 Cc.154 (b) Substrate recognition sites (SRSs) in P450 enzyme (CYP2C9, PDB ID: 1R9O)155 shown by arrows: SRS1 (red), SRS2 (green), SRS3 (blue), SRS4 (yellow), SRS5 (orange), SRS6 (magenta).156
In order to impart peroxidase-like activity in Cc, the M80V, T67H, and T67H/M80V mutants have been employed to examine the effects of removing the distal Met80 as well as of adding a His residue to the distal pocket.152 The M80V mutation was made in order to displace the native distal Met80 and generate an open coordination site at the heme iron, while the T67H mutation adds a distal His residue with an Nε(His67)-to-Fe distance resembling that found in HRP and CcP (5.84 and 5.55 Å, respectively). Interestingly, the single mutant T67H Cc showed the highest peroxidase activity, even higher than that of T67H/M80V Cc, suggesting that the presence of a distal His residue is more important than eliminating the axial Met ligand for the enhancement of the peroxidase activity in Cc. Further modeling studies suggest that the additional M80V mutation leads to improper positioning of the His67 side chain, thus negatively impacting its function as an acid–base catalyst in the peroxidase cycle.
In addition to SDM, new routes have been explored to design novel metalloenzymes by replacing a full segment of the template protein, such as a loop, with another segment from a different protein. In Cc, residues of Ω-loop D (including Met80) constitute the distal coordination environment of the heme iron. Replacing these residues with key protein segments from other heme-containing enzymes that are responsible for substrate recognition and binding offers a great opportunity for protein engineering. In 2011, Ying et al. reported a P450-mimetic system in Cc by constructing hybrid proteins in which the segment of the coordination loop (residues 78–85) of yeast iso-1 Cc was substituted by the “substrate recognition sites” (SRSs) of P450 (Figure 9b).153 Two constructs, HY1 and HY2, which possess SRS-1 and SRS-2 of P450, respectively, were successfully expressed and further exhibited P450-like function by catalyzing the oxidation of styrene while maintaining the high stability of WT-Cc. In this manner, these hybrids inherited the advantages of both parent proteins. Further enhancement of the peroxidation activity was accomplished by incorporation of the heme pocket of a peroxidase into Cc by structure-assisted mutagenesis in the distal-ligand-containing loop (Tyr67, Asn70, Pro71, and Met80) of Cc along with a highly conserved amino acid sequence.154 The enhanced activities in these variants were primarily ascribed to the stabilization of compound I through the H-bonding interactions involving His and Arg residues. Taken together, these results indicate that loop residues 71–85 are critical for the function of Cc and that protein engineering targeting this region is a promising strategy for generating novel biocatalysts.
2.4. Rational Design Using c-Type Cytochrome Scaffolds
Although peroxidases are ubiquitous enzymes that catalyze the oxidation of many organic compounds, their poor thermal and environmental stability limits their range of practical applications.157 Therefore, engineering peroxidases with improved thermal stability offers a promising route to broadening their industrial application. In 2008, Watanabe and co-workers presented the first design of a thermally stable artificial peroxidase in cytochrome c552 (Cc552) from Thermus thermophilus (Figure 10).158 In these studies, mutations were made to remove the sixth Fe-coordinating ligand (M69A) and to introduce a general acid–base catalyst (V49D). Although V49D/M69A Cc552 displayed an increase in peroxidase activity, significant heme degradation prevented this mutant from achieving sustained catalysis over long periods of time. Shortly after, an improved design of this artificial peroxidase was reported by the additional replacement of Tyr45 with phenylalanine (Y45F) or tryptophan (Y45W) in the basal mutant (V49D/M69A), which resulted in both higher peroxidase activity and thermal tolerance.159 Many class I peroxidases, including CcP and APX, contain a Trp residue in the vicinity of the heme iron, acting as a functional receiver of the oxo-ferryl heme-π-cation radical. EPR studies of the V49D/M69A mutant of Cc552 indicate weak magnetic coupling between the oxo-ferryl heme and Tyr45 radical. Replacing Tyr45 with Trp or Phe resulted in delocalization of the radical and increased magnetic coupling between the oxo-ferryl heme and the protein radical, which helped to suppress heme degradation while increasing enzymatic activity. These results suggest that engineering proteins from thermophiles could be a promising approach to generating artificial enzymes for practical applications. Moreover, this work demonstrates the importance of fine-tuning the microenvironment of redox active protein residues in improving catalytic performance.
Figure 10.

Crystal structure of Cc552 (PDB ID: 1C52).160 The key residues for mutations are shown in light yellow.
2.5. Rational Design Using Cytochrome P450 as a Scaffold
Cytochrome P450s (P450) have enormous biotechnological potential due to their unparalleled activity and selectivity toward oxidative chemistry. Members of the class VIII subgroup of this superfamily, and particularly its’ first discovered member, P450BM3, are commonly employed as scaffolds for redesign as they are fused with their complimenting reductase, forgoing the need for purification and assaying of an additional protein component.161 Furthermore, the high turnover rate and bountiful structural data available for P450BM3 have solidified its instrumental role in the advancement of protein engineering using P450 enzymes. Although numerous studies using both rational design and directed evolution approaches have unearthed common residues that can be altered to expand the scope of the reactivity, the amount of structural evidence capable of providing insight into how mutations guide the binding of new substrates or enhance activity is relatively limited. In 2013, Butler et al. reported the crystal structures of A82F and F87V/A82F mutants of P450BM3 with and without substrate bound (Figure 11a), showing for the first time how the A82F mutation can serve as a “gatekeeper” to alter the substrate binding pocket toward the nonnative omeprazole substrate.162 The enhancement of ligand binding affinity in the A82F mutant was attributed to the large change in free energy between the ligand-free and ligand-bound conformations. Notably, the A82F mutation significantly destabilizes the ligand-free state, thus favoring access of the substrate to the binding site.
Figure 11.

(a) Mode of binding of omeprazole shown for the F87V/A82F P450BM3 double mutant. Reproduced from ref 162 under the Creative Commons CC-BY license. (b) Active site of substrate-free P450BM3 (PDB ID: 2IJ2).168 Conserved residues Thr268, Phe393, and Cys400 are shown in green, and the heme cofactor is shown in purple. (c) From left to right are the crystal structures of WT-P450BM3h (PDB ID: 1BU7),169 the F393A mutant (PDB ID: 1P0V),170 and the F393H mutant (PDB ID: 1JME)171 with the side chain of F393H orienting in two fashions. In the structure of F393A, a hydrogen bond is formed between the side chain amide nitrogen of Q403 and the thiolate of the heme ligating Cys400. For the two orientations of the His side chain in F393H, in one orientation the His side chain hydrogen bonds to an exterior water molecule. In a ring-flipped conformation, the side chain may form a weak hydrogen bond with the axial Cys.167
Engineering efforts in repurposing P450BM3 for cyclopropanation reactions have resulted in highly active, trans selective enzymes. However, engineering in P450BM3 for cis-diastereomers has had limited success, requiring a large number of mutations.163 To access stereoselective cyclopropanation catalysts using simpler variants, Gober et al. applied a strategic mutation, T268A, into a panel of 13 diverse P450 scaffolds and identified T286A P450BM3 as having extraordinary trans-selectivity.164 Although the detailed function of this mutation is not well understood, the crystal structure of T286A P450BM3 suggests that this mutation has little impact on either secondary or tertiary protein structure but rather serves to alter protein dynamics and/or H-bonding in the active site to disfavor the O2 binding and enable nitrene/carbene precursor binding156
Mutation of the conserved heme ligating Cys residue (Figure 11b) to Ser or His in the PCS has provided a basis for P450BM3 variants with substantially increased cyclopropanation activity.165,166 However, these changes in the PCS can result in the stability of the protein itself being compromised. A complementary route toward engineering P450-based biocatalysts for highly active and selective cyclopropanation reactions is through modulation of the SCS.167 It has been demonstrated that mutation of the conserved Phe393 residue to His or Ala increased the heme reduction potential and conversion of the cyclopropanation reaction to comparable levels of the PCS mutants without sacrificing protein stability. Structural analysis (Figure 11c) of the Phe393 variants suggests that the F393A and F393H mutations led to the formation of an electron-withdrawing H-bond to the Cys ligand via Gln403 or His393, thus increasing the heme reduction potential, which is the key to increasing catalytic reactivity.
2.6. Development of Heme Enzymes beyond the Primary Coordination Sphere by Directed Evolution
While the rational design of enzymes based on structure and calculations can offer insights into the role of residues beyond the PCS in promoting catalysis, this approach can still be limited by human intuition or computer programs that may not be able to capture or reproduce the functional properties of PCS and SCS features, especially subtle differences of metal ions and weak SCS interactions. Directed evolution of enzymes allows a route of overcoming these limitations to not only improve enzymatic activity but also unveil new structure–function relationships that are not readily intuited from a rational design approach, including residues far away from the PCS. The general approach of directed evolution uses various techniques to generate a random library of protein mutants, which is then screened based on desired activity.59,130,172 The main requirements for this process to work are (1) starting from an enzyme with nonzero target activity, (2) sufficient stability of the target enzyme to undergo several rounds of mutagenesis, and (3) an effective screening process for assaying the target activity.173 Several ensuing directed evolution studies focused on the P450BM3 enzyme to enhance its substrate or product scope.174-178 In one variant, the positions of several activity enhancing mutations were found to be surprisingly distant from the active site, with less than half of mutations appearing in the substrate binding region.174 Notably, mutations that have been employed by rational design were not found in the selection process of these enzymes.179 Based on activity and binding assays, it was concluded that the electronic environment around the heme binding site was similar to that of the native enzyme. However, despite the directed evolution engineered variant having a higher turnover with native substrate than its native enzyme counterpart, it did not exhibit the same regio- or enantioselectivity, demonstrating the importance of the screening method in acquiring the desired activity.179 In such cases where the development of an appropriate assay is not possible, rational design can be used complementarily. For example, the addition of an A238V mutation to another engineered variant, 9-10A, results in the positioning of a residue close to the alkane methyl group at the active site, drastically improving the regioselectivity for alkane oxidation.
While directed evolution has successfully tailored a number of enzymes for specific activity with target substrates, the roles of many of the resulting mutations are not particularly well understood. However, based on previous rational design studies, a number of mutations in particular scaffolds can be rationalized. P450 has been particularly thoroughly studied, and the reader is referred to ref 180 for an in-depth discussion of the enzyme itself. For unnatural reaction landscapes explored by directed evolution, the reader is referred to refs 77 and 180-187, which cover the most recent advancements in the field, specifically for heme-enzymes, and also focus on the mechanistic aspect of these unnatural transformations.
2.6.1. Discovery of Noncanonical Reactions for Cytochrome P450.
The main driving force behind the P450 mediated oxidation reaction is the formation of an FeIV=O− (heme b•+) intermediate termed as compound I. The core of this system and the subsequent reduced system termed compound II is isoelectronic with iron carbenes and nitrenes, the former of which is well studied (Figure 12).188 Inspired by the similarities between synthetic Fe-porphyrin carbene complexes and compound I, Arnold and co-workers employed P450BM3 for the study of carbene transfer reactions, starting with cyclopropanation of styrene.163
Figure 12.

Structural comparison of the free heme cofactor bound to the cysteine thiolate compared with compound I, iron-carbene, and iron-nitrene intermediates. Reproduced with permission from ref 186. Copyright 2021 American Chemical Society.
To identify an optimal starting candidate for further reaction optimization via directed evolution, native P450BM3 (Figure 13) and 92 other variants were included in an initial selection pool.163 Native P450 is capable of catalyzing the cyclopropanation of styrene; albeit, very low yield numbers (TONs) above 100 have been found. Notably, a number of these active variants, including H2A10, H2-5-F10, and H2-4-D4, contain several Ala residues in the active site across positions Leu75, Met177, Leu181, Ile263, Thr268, and Leu437. To elucidate the effects of each, a series of SDM studies with respect to each active site residue was performed on different select variants. As a result, the T268A mutation was identified as the most important factor for modulating cyclopropanation activity, with a 60-fold increase in activity observed for the single mutant relative to WT-P450BM3. This residue has also been identified as fundamental in native P450 activity, in which its elimination results in almost complete deactivation.180 Based on these observations, it can be inferred that one of the main factors in improving selectivity for newly introduced non-native reactivity is the suppression of the native reaction pathway. These findings concluded on the development of a highly reactive and cis-selective variant termed P450BM3-CIS containing 13 mutations. Next, site saturation mutagenesis (SSM) on the active site residues Leu181, Ile263, Ala328, Leu437, and Thr438 in this variant helped to elucidate the influence of each on reactivity. Out of these residues, only mutations at Ala328 and Thr438 improved the cis-selectivity. The A328G, T438A, T438S, and T438P mutations were found to be the most beneficial without significantly reducing TON.180
Figure 13.

Structure of the active site of WT-P450BM3 (PDB ID: 4ZFA) shown (a) as the full domain and (b) at the enzyme active site.189 Commonly targeted residues for mutagenesis are highlighted in purple, Thr268 is displayed in green, and heme b is in orange. Some residues are not labeled for clarity.
P450 enzymes exhibit redox switching behavior in which the FeIII/FeII potential is decreased upon binding of substrate. Importantly, this behavior only enables reduction of the heme cofactor (and subsequent O2 binding) in the presence of substrate, helping to mitigate the generation of reactive oxygen species (ROS) which could potentially degrade the enzyme or waste reductant via direct formation of compound I or by peroxide-to-water reduction (the oxidase and peroxide shunts, which have been extensively studied).190-192 In ArMs, however, this redox switch is no longer intrinsically present, requiring both stronger reductants to effectively reduce ferric heme and raising potential concerns regarding both inefficiency and protein degradation. Therefore, in the absence of such a mechanism, practical applications of designed P450 mimics as carbene and nitrene transfer catalysts would be limited.
To overcome this barrier, a series of varying proximal ligands in P450 were assayed to optimize activity under in vivo conditions in which C400H and C400S variants were found to be the most active. The latter resulted in a shift of the Soret band for the ferrous-CO bound state to 411 nm, and therefore, the C400S P450BM3 variant is referred to as P411BM3.165 The increased activity of this variant was attributed to an increase in the FeIII/FeII redox potential of the active site and subsequent stabilization of the carbene intermediate. Interestingly, this also drastically increases the cis-selectivity of the enzyme, which could allude to enhanced bond rigidity of the carbene intermediate. Based on these findings, other groups utilized the T268A and C400S mutations on homologous P450 enzymes to achieve similar reactivity with other substrates.193
Combining chemical intuition with directed evolution has proven fruitful for advancing the reaction landscape of these developed enzymes. Wang et al. developed a new P450BM3 cyclopropanation variant with only five mutations relative to the wild-type enzyme (C400H, T268A, L437W, V78M, and L181V) capable of reacting with N,N-diethyl-2-phenylacrylamide in very high yields, as well as high enantio- and diastereoselectivity.194 This showcases the importance of the evolution pathway in reaction optimization.77 In contrast to P450BM3-CIS, this variant was able to achieve higher reactivity with only five mutations, albeit with different substrates. Furthermore, this variant has shown wide alkene selectivity.166
Direct evolution has also been used to expand the substrate scope of cyclopropanation. Brandenberg et al. developed two sets of new variants, P411-VACcis and P411-VACtrans, which were optimized for the catalysis of N-vinylphthalimide cyclopropanate to cis- and trans-cyclopropanes, respectively.195 Previous studies have demonstrated the activity of P450 variants toward N-H insertion,196 and this work further highlighted the challenges involved in designing enzymes with new reactivity while suppressing possible side reactions. Interestingly, the P411-VACcis and P411-VACtrans constructs were also capable of catalyzing O- and S-vinyl compounds, albeit with one additional mutation. The substrate scope of this system was further diversified by the development of a new P411BM3 variant capable of accepting nonaromatic substrates for cyclopropanation,197 and a subsequent study found additional new variants capable of catalyzing C3 or C2 selective pyrrole alkylation of indoles and cyclopropanate cyclic hydrocarbons.198 The key to enabling reactivity has been the utilization of two generations of directed evolution, initially for the development of indole reactivity and subsequently for diversification toward either hydrocarbons or pyrroles.
Directed evolution has also been used to develop P411BM3 into a C-H insertion carbene transfer enzyme.199 Specifically, P411-CHF was developed by directed evolution of P411BM3 and shown capable of sp3 C-H activation with aromatic substrates with high enantio- and regioselectivity for α-carbon over aryl or allylic carbons in alkene substrates. It was also found to be highly selective for C-H insertion over cyclopropanation. Further development of P411-CHF also led to selectivity for highly fluorinated alkanes.200
The scope of the insertion activity of P450 has also been expanded further to include tandem cyclopropanation of bicyclobutanes.201 Previously developed variants of P411 and other heme proteins with nitrene insertion activity were screened for carbene transfer activity with alkyne substrate to provide a stepping stone for subsequent directed evolution of the most active construct (P411-E10). Structural analysis suggested the Trp263 acted to obscure the active site, inhibiting its ability to perform tandem cyclopropanation. Additionally, a second P411BM3 variant was separately developed for cycloprenation catalysis, starting from a less reactive alkyne substrate. The mutations incorporated to achieve this reactivity, however, have not been rationalized. In another study, cyclopropene formation was developed further to incorporate less reactive internal alkynes.202
In most studies of P450-based carbene transfer activity, the carbene precursor, with a few exceptions,198-200 has been ethyl diazoacetate (EDA). Recently, a new P411-CIS variant has been developed which binds α-diazo lactone rather than EDA to perform enantio- and diastereoselective cyclopropanation reactions as well as S─H bond insertion with aromatic substrates.203 The activity of this precursor with the S400C variant (reinsertion of the native Cys ligand) has been studied as well, demonstrating that the proximal ligand affects reaction TON but not selectivity. This reaction has also been independently studied using another variant of P411BM3, which was also capable of binding the same lactone and performing enantioselective C─C bond formation.204
Nitrene transfer reactivity has also been explored using the same strategy employed for carbene transfer, and the same mutants optimized for cyclopropanation also exhibit activity toward intramolecular amination.205 To this end, P411BM3 variants have been commonly employed, including one based on the CIS variant163 and another a double mutant of P411BM3.205 A similar cyclization study performed by Singh et al. showcased the radical character of the aziridation reaction.206 In this study, different stereoisomers reacted with the enzyme to yield the same product, indicating a common intermediate and ruling out a concerted insertion mechanism. Furthermore, comparison of the kinetic isotope effect (KIE) for the cleaved C─H bonds to those of known, concerted, C─H insertion reactions and the correlation of the reaction rate with the C─H bond strength further suggested an intramolecular hydrogen abstraction by the nitrene as the rate-limiting step. Comparisons with previous research207 and analysis of undesired side products demonstrated that the main side reaction for the putative nitrene intermediate is an enzyme mediated reduction similar to native hydroxylation reactions.
In 2016, Arnold and co-workers utilized the sigmatropic rearrangement of allylic sulfimides to form an N─C bond following the initial enzyme-catalyzed nitrene transfer to the sulfur atom.208 The finalized variant, developed through five rounds of SSM, was capable of reacting with a variety of different phenyl thioethers. The developed mutant (P5) was developed to target allylic substrates and was also found to react with nonallylic substrates with high enantioselectivity and TON.
The first enzymatic olefin aziridation catalyst was constructed by Farwell et al. based on P411-CIS, which produced significant quantities of amido-alcohol products due to the styrene-based skeleton of the substrate.209 In 2017, one of the first cases of C-H amination by P411BM3 variants was reported.210 In this variant, SSM of the previously developed P411-CIS variant culminated in a variant termed P411-CHA This variant contains a “reversal” of Phe263 to Leu in addition to three other mutations which activate the enzyme toward enantioselective reactions with different aromatic substrates. Using this approach, Brandenberg and co-workers engineered a P411BM3 variant capable of selective indole amination.211 Initial rounds of SSM revealed a variant capable of [3 + 2] cycloaddition reaction, showcasing the possibility of substrate binding without nitrene generation. Interestingly, this behavior was enabled by a W1046F mutation at the C-terminus of the enzyme. The motivation behind including this region for SSM was to reduce the rate of substrate reduction, and it was initially attempted by truncating the enzyme after its reduction domain to leave only the P450 region. After observing the adverse effects of truncation, SSM of the NADPH binding site was attempted. As Trp1046 stacks on top of the NADPH ring, the mutation to Phe was speculated to disrupt binding, which is supported by the resulting 10-fold decrease in electron transfer rate.
Another study in 2020 expanded the substrate scope to allylic and benzylic amines.212 The nitrene precursor has been changed from tosylazide to pivaloylhydroxyamine, which was based on a previous study213 inspired by the findings of Tsutsumi et al.214 and Morandi et al.215 This serves to disable the direct reduction of the substrate and provides a final primary amine as the product instead of a secondary tosylamine. For benzylic amines, P411-BPA has been engineered, which acts with high selectivity toward them. Another variant, termed P411-APA, has been found to be successful at allylic aminations. The difference between these variants is the addition of two mutations at N395R and S438A.
In 2021, Athavale et al. reported a P411 variant developed through nine rounds of directed evolution which is capable of nitrene transfer of pivaloxyl-amides.216 In contrast to the P411 enzymes designed for tosylazide as the substrate, the rate-determining step (RDS) in this variant is not C-H activation but nitrene formation as shown by KIE studies. These variants extend the substrate scope of the benzylic amines while also increasing the TON compared to that of P411-BPA.
As discussed vide supra, the redox switch mechanism is not intrinsically present in engineered enzymes, the O2 binding capacity has been drastically altered via numerous mutations, and the active intermediate formation and substrate binding steps can be at least partially uncoupled. As a result, for the carbene intermediate, side reactions with nearby residues are one of the main factors limiting the enzyme turnover number (TON).217 For nitrene transfer, the main barrier has been the reduction of the nitrene intermediate to its corresponding amine prior to reaction with the substrate. These side reactions could be potentially suppressed by either mutation of the residues responsible for the side reaction or optimization of the enzyme reactivity to increase the reaction rate of the generated compound one-like intermediate.218
2.6.2. Myoglobin as a Scaffold.
One of the first reports of directed evolution in a heme protein was presented by Wan et al., who enhanced the peroxidase activity of Mb 25-fold through a quadruple mutant without sacrificing the O2-binding affinity.219 This same mutant was later used as a basis for the development of Mb variants active toward carbene and nitrene transfer. Several key amino acids that define the distal pocket in Mb significantly influence the activity and selectivity of this artificial biocatalyst in carbene- or nitrene transfer reactions.124-127,131 In particular, mutations at the “gating” residue His64 lead to a general activity enhancement for many carbene-mediated reactions. SSM at the active residues Leu29, Phe43, His64, Val68, and Ile107 (Figure 14a) has proven to be a valuable strategy in generating biocatalysts with high proficiency and superior selectivity.
Figure 14.

(a) Crystal structure of WT-swMb (PDB ID: 5YCE)89 with key residues in the distal heme pocket for directed evolution shown in magenta. (b) Stereochemical model for intramolecular cyclopropanation catalyzed by the stereodivergent Mb variants. Reproduced from ref 220. Copyright 2019 American Chemical Society.
By screening the active site mutation landscape library, in which active positions are subject to substitution by all other possible 19 amino acids, two triple mutants of Mb, L29A/H64V/V68A and F43A/H64W/V68F, were developed that displayed high but complementary stereoselectivity toward intramolecular cyclopropanation (Figure 14).220 Although detailed structural evidence for this stereodivergence is currently lacking, it is clear from mapping these mutations onto the Mb structure that the active site configurations of these two mutants are distinct; smaller residues in positions 29 and 68 in L29A/H64V/V68A render an expanded cavity in the upper side of ring A/D of the heme, while increased steric bulk in the F43A/H64W/V68F variant enlarged the cavity at the opposite side of the cofactor (Figure 14b). Therefore, by fine-tuning the distribution and expansion of the distal heme pocket by active site mutagenesis, one can modulate the stereoselectivity of Mb-based biocatalysts.
The active site of Mb has also been configured using directed evolution to enhance opposite enantioselectivities for intramolecular cyclopropanation via the quadruple mutants F43L/H64A/V68G/I107V Mb and F43Y (or F43H)/H64A/V68G/I107F Mb.221 Notably, R-selectivity was achieved by increasing the steric bulk at position 107 (I107F) together with addition of an aromatic group with hydrogen bonding abilities (F43Y or F43H) at position 43. In contrast, S-selectivity could be made favorable by reduced the steric bulk at both positions 43 and 107 (F43L and I107V). To expand the substrate scope to more sterically demanding intramolecular cyclopropanation reactions, SSM was used to target active site residues to further expand the distal heme pocket, resulting in an optimized F43Y/H64V/V68A/I107V Mb mutant.222 These results also supported that positions Phe43 and Ile107 are especially critical for the stereoinduction of substrate when large (i.e., aryl) substituents on the olefinic group are present.
Inspired by previous work investigating mutations at His64 and Val68 to tune the stereoselectivity of the cyclopropanation reactions,124,135 Hernandez et al. mutated the analogous residues (T45 and Q49) in B. subtilis truncated globin to one of three nonpolar residues (Leu, Phe, and Ala) in an effort to enhance the performance toward cyclopropanation.223 The double mutant T45A/Q49A exhibited high selectivity, possibly due to the increased volume of the active site.
It is notable that the structure-guided rational design and directed evolution methods are not alone in guiding protein engineering for developing artificial metalloenzymes. Although these two approaches have provided fruitful results, both methods are time consuming and labor intensive. The efficiency of protein engineering can be significantly improved by employing calculations to identify promising target residues and mutations. Thanks to advances in computational tools, calculation-guided methods have emerged as effective comple-mentary tools for protein engineering. For the practical use of artificial enzymes, protein stabilization against thermal and chemical denaturation is a long-standing goal in enzyme engineering. In 2017, Moore et al. reported the development of a Rosetta-guided protein design to optimize protein stability for better performance in enzymatic catalysis.224 Starting from the H64V/V68A Mb construct, which has been found to perform carbene-mediated cyclopropanation reactions with enhanced activity and high diastereo- and enantioselectivities, the authors utilized the Rosetta enzyme design framework to identify the optimal positions for incorporating a thioether covalent staple to improve its robustness for synthetic applications, obtaining nine designs that were further examined for stability and catalytic activity in cyclopropanation reactions. This methodology allowed them to uncover stabilized variants with superior catalytic performance compared to the parent enzyme when exposed to organic solvents.
2.6.3. Rma Cytochrome c as a Scaffold.
Protein machinery promiscuity is observed not only in the P450 enzymes but also in bacterial cytochrome c from Rhodothermus marinus (Rma-Cc, Figure 15a), which is an electron transfer protein and has no catalytic functions in living systems. However, Arnold and co-workers have uncovered activity in Rma-Cc toward carbene-mediated C─Si bond formation and thereafter pursued the laboratory evolution of Rma-Cc variants to perform many new-to-nature functions with high activity and selectivity.225
Figure 15.

(a) Crystal structure of WT-Rma-Cc (PDB ID: 3CP5)230 with the key residues involved in directed evolution shown in purple. (b) Crystal structure of carbene-bound TDE-Rma-Cc (PDB ID: 6CUN) with the carbene species in cyan. (Inset) Interactions between the carbene and amino acid residues are shown with yellow lines: (i) Thr75, 3.7 Å; (ii) Met76, 4.3 Å; (iii) Pro79, 3.5 Å; (iv) Ile83, 3.6 Å; (v) Met89, 3.5 Å.226
In 2018, Kan et al. generated a triple mutant, V75T/M100D/M103E (TDE-Rma-Cc), by SSM at three positions near the distal heme pocket to perturb the distal ligand (M100) and the two residues that are close to the heme iron (Val75 and Met103).226 The evolved variant catalyzed the C─Si bond formation with more than 15-fold higher turnover than state-of-art synthetic catalysts and extremely high enantioselectivity (>99% ee). In 2018, Lewis and co-workers resolved the high-resolution crystal structure of the TDE-Rma-Cc mutant, which revealed a mutation-generated pocket in the distal side of the heme.226 Molecular dynamics (MD) simulations indicated that the front loop (Thr98-Glu103) of the active site is quite flexible, adopting an “open” conformation which favors the accessibility of substrate to the iron porphyrin carbene (IPC) intermediate with pro-R facial selectivity. The more recently solved high resolution crystal structure of carbene-bound TDE-Rma-Cc reveals how the laboratory-evolved enzyme achieved perfect carbene transfer by holding the iron porphyrin carbene in a single orientation (Figure 15b).226
In addition to C─Si bond formation, the Rma-Cc-based biocatalyst has been evolved for C─B bond formation by multiple rounds of SSM.227 By incorporating different substitutions at Val75 and Met103, the borylation reactivity was directed to diverse diazo ester substituents. The substrate scope of these biocatalysts was later expanded to perform C─B bond-forming chemistry with more rigid, cyclic, lactone-based carbenes by creating a V75R/M99/M100D/T101Y/M103V mutant (BORLAC).228
Encouraged by the beneficial mutations targeting the active site front loop, Cho et al. continued to explore the new-to-nature functions of the thermostable Rma-Cc platform by generating a biocatalyst for enantioselective aminohydroxylation of styrenyl olefins.213 Apart from the SSM targeted residues (Thr98, Met99, Thr101, and Met103) within the same loop as the Met100 ligand, mutations at Met76 and Tyr44, which are relatively far from the active site, also improved the catalytic performance. Similarly, a Y44I mutation enhanced the borylation activity and enantioselectivity of an Rma-Cc-based biocatalyst for the synthesis of chiral α-CF3 organoboranes.229 MD simulations revealed that the -CF3 group pointed toward the active site, whereas the bulkier phenyl group lay between the side chains of the M103D and Y44I substituted residues, implying that the primary roles of these mutations were to facilitate the binding of the diazo substrate. Therefore, residue 44 could be a key to modulating substrate selectivity in this artificial enzyme.
The TDE-Rma-Cc mutant was revisited in 2021 by Garcia-Borràs et al. to study the origin of the chemoselectivity of this variant toward C─Si over C─N bond formation.231 A combination of computational and experimental methods suggests that the protein front loop conformation and dynamics dictate silane binding, which is a major contributor to the rate-limiting step. The more favorable silane binding affinity in the TDE-Rma-Cc mutant may result from the increased flexibility of the front loop, induced by the hydrophilicity of the Asp100 and Glu103 side chains. Redesign of several mutations in the front loop led to the generation of a new protein (N80F/M99P/E103I TD-Rma-Cc) in which chemoselectivity was switched from silylation to amination.
2.7. DNA as a Scaffold for Heme-Based Biosynthetic Models
While proteins have long been known to serve enzymatic functions, it has also been discovered in recent decades that both RNA and DNA can function as enzymes.232-234 Naturally occurring oligonucleotides are composed of a much narrower range of building blocks in comparison to proteins, and therefore, recruiting metallocofactors such as heme can help oligonucleotides to expand their enzymatic capabilities. Based on this hypothesis, Li, Geyer, and Sen used in vitro selection to obtain single-stranded DNA aptamers that specifically bound N-methylmesoporphyrin IX (NMM), a stable transition-state analogue for porphyrin-metalation reactions.235 A library of 1.8 × 1015 DNA molecules was screened for NMM binding affinity using NMM-bound acrylic beads through HPLC, subjected to both positive and negative selection in 12 rounds. In the DNA aptamers that bound strongly, sequence analysis revealed a common guanine-rich motif. Further characterization of the resulting DNA aptamers showed that hemin could form complexes with guanine-rich, short single-stranded DNA molecules with submicromolar affinities. The authors presented spectroscopic evidence that this complex more closely resembled a peroxidase than it did uncoordinated hemin, consistent with the observed increase in peroxidase activity from the hemin/DNA complex when compared to hemin alone.236
Shortly thereafter, the electron paramagnetic resonance (EPR) and UV/visible spectra of a folded guanine-rich oligonucleotide and hemin complex (wholly referred to as PS2.M-hemin) were shown to be similar to the spectra observed for high-spin ferric heme with axial symmetry, as observed with myoglobin.237,238 The DNA component of PSM2.M-hemin was known previously to form a G-quadruplex structure, in which tetrads of guanosine stack atop each other to form a four-stranded supermolecule under physiological salt conditions.239 The initial work characterizing the peroxidase activity of a G-quadruplex/heme complex used ABTS as a substrate.236 A little more than a decade later, the G-quadruplex/heme complex was demonstrated to act as an NADH oxidase and NADH peroxidase mimic, oxidizing NADH via O2 to generate H2O2.240 The complex of G-quadruplexed DNA and heme has since been characterized as a peroxidase-like DNAzyme, able to accommodate ligands in the distal sixth coordination site such as CO, O2, and imidazole.241,242 Heme-G-quadruplexed-DNA-zymes, however, have a major difference from protein peroxidases, as they lack a proximal imidazoyl ligand from a histidine amino acid residue.
Ten years after the spectroscopic investigation on PS2.M-hemin, the same complex was shown to catalyze two-electron oxidation reactions with substrates such as thioanisoles, indole, and styrene, and 18O labeling demonstrated that the transferred oxygen atom originates from hydrogen peroxide through what is presumably a ferryl moiety.243 Docking simulations of heme to a G-quadruplex structure suggested a lack of a structured environment distal to the heme, and it was suggested this is linked to the lack of enantioselectivity observed with oxidation reactions catalyzed by PS2.M-hemin, as the oxygen transfer reaction to thioanisole produces a racemic mixture and indole oxidation yields several products.243
Although no additional complexes of oligonucleotides with heme have been reported since, several other interactions do exist with other porphyrin molecules in complex with DNA.244 In G-quadruplex DNA formed from the repeating human telomere sequence d(TTAGGG), ferrous heme binds to the 3′-terminal G-quartet through π−π stacking interactions.245-247 Chemical modifications of the heme in the d(TTAGGG)-heme complex to form different electron densities at the catalytic iron site altered the peroxidase activity of the complex, with decreasing peroxidase activity observed with decreasing ρFe.248 Resonance Raman spectroscopy has revealed that the vibrational feature νCO for the [d(TTAGGG)]4-heme complex with a bound CO adduct appears at a lower frequency than in myoglobin, suggesting a weaker donor strength of the proximal ligand and slightly longer Fe─C bond.248 This agrees well with earlier NMR work of chemically modified hemes in complex with G-quadruplex DNA, where the fifth (proximal) ligand appears to be a water molecule.247
In 2019, a different G-quadruplex/heme complex (called G4) was shown to work as a catalyst for carbene insertion into styrene, expanding the range of heme-DNAzymes beyond oxygen transfer reactions.249 As the heme molecule is situated above the 3′ G-quartet, the 3′ end of the oligonucleotide is free to interact with the cofactor—the authors report that the presence of 3′-terminal AAA or TTT results in indistinguishable effects upon carbene insertion, while AAA shows higher oxidation rates relative to TTT.249 The addition of multiple pendant nucleotides shows increasing stereoselectivity compared to porphyrin/DNAzymes that do not possess nucleotides on the distal side of the porphyrin. Figure 16 shows the “active site” of the heme-DNAzymes studied. Reactions were limited to oxene and carbene insertions, but they are mechanistically distinct from the FeIV-based, radical-dependent mechanism of heme monooxygenases.183
Figure 16.

Active site of heme/DNAzymes (GQ-heme complexes), where the heme end-stacks on the 3′-most guanine-quartet (gray squares): (a) “G4-AAA” and “G4-TTT” oligonucleotides both form intramolecular, propeller looped GQs, with the 3′-terminal AAA or TTT overhanging (shown as blue rectangles). (b) An intermolecularly assembled GQ such as “(dA4G5A4)4” is expected to have a complex “distal” environment above the stacked heme, potentially imparting stereoselectivity to reactions catalyzed by the DNA-bound heme. Reproduced with permission from ref 249. Copyright 2019 American Chemical Society.
While there is a lack of crystal structure data for a G-quadruplex/hemin complex, efforts from the Mergny and Petitjean groups produced a planar PtII complex that binds in the same proposed position where hemin binds atop the G-tetrads at the 3′ end.250 This serves as a good structural model for the heme binding position on G-complexes in general, but the Pt/DNA complex does not display the proximal water ligand that has been demonstrated in peroxidase-type heme/DNAzymes.
3. CATALYSIS BEYOND THE PRIMARY COORDINATION SPHERE BY DESIGNED NON-HEME IRON ENZYMES
Non-heme Fe enzymes constitute a broad class of proteins that bind Fe to promote a variety of reactions, many of which parallel those of heme enzymes. Many non-heme Fe enzymes employ mono- or dinuclear Fe sites to promote O2 activation and incorporation into organic substrates.251,252 Additionally, non-heme Fe enzymes have been found to promote reactions including nitric oxide reduction, the reversible of oxidation of H2, and even the reduction of atmospheric nitrogen to ammonia.56,253-256 In many O2-activating mononuclear non-heme Fe enzymes, the active site is composed of a (His)2-carboxylate binding motif arranged to facially coordinate FeII, commonly referred to as the “facial triad”, with one or two labile ligands (such as H2O) that allow facile binding of O2.257-260 Dinuclear non-heme Fe enzymes that serve to activate O2 or reduce NO typically exhibit mixed His and carboxylate-coordinated active sites, often with one or more carboxylates (Asp or Glu) bridging the two irons that can appear in both the terminal and bridging positions and additional non-amino acid coordinating atoms such as H2O, OH−, or O2−.252,261 Further non-heme Fe enzymes may offer more exotic coordination environments, such as the 5,10-methlenetetrahydromethanopterin cofactor in Fe hydrogenase (H2ase),262 the bridging dithiolate/pendant amine cofactor and CO/CN− ligands of FeFe H2ase,263 and hetero iron-sulfur-based clusters of nitrogenases and carbon monoxide dehydrogenase (CODH).264-267
The broad nature of non-heme iron systems naturally affords a high degree of modularity in the PCS and SCS. We have organized our discussion of these systems first in terms of mononuclear vs dinuclear systems and then further between approaches toward the active site, namely, (1) the use of amino acid residues to provide the PCSs of metal centers, (2) those which employ unnatural amino acids for metal coordination and those which incorporate a guest organometallic complex in a host protein scaffold (also referred to as the “Trojan horse” approach) using (3) covalent interactions and (4) supra-molecular interactions.
3.1. Design of Mononuclear Non-heme Iron Proteins
3.1.1. Design Using Natural Amino Acids.
One of the earliest studies to rationally design a catalytically active mononuclear non-heme Fe site into a non-native protein scaffold was performed by Pinto, Hellinga, and Caradonna.268 Utilizing the scaffold thioredoxin (Trx), a facial triad binding site was designed with the assistance of the program DEZYMER via Leu7His, Phe27Asp, Ile60His, and Asn63His mutations (Figure 17). Fe binding was observed via the appearance of a new absorption feature at 350 nm, assigned to a His → FeIII ligand-to-metal charge transfer transition. When tested via colorimetric assay, the Fe-Trx ArM exhibited significant superoxide dismutase (SOD) activity, with a dismutation reaction rate of k ~ 105 M−1 s−1.
Figure 17.

Crystal structure of Trx from E. coli (PDB ID: 2TRX).269 First coordination residues in designed Trx-SOD were made to residues highlighted in light gray, and SCS mutations are highlighted in blue.268 Atom coloring: S (yellow); O (red); N (blue).
Azurin (Az) has also served as an effective scaffold for the design of low-coordinate mononuclear non-heme Fe binding sites. Az is a type 1 copper metalloprotein but has garnered attention for protein engineering efforts due to its stability and ease of expression. The use of Az to bind Fe was first demonstrated by Holland and co-workers,270 who were able to incorporate FeII into both WT-Az and the M121A Az variant. Surprisingly, WT-FeII-Az appeared to resist oxidation and reduction by a variety of redox agents, suggesting that the accessible iron reduction potentials are outside the physiological potential range from approximately −1 to + 1 V vs SHE. Characterization by a combination of 1H NMR and 57Fe Mossbauer spectroscopies strongly indicates that there is little structural difference between WT-FeII-Az and M121A FeII-Az, implying the absence of Met121 does not significantly affect the coordination geometry. X-ray crystallography reveals clear coordination of Fe by His46, Cys112, and His117 and likely Gly45 as well. Further N3− binding experiments reveal that replacement of Met121 for Ala opens an additional binding pocket without perturbing metal binding. Additional binding experiments demonstrated the ability of CN− to coordinate Fe but that alternative ligands such as N-methylformamide, hydrazine, and CO are incapable of binding.
Following the initial discovery of Fe binding in Az, the Liu group further engineered the binding pocket to impart superoxide reductase activity via a M121E substitution (Figure 18b).271 FeII incorporation into M121E Az was supported by a combination of absorption spectroscopy, inductively coupled plasma (ICP) analysis, and electrospray ionization mass spectrometry (ESI-MS). Successful crystallization of M121E FeII-Az allowed structural determination at 2.0 Å resolution. Interestingly, M121E FeII-Az crystallizes as a tetramer of two heterodimers, with subunits A and C in conformation 1 and subunits B and D in conformation 2. No observable occupancy was found in conformation 1, while 100% occupancy was found in conformation 2. In addition to Gly45, His46, C112, and His117, Glu121 was found to coordinate FeII through the extended carboxylate side chain. Unlike WT-FeII-Az or M121A FeII-Az, the FeII → FeIII redox couple of M121E FeII-Az was found to be readily accessible, with a reduction potential of +0.327 V. Further testing for superoxide reductase (SOR) activity using the Fridovich method273 revealed an SOR activity, with a reduction rate of k = 1.8 × 104 M−1 s−1. Based on observations for native SOR, a second substitution was made exchanging Met44 for Lys. In the native system, it is believed this SCS residue aids in guiding superoxide to the metal center or even potentially stabilizes binding of the negatively charged superoxo via its positively charged side chain. This rational modification further only slightly lowered the FeII/FeIII redox couple to +0.320 V but boosted the SOR activity of M121E/M44L FeII-Az by 2 orders of magnitude to 1.1 × 106 M−1 s−1 relative to M121E FeII-Az, supporting the importance of this SCS interaction in promoting the SOR activity.
Figure 18.

(a) Crystal structure of M121E Az (PDB ID: 4QLW).271 (b) Fe-bound metal binding site of M121E Az. (c) Cu-bound metal binding site of M121H/H46GE (PDB ID: 4WKX).272 PCS atoms are highlighted in light gray, while SCS target residues are highlighted in blue. Atom coloring: S (yellow); O (red); N (blue); Fe (orange); Cu (copper).
More recently, the Lu group has further utilized Az to engineer the formation of a dinitrosyl iron complex (DNIC).274 Nitric oxide (NO) plays an important role in numerous processes ranging from cellular signaling to immune defense, and understanding how these processes are regulated by non-heme Fe proteins remains an intense subject of investigation. Note that in the following we will use Enemark–Feltham notation, {MNO}n, to describe the overall oxidation state of NO-bound metal (M) systems, where n denotes the number of valence electrons (metal d plus NO π* electrons).275 Using the M121H/H46E Az scaffold previously developed for CuII binding aimed at S-nitrosylation276 (Figure 18c), FeII binding was observed via a combination of absorption and 57Fe Mössbauer spectroscopy. Further addition of 1 equiv of NO produced significant spectroscopic changes in the absorption spectrum, as well as the appearance of a new S = 3/2 signal as observed by EPR, indicative of {FeNO}7 formation. This new S = 3/2 species was actually a convolution of two signals with unique zero-field splitting (ZFS) properties, representing two different conformations of the {FeNO}7 species at low temperature, with one slightly more axial and the second more rhombic. By combining EPR and field-dependent 57Fe Mössbauer spectroscopic measurements with density functional theory (DFT) calculations, it was proposed that these species represented two different PCSs. Specifically, the more axial species was proposed to arise from {FeNO}7(His)(Glu)(Cys) coordination, while the more rhombic from {Fe-NO}7(His)2(Glu). Interestingly, room temperature resonance Raman (rR) measurements revealed a single set of Fe-NO and NO vibrations, indicating that the appearance of two conformers as observed by EPR and 57Fe Mössbauer spectroscopy may only arise at low temperatures.
Exposure of M121H/H46E {FeNO}7-Az to an excess of NO produced a new novel species, which exhibited an axial S = 1/2 signal as observed by EPR. Using 14N/15N Q-band electron double nuclear resonance (ENDOR) spectroscopy, two isotope-dependent signals with relatively weak hyperfine interactions (3.6 and ~ 7 MHz in 15NO) were observed, supporting the formation of a {Fe(NO)2}9 DNIC species. This hypothesis was further bolstered by the presence of two unique sets of Fe-NO/NO stretching frequencies as observed by Fourier transform infrared (FT-IR) and rR spectroscopies. Further 1H Q-band ENDOR measurements supported Cys coordination, and by combining these results with 57Fe Mössbauer measurements and DFT calculations, a {Fe(NO)2}9(His)(Cys) coordination environment was proposed. Finally, investigation of the mechanism of {Fe(NO)2}9 formation from {FeNO}7 revealed the presence of an additional S = 1 intermediate species assigned as {FeNO}8. Single-value decomposition of time-dependent absorption measurements shows this species is on-path to {Fe(NO)2}9 formation, and researchers further propose this species to be {FeNO}8(His)(Glu)(Cys) coordinated based again on 57Fe Mössbauer measurements and DFT calculations. This study not only provides insight into the stepwise nitrosylation of Fe in a protein scaffold and the resulting coordination environments but strongly illustrates the deep structural insight that can be obtained by applying a combination of multiple orthogonal spectroscopic and computational methods.
In addition to Az, reengineering of rubredoxin (Rd) has also allowed for successful {FeNO}7 formation. Rd is a low-molecular-weight electron transfer protein which natively binds Fe through the coordination of 4Cys (Figure 19). Substituting either Cys8 or Cys41 for Ala in Clostridium pasteurianum (Cp) Rd it has been shown to open a binding site, forming Fe(Cys)3(OH) coordination.278 Using this approach, Lin and He were able to generate an open binding site for FeIII in Rd from Pyrococcus furiosus (Pf) and subsequently bind NO following reduction with sodium dithionite (DT) to form an air-stable C9A {FeNO}7-Rb (and equivalently C41A {FeNO}7-Rb).272 The dominant S = 3/2 species was characterized by EPR and absorption spectroscopies; interestingly, an additional S = 1/2 signal was also observed and assigned to a nonspecifically bound {FeNO}7 species. Irradiation with red light (625–650 nm) resulted in the stoichiometric release of NO from both C8A {FeNO}7-Rb and C41A {FeNO}7-Rb systems under either aerobic or anaerobic conditions. Under anaerobic conditions, photolysis of NO resulted in the FeII precursor, and further air exposure produced the starting FeIII complex, which in turn was capable of binding NO to reform the {FeNO}7 complex. Meanwhile, photolysis of C8A {FeNO}7-Rb under aerobic conditions showed similar properties to the FeIII starting material, but with a ~16 Da increased molecular weight. Based on this difference in molecular weight, it was hypothesized that oxygenation of either a binding Cys or Fe itself may occur. This species further decomposed to C8A FeIII-Rd, which then could be used to also reform the {FeNO}7 complex. Further incubation with plasmid pBR322 DNA demonstrated photolysis of C8A {FeNO}7-Rb promoted DNA cleavage, demonstrating the practical biological applications of this system.
Figure 19.

Crystal structure of Pf Rd (PDB ID: 1VCX).277 Atom coloring: S (yellow); Fe (orange).
3.1.2. Design Using Unnatural Amino Acids.
One of the first examples utilizing an unnatural amino acid (UAA) to generate a biosynthetic non-heme iron binding pocket was accomplished by Schultz and co-workers, who genetically encoded E. coli to incorporate (2,2′-bipyridin-5-yl)alanine (AlaBpy) into the sequence of a DNA-binding protein).279,280 Earlier studies have shown that incorporation of FeII-EDTA in DNA binding enzymes at the DNA interface induces oxidative cleavage at deoxyribose,281,282 which has inspired efforts to directly bind FeII using bipyridyl as a high-affinity ligator. The homodimeric catabolite activator protein (CAP) was chosen as a scaffold due to its specificity for a 2-fold symmetric 22 base pair binding site. Inspired by the crystal structure of the CAP-DNA complex,283,284 Lys26 was chosen for substitution to form the L26ABpy CAP variant (Figure 20a). Determination of the Kd of L26ABpy CAP vs WT-CAP for a 50 base pair DNA strand containing the 22 base pair binding site revealed similar affinities for both proteins, demonstrating that incorporation of AlaBpy did not significantly perturb the CAP-DNA interaction. Incubation of L26ABpy CAP with the same sequence in the presence of FeII, cyclic adenosine monophosphate (cAMP), and a reducing agent such as ascorbic acid or 3-mercaptopropionic acid resulted in DNA cleavage. Analysis of the products revealed cleavage primarily at either end of the recognition sequence, near the anticipated location of the AlaBpy. As cleavage was not observed in the absence of either AlaBpy or FeII (or equivalently CuII), it was inferred that the formation of an FeII-Bpy complex was necessary for activity. Continuing their work in DNA binding and regulation, Schultz and co-workers utilized the Zif268 transcription factor protein to design a novel FeII binding site, again using AlaBpy as a chelating ligand.285 Zif268 is a zinc finger protein, which uses ZnII to stabilize the protein fold through ZnII(Cys)2(His)2 coordination (Figure 20b). While Zif268 natively binds a specific nine base pair double-stranded DNA sequence, modification of the finger domain can lead to binding of both alternative sequences and other metals. Following successful incorporation of AlaBpy into Zif268 at three different positions (26, 28, and 29), it was found that all three mutants were capable of binding DNA with comparable affinities and with the same site specificity as WT-Zif268. Furthermore, substitution of AlaBpy for AlaBph (Bph = biphenyl) eliminated DNA binding affinity, as did the presence of EDTA, together demonstrating a requirement for both AlaBpy and a metal ion. Using a combination of ESI-MS along with absorption and EPR spectroscopies, it was elucidated that DNA binding by Zif268 AlaBpy is FeII-dependent. Comparison of the rR spectra from FeII-Zif268 AlaBpy and [FeII(bpy)(CN)4] was indicative of FeII-bpy coordination. Further 57Fe Mössbauer measurements revealed a single species with d = +0.41 mm/s and ∣ΔEQ∣ = 0.34 mm/s, consistent with low-spin FeII. This result was further corroborated by temperature-dependent magnetic circular dichroism (VT-MCD) measurements, where the similarity of the low- and high-temperature spectra revealed a diamagnetic (S = 0) system. Together, these results support that incorporation of the UAA AlaBpy to Zif268 results in a similar fold to WT-Zif268, but with alternative metal selectivity.
Figure 20.

Crystal structures of target proteins for AlaBpy incorporation. (a) Catabolite activator protein (CAP) with the Lys26 target location of AlaBpy incorporation highlighted (PDB ID: 1J59).280,284 (b) Transcription factor protein Zif268 with target residues for randomization highlighted. Native metal binding residues are highlighted in light gray, while additional target residues are highlighted in blue (PDB ID: 1ZAA).288,285 (c) LmrR with target residues for AlaBpy incorporation highlighted (d) in light gray, with additional SCS target residues highlighted in light blue (PDB ID: 3F8B).286,287 (e) Designed AlaBpy-incorporated protein scaffold based on indole-3-glycerolphosphate synthase containing bound Ni2+ (PDB ID: 4IXO) and (f) Co2+ (PDB ID: 4IWW). PCS residues are highlighted in light gray, while SCS target residues are highlighted in light blue.289 Atom coloring: N (blue); O (red); S (yellow); Fe (orange); Co (salmon); Ni (green).
Incorporation of UAAs has also been used to design metal binding sites for radical intermediate stabilization. Continuing from their previous work using a AlaBpy-incorporated lactococcal multidrug resistance regulator (M89X LmrR) protein (Figure 20c and d), Roelfes and co-workers probed the influence of varying the surrounding AA residues on the binding affinity for 3d transition metals ranging from MnII to ZnII.286,287 In order to bind FeII (as evidenced by the appearance of a 310 nm LMCT absorption band), additional substitutions of F93D and V15G were required, evidencing the need for available carboxyl ligands to induce FeII binding to bpy. Furthermore, FeII was found to bind and stabilize 3,5-di-tert-butylsemiquinone (DTB-SQ).
A significant challenge in designing competent metal catalysts in non-native scaffolds is understanding how each substitution will impact both structure and function. This problem can be even greater when employing unnatural amino acids, as most studies have focused around the use of natural AAs. Using the program Rosetta,290,291 Baker and co-workers computationally designed a series of potential divalent metal binding sites utilizing AlaBpy, with the goal of forming a catalytic site mimicking catechol dioxygenase.289 Potential models were screened around a PCS involving AlaBpy and dopamine (a catechol). Genes for the best 13 candidates were synthesized and expressed in E. coli, of which five successfully produced soluble protein and two (CB_02 and CB_12) were capable of binding FeII. A successful crystallization of CB_02, a scaffold based on bacterial sialidase, revealed the AlaBpy side chain to be oriented outward toward solvent rather than aiding formation of a well-organized metal binding site. To overcome this, a second round of computational design was undertaken with a more constrained metal binding site geometry involving AlaBpy, Asp, Glu, and His, as well as two H2O molecules replacing the catechol moiety. Of the 28 designs investigated, eight appeared to bind FeII in a buried fashion. Model MB_07 (based on indole-3-glycerolphosphate synthase) was successfully crystallized with both bound CoII and NiII (Figure 20e and f) showing good homology with the respective computational designs. Although FeII-MB_07 could not be crystallized for direct comparisons, this pioneering work demonstrates the power of computational approaches in rationally designing PCS and SCS interactions to form novel metal binding sites.
3.1.3. Design via Covalent Interactions.
In addition to direct natural AA or UAA coordination, protein–metal binding sites can be generated via direct incorporation of an exogeneous organometallic complex into a host scaffold, commonly referred to as the “Trojan horse” method. Early applications of this approach were pioneered in the late 1990s by Distefano and co-workers by incorporation of a metal-binding 1,10-phenanthroline and pyridoxamine derivatives into adipocyte lipid binding protein.292-295 This method is less commonly used to investigate the function of natural metalloenzymes and instead imparts altogether new catalytic activity. Here, the protein serves to impart SCS effects, such as controlling solvent access and hydrophilicity/hydrophobicity, charge screening, hydrogen bonding, enforcing conformers, and even imparting stability. Anchoring of these complexes can be accomplished either directly through covalent linking or via supramolecular interactions.
Maleimide reacts facilely with thiols via Michael addition to form thiosuccinimide under very mild basic conditions (pH ~ 7.5), providing an excellent route to forming a covalent linkage with Cys side chains (Scheme 2). Although use of this route requires careful consideration of cysteine accessibility, it offers a powerful tool for the incorporation of guest complexes into a wide range of protein scaffolds.
Scheme 2.

Banse and co-workers prepared one of the first examples of a mononuclear nonheme Fe site generated using a thiosuccini-mide anchor by incorporation of [FeII(L52maleimide)Cl]+ into isoform B of bovine β-lactoglobulin (β-LG, Figure 21a) (L52maleimide = 1-(2-{[2-(Bis-pyridin-2-ylmethylamino)ethyl]-pyridin-2-ylmethylamino}ethyl)pyrrole-2,5-dione)).296 The [FeII(L52maleimide)Cl]+ complex was specifically chosen based on its reactivity with thioanisole and H2O2 to form phenyl-methylsulfoxide. Interestingly, incorporation into β-LG resulted in a 5-fold decrease in reactivity while simultaneously producing a 20% enantiomeric excess (ee). In the absence of thioanisole substrate, time-dependent absorption measurements revealed a transient signal at 660 nm maximizing around 4 min; further EPR measurements of the reaction mixture revealed this to be an S = 1/2 species, consistent with the formation of a low-spin FeIII intermediate. Based on the energy of the 660 nm transition, this intermediate was further proposed to be FeIII(H2O2) at room temperature, undergoing an acid/base equilibrium to form FeIII(OOH) at low temperatures.
Figure 21.

Crystal structures of target proteins for Cys-maleimide linkages. (a) β-Lactoglobulin isoform B (β-LG) with the target Cys121 for thiosuccinimide formation highlighted in light gray (PDB ID: 4IB9).296,297 (b) Steroid carrier protein SL (SCP-2L) containing an Ala100Cys mutation as the target for thiosuccinimide formation (highlighted in light gray). An additional target residue, Phe94, targeted for SCS modifications, is highlighted in light blue (PDB ID: 6Z1W).298,299 (c) Xylanase A (Xln) with target residue for Ser212 (for the Ser212Cys incorporation highlighted in light gray, with additional SCS target residues highlighted in light blue) (PDB ID: 1E0W).300,301 Atom coloring: O (red); S (yellow).
Further work by Jarvis and co-workers looked to utilize the oxidizing power of iron-peroxo to attack the β-O-4 linkage of lignin, a notoriously difficult to process material with important consequences as a potential renewable resource.299 A series of organic ligands were designed to enable both iron binding and covalent linkage via thiosuccinimide formation to the A100C variant of steroid carrier protein 2L (SCP-2L, Figure 21b). Of the explored organometallic complexes, only the maleimide-substituted tris(2-pyridylmethyl)amine exhibited activity to activate a lignin model complex via hydroxide to ketone conversion, which was improved by incorporation of the complex into SCP-2L. In an attempt to further stabilize the metal site (via either hydrogen bonding or direct dative bonding), F94E and F94H substitutions were introduced on the basis of MD simulations. While no effect was observed with histidine, glutamate resulted in improved conversion and yield. It is still uncertain whether direct coordination of Fe by Glu occurs, but it is possible the carboxylate side chain serves to stabilize the Fe center.
Counter to the pyridyl-based complexes used to impart oxygenase activity in studies with β-LG and SCP-2L, Mahy and co-workers combined the S212C variant of xylanase (Ser212Cys Xln, Figure 21c) with a maleimide Knölker’s Fe complex to generate a biohybrid with hydrogenation activity.301 Using Rosetta,290,291 the backbone perturbations arising from Michael addition of the maleimide-Knölker complex at S212C were modeled using a combination of MD and normal-mode analysis (NMA) which indicated some conformational rearrangement may be necessary for favorable binding to occur. However, successful incorporation was achieved, and hydrogenation of trifluoroacetophenone (TFAC) to its alcohol complement was boosted approximately 2.5-fold relative to Knölker’s complex in ethanol solution.
3.1.4. Design via Supramolecular Interactions.
While covalent attachment is one method of inserting a guest complex into a host scaffold, supramolecular interactions provide an equally powerful approach. Several systems have been employed to generate mononuclear non-heme Fe active sites via, for example, salt bridge, hydrogen bonding, and/or π-stacking interactions.
Ménage and co-workers have extensively employed the protein scaffold NikA (nickel-binding periplasmic protein, Figure 22a) to incorporate mononuclear non-heme Fe organometallic complexes to study the impact of the surrounding protein scaffold on oxygenation and hydroxylation activity. NikA natively serves as a NiII transporter protein which is essential for the maturation of a variety of microbial energy-converting enzymes.303 Interestingly, initial studies reinvestigating the structure of recombinantly expressed NikA resulted in the unexpected coordination of [FeIIIEDTA(H2O)]− (EDTA = ethylenediaminetetraacetic acid).304 Binding of this complex was proposed to be stabilized through several possible salt bridges, most importantly via Arg137 to the apical carboxylate of EDTA. Spurred on by these results, Ménage and co-workers generated an EDTA-inspired organometallic ligand L0 (L0 = N-benzyl-N′-(2-hydroxybenzyl)-N,N′-ethylenediaminediacetic acid, Scheme 3) as a mimic of Fe oxygenase and further incorporated the FeIIIL0 complex into the NikA scaffold to form FeIIIL0-NikA.302 Crystallography revealed stabilization via the previously observed carboxylate-Arg137 salt bridge and further π-stacking between an equatorial phenol and Trp398 (Figure 22b). Exposure of FeIIIL-NikA crystals to dithiothreitol (DTT) induced a transformation of the equatorial ligand phenol → catechol and of benzyl → phenol, and further exposure to O2 intramolecular resulted in dihydroxylation of the L0 scaffold. Further combination of crystallography and rR allowed the characterization of two O2-bound intermediates, leading to the proposal that homolytic O2 cleavage occurs to form an FeIV═O/FeIII─O• intermediate (Figure 22c-f).
Figure 22.

(a) Crystal structure of NikA with bound FeIIIL0. (b) FeIIIL0 binding site of NikA with interacting SCS residues Arg137 and Trp398 highlighted in blue. (c) Dihydroxylated product formed following exposure of NikA/FeL0 to 50 mM DTT and O2. (d) O2-bound intermediate formed following four successive back soaks of DTT-reduced NikA/FeL0 in DTT-free mother liquor, followed by O2 exposure. (e) NikA/FeL0 soaked in 50 mM DTT. (f) Partially hydroxylated O2-bound intermediate formed following three successive back soaks of DTT-reduced NikA/FeL0 in DTT-free mother liquor, followed by O2 exposure. Respective PDB IDs for parts a—f: 3MVW, 3MVW, 3MW0, 3MVY, 3MVX, 3MVZ.302 Atom coloring: N (blue); O (red); Fe (orange).
Scheme 3.

While the dioxygenase activity of FeIIIL0-NikA is promising, the dihydroxylated product results in coordination of Fe by the newly formed phenol, hindering any further turnover.302 This inspired further efforts to deter the intramolecular reaction via two approaches. Under the first approach, substitutions on the benzyl and phenol groups of L0 inhibit the intramolecular reaction, while the second employed organic ligand was modified to provide N2Py2 coordination (N = substituted amine, Py = pyridyl).
To inhibit intramolecular hydroxylation, Cavazza, Ménage, and co-workers modified the origin L ligand by blocking the sites of aromatic hydroxylation observed previously.302,305 To do so, a thiomethyl substituent was introduced at the para position of the benzyl group (complex L1, Scheme 3), and an additional methoxy substituent was placed at the meta position of the phenol (complex L2). Crystal structures were obtained for the FeIIILY-NikA and FeIILY-NikA systems (Y = 1, 2), as well as FeIILY-NikA following exposure to O2 (FeLY-NikA-O2). This structural characterization revealed that, in addition to the previously observed Arg137 salt bridge, H-bonding from Tyr402 and π-stacking from Trp398 provide important stabilizing interactions with FeIIILY-NikA. Upon reduction to form FeIILY-NikA, the conformational change resulting from rotation at the thiomethyl-substituted benzylic group results in substitution of H2O for thioether coordination and the formation of a new salt bridge with Arg97. While O2 exposure of FeIIL1-NikA resulted in intramolecular hydrogenation to form a catechol, FeIIL2-NikA appeared unimpacted. Through this process, products of both still preserve an open coordination site. Activity studies demonstrated some monooxygenase activity in FeLY-NikA, which was also greatly improved by cross-linking NikA to form enzymatic crystals (CLECs).305
To explore N2Py2 coordination, a series of Fe-EDTA derivatives FeLX (X = 3–7) were generated with pyridyl-substituted amines and a varying number of carboxylate groups, and their interactions with NikA were subsequently investigated (Scheme 3).308 The presence of a carboxylate moiety was found to be necessary for binding, and the affinity was significantly boosted by the presence of two carboxylates. Addition of steric hindrance by substitution of the 1,2-diaminoethane backbone for 1,2-diaminocyclohexane additionally decreased the binding affinity. Comparing complex charges, it was found that binding was also influenced by charge screening, where negatively charged complexes generally exhibited higher affinity. This effect was partially overcome in complexes that were capable of forming CH/π interactions. Further catalytic studies for these complexes were performed to probe their oxygenase activities toward sulfide complexes.309 Substrates were screened using molecular docking calculations and chosen on the basis of probing the presence/absence of H-bonding interactions with Glu247 and Gln385 residues. With a select substrate, the activities for sulfoxidation in the presence of NaOCl were monitored, demonstrating that the presence of the NikA host around FeLX-NikA is critical for catalysis. To further boost the catalytic capabilities of the NikA-hosted N2Py2 coordination complexes, CLECs of NikA were grown and substituted with FeLX-NikA (X = 8, 9) containing ortho methyl substituents on either pyridyl group.310 The sulfoxidation activity was significantly improved in the FeLX-NikA/CLEC system relative to solution FeLX-NikA, promoting both higher yields and significantly greater turnover numbers (TONs). The harsh conditions used in the sulfoxidation reaction (including the presence of NaOCl as oxidant) were suggested to degrade the catalyst and protein during turnover, and the stability imparted by the CLEC likely mitigates this issue to some degree.
Ménage and co-workers have also explored sulfoxidation via incorporation of a Fe-N2Py2 complex in human serum albumin (HSA) by taking advantage of this protein’s ibuprofen recognition site.306 An ibuprofen-substituted N,N′-bis(2-pyridylmethyl)-N-methyl-1,2-ethanediamine complex (Libu) was used to coordinate FeIICl2, forming the FeIILibuCl2 complex, which was further successfully incorporated into HSA as supported by ICP, absorption, and fluorescence measurements (Figure 23). While FeIILibuCl2 was found to completely oxidize thioanisole to sulfone in the presence of H2O2, product formation was limited predominately to sulfoxide in the FeIILibu-HSA complex. Significant activity was also observed for HSA alone, although both selectivity and TON were improved by the presence of FeIILibu.
Figure 23.

(a) Fe-chelating complex Libu.306 (b) Crystal structure of human serum albumin (HSA) with bound ibuprofen (abbreviated as “ibu”) highlighted in light gray (PDB ID: 2BXG).307 Atom coloring: N (blue); O (red).
In addition to NikA and HSA, the streptavidin (Sav)-biotin system has been effectively used to generate a wide range of artificial metalloenzymes, including several examples of mononuclear non-heme Fe sites. The first example incorporating a biotinylated Fe complex into Sav comes from Renaud, Ward, and co-workers, who utilized a series of modified Knölker’s complexes to promote the enantioselective hydrogenation activity (Figure 24).311 While most exhibited low catalytic activity, the use of an intermediate length linker was found to promote ketone hydrogenation with 26% conversion and 34% ee. Ward and co-workers have also incorporated a biotinylated Fe-TAML complex (Fe-TAMLbiot, TAML = tetraamido macrocyclic ligand) into Sav to impart hydroxylation activity into the scaffold.312 While the Fe-TAMLbiot exhibits similar activity independently of the Sav scaffold, incorporation of a S112R mutation resulted in >40% ee in the conversion of ethylbenzene to 1-phenylethanol, favoring the R enantiomer. Meanwhile, the L121R and S112R/L121E variants favored formation of the (S)-1-phenylethanol enantiomer. Structural characterization via crystallography of the S112R Sav and S112R/L121E Sav variants revealed salt bridge formation between Arg112 and the TAML ligand, forcing the Fe-TAML complex into a conformation allowing for Lys121 of the neighboring subunit (Lys121′) to bind Fe. In the S112R/L121E variant, the Arg112 side chain is turned away from TAML, with the side chain of Glu121′ coordinating Fe in an h2 conformation. These changes in coordination are believed to impact the resulting product distribution and ee of these systems.
Figure 24.

(a) Fe-TAMLbiot with a C4 linker. (b) Fe-TAMLbiot with a C4 linker bound in Ser112Arg/Lys121Glu Sav. SCS mutations are highlighted in light blue (PDB ID: 6Y25).311 Atom coloring: N (blue); O (red); Fe (orange).
The Sav-biotin system has also been employed to simulate the classic facial triad present in many mononuclear non-heme Fe enzymes. To accomplish such a binding site, Borovik and co-workers designed a biotinylated bis(2-pyridylmethyl)amine (dpa) which was bound to a reengineered Sav scaffold with S112E, L121A, and E101Q mutations (2xm-S112E-Sav), each respectively intended to provide direct metal coordination, reduce sterics, and prevent exogeneous metal binding.313 The crystal structure of FeIIdpa-(2xm-S112E-Sav) revealed an unusual binding mode in which only one of the two pyridines of dpa coordinates FeII, resulting in N2O3 coordination (Figure 25). Meanwhile, structural characterization of FeIIdpa-(2xm-S112E-Sav) showed facial coordination of all three N-donors of dpa, forming an N3O3 coordination set. In both FeII- and FeIIIdpa-(2xm-S112E-Sav), Glu121 serves to coordinate Fe in a k1 fashion.
Figure 25.

(a) FeIII-biot-et-dpa. (b) Crystal structure of Ser112Glu Sav-bound FeIII-biot-et-dpa. Two configurations were observed (over-lapping, highlighted as light gray and blue) (PDB ID: 6UIY).313 Atom coloring: N (blue,) O (red), Fe (orange).
3.2. Designed Dinuclear Non-heme Iron Enzymes
Catalytic nonheme diiron enzymes can be broadly separated into two categories based on the nature of their bridging moieties, namely, those involving μ-carboxylate and those involving μ-thiolate. The former represents a large family with a diverse array of functionality, including methane and toluene monooxygenases, ribonucleotide reductases, flavodiiron nitric oxide reductases, rubrerythrin, and stearyl-ACP desaturase, to name a few. Meanwhile, the primary example of a catalytic (μ-thiolato)diiron site is FeFe hydrogenase (FeFe H2ase).
Despite the ubiquitous nature of natural (μ-carboxylato)-diiron enzymes, there are few examples of incorporating such an active site into an ArM due to the difficulty involved in tailoring a binding environment for not one but two metal centers. The de novo Due Ferri (DF) scaffold designed by Lombardi, DeGrado, and co-workers has proven to be extremely valuable for the generation and study of such active sites and is discussed in depth by Pecoraro and co-workers.314-316 Recently, the Sav-biotin system was reengineered to form a unique (μ-carboxylato)diiron binding site, employing a combination of Trojan Horse and direct AA coordination approaches.317 To do so, Borovik and co-workers took advantage of the quaternary structure of Sav, which positions two biotin-binding sites in close proximity and with an intermediary pocket. Utilizing variants of biot-et-dpa, a series of ligands (biot-x-dpa) with varying linker length (x = ethyl, propyl, and butyl) were cross-correlated with a series of Sav variants, namely, WT, S112Y, L121Y, and L121A/L124Y.313 Tyr was chosen due to the intense CT absorption feature that arises from FeIII-OTyr coordination,318 which allowed for the design of a visual assay testing for Fe─O coordination. Successful binding was observed when employing a combination of the biot-bu-dpa ligand (bu = butyl) with the L121A/L124Y Sav variant. Using a combination of crystallography, 57Fe Mössbauer spectroscopy, and DFT calculations, it was demonstrated that the newly formed site contained a [FeIII-(μ-OH)-FeIII] core with a labile proximal position at either Fe (Figure 26). Interestingly, an unusually long Fe─Fe distance of 3.96 Å and a small OTyr─Fe─Oμ-OH angle of 65° were observed, presumedly due to the strain imposed by the rigid Sav host, serving as an example of how deeply the SCS structure can impact the PCS.
Figure 26.

FeIII-biot-bu-dpa bound in Lys121Ala/Leu124Tyr Sav forming a [FeIII-(μ-OH)-FeIII] core. PCS residues are highlighted as light gray, and SCS in light blue (PDB ID: 6VP1).317 Atom coloring: N (blue); O (red); S (yellow); Fe (orange).
Although (μ-thiolato)diiron enzymes are found in a much smaller family of enzymes, the most prominent member, [FeFe] H2ase, has been the subject of intense interest for several decades due to its ability to efficiently and reversibly catalyze the production of H2. The catalytic H-cluster of [FeFe] H2ase, [Fe2(CO)3(CN)2(adt)] (adt = −SCH2NHCH2S−), is unto itself an organometallic complex covalently tethered to a neighboring [Fe4S4] cluster via a cysteine-thiol bridge, making it an attractive target for incorporation in biosynthetic designs.
Given the H-cluster is a stable entity unto its own, one of the critical questions posed for the development of biologically inspired H2ase catalysts is the extent of the SCS influence on H2 production. As a result, many studies have utilized minimal oligopeptide maquettes to either form or bind H-cluster mimics. One of the first attempts was made by Jones et al., who incorporated a [Fe(CO)3-(μ-SCys)2-Fe(CO)3] complex via a CXXC motif in a single-stranded 36-mer oligopeptide maquette.319 Based on this work, Hayashi and co-workers provided one of the first examples attempting to incorporate an H-cluster mimic into a non-native protein scaffold.320 Inspired by the success of Jones et al., apo-cytochrome c (apo-Cc) was chosen due to its native CXXC motif (Figure 27a and b). Formation of a [Fe(CO)3-(μ-SCys)2-Fe(CO)3] complex in apo-Cc to form H-apo-Cc was evidenced by a combination of ESI-TOF MS, absorption spectroscopy, and IR spectroscopy. Addition of the photosensitizer [Ru(bpy)3]2+ and excess ascorbate to H-apo-Cc led to visible light-controlled H2 formation, with a TON reaching ~80 after 3 h. The drop in efficiency observed with decreasing amounts of [Ru(bpy)3]2+ inspired further investigation of an alternative scaffold that could prospectively also bind a photosensitizer close in proximity to the CXXC motif. Using a single-stranded α-helical octadecapeptide maquette (Pep18) based on the sequence of cytochrome c556, a CXXCH motif was employed (Figure 27c and d).321 Similar to the case of Cc, ESI-TOF MS, absorption spectroscopy, and IR spectroscopy were used to evidence both formation of [Fe(CO)3-(μ-SCys)2-Fe(CO)3] and binding of [Ru(bpy)3]2+ via His. Importantly, this maquette was able to evolve H2 in the presence of light and excess ascorbate, which was otherwise not found without datively tethered [Ru(bpy)3]2+.
Figure 27.

Summary of H2ase-inspired (μ-thiolato)diiron enzymes. (a) Synthetic process for the formation of Cc-bound [Fe(CO)3-(μ-SCys)2-Fe(CO)3] and (b) ensuing H2 production system.320 Reproduced with permission from ref 320. Copyright 2011 Royal Society of Chemistry. (c) Synthetic process for the formation of Pep-18 [Fe(CO)3-(μ-SCys)2-Fe(CO)3] with bound Ru(bpy)3 and (d) ensuing H2 production assay system.321 Reproduced with permission from ref 321. Copyright 2012 Elsevier. (e) Model of the [Fe(CO)3-(μ-SCys)2-Fe(CO)3] binding single-stranded α-helical maquette containing the dithiol UAA “Dt”.322 Reproduced with permission from ref 322. Copyright 2012 Royal Society of Chemistry. (f) Proposed schematic for [Fe4S4]-[Fe2(CO)3(CN)2(adt)] bridge formation in the FdM maquette.323 Reproduced with permission from ref 323. Copyright 2019 Royal Society of Chemistry. (g) Binding of a [Fe(CO)3-(μ-SCys)2-Fe(CO)3]-maleimide complex in the β-barrel protein nitrobindin.324 Reproduced with permission from ref 324. Copyright 2014 American Chemical Society.
Alternative to the CXXC motif, Ghirlanda and co-workers explored the formation of a [Fe(CO)3-(μ-SCys)2-Fe(CO)3] complex with a single-stranded α-helical maquette using a UAA containing a dithiol side chain (Figure 27e).322 Interestingly, reaction of this system with [Ru(bpy)3]2+, citrate buffer, and light produced a H2 TON of 84 after 2.3 h, comparable to that observed by Sano et al. in their H-apo-cyt c system.320
Berggren and co-workers have recently expanded on the maquette-binding H-cluster model by design of a [Fe4S4]-binding oligopeptide (FdM) capable of binding [Fe2(CO)3(CN)2(adt)] via a bridging Cys to generate a more comprehensive H-cluster structural model (Figure 27f).323 While [Fe2(CO)4(CN)2(adt)] or [Fe4S4]-FdM was not observed to evolve H2 as isolated in the presence of methyl viologen, the [Fe2(CO)3(CN)2(adt)]-(μ-SCys-[Fe4S4]-FdM complex was capable of producing H2 with a TON = 11 over the course of 1 h. Together with the maquette studies of Hayashi and Ghirlanda,320-322 these results emphasize the importance of an electron transfer site in the proximity of the H-cluster.
Further studies have incorporated mimics of the H-cluster into larger scaffolds. Hayashi and co-workers have employed the thiosuccinimide-anchor method to generate a covalently bound H2ase model using a modified nitrobindin (NB) β-barrel peptide scaffold with a Q96C substitution in combination with a maleimide-containing synthetic [Fe(CO)3-(μ-S)2-Fe(CO)3] complex (Figure 27g).324 This combination yielded up to a TON of 130 over the course of 6 h in the presence of [Ru(bpy)3]2+, ascorbate, and light. Interestingly, in the absence of NB this complex still is capable of reaching a TON ~ 130 over the course of just 2 h. In this case, it appears that the limitations of [Ru(bpy)3]2+ access to the catalytic site imposed by NB hinder the reaction as opposed to promoting it.
Most recently, Ghirlanda and co-workers have employed the Sav-biotin system to generate a synthetic H2ase using a biotinylated [Fe(CO)3-(μ-S)2-Fe(CO)3] complex.325 Use of [Ru(bpy)3]2+ to drive catalysis in the presence of visible light and ascorbate resulted in slow H2 production, reaching ~48 TON over the course of 11 h, significantly greater than the TON = 6 found in the absence of the Sav scaffold, demonstrating that the presence of a surrounding protein scaffold significantly improves the catalyst integrity. This hypothesis was further supported by transient absorption measurements, which evidenced an increase in the lifetime of the catalytically competent intermediate in the presence of Sav.
4. ACTIVITY BEYOND THE FIRST COORDINATION SPHERE IN ENGINEERED COPPER ARMS
Like Fe, Cu serves as an important cofactor for a variety of protein functions, including electron transfer (ET) and catalysis.326-332 These functions can be regulated by tuning both the PCS and SCS. Cu proteins can function as ET proteins using one of two types of copper centers, known as type 1 Cu (T1Cu) (Figure 28a) and copper A (CuA) (Figure 28b); structurally homologous proteins (or protein subunits) containing either type of the copper site are referred to as cupredoxins.333-335 The T1Cu center is coordinated by one cysteine and two histidine residues in a trigonal plane; for many T1Cu cupredoxins, another interaction from the axial position above the trigonal plane can form a distorted tetrahedral geometry that is between the coordination geometries preferred by the CuII and CuI states, thus facilitating ET.336 Meanwhile, the CuA center is more complicated, involving a spatially close, electronically delocalized dicopper core bridged by two cysteines together with one histidine and one variable axial ligand on either side.337,338 The geometry of CuA is more rigid than T1Cu with a comparably lower reorganization energy for the Cu1.5+-Cu1.5+/Cu2+-Cu2+ redox pair. CuA sites are often found in conjunction with metalloenzymes containing catalytically active sites, including heme-copper oxidase (HCO),339 nitrous oxide reductase (N2OR),340 and non-heme nitric oxide reductase.341 In HCO, the CuA center serves to transfer electrons from the periplasm to the buried heme-CuB site for the reduction of O2. Meanwhile, the CuA site of N2OR shuttles electrons to the copper Z (CuZ) site to catalyze the reduction of N2O to N2.342 A CuA center with a unique PCS was recently identified in the protein PmoD.343,344 Although this protein is related to two copper enzymes, ammonia monooxygenase (AMO) and particulate methane monooxygenase (pMMO), the function of this CuA center is not yet well understood.
Figure 28.

Active site structures of several representative natural copper binding proteins. (a) T1Cu from Pseudomonas aeruginosa (PDB ID: 4AZU).345 (b) CuA site of the cupredoxin domain from Thermus thermophilus cytochrome ba3 oxidase (PDB ID: 2CUA).346 (c) Substrate-bound (O2) T3Cu site of laccase from Melanocarpus albomyces (PDB ID: 1GW0).347 (d) Substrate-bound (nitrite) T2Cu site of Alcaligenes xylosoxidans nitrite reductase (PDB ID: 2XWZ).319,348 (e) Cuz site of the Pseudomonas stutzeri N2OR (PDB ID: 3SBR).349 Atom coloring: N (blue); C (cyan); O (red); S (yellow); Cu (copper).
While the T1Cu and CuA sites serve to transfer electrons, type 2 copper (T2Cu) is directly involved in biocatalysis as part of a catalytic active site.350 The ligand set of T2Cu is typically composed of nitrogen- and oxygen-based ligands and, in special cases, methionine as well. Unlike the trigonal or distorted tetrahedral geometry of the PCS of T1Cu sites, T2Cu is in a distorted square-planar geometry in the CuII resting state with one or more empty sites allowing for the binding of small molecular oxidants such as O2 or superoxide and enzyme substrates (Figure 28d). T2Cu-containing enzymes are generally oxidoreductases with vastly different structures, including nitrite reductase (NiR),351,352 SOD,353 and numerous O2-utilizing enzymes known as oxygenases and oxidases.354-356 Oxygenases function to incorporate an oxygen atom from molecular oxygen into the substrate. For example, lytic polysaccharide monooxygenase (LPMO) catalyzes the hydroxylation of the C─H bond in the glycosidic linkage using molecular O2.357,358 Meanwhile, oxidases use O2 to reoxidize copper in the catalytic cycle without incorporation of O into the substrate. For example, laccase catalyzes the oxidative cross-linking of phenolic substrates while reducing O2 to water.359,360 For T2Cu enzymes, secondary coordination sphere (SCS) interactions function to not only tune the copper center but more importantly stabilize and/or activate substrates by hydrophobic interactions or hydrogen bonds or directly participate in the formation of reaction intermediates.
Beyond native functionality, the development of novel reactivity is an important perspective of artificial metalloenzymes. Copper complexes are important catalysts in organometallic chemistry, a topic covered by several in-depth reviews.361-366 By combining a copper cofactor with a protein, one can take further advantage of the SCS environment imposed by the surrounding scaffold while maintaining the versatile chemistry of the organometallic copper compounds. There have been emerging examples using proteins or peptides as hosts for copper cofactors to achieve nonbiological reactivity, including Diels–Alder coupling,367 Friedel–Crafts alkylation,368 the Michael addition reaction,369 and atom transfer radical polymerization (ATRP).370 In many of these cases, the interactions between the SCS of the protein and the substrate molecules were shown to have a significant impact on the activity and selectivity.
The following section introduces a series of case studies on the SCS tuning effect of the two essential aspects of copper protein function—the electron transfer properties of T1Cu and CuA and the catalytic behaviors of T2Cu sites—in biosynthetic proteins or peptides. This section also covers nonbiological coppers catalysts including protein-fused synthetic copper complexes and the use of natural copper centers to catalyze nonbiological reactions. Due to their scarcity in nature and high complexity, other copper centers, such as type 3 copper (T3Cu)371-373 and CuZ,349 are rarely studied using biosynthetic models and, therefore, are not discussed in this section. Despite their significance, the coverage of de novo designed copper proteins will be limited in this article, and we refer the reader to the review by Pecoraro and coauthors in this same issue for an in-depth discussion.
4.1. Designed Copper Proteins with Electron Transfer Functionality
The ET functions of T1Cu and CuA are essential for many biological processes requiring the transfer of electrons to active sites that are not close in contact with electron donors or acceptors. T1Cu can carry out efficient ET across long (>10 Å) distances, which is rationalized by the optimization of the driving force (ΔG) and reorganization energy (λ) according to Marcus theory.333,374 CuA centers have been reported to perform even faster electron transfer than T1Cu in a comparative study.375,376 Therefore, tuning the driving force—the reduction potential of copper (E°′)—is essential to understand electron transfer processes in nature and to build better systems with improved ET properties.
To avoid the complexity of many native enzymes, the electron transfer properties of T1Cu have been studied using simple cupredoxins such as Az.378-381 The E°′ values of the native T1Cu proteins typically exhibit a wide (500 mV) range despite their similar structures.382-384 Three main factors are considered responsible for such a large variarion in E°′, namely, the identity of the axial ligand, hydrophobicity, and H-bonding (Figure 29). Axial ligation is not conserved among T1Cu proteins, taking varying identities such as Met in Az, Gln in stellacyanin, and a variety of hydrophobic residues in fungal laccases, etc.385 To exclude other structural variables, the relationship between the axial ligand and E°′ has been demonstrated in Az, in which the native Met has been mutated into nearly every other natural amino acid, as well as a series of unnatural amino acids.386-391 A positive correlation was found between the hydrophobicity of the axial ligand and the E°′ value of the T1Cu copper site, yet the range of observed potentials (150 mV) was not nearly as high as that observed between natural T1Cu cupredoxins. The influence of hydrophobicity in the T1Cu site SCS environment was examined in another study in which substitution of three more distant residues (Leu33, Met44, and Leu86) with Phe resulted in an increase of the reduction potential by 80 mV, which is additive to the tuning effect established by varying the axial residue.392 Additionally, Lu and co-workers have demonstrated that the hydrogen bonding network may play a more subtle but equally important role.377 The F114P mutation disrupts H-bonding between a backbone amide nitrogen and the S(Cys), increasing S-Cu covalency and causing a 110 mV decrease in E°′. The N47S mutation further alters the H-bonding interaction with Cys112, leading to a 140 mV increase in E°′ without significantly changing the PCS. The effects of H-bonding are additive to those from axial ligand modifications, producing a cumulative tuning range of ~800 mV.377 SCS mutations also play a role in tuning the reorganization energy of the copper center. Interestingly, most of the mutants investigated were found to have lower reorganization energies and, hence, higher intra-molecular electron transfer rates than WT-Az, with the N47S/F114N Az mutant exhibiting the fastest electron transfer rate.393 A following study of electron transfer in these mutants suggested a Marcus-inverted trend in which a higher reduction potential may lead to slower electron transfer,394 therefore supporting the hypothesis that maximizing the rate of electron transfer requires optimizing the reduction potential.
Figure 29.

Three SCS targets of reduction potential tuning in Cu-Az: the axial residue hydrophobicity, hydrogen bonding network, and distal residue hydrophobicity. The plot shows the cumulative effect when different strategies are combined (PDB ID: 4AZU).345 Reprinted with permission from ref 377. Copyright 2011 Elsevier.
Besides homology-based biosynthetic modeling, de novo design emerged in the 1990s as a powerful alternative approach to building metal binding sites in simple protein scaffolds.397,398 Hildebrandt and co-workers reported a series of designed four-helix bundle proteins with Cys and His residues in the core of the protein. The position of these residues was shuffled throughout helices, among which 30 candidates displayed copper binding affinity.399 The design was improved in a subsequent study by optimization of SCS interactions. By varying core residues with different hydrophobicity and bulkiness, the copper binding site was shown to be between T1Cu and T2Cu based on electronic absorption spectroscopy (color) and EPR (A∥).400 A more recent report by Shiga et al. showcased an exact blue T1Cu center in a four-helix protein (Figure 30a).395,401 The reduction potential of copper was determined to be 328 mV, close to the reduction potential of Az (~280 mV). The Ala residues in the SCS were proposed to be replaceable, allowing for enhanced packing of the core and, thereby, tuning of the configuration of the T1Cu, although this is yet to be experimentally verified. Quaranta, Pecoraro, and co-workers reported a three-helix T1Cu protein with spectral features resembling the red copper nitrosocyanin.402,403 Recently, the electronic properties of red copper were found to depend on Glu41 in the SCS that lengthens both the Cu─S(Cys) and the Cu─N(His) bonds (Figure 30b).396,404 This residue was also found to tune the T1Cu reduction potential, as a E41D substitution was shown to increase the reduction potential by 50 mV. Two additional designs were made from the initial red copper construct to mimic the T1Cu center of enzymes such as plastocyanin and nitrite reductase, which are blue and green copper, respectively. The primary difference between these two proteins is the absence or presence of a SCS axial methionine.404 Interestingly, the reduction potentials of the three mutants did not vary significantly, which opened up opportunities to further study the influence of the SCS in these three helical proteins to tune reduction potential.
Figure 30.

(a) De novo designed T1Cu protein by Shiga et al. with SCS Ala residues. (b) De novo designed T1Cu protein by Koebke et al. with an axial Glu residue. Reprinted with permission from refs 395 and 396. Copyright 2010 and 2018 American Chemical Society.
The necessity of redox potential tuning has been demonstrated in several T1Cu-containing enzymes, where a fine-tuning of the T1Cu potential can significantly affect the enzymatic activity. Multicopper oxidases (MCOs) are a class of enzymes that catalyze the oxidation of organic substrates while reducing O2 to H2O, such as ascorbic oxidase, bilirubin oxidase, copper efflux oxidase, and laccase (Figure 31a).326,354 Despite the different structures and functionalities, all MCOs require efficient electron transfer from T1Cu to the active site, which is directly dependent on the redox potential of the T1Cu.410-412 The strategies of redox potential tuning in single T1Cu protein have been applied in MCOs.413 Mutation of the axial T1Cu ligand from Met to either Leu or Phe in Bacillus subtilis laccase resulted in a 100 mV increase in reduction potential, which, in turn, decreased the laccase activity by 2–4-fold in the Leu mutant, with a near complete loss of activity in the Phe mutant (Figure 31b).414 This trend was corroborated by another study in which mutation of the native axial Phe of Thapsia villosa laccase to Met lowered the T1Cu potential by 100 mV.384 This mutation increased kcat and KM while shifting the optimal pH to a more basic regime. Kamitaka et al. reported a M467Q mutant of Myrothecium verrucaria bilirubin oxidase which lowered the formal potential of T1Cu by 230 mV while improving the kinetics of the electrocatalytic oxygen reduction reaction (Figure 31c).415 Similar to the case of cupredoxins, introduction of an additional hydrogen bond donor can pose a significant impact on the T1Cu potential in MCO. Kakaota et al. studied the effect of the SCS hydrogen bonds of the T1Cu in E. coli copper efflux oxidase (CueO).416,417 The conserved Pro444 was mutated to Ala, Leu, or Ile to introduce a second hydrogen bond to Cu-bound S(Cys) (Figure 31d). Similar to its Az counterpart, this modification decreased the covalency of the S(Cys)─Cu bond while increasing the T1Cu reduction potential by 40–70 mV. As a result, kcat was observed to decrease by 40–60% among these mutants. Meanwhile, the P444G mutant showed higher activity than the native enzyme; however, the T1Cu reduction potential in this variant was not reported.
Figure 31.

(a) Structure of wild-type laccase from B. subtilis (PDB ID: 1GSK). The black arrow shows the electron transfer from the T1Cu to the T2/T3Cu site to reduce O2. (b) Axial mutants of B. subtilis laccase modeled from the wild-type (PDB ID: 1GSK)405 using UCSF Chimera.406 (c) M467Q axial mutant of bilirubin oxidase from M. verrucaria (PDB ID: 6IQX).407 (d) Modifying Pro444, the H-bond donor to the Cys residue of T1Cu in E. coli CueO (PDB ID: 1KV7).408 (e) Modifying Glu506, a proton transfer mediator residue in E. coli CueO. Reprinted with permission from ref 409. Copyright 2012 Elsevier.
Another study by the same group focused on proton transfer in the SCS between the T1Cu and T2/T3Cu sites in E. coli CueO.409 Mutation of the proton donor Glu506 to Ile resulted in complete loss of activity, whereas a less bulky Ala mutation led to a compensated proton transfer pathway bridged by two water molecules. The E506N mutant also showed no activity, demonstrating the importance of a dissociable proton. The correlation between the T1Cu redox potential and the MCO activity has been previously reviewed411 and recently demonstrated in a comparative study by Solomon and co-workers.418 The authors compared the high-potential fungal laccase of Trametes versicolor (axial Phe) with the low-potential plant laccase (axial Met) from Rhus vernicifera. The higher potential was found to result in an 8 kcal/mol decrease in the intramolecular electron transfer driving force. While electron transfer from T1Cu to the trinuclear Cu active site is the rate-limiting step of fungal laccase, this slow electron transfer is partially offset by the higher reduction potential of the trinuclear center (TNC, the T2Cu and T3Cu sites) due to a more positive protein environment and is still faster than the decay of the native intermediate during catalytic turnover.
In addition to laccases, the tuning of T1Cu and correlation with enzymatic activity have been studied in copper-dependent nitrite reductase (CuNiR), a more complex enzyme that catalyzes the reduction of nitrite (NO2−) to nitric oxide (NO), a process essential for denitrification as part of the nitrogen cycle.422 The reduction of NO2− at the T2Cu of CuNiR requires the injection of one electron and two protons, the former of which is mediated by T1Cu through a Cys-His bridge (Figure 32a).348,423 A subtle balance exists between the two copper centers that their reduction potentials are only slightly different to prevent premature reduction of T2Cu in the rest state.424 Inspired by the studies on individual cupredoxins, the axial ligands of T1Cu in CuNiR were subjected to mutagenesis. The M144A mutation of the Alcaligenes xylosoxidans NiR (AxNir) profoundly increased the reduction potential of the T1Cu from 255 mV to 314 mV and, therefore, reversed electron transfer from the low-potential T2Cu (230 mV) (Figure 32b).421 An even greater increase of the T1Cu potential was observed in the more hydrophobic M144L mutant (336 mV), whereas incorporation of the hydrophilic M144N mutation decreased the reduction potential to 173 mV.420 Interestingly, the M144L mutants showed higher NO2− reduction activity than the native enzyme while the Gln mutants exhibited no activity, suggesting that an artificially tuned, moderately uphill electron transfer is beneficial to the CuNiR activity (Figure 32c). In another study by Sato et al. the T1Cu binding loop of NiR was replaced with that from amicyanin, which maintains the PCS ligand sets but with a much shorter length (nine residues) (Figure 32d and e).419 The swap caused a significant increase of 200 mV in the T1Cu reduction potential together with diminishing NiR activity due to the excessively negative driving force of electron transfer. This effect does not appear to corroborate with the trend observed between the decreased hydrophobicity and redox potential, as indicated by the coordinating water molecule, suggesting a profound but unknown influence from the SCS of the binding loop.
Figure 32.

(a) Structure of the T1Cu (right, cyan) and the T2Cu (left, tan) of AxNiR (PDB ID: 2XWZ).348 (b and c) Axial residue engineering of the T1Cu in AxNiR. (d and e) NiR with the T1Cu loop exchanged with the loop from amicyanin. Reprinted with permission from refs 419-421. Copyright 2008, 2005, and 2002 Wiley-VCH, Elsevier, and Elsevier.
Likewise, CuA has been studied in both native cupredoxins and biosynthetic models using similar tuning strategies as employed to study the electron transfer properties of T1Cu. For example, mutation of the axial Met227 to Ile in Paracoccus denitrificans cytochrome c oxidase (CcO), was shown to result in the loss of electron delocalization in the Cu-Cu core and impair electron transfer to heme a.429 A study of Rhodobacter sphaeroides CcO showed that the M263L mutation maintained the electron delocalization between the copper atoms but caused a 120 mV increase in the reduction potential and, correspondingly, a 2.5-fold decrease in the electron transfer rate and 90% diminishment of the CcO enzyme activity.430,431 Other studies have investigated CuA by truncating native proteins, effectively separating the electron transfer and catalytic components.346 In a series of studies on the CuA domain of Thermus thermophilus cytochrome ba3 oxidase (TtCuA), the weakly bound axial Met160 was mutated to Glu and Gln (Figure 33b).432 In both cases, the mutated axial residues showed strong interactions with Cu possessing features similar to the PCS residues. Early EPR studies suggested an elongated Cu─Cu distance and change in geometry of the Cu2S2 core,433 while recent results from X-ray spectroscopy indicated that the Cu─Cu distance remains mostly unchanged in the M160Q mutant.428 The M160Q mutation resulted in a change of redox potential by −170 mV at neutral pH, with a Cu─Cu distance nearly identical to that of the wild-type (2.45 Å). An even greater difference of −190 mV was observed in another M160H mutant. Despite similar reduction potentials, the electronic structures of M160H and M160Q were found to differ significantly. The M160Q mutant has a significantly higher energy gap between the two accessible ground states πu and σu* (900 cm−1, pH = 6.0) than the wild-type (600 cm−1) and the M160H mutant (200 cm−1) (Figure 33f). A broader set of axial mutations were studied by Ledesma et al. with M160S, M160L, and M160Y mutations.434 The hydrophobic M160Y and M160L mutants exhibited similarly high reduction potentials (348 and 346 mV, +55 and +53 mV vs WT), while the hydrophilic M160S mutant exhibited lower reduction potentials (209 mV, −84 mV vs WT).434 In a study by Blackburn et al., the axial Met was replaced by MetSe. The electrochemical properties were mostly identical between Met and MetSe, and the EXAFS data indicated very similar geometries at the CuA core.435
Figure 33.

(a) Schematic of the O2 reduction reaction catalyzed by heme-copper oxidase (cytochrome ba3 oxidase of T. thermophilus, PDB ID: 1XME).425 The electron transfer from CuA through heme b to heme a3 and the proton pumping pathway are shown with black arrows. (b) Mutation of the axial Met of the CuA from T. thermophilus cytochrome ba3 oxidase (PDB ID: 2CUA).346,426 (c) Loop swapping strategy to tune the CuA center.427 (d) Hydrogen bonding network surrounding the artificial CuA site in engineered azurin. (e) Mutation of the axial Met of the artificial CuA in engineered azurin (PDB ID: 1CC3).426 (f) Tuning of the πu and <u* energy gap by axial ligand mutation of T. thermophilus CuA cupredoxin.428 Parts c and f were reprinted with permission from ref 428. Copyright 2019 American Chemical Society.
CuA centers have also been introduced into the homologous cupredoxins to explore the influence of the SCS. Biosynthetic CuA centers have been built in amicyanin436,437 and Az438,439 by replacing the original Cu binding loop with a CuA binding loop. Similar to studies of T1Cu, the axial Met has been mutated to hydrophilic Asp and Glu and hydrophobic Leu residues, altering the CuA redox potential by +16, −5, and −8 eV (Figure 33e).440 To modulate the H-bonding interaction with Cys112, N47S and E114P mutations were made in CuA azurin (CuAAz) (Figure 33d), leading to + 30 and −42 mV changes in the CuA reduction potential, which are modest in comparison to the + 120 and −46 mV differences seen in T1Cu.441 Both strategies resulted in a smaller tuning effect compared to the T1Cu counterpart, which was attributed to either the higher robustness of the CuA diamond core or the fact that these alterations produced more subtle changes in the coordination environment, such as the possible presence of a water ligand in M123L CuAAz.442
The loop replacement strategy has also been applied in the native CuA cupredoxins, where the use of different loops allows for the PCS of CuA to be kept consistent while modifying the SCS. The three loops of TtCuA, referred to as the His loop, entry loop, and ligand loop, were replaced by the loops from the native CuA subunits of human or plant CcO (Figure 33c). A moderate ±30 mV tuning of the reduction potential was observed in the loop-exchanged mutants compared to wild-type TtCuA.427 Other electronic properties, including the ground state (πu/σu*) energy and delocalized spin density on axial Met, were also tuned by the loop replacements.443,444
In addition to cupredoxins, CuA has been constructed in both natural nonhomologous proteins and de novo designed proteins. Mirts et al. designed a CuA site in a cytochrome c peroxidase (CcP) scaffold (Figure 34).446 The new CuA center resides in the buried α-helical pocket where the heme is docked in the unmodified CcP. The reduction potential of the CuA center was found to be 251 mV as measured by redox titration, comparable to the case of CuA in native CcO (240 mV). Electron transfer between this CuACcP protein and cytochrome c was observed, albeit at a significantly lower rate compared to the case of native CuA (10−3 s−1 vs 105 s−1). CuA has also been incorporated into de novo designed helical bundles by Hildebrandt and co-workers, as evidenced by EPR, MCD, and UV–vis absorption spectroscopies.400 A later study by Tanaka and co-workers reported another example of CuA in a coiled-coil protein in a rationally designed Cu2(Cys)2(His)2 core, exhibiting a Cu─Cu distance of 2.55 Å, longer than in native CuA.445 Unfortunately, no electrochemical characterization was carried out in either of these studies, and the ET activities of these constructs remain unknown. Studies using nonhomologous scaffolds have shown that the CuA structure can be well mimicked by transplanting the PCS ligand set. While full functionality is dependent on outer sphere interactions, it can be partially restored in these biosynthetic models.
Figure 34.

(a) CuA site of TtCuA. (b) Original heme pocket of CcP. The three residues Trp51, His52, and Arg48 were engineered as the PCS core for CuA. (c) De novo designed CuA protein and (d) CuA structure. Reprinted with permission from references 445 and 446. Copyright 2012 and 2020 American Chemical Society.
4.2. Designed T2Cu Proteins with Catalytic Functions
Compared to the electron transfer copper proteins, modeling the catalytic sites of T2Cu proteins requires more detailed consideration. T2Cu proteins are, in general, more complex due to the numerous auxiliary units required to promote native catalytic activity. It is also difficult to achieve functional interactions with the substrates, as well as to stabilize redox-active intermediates during enzymatic turnover, roles which are fulfilled by well-positioned amino acid residues in the native protein. Three approaches, de novo design,83,447-449 incorporation of T2Cu into natural proteins by coordination with amino acid side chains, and bioconjugation of organometallic complexes, have been applied to mimic the active site of catalytic Cu enzymes, and each of these approaches has provided insights into the role of SCS interactions.
The functionalities of several Cu enzymes, such as catechol oxidase (COx),451 superoxide dismutase (SOD),452 and nitrite reductase (NiR),453 have been applied in artificial protein models, among which models of NiR are the most well studied with respect to probing the influence of the SCS. The first functional model of the catalytic T2Cu was reported by Pecoraro and co-workers.450,453 A tri-His copper binding site was introduced into the core of the three-helical peptide TRI-W. The NiR activity was observed by generation of N2O without side reactions, albeit with lower activity than that of native NiR (Figure 35a). The activity was improved by tuning the SCS through electrostatic interactions, for which several solvent-exposed Lys and Glu residues were mutated to their counterparts with opposite charge.454 A 4-fold increase in the activity was observed between the most positively charged E27K and the most negatively charged K24E mutants. The reduction potential also decreased with increasing negative charge in the SCS by a maximum of 100 mV. Meanwhile, the influence of the steric hindrance was tuned by mutating the Leu residues below or above the active site into Ala or Asp.455 All variants showed elevated activity, with an increase in reaction rate up to 75-fold. These changes were attributed to an interplay between the coordination geometry of the CuI state and the electronic structure of the transition states. Recently, similar designs have been applied to construct Cu-bound α-helical peptides for other activities, including superoxide dismutation452 and peroxide activation,456 with the effects beyond the PCS unexplored but promising for future studies.
Figure 35.

(a) De novo designed NiR peptide and the active site structure. Reprinted with permission from ref 450. Copyright 2018 American Chemical Society. (b and c) Structures of two AzNiR mutants modeled by UCSF Chimera using PDB ID 4AZU.345,406
In addition to de novo design, strategies employing natural protein scaffolds have also been reported for NiR. Berry and co-workers have incorporated a His3 or a His2Asp site on the surface of Az (AzNiR), producing a system that includes both the T1Cu and T2Cu sites of CuNiR (Figure 35b and c).457 Both UV–vis and EPR spectra showed a new T2Cu species in addition to the native T1Cu. The incorporated T2Cu is solvent-exposed, leading to a moderately high potential (350 mV), which is between those of the previous model and native NiR, which are both hydrophobically buried. The NiR reaction rate (0.34–0.59 min−1) in this Az model is lower than that of the native NiR yet comparable with those of the previously mentioned de novo designed peptides. The native T1Cu site serves as an optional electron donor which may participate in NiR catalysis directly, although the T2Cu was also found to independently catalyze nitrite reduction when the T1 site is blocked by Hg2+.
Similar cupredoxin scaffolds have been adopted to model catalytic Cu sites other than NiR. In Az, the substitution of the chromogenic Cys residue with a hard Asp ligand resulted in a typical (His)2Asp T2Cu center, with large A∥ as measured by EPR and a weak absorption feature at 600 nm.460,461 Surprisingly, it has been found in a more recent study that further mutation of the axial Met residue to a hydrophobic, noncoordinating Leu, Phe, or Ile residue results in a new type of copper center that resembles neither T1Cu nor T2Cu.458 The new copper center, called “Type 0 Cu” (T0Cu, Figure 36), showed a weak d─d transition band at 800 nm but no absorption in the visible region. The A∥ splitting falls into the low 300 MHz range, which is between the values from T1 (<200 MHz) and T2 Cu (>400 MHz). The reduction potential of T0Cu is higher than that of T2Cu and closer to that of the native T1Cu. The authors mutated the axial ligands to Glu in a following study, which reverted the spectroscopic features back to T2Cu with a comparably high potential (300 mV).459 The axial Glu exerts only an electrostatic effect and does not bind directly to copper at lower pH. At higher pH (9.0), the H-bonding network in the SCS caused a structural rearrangement of the T2Cu site as indicated by XANES and X-ray crystallography, leading to binding of the deprotonated Glu to Cu and a down-tuning of the reduction potential to 120 mV. Both modified T0Cu and T2Cu possess promising features, such as the low coordination number, high reduction potential, and flexible coordination pattern, for mimicking the catalytic T2Cu sites in nature.462
Figure 36.

(a) C112D Az (PDB ID: 3FQY). (b) C112D/M121L Az (PDB ID: 3FPY).458 (c) C112D/M121E Az at neutral pH. The SCS residues enforcing the binding configuration of the T0Cu site are circled in red. (d) C112D/M121E Az crystallized at basic pH. Reprinted with permission from ref 459. Copyright 2010 American Chemical Society.
Starting from the same Az model, the Lu group turned to investigate Cu-mediated post-translational modifications of S(Cys). A report in 2014 demonstrated that the M121G mutation decreased the hydrophobicity of the T1Cu pocket while opening an axial binding site (Figure 37a).463 Reduction to CuI enabled the interaction of the T1Cu site with hydrogen peroxide to yield a species with an absorption maximum at 400 nm that decays quickly in air to form the starting CuII-M121G Az. Further investigation of this intermediate by resonance Raman spectroscopy revealed an S-O vibration which could be attributed to the formation of sulfenic acid (Cys-SOH) formed at the Cys112 as further supported by proteomic analysis. The formation of sulfenic acid was attributed to the Cu-activated H2O2 based on 18O-isotope labeling studies. A computational study suggested the SCS served to stabilize the sulfenate by removing a negatively oriented dipole near Cu. This study was one of the first to demonstrate metal-sulfenate coordination in an artificial metalloenzyme system, whose natural presence was considered essential for the activity of nitrile hydratase and thiocyanate hydrolase. Although this process was accomplished using a different metal cofactor, this Az model demonstrated a plausible reaction mechanism for this post-translational modification in nature. Furthermore, the Lu group investigated the intramolecular nitrosylation of the Cys residue to form nitrosothiol (Cys-SNO), which serves as one of the most important biological reactions in NO regulation.276 To accomplish this, a H46E/M121H Az mutant was created to mimic the T1Cu of nitrosocyanin (Figure 37b). An additional F114P SCS mutation was introduced to eliminate H-bonding with S(Cys) and decrease the Cu potential. Under anoxic conditions, the T1Cu of the triple mutant catalyzed the formation of a Cu-Cys-SNO species on the ligating Cys residue, while a facile and complete release of NO could be achieved in vacuum or air. The Cu-mediated nitrosylation reaction displayed rapid kinetics (k = 6.7 × 105 M−1 s−1), high yields (>90%), and efficient scavenging of NO at micromolar concentration.
Figure 37.

(a) M121G azurin and the oxidative modification (PDB ID: 4MFH).103 The first step could only be achieved by H2O2 supported by the experiment with isotope labeled agents. The sulfenate could no longer be oxidized by H2O2, but further oxidation by O2 from air was observed. (b) Active site structure of H46E/F114P CuII-Az. (c–e) EXAFS results for (c) M121H/H46E/F114P CuII-Az, (d) M121H/H46E/F114P CuI-Az, and (e) NO-treated M121H/H46E/F114P CuII-Az. Reprinted with permission from ref 276. Copyright 2016 Springer Nature.
Ward and co-workers have also designed a copper peroxidase using 6-phosphogluconolactonase, a natural hydrolase scaffold, that has a potential (His)2Asp copper binding motif discovered by the STAMPS algorithm (Figure 38).464 The protein was expressed and cocrystallized with CuSO4, showing that the two predicted His residues bound Cu as anticipated, although the third ligand was found to be H2O stabilized by H-bonding with a side chain carbonyl moiety rather than the anticipated Asp131 residue. The peroxidase activity was found through the Cu-catalyzed oxidation of o-dianisidine using tert-butyl-hydroperoxide (t-BuOOH) as oxidant, with a maximum turnover number of ~25 achieved over the course of 1 h. Computational docking simulations revealed a possible hydrogen bond between the substrate and residues Asp131 and Tyr69, leading to the arrangement of the aromatic rings in-plane with the copper ion. A mutagenesis study of these two residues did not result in a significant change of catalytic efficiency when varied, although one mutant, Y69L, showed a 3-fold increase in turnover frequency.
Figure 38.

(a) Copper binding form of engineered 6-phosphogluconolactonase (PDB ID: 4TM8).464 (b) Peroxidation of o-dianisidine catalyzed by the Cu enzyme and the computationally docked substrate in the active site. The Asp131 and Tyr69 residues were believed to align the substrate. Reprinted with permission from ref 464. Copyright 2015 Royal Society of Chemistry.
In addition to natural proteins, Borovik and co-workers have used the Sav scaffold together with a biotinylated CuN3 synthetic complex to study Cu-H2O2 interactions (Figure 39),465 a similar strategy previously adopted to create a T1Cu in the same scaffold.466 The protein/copper complex was used to model lytic polysaccharide monooxygenase (LPMO), an enzyme with a special Cu-His-brace active site to catalyze the oxidative degradation of long-chain polysaccharides.358,467 The SCS of LPMO is known to stabilize the transient Cu-hydroperoxo (Cu-OOH) complexes but eventually lead to the cleavage of the O─O bond and the oxidation of the substrate.468,469 Such interaction was mimicked by this Sav-Cu model but resulted in a room-temperature-stable Cu-OOH complex. Upon addition of hydrogen peroxide, an intense shoulder around 380 nm was observed and attributed as an −OOH → Cu LMCT transition. The Sav-Cu protein crystal was soaked in H2O2-containing buffer, and the structure was determined with X-ray crystallography. The structure showed an electron density of two oxygen atoms on the axial site of copper, which was assigned as the axial hydroperoxo ligand. A hydrogen bond network was depicted in the crystal structure. The distal oxygen of the hydroperoxo anion formed a hydrogen bond with the amide nitrogen of the Asn49 residue. The proximal oxygen was involved in a more complicated hydrogen bond network consisting of Ala86, Ser112, Lys121, and two water molecules. The latter interaction was proposed to be important for the stability of the Cu-OOH complex as it offsets the push–pull effect from the distal hydrogen bond and avoids breaking the O─O bond, similar to the His-O2 interaction in myoglobin.470 The study provided key insight into how a well-positioned hydrogen bond alters the stabilization or activation of a Cu-oxygen adduct.
Figure 39.

(a) Crystal structure of the H2O2-bound Sav-Cu protein (PDB ID: 6ANX). (b) Structure of the copper complex conjugated to Sav. (c) Proposed mechanism to control Cu-OOH stability by different positioning of hydrogen bonds. Reprinted with permission from ref 465. Copyright 2017 American Chemical Society.
Aside from the α-helical bundle proteins mentioned previously, another type of de novo designed peptides was reported by Rufo et al. as copper-based oxidases (Figure 40). These β-sheet peptides self-assemble to form an amyloid fibril, serving as robust heterogeneous biocatalysts. A tri-His motif was built on the outer face between the β-strands to bind metals such as Zn and Cu.471 These solvent-exposed metal binding sites may be able to interact with substrates that are incompatible with hydrophobic environments in common enzymes. A report in 2016 showed that such amyloid assembly catalyzed the oxidative C─C coupling of 2,6-dimethoxyphenol using molecular oxygen.472 In a following study, the Cu-amyloid assembly showed versatile activities, catalyzing the cascade hydrolysis and oxidation of 2′,7′-dichlorofluorescin diacetate (DCFH-DA).473 It also showed that pure hydrolase activity could be achieved by catalyzing the hydrolysis and detoxification of paraoxon, a toxic organophosphate insecticide.
Figure 40.

Schematics of (a) Cu-bound amyloid assembly protein catalyzing the oxidation of DMP and (b) Cu-bound amyloid assembly protein catalyzing the cascade hydrolysis/oxidation of a fluorescence derivative and the hydrolysis of paraoxon. Reprinted with permission from refs 472 and 473. Copyright 2016 and 2018 Wiley-VDH and American Chemical Society.
4.3. Designed Copper Enzymes with New-to-Nature Activities
The concept to combine artificial organometallic complexes with protein scaffolds was first raised in the 1970s by Whitesides, Kaiser, and co-workers.474,475 Not limited by the natural amino acid ligand set, this bioconjugation-based strategy takes advantage of the non-natural reactivities of artificial cofactors while using the outer coordination spheres imposed by the surrounding protein matrix to impart unique properties on reactions, including enhanced rates, yields, stability, and selectivity. Various transformations have been reported in the past decades and reviewed extensively.69,476 This section will focus on the protein-conjugated copper catalysts that specifically utilize the outer coordination spheres of the protein hosts.
Copper, known for its Lewis acidity, has been used as a catalyst for the Diels–Alder (D-A) reaction since the 1960s.486 To mimic this reactivity, Reetz et al. used commercially available Cu-phthalocyanine imbedded in serum albumin (SA).487 The Cu macrocycle was proposed to be noncovalently anchored in the IB subdomain, analogous to the natural hemin. Among a series of substrates, an endo/exo product ratio of up to 95:5 with an ee value up to 98% could be accomplished. The high enantioselectivity was not experimentally probed beyond product analysis, but it was proposed that protonation and/or hydrogen bonding with the substrate nitrogen atom was relevant. A later study by the same group created a Cu-(His)2Asp facial triad site in a β-barrel core of the tHisF protein (Figure 41a), which was found to catalyze the D─A reaction on the same set of substrates but with lower enantioselectivity.477 The same Cu-(His)2Asp core was later adopted and improved by Ghattas et al. in a 1-aminocyclopropane carboxylic acid oxidase (ACAO) scaffold by substituting the native Fe center with Cu (Figure 41b).478 Yields over 85% for all substrates were accomplished, with a maximum endo/exo ratio of 99:1 and ee of 95%. A computational docking study indicated that the substrate is flanked and orientated by Lys158 and Ile184, while Phe250 packs with the substrate from the noncoordinating side. These SCS restraints only enable one possible direction for the attack by cyclopentadiene, explaining why such high enantioselectivity was achievable.
Figure 41.

Examples for artificial Diels—Alderase. (a) tHisF protein with the copper binding site. (b) ACCO protein with the copper-bound cavity and docked substrate. (c) FhuA protein and the copper binding site. (d) Oligonucleotide and the conjugated copper complexes. (e) LmrR protein with the conjugation sites for copper complexes shown in red. (f) αRep A3 protein with the central pocket to conjugate copper complexes. (g) Neocarzinostatin protein with the copper complex noncovalently docked in the pocket via the testosterone tail. (h) FhuA protein with the copper complex conjugated. (i) apo-nitrobindin protein with the covalently conjugated copper complex. (j) Schematic representation that the copper complex with the adenosine tail may bind to the cell-surface receptor and catalyze the D—A reaction. (k) Bovine pancreatic polypeptide and the unnatural amino acid residues introduced to form a copper binding site. Reprinted from references 367, 369, 477-485. Copyright 1021, 2009, 2010, 2019, 2019, 2011, 2017, 2016, 2016, 2016, and 2018 Wiley-VCH and American Chemical Society.
Meanwhile, a series of artificial enzymes were reported using covalently fused Cu-phenanthroline or Cu-dipyridine compounds and scaffolds including polynucleotides,480,488 LmrR,367,489 αRep A3,481 neocarzinostatin,482 FhuA,483 aponitrobindin,484 adipocyte lipid binding protein, and human adenosine receptor (Figure 41c-j).485 Most of these examples adopt a hydrophobic SCS, either inside the protein or formed between dimer surfaces, to stabilize the aromatic copper complex, facilitate substrate binding, and accelerate the reaction. Another strategy using amino acid residues as ligands has also been adopted to design a Diels–Alderase in bovine-pancreatic polypeptide (BPP)369 (Figure 41k), LmrR490 and mTFP.479 Nonetheless, many of the enzymes in these early works have slow catalytic rates and relatively low conversion. This issue was addressed in a very recent study by Basler et al.491 An α-helical protein binding Zn (a structural analogue of Cu with different acidity) was designed and developed through several rounds of directed evolution. The catalytic efficiency of this construct was improved by over 105-fold between the starting and final designs. The final design, DA7, has a catalytic efficiency of 2.9 × 1010 M−1, a TON of 104, and >99% endo selectivity. Examination of the crystal structure and a docking model revealed the alignment of the diene substrate by the hydrogen bond donor Arg28 from the carboxylate side and by Gln31, Gln80, and Tyr84 from the pyridyl side. The dienophile substrate was packed underneath the diene by the aromatic ring of Trp68 with the two dienedienophile moieties closely facing each other. The precise orientation of the substrate explained how these high numbers were accomplished, the most efficient currently reported for a Diels–Alderase.
Another application of Cu-protein hybrid catalysts is the Friedel–Crafts (F-C) alkylation reaction. To this end, the Roelfes group used a LmrR protein that forms a homodimer with a large hydrophobic pocket.368 The two Trp residues at the interface exert strong π-π stacking to allow tight binding to an aromatic CuII-phenanthroline (CuII(phen)) or CuII-2,2′-bipyridine (CuII(bpy)) complex with micromolar affinity (Figure 42a). These artificial metalloenzymes catalyzed the site selective alkylation of an indole substrate with near full conversion and ee up to 93%. Interestingly, the reactivity of this artificial enzyme could be tuned by mutation of two SCS residues. The A92E mutation altered the side chain orientation toward an outer Asn14 residue, enhancing binding of the CuII(phen) cofactor between the two Trp residues according to MD simulations and eliminating the minor reactivity of the tandem F-C alkylation/enantioselective protonation reaction (FC-EP).492 Removal of the packing Trp96 residue, however, switched the reactivity toward the FC-EP reaction with moderate enantioselectivity due to the binding dynamics of the cofactor. The hybrid catalyst was further applied in whole-cell biocatalysis, and directed evolution was applied to the outer sphere residues to enhance the yield and selectivity.494 A similar noncovalent cofactor docking method has also been applied in other proteins from the TetR family to catalyze the same Friedel–Crafts reaction (Figure 42b).493 An alternative strategy of genetic unnatural amino acid incorporation was adopted by the same group to introduce the AlaBpy unnatural amino acid residue as Cu ligand.490 Mutation of three SCS residues, Asn19, His86, and Phe93, had a moderate impact on the conversion rate, enantioselectivity, and substrate preference, which was attributed to π-stacking interactions and the steric freedom of the hydrophobic pocket. This AlaBpy LmrR mutant was further used to stabilize a semiquinone radical (Figure 42c), reminiscent of the chemistry of COx.286
Figure 42.

(a) LmrR protein with two tryptophans that docks the aromatic ligand of the copper complex. The active site structure at the bottom right shows the targets for SCS mutagenesis studies. (b) Different proteins from the TetR family that were engineered for F-C catalysis. The proteins are LmrR (top left), CgmR (top right), RamR (bottom left), and QacR (bottom right). (c) The AlaBpy LmrR mutant that stabilizes a semiquinone radical. Reprinted with permission from references 286, 368, 492, and 493. Copyright 2017, 2015, and 2020 American Chemical Society and Wiley-VCH.
The Michael addition reaction, another type of C─C bond formation reaction, can be catalyzed by artificial copper enzymes. In the late 2000s, Roelfes and co-workers reported the enantioselective catalysis of the Michael addition reaction by a Cu binding oligonucleotide488 and bovine pancreatic peptide scaffold,369 respectively. Recently, the group expanded the previously mentioned CuII(phen)-Trp docking strategy with an unnatural adjacent p-aminophenylalanine (pAF) residue.495 The two active centers work in a synergistic manner, in which Cu binds and activates the Michael donor while the pAF residue activates the Michael acceptor substrate by forming a Schiff base (Figure 43a). An improved yield (36%) and ee (86%) were observed in the dual-functional catalyst versus controls. The M8L mutation provided moderately improved activity and selectivity (99%), which were correlated to a hydrophobic interaction with Trp96, as described in a previous study vide supra. The Itoh group has reported a cupin-based catalyst using only natural amino acids (Figure 43b).496 Starting from a natural (His)4 binding motif, a ligand set library was screened by decreasing numbers of His residues. Both the tris-His mutant H52A (S product) and the bis-His mutant H54A/H58A (R product) were found capable of catalyzing the Michael addition reaction with both high yield and selectivity, but interestingly, their enantioselectivities were reversed relative to one another. A docking study revealed that the hydrogen bond from Cys106 (sulfinic acid form) may swing to the Si-face of the azachalcone substrate in the H52A mutant, favoring the attack by nitromethane from this side. On the other hand, the H54A/H58A mutant appeared to have an open Re-face with the Phe104 residue blocking the Si-face, and as a result the Cys106 swings to the Re-face to enable attack from the Re side. This effect was supported by a subsequent mutagenesis study, in which the H52A/C106A or H52A/C106S mutation removes or disori-entates the H-bond and reduces the Si selectivity, whereas the H54A/H58A/F104W mutation enhances the steric hindrance from residue 104 to increase the Re selectivity. Another recent study by Rimoldi et al. used two simple peptides derived from the methionine-rich motif in hCtr-1 transporter to catalyze the Michael addition reaction.497 The yield was comparable to those of previous studies with a relatively lower enantioselectivity, possibly due to the absence of the SCS interactions of more complex proteins.
Figure 43.

(a) LmrR variant catalyzing the Michael addition reaction with an aniline moiety on an unnatural amino acid residue to activate the ketone Michael acceptor. The structure was modeled from PDB ID 6R1L,492 and the scheme was adapted from ref 464. (b) Two cupin variants with different enantioselectivities of the Michael addition reaction. The insets at the bottom demonstrate how the hydrogen bond with Cys106 changes the orientation of the substrate and thereby alters the product enantioselectivity. Reprinted with permission from refs 495 and 496. Copyright 2020 Nature Publishing Group and Wiley-VCH.
Artificial Cu enzymes have also been reported for hydration reactions and small molecule polymerization. Both covalent conjugation and noncanonical amino acid incorporation strategies were used in two studies to introduce phenanthroline or AlaBpy Cu complexes into the LmrR scaffold (Figure 44).498,499 Hydration of α,β-unsaturated ketone substrates was achieved in both cases with moderate enantioselectivity (60–80%). Two residues, Phe93 and Asp100, were found to serve as important SCS residues in both cases. The Phe residue provided a π−π stacking interaction to support binding of aromatic copper complexes, while the Asp residue acts as a general Lewis base, forming hydrogen bonds and activating the water substrate. In the latter case (Cu-AlaBpy), adding an alternative general base such as V15E significantly increased both the conversion rate and enantioselectivity due to better positioning of the carboxylate. As for polymerization, the Bruns group reported a protein cage nanoreactor system using thermosome (THS).370 This protein is a group II archaea chaperonin with a large (130 nm3) cavity, that was used to confine the Cu complex and substrate, as well as wide pores, to release polymeric products. The nanoreactor produced poly(N-isopropyl acrylamide) by ATRP with an average molecular weight of 1500 g/mol and a polydispersity index of 1.11. Other catalysts, such as horseradish peroxidase, have also been used in this nanoreactor system.500
Figure 44.

(a) LmrR variant as a hydratase with important SCS residues shown as sticks. (b) LmrR variant with an AlaBpy residue to bind copper and carry out hydration of alkene. Reprinted with permission from refs 498 and 499. Copyright 2013 and 2017 Royal Society of Chemistry.
5. CATALYSIS BEYOND THE PRIMARY COORDINATION SPHERE BY RATIONALLY DESIGNED MULTINUCLEAR METALLOENZYMES CONTAINING HEME
By incorporating another metallocofactor next to the heme, nature evolved a number of multinuclear metalloenzymes to fulfill different and more challenging functions. One example is manganese peroxidase (MnP), in which a MnII-binding site next to the heme macrocycle can oxidize MnII to MnIII upon reaction with H2O2. The generated MnIII is then used to oxidize other organic substrates, in some instances.501 On the other hand, heme-copper oxidase (HCO) consists of a heme-CuB heteronuclear center. It functions as a terminal oxidase catalyzing the reduction of O2 to water to generate the transmembrane proton gradient required for ATP synthesis.41,502 Interestingly, bacterial nitric oxide reductase (NOR) has a non-heme iron center (FeB site) at the corresponding CuB position of HCO and catalyzes the one-step, two-electron reduction of NO into N2O, which is an important step in denitrification.422,503 Dissimilatory sulfite reductase (SiR) catalyzes a six-electron reduction of sulfite into sulfide using a combination of a heme macrocycle (siroheme) and a [4Fe-4S] cluster as cofactor. It makes up one of the important steps in sulfate respiration in sulfate-reducing bacteria.504 In order to elucidate how nature evolved these different multinuclear heme-containing metalloenzymes to catalyze different reactions, efforts have been made using synthetic505 and biosynthetic models69,476,506 to provide more insights. Moreover, inspired by the water-bridged homodinuclear magnesium mechanism for some natural nucleases,507 an artificial nuclease was made by the design of a Mg2+-heme heteronuclear Mb system.508 In this section, we will focus on how SCS affects the performance of these multinuclear heme-containing ArMs.
5.1. Manganese Peroxidase Using Cytochrome c Peroxidase as a Scaffold
To engineer a functional mimic of MnP, the Lu group successfully designed a Mn binding site into CcP through the introduction of G41E, V45E, and H181D mutations (Figure 45a and b).509 The resulting triple mutant (MnCcP) could oxidize FeII at least five times more efficiently than the native CcP. A deeper structural analysis and comparison of the secondary coordination spheres of CcP and MnP uncovered two residues of interest, namely Trp51 and Trp191 of CcP, which sit in the same positions occupied by Phe in MnP (Figure 45c and d). Hence, the W51F and W191F mutations were made to further tune the reactivity of MnCcP. While no significant change in activity on MnII oxidation was observed for W191F MnCcP, W51F MnCcP was found to have a 4.3-fold increase in Vmax compared to MnCcP, and the W51F/W191F MnCcP double mutant displayed a further 14.3-fold increase in Vmax over that of W191F MnCcP. This compounding effect may be rationalized if one assumes there is an equilibrium between the FeIIITrp+. state (the radical of compound I in CcP is believed to preside on Trp191 rather than the porphyrin ring, as Trp is readily oxidized) and the ferryl state of CcP compound II. Both Trp51 and Trp191 are capable of stabilizing compound II, with either Trp51 through H-bonding to the ferryl oxygen or Trp191 through the proposed equilibrium. Although W191F MnCcP cannot stabilize compound II through radical delocalization, it can still be stabilized by H-bonding interactions with Trp51; therefore, no increase in activity is observed. For W51F MnCcP, the delocalization from Trp191 can stabilize compound II, but the H-bonding interaction is missing, so an increase in activity is observed. By mutating both Trp to Phe, both stabilizing effects are removed, producing the largest increase in activity.510,511 The resonance Raman spectra of these mutants also reveal that the W51F mutant lacks the H-bonding interactions between tryptophan and a weakly bound distal water. The loosening of this water may lead to quicker formation of compound I and an increase in activity.512 However, all the MnCcP and its mutant mentioned did not match the activity of native MnP. The kcat/KM for W51F/W191F MnCcP was 0.599 s−1 mM−1, while the one for native MnP was 3952 s−1 mM−1.510
Figure 45.

(a) Mn-binding site in MnP (PDB ID: 1MNP).513 (b) Putative Mn-binding site in CcP, forming MnCcP (PDB ID: 2CCP).139 (c) Position of Phe45 and Phe190 around the active site of MnP. (d) Position of Trp51 and Trp191 around the active site of CcP. Atom coloring: N (blue); O (red); CMnP (magenta); CCcP (green); Fe (orange); Mn (lilac).
To enhance the activity of MnCcP and investigate the roles of other secondary sphere residues, MnCcP was further redesigned by replacing Asp181 with Glu, reverting the G41E mutation, and further mutating Asp37 to Glu. The resulting construct is referred to here as MnCcP.1. MnCcP.1 was found to have a 2.5-fold higher catalytic efficiency (kcat/KM) than MnCcP.514 Based on MnCcP.1, several additional alterations to the secondary coordination sphere were made to further tune its activity.515
First, comparison of the crystal structures of MnP and MnCcP revealed that Glu39 in MnP is very flexible and can adapt different rotamers. As a result, binding of MnII can occur with a relatively short distance of 2.10 Å. Meanwhile, the correspond-ing Glu45 of MnccP.1 is rather rigid, retaining its position in either MnII-free form or MnII-bound form with a distance of 3.94 Å in the latter. This results in a different metal center geometry and may be responsible for the different MnII binding affinities and MnP activities. Tyr36 was found to be within H-bonding distance of the carboxylate group of Glu45 in MnCcP.1 (Figure 46), an interaction that is otherwise missing in MnP. This H-bond may compete with MnII coordination by stabilizing rotamers that are unfavorable for coordination. To address this, a Y36F mutation was introduced into MnccP.1 and was found to have a 2.8-fold improvement of KM, indicating the binding affinity of MnII was increased.
Figure 46.

Different SCS residues included to MnCcP.1 and their effects on the catalytic efficiency. Reprinted with permission from ref 515. Copyright 2016 American Chemical Society.
Second, residues Glu37 and Glu181 were found to closely neighbor MnII in MnCcP.1, with distances of 3.23 Å and 4.08 Å, respectively. However, these distances are considerably longer than the corresponding coordination bonds of MnII─Glu35 and MnII─Asp179 in MnP, which are 2.4 Å and 2.41 Å, respectively. A structure-based sequence alignment of MnPs from different organisms revealed that a conserved Arg was located below Glu35 in MnP. Bearing in mind that this arginine might play a key role in the positioning of Glu35 and Asp179 in MnP through the formation of a salt bridge, a K179R mutation was made in MnCcP.1 (Figure 46). MD simulations showed that Arg179 moved farther from Glu181, causing a reorientation of Glu181 and, consequently, reducing the interatomic distance of the carboxylate side chain to MnII by nearly 2 Å. Activity assays showed that, as a result, a 2.6-fold improvement in KM was observed.
Additionally, it was discovered that Ile40 in MnCcP.1 may sterically clash with Glu37 in MnccP.1, causing the Glu side chain to reorient away from the MnII center. In most MnPs, the corresponding position is occupied by glycine, and therefore, an I40G mutation was introduced in MnCcP.1 (Figure 46). The corresponding MD simulations showed a decrease in bond length between the ligands and MnII. That for Glu37 decreased from 3.23 Å to 2.54 Å, and that for Glu45 decreased from 3.94 Å to 2.34 Å. This resulted in a 3.6-fold improvement for KM. For this mutant, a 2.4-fold increase for kcat is also observed and resulted in an overall 8.6-fold increase in catalytic efficiency.
5.2. Heme-Copper Oxidase Using Myoglobin as a Scaffold
5.2.1. Design Using Natural Amino Acids.
The practical application of enzymes such as HCO outside of the cellular environment is often challenging due to their large size and complexity. Therefore, designing ArMs based on small and robust protein scaffolds is an attractive solution to this problem.
While Mb contains a single heme center, HCOs contain a high-spin heme-CuB center and a low-spin heme dinuclear center (Figure 47). As a subclass of HCO, CcO also possesses a dinuclear Cu center called CuA.331 To understand the role of the CuB site for the function of HCOs, Lu and co-workers reported their first design of the HCO biomimetic system using the Mb scaffold in 2000.518 To accomplish this, a Cu-binding site (CuB site) was engineered in the vicinity of the heme Fe by introduction of two His residues, L29H and F43H, in the distal pocket of the heme site, closely resembling the heterobinuclear active site found in HCOs (Figure 47c). Herein, the L29H/F43H Mb mutant is referred to as CuBMb. Initial studies on CuBMb suggest that the copper ion in the CuB center is essential for the O2 reduction activity.519 However, CuBMb generates verdoheme, instead of ferryl-heme, during the O2 reduction due to the lack of the H-bonding network that delivers protons to promote the heterolytic O─O cleavage. Since the Tyr residue in the active site of HCOs has been thought to be critical to the enzymatic functions,520 Miner et al. later found that introduction of Tyr to the active site of CuBMb (F33Y-CuBMb and G65Y-CuBMb) enabled this artificial enzyme to convert O2 to water with minimal release of reactive oxygen species (ROS), which in turn prevented the degradation of heme to verdoheme (Figure 48).517 Repositioning of Tyr in the G65Y CuBMb construct further improved the rate of H2O production relative to that of F33Y CuBMb, illustrating for the first time that the positioning of the Tyr is critical for modulating the catalytic rate. Moreover, the crystal structure of apo-F33Y CuBMb revealed additional water molecules in the distal heme pocket, suggesting that Tyr may be involved in the H-bonding network that activates the ferric-superoxo intermediate for water generation. Two years later, Yu et al. reported the direct observation of the Tyr33 radical in F33Y CuBMb by EPR (Figure 48b), providing firm support for the presence of a Tyr radical and the role of Tyr as an electron and proton donor in the enzymatic mechanism of HCO.521 Moreover, in 2016, the EPR spectra of cryoreduced oxy-F33Y CuBMb and the crystal structure (Figure 48c) of this mutant provided further direct evidence of the importance of H-bonding networks in activating the enzyme to reduce O2 to water.522
Figure 47.

(a) Crystal structure of ba3 CcO (PDB ID: 3S8F).516 (b) Structure of the catalytic site. The low-spin heme b and heme a3 are depicted in cyan; CuB is indicated by a yellow sphere. The cross-linked residues His233 and Tyr237, located in proximity of the catalytic site, are shown. (c) Crystal structure of L29H/F43H Mb (PDB ID: 4FWZ).517
Figure 48.

(a) Structures and computer models of native and designed oxidases. Left panel: overlay of the crystal structures of bovine CcO(black) and E-F33Y-CuBMb (cyan). Right panel: overlay of the crystal structures of cbb3 HCO from Pseudomonas stutzeri (tan) and the E-G65Y-CuBMb computer model (yellow). The CuB copper is represented as an orange sphere. N (blue); O (red); Fe (green). Reproduced with permission from ref 517. Copyright 2012 John Wiley & Sons. (b) EPR spectra of ferric F33Y-CuBMb reacted with 1 equiv of H2O2 (red) and ferrous F33Y-CuBMb with O2 (black). Reprinted with permission from ref 521. Copyright 2014 American Chemical Society. (c) Crystal structure of oxy-F33Y-CuBMb (PDB ID: 5HAV, left panel) compared with that of oxy-WT-Mb (PDB ID: 1A6M).523 Reprinted with permission from ref 522. Copyright 2016 American Chemical Society.
Taking advantage of the enhanced O2-reduction activity of G65Y CuBMb, Mukherjee et al. reported the Mb-based biosynthetic model CcO, capable of electrochemically catalyzing O2 reduction with a reaction rate higher than that of either any known synthetic analogue or the native CcO.524 The electrons required for the O2 reduction reaction were directly injected into the heme cofactor via tethering by click reaction of the functionalized hemin-yne to azide terminated thiols bound to a gold electrode. This approach to direct electron transfer circumvents the rate-limiting dissociation of ferric hydroxide and the presence of the protecting Tyr65, resulting in an O2 reduction activity that is an order of magnitude faster than that of CcO fixed by immobilization on an electrode.
In 2015, Yu et al. further developed a Mb variant (D44K/D60K/E85K) based on the G65Y-CuBMb platform by incorporating residues to promote favorable surface electrostatic interactions with the native redox partner of HCO, cyt b5 (Figure 49).525 Using this approach increased the activity of the CuBMb model to a level comparable to that of the native enzyme.
Figure 49.

(a) Structures of G65Y-CuBMb(+6), showing the engineered Lys in blue, and cyt b5 (PDB ID: 1CYO).526 (b) Oxidase activity of G65Y-CuBMb(+6) in comparison with that of native cyt cbb3 oxidase. The black arrows indicate the addition of reductant, and the double arrow shows the injection of native cyt cbb3 oxidase. Reprinted with permission from ref 525. Copyright 2015 American Chemical Society.
In addition to serving as functional models of HCOs, engineered Mb oxidases have also proven to be excellent models for studying the relationship between the reduction potential and O2-reducing activity. In 2014, Bhagi-Damodaran et al. introduced an S92A mutation to F33Y-CuBMb, aiming to remove the H-bonding interactions between Ser92, the proximal His ligand, and a heme propionate as well as increasing the hydrophobicity in the heme pocket (Figure 50).527 The S92A mutation led to an increase in the reduction potential by about 30 mV, resulting in a higher driving force and faster O2 reduction.
Figure 50.

Overlay of the crystal structures of F33Y-CuBMb (PDB ID: 4FWX, cyan)517 and S92A-F33Y-CuBMb (PDB ID: 4TYX, orange).527 Heme b and the side chains of His29, His43, His64, Tyr33, His93, and Ser/Ala92 are shown in licorice. Ser92 forms H-bonds with heme propionate and His93. Reprinted with permission from ref 527. Copyright 2014 American Chemical Society.
In 2018, Mukherjee et al. incorporated one or two proton transfer glutamate residues (V68E and V68E/I107E) in CuBMb to mimic the proton donor acidic residues in HCOs (Figure 51a).524,528 Although V68E and V68E/I107E-CuBMb showed O2 reduction rates comparable to that of G65Y-CuBMb, which does not contain a glutamate residue in the active site, the presence of Glu resulted in a significantly reduced solvent kinetic isotope effect (SKIE), with V68E/I107E-CuBMb bearing the lowest SKIE of ~2.4 (Figure 51b). The dramatic difference in SKIE indicates a change in the rate-limiting step in the reaction mechanisms, with Glu mutations presumably providing a preorganized proton transfer channel that speeds up proton translocation.
Figure 51.

(a) Crystal structure of FeBII-V68E/I107ECuBMb (PDB ID: 3M39).529 (b) Cartoon showing that incorporation of the Glu residue near the active site facilitates the proton transfer from the external solvent to the active site, which results in lower KSIE. (c) Structure of the active site of oxy-I107E-CuBMb (PDB ID: 7L3Y).530 (d) Rates of O2 consumption yielding water (blue) or reactive oxygen species (ROS) (red). Parts c and d are reprinted with permission from ref 530. Copyright 2021 American Chemical Society.
The crucial role of the active site Glu residue in regulating oxidase activity was further reinforced by Petrik et al. in 2021.530 An I107E mutation not only promoted the O2 reduction rate but almost completely eradicated ROS formation. A combination of crystallographic and EPR studies revealed an extensive H-bonding network involving glutamate-facilitated reaction intermediate protonation (Figure 51).
5.2.2. Design Using Unnatural Amino Acids.
Although the Tyr-His cross-link is a well-established feature in native HCOs (Figure 52a), the mechanistic role of this cross-link is still unclear.531 To better understand the impact of this feature in HCOs, Liu et al. reported the genetic incorporation of an UAA, imiTyr, into CuBMb at position 33 to mimic the Tyr-His cross-link of HCOs (Figure 52b).532 Compared to the F33Y CuBMb mutant, the Mb model bearing imiTyr exhibited an 8-fold increase in selectivity and 3-fold increase in catalytic turnover (Figure 52c). It was postulated that the decrease in the pKa of the phenol in imiTyr is key to facilitating proton delivery and Tyr radical formation.
Figure 52.

(a) CuB site of CcO (bovine numbering). (b) Structural model overlay of imiTyrCuBMb (cyan) and F33Y CuBMb (yellow). (Inset) Chemical structure of imiTyr. (c) O2 reduction turnover number measured during the stepwise addition of O2. The reduction was catalyzed by imiTyrCuBMb or F33Y CuBMb. Reprinted with permission from ref 532. Copyright 2012 Wiley-VCH. (d) Crystal structure of F33Y/L29H/F43H swMb (F33Y CuBMb, PDB ID: 4FWX)517 and pKa values of the Tyr and Tyr analogs. From top to bottom: Tyr, 3-chlorotyrosine (ClTyr), 3,5-difluorotyrosine (F2Tyr), and 2,3,5-trifluorotyrosine (F3Tyr). The red arrow indicates increased activity with the increased pKa values of Tyr and Tyr analogs. Reprinted with permission from ref 533. Copyright 2015 American Chemical Society. (e) Structural model of the OMeY Mb mutant, constructed based on the crystal structure of F33Y CuBMb (PDB ID: 4FWX).517 Reprinted from ref 534 with permission from The Royal Society of Chemistry.
Since Tyr has been suggested to function as both a proton and electron donor in the enzymatic cycle of HCOs, both the pKa and reduction potential of Tyr can potentially affect its role in modulating the enzymatic activity. To analyze the correlations between these factors and the oxidase activity, Yu and co-workers established a linear relationship between the pKa of the phenol group at position 33 and the O2 reduction activity using a series of unnatural analogues of Tyr with varying pKa values of the phenol ring (Figure 52d).533 However, the dependence of the oxidase activity on the reduction potential of Tyr is convoluted, since the pKa and the reduction potential of Tyr are closely correlated. To tackle this problem, Yu et al. incorporated a Tyr analog, 3-methoxy tyrosine (OMeY), that has a comparable pKa to that of Tyr but a lower reduction potential (Figure 52e).534 The F33OMeY CuBMb exhibited a higher O2 consumption rate with a lower generation of ROS compared to the case of F33Y CuBMb. In this manner, they demonstrated that the electron donating ability of Tyr in the active site is a critical factor for the function of HCOs.
5.3. Bacterial Nitric Oxide Reductase Using Myoglobin as a Scaffold
NOR is structurally homologous to the largest subunit of HCO; rather than a three-histidine-coordinated CuB site in the distal position of the heme, the FeB site is incorporated into this position instead. Aside from the three His residues, two additional Glu residues were found to be essential for NOR activity. To mimic the structure and activity of NOR, the Lu group engineered a three His/one Glu site in the distal pocket of swMb, successfully established NOR activity.535 To make this model more similar to the native NOR and to investigate the role of the second Glu, the second Glu was introduced into FeBMb by a I107E mutation. The resulting crystal structure showed a bridging water molecule between the OE2 atom of Glu107 and the non-heme iron center (Figure 53). The NO reduction activity of FeII-I107E FeBMb was found to be higher than that of FeII-FeBMb, with ~24% N2O for the former and ~10% for the latter over the course of ~20 h under single-turnover conditions. This suggests that unlike the initial three His residues and one Glu, which directly coordinate to the FeB site, the second Glu may potentially play a role in the proton transfer pathway during the reduction of NO.529 This hypothesis was further supported by making a I107F mutation in FeBMb, which inhibited N2O formation.536
Figure 53.

Overlay of FeII-FeBMb and FeII-I107E FeBMb: FeII-I107E FeBMb, PDB ID: 3M39 (cyan); FeII-I107E FeBMb, PDB ID: 3K9Z (orange).529 Reprinted with permission from ref 529. Copyright 2010 National Academy of Sciences.
By utilizing this model FeBMb NOR enzyme, an important question as to why Cu is preferred over Fe for O2 activation and reduction in HCO was resolved. The role of different metal ions in the distal pocket was explored by titration of the non-heme FeB center with CuI (CuI-FeBMb, mimic of HCO), FeII (FeII-FeBMb, mimic of NOR), and ZnII (ZnII-FeBMb, control).537 While the redox potential values of the heme center were similar (−64 ± 4 mV, − 58 ± 3 mV, and − 45 ± 2 mV, respectively), the redox potential values of the non-heme metal center were different, with +259 ± 20 mV for non-heme FeIII/FeII and +387 ± 25 mV for CuII/CuI. Resonance Raman (rR) spectroscopy showed that, in the O2-bound form, ZnII-FeBMb exhibited a stronger Fe─O2 bond, which, in turn, implies greater O─O bond activation compared to that of FeBMb in the absence of a distal metal ion. DFT calculations indicated that the O─O bond in Fe-FeBMb was longer than that in Zn-FeBMb and even longer in Cu-FeBMb. One possible explanation for this behavior is that FeII and CuI are redox active and capable of electron donation to O2. Additionally, CuII may provide a higher degree of d-electron density (d9) than FeIII (d5) to O2 through dπ-pπ backbonding, further weakening the O─O bond. In fact, copper is the only 3d transition metal that is redox active and have maximum number of d-electrons for O2 activation.537
The redox potential of heme can play an important role in tuning NOR reactivity. Bhagi-Damodaran et al. tuned FeBMb by exchanging heme b for monoformyl (MF-) or diformyl (DF-) heme, as well as by mutating Leu89, a residue close to the proximal His, into Ser. The exchange of heme increased the redox potential of heme center, while the mutation increased the hydrophilicity around heme and lowered the redox potential by stabilizing the higher oxidation states. This work found that a lower redox potential facilitated fast NO binding and subsequent decay of dinitrosyl species, while higher redox potentials facilitate faster electron transfer. Accomplishing multiple turnovers of NOR reactivity requires balancing optimizing the redox potential to balance these two factors.538
5.4. Sulfite Reductase (SiR) Using Cytochrome c Peroxidase as a Scaffold
In 2018, the Lu group successfully engineered a [4Fe-4S] binding site into CcP using six mutations (H175C, T180C, W191C, L232C, M230A, and D235V), which will be referred to as SiRCcP.1, with the purpose of creating a biosynthetic model of SiR.539 Following incorporation of heme b and the [4Fe-4S] cluster into SiRCcP.1, the resulting heme-FeS-SiRCcP.1 exhibited notable sulfite reducing activity, forming a basis for further work tuning the SCS to enhance its activity. A series of mutations were incorporated to form a positively charged cavity reminiscent of native SiR, including W51K, H52R, and P145K, mimicking Lys217, Arg153, and Lys215 in native SiR. The position of native Arg48 in CcP is similar to that of Arg83 in native SiR (Figure 54a). This W51K/H52R/P145K-SiRCcP.1 variant exhibited a 5.3-fold increase in activity over that of SiRCcP.1. Residue Asn481 in native SiR was oriented towards the iron sulfur cluster and may increase activity. Therefore, a D235N mutation at a similar position as SiR Asn481 was made to replace the original D235V mutation, with the resulting construct named SiRCcP.2 (Figure 54b). This new SiRCcP.2 construct was found to be 17-fold more active than SiRCcP.1. Further exchange of residue 235 for Cys in the D235C variant (SiRCcP.3) (Figure 54b) resulted in even higher activity. In combination with the W51K/H52R/P145K substitutions, W51K/H52R/P145K-SiRCcP.3 displayed a 63-fold increase in activity relative to the original SiRCcP.1 construct, achieving ~18% of the activity observed for native SiR from Mycobacterium tuberculosis.539
Figure 54.

(a) Computational model of the SiRCcP substrate-binding site with mutations W51K, H52R, P145K (cyan), and Arg48 (orange). (b) Computational model of the mutations D235V (SiRCcP.1, cyan), D235N (SiRCcP.2, green), and D235C (SiRCcP.3, magenta) in SiRCcP. Reproduced with permission from ref 539. Copyright 2018 American Association for the Advancement of Science.
5.5. An Artificial Nuclease Using Myoglobin as a Scaffold
By incorporating a Mg2+ into L29E Mb, an efficient artificial nuclease Mg2+-L29E Mb was made. Through molecular docking simulations of double-stranded B-DNA with the enzyme (Figure 55), it was determined that the distal Arg45 can interact with the sugar/phosphate group of DNA through H-bonding interactions. This basic residue was found to be conserved in the native nuclease, possibly enhancing the DNA binding affinity and further promoting cleavage. Introduction of a R45S mutation was introduced to eliminate the interaction and the DNA cleavage, and it was found to successfully inhibit nuclease activity by ~55–60%.508
Figure 55.

Docking structure of the Mg2+-L29E Mb-DNA complex (left) and enlarged view of the Mg2+-heme heterodinuclear center (right), highlighting the coordination of the Mg2+ ion and the H-bonding interactions with the sugar phosphate group of DNA. Reprinted with permission from ref 508. Copyright 2020 American Chemical Society.
6. BEYOND FE AND CU: INSIGHTS INTO THE SECONDARY COORDINATION SPHERE OF ENGINEERED METALLOENZYMES INCORPORATING NON-NATIVE METAL IONS
6.1. Replacement of Native Metal Ions and Metallocofactors
Besides mimicking the diverse array of metalloenzymes found in nature, the protein reengineering approach can be used to generate novel active sites to impart new reactivity with non-native metal ions and cofactors. However, doing so presents its own unique challenges, particularly in finding a scaffold capable of tightly binding the desired metal center or organometallic complex. To solve this problem, noncovalent, single-point covalent attachments, and dual covalent anchoring strategies can be employed. The Watanabe group has demonstrated that enantioselective enzymatic sulfoxidation can be regulated by unnatural metal complexes in a redesigned protein active site.540 Oxidative CrIII and MnIII Schiff base catalysts were inserted to Mb using His93 as an anchor, forming a metal–His93 dative bond. In addition to the noncovalent interactions, including π-stacking between the benzene ring of the salophen ligand and the Phe43 side chain, and C-H/π-interactions between the side chains of Leu89 and Ile99 and the ligand (Figure 56a), the two methyl groups of the salophen ligand were found to interact with Ile107, allowing the Mn complex to be inserted deep in the pocket (Figure 56b). The rate of sulfoxidation catalyzed by these complexes was accelerated by inserting into apo-Mb, and CrIII-Mb and MnIII-Mb exhibited 15- and 6-fold higher reactivities than that of the CrIII Schiff base complex in buffer, respectively. Further modification of the ligand sterics regulated the position of the inserted synthetic metal complex, helping to tune the enantioselectivity, and additional mutation of H64D increased the substrate accessibility, enabling a larger substrate scope.
Figure 56.

Active site structure of Mn-1-apo-A71G Mb: (a) side and (b) top views. The heme of met-Mb128 (blue) is superimposed on Mn-1-apo-A71GMb. (c) Metal-salophen complex (1) Reprinted with permission from ref 540. Copyright 2005 American Chemistry Society.
To further enhance both the rate and enantioselectivity of the sulfoxidation reaction, the Lu group constructed an artificial metalloenzyme by incorporating manganese salen complex into Mb using a dual covalent anchoring approach, generating MnSalen-Mb(T39C/L72C) (Figure 57).541,542 Mb(T39C/L72C) was found to exclusively catalyze the formation of sulfoxide. To account for such high chemoselectivity, it was hypothesized that the polarity of the residue near the entrance of the protein pocket may play an important role in regulating substrate access. A single mutation, A71S, near the entrance of the protein pocket was made to increase hydrophilicity and, as a result, led to the further oxidized product sulfone. This demonstrates the delicate balance of the secondary coordination sphere in tuning selectivity.
Figure 57.

(a) Computer model of Mb(L72C/Y103C) with 1 covalently attached overlaid with heme. (b) Complex 1. Reprinted with permission from ref 541. Copyright 2004 American Chemistry Society.
To further enhance reactivity, the distal His64 residue was modulated by substitution with Phe to remove the H-bonding interaction with the active Mn-hydroperoxo species.543 Such mutation did not increase in the turnover numbers; however, substitution of His64 with Arg resulted in increased oxidation activity. This observation strongly suggested that the distal ligand His64 plays a key role in tuning the reactivity of the Mn-salen complex through H-bonding interactions.
Superoxide dismutases (SODs) serve to detoxify superoxides produced by aerobic metabolism and by the mammalian immune system in response to infection.544 Different SOD families are defined by the cofactor they utilize, including Ni SODs, Cu and Zn SODs, and Mn or Fe SODs. All members of the most common Mn/Fe-dependent SODs are related in sequence. They exhibit identical protein folds and have identical ligands coordinating the metal ion. Therefore, it is unclear why some enzymes require exclusively Mn for catalysis (MnSOD), while others require Fe (FeSOD) and yet others, such as camSOD, show metal cofactor flexibility.
The metal ion reduction midpoint potential (Em) plays a critical role in which reactions an enzyme can participate and, therefore, must be carefully controlled. Miller and co-workers successfully tuned Fe- and Mn-containing SODs, generating FeSOD variants with Em values spanning 900 mV (Figure 58a).545 The protonation state of the coordinated solvent accounted for the significant differences observed in both the redox potential and metal ion specificity. It was postulated that protonation of the coordinated solvent was suppressed in MnSOD by a strong H-bonding interaction with Gln69, leading to drastic differences in the Em values relative to FeSOD (Figure 58b). Mutations Q69H and Q69E in FeSOD were designed to decrease the strength of this H-bond interaction with the coordinated solvent (Q69H) or to reverse the polarity of the H-bond (Q69E). The Em was observed to increase moving from WT-FeSOD to Q69H to Q69E, supporting this hypothesis (Figure 58).
Figure 58.

(a) Redox tuning over almost 1 V of Fe-containing superoxide dismutase. (b) Hydrogen bonds present in the active sites of some FeSOD variants. Reprinted with permission from ref 545. Copyright 2008 American Chemistry Society.
To further explain the metal specificity of MnSOD and camSOD, Waldron and co-workers identified three residues (19, 159, and 160) that are spatially close to the metal binding site and vary between MnSOD and camSOD (Figure 59).546 Modification of position 159 in the G159L MnSOD increased its Fe-dependent activity while decreasing its Mn-dependent activity by ~2-fold. On the contrary, camSOD L159G diminished its Fe-dependent activity by >10-fold while increasing its Mn-dependent activity by >3-fold. Interconversion of activity and metal specificity could also be achieved by varying position 160, although alterations at position 19 seemed to have little impact on these properties. Surprisingly, although mutations at these positions change the metal specificity, there are essentially no changes observable in the protein backbone structure, at least within the resolution of the crystal structures. The primary coordination spheres, substrate access channels, and H-bonding networks all appear otherwise identical. However, such mutations had direct effects on the electronic properties of the Mn center, as demonstrated by high-field EPR. Different reactivities toward auto-oxidation and dithionite reduction suggested that each of the two WT-SODs have different redox potentials. These differences were inverted in the double mutants (MnSOD G159L/L160F and camSOD L159G/F160L), correlating also to an inversion of the Mn-dependent activity in these variants. Together, these results provide important insight into the role of secondary coordination sphere residues in modulating SODs and the factors that contribute to both altered metal specificity and reduction potential.
Figure 59.

Structure overlay of MnSOD (PDB ID: 5N56, yellow, F19/G159/L160) and CamSOD (PDB ID: 5N57, blue, I19/L159/F160).547
Cobalamin-dependent methionine synthase (MS) catalyzes the conversion of homocysteine to methionine in most mammals and bacteria. This large, modular protein (ca. 146 Da) consists of four distinct functional domains. Two kinds of methyl group transfers occur during the catalytic cycle. Namely, N-methyl tetrahydrofolate first acts as a methyl group donor to methylate cobalamin. This methyl-cobalamin is then used for methionine synthesis from homocysteine. Mechanistic studies of the native enzyme are challenging due to the structural complexities and the dynamic conformational changes that occur in MS during the reaction.548 Despite the importance of this enzyme, the number of protein model systems developed to investigate how MS initiates the Sn2-like transmethylation reaction and stabilizes reaction intermediates is limited.548 Using Mb reconstituted with a Co tetrahydrocorrin derivate (Co(TDHC)Mb), Hayashi and co-workers investigated the reaction catalyzed by methionine synthase.549 It was found that CoI(TDHC)Mb could react with methyl iodide to form a methylated cobalt complex, a process that did not efficiently proceed for CoI(TDHC) in organic solvents. These initial results highlight the importance of the heme pocket of Mb, which may serve to enhance the nucleophilicity of the CoI species while simultaneously stabilizing the methylated CoIII species. Interestingly, transmethylation from the methylated cobalt complex to one of the nitrogen atoms of the His64 imidazole ring was observed. DFT calculations exploring this process supported a reduced barrier for His64 methylation together with significant stabilization of the methylated His product through the dielectric effect of the surrounding protein environment. In the crystal structure, the Nδ atom of His64 was found to be hydrogen bonded to a water molecule (Figure 60), suggesting barriers can be further reduced by the partial or full deprotonation of the methylated histidine.
Figure 60.

Crystal structure of horse heart myoglobin reconstituted with CoII-tetradehydrocorrin (PDB ID: 3WFT).550 The Nδ1 atom of His64 hydrogen bonded to a water molecule is shown.
Ghirlanda and co-workers have also investigated the influence of the protein second coordination sphere on Mb substituted with cobalt-protoporphyrin XI (CoMb) (Figure 61).551 Compared to free cobalt-protoporphyrin IX (CoPP(IX)), a 4-fold increase in the photoinduced hydrogen production activity was observed for CoMb. Substitution of the distal His residues in the engineered H64A/H97A CoMb mutant displayed a 2.5-fold higher reactivity relative to that of CoMb. It is hypothesized that the H64A substitution removes a slightly positive charge from His at physiological pH as well as a potential competitor for proton binding in the proximity of the CoPP(IX) active site. A hydrogen bond formed between His97 and one of the propionic acid groups of CoPP(IX) was also observed based on analysis of the crystal structure. Meanwhile, the H97A mutation was found to destabilize binding of CoPP(XI), alter the steric constraints on the porphyrin, and increase the degree of freedom between the scaffold and the CoPP(XI), effectively lowering the catalytic ability. However, the H97A substitution also increased the solvent accessibility of the active site, which may favor interaction between the photosensitizer and CoPP(XI), as the TON of H64A/H97A was found to be higher than that of the H64A mutant.
Figure 61.

Crystal structure of Co-Mb highlighting residues His64/93/97 (PDB ID: 1YOI).552
In 2016, Hartwig and co-workers constructed an artificial metalloenzyme (ArM) by incorporation of an Ir(Me)-heme cofactor into myoglobin (IrMb), capable of carbene insertion activity with up to 50% yields for a model reaction,553 Unlike many other anchoring strategies, the Ir(Me)-heme cofactor is embedded within the protein cavity, allowing SCS interactions to be readily employed to tune reactivity.554 Directed evolution of this IrMb was conducted by mutating a variety of residues, including the axial ligand and those in the substrate-binding region, to achieve substrate specificity. Aside from the axial and proximal ligands (His64 and His93, respectively), Phe43 and Val68 were mutated into hydrophobic or uncharged residues to maintain the hydrophobicity of the porphyrin/substrate binding site. Four additional residues around the active site (Leu32, Phe33, His97, and Ile99) were also investigated (Figure 62a). Different selectivities and yields were achieved for varying carbene insertion reactions across the range of designed mutants, with selectivities reaching up to a 92:8 enantiomeric ratio, yields up to 97%, and TONs up to 7260.553 The Ir(Me)-heme catalyst was also incorporated into the CYP119 scaffold (a variant of P450 enzymes), producing an ArM with better thermal stability and reactivity toward carbene insertion,555 cyclopropanation,556 and (sp3)C-H site selective functionalization reactions.557 Upon directed evolution of the residues near the active site, the authors achieved ArMs with improved yields, enantiomeric excess, chemoselectivity, total turnovers and turnover frequencies relative to those of IrMb. These mutations mainly focused around varying the heme axial residue (Cys317), residues lining the substrate binding pocket (Leu69, Ala209, Thr213, and Val254), and residues toward the distal side of the substrate binding site (Ala152, Leu155, Phe310, and Leu318) (Figure 62b).
Figure 62.

(a) Inner sphere (red), middle sphere (cyan), and outer sphere (orange) residues of the active site studied in Mb-based ArM for carbene insertion reactions (PDB ID: 1MBN).558 (b) Residues studied in CYP119-based ArMs: axial ligand for heme (green); residues lining the substrate binding pocket (cyan); residues to the distal sites of the substrate binding pocket (orange) (PDB ID: 1IO7).559
6.2. Design of New Metal Binding Sites
Nickel is intimately involved at the active sites of several important energy-converting enzymes, including NiFe H2ases, carbon monoxide dehydrogenase (CODH), and acetyl coenzyme-A synthetase (ACS), all of which coordinate Ni in a S-rich environment. To better understand the nature of these enzymes and to mimic their unique activities, a number of protein reengineering efforts have been put forth.
Chakraborty and co-workers reported the rational redesign of a copper storage protein (Csp1) into a nickel binding protein as a novel mimic of [NiFe] H2ase (Figure 63).560 The native active site of NiFe employs two Fe-bridging and two terminal Cys residues to coordinate Ni, making the already thiol-rich Csp1 an intuitive starting point. The 13 Cys residues along the central helical core of native Csp1 are capable of binding a total of 13 equiv of CuI. The Cu4 binding site (Cys26/62/87/113), which already enables tetrathiolate ligation, was chosen as the target location for the formation of a Ni(Cys)4 site. During the redesign process, seven Cys residues away from the Cu4 site were mutated to Ala due to the similar side chain size of these residues in an effort to maintain a compact structure. Additionally, two bulky hydrophobic residues, Val (Cys94) and Leu (Cys103), were introduced to provide hydrophobic interactions at the exposed ends of the protein, aiding in folding. Spectroscopic and computational data supported Ni binding to occur in this redesigned construct, adapting a distorted square planar geometry. More strikingly, the NiII-incorporated protein was found to catalyze proton reduction to H2 under photochemical and electrochemical conditions, with a TOF of ~210 under photochemical conditions, comparable to those of reported biomolecular catalysts.561
Figure 63.

Redesign approach for the conversion of Csp1 (a and b) to a Ni-binding protein (NBP). Copyright NAMD model of apo NBP. (d) Overlay of the X-ray structures of apo Csp1 (purple, PDB ID: 5FJD) and CuI-Csp1 (cyan, PDB ID: 5FJE) and the minimized structure of apo NBP (green). Ball and stick model of the tetrathiolate site of minimized NBP viewed perpendicular to the helical axis (e) and from the top of the helical axis (f) with N-terminus at the top. Modeling performed using CHARMM22 force fields. Reprinted with permission from refs 560 and 562. Copyright 2019 and 2015 American Chemistry Society and Nature Publishing Group.
Also taking advantage of a naturally Cys-rich scaffold, the Shafaat group has developed a nickel-substituted rubredoxin (NiRd) and further probed the secondary coordination sphere of this construct to shed light on how the secondary sphere can modulate the hydrogenase activity.563 A rationally designed library of 30 mutants were generated in the metal-binding loops of Rd targeting residues V08, V34, and V37 (Figure 64), which exhibit H-bonding interactions with the Ni-coordinated cysteines through the amide backbone. By varying the identity of these residues, it was found that the strength of these H-bonding interactions is reflected in changes of the NiIII/II reduction potential, in which stronger hydrogen bonds between the amide proton and sulfur atom of Cys were correlated with higher reduction potentials, likely due to the increased electronic delocalization and stabilization of the reduced state. Further-more, it was found that the steric constraints imposed by certain side chain residues are capable of influencing both H-bonding with the amide backbone and solvent accessibility. Solvent accessibility is a key factor in proton-dependent H2 evolution reactions. Specifically, the greatest increases in activity correlated with mutations of the V08 residue. The TOF and solvent accessibility of V08X mutants increased relative to WT-NiRd due to the rotation of side chains away from Cys35, improving the solvent accessibility of the active site. Meanwhile, the solvent accessibility and catalytic performance of V34X mutants were comparable to those of WT-NiRd, with the exceptions of V34G and V34H, which reduced the degree of H-bonding between the amide backbone and nearby Cys32 due to substantial movement of the backbone. The dependence of the TOF on pH was found to correlate with secondary sphere perturbations as well. The V34H mutant showed a logarithmic dependence of reactivity on pH. MD simulations demonstrated that protonation of His34 at low pH resulted in a rotation of the His side chain away from the metal binding site, enabling direct access of water to the active site. This extensive work from the Shafaat group with a simple NiRd scaffold demonstrates the impact of the secondary coordination sphere in tuning both catalysis and reaction mechanism.563
Figure 64.

Structures of the (a) Dv MF NiFe H2ase and (b) Dd NiRd active sites including key secondary sphere interactions. Reprinted with permission from ref 563. Copyright 2019 American Chemistry Society.
Besides H2ase mimics, the Shafaat group recently reconstructed ferrodoxin using a [NiFe3S4] cluster to form a protein model of CODH.564 While native ferrodoxin is capable of binding CN−, only the [NiFe3S4] reconstituted enzyme was found capable of binding CO, as supported by an extensive spectroscopy study. Further systematic studies of the PCS and SCS residues of surrounding this ferredoxin-based model system will provide even greater insights into the mechanism of native CO2 reduction by CODH.
The Baker lab has also successfully redesigned a mononuclear Zn metalloenzyme for organophosphate hydrolysis with the assistance of applied computational tools.565 Starting from an adenosine deaminase scaffold (PT3), eight mutations were introduced to help establish a Zn coordination site as well as stabilize a postulated transition state geometry for hydrolysis based on an extracted set of mononuclear Zn enzyme scaffolds. Based on these calculations, residue Glu217 was suggested to be crucial for proton shuttling in the deamination reaction. These calculations were supported by the low activity observed for the E217Q variant, highlighting the importance of Glu217 for organophosphate hydrolysis, which was suggested to aid in fine-tuning substrate alignment at the active site. Directed evolution was further performed to enhance the hydrolysis reactivity toward organophosphate substrates, resulting in the develop-ment of variant PT3.1, with approximately 2500-fold higher reactivity than the initial PT3 variant. Based on these results, it was postulated that substitutions in the vicinity of Glu217, such as V218F and Q58V, may increase the hydrophobicity around the putative catalytic base Glu217 (Figure 65). A change in pH of 1.6 accounts for the catalytic increase observed in PT3.1, containing V218F mutation relative to PT3, which suggests that the basicity of Glu217 is related to the activity enhancement obtained through the directed evolution pathway.
Figure 65.

Structure of engineered organophospate hydrolase PT3.1. Residues 58 and 218 influencing the hydrophobicity of Glu217 are shown in sticks (PDB ID: 3T1G).565
6.3. Design Using Strong Noncovalent Interactions
The biotin–avidin interaction is known to be one of the strongest noncovalent interactions in nature,566 and the use of this strong interaction to construct ArMs by connecting avidin-based protein scaffolds with biotin-substituted artificial metal-locofactors can be dated back to the last century.475,567 In 2003, the Ward group reported an artificial metalloenzyme (ArM) using Sav and a Rh complex with a biotinylated amino-diphosphine ligand Biot-1 ([Rh(COD)(Biot-1)]+⊂Sav, Scheme 4, bottom) capable of achieving 92% ee (R) when catalyzing the enantioselective hydrogenation of α-acetamidoacrylic acid (Scheme 4, top). When mutating the Ser112 of the L7,8 loop of Sav (Figure 66), which is in the proximity of the metal active center, into glycine, an increase in enantioselectivity for the ArM [Rh(COD)(Biot-1)]+⊂Sav S112G to 96% (R) was observed.568 In addition to S112G, the group also studied the V47G, K80G, and P64G mutations. Val47 forms a close contact with Ser45, which interacts with the H-N group of biotin through H-bonding. Meanwhile, Lys80 is a neighboring residue of Trp79, which is a SCS residue of biotin, forming a hydrogen bond with Thr90, which further interacts with the thioether of biotin (Figure 66). Although Pro64 is located on a loop that is relatively remote from the biotin, mutations at this residue may have an impact on the loop geometry. It was found that mutations closer to the active site (V47G and S112G) had a more significant effect on the enantioselectivity compared to those that are relatively remote (K80G and P64G).569 Site saturation mutagenesis (SSM) on Ser112 in the same ArM system catalyzing α-acetamidoacrylic acid or α-acetamidophe-nylacrylic acid provided more insights into this site. Generally, the ArMs having S112A and S112G mutations more heavily favor the R products, while those with S112H, S112K, and S112R mutations favor producing S products. By modifying the artificial cofactor itself, a further increase in the ee of both R and S products was achieved. The authors speculated that the cationic side chains of residue 112 can interact with the carboxylate moiety of the substrate through hydrogen bonds, making one of the prochiral faces of the substrate more favorable for coordination.570 A directed evolution approach using the same Biot-1 ArM system for catalysis of the α-acetamidoacrylic acid ester substrate found that several residues ~4–6 Å away from the metal center can change the ee to favor either R or S isomers, possibly by directly affecting the conformation of the catalyst or catalyst/substrate complex. Among these, N49V increased the ee from 23% (R) to 65% (R), and N49H/L124F decreased the ee from 23% (R) to −7% (S).571
Scheme 4. Enantioselective Hydrogenation of α-Acetamidoacrylic Acid and Structure of Biot-1568.

Figure 66.

Crystal structure of a wild-type Sav with bound biotin with the relevant residues show (PDB ID: 1STP).572 The protein backbone is shown in green, and biotin C atoms are shown in white, relevant residues with direct interaction with biotin in cyan, and relevant residues distal to biotin in magenta.
Incorporation of a racemic biotinylated three-legged d5/d6-piano stool complex (Scheme 5) into Sav enabled the formation of a series of artificial transfer hydrogenases (ATHases). Starting from the Ru d6-piano stool complex, several mutations were made to study the influence of the secondary coordination sphere on ketone hydrogenation. S112G, a mutation close to the catalytic site, resulted in the highest conversion (90%) but lowest enantioselectivity (28% ee (R)). P64G, a mutation at a relatively remote site, has the greatest enhancement on enantioselectivity (72% ee (R)) but moderate conversion (54%). The double mutant P64G/S112G combined the positive effects of both single mutants, resulting in excellent conversion (95%) and reasonable enantioselectivity (58% ee (R)).573 Later, a series of alternative d5/d6-piano stool complexes coordinating Rh, Ru, and Ir, respectively, were screened for activity toward the reduction of ketones or cyclic imines. Select complexes were further developed by SSM to generate more advanced ArMs. Variants of Sav containing cationic residues at position 112 (S112R and S112K) generally have higher S-enantioselectivity. It was also found that the introduction of aromatic residues (i.e., S112F and S112Y) or using small amino acids at the same site (i.e., S112G and S112A) leads to higher R-enantioselectivity.574,575 Based on the S112A and S112K mutations, a follow-up study was performed focusing on the SSM of the Lys121 and Leu124 sites. Sav is a homotetrameric protein, in which the Lys121 of one monomer interacts with the η6-arene ring of the complex, while the Lys121 from the adjacent monomer interacts with the incoming substrate (Figure 67a). Therefore, this residue may play a role in favoring certain prochiral substrate interactions. Leu124 may influence the biotinylated catalyst slightly because its methyl group is in close proximity (3.53 Å) with the sulfone group of the catalyst. Overall, SSM of Lys121 proved to be more effective for the optimization of enantioselectivity than when applied to Leu124.576 The crystal structure of S112K Sav containing Ru-cofactor clearly showed interaction between Lys112 and Leu124. Also, several other residues close to the active site, including Thr114 and Pro64, were investigated, where mutation of either to Gly was found beneficial in enhancing the enantioselectivity. Thr114 is close to the η6-arene ring, while Pro64 is located in the L4,5 loop, which neighbors the L7,8 loop from another monomer, the latter of which is close to the catalytic metal center (Figure 67a). Mutation of the Pro64 to Gly hypothetically perturbs the location of the L4,5 loop, which in turn should influence the positioning of the L7,8 loop and further influence the Ru complex.577
Scheme 5.

Formation of a Series of Artificial Transfer Hydrogenases574,575
Figure 67.

(a) Crystal structure of a Ru complex-based Sav ArM tetramer showing the positions of relevant protein residues and loops (PDB ID: 2QCB):576 monomer A (orange); monomer B (cyan); monomer C (green); monomer D (magenta); Ru complex (white). (b) Crystal structure of Ir complex-based Sav-FPD S112V-K121A (PDB ID: 6GMI) overlaid with Ir complex-based Sav S112A (PDB ID: 3PK2): monomer A of Sav-FPD S112V-K121A (blue); monomer B (green); monomer C (lilac); monomer D (cyan); Sav S112A (gray); mutated residues S112V and K121A (magenta). Reprinted with permission from ref 579. Copyright 2018 American Chemical Society.
There are also some ATHases that have been constructed based on similar Sav-bound biotinylated Ir complexes. Several cationic and anionic residues around the metal complex were investigated using imine reduction as a model reaction. Both S112A/K121A and S112A/K121L displayed excellent catalytic efficiency (kcat/KM). An additional D67V mutation to the S112A/K121L construct was made to remove a negative charge near the catalytic site, resulting in a lowering of the catalytic efficiency due to a lower kcat. A possible explanation for this effect is that the reduction of imine may proceed through an iminium ion intermediate, and removal of the cationic residue (K121A or K121L) next to the catalytic site would help with the catalytic process. The presence of an anionic residue (Asp67) would stabilize the cationic transition state and increase the kcat.578
To further modulate the ATHase activity in the Ir-complex-based Sav, an additional 24-residue helix-turn-helix motif (FPD) was incorporated into the L7,8 loop and combined with other previously investigated point mutations to tune the catalytic performance of the ArM. The two best-performing mutants found for the reduction of a quinolium substrate were S112A/K121P Ir-Sav-FPD and S112V/K121A Ir-Sav-FPD. The crystal structure of S112V/K121A Ir-Sav-FPD revealed that the L4,5 loop was shifted 5.6 Å compared with that of Sav S112A, and the directionality of the L7,8 loop had rearranged to point toward the Ir cofactor, although the FPD-loop was not resolvable (Figure 67b). The cofactor occupancy for S112V/K121A Sav-FPD was also found to increase relative to that for S112V/K121A Sav (from 50% to 80%, indicating that the FPD-loop shielded the biotin-binding vestibule and helped the binding of the complex.579 Since Sav is a tetramer hypothetically capable of binding up to four biotinylated cofactors, the roles for different monomeric units were also investigated. To this end, two Sav subunits were fused together and mutated separately at the respective Ser112 and Lys121 positions. It was found that mutations made in the first subunit (SavA) had a greater impact on both substrate conversion and product enantioselectivity. Meanwhile, those made in the second subunit (SavB) do not show a clear trend for the influence on the reactivity; however, these residues do still appear to play a role in fine-tuning substrate interactions, as well as conversion and enantioselectivity. This likely arises from different conformations of the Ir complex in each subunit, with SavA serving as the predominately active form.580
The [Cp*RhCl2]2 (Cp* = pentamethylcyclopentadienyl) complex is a versatile catalyst for electrophilic aromatic C-H activation.581 With the aim of introducing asymmetric activity and improving conversion, Hyster et al. synthesized a biotinylated [Cp*RhCl2]2 complex ([Cp*RhCl2]2-biot) and successfully incorporated the complex into Sav.582 The low activity observed in the resulting ([Cp*RhCl2]2-biot)-Sav construct inspired further modification of the secondary coordination sphere based on molecular docking simulations. Initial efforts introduced a basic residue in the vicinity of the metal center through S112E and K121E mutations. Similar to previous reports,570,574,575 both of these variants exhibited low conversion for the reaction (10% and 7%, respectively). However, mutation of K121 to Asp resulted in a significantly improved reaction conversion of up to 89%, demonstrating that both the presence of a carboxylate and its precise positioning can significantly impact aromatic C-H activation. Inspired by these results, the authors further added a N118K mutation to K121E ([Cp*RhCl2]2-biot)-Sav (Asn118 is adjacent to Lys121), and they observed a further increase in conversion to 99%, as well as an excellent regioisomeric ratio (rr) (15:1) and enantiomeric ratio (er) (82:18). By screening different mutations at the Ser112 site, S112Y was found to have the best enantioselectivity, with an er of 88:12. The synergistic effect of combining S112Y and K121E mutations also led to a total conversion of up to 95%, rr of 19:1, and er of 91:9. Through kinetic isotope effect (KIE) and competition studies, the authors found that the introduction of the carboxylate had the most dominant effect on improving the activity and selectivity.582
Further expanding the scope of the biotin-Sav guest–host scaffold, Ward and co-workers incorporated a biotinylated second-generation Hoveyda–Grubbs catalyst into Sav to generate a novel artificial metathase. Importantly, this ArM system was developed to function in whole cells as a catalyst for in cellulo abiotic transformations. Directed evolution was applied to enhance the activity of this ArM, and a quintuple mutant (V47A/N49K/T114Q/A119G/K121R) was found to increase the cell-specific activity by 5.4-fold. According to the B factors associated with the crystal structures of the wild-type and quintuple mutant, the biotin-vestibule loop of the quintuple mutant exhibited greater flexibility, with mutations T114Q, A119G, and V47A serving as the key mutations for this enhanced flexibility.583
Ward and co-workers further explored the use of ArMs as in vivo new-to-nature catalysts by developing a method for displaying Sav on the surface of E. coli. It was reasoned that a surface display would more readily bind designed biotinylated metallocofactors, as diffusion of the complex through the periplasm is not required. A biotinylated CpRu (2-quinoline-carboxylate) complex was successfully incorporated into surface-displayed Sav with the intention of catalyzing the uncaging of allylcarbamate-protected substrates, generating a functional allylic deallylase. SSM at positions in close proximity to the Ru complex (S112 and K121, vide supra) revealed two double mutants, S112Y/K121S and S112M/K121A, with activities 25- and 24-fold higher than that of the wild-type, respectively. The crystal structure of the S112M/K121A mutant showed that the aromatic quinoline ring was stabilized by a methionine CH3/π-interaction in S112M. Also, the carbonyl groups from K121A and Ser122 helped the binding of the quinoline ligand.584
Beyond Ru, Rh, and Ir, the biotin–avidin method has also been used to create ArMs with many other different metal complexes, including Pd for asymmetric allylic alkylation585 and Suzuki-Miyaura cross-coupling,586 Au for hydroamination and hydroarylation,587 and OsO4 for cis-dihydroxylation of olefins.588 Mutagenesis investigations have been conducted for all of the examples illustrated above, focusing predominately on varying residues Ser112 and Lys121. In the Suzuki-ase ArM generated by incorporation of a Pd complex (Scheme 6) into Sav, the crystal structure of the S112Y/K121E mutant was acquired, revealing that S112Y provided a significant steric barrier on the lower face of Pd, likely accounting for the modest improvement observed in the enantioselectivity. Additionally, an H-bonding interaction was found between Tyr112 and Glu121, further locking the conformation of Tyr.586
Scheme 6.

Suzuki-ase ArM Generated by Incorporation of a Pd Complex into Sav586
Aside from the strong biotin–avidin interaction for constructing ArMs, vanadium pentahydrate was found to interact with the Sav host in acidic media through H-bonding with H-donor residues that are also critical for biotin binding (e.g., Asn23, Ser27, Ser45, and Asp128). An ArM catalyzing enantioselective sulfoxidation reaction was, hence, constructed (Figure 68), and this artificially generated vanadyl enzyme successfully catalyzed the oxidation of prochiral sulfides with good enantioselectivities of up to 93% ee.589
Figure 68.

Superimposition of the docked structure [VO(H2O)5]2+ (ball-and-stick representation) with WT-Sav (monomers A and D, light blue schematic secondary structure) with the structure of biotin with WT-Sav (PDB ID: 2IZG): biotin (yellow stick and yellow transparent surface); monomers A and D (yellow schematic secondary structure). Reprinted with permission from ref 589. Copyright 2008 American Chemistry Society.
6.4. Engineered Metalloenzymes by Covalent Attachment of Metallocofactors
In addition to the noncovalent anchoring approaches of scaffolds, such as the biotin–avidin method, ArMs can also be generated through covalent anchoring of synthetic catalysts. For example, azide-alkyne cycloaddition (the classic “click” reaction) has been employed to anchor an alkyne-bearing dirhodium complex in propyl oligopeptidase containing the UAA 4-azidophenylalanine (POP-Z). Although initial reactions of otherwise unmodified POP-Z failed to incorporate the cofactor, mutations aimed at reducing the sterics at the end of the β-barrel were found to enable bioconjugation in POP-ZA4. Further mutations of residues 99 and 594 to Phe, which neighbor the distal Rh based on homology modeling, were found to increase the yields from 61% to 74% and the ee from 85% to 92%,590 despite later crystal structures that showed the Phe99 was not positioned as suggested by homology modeling.591 Random mutagenesis was also applied to POP-ZA4 to generate several mutations which improved the enzymatic activity. Among these, only three mutations, G99S/S301G/Y326H (1-SGH-POP-ZA4), were required to reach optimal selectivity. Based on crystallographic analysis of 1-SGH-POP-ZA4, Ser99 and His326 were found in the active site while Gly301 was farther away.591 1-SGH-POP-ZA4 was further subjected to directed evolution targeted at improving the catalytic performance of diazo coupling cascade reactions. Site saturating mutations were introduced in the β-strand across from the putative Rh-binding His to examine mutations that could significantly impact the product yield and selectivity. From this effort, mutations Q98P and S99H were identified, and a further combination of combinatorial codon mutagenesis across 25 active site residues followed by another round of SSM identified residues V71G and E283G to generate a final mutant with product yields as high as 76%.592
Thiol-maleimide bioconjugation has also been used to generate 4d/5d metal containing ArMs. C96 nitrobindin was employed to generate an ArM capable of C(sp2)─H bond functionalization via concerted metalation deprotonation (CMD) with 15% yield and 92:8 regioselectivity through the covalent binding of a maleimide-tagged Rh complex. Carboxylates are known to facilitate the CMD-type C(sp2)─H bond functionalization, and therefore, glutamate residues were introduced near the active site in a L100E/A125E mutant, which increased the yields to 40% with similar regioselectivity (90/10).593 Directed evolution was later applied to this system, and the T98H/L100K/K127E mutant was identified to increase the activity 2.2-fold. Interestingly, MD simulations suggested that all three of these residues are located opposite to the Rh metal center.594
Human carbonic anhydrase II (hCA II) exhibits a high binding affinity toward para-substituted aryl-sulfonamides at the Zn binding site, providing another tool for the development of ArMs. An {IrCp*}2+ moiety was introduced into hCA II to generate a novel ATHase. Structural analysis based on the resulting X-ray structure showed that Ile91 and Lys170 occupy a potential substrate-binding site. Therefore, I91A and K170A mutations were incorporated, with the former showing some increased activity for specific substrates; however, the apparent low occupancy of the catalytic complex precluded significant improvements.595 Computational redesign using the program Rosetta was applied to tackle the problem of low cofactor occupancy.596 It was speculated that the active site of WT-hCA II was not well defined toward the Ir complex. To solve this problem, a variety of hydrophobic residues were introduced to tune the shape of the binding site and optimize the packing interactions between the protein and cofactor. N67W, E69Y, and Q92F mutations were made to β-strands 2 and 3 to allow interactions between the biaryl and phenylsulfonamide groups. An L140 M substitution on β-strand 5 and L197 M on loop 7,8 were also made to pack against the coplanar biaryl moiety of the ligand, and several additional mutations were made to stabilize the protein backbone. L60V on β-strand 1 was introduced for stabilization, A65T was made on β-strand 2 to enable H-bonding with the carbonyl oxygen of Phe95, and C205S was introduced at the end of loop 7,8 to add rigidity to the loop via H-bonding with the carbonyl oxygen of Val134, maintaining the orientation of the cofactor (Figure 69). Based on these mutations, one of the variants d3 (containing L60V, A65T, N67W, E69Y, Q92F, L197M, and C205S) displayed a 64-fold increase in binding affinity for the Ir cofactor. Reverting selected residues such as E69Y (d2) or L197 M (d1) resulted in some reduction in the binding affinity, although still higher than that in the WT. All three mutants (d1, d2, and d3) showed improved ee and higher TONs than with the wild-type protein.597 This ArM system was later applied to another similar IrCp cofactor using a dual-anchoring system to further increase the occupancy and stabilize the geometric configuration.
Figure 69.

Model of Rosetta design on the crystal structure of the Ir complex in WT-hCA II (PDB ID: 3ZP9). Designed backbone-stabilizing H-bonds are represented as green dashed lines. Reprinted with permission from ref 597. Copyright 2015 American Chemical Society.
Directed evolution based on the dual-anchoring mutant I91C was conducted at the Leu60, Asn62, Asn67, Glu69, and Gln92 sites, all of which neighbor the terminal chloride of the Ir complex. All mutations at Gln92 led to decreases in activity, possibly due to the elimination of an optimal H-bonding interaction between Gln92 and the amide oxygen of the cofactor, which serves a crucial role in fixing the Ir cofactor in the active site and maintaining the pyridine–sulfonamide linkage. For purified protein, N67G/E69R/I91C displayed the highest activity toward the R-enantioselectivity of the substrate with a TON of 451 and ee of 96%. For S-enantioselectivity, N67L/E69Y/I91C and L60W/N67L/E69Y/I91C reached moderate ee values of −62% and −49%, respectively (TONs 458 and 221, respectively). In comparison, the starting I91C construct exhibited a TON of 265 and ee of −39%. The L60W/N67L/E69Y variant alone enhanced the activity by 4.3-fold as well as improved the S-enantioselectivity to 59% ee. From the crystal structures of N67G/E69R/I91C and I91C, it was surmised that N67G provided more space for the substrate, allowing a more facile approach to Ir-H, while E69R may serve to introduce a cation–π interaction between the residue and the substrate. As a result, the above two mutations together may aid in substrate binding to the active site, leading to the increase of the TON and enantioselectivity.598
The HaloTag (HT) protein utilizes a reactive aspartic acid in its binding pocket to react with haloalkanes, forming a covalent linkage. Taking advantage of this, Fischer and co-workers developed a metathase by linking a Ru complex to HT, which was shown to exhibit enhanced TONs when compared to the catalyst alone. Directed evolution was applied to residues around the binding pocket; however, no significant improve-ments in the activity were observed. This is likely due to the long distance between the cofactor and the protein scaffold caused by the length of the linker or possibly due to the minimal interactions between the SCS and the catalytic cofactor transition state.599
7. SUMMARY AND OUTLOOK
In this review, we have showcased how catalysis can be modulated in a wide range of designed ArMs by focusing on the roles of the environment beyond the PCS. While we have covered many examples involving the generation of ArMs through the redesign of existing PCSs or the introduction of a new PCS into a native protein scaffold, most of these ArMs display much lower activities in comparison to their native enzyme counterparts. The most impressive examples with high catalytic activities, including several that approach or exceed those of native enzymes, have arisen from those that have further employed SCS modifications to optimize properties such as the reactivity and substrate selectivity.
Throughout this review, we have seen both rational design and directed evolution used as effective tools to impart and enhance new reactivities in ArMs. At face value, the ethoses of rational design and directed evolution appear at odds, in-so-far as the first insists on the question “How?”, while the second avoids it. However, these two strategies can be powerfully used to complement one another and overcome their own respective limitations. In particular, the further optimization of rationally designed ArMs by directed evolution can uncover subtle SCS roles that may otherwise be easily overlooked. Likewise, the use of rational design to analyze ArMs directed by evolution can provide new insight to overcome barriers associated with limitations in screening methods and to design smaller, higher quality libraries.600 When synergized with computational methods, particularly those available for initial scaffold design, such as Rosetta61,62 and AlphaFold,63,64 and those for analysis, such as QM/MM methods, both our understanding of the roles of SCS residues and the practical development of ArMs can be greatly advanced. Doing so may not only enable further development of the systems discussed in the present review but also open doors to the design of ArMs modeling metalloenzymatic systems with more complex cofactors and more challenging reactions, such as photosystem II, nitrogenases, and methane monooxygenase.
Due to the prevalence, long history of study, and biological significance of heme enzymes, heme-based ArMs represent one of the most thoroughly studied classes in this review, particularly regarding the roles of SCS residues. Engineered heme ArMs generally fall into one of two groups: (1) nonenzymatic native scaffolds, such as Mb and Cc, that are engineered to generate new activities and (2) enzymatic scaffolds modified to exhibit activity toward non-native substrates or non-native functions. Additionally, three strategies have been employed to study and enhance catalysis in these systems. The first approach is to mimic the structural features of a desired native enzyme, such as the His-Arg pair or the positioning of the distal His in peroxidases.101,122 The second is to employ SDM at SCS residues surrounding the active site to vary structural features such as sterics, polarization, and H-bonding to tune the activity. The third entails using directed evolution to repurpose a native heme protein for new, non-native functions. This is often accomplished through SDM of residues identified as “hot spots” near the active site, although it is worthwhile to note that directed evolution is also effective at identifying beneficial mutations that are surprisingly remote.569,573 These strategies have resulted in the generation of several highly competent catalytic heme ArMs, with some of them approaching,539 rivaling,525 or even exceeding524 those of native enzymes. Through this process, we have learned not only how the presence of SCS features, such as H-bonding, influences catalysis but also how the positioning of the same SCS residue in different scaffolds (e.g., distal His in Mb and CcP) can play a key role in both conferring and fine-tuning activity. However, the practical development of these enzymes would benefit from further developments in terms of scaffold stability, and further optimization of these systems for varying activity stands to benefit from the more extensive application of computationally aided design and screening.
It is interesting that DNA has been used as an alternative scaffold for designing artificial heme enzymes. However, current DNA models do not support proximal ligation to the heme by the scaffold molecule, which limits the reactivity of heme/DNAzymes compared to heme enzymes. Extending the oligonucleotide beyond the G-quartet/heme interface provides a structure analogous to the active site of heme proteins;249 however, the much more disordered distal environment of a G-quadruplex scaffold is very different from typical protein scaffolds. This provides an opportunity for developing an active site de novo—importantly, as the DNA scaffold can be developed by in vitro selection, active sites made from pendant nucleotides can be selected against one another without the issues of expression or purification that can otherwise arise when employing directed evolution for proteins.
The PCSs of native mononuclear non-heme Fe enzymes predominately utilize the facial (His)2-carboxylate triad, which has served as the major inspiration for initial designs of non-heme Fe ArMs. However, despite their prevalence in nature, relatively few non-heme Fe ArMs have been designed which employ natural amino acid side chains/protein backbone for metal ion coordination,268,246-248,274 at least when compared to those for other metals such as Cu, Ni, and Zn. As a result, the study of SCS interactions in non-heme Fe ArMs has not advanced to the extent discussed in some other sections, underlining the need for continued developments in this field, particularly toward mono- and dioxygenase activity. Similarly, the employment of the UAA AlaBpy has been effective in forming FeII binding sites (especially when combined with computational design), but UAA-based systems have yet to be turned into efficient catalysts.255-258,286,287 Meanwhile, non-heme Fe ArMs formed using a protein scaffold to host an organometallic cofactor have proven to be the most well-characterized in terms of catalytic competence, particularly through the efforts of Ménage and co-workers.302,305,306,308 In these systems, SCS residues have primarily been investigated to support fixation and positioning of the organometallic scaffold through π-stacking and salt bridge interactions to enhance the specificity and selectivity. Lastly, the development of dinuclear non-heme Fe sites in native scaffold-based ArMs has largely been inspired by FeFe H2ase,291-295 although little further development has been made in these systems to understand the role of the peptide scaffold beyond the PCS. Borovik and co-workers have developed a notable exception, forming a non-heme diiron site via bridging of Sav active sites that is reminiscent of native oxygenases, although further catalytic studies are yet to be presented.317 Taken together, non-heme Fe ArMs present a promising field for continued study, particularly through more expansive investigations of the roles of SCS residues in modulating the properties of Fe.
Cu proteins serve diverse roles in nature, including ET and various catalytic roles. Two types of Cu sites natively serve in ET (T1Cu and CuA), both of which have been studied by the development of cupredoxin units that are isolated from native ET systems, as well as by incorporation into nonhomologous scaffolds. By modulating the SCS to vary factors such as hydrophobicity and hydrogen bonding, the ET properties of these centers can be effectively optimized by balancing E° with reorganization energy.349-355,392,393 The next development of these centers requires consideration of how to effectively couple these ET active proteins with catalytic ArMs to enhance enzymatic activity, including the realization of specific surface interactions. Additionally, SCS development optimizing pathways for long-range electron transfer offer a tantalizing challenge, requiring not only a well-balanced electrochemical gradient but also possible involvement of proton donor/acceptors.
Catalytic Cu centers mimicking natural reactivities present a greater challenge. Several reactions with small inorganic molecules have been demonstrated in Cu-based ArMs, including nitrite reduction,457 superoxide dismutation,452 S(Cys) peroxidation,456,464 and S-nitrosylation.276 In these examples, studies of the SCS have allowed the electrostatic environment and E° of the Cu active site to be tuned. The oxidation of organic substrates has also been reported, where SCS interactions have been employed to control protein–substrate interactions. Furthermore, artificial Cu proteins have been repurposed to catalyze a number of unnatural reactions including the D-A reaction,478,487 F-C alkylation,368 Michael addition,497 and alkene hydration.468 In these cases, the SCS has been employed to enforce product selectivity via substrate–SCS interactions. It is worth noting that Cu centers have often been treated as Lewis acids, and their natural roles as oxidoreductases are surprisingly less studied. In particular, oxidative reactivity has been more difficult to achieve than reductive chemistry (in large part due to the damage that can occur to the protein scaffold from ROSs), despite numerous examples of Cu-based oxidases and oxygenases in nature. Therefore, much remains to be explored in the area of Cu ArMs by further consideration of SCS effects.
In addition to Fe and Cu metalloenzymes, there are many ArMs that have employed other biologically relevant metal cofactors, such as V,589 Cr,540 Mn,540,541,547 Co,549,551 Ni,560,563 Zn,565 and Mg,508 as well as those with abiological metal centers, including Ru,577 Rh,568,581,590,594 Pd,586 Os,588 Ir,558,559,579,597 and Au.587 These studies can be divided into two categories, namely those that target understanding native metalloenzyme functionality and those that aim to utilize the ArM scaffold to enhance the catalytic properties of organometallic complexes. The role of the SCS is key to both of these aims, functioning both to tune the metal site and control substrate–metal interactions through varying factors including H-bonding, steric hindrance, hydrophobicity, and pKa. Future studies using directed evolution to investigate the SCSs of novel ArMs could benefit from more thorough structural and mechanistic studies of the evolved enzymes to rationalize the specific influence of varying residues on ArM performance for each sequential round of selection. By tracing the evolutionary path, greater insight could be gained on how to further engineer ArMs with enhanced performance through tuning the environment beyond the PCS.
SCS interactions are often weak and difficult to pinpoint, with many effects remaining nonobvious even for experienced ArM designers and too subtle to be reliably predicted by current computational methods. To overcome these limitations, the study and development of ArMs stand to benefit from machine learning approaches. To achieve such goals, a large and well-defined set of structural and activity data for each target metalloenzyme is required. Since it is much easier to produce these data sets using ArMs, designed ArMs can play a key role in advancing this field. Given the rapid progress made in data collection and improvement of machine learning algorithms, it would be possible to routinely incorporate the best combinations of PCS and SCS features to design any metalloenzyme with desired activity.
ACKNOWLEDGMENTS
The research in the Lu group was supported by grants from the National Science Foundation (CHE-2108837) and the National Institutes of Health (GM141931). We also thank the Robert A. Welch Foundation (Grant F-0020) for support of the Lu group research program at the University of Texas at Austin.
ABBREVIATIONS
- AA
natural amino acid
- ABTS
2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid diammonium salt
- ACCO
1-aminocyclopropane-1-carboxylate oxidase
- ACS
coenzyme-A synthetase
- adt
-SCH2NHCH2S-
- AlaBpy
(2,2′-bipyridin-5-yl)alanine
- AMO
ammonia monooxygenase
- apo-Cc
apo-cytochrome c
- APX
ascorbate peroxidase
- ArM
artificial metalloenzyme
- ATHase
artificial transfer hydrogenase
- ATRP
atom transfer radical polymerization
- AxNiR
Alcaligenes xylosoxidans nitrite reductase
- Az
azurin
- β-LG
β-lactoglobulin
- biot
biotin
- Bph
biphenyl
- BPP
bovine-pancreatic polypeptide
- bpy
bipyridine
- CA
carbonic anhydrase
- cAMP
cyclic adenosine monophosphate
- camSOD
cofactor-flexible superoxide dismutase
- CAP
catabolite activator protein
- Cc
cytochrome c
- Cc552
cytochrome c552
- CcP
cytochrome c peroxidase
- CcO
cytochrome c oxidase
- CiP
Coprinus cinereus peroxidase
- CLEC
cross-linked enzymatic crystal
- ClTyr
3-chlorotyrosine
- CMD
concerted metalation deprotonation
- CODH
carbon monoxide dehydrogenase
- CoPP(IX)
cobalt-protoporphyrin IX
- COx
catechol oxidase
- Cp
Clostridium pasteurianum
- Csp1
copper storage protein
- CuAAz
CuA azurin
- CuBMb
myoglobin containing a designed CuB binding site
- CueO
copper efflux oxidase
- CuNiR
copper-dependent nitrite reductase
- Cgb
cytoglobin
- D-A
Diels–Alder reaction
- DF
diformyl
- DFT
density functional theory
- DCFH-DA
2′,7′-dichlorofluorescin diacetate
- DHP
dehaloperoxidase
- DNA
DNA
- DNIC
dinitrosyl iron complex
- dpa
bis(2-pyridylmethyl)amine
- DTB-SQ
3,5-di-tert-butylsemiquinone
- DTT
dithiothreitol
- DyP
decolorizing peroxidases
- E°′
reduction potential
- EDA
ethyl diazoacetate
- EDTA
ethylenediaminetetraacetic acid
- ee
enantiomeric excess
- E m
midpoint potential
- ENDOR
electron double nuclear resonance spectroscopy
- ESI-MS
electrospray ionization mass spectrometry
- ET
electron transfer
- EXAFS
extended X-ray absorption fine structure
- EPR
electron paramagnetic resonance spectroscopy
- F2Tyr
3,5-difluorotyrosine
- F3Tyr
2,3,5-trifluorotyrosine
- F-C
Friedel–Crafts alkylation
- FdM
[Fe4S4]-binding oligopeptide
- FeSOD
iron superoxide dismutase
- FT-IR
Fourier transform infrared spectroscopy
- GQ
G-quadruplex
- H-bonding
hydrogen bonding
- H-cluster
[Fe2(CO)3(CN)2(adt)]
- H2ase
hydrogenase
- hCA II
Human carbonic anhydrase II
- HCO
heme-copper oxidase
- HRP
horseradish peroxidase
- HSA
human serum albumin
- HT
HaloTag
- IR
infrared
- ICP
inductively coupled plasma
- imiTyr
Tyr-His cross-link
- IrMb
myoglobin containing the Ir(Me)-heme cofactor
- KIE
kinetic isotope effect
- L0
N-benzyl-N′- (2-hydroxybenzyl)-N,N′-ethylenediaminediacetic acid
- L52maleimide
1-(2-{[2-(bispyridin-2-ylmethylamino)ethyl]-pyridin-2-ylmethylamino}ethyl)pyrrole-2,5-dione
- L ibu
N,N′-bis(2-pyridylmethyl)-N-methyl-1,2-ethanediamine complex
- LiP
lignin peroxidase
- LMCT
ligand-to-metal charge transfer
- LmrR
Lactococcal multidrug resistance regulator
- LPMO
lytic polysaccharide monooxygenase
- Mb
myoglobin
- MCO
multicopper oxidase
- MD
molecular dynamics
- MF
monoformyl
- MG
malachite green
- MnCcP
Mn-bound cytochrome c peroxidase
- MnP
manganese peroxidase
- MnSOD
manganese superoxide dismutase
- MtTyr
2-amino-3-(4-hydroxy-3-(methylthio)phenyl)-propanoic acid
- N2OR
nitrous oxide reductase
- NB
nitrobindin
- Ngb
neuroglobin
- NiR
nitrite reductase
- NiRd
nickel rubredoxin
- NMM
N-methylmesoporphyrin IX
- NMR
nuclear magnetic resonance
- NO
nitric oxide
- NOR
nitric oxide reductase
- OMeY
3-methoxytyrosine
- P411
cytochrome P411
- P450
cytochrome P450
- Pf
Pyrococcus furiosus
- pMMO
particulate methane monooxygenase
- pAF
p-aminophenylalanine
- phen
phenylalanine
- PT3
adenosine deaminase protein scaffold
- POP
propyl oligopeptidase
- POP-Z
propyl oligopeptidase containing the UAA 4-azidophenylalanine
- PCS
primary coordination sphere
- RDS
rate-determining step
- Rma-Cc
bacterial cytochrome c from Rhodothermus marinus
- Rd
rubredoxin
- ROS
reactive oxygen species
- RMSD
root mean squared deviation
- RNA
ribonucleic acid
- rR
resonance Raman spectroscopy
- SA
serum albumin
- Sav
streptavidin
- SCP-2L
steroid carrier protein 2L
- SCS
secondary coordination sphere
- SDM
side-directed mutagenesis
- SHE
standard hydrogen electrode
- SKIE
solvent kinetic isotope effect
- SNO
nitrosothiol
- SOD
superoxide dismutase
- SOH
sulfenic acid
- SOR
superoxide reductase
- SRS
substrate recognition site
- SSM
site-saturation mutagenesis
- STAMPS
synchronous, transactional, and asynchronous multiprocessing
- swMb
sperm whale myoglobin
- t-BuOOH
t-butyl-hydroperoxide
- TAML
tetraamido macrocyclic ligand
- TCP
2,4,6-trichlorophenol
- TDHC
tetrahydrocorrin
- TFAC
trifluoroacetophenone
- THS
thermosome
- TOF
turnover frequency
- TOF MS
time-of-flight mass spectrometry
- TON
turnover number
- Trx
thioredoxin
- TvNiR
T. nitratireducens cytochrome c nitrite reductase
- TtCuA
Thermus thermophilus cytochrome ba3 oxidase
- UAA
unnatural amino acid
- UV
ultraviolet
- VT-MCD
variable temperature magnetic circular dichroism
- WT
wild-type
- XANES
X-ray absorption near-edge spectroscopy
- Xln
xylanase
- ZFS
zero-field splitting
Biographies
Casey Van Stappen completed his B.Sc. (2011) and M.Sc. (2013) degrees in Chemistry at the University of Minnesota, Duluth, under the supervision of Prof. Viktor Nemykin. Following several years of postgraduate study at the University ofMichigan, Ann Arbor, under the advisement of Profs. Nicolai Lehnert and Vincent Pecoraro, he transferred his studies to the Max Planck Institute for Chemical Energy Conversion (MPI-CEC, 2015), as a member of the IMPRS-RECHARGE, and completed his D.Sc. (2019) in Chemistry via enrollment at Ruhr-Universität Bochum under the advisement of Prof. Dr. Serena DeBeer, Dr. Eckhard Bill, and Prof. Dr. Frank Neese. Following a short period as a postdoctoral research associate, also at the MPI-CEC, he joined the group of Prof. Yi Lu at the University of Texas at Austin as a postdoctoral research fellow. His research interests lie in combining spectroscopic, biochemical, and computational methods to investigate small molecule activation and catalysis, particularly in biological and biologically inspired systems.
Yunling Deng grew up in Changchun, China, and received her B.Sc. (2014) in Chemistry from Nankai University, China (2014). She completed her Ph.D. (2020) in Bioinorganic Chemistry at Dartmouth College investigating the effects of heme propionates on the structural and redox properties of c-type cytochromes under the guidance of Prof. Ekaterina Pletneva. She next moved to the University of Illinois at Urbana-Champaign (and recently to the University of Texas at Austin) as a postdoctoral research fellow to work with Prof. Yi Lu, working on developing artificial metalloenzymes for CO2 reduction.
Yiwei Liu was born in Zhengzhou, China, and received his B.Sc. (2018) from Peking University, China, under the supervision of Prof. Jun-Long Zhang. He joined Prof. Yi Lu’s lab at the University of Illinois at Urbana-Champaign (now at the University of Texas at Austin) in the same year. As a Ph.D. candidate in Inorganic Chemistry, his research focuses on engineering simple, noncatalytic proteins such as cupredoxins into artificial metalloenzymes to mimic the coordination chemistry and reactivity of natural enzymes.
Hirbod Heidari was born in Hamedan, Iran, and completed his Chemistry B.Sc. (2018) under the supervision of Prof. Sirus Jamali at the Sharif University of Technology, Tehran, Iran, along with a minor degree in Physics. In 2017, he conducted a summer-long internship under the supervision of Prof. Truhlar and the late Dr. Steven Mielke on Molecular Partition Function Computations. In 2019, he joined the lab of Prof. Yi Lu at the University of Illinois at Urbana-Champaign (now at the University of Texas at Austin) as a Ph.D. candidate. His research interests lie at the interface of computational chemistry and artificial protein engineering.
Jing-Xiang Wang obtained his B.Sc. (2019) from Peking University, Beijing, China, under the supervision of Prof. Jun-Long Zhang. After he joined Dr. Yi Lu’s lab at the University of Illinois at Urbana-Champaign in the same year, he moved to the University of Texas at Austin in 2021 with Dr. Yi Lu, continuing to pursuit his doctorate degree. His research interests lie in metalloprotein engineering and constructing artificial metalloenzymes. His current research is focusing on enhancing activities for metalloenzymes for further applications.
Yu Zhou was born in Jiangxi, China. He received his B.Sc. degree (2019) from Zhiyuan College, Shanghai Jiao Tong University, under the supervision of Prof. Xiaomin Xie and Prof. Zhaoguo Zhang and conducted a half-year internship with Prof. David Sarlah in 2018 (University of Illinois at Urbana-Champaign, UIUC) as a visiting undergraduate. After a three-year undergraduate research experience in copper- and palladium-catalyzed organic reaction development, he decided to join Prof. Yi Lu’s lab at UIUC (now at the University of Texas at Austin) and study biocatalysis. His current research aims at designing novel, efficient, and highly selective biocatalytic systems and metabolic networks.
Aaron P. Ledray received a B.S. (2015) in Biotechnology and Biomedical Sciences from the University of Central Florida under the supervision of Prof. Stephen J. King, where he studied neuro-degenerative disease. He completed his Ph.D. (2021) in Biological Sciences from the University of California, Irvine, investigating reactive heme intermediates under the advisement of Prof. Michael T. Green. He joined Prof. Yi Lu’s group at the University of Texas at Austin as a postdoctoral research fellow in 2021, shortly after completing his doctorate. Aaron’s research interests include bioinorganic chemistry, enzyme design, and mechanisms of human disease.
Yi Lu received his B.S. degree from Peking University in 1986 and his Ph.D. degree from the University of California at Los Angeles in 1992. After two years of postdoctoral research in Professor Harry B. Gray’s group at Caltech, Dr. Lu started his own independent career in the Department of Chemistry at the University of Illinois at Urbana-Champaign in 1994. He was promoted to Associate Professor with tenure in 2000, full Professor in 2004, and endowed Jay and Ann Schenck Professor in 2010. In August of 2021, Dr. Lu moved to the University of Texas at Austin, becoming the Robert J. V. Johnson-Welch Regents Chair in Chemistry. Dr. Lu’s research interests lie at the interface between chemistry and biology. Specific areas of current interest include (a) design and engineering of functional metalloproteins as environmentally benign catalysts in renewable energy generation and pharmaceuticals; (b) fundamental understanding of DNAzymes and their applications in environmental monitoring, medical diagnostics, and targeted drug delivery; (c) employing principles from biology for directed assembly of nanomaterials with controlled morphologies and its applications in imaging and medicine and (d) engineer biocatalysts to address challenges in synthetic organic chemistry and applications of novel biocatalysts in synthetic biology for biomass conversion, valuable products generation in high yield and good selectivity.
Contributor Information
Casey Van Stappen, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States.
Yunling Deng, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States.
Yiwei Liu, Department of Chemistry, University of Illinois, Urbana-Champaign, Urbana, Illinois 61801, United States.
Hirbod Heidari, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States.
Jing-Xiang Wang, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States.
Yu Zhou, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States.
Aaron P. Ledray, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States
Yi Lu, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States; Department of Chemistry, University of Illinois, Urbana-Champaign, Urbana, Illinois 61801, United States.
REFERENCES
- (1).Andreini C; Bertini I; Cavallaro G; Holliday GL; Thornton JM Metal Ions in Biological Catalysis: From Enzyme Databases to General Principles. JBIC, J. Biol. Inorg. Chem 2008, 13, 1205–1218. [DOI] [PubMed] [Google Scholar]
- (2).Cvetkovic A; Menon AL; Thorgersen MP; Scott JW; Poole FL II; Jenney FE Jr.; Lancaster WA; Praissman JL; Shanmukh S; Vaccaro BJ; et al. Microbial Metalloproteomes Are Largely Uncharacterized. Nature 2010, 466, 779–782. [DOI] [PubMed] [Google Scholar]
- (3).Encyclopedia of Geobiology; Reitner J, Thiel V, Eds.; Encyclopedia of Earth Sciences Series; Springer Netherlands: Dordrecht, 2011; pp 558–562. [Google Scholar]
- (4).Rajakovich LJ; Balskus EP Metabolic Functions of the Human Gut Microbiota: The Role of Metalloenzymes. Nat. Prod. Rep 2019, 36, 593–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Nitschke W; McGlynn SE; Milner-White EJ; Russell MJ On the Antiquity of Metalloenzymes and Their Substrates in Bioenergetics. Biochim. Biophys. Acta, Bioenerg 2013, 1827, 871–881. [DOI] [PubMed] [Google Scholar]
- (6).Froese DS; Fowler B; Baumgartner MR Vitamin B12 Folate, and the Methionine Remethylation Cycle—Biochemistry, Pathways, and Regulation. J. Inherited Metab. Dis 2019, 42, 673–685. [DOI] [PubMed] [Google Scholar]
- (7).Persson B; Hedlund J; Jörnvall H Medium- and Short-Chain Dehydrogenase/Reductase Gene and Protein Families. Cell. Mol. Life Sci 2008, 65, 3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gonzalez FJ; Gelboin HV Human Cytochromes P450: Evolution and CDNA-Directed Expression. Environ. Health Perspect 1992, 98, 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Danielson PB The Cytochrome P450 Superfamily: Biochemistry, Evolution and Drug Metabolism in Humans. Curr. Drug. Metab 2002, 3, 561–597. [DOI] [PubMed] [Google Scholar]
- (10).Nelson DR Cytochrome P450 Diversity in the Tree of Life. Biochim. Biophys. Acta, Proteins Proteomics 2018, 1866, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Adam PS; Borrel G; Gribaldo S Evolutionary History of Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase, One of the Oldest Enzymatic Complexes. Proc. Natl. Acad. Sci. U.S.A 2018, 115, No. E1166–E1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Can M; Armstrong FA; Ragsdale SW Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase. Chem. Rev 2014, 114, 4149–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kumar M; Lu W-P; Smith A; Ragsdale SW; McCracken J Azide Binding to Carbon Monoxide Dehydrogenase from Clostridium Thermoaceticum. J. Am. Chem. Soc 1995, 117, 2939–2940. [Google Scholar]
- (14).Wittenborn EC; Cohen SE; Merrouch M; Léger C; Fourmond V; Dementin S; Drennan CL Structural Insight into Metallocofactor Maturation in Carbon Monoxide Dehydrogenase. J. Biol. Chem 2019, 294, 13017–13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Eklund H; Uhlin U; Färnegårdh M; Logan DT; Nordlund P Structure and Function of the Radical Enzyme Ribonucleotide Reductase. Prog. Biophys. Mol. Biol 2001, 77, 177–268. [DOI] [PubMed] [Google Scholar]
- (16).Nordlund P; Reichard P Ribonucleotide Reductases. Annu. Rev. Biochem 2006, 75, 681–706. [DOI] [PubMed] [Google Scholar]
- (17).Hofer A; Crona M; Logan DT; Sjöberg B-M DNA Building Blocks: Keeping Control of Manufacture. Crit. Rev. Biochem. Mol. Biol 2012, 47, 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jordan A; Reichard P Ribonucleotide Reductases. Annu. Rev. Biochem 1998, 67, 71–98. [DOI] [PubMed] [Google Scholar]
- (19).Vinyard DJ; Ananyev GM; Charles Dismukes G Photosystem II: The Reaction Center of Oxygenic Photosynthesis. Annu. Rev. Biochem 2013, 82, 577–606. [DOI] [PubMed] [Google Scholar]
- (20).McEvoy JP; Brudvig GW Water-Splitting Chemistry of Photosystem II. Chem. Rev 2006, 106, 4455–4483. [DOI] [PubMed] [Google Scholar]
- (21).Boyd ES; Anbar AD; Miller S; Hamilton TL; Lavin M; Peters JW A Late Methanogen Origin for Molybdenum-Dependent Nitrogenase. Geobiology 2011, 9, 221–232. [DOI] [PubMed] [Google Scholar]
- (22).Bulen WA; LeComte JR The Nitrogenase System from Azotobacter: Two-Enzyme Requirement for N2 Reduction, ATP-Dependent H2 Evolution, and ATP Hydrolysis. Proc. Natl. Acad. Sci. U.S.A 1966, 56, 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dervartanian DV; Shethna YI; Beinert H Purification and Properties of Two Iron-Sulfur Proteins from Azotobacter vinelandii. Biochim. Biophys. Acta, Protein Struct 1969, 194, 548–563. [DOI] [PubMed] [Google Scholar]
- (24).Burns RC; Holsten RD; Hardy RWF Isolation by Crystallization of the Mo-Fe Protein of Azotobacter Nitrogenase. Biochem. Biophys. Res. Commun 1970, 39, 90–99. [DOI] [PubMed] [Google Scholar]
- (25).Crichton RR Biological Ligands for Metal Ions. In Biological Inorganic Chemistry, 2nd ed.; Crichton RR, Ed.; Elsevier: Oxford, 2012; Chapter 4, pp 69–89. [Google Scholar]
- (26).Poulos TL Heme Enzyme Structure and Function. Chem. Rev 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Keller TCS; Lechauve C; Keller AS; Brooks S; Weiss MJ; Columbus L; Ackerman H; Cortese-Krott MM; Isakson BE The Role of Globins in Cardiovascular Physiology. Physiol. Rev 2022, 102, 859–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wajcman H; Kiger L; Marden MC Structure and Function Evolution in the Superfamily of Globins. C.R. Biol 2009, 332, 273–282. [DOI] [PubMed] [Google Scholar]
- (29).Lecomte JTJ; Vuletich DA; Lesk AM Structural Divergence and Distant Relationships in Proteins: Evolution of the Globins. Curr. Opin. Struct. Biol 2005, 15, 290–301. [DOI] [PubMed] [Google Scholar]
- (30).Derbyshire ER; Marletta MA Structure and Regulation of Soluble Guanylate Cyclase. Annu. Rev. Biochem 2012, 81, 533–559. [DOI] [PubMed] [Google Scholar]
- (31).Poulos TL Soluble Guanylate Cyclase. Curr. Opin. Struct. Biol 2006, 16, 736–743. [DOI] [PubMed] [Google Scholar]
- (32).Horst BG; Marletta MA Physiological Activation and Deactivation of Soluble Guanylate Cyclase. Nitric Oxide 2018, 77, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Shuvalov VA Composition and Function of Cytochrome b559 in Reaction Centers of Photosystem II of Green Plants. J. Bioenerg. Biomembr 1994, 26, 619–626. [DOI] [PubMed] [Google Scholar]
- (34).Pospíšil P Enzymatic Function of Cytochrome b559 in Photosystem II. J. Photochem. Photobiol. B 2011, 104, 341–347. [DOI] [PubMed] [Google Scholar]
- (35).Shinopoulos KE; Brudvig GW Cytochrome b559 and Cyclic Electron Transfer within Photosystem II. Biochim. Biophys. Acta, Bioenerg 2012, 1817, 66–75. [DOI] [PubMed] [Google Scholar]
- (36).Maté MJ; Murshudov G; Bravo J; Melik-Adamyan W; Loewen PC; Fita I Heme-Catalases. In Handbook of Metalloproteins; John Wiley & Sons, Ltd, 2006; pp 1–17. [Google Scholar]
- (37).Yuan F; Yin S; Xu Y; Xiang L; Wang H; Li Z; Fan K; Pan G The Richness and Diversity of Catalases in Bacteria. Front. Microbiol 2021, 12, 645477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Díaz A; Loewen PC; Fita I; Carpena X Thirty Years of Heme Catalases Structural Biology. Arch. Biochem. Biophys 2012, 525, 102–110. [DOI] [PubMed] [Google Scholar]
- (39).Vlasits J; Jakopitsch C; Bernroitner M; Zamocky M; Furtmüller PG; Obinger C Mechanisms of Catalase Activity of Heme Peroxidases. Arch. Biochem. Biophys 2010, 500, 74–81. [DOI] [PubMed] [Google Scholar]
- (40).Anjum NA; Sharma P; Gill SS; Hasanuzzaman M; Khan EA; Kachhap K; Mohamed AA; Thangavel P; Devi GD; Vasudhevan P; et al. Catalase and Ascorbate Peroxidase-Representative H2O2-Detoxifying Heme Enzymes in Plants. Environ. Sci. Pollut. Res. Int 2016, 23, 19002–19029. [DOI] [PubMed] [Google Scholar]
- (41).García-Horsman JA; Barquera B; Rumbley J; Ma J; Gennis RB The Superfamily of Heme-Copper Respiratory Oxidases. J. Bacteriol 1994, 176, 5587–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).van der Oost J; De Boer APN; de Gier J-WL; Zumft WG; Stouthamer AH; Van Spanning RJM The Heme-Copper Oxidase Family Consists of Three Distinct Types of Terminal Oxidases and Is Related to Nitric Oxide Reductase. FEMS Microbiol. Lett 1994, 121, 1–9. [DOI] [PubMed] [Google Scholar]
- (43).Ortiz de Montellano PR The Mechanism of Heme Oxygenase. Curr. Opin. Chem. Biol 2000, 4, 221–227. [DOI] [PubMed] [Google Scholar]
- (44).Kikuchi G; Yoshida T; Noguchi M Heme Oxygenase and Heme Degradation. Biochem. Biophys. Res. Commun 2005, 338, 558–567. [DOI] [PubMed] [Google Scholar]
- (45).Gozzelino R; Jeney V; Soares MP Mechanisms of Cell Protection by Heme Oxygenase-1. Annu. Rev. Pharmacol. Toxicol 2010, 50, 323–354. [DOI] [PubMed] [Google Scholar]
- (46).Poulos TL Thirty Years of Heme Peroxidase Structural Biology. Arch. Biochem. Biophys 2010, 500, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Davies MJ; Hawkins CL; Pattison DI; Rees MD Mammalian Heme Peroxidases: From Molecular Mechanisms to Health Implications. Antioxid. Redox. Signal 2008, 10, 1199–1234. [DOI] [PubMed] [Google Scholar]
- (48).Smith AT; Veitch NC Substrate Binding and Catalysis in Heme Peroxidases. Curr. Opin. Chem. Biol 1998, 2, 269–278. [DOI] [PubMed] [Google Scholar]
- (49).Watmough NJ; Field SJ; Hughes RJL; Richardson DJ The Bacterial Respiratory Nitric Oxide Reductase. Biochem. Soc. Trans 2009, 37, 392–399. [DOI] [PubMed] [Google Scholar]
- (50).Daiber A; Shoun H; Ullrich V Nitric Oxide Reductase (P450nor) from Fusarium oxysporum. J. Inorg. Biochem 2005, 99, 185–193. [DOI] [PubMed] [Google Scholar]
- (51).Zumft WG Nitric Oxide Reductases of Prokaryotes with Emphasis on the Respiratory, Heme-Copper Oxidase Type. J. Inorg. Biochem 2005, 99, 194–215. [DOI] [PubMed] [Google Scholar]
- (52).Murphy MJ; Siegel LM Siroheme and Sirohydrochlorin: The Basis for a New Type of Porphyrin-Related Prosthetic Group common to Both Assimilatory and Dissimilatory Sulfite Reductases. J. Biol. Chem 1973, 248, 6911–6919. [PubMed] [Google Scholar]
- (53).Trüper HG [29] Reverse Siroheme Sulfite Reductase from Thiobacillus denitrificans. In Inorganic Microbial Sulfur Metabolism; Methods in Enzymology, Vol. 243; Abelson JN, Simon MI, Peck HD Jr., LeGall J, Eds.; Academic Press, 1994; pp 422–426. [Google Scholar]
- (54).Nakayama M; Akashi T; Hase T Plant Sulfite Reductase: Molecular Structure, Catalytic Function and Interaction with Ferredoxin. J. Inorg. Biochem 2000, 82, 27–32. [DOI] [PubMed] [Google Scholar]
- (55).Van Stappen C; Decamps L; Cutsail GE; Bjornsson R; Henthorn JT; Birrell JA; DeBeer S The Spectroscopy of Nitrogenases. Chem. Rev 2020, 120, 5005–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Lubitz W; Ogata H; Rüdiger O; Reijerse E Hydrogenases. Chem. Rev 2014, 114, 4081–4148. [DOI] [PubMed] [Google Scholar]
- (57).Feynman R What I cannot create I do not understand. https://archives.caltech.edu/pictures/1.10-29.jpg (accessed 2022-02-07). [Google Scholar]
- (58).Hibbert EG; Dalby PA Directed Evolution Strategies for Improved Enzymatic Performance. Microb. Cell Fact 2005, 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Cobb RE; Chao R; Zhao H Directed Evolution: Past, Present, and Future. AIChE J 2013, 59, 1432–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Voigt CA; Kauffman S; Wang ZG Rational Evolutionary Design: The Theory of in Vitro Protein Evolution. Adv. Protein Chem 2000, 55, 79–160. [DOI] [PubMed] [Google Scholar]
- (61).Das R; Baker D Macromolecular Modeling with Rosetta. Annu. Rev. Biochem 2008, 77, 363–382. [DOI] [PubMed] [Google Scholar]
- (62).Leaver-Fay A; Tyka M; Lewis SM; Lange OF; Thompson J; Jacak R; Kaufman K; Renfrew PD; Smith CA; Sheffler W; et al. ROSETTA3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol. 2011, 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Senior AW; Evans R; Jumper J; Kirkpatrick J; Sifre L; Green T; Qin C; Žídek A; Nelson AWR; Bridgland A; et al. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577, 706–710. [DOI] [PubMed] [Google Scholar]
- (64).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Korendovych IV; DeGrado WF De Novo Protein Design, a Retrospective. Q. Rev. Biophys 2020, 53, No. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Huang P-S; Boyken SE; Baker D The Coming of Age of De Novo Protein Design. Nature 2016, 537, 320–327. [DOI] [PubMed] [Google Scholar]
- (67).Chalkley MJ; Mann SI; DeGrado WF De Novo Metalloprotein Design. Nat. Rev. Chem 2022, 6, 31–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Pinter TBJ; Koebke KJ; Pecoraro VL Catalysis and Electron Transfer in De Novo Designed Helical Scaffolds. Angew. Chem., Int. Ed. Engl 2020, 59, 7678–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Schwizer F; Okamoto Y; Heinisch T; Gu Y; Pellizzoni MM; Lebrun V; Reuter R; Köhler V; Lewis JC; Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- (70).Steinreiber J; Ward TR Artificial Metalloenzymes as Selective Catalysts in Aqueous Media. Coord. Chem. Rev 2008, 252, 751–766. [Google Scholar]
- (71).Mao J; Ward TR Artificial Metalloenzymes for Enantiose-lective Catalysis Based on the Biotin-Avidin Technology. CHIMIA International Journal for Chemistry 2008, 62, 956–961. [Google Scholar]
- (72).Creus M; Ward TR Designed Evolution of Artificial Metalloenzymes: Protein Catalysts Made to Order. Org. Biomol. Chem 2007, 5, 1835–1844. [DOI] [PubMed] [Google Scholar]
- (73).Thomas CM; Ward TR Artificial Metalloenzymes: Proteins as Hosts for Enantioselective Catalysis. Chem. Soc. Rev 2005, 34, 337–346. [DOI] [PubMed] [Google Scholar]
- (74).Creus M; Ward TR Design and Evolution of Artificial Metalloenzymes: Biomimetic Aspects. In Progress in Inorganic Chemistrty, Vol. 4; Karlin KD, Ed.; John Wiley & Sons, Ltd, 2011; Chapter 4, pp 203–253. [Google Scholar]
- (75).Nastri F; D’Alonzo D; Leone L; Zambrano G; Pavone V; Lombardi A Engineering Metalloprotein Functions in Designed and Native Scaffolds. Trends Biochem. Sci 2019, 44, 1022–1040. [DOI] [PubMed] [Google Scholar]
- (76).Bos J; Roelfes G Artificial Metalloenzymes for Enantioselective Catalysis. Curr. Opin. Chem. Biol 2014, 19, 135–143. [DOI] [PubMed] [Google Scholar]
- (77).Leveson-Gower RB; Mayer C; Roelfes G The Importance of Catalytic Promiscuity for Enzyme Design and Evolution. Nat. Rev. Chem 2019, 3, 687–705. [Google Scholar]
- (78).Springer BA; Sligar SG; Olson JS; Phillips GN Jr. Mechanisms of Ligand Recognition in Myoglobin. Chem. Rev 1994, 94, 699–714. [Google Scholar]
- (79).Antonini E; Brunori M Hemoglobin and Myoglobin in Their Reactions with Ligands. In Frontiers of Biology, Vol. 21; North-Holland, 1971; pp 296. [Google Scholar]
- (80).Lin Y; Wang J; Lu Y Functional Tuning and Expanding of Myoglobin by Rational Protein Design. Sci. China: Chem 2014, 57, 346–355. [Google Scholar]
- (81).Bhagi-Damodaran A; Petrik I; Lu Y Using Biosynthetic Models of Heme-Copper Oxidase and Nitric Oxide Reductase in Myoglobin to Elucidate Structural Features Responsible for Enzymatic Activities. Isr. J. Chem 2016, 56, 773–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Lin Y-W Rational Design of Heme Enzymes for Biodegradation of Pollutants toward a Green Future. Biotechnol. Appl. Biochem 2020, 67, 484–494. [DOI] [PubMed] [Google Scholar]
- (83).Rittle J; Field MJ; Green MT; Tezcan FA An Efficient, Step-Economical Strategy for the Design of Functional Metalloproteins. Nat. Chem 2019, 11, 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Lu Y; Berry SM; Pfister TD Engineering Novel Metalloproteins: Design of Metal-Binding Sites into Native Protein Scaffolds. Chem. Rev 2001, 101, 3047–3080. [DOI] [PubMed] [Google Scholar]
- (85).Ozaki S; Matsui T; Roach MP; Watanabe Y Rational Molecular Design of a Catalytic Site: Engineering of Catalytic Functions to the Myoglobin Active Site Framework. Coord. Chem. Rev 2000, 198, 39–59. [Google Scholar]
- (86).Hayashi T; Hisaeda Y New Functionalization of Myoglobin by Chemical Modification of Heme-Propionates. Acc. Chem. Res 2002, 35, 35–43. [DOI] [PubMed] [Google Scholar]
- (87).Lu Y Design and Engineering of Metalloproteins Containing Unnatural Amino Acids or Non-Native Metal-Containing Cofactors. Curr. Opin. Chem. Biol 2005, 9, 118–126. [DOI] [PubMed] [Google Scholar]
- (88).Lu Y Biosynthetic Inorganic Chemistry. Angew. Chem., Int. Ed. Engl 2006, 45, 5588–5601. [DOI] [PubMed] [Google Scholar]
- (89).Isogai Y; Imamura H; Nakae S; Sumi T; Takahashi K; Nakagawa T; Tsuneshige A; Shirai T Tracing Whale Myoglobin Evolution by Resurrecting Ancient Proteins. Sci. Rep 2018, 8, 16883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Bonagura CA; Bhaskar B; Shimizu H; Li H; Sundaramoorthy M; McRee DE; Goodin DB; Poulos TL High-Resolution Crystal Structures and Spectroscopy of Native and Compound I Cytochrome c Peroxidase. Biochemistry 2003, 42, 5600–5608. [DOI] [PubMed] [Google Scholar]
- (91).Patterson WR; Poulos TL Crystal Structure of Recombinant Pea Cytosolic Ascorbate Peroxidase. Biochemistry 1995, 34, 4331–4341. [DOI] [PubMed] [Google Scholar]
- (92).LaCount MW; Zhang E; Chen YP; Han K; Whitton MM; Lincoln DE; Woodin SA; Lebioda L The Crystal Structure and Amino Acid Sequence of Dehaloperoxidase from Amphitrite ornata Indicate Common Ancestry with Globins. J. Biol. Chem 2000, 275, 18712–18716. [DOI] [PubMed] [Google Scholar]
- (93).Ortiz de Montellano PR; Catalano CE Epoxidation of Styrene by Hemoglobin and Myoglobin. Transfer of Oxidizing Equivalents to the Protein Surface. J. Biol. Chem 1985, 260, 9265–9271. [PubMed] [Google Scholar]
- (94).Rao SI; Wilks A; Ortiz de Montellano PR The Roles of His-64, Tyr-103, Tyr-146, and Tyr-151 in the Epoxidation of Styrene and Beta-Methylstyrene by Recombinant Sperm Whale Myoglobin. J. Biol. Chem 1993, 268, 803–809. [PubMed] [Google Scholar]
- (95).Adachi S; Nagano S; Ishimori K; Watanabe Y; Morishima I; Egawa T; Kitagawa T; Makino R Roles of Proximal Ligand in Heme Proteins: Replacement of Proximal Histidine of Human Myoglobin with Cysteine and Tyrosine by Site-Directed Mutagenesis as Models for P-450, Chloroperoxidase, and Catalase. Biochemistry 1993, 32, 241–252. [DOI] [PubMed] [Google Scholar]
- (96).Yonetani T; Schleyer H Studies on Cytochrome c Peroxidase. J. Biol. Chem 1967, 242, 1974–1979. [PubMed] [Google Scholar]
- (97).Finzel BC; Poulos TL; Kraut J Crystal Structure of Yeast Cytochrome c Peroxidase Refined at 1.7-Å Resolution. J. Biol. Chem 1984, 259, 13027–13036. [PubMed] [Google Scholar]
- (98).Gajhede M; Schuller DJ; Henriksen A; Smith AT; Poulos TL Crystal Structure of Horseradish Peroxidase C at 2.15 Å Resolution. Nat. Struct. Mol. Biol 1997, 4, 1032–1038. [DOI] [PubMed] [Google Scholar]
- (99).Matsui T; Ozaki S; Watanabe Y On the Formation and Reactivity of Compound I of the His-64 Myoglobin Mutants. J. Biol. Chem 1997, 272, 32735–32738. [DOI] [PubMed] [Google Scholar]
- (100).Ozaki S; Matsui T; Watanabe Y Conversion of Myoglobin into a Highly Stereo- Specific Peroxygenase by the L29H/H64L Mutation. J. Am. Chem. Soc 1996, 118, 9784–9785. [Google Scholar]
- (101).Wu L-B; Du K-J; Nie C-M; Gao S-Q; Wen G-B; Tan X; Lin Y-W Peroxidase Activity Enhancement of Myoglobin by Two Cooperative Distal Histidines and a Channel to the Heme Pocket. J. Mol. Cat. B: Enzym 2016, 134, 367–371. [Google Scholar]
- (102).Matsui T; Ozaki S; Liong E; Phillips GN; Watanabe Y Effects of the Location of Distal Histidine in the Reaction of Myoglobin with Hydrogen Peroxide. J. Biol. Chem 1999, 274, 2838–2844. [DOI] [PubMed] [Google Scholar]
- (103).Poulos TL; Finzel BC; Howard AJ High-Resolution Crystal Structure of Cytochrome P450cam. J. Mol. Biol 1987, 195, 687–700. [DOI] [PubMed] [Google Scholar]
- (104).Hara I; Ueno T; Ozaki S; Itoh S; Lee K; Ueyama N; Watanabe Y Oxidative Modification of Tryptophan 43 in the Heme Vicinity of the F43W/H64L Myoglobin Mutant. J. Biol. Chem 2001, 276, 36067–36070. [DOI] [PubMed] [Google Scholar]
- (105).Quillin ML; Li T; Olson JS; Phillips GN Jr; Duo Y; Ikeda-Saito M; Regan R; Carlson M; Gibson QH; Li H; et al. Structural and Functional Effects of Apolar Mutations of the Distal Valine in Myoglobin. J. Mol. Biol 1995, 245, 416–436. [DOI] [PubMed] [Google Scholar]
- (106).Yang H-J; Matsui T; Ozaki S; Kato S; Ueno T; Phillips GN; Fukuzumi S; Watanabe Y Molecular Engineering of Myoglobin: Influence of Residue 68 on the Rate and the Enantioselectivity of Oxidation Reactions Catalyzed by H64D/V68X Myoglobin. Biochemistry 2003, 42, 10174–10181. [DOI] [PubMed] [Google Scholar]
- (107).Quillin ML; Arduini RM; Olson JS; Phillips GN High-Resolution Crystal Structures of Distal Histidine Mutants of Sperm Whale Myoglobin. J. Mol. Biol 1993, 234, 140–155. [DOI] [PubMed] [Google Scholar]
- (108).Tiso M; Tejero J; Basu S; Azarov I; Wang X; Simplaceanu V; Frizzell S; Jayaraman T; Geary L; Shapiro C; et al. Human Neuroglobin Functions as a Redox-Regulated Nitrite Reductase. J. Biol. Chem 2011, 286, 18277–18289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Beckerson P; Reeder BJ; Wilson MT Coupling of Disulfide Bond and Distal Histidine Dissociation in Human Ferrous Cytoglobin Regulates Ligand Binding. FEBS Lett. 2015, 589, 507–512. [DOI] [PubMed] [Google Scholar]
- (110).Wu L-B; Yuan H; Zhou H; Gao S-Q; Nie C-M; Tan X; Wen G-B; Lin Y-W An Intramolecular Disulfide Bond Designed in Myoglobin Fine-Tunes Both Protein Structure and Peroxidase Activity. Arch. Biochem. Biophys 2016, 600, 47–55. [DOI] [PubMed] [Google Scholar]
- (111).Yin L-L; Yuan H; Du K-J; He B; Gao S-Q; Wen G-B; Tan X; Lin Y-W Regulation of Both the Structure and Function by a De Novo Designed Disulfide Bond: A Case Study of Heme Proteins in Myoglobin. Chem. Commun 2018, 54, 4356–4359. [DOI] [PubMed] [Google Scholar]
- (112).Chen YP; Woodin SA; Lincoln DE; Lovell CR An Unusual Dehalogenating Peroxidase from the Marine Terebellid Polychaete Amphitrite ornata. J. Biol. Chem 1996, 271, 4609–4612. [PubMed] [Google Scholar]
- (113).Osborne RL; Taylor LO; Han KP; Ely B; Dawson J Amphitrite ornata Dehaloperoxidase: Enhanced Activity for the Catalytically Active Globin Using MCPBA. Biochem. Biophys. Res. Commun 2004, 324, 1194. [DOI] [PubMed] [Google Scholar]
- (114).Du J; Huang X; Sun S; Wang C; Lebioda L; Dawson JH Amphitrite Ornata Dehaloperoxidase (DHP): Investigations of Structural Factors That Influence the Mechanism of Halophenol Dehalogenation Using “Peroxidase-like” Myoglobin Mutants and “Myoglobin-like” DHP Mutants. Biochemistry 2011, 50, 8172–8180. [DOI] [PubMed] [Google Scholar]
- (115).Yan D-J; Li W; Xiang Y; Wen G-B; Lin Y-W; Tan X A Novel Tyrosine-Heme C-O Covalent Linkage in F43Y Myoglobin: A New Post-Translational Modification of Heme Proteins. ChemBioChem 2015, 16, 47–50. [DOI] [PubMed] [Google Scholar]
- (116).Liao F; He B; Du K-J; Gao S-Q; Wen G-B; Lin Y-W Enhanced Dehaloperoxidase Activity of F43Y Myoglobin with a Novel Thyrosine-Heme Crosslink. Chem. Lett 2016, 45, 1087–1089. [Google Scholar]
- (117).Li L-L; Yuan H; Liao F; He B; Gao S-Q; Wen G-B; Tan X; Lin Y-W Rational Design of Artificial Dye-Decolorizing Peroxidases Using Myoglobin by Engineering Tyr/Trp in the Heme Center. Dalton Trans. 2017, 46, 11230–11238. [DOI] [PubMed] [Google Scholar]
- (118).Yin L; Yuan H; Liu C; He B; Gao S-Q; Wen G-B; Tan X; Lin Y-W A Rationally Designed Myoglobin Exhibits a Catalytic Dehalogenation Efficiency More than 1000-Fold That of a Native Dehaloperoxidase. ACS Catal. 2018, 8, 9619–9624. [Google Scholar]
- (119).Sundaramoorthy M; Terner J; Poulos TL The Crystal Structure of Chloroperoxidase: A Heme Peroxidase-Cytochrome P450 Functional Hybrid. Structure 1995, 3, 1367–1378. [DOI] [PubMed] [Google Scholar]
- (120).Matsui T; Ozaki S; Watanabe Y Formation and Catalytic Roles of Compound I in the Hydrogen Peroxide-Dependent Oxidations by His64 Myoglobin Mutants. J. Am. Chem. Soc 1999, 121, 9952–9957. [Google Scholar]
- (121).Liu C; Xu J; Gao S-Q; He B; Wei C-W; Wang X-J; Wang Z; Lin Y-W Green and Efficient Biosynthesis of Indigo from Indole by Engineered Myoglobins. RSC Adv. 2018, 8, 33325–33330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Liao F; Xu J-K; Luo J; Gao S-Q; Wang X-J; Lin Y-W Bioinspired Design of an Artificial Peroxidase: Introducing Key Residues of Native Peroxidases into F43Y Myoglobin with a Tyr-Heme Cross-Link. Dalton Trans. 2020, 49, 5029–5033. [DOI] [PubMed] [Google Scholar]
- (123).Xiang H-F; Xu J-K; Liu J; Yang X-Z; Gao S-Q; Wen G-B; Lin Y-W Efficient Biodegradation of Malachite Green by an Artificial Enzyme Designed in Myoglobin. RSC Adv. 2021, 11, 16090–16095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Bordeaux M; Tyagi V; Fasan R Highly Diastereoselective and Enantioselective Olefin Cyclopropanation Using Engineered Myoglobin-Based Catalysts. Angew. Chem., Int. Ed 2015, 54, 1744–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Tyagi V; Bonn RB; Fasan R Intermolecular Carbene S-H Insertion Catalysed by Engineered Myoglobin-Based Catalysts. Chem. Sci 2015, 6, 2488–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Tyagi V; Sreenilayam G; Bajaj P; Tinoco A; Fasan R Biocatalytic Synthesis of Allylic and Allenyl Sulfides through a Myoglobin-Catalyzed Doyle-Kirmse Reaction. Angew. Chem., Int. Ed 2016, 55, 13562–13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Tinoco A; Steck V; Tyagi V; Fasan R Highly Diastereo- and Enantioselective Synthesis of Trifluoromethyl-Substituted Cyclopropanes via Myoglobin-Catalyzed Transfer of Trifluoromethylcarbene. J. Am. Chem. Soc 2017, 139, 5293–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Tyagi V; Fasan R Myoglobin-Catalyzed Olefination of Aldehydes. Angew. Chem., Int. Ed 2016, 55, 2512–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Bourissou D; Guerret O; Gabbaï FP; Bertrand G Stable Carbenes. Chem. Rev 2000, 100, 39–92. [DOI] [PubMed] [Google Scholar]
- (130).Wang Y; Xue P; Cao M; Yu T; Lane ST; Zhao H Directed Evolution: Methodologies and Applications. Chem. Rev 2021, 121, 12384–12444. [DOI] [PubMed] [Google Scholar]
- (131).Sreenilayam G; Fasan R Myoglobin-Catalyzed Intermolecular Carbene N-H Insertion with Arylamine Substrates. Chem. Commun 2015, 51, 1532–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Vargas DA; Tinoco A; Tyagi V; Fasan R Myoglobin-Catalyzed C-H Functionalization of Unprotected Indoles. Angew. Chem., Int. Ed 2018, 57, 9911–9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Vargas DA; Khade RL; Zhang Y; Fasan R Biocatalytic Strategy for Highly Diastereo- and Enantioselective Synthesis of 2,3-Dihydrobenzofuran-Based Tricyclic Scaffolds. Angew. Chem., Int. Ed 2019, 58, 10148–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Nam D; Steck V; Potenzino RJ; Fasan R A Diverse Library of Chiral Cyclopropane Scaffolds via Chemoenzymatic Assembly and Diversification of Cyclopropyl Ketones. J. Am. Chem. Soc 2021, 143, 2221–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Tinoco A; Wei Y; Bacik J-P; Carminati DM; Moore EJ; Ando N; Zhang Y; Fasan R Origin of High Stereocontrol in Olefin Cyclopropanation Catalyzed by an Engineered Carbene Transferase. ACS Catal. 2019, 9, 1514–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Zhou Q; Hu M; Zhang W; Jiang L; Perrett S; Zhou J; Wang J Probing the Function of the Tyr-Cys Cross-Link in Metalloenzymes by the Genetic Incorporation of 3-Methylthiotyrosine. Angew. Chem., Int. Ed 2013, 52, 1203–1207. [DOI] [PubMed] [Google Scholar]
- (137).Meharenna YT; Oertel P; Bhaskar B; Poulos TL Engineering Ascorbate Peroxidase Activity into Cytochrome c Peroxidase. Biochemistry 2008, 47, 10324–10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Erman JE; Kilheeney H; Bidwai AK; Ayala CE; Vitello LB Peroxygenase Activity of Cytochrome c Peroxidase and Three Apolar Distal Heme Pocket Mutants: Hydroxylation of 1-Methoxynaphthalene. BMC Biochemistry 2013, 14, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Wang J; Mauro JM; Edwards SL; Oatley SJ; Fishel LA; Ashford VA; Xuong NH; Kraut J X-Ray Structures of Recombinant Yeast Cytochrome c Peroxidase and Three Heme-Cleft Mutants Prepared by Site-Directed Mutagenesis. Biochemistry 1990, 29, 7160–7173. [DOI] [PubMed] [Google Scholar]
- (140).Stubbe J; van der Donk WA Protein Radicals in Enzyme Catalysis. Chem. Rev 1998, 98, 705–762. [DOI] [PubMed] [Google Scholar]
- (141).Seyedsayamdost MR; Yee CS; Reece SY; Nocera DG; Stubbe J pH Rate Profiles of FnY356-R2s (n = 2, 3, 4) in Escherichia coli Ribonucleotide Reductase: Evidence That Y356 Is a Redox-Active Amino Acid along the Radical Propagation Pathway. J. Am. Chem. Soc 2006, 128, 1562–1568. [DOI] [PubMed] [Google Scholar]
- (142).Schoemaker HE; Piontek K On the Interaction of Lignin Peroxidase with Lignin. Pure Appl. Chem 1996, 68, 2089–2096. [Google Scholar]
- (143).Choinowski T; Blodig W; Winterhalter KH; Piontek K The Crystal Structure of Lignin Peroxidase at 1.70 Å Resolution Reveals a Hydroxy Group on the Cβ of Tryptophan 171: A Novel Radical Site Formed during the Redox Cycle. J. Mol. Biol 1999, 286, 809–827. [DOI] [PubMed] [Google Scholar]
- (144).Blodig W; Smith AT; Doyle WA; Piontek K Crystal Structures of Pristine and Oxidatively Processed Lignin Peroxidase Expressed in Escherichia Coli and of the W171F Variant That Eliminates the Redox Active Tryptophan 171. Implications for the Reaction Mechanism. J. Mol. Biol 2001, 305, 851–861. [DOI] [PubMed] [Google Scholar]
- (145).Smith AT; Doyle WA; Dorlet P; Ivancich A Spectroscopic Evidence for an Engineered, Catalytically Active Trp Radical That Creates the Unique Reactivity of Lignin Peroxidase. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 16084–16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Field MJ; Bains RK; Warren JJ Using an Artificial Tryptophan “Wire” in Cytochrome c Peroxidase for Oxidation of Organic Substrates. Dalton Trans. 2017, 46, 11078–11083. [DOI] [PubMed] [Google Scholar]
- (147).Berghuis AM; Brayer GD Oxidation State-Dependent Conformational Changes in Cytochrome c. J. Mol. Biol 1992, 223, 959–976. [DOI] [PubMed] [Google Scholar]
- (148).Yin V; Shaw GS; Konermann L Cytochrome c as a Peroxidase: Activation of the Precatalytic Native State by H2O2-Induced Covalent Modifications. J. Am. Chem. Soc 2017, 139, 15701–15709. [DOI] [PubMed] [Google Scholar]
- (149).Xu Y; Mayne L; Englander SW Evidence for an Unfolding and Refolding Pathway in Cytochrome c. Nat. Struct. Biol 1998, 5, 774–778. [DOI] [PubMed] [Google Scholar]
- (150).Krishna MMG; Maity H; Rumbley JN; Lin Y; Englander SW Order of Steps in the Cytochrome c Folding Pathway: Evidence for a Sequential Stabilization Mechanism. J. Mol. Biol 2006, 359, 1410–1419. [DOI] [PubMed] [Google Scholar]
- (151).Tezcan FA; Winkler JR; Gray HB Effects of Ligation and Folding on Reduction Potentials of Heme Proteins. J. Am. Chem. Soc 1998, 120, 13383–13388. [Google Scholar]
- (152).Wang Z-H; Lin Y-W; Rosell FI; Ni F-Y; Lu H-J; Yang P-Y; Tan X-S; Li X-Y; Huang Z-X; Mauk AG Converting Cytochrome c into a Peroxidase-like Metalloenzyme by Molecular Design. ChemBioChem 2007, 8, 607–609. [DOI] [PubMed] [Google Scholar]
- (153).Ying T; Zhong F; Wang Z-H; Li W; Tan X; Huang Z-X A Route to Novel Functional Metalloproteins via Hybrids of Cytochrome P450 and Cytochrome c. ChemBioChem 2011, 12, 707–710. [DOI] [PubMed] [Google Scholar]
- (154).Ying T; Zhong F; Wang Z-H; Xie J; Tan X; Huang Z-X Generation of Novel Functional Metalloproteins via Hybrids of Cytochrome c and Peroxidase. Protein Eng., Des. Sel 2013, 26, 401–407. [DOI] [PubMed] [Google Scholar]
- (155).Wester MR; Yano JK; Schoch GA; Yang C; Griffin KJ; Stout CD; Johnson EF The Structure of Human Cytochrome P450 2C9 Complexed with Flurbiprofen at 2.0-A Resolution. J. Biol. Chem 2004, 279, 35630–35637. [DOI] [PubMed] [Google Scholar]
- (156).Nair PC; McKinnon RA; Miners JO Cytochrome P450 Structure-Function: Insights from Molecular Dynamics Simulations. Drug Metab. Rev 2016, 48, 434–452. [DOI] [PubMed] [Google Scholar]
- (157).Ayala M; Verdin J; Vazquez-Duhalt R The Prospects for Peroxidase-Based Biorefining of Petroleum Fuels. Biocatal. Biotrans 2007, 25, 114–129. [Google Scholar]
- (158).Nakajima H; Ichikawa Y; Satake Y; Takatani N; Manna SK; Rajbongshi J; Mazumdar S; Watanabe Y Engineering of Thermus Thermophilus Cytochrome C552: Thermally Tolerant Artificial Peroxidase. ChemBioChem 2008, 9, 2954–2957. [DOI] [PubMed] [Google Scholar]
- (159).Nakajima H; Ramanathan K; Kawaba N; Watanabe Y Rational Engineering of Thermus Thermophilus Cytochrome C552 to a Thermally Tolerant Artificial Peroxidase. Dalton Trans. 2010, 39, 3105–3114. [DOI] [PubMed] [Google Scholar]
- (160).Than ME; Hof P; Huber R; Bourenkov GP; Bartunik HD; Buse G; Soulimane T Thermus thermophilus Cytochrome c552: A New Highly Thermostable Cytochrome c Structure Obtained by MAD Phasing. J. Mol. Biol 1997, 271, 629–644. [DOI] [PubMed] [Google Scholar]
- (161).Finnigan JD; Young C; Cook DJ; Charnock SJ; Black GW Cytochromes P450 (P450s): A Review of the Class System with a Focus on Prokaryotic P450s. In Advances in Protein Chemistry and Structural Biology, Vol. 122; Karabencheva-Christova T, Christov C, Eds.; Academic Press, 2020; Chapter 9, pp 289–320. [DOI] [PubMed] [Google Scholar]
- (162).Butler CF; Peet C; Mason AE; Voice MW; Leys D; Munro AW Key Mutations Alter the Cytochrome P450 BM3 Conformational Landscape and Remove Inherent Substrate Bias. J. Biol. Chem 2013, 288, 25387–25399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Coelho PS; Brustad EM; Kannan A; Arnold FH Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307–310. [DOI] [PubMed] [Google Scholar]
- (164).Gober JG; Rydeen AE; Gibson-O’Grady EJ; Leuthaeuser JB; Fetrow JS; Brustad EM Mutating a Highly Conserved Residue in Diverse Cytochrome P450s Facilitates Diastereoselective Olefin Cyclopropanation. ChemBioChem 2016, 17, 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Coelho PS; Wang ZJ; Ener ME; Baril SA; Kannan A; Arnold FH; Brustad EM A Serine-Substituted P450 Catalyzes Highly Efficient Carbene Transfer to Olefins in Vivo. Nat. Chem. Biol 2013, 9, 485–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Renata H; Wang ZJ; Kitto RZ; Arnold FH P450-Catalyzed Asymmetric Cyclopropanation of Electron-Deficient Olefins under Aerobic Conditions. Catal. Sci. Technol 2014, 4, 3640–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Gober JG; Rydeen AE; Schwochert TD; Gibson-O’Grady EJ; Brustad EM Enhancing Cytochrome P450-Mediated Non-Natural Cyclopropanation by Mutation of a Conserved Second-Shell Residue. Biotechnol. Bioeng 2018, 115, 1416–1426. [DOI] [PubMed] [Google Scholar]
- (168).Girvan HM; Seward HE; Toogood HS; Cheesman MR; Leys D; Munro AW Structural and Spectroscopic Characterization of P450 BM3 Mutants with Unprecedented P450 Heme Iron Ligand Sets: New Heme Ligation States Influence Conformational Equilibria in P450 BM3. J. Biol. Chem 2007, 282, 564–572. [DOI] [PubMed] [Google Scholar]
- (169).Sevrioukova IF; Li H; Zhang H; Peterson JA; Poulos TL Structure of a Cytochrome P450-Redox Partner Electron-Transfer Complex. Proc. Natl. Acad. Sci. U.S.A 1999, 96, 1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Ost TWB; Clark J; Mowat CG; Miles CS; Walkinshaw MD; Reid GA; Chapman SK; Daff S Oxygen Activation and Electron Transfer in Flavocytochrome P450 BM3. J. Am. Chem. Soc 2003, 125, 15010–15020. [DOI] [PubMed] [Google Scholar]
- (171).Ost TWB; Munro AW; Mowat CG; Taylor PR; Pesseguiero A; Fulco AJ; Cho AK; Cheesman MA; Walkinshaw MD; Chapman SK Structural and Spectroscopic Analysis of the F393H Mutant of Flavocytochrome P450 BM3. Biochemistry 2001, 40, 13430–13438. [DOI] [PubMed] [Google Scholar]
- (172).Behrendorff JBYH; Huang W; Gillam EMJ Directed Evolution of Cytochrome P450 Enzymes for Biocatalysis: Exploiting the Catalytic Versatility of Enzymes with Relaxed Substrate Specificity. Biochem. J 2015, 467, 1–15. [DOI] [PubMed] [Google Scholar]
- (173).Arnold FH Design by Directed Evolution. Acc. Chem. Res 1998, 31, 125–131. [Google Scholar]
- (174).Glieder A; Farinas ET; Arnold FH Laboratory Evolution of a Soluble, Self-Sufficient, Highly Active Alkane Hydroxylase. Nat. Biotechnol 2002, 20, 1135–1139. [DOI] [PubMed] [Google Scholar]
- (175).Peters MW; Meinhold P; Glieder A; Arnold FH Regio- and Enantioselective Alkane Hydroxylation with Engineered Cytochromes P450 BM-3. J. Am. Chem. Soc 2003, 125, 13442–13450. [DOI] [PubMed] [Google Scholar]
- (176).Farinas ET; Alcalde M; Arnold F Alkene Epoxidation Catalyzed by Cytochrome P450 BM-3 139–3. Tetrahedron 2004, 60, 525–528. [DOI] [PubMed] [Google Scholar]
- (177).Li QS; Schwaneberg U; Fischer P; Schmid RD Directed Evolution of the Fatty-Acid Hydroxylase P450 BM-3 into an Indole-Hydroxylating Catalyst. Chemistry 2000, 6, 1531–1536. [DOI] [PubMed] [Google Scholar]
- (178).Quéméneur E; Moutiez M; Charbonnier J-B; Menez A Engineering Cyclophilin into a Proline-Specific Endopeptidase. Nature 1998, 391, 301–304. [DOI] [PubMed] [Google Scholar]
- (179).Appel D; Lutz-Wahl S; Fischer P; Schwaneberg U; Schmid RD A P450 BM-3 Mutant Hydroxylates Alkanes, Cycloalkanes, Arenes and Heteroarenes. J. Biotechnol 2001, 88, 167–171. [DOI] [PubMed] [Google Scholar]
- (180).Whitehouse CJC; Bell SG; Wong L-L P450BM3 (CYP102A1): Connecting the Dots. Chem. Soc. Rev 2012, 41, 1218–1260. [DOI] [PubMed] [Google Scholar]
- (181).Kaur P; Tyagi V Recent Advances in Iron-Catalyzed Chemical and Enzymatic Carbene-Transfer Reactions. Adv. Synth. Catal 2021, 363, 877–905. [Google Scholar]
- (182).Coin G; Latour J-M Nitrene Transfers Mediated by Natural and Artificial Iron Enzymes. J. Inorg. Biochem 2021, 225, 111613. [DOI] [PubMed] [Google Scholar]
- (183).Wei Y; Tinoco A; Steck V; Fasan R; Zhang Y Cyclopropanations via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution. J. Am. Chem. Soc 2018, 140, 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Su H; Ma G; Liu Y Theoretical Insights into the Mechanism and Stereoselectivity of Olefin Cyclopropanation Catalyzed by Two Engineered Cytochrome P450 Enzymes. Inorg. Chem 2018, 57, 11738–11745. [DOI] [PubMed] [Google Scholar]
- (185).Hammer SC; Knight AM; Arnold FH Design and Evolution of Enzymes for Non-Natural Chemistry. Curr. Opin Green Sustainable Chem 2017, 7, 23–30. [Google Scholar]
- (186).Yang Y; Arnold FH Navigating the Unnatural Reaction Space: Directed Evolution of Heme Proteins for Selective Carbene and Nitrene Transfer. Acc. Chem. Res 2021, 54, 1209–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Miller DC; Athavale SV; Arnold FH Combining Chemistry and Protein Engineering for New-to-Nature Biocatalysis. Nat. Synth 2022, 1, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Batista VF; Pinto DCGA; Silva AMS Iron: A Worthy Contender in Metal Carbene Chemistry. ACS Catal. 2020, 10, 10096–10116. [Google Scholar]
- (189).Geronimo I; Denning CA; Rogers WE; Othman T; Huxford T; Heidary DK; Glazer EC; Payne CM Effect of Mutation and Substrate Binding on the Stability of Cytochrome P450BM3 Variants. Biochemistry 2016, 55, 3594–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Denisov IG; Makris TM; Sligar SG; Schlichting I Structure and Chemistry of Cytochrome P450. Chem. Rev 2005, 105, 2253–2278. [DOI] [PubMed] [Google Scholar]
- (191).Shaik S; Kumar D; de Visser SP; Altun A; Thiel W Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev 2005, 105, 2279–2328. [DOI] [PubMed] [Google Scholar]
- (192).Luthra A; Denisov IG; Sligar SG Spectroscopic Features of Cytochrome P450 Reaction Intermediates. Arch. Biochem. Biophys 2011, 507, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Gober JG; Ghodge SV; Bogart JW; Wever WJ; Watkins RR; Brustad EM; Bowers AA P450-Mediated Non-Natural Cyclopropanation of Dehydroalanine-Containing Thiopeptides. ACS Chem. Biol 2017, 12, 1726–1731. [DOI] [PubMed] [Google Scholar]
- (194).Wang ZJ; Renata H; Peck NE; Farwell CC; Coelho PS; Arnold FH Improved Cyclopropanation Activity of Histidine-Ligated Cytochrome P450 Enables the Enantioselective Formal Synthesis of Levomilnacipran. Angew. Chem., Int. Ed 2014, 53, 6810–6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Brandenberg OF; Prier CK; Chen K; Knight AM; Wu Z; Arnold FH Stereoselective Enzymatic Synthesis of Heteroatom-Substituted Cyclopropanes. ACS Catal. 2018, 8, 2629–2634. [Google Scholar]
- (196).Wang ZJ; Peck NE; Renata H; Arnold FH Cytochrome P450-Catalyzed Insertion of Carbenoids into N-H Bonds. Chem. Sci 2014, 5, 598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Knight AM; Kan SBJ; Lewis RD; Brandenberg OF; Chen K; Arnold FH Diverse Engineered Heme Proteins Enable Stereodivergent Cyclopropanation of Unactivated Alkenes. ACS Cent. Sci 2018, 4, 372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Brandenberg OF; Chen K; Arnold FH Directed Evolution of a Cytochrome P450 Carbene Transferase for Selective Functionalization of Cyclic Compounds. J. Am. Chem. Soc 2019, 141, 8989–8995. [DOI] [PubMed] [Google Scholar]
- (199).Zhang RK; Chen K; Huang X; Wohlschlager L; Renata H; Arnold FH Enzymatic Assembly of Carbon-Carbon Bonds via Iron-Catalysed sp3 C-H Functionalization. Nature 2019, 565, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Zhang J; Huang X; Zhang RK; Arnold FH Enantiodivergent α-Amino C-H Fluoroalkylation Catalyzed by Engineered Cytochrome P450s. J. Am. Chem. Soc 2019, 141, 9798–9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Chen K; Huang X; Kan SBJ; Zhang RK; Arnold FH Enzymatic Construction of Highly Strained Carbocycles. Science 2018, 360, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Chen K; Arnold FH Engineering Cytochrome P450s for Enantioselective Cyclopropenation of Internal Alkynes. J. Am. Chem. Soc 2020, 142, 6891–6895. [DOI] [PubMed] [Google Scholar]
- (203).Chen K; Zhang S-Q; Brandenberg OF; Hong X; Arnold FH Alternate Heme Ligation Steers Activity and Selectivity in Engineered Cytochrome P450-Catalyzed Carbene-Transfer Reactions. J. Am. Chem. Soc 2018, 140, 16402–16407. [DOI] [PubMed] [Google Scholar]
- (204).Zhou AZ; Chen K; Arnold FH Enzymatic Lactone-Carbene C-H Insertion to Build Contiguous Chiral Centers. ACS Catal. 2020, 10, 5393–5398. [Google Scholar]
- (205).Hyster TK; Farwell CC; Buller AR; McIntosh JA; Arnold FH Enzyme-Controlled Nitrogen-Atom Transfer Enables Regiodivergent C-H Amination. J. Am. Chem. Soc 2014, 136, 15505–15508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Singh R; Kolev JN; Sutera PA; Fasan R Enzymatic C(sp3)-H Amination: P450-Catalyzed Conversion of Carbonazidates into Oxazolidinones. ACS Catal. 2015, 5, 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Farwell CC; McIntosh JA; Hyster TK; Wang ZJ; Arnold FH Enantioselective Imidation of Sulfides via Enzyme-Catalyzed Intermolecular Nitrogen-Atom Transfer. J. Am. Chem. Soc 2014, 136, 8766–8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Prier CK; Hyster TK; Farwell CC; Huang A; Arnold FH Asymmetric Enzymatic Synthesis of Allylic Amines: A Sigmatropic Rearrangement Strategy. Angew. Chem., Int. Ed 2016, 128, 4789–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Farwell CC; Zhang RK; McIntosh JA; Hyster TK; Arnold FH Enantioselective Enzyme-Catalyzed Aziridination Enabled by Active-Site Evolution of a Cytochrome P450. ACS Cent. Sci 2015, 1, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Prier CK; Zhang RK; Buller AR; Brinkmann-Chen S; Arnold FH Enantioselective, Intermolecular Benzylic C-H Amination Catalysed by an Engineered Iron-Haem Enzyme. Nat. Chem 2017, 9, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Brandenberg OF; Miller DC; Markel U; Ouald Chaib A; Arnold FH Engineering Chemoselectivity in Hemoprotein-Catalyzed Indole Amidation. ACS Catal. 2019, 9, 8271–8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Jia Z-J; Gao S; Arnold FH Enzymatic Primary Amination of Benzylic and Allylic C(sp3)-H Bonds. J. Am. Chem. Soc 2020, 142, 10279–10283. [DOI] [PubMed] [Google Scholar]
- (213).Cho I; Prier CK; Jia Z-J; Zhang RK; Görbe T; Arnold FH Enantioselective Aminohydroxylation of Styrenyl Olefins Catalyzed by an Engineered Hemoprotein. Angew. Chem., Int. Ed 2019, 58, 3138–3142. [DOI] [PubMed] [Google Scholar]
- (214).Tsutsumi H; Katsuyama Y; Izumikawa M; Takagi M; Fujie M; Satoh N; Shin-ya K; Ohnishi Y Unprecedented Cyclization Catalyzed by a Cytochrome P450 in Benzastatin Biosynthesis. J. Am. Chem. Soc 2018, 140, 6631–6639. [DOI] [PubMed] [Google Scholar]
- (215).Legnani L; Morandi B Direct Catalytic Synthesis of Unprotected 2-Amino-1-Phenylethanols from Alkenes by Using Iron(II) Phthalocyanine. Angew. Chem., Int. Ed 2016, 55, 2248–2251. [DOI] [PubMed] [Google Scholar]
- (216).Athavale SV; Gao S; Liu Z; Mallojjala SC; Hirschi JS; Arnold FH Biocatalytic, Intermolecular C-H Bond Functionalization for the Synthesis of Enantioenriched Amides. Angew. Chem., Int. Ed 2021, 60, 24864–24869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Renata H; Lewis RD; Sweredoski MJ; Moradian A; Hess S; Wang ZJ; Arnold FH Identification of Mechanism-Based Inactivation in P450-Catalyzed Cyclopropanation Facilitates Engineering of Improved Enzymes. J. Am. Chem. Soc 2016, 138, 12527–12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Brandenberg OF; Fasan R; Arnold FH Exploiting and Engineering Hemoproteins for Abiological Carbene and Nitrene Transfer Reactions. Curr. Opin. Biotechnol 2017, 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Wan L; Twitchett MB; Eltis LD; Mauk AG; Smith M In Vitro Evolution of Horse Heart Myoglobin to Increase Peroxidase Activity. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 12825–12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Chandgude AL; Ren X; Fasan R Stereodivergent Intramolecular Cyclopropanation Enabled by Engineered Carbene Transferases. J. Am. Chem. Soc 2019, 141, 9145–9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Ren X; Liu N; Chandgude AL; Fasan R An Enzymatic Platform for the Highly Enantioselective and Stereodivergent Construction of Cyclopropyl-δ-Lactones. Angew. Chem., Int. Ed 2020, 59, 21634–21639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Ren X; Chandgude AL; Fasan R Highly Stereoselective Synthesis of Fused Cyclopropane-γ-Lactams via Biocatalytic Iron-Catalyzed Intramolecular Cyclopropanation. ACS Catal. 2020, 10, 2308–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Hernandez KE; Renata H; Lewis RD; Kan SBJ; Zhang C; Forte J; Rozzell D; McIntosh JA; Arnold FH Highly Stereoselective Biocatalytic Synthesis of Key Cyclopropane Inter-mediate to Ticagrelor. ACS Catal. 2016, 6, 7810–7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Moore EJ; Zorine D; Hansen WA; Khare SD; Fasan R Enzyme Stabilization via Computationally Guided Protein Stapling. Proc. Natl. Acad. Sci. U.S.A 2017, 114, 12472–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Kan SBJ; Lewis RD; Chen K; Arnold FH Directed Evolution of Cytochrome c for Carbon-Silicon Bond Formation: Bringing Silicon to Life. Science 2016, 354, 1048–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Lewis RD; Garcia-Borràs M; Chalkley MJ; Buller AR; Houk KN; Kan SBJ; Arnold FH Catalytic Iron-Carbene Intermediate Revealed in a Cytochrome c Carbene Transferase. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 7308–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Kan SBJ; Huang X; Gumulya Y; Chen K; Arnold FH Genetically Programmed Chiral Organoborane Synthesis. Nature 2017, 552, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Chen K; Huang X; Zhang S-Q; Zhou AZ; Kan SBJ; Hong X; Arnold FH Engineered Cytochrome c-Catalyzed Lactone-Carbene B-H Insertion. Synlett. 2019, 30, 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Huang X; Garcia-Borràs M; Miao K; Kan SBJ; Zutshi A; Houk KN; Arnold FH A Biocatalytic Platform for Synthesis of Chiral α-Trifluoromethylated Organoborons. ACS Cent. Sci 2019, 5, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Stelter M; Melo AMP; Pereira MM; Gomes CM; Hreggvidsson GO; Hjorleifsdottir S; Saraiva LM; Teixeira M; Archer M A Novel Type of Monoheme Cytochrome c: Biochemical and Structural Characterization at 1.23 Å Resolution of Rhodothermus marinus Cytochrome c. Biochemistry 2008, 47, 11953–11963. [DOI] [PubMed] [Google Scholar]
- (231).Garcia-Borràs M; Kan SBJ; Lewis RD; Tang A; Jimenez-Osés G; Arnold FH; Houk KN Origin and Control of Chemoselectivity in Cytochrome c Catalyzed Carbene Transfer into Si-H and N-H Bonds. J. Am. Chem. Soc 2021, 143, 7114–7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Cech TR; Zaug AJ; Grabowski PJ In Vitro Splicing of the Ribosomal RNA Precursor of Tetrahymena: Involvement of a Guanosine Nucleotide in the Excision of the Intervening Sequence. Cell 1981, 27, 487–496. [DOI] [PubMed] [Google Scholar]
- (233).Breaker RR; Joyce GF A DNA Enzyme That Cleaves RNA. Chem. Biol 1994, 1, 223–229. [DOI] [PubMed] [Google Scholar]
- (234).Altman S The Road to RNase P. Nat. Struct. Biol 2000, 7, 827–828. [DOI] [PubMed] [Google Scholar]
- (235).Li Y; Geyer R; Sen D Recognition of Anionic Porphyrins by DNA Aptamers. Biochemistry 1996, 35, 6911–6922. [DOI] [PubMed] [Google Scholar]
- (236).Travascio P; Li Y; Sen D DNA-Enhanced Peroxidase Activity of a DNA Aptamer-Hemin Complex. Chem. Biol 1998, 5, 505–517. [DOI] [PubMed] [Google Scholar]
- (237).Travascio P; Witting PK; Mauk AG; Sen D The Peroxidase Activity of a Hemin-DNA Oligonucleotide Complex: Free Radical Damage to Specific Guanine Bases of the DNA. J. Am. Chem. Soc 2001, 123, 1337–1348. [DOI] [PubMed] [Google Scholar]
- (238).Mikuma T; Terui N; Yamamoto Y; Hori H A Novel Heme-DNA Coordination Complex and Its Stability. Nucleic Acids Symp. Ser 2002, 2, 285–286. [DOI] [PubMed] [Google Scholar]
- (239).Sen D; Gilbert W Formation of Parallel Four-Stranded Complexes by Guanine-Rich Motifs in DNA and Its Implications for Meiosis. Nature 1988, 334, 364–366. [DOI] [PubMed] [Google Scholar]
- (240).Golub E; Freeman R; Willner I A Hemin/G-Quadruplex Acts as an NADDH Oxidase and NADH Peroxidase Mimicking DNAzyme. Angew. Chem., Int. Ed 2011, 50, 11710–11714. [DOI] [PubMed] [Google Scholar]
- (241).Ohyama T; Kato Y; Mita H; Nagatomo S; Yamamoto Y Structural and Functional Characterization of Novel G-Quadruplexed DNA-Heme Coordination Complex. Nucleic Acids Symp. Ser 2005, 49, 245–246. [DOI] [PubMed] [Google Scholar]
- (242).Saito K; Nakano Y; Tai H; Nagatomo S; Hemmi H; Mita H; Yamamoto Y Characterization of Heme Coordination Structure in Heme-DNA Complex Possessing Gaseous Molecule as an Exogenous Ligand. Nucleic Acids Symp. Ser 2009, 53, 241–242. [DOI] [PubMed] [Google Scholar]
- (243).Poon LC-H; Methot SP; Morabi-Pazooki W; Pio F; Bennet AJ; Sen D Guanine-Rich RNAs and DNAs That Bind Heme Robustly Catalyze Oxygen Transfer Reactions. J. Am. Chem. Soc 2011, 133, 1877–1884. [DOI] [PubMed] [Google Scholar]
- (244).Pratviel G Porphyrins in Complex with DNA: Modes of Interaction and Oxidation Reactions. Coord. Chem. Rev 2016, 308, 460–477. [Google Scholar]
- (245).Saito K; Tai H; Fukaya M; Shibata T; Nishimura R; Neya S; Yamamoto Y Structural Characterization of a Carbon Monoxide Adduct of a Heme-DNA Complex. JBIC, J. Biol. Inorg. Chem 2012, 17, 437–445. [DOI] [PubMed] [Google Scholar]
- (246).Shibata T; Nakayama Y; Katahira Y; Tai H; Moritaka Y; Nakano Y; Yamamoto Y Characterization of the Interaction Between Heme and a Parallel G-Quadruplex DNA Formed from d(TTGAGG). Biochim. Biophys. Acta, General Subj 2017, 1861, 1264–1270. [DOI] [PubMed] [Google Scholar]
- (247).Yamamoto Y; Kinoshita M; Katahira Y; Shimizu H; Di Y; Shibata T; Tai H; Suzuki A; Neya S Characterization of Heme-DNA Complexes Composed of Some Chemically Modified Hemes and Parallel G-Quadruplex DNAs. Biochemistry 2015, 54, 7168–7177. [DOI] [PubMed] [Google Scholar]
- (248).Shinomiya R; Katahira Y; Araki H; Shibata T; Momotake A; Yanagisawa S; Ogura T; Suzuki A; Neya S; Yamamoto Y Characterization of Catalytic Activities and Heme Coordination Structures of Heme-DNA Complexes Composed of Some Chemically Modified Hemes and an All Parallel-Stranded Tetrameric G-Quadruplex DNA Formed from d(TTAGGG). Biochemistry 2018, 57, 5930–5937. [DOI] [PubMed] [Google Scholar]
- (249).Ibrahim H; Mulyk P; Sen D DNA G-Quadruplexes Activate Heme for Robust Catalysis of Carbene Transfer Reactions. ACS Omega 2019, 4, 15280–15288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Miron CE; Staalduinen L; Rangaswamy AM; Chen M; Liang Y; Jia Z; Mergny J; Petitjean A Going Platinum to the Tune of a Remarkable Guanine Quadruplex Binder: Solution- and Solid-State Investigations. Angew. Chem., Int. Ed 2021, 60, 2500–2507. [DOI] [PubMed] [Google Scholar]
- (251).Feig AL; Lippard SJ Reactions of Non-Heme Iron(II) Centers with Dioxygen in Biology and Chemistry. Chem. Rev 1994, 94, 759–805. [Google Scholar]
- (252).Wallar BJ; Lipscomb JD Dioxygen Activation by Enzymes Containing Binuclear Non-Heme Iron Clusters. Chem. Rev 1996, 96, 2625–2658. [DOI] [PubMed] [Google Scholar]
- (253).Ferousi C; Majer SH; DiMucci IM; Lancaster KM Biological and Bioinspired Inorganic N-N Bond-Forming Reactions. Chem. Rev 2020, 120, 5252–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Khatua S; Majumdar A Flavodiiron Nitric Oxide Reductases: Recent Developments in the Mechanistic Study and Model Chemistry for the Catalytic Reduction of NO. J. Inorg. Biochem 2015, 142, 145–153. [DOI] [PubMed] [Google Scholar]
- (255).Gardner AM; Helmick RA; Gardner PR Flavorubredoxin, an Inducible Catalyst for Nitric Oxide Reduction and Detoxification in Escherichia Coli. J. Biol. Chem 2002, 277, 8172–8177. [DOI] [PubMed] [Google Scholar]
- (256).Bulen WA; Burns RC; LeComte JR Nitrogen Fixation: Cell-Free System with Extracts of Azotobacter. Biochem. Biophys. Res. Commun 1964, 17, 265–271. [DOI] [PubMed] [Google Scholar]
- (257).Hegg EL; Que L Jr. The 2-His-1-Carboxylate Facial Triadan Emerging Structural Motif in Mononuclear Non-Heme Iron(II) Enzymes. Eur. J. Biochem 1997, 250, 625–629. [DOI] [PubMed] [Google Scholar]
- (258).Kal S; Que L Dioxygen Activation by Nonheme Iron Enzymes with the 2-His-1-Carboxylate Facial Triad That Generate High-Valent Oxoiron Oxidants. JBIC, J. Biol. Inorg. Chem 2017, 22, 339–365. [DOI] [PubMed] [Google Scholar]
- (259).Que L; Ho RYN Dioxygen Activation by Enzymes with Mononuclear Non-Heme Iron Active Sites. Chem. Rev 1996, 96, 2607–2624. [DOI] [PubMed] [Google Scholar]
- (260).Abu-Omar MM; Loaiza A; Hontzeas N Reaction Mechanisms of Mononuclear Non-Heme Iron Oxygenases. Chem. Rev 2005, 105, 2227–2252. [DOI] [PubMed] [Google Scholar]
- (261).Tshuva EY; Lippard SJ Synthetic Models for Non-Heme Carboxylate-Bridged Diiron Metalloproteins: Strategies and Tactics. Chem. Rev 2004, 104, 987–1012. [DOI] [PubMed] [Google Scholar]
- (262).Shima S; Pilak O; Vogt S; Schick M; Stagni MS; Meyer-Klaucke W; Warkentin E; Thauer RK; Ermler U The Crystal Structure of [Fe]-Hydrogenase Reveals the Geometry of the Active Site. Science 2008, 321, 572–575. [DOI] [PubMed] [Google Scholar]
- (263).Berggren G; Adamska A; Lambertz C; Simmons TR; Esselborn J; Atta M; Gambarelli S; Mouesca J-M; Reijerse E; Lubitz W; Happe T; Artero V; Fontecave M Biomimetic Assembly and Activation of [FeFe]-Hydrogenases. Nature 2013, 499, 66–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Lancaster KM; Roemelt M; Ettenhuber P; Hu Y; Ribbe MW; Neese F; Bergmann U; DeBeer S X-Ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334, 974–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Spatzal T; Aksoyoglu M; Zhang L; Andrade SLA; Schleicher E; Weber S; Rees DC; Einsle O Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Sippel D; Einsle O The Structure of Vanadium Nitrogenase Reveals an Unusual Bridging Ligand. Nat. Chem. Biol 2017, 13, 956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Dobbek H; Svetlitchnyi V; Gremer L; Huber R; Meyer O Crystal Structure of a Carbon Monoxide Dehydrogenase Reveals a [Ni-4Fe-5S] Cluster. Science 2001, 293, 1281–1285. [DOI] [PubMed] [Google Scholar]
- (268).Pinto AL; Hellinga HW; Caradonna JP Construction ofa Catalytically Active Iron Superoxide Dismutase by Rational Protein Design. Proc. Natl. Acad. Sci. U.S.A 1997, 94, 5562–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Katti SK; LeMaster DM; Eklund H Crystal Structure of Thioredoxin from Escherichia coli at 1.68 Å Resolution. J. Mol. Biol 1990, 212, 167–184. [DOI] [PubMed] [Google Scholar]
- (270).McLaughlin MP; Retegan M; Bill E; Payne TM; Shafaat HS; Peña S; Sudhamsu J; Ensign AA; Crane BR; Neese F; Holland PL Azurin as a Protein Scaffold for a Low-Coordinate Nonheme Iron Site with a Small-Molecule Binding Pocket. J. Am. Chem. Soc 2012, 134, 19746–19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Liu J; Meier KK; Tian S; Zhang J; Guo H; Schulz CE; Robinson H; Nilges MJ; Münck E; Lu Y Redesigning the Blue Copper Azurin into a Redox-Active Mononuclear Nonheme Iron Protein: Preparation and Study of Fe(II)-M121E Azurin. J. Am. Chem. Soc 2014, 136, 12337–12344. [DOI] [PubMed] [Google Scholar]
- (272).Lin S; He C Development ofNonheme {FeNO}7 Complexes Based on the Pyrococcus furiosus Rubredoxin for Red-Light-Control-lable Nitric Oxide Release. Inorg. Chem 2021, 60, 14364–14370. [DOI] [PubMed] [Google Scholar]
- (273).Beauchamp C; Fridovich I Superoxide Dismutase: Improved Assays and an Assay Applicable to Acrylamide Gels. Anal. Biochem 1971, 44, 276–287. [DOI] [PubMed] [Google Scholar]
- (274).Tian S; Fan R; Albert T; Khade RL; Dai H; Harnden K ; Hosseinzadeh P; Liu J; Nilges MJ; Zhang Y; Moënne-Loccoz P; Guo Y; Lu Y Stepwise Nitrosylation of the Nonheme Iron Site in an Engineered Azurin and a Molecular Basis for Nitric Oxide Signaling Mediated by Nonheme Iron Proteins. Chem. Sci 2021, 12, 6569–6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Enemark JH; Feltham RD Principles of Structure, Bonding, and Reactivity for Metal Nitrosyl Complexes. Coord. Chem. Rev 1974, 13, 339–406. [Google Scholar]
- (276).Tian S; Liu J; Cowley RE; Hosseinzadeh P; Marshall NM; Yu Y; Robinson H; Nilges MJ; Blackburn NJ; Solomon EI; et al. Reversible S-Nitrosylation in an Engineered Azurin. Nat. Chem 2016, 8, 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Kurihara K; Tanaka I; Chatake T; Adams MWW; Jenney FE; Moiseeva N; Bau R; Niimura N Neutron Crystallographic Study on Rubredoxin from Pyrococcus Furiosus by BIX-3, a Single-Crystal Diffractometer for Biomacromolecules. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 11215–11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Cross M; Xiao Z; Maes EM; Czernuszewicz RS; Drew SC; Pilbrow JR; George GN; Wedd AG Removal of a Cysteine Ligand from Rubredoxin: Assembly of Fe2S2 and Fe(S-Cys)3(OH) Centres. JBIC, J. Biol. Inorg. Chem 2002, 7, 781–790. [DOI] [PubMed] [Google Scholar]
- (279).Xie J; Liu W; Schultz PG A Genetically Encoded Bidentate, Metal-Binding Amino Acid. Angew. Chem., Int. Ed 2007, 46, 9239–9242. [DOI] [PubMed] [Google Scholar]
- (280).Lee HS; Schultz PG Biosynthesis of a Site-Specific DNA Cleaving Protein. J. Am. Chem. Soc 2008, 130, 13194–13195. [DOI] [PubMed] [Google Scholar]
- (281).Schultz PG; Dervan PB Sequence-Specific Double-Strand Cleavage of DNA by Penta-N-Methylpyrrolecarboxamide-EDTA X Fe(II). Proc. Natl. Acad. Sci. U.S.A 1983, 80, 6834–6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Sluka JP; Horvath SJ; Bruist MF; Simon MI; Dervan PB Synthesis of a Sequence-Specific DNA-Cleaving Peptide. Science 1987, 238, 1129–1132. [DOI] [PubMed] [Google Scholar]
- (283).Schultz SC; Shields GC; Steitz TA Crystal Structure of a CAP-DNA Complex: The DNA Is Bent by 90 Degrees. Science 1991, 253, 1001–1007. [DOI] [PubMed] [Google Scholar]
- (284).Parkinson G; Wilson C; Gunasekera A; Ebright YW; Ebright RE; Berman HM Structure of the CAP-DNA Complex at 2.5 Å Resolution: A Complete Picture of the Protein-DNA Interface. J. Mol. Biol 1996, 260, 395–408. [DOI] [PubMed] [Google Scholar]
- (285).Kang M; Light K; Ai H; Shen W; Kim CH; Chen PR; Lee HS; Solomon EI; Schultz PG Evolution of Iron(II)-Finger Peptides by Using a Bipyridyl Amino Acid. ChemBioChem 2014, 15, 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Ségaud N; Drienovská I; Chen J; Browne WR; Roelfes G Artificial Metalloproteins for Binding and Stabilization of a Semi-quinone Radical. Inorg. Chem 2017, 56, 13293–13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Madoori PK; Agustiandari H; Driessen AJM; Thunnissen A-MWH Structure of the Transcriptional Regulator LmrR and Its Mechanism of Multidrug Recognition. EMBO J. 2009, 28, 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Pavletich NP; Pabo CO Zinc Finger-DNA Recognition: Crystal Structure of a Zif268-DNA Complex at 2.1 Å. Science 1991, 252, 809–817. [DOI] [PubMed] [Google Scholar]
- (289).Mills JH; Khare SD; Bolduc JM; Forouhar F; Mulligan VK; Lew S; Seetharaman J; Tong L; Stoddard BL; Baker D Computational Design of an Unnatural Amino Acid Dependent Metalloprotein with Atomic Level Accuracy. J. Am. Chem. Soc 2013, 135, 13393–13399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Kuhlman B; Baker D Native Protein Sequences Are Close to Optimal for Their Structures. Proc. Natl. Acad. Sci. U.S.A 2000, 97, 10383–10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Zanghellini A; Jiang L; Wollacott AM; Cheng G; Meiler J; Althoff EA; Röthlisberger D; Baker D New Algorithms and an In Silico Benchmark for Computational Enzyme Design. Protein Sci. 2006, 15, 2785–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Davies RR; Distefano MD A Semisynthetic Metalloenzyme Based on a Protein Cavity That Catalyzes the Enantioselective Hydrolysis of Ester and Amide Substrates. J. Am. Chem. Soc 1997, 119, 11643–11652. [Google Scholar]
- (293).Ory JJ; Mazhary A; Kuang H; Davies RR; Distefano MD; Banaszak LJ Structural Characterization of Two Synthetic Catalysts Based on Adipocyte Lipid-Binding Protein. Protein Eng. Des. Sel 1998, 11, 253–261. [DOI] [PubMed] [Google Scholar]
- (294).Davies RR; Kuang H; Qi D; Mazhary A; Mayaan E; Distefano MD Artificial Metalloenzymes Based on Protein Cavities: Exploring the Effect ofAltering the Metal Ligand Attachment Position by Site Directed Mutagenesis. Bioorg. Med. Chem. Lett 1999, 9, 79–84. [DOI] [PubMed] [Google Scholar]
- (295).Qi D; Kuang H; Distefano MD Effects of Metal Ions on the Rates and Enantioselectivities of Reactions Catalyzed by a Series of Semisynthetic Transaminases Created by Site Directed Mutagenesis. Bioorg. Med. Chem. Lett 1998, 8, 875–880. [DOI] [PubMed] [Google Scholar]
- (296).Buron C; Sénéchal-David K; Ricoux R; Le Caër J-P; Guérineau V; Méjanelle P; Guillot R; Herrero C; Mahy J-P; Banse F An Artificial Enzyme Made by Covalent Grafting of an FeII Complex into β-Lactoglobulin: Molecular Chemistry, Oxidation Catalysis, and Reaction-Intermediate Monitoring in a Protein. Chem. Eur. J 2015, 21, 12188–12193. [DOI] [PubMed] [Google Scholar]
- (297).Loch JI; Bonarek P; Polit A; Świątek S; Dziedzicka-Wasylewska M; Lewiński K The Differences in Binding 12-Carbon Aliphatic Ligands by Bovine β-Lactoglobulin Isoform A and B Studied by Isothermal Titration Calorimetry and X-Ray Crystallography. J. Mol. Recog 2013, 26, 357–367. [DOI] [PubMed] [Google Scholar]
- (298).Doble MV; Obrecht L; Joosten H-J; Lee M; Rozeboom HJ; Branigan E; Naismith JH; Janssen DB; Jarvis AG; et al. Engineering Thermostability in Artificial Metalloenzymes to Increase Catalytic Activity. ACS Catal. 2021, 11, 3620–3627. [Google Scholar]
- (299).Doble MV; Jarvis AG; Ward ACC; Colburn JD; Gotze JP; Buhl M; Kamer PCJ Artificial Metalloenzymes as Catalysts for Oxidative Lignin Degradation. ACS Sustainable Chem. Eng 2018, 6, 15100–15107. [Google Scholar]
- (300).Ducros V; Charnock SJ; Derewenda U; Derewenda ZS; Dauter Z; Dupont C; Shareck F; Morosoli R; Kluepfel D; Davies GJ Substrate Specificity in Glycoside Hydrolase Family 10. Structural and Kinetic Analysis of the Streptomyces Lividans Xylanase 10A. J. Biol. Chem 2000, 275, 23020–23026. [DOI] [PubMed] [Google Scholar]
- (301).Kariyawasam K; Ghattas W; De Los Santos YL; Doucet N; Gaillard S; Renaud J-L; Avenier F; Mahy J-P; Ricoux R Artificial Iron Hydrogenase Made by Covalent Grafting of Knölker’s Complex into Xylanase: Application in Asymmetric Hydrogenation of an Aryl Ketone in Water. Biotechnol. Appl. Biochem 2020, 67, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Cavazza C; Bochot C; Rousselot-Pailley P; Carpentier P; Cherrier MV; Martin L; Marchi-Delapierre C; Fontecilla-Camps JC; Menage S Crystallographic Snapshots of the Reaction of Aromatic C-H with O2 Catalysed by a Protein-Bound Iron Complex. Nat. Chem 2010, 2, 1069–1076. [DOI] [PubMed] [Google Scholar]
- (303).de Pina K; Navarro C; McWalter L; Boxer DH; Price NC; Kelly SM; Mandrand-Berthelot MA; Wu LF Purification and Characterization of the Periplasmic Nickel-Binding Protein NikA of Escherichia Coli K12. Eur. J. Biochem 1995, 227, 857–865. [DOI] [PubMed] [Google Scholar]
- (304).Cherrier MV; Martin L; Cavazza C; Jacquamet L; Lemaire D; Gaillard J; Fontecilla-Camps JC Crystallographic and Spectroscopic Evidence for High Affinity Binding of FeEDTA(H2O)− to the Periplasmic Nickel Transporter NikA. J. Am. Chem. Soc 2005, 127, 10075–10082. [DOI] [PubMed] [Google Scholar]
- (305).Lopez S; Rondot L; Leprêtre C; Marchi-Delapierre C; Ménage S; Cavazza C Cross-Linked Artificial Enzyme Crystals as Heterogeneous Catalysts for Oxidation Reactions. J. Am. Chem. Soc 2017, 139, 17994–18002. [DOI] [PubMed] [Google Scholar]
- (306).Rondot L; Girgenti E; Oddon F; Marchi-Delapierre C; Jorge-Robin A; Ménage S Catalysis without a Headache: Modification of Ibuprofen for the Design of Artificial Metalloenzyme for Sulfide Oxidation. J. Mol. Cat. A: Chem 2016, 416, 20–28. [Google Scholar]
- (307).Ghuman J; Zunszain PA; Petitpas I; Bhattacharya AA; Otagiri M; Curry S Structural Basis of the Drug-Binding Specificity of Human Serum Albumin. J. Mol. Biol 2005, 353, 38–52. [DOI] [PubMed] [Google Scholar]
- (308).Cherrier MV; Girgenti E; Amara P; Iannello M; Marchi-Delapierre C; Fontecilla-Camps JC; Ménage S; Cavazza C The Structure of the Periplasmic Nickel-Binding Protein NikA Provides Insights for Artificial Metalloenzyme Design. JBIC, J. Biol. Inorg. Chem 2012, 17, 817–829. [DOI] [PubMed] [Google Scholar]
- (309).Esmieu C; Cherrier MV; Amara P; Girgenti E; Marchi-Delapierre C; Oddon F; Iannello M; Jorge-Robin A; Cavazza C; Ménage S An Artificial Oxygenase Built from Scratch: Substrate Binding Site Identified Using a Docking Approach. Angew. Chem., Int. Ed 2013, 52, 3922–3925. [DOI] [PubMed] [Google Scholar]
- (310).Lopez S; Marchi-Delapierre C; Cavazza C; Ménage S A Selective Sulfide Oxidation Catalyzed by Heterogeneous Artificial Metalloenzymes Iron@NikA. Chem. Eur. J 2020, 26, 16633–16638. [DOI] [PubMed] [Google Scholar]
- (311).Mérel DS; Gaillard S; Ward TR; Renaud J-L Achiral Cyclopentadienone Iron Tricarbonyl Complexes Embedded in Streptavidin: An Access to Artificial Iron Hydrogenases and Application in Asymmetric Hydrogenation. Catal. Lett 2016, 146, 564–569. [Google Scholar]
- (312).Serrano-Plana J; Rumo C; Rebelein JG; Peterson RL; Barnet M; Ward TR Enantioselective Hydroxylation of Benzylic C(sp3)-H Bonds by an Artificial Iron Hydroxylase Based on the Biotin-Streptavidin Technology. J. Am. Chem. Soc 2020, 142, 10617–10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (313).Miller KR; Paretsky JD; Follmer AH; Heinisch T; Mittra K; Gul S; Kim I-S; Fuller FD; Batyuk A; Sutherlin KD; et al. Artificial Iron Proteins: Modeling the Active Sites in Non-Heme Dioxygenases. Inorg. Chem 2020, 59, 6000–6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (314).Betz SF; Liebman PA; DeGrado WF De Novo Design of Native Proteins: Characterization of Proteins Intended To Fold into Antiparallel, Rop-like, Four-Helix Bundles. Biochemistry 1997, 36, 2450–2458. [DOI] [PubMed] [Google Scholar]
- (315).Kaplan J; DeGrado WF De Novo Design of Catalytic Proteins. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 11566–11570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Faiella M; Andreozzi C; de Rosales RTM; Pavone V; Maglio O; Nastri F; DeGrado WF; Lombardi A An Artificial Di-Iron Oxo-Protein with Phenol Oxidase Activity. Nat. Chem. Biol 2009, 5, 882–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Miller KR; Biswas S; Jasniewski A; Follmer AH; Biswas A; Albert T; Sabuncu S; Bominaar EL; Hendrich MP; Moënne-Loccoz P; et al. Artificial Metalloproteins with Dinuclear Iron-Hydroxido Centers. J. Am. Chem. Soc 2021, 143, 2384–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (318).Que L Metalloproteins with Phenolate Coordination. Coord. Chem. Rev 1983, 50, 73–108. [Google Scholar]
- (319).Jones AK; Lichtenstein BR; Dutta A; Gordon G; Dutton PL Synthetic Hydrogenases: Incorporation of an Iron Carbonyl Thiolate into a Designed Peptide. J. Am. Chem. Soc 2007, 129, 14844–14845. [DOI] [PubMed] [Google Scholar]
- (320).Sano Y; Onoda A; Hayashi T A Hydrogenase Model System Based on the Sequence of Cytochrome c: Photochemical Hydrogen Evolution in Aqueous Media. Chem. Commun 2011, 47, 8229–8231. [DOI] [PubMed] [Google Scholar]
- (321).Sano Y; Onoda A; Hayashi T Photocatalytic Hydrogen Evolution by a Diiron Hydrogenase Model Based on a Peptide Fragment of Cytochrome c556 with an Attached Diiron Carbonyl Cluster and an Attached Ruthenium Photosensitizer. J. Inorg. Biochem 2012, 108, 159–162. [DOI] [PubMed] [Google Scholar]
- (322).Roy A; Madden C; Ghirlanda G Photo-Induced Hydrogen Production in a Helical Peptide Incorporating a [FeFe] Hydrogenase Active Site Mimic. Chem. Commun 2012, 48, 9816–9818. [DOI] [PubMed] [Google Scholar]
- (323).Esmieu C; Guo M; Redman HJ; Lundberg M; Berggren G Synthesis of a Miniaturized [FeFe] Hydrogenase Model System. Dalton Trans. 2019, 48, 2280–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Onoda A; Kihara Y; Fukumoto K; Sano Y; Hayashi T Photoinduced Hydrogen Evolution Catalyzed by a Synthetic Diiron Dithiolate Complex Embedded within a Protein Matrix. ACS Catal. 2014, 4, 2645–2648. [Google Scholar]
- (325).Roy A; Vaughn MD; Tomlin J; Booher GJ; Kodis G; Simmons CR; Allen JP; Ghirlanda G Enhanced Photocatalytic Hydrogen Production by Hybrid Streptavidin-Diiron Catalysts. Chemistry 2020, 26, 6240–6246. [DOI] [PubMed] [Google Scholar]
- (326).Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (327).Oka T; Simpson FJ Quercetinase, a Dioxygenase Containing Copper. Biochem. Biophys. Res. Commun 1971, 43, 1–5. [DOI] [PubMed] [Google Scholar]
- (328).Crabbe MJ; Waight RD; Bardsley WG; Barker RW; Kelly ID; Knowles PF Human Placental Diamine Oxidase. Improved Purification and Characterization of a Copper- and Manganese-Containing Amine Oxidase with Novel Substrate Specificity. Biochem. J 1976, 155, 679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Kosman DJ; Ettinger MJ; Weiner RE; Massaro EJ The Molecular Properties of the Copper Enzyme Galactose Oxidase. Arch. Biochem. Biophys 1974, 165, 456–467. [DOI] [PubMed] [Google Scholar]
- (330).Makino N; McMahill P; Mason HS; Moss TH The Oxidation State of Copper in Resting Tyrosinase. J. Biol. Chem 1974, 249, 6062–6066. [PubMed] [Google Scholar]
- (331).Beinert H Copper A of Cytochrome c Oxidase, A Novel, Long-Embattled, Biological Electron-Transfer Site. Eur. J. Biochem 1997, 245, 521–532. [DOI] [PubMed] [Google Scholar]
- (332).Malmström BG; Vänngård T Electron Spin Resonance of Copper Proteins and Some Model Complexes. J. Mol. Biol 1960, 2, 118–124. [Google Scholar]
- (333).Rorabacher DB Electron Transfer by Copper Centers. Chem. Rev 2004, 104, 651–698. [DOI] [PubMed] [Google Scholar]
- (334).Solomon EI; Randall DW; Glaser T Electronic Structures of Active Sites in Electron Transfer Metalloproteins: Contributions to Reactivity. Coord. Chem. Rev 2000, 200–202, 595–632. [Google Scholar]
- (335).Dennison C Investigating the Structure and Function of Cupredoxins. Coord. Chem. Rev 2005, 249, 3025–3054. [Google Scholar]
- (336).Gray HB; Malmström BG; Williams RJP Copper Coordination in Blue Proteins. JBIC, J. Biol. Inorg. Chem 2000, 5, 551–559. [DOI] [PubMed] [Google Scholar]
- (337).Malmström BG; Aasa R The Nature of the CuA Center in Cytochrome c Oxidase. FEBS Lett. 1993, 325, 49–52. [DOI] [PubMed] [Google Scholar]
- (338).Savelieff MG; Lu Y CuA Centers and Their Biosynthetic Models in Azurin. JBIC, J. Biol. Inorg. Chem 2010, 15, 461–483. [DOI] [PubMed] [Google Scholar]
- (339).Wikström M Cytochrome c Oxidase: 25 Years of the Elusive Proton Pump. Biochim. Biophys. Acta, Bioenerg 2004, 1655, 241–247. [DOI] [PubMed] [Google Scholar]
- (340).Brown K; Tegoni M; Prudêncio M; Pereira AS; Besson S; Moura JJ; Moura I; Cambillau C A Novel Type of Catalytic Copper Cluster in Nitrous Oxide Reductase. Nat. Struct. Biol 2000, 7, 191–195. [DOI] [PubMed] [Google Scholar]
- (341).Suharti; Strampraad MJF; Schröder I; de Vries S A Novel Copper A Containing Menaquinol NO Reductase from Bacillus azotoformans. Biochemistry 2001, 40, 2632–2639. [DOI] [PubMed] [Google Scholar]
- (342).Bertagnolli H; Kaim W The Dinuclear CuA Center in Cytochrome c Oxidase and N2O Reductase–A Metal-Metal Bond in Biology? Angew. Chem, Int. Ed. Engl 1995, 34, 771–773. [Google Scholar]
- (343).Fisher OS; Kenney GE; Ross MO; Ro SY; Lemma BE; Batelu S; Thomas PM; Sosnowski VC; DeHart CJ; Kelleher NL; et al. Characterization of a Long Overlooked Copper Protein from Methane- and Ammonia-Oxidizing Bacteria. Nat. Commun 2018, 9, 4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (344).Ross MO; Fisher OS; Morgada MN; Krzyaniak MD; Wasielewski MR; Vila AJ; Hoffman BM; Rosenzweig AC Formation and Electronic Structure of an Atypical CuA Site. J. Am. Chem. Soc 2019, 141, 4678–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (345).Nar H; Messerschmidt A; Huber R; van de Kamp M; Canters GW Crystal Structure Analysis of Oxidized Pseudomonas aeruginosa Azurin atpH 5·5 andpH 9·0: ApH-Induced Conformational Transition Involves a Peptide Bond Flip. J. Mol. Biol 1991, 221, 765–772. [DOI] [PubMed] [Google Scholar]
- (346).Williams PA; Blackburn NJ; Sanders D; Bellamy H; Stura EA; Fee JA; McRee DE The CuA Domain of Thermus thermophilus Ba3-Type Cytochrome c Oxidase at 1.6 Å Resolution. Nat. Struct. Bio 1999, 6, 509–516. [DOI] [PubMed] [Google Scholar]
- (347).Hakulinen N; Kiiskinen L-L; Kruus K; Saloheimo M; Paananen A; Koivula A; Rouvinen J Crystal Structure of a Laccase from Melanocarpus albomyces with an Intact Trinuclear Copper Site. Nat. Struct. Mol. Biol 2002, 9, 601–605. [DOI] [PubMed] [Google Scholar]
- (348).Leferink NGH; Han C; Antonyuk SV; Heyes DJ; Rigby SEJ; Hough MA; Eady RR; Scrutton NS; Hasnain SS Proton-Coupled Electron Transfer in the Catalytic Cycle of Alcaligenes xylosoxidans Copper-Dependent Nitrite Reductase. Biochemistry 2011, 50, 4121–4131. [DOI] [PubMed] [Google Scholar]
- (349).Pomowski A; Zumft WG; Kroneck PMH; Einsle O N2O Binding at a [4Cu:2S] Copper-Sulphur Cluster in Nitrous Oxide Reductase. Nature 2011, 477, 234–237. [DOI] [PubMed] [Google Scholar]
- (350).McGuirl MA; Dooley DM Copper Proteins with Type 2 Sites. In Encyclopedia of Inorganic Chemistry; King RB, Crabtree RH, Lukehart CM, Atwood DA, Scott RA, Eds.; John Wiley & Sons, Ltd: Chichester, UK, 2006; pp 294. [Google Scholar]
- (351).Godden JW; Turley S; Teller DC; ADMAr ET; Liu MY; Payne WJ; Legall J The 2.3 Angstrom X-Ray Structure of Nitrite Reductase from Achromobacter cycloclastes. Science 1991, 253, 438–442. [DOI] [PubMed] [Google Scholar]
- (352).Tavares P; Pereira AS; Moura JJG; Moura I Metalloenzymes of the Denitrification Pathway. J. Inorg. Biochem 2006, 100, 2087–2100. [DOI] [PubMed] [Google Scholar]
- (353).Sheng Y; Abreu IA; Cabelli DE; Maroney MJ; Miller A-F; Teixeira M; Valentine JS Superoxide Dismutases and Superoxide Reductases. Chem. Rev 2014, 114, 3854–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Solomon EI; Sundaram UM; Machonkin TE Multicopper Oxidases and Oxygenases. Chem. Rev 1996, 96, 2563–2606. [DOI] [PubMed] [Google Scholar]
- (355).Solomon EI; Chen P; Metz M; Lee S-K; Palmer AE Oxygen Binding, Activation, and Reduction to Water by Copper Proteins. Angew. Chem, Int. Ed 2001, 40, 4570–4590. [DOI] [PubMed] [Google Scholar]
- (356).Solomon EI; Ginsbach JW; Heppner DE; Kieber-Emmons MT; Kjaergaard CH; Smeets PJ; Tian L; Woertink JS Copper Dioxygen (Bio)Inorganic Chemistry. Faraday Discuss. 2011, 148, 11–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (357).Hemsworth GR; Johnston EM; Davies GJ; Walton PH Lytic Polysaccharide Monooxygenases in Biomass Conversion. Trends Biotechnol. 2015, 33, 747–761. [DOI] [PubMed] [Google Scholar]
- (358).Ciano L; Davies GJ; Tolman WB; Walton PH Bracing Copper for the Catalytic Oxidation of C-H Bonds. Nat. Catal 2018, 1, 571–577. [Google Scholar]
- (359).Riva S Laccases: Blue Enzymes for Green Chemistry. Trends Biotechnol. 2006, 24, 219–226. [DOI] [PubMed] [Google Scholar]
- (360).Giardina P; Faraco V; Pezzella C; Piscitelli A; Vanhulle S; Sannia G Laccases: A Never-Ending Story. Cell. Mol. Life Sci 2010, 67, 369–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Monnier F; Taillefer M Catalytic C-C, C-N, and C-O Ullmann-Type Coupling Reactions. Angew. Chem., Int. Ed 2009, 48, 6954–6971. [DOI] [PubMed] [Google Scholar]
- (362).Alexakis A; Bäckvall JE; Krause N; Pàmies O; Diéguez M Enantioselective Copper-Catalyzed Conjugate Addition and Allylic Substitution Reactions. Chem. Rev 2008, 108, 2796–2823. [DOI] [PubMed] [Google Scholar]
- (363).Wendlandt AE; Suess AM; Stahl SS Copper-Catalyzed Aerobic Oxidative C-H Functionalizations: Trends and Mechanistic Insights. Angew. Chem., Int. Ed 2011, 50, 11062–11087. [DOI] [PubMed] [Google Scholar]
- (364).Allen SE; Walvoord RR; Padilla-Salinas R; Kozlowski MC Aerobic Copper-Catalyzed Organic Reactions. Chem. Rev 2013, 113, 6234–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Guo X-X; Gu D-W; Wu Z; Zhang W Copper-Catalyzed C-H Functionalization Reactions: Efficient Synthesis of Heterocycles. Chem. Rev 2015, 115, 1622–1651. [DOI] [PubMed] [Google Scholar]
- (366).Trammell R; Rajabimoghadam K; Garcia-Bosch I Copper-Promoted Functionalization of Organic Molecules: From Biologically Relevant Cu/O2 Model Systems to Organometallic Transformations. Chem. Rev 2019, 119, 2954–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (367).Bos J; Fusetti F; Driessen AJM; Roelfes G Enantioselective Artificial Metalloenzymes by Creation of a Novel Active Site at the Protein Dimer Interface. Angew. Chem., Int. Ed 2012, 51, 7472–7475. [DOI] [PubMed] [Google Scholar]
- (368).Bos J; Browne WR; Driessen AJM; Roelfes G Supramolecular Assembly of Artificial Metalloenzymes Based on the Dimeric Protein LmrR as Promiscuous Scaffold. J. Am. Chem. Soc 2015, 137, 9796–9799. [DOI] [PubMed] [Google Scholar]
- (369).Coquière D; Bos J; Beld J; Roelfes G Enantioselective Artificial Metalloenzymes Based on a Bovine Pancreatic Polypeptide Scaffold. Angew. Chem, Int. Ed 2009, 48, 5159–5162. [DOI] [PubMed] [Google Scholar]
- (370).Renggli K; Nussbaumer MG; Urbani R; Pfohl T; Bruns N A Chaperonin as Protein Nanoreactor for Atom-Transfer Radical Polymerization. Angew. Chem., Int. Ed 2014, 53, 1443–1447. [DOI] [PubMed] [Google Scholar]
- (371).Magnus KA; Hazes B; Ton-That H; Bonaventura C; Bonaventura J; Hol WGJ Crystallographic Analysis of Oxygenated and Deoxygenated States of Arthropod Hemocyanin Shows Unusual Differences. Proteins 1994, 19, 302–309. [DOI] [PubMed] [Google Scholar]
- (372).Volbeda A; Hol WGJ Crystal Structure of Hexameric Haeqocyanin from Panulirus interruptus Refined at 3.2 Å Resolution. J. Mol. Biol 1989, 209, 249–279. [DOI] [PubMed] [Google Scholar]
- (373).Matoba Y; Kumagai T; Yamamoto A; Yoshitsu H; Sugiyama M Crystallographic Evidence That the Dinuclear Copper Center of Tyrosinase Is Flexible during Catalysis. J. Biol. Chem 2006, 281, 8981–8990. [DOI] [PubMed] [Google Scholar]
- (374).Solomon EI; Szilagyi RK; DeBeer George S; Basumallick L Electronic Structures of Metal Sites in Proteins and Models: Contributions to Function in Blue Copper Proteins. Chem. Rev 2004, 104, 419–458. [DOI] [PubMed] [Google Scholar]
- (375).Farver O; Lu Y; Ang MC; Pecht I Enhanced Rate of Intramolecular Electron Transfer in an Engineered Purple CuA Azurin. Proc. Natl. Acad. Sci. U.S.A 1999, 96, 899–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (376).Wilson TD; Yu Y; Lu Y Understanding Copper-Thiolate Containing Electron Transfer Centers by Incorporation of Unnatural Amino Acids and the CuA Center into the Type 1 Copper Protein Azurin. Coord. Chem. Rev 2013, 257, 260–276. [Google Scholar]
- (377).Marshall NM; Garner DK; Wilson TD; Gao Y-G; Robinson H; Nilges MJ; Lu Y Rationally Tuning the Reduction Potential of a Single Cupredoxin beyond the Natural Range. Nature 2009, 462, 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Solomon EI; Hadt RG Recent Advances in Understanding Blue Copper Proteins. Coord. Chem. Rev 2011, 255, 774–789. [Google Scholar]
- (379).Liu J; Chakraborty S; Hosseinzadeh P; Yu Y; Tian S; Petrik I; Bhagi A; Lu Y Metalloproteins Containing Cytochrome, Iron-Sulfur, or Copper Redox Centers. Chem. Rev 2014, 114, 4366–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (380).Mirts EN; Bhagi-Damodaran A; Lu Y Understanding and Modulating Metalloenzymes with Unnatural Amino Acids, Non-Native Metal Ions, and Non-Native Metallocofactors. Acc. Chem. Res 2019, 52, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (381).Petrik ID; Liu J; Lu Y Metalloenzyme Design and Engineering through Strategic Modifications of Native Protein Scaffolds. Curr. Opin. Chem. Biol 2014, 19, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (382).Nersissian AM; Valentine JS; Immoos C; Hill MG; Hart PJ; Williams G; Herrmann RG Uclacyanins, Stellacyanins, and Plantacyanins Are Distinct Subfamilies of Phytocyanins: Plant-Specific Mononuclear Blue Copper Proteins. Protein Sci. 1998, 7, 1915–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Lappin AG; Lewis CA; Ingledew WJ Kinetics and Mechanisms of Reduction of Rusticyanin, a Blue Copper Protein from Thiobacillus ferrooxidans, by Inorganic Cations. Inorg. Chem 1985, 24, 1446–1450. [Google Scholar]
- (384).Xu F; Palmer AE; Yaver DS; Berka RM; Gambetta GA; Brown SH; Solomon EI Targeted Mutations in a Trametes villosa Laccase. J. Biol. Chem 1999, 274, 12372–12375. [DOI] [PubMed] [Google Scholar]
- (385).Hosseinzadeh P; Lu Y Design and Fine-Tuning Redox Potentials of Metalloproteins Involved in Electron Transfer in Bioenergetics. Biochim. Biophys. Acta, Bioenerg 2016, 1857, 557–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Karlsson BG; Nordling M; Pascher T; Tsai L-C; Sjolin L; Lundberg LG Cassette Mutagenesis of Met121 in Azurin from Pseudomonas aeruginosa. Protein Eng. 1991, 4, 343–349. [DOI] [PubMed] [Google Scholar]
- (387).Kroes SJ; Hoitink CWG; Andrew CR; Ai J; Sanders-Loehr J; Messerschmidt A; Hagen WR; Canters GW The Mutation Met121His Creates a Type-1.5 Copper Site in Alcaligenes denitrificans Azurin. Eur. J. Biochem 1996, 240, 342–351. [DOI] [PubMed] [Google Scholar]
- (388).Pascher T; Karlsson BG; Nordling M; Malmstrom BG; Vanngard T Reduction Potentials and Their pH Dependence in Site-Directed-Mutant Forms of Azurin from Pseudomonas aeruginosa. Eur. J. Biochem 1993, 212, 289–296. [DOI] [PubMed] [Google Scholar]
- (389).Karlsson BG; Aasa R; Malmström BG; Lundberg LG Rack-Induced Bonding in Blue Copper Proteins: Spectroscopic Properties and Reduction Potential of the Azurin Mutant Met-121 → Leu. FEBS Lett 1989, 253, 99–102. [Google Scholar]
- (390).Murphy LM; Strange RW; Karlsson BG; Lundberg LG; Pascher T; Reinhammar B; Hasnain SS Structural Characterization of Azurin from Pseudomonas aeruginosa and Some of Its Methionine-121 Mutants. Biochemistry 1993, 32, 1965–1975. [DOI] [PubMed] [Google Scholar]
- (391).Garner DK; Vaughan MD; Hwang HJ; Savelieff MG; Berry SM; Honek JF; Lu Y Reduction Potential Tuning of the Blue Copper Center in Pseudomonas aeruginosa Azurin by the Axial Methionine as Probed by Unnatural Amino Acids. J. Am. Chem. Soc 2006, 128, 15608. [DOI] [PubMed] [Google Scholar]
- (392).Berry SM; Baker MH; Reardon NJ Reduction Potential Variations in Azurin through Secondary Coordination Sphere Phenylalanine Incorporations. J. Inorg. Biochem 2010, 104, 1071–1078. [DOI] [PubMed] [Google Scholar]
- (393).Farver O; Marshall NM; Wherland S; Lu Y; Pecht I Designed Azurins Show Lower Reorganization Free Energies for Intraprotein Electron Transfer. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 10536–10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Farver O; Hosseinzadeh P; Marshall NM; Wherland S; Lu Y; Pecht I Long-Range Electron Transfer in Engineered Azurins Exhibits Marcus Inverted Region Behavior. J. Phys. Chem. Lett 2015, 6, 100–105. [DOI] [PubMed] [Google Scholar]
- (395).Shiga D; Nakane D; Inomata T; Funahashi Y; Masuda H; Kikuchi A; Oda M; Noda M; Uchiyama S; Fukui K; et al. Creation of a Type 1 Blue Copper Site within a de Novo Coiled-Coil Protein Scaffold. J. Am. Chem. Soc 2010, 132, 18191–18198. [DOI] [PubMed] [Google Scholar]
- (396).Koebke KJ; Ruckthong L; Meagher JL; Mathieu E; Harland J; Deb A; Lehnert N; Policar C; Tard C; Penner-Hahn J; et al. Clarifying the Copper Coordination Environment in a De Novo Designed Red Copper Protein. Inorg. Chem 2018, 57, 12291–12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (397).Lovejoy B; Choe S; Cascio D; McRorie DK; DeGrado WC; Eisenberg D Crystal Structure of a Synthetic Triple-Stranded Ac-Helical Bundle. Science 1993, 259, 1288–1293. [DOI] [PubMed] [Google Scholar]
- (398).Woolfson DN A Brief History of De Novo Protein Design: Minimal, Rational, and Computational. J. Mol. Biol 2021, 433, 167160. [DOI] [PubMed] [Google Scholar]
- (399).Schnepf R; Horth P; Bill E; Wieghardt K; Hildebrandt P; Haehnel W De Novo Design and Characterization of Copper Centers in Synthetic Four-Helix-Bundle Proteins. J.Am. Chem. Soc 2001, 123, 2186–2195. [DOI] [PubMed] [Google Scholar]
- (400).Schnepf R; Haehnel W; Wieghardt K; Hildebrandt P Spectroscopic Identification of Different Types of Copper Centers Generated in Synthetic Four-Helix Bundle Proteins. J. Am. Chem. Soc 2004, 126, 14389–14399. [DOI] [PubMed] [Google Scholar]
- (401).Shiga D; Hamano Y; Kamei M; Funahashi Y; Masuda H; Sakaguchi M; Ogura T; Tanaka T Tuning the Geometries of a De Novo Blue Copper Protein by Axial Interactions. JBIC, J. Biol. Inorg. Chem 2012, 17, 1025–1031. [DOI] [PubMed] [Google Scholar]
- (402).Plegaria JS; Duca M; Tard C; Friedlander TJ; Deb A; Penner-Hahn JE; Pecoraro VL De Novo Design and Characterization of Copper Metallopeptides Inspired by Native Cupredoxins. Inorg. Chem 2015, 54, 9470–9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (403).Plegaria JS; Herrero C; Quaranta A; Pecoraro VL Electron Transfer Activity of a De Novo Designed Copper Center in a Three-Helix Bundle Fold. Biochim. Biophys. Acta, Bioenerg 2016, 1857, 522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (404).Koebke KJ; Alfaro VS; Pinter TBJ; Deb A; Lehnert N; Tard C; Penner-Hahn JE; Pecoraro VL Traversing the Red-Green-Blue Color Spectrum in Rationally Designed Cupredoxins. J. Am. Chem. Soc 2020, 142, 15282–15294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (405).Enguita FJ; Martins LO; Henriques AO; Carrondo MA Crystal Structure of a Bacterial Endospore Coat Component: A Laccase with Enhanced Thermostability Properties. J. Biol. Chem 2003, 278, 19416–19425. [DOI] [PubMed] [Google Scholar]
- (406).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera-a Visualization System for Exploratory Research and Analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (407).Akter M; Tokiwa T; Shoji M; Nishikawa K; Shigeta Y; Sakurai T; Higuchi Y; Kataoka K; Shibata N Redox Potential-Dependent Formation of an Unusual His-Trp Bond in Bilirubin Oxidase. Chem. Eur. J 2018, 24, 18052–18058. [DOI] [PubMed] [Google Scholar]
- (408).Roberts SA; Weichsel A; Grass G; Thakali K; Hazzard JT; Tollin G; Rensing C; Montfort WR Crystal Structure and Electron Transfer Kinetics of CueO, a Multicopper Oxidase Required for Copper Homeostasis in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (409).Kajikawa T; Kataoka K; Sakurai T Modifications on the Hydrogen Bond Network by Mutations of Escherichia coli Copper Efflux Oxidase Affect the Process of Proton Transfer to Dioxygen Leading to Alterations of Enzymatic Activities. Biochem. Biophy. Res. Comm 2012, 422, 152–156. [DOI] [PubMed] [Google Scholar]
- (410).Sakurai T; Kataoka K Structure and Function of Type I Copper in Multicopper Oxidases. Cell. Mol. Life Sci 2007, 64, 2642–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (411).Jones SM; Solomon EI Electron Transfer and Reaction Mechanism of Laccases. Cell. Mol. Life Sci 2015, 72, 869–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Vilbert AC; Liu Y; Dai H; Lu Y Recent Advances in Tuning Redox Properties of Electron Transfer Centers in Metal-loenzymes Catalyzing the Oxygen Reduction Reaction and H2 Oxidation Important for Fuel Cell Design. Curr. Opin. Electrochem 2021, 30, 100780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (413).Pardo I; Camarero S Laccase Engineering by Rational and Evolutionary Design. Cell. Mol. Life Sci 2015, 72, 897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (414).Durão P; Bento I; Fernandes AT; Melo EP; Lindley P; Martins LO Perturbations of the T1 Copper Site in the CotA Laccase from Bacillus subtilis: Structural, Biochemical, Enzymatic and Stability Studies. J. Biol. Inorg. Chem 2006, 11, 514–526. [DOI] [PubMed] [Google Scholar]
- (415).Kamitaka Y; Tsujimura S; Kataoka K; Sakurai T; Ikeda T; Kano K Effects of Axial Ligand Mutation of the Type I Copper Site in Bilirubin Oxidase on Direct Electron Transfer-Type Bioelectrocatalytic Reduction of Dioxygen. J. Electroanal. Chem 2007, 601, 119–124. [Google Scholar]
- (416).Kataoka K; Hirota S; Maeda Y; Kogi H; Shinohara N; Sekimoto M; Sakurai T Enhancement ofLaccase Activity through the Construction and Breakdown of a Hydrogen Bond at the Type I Copper Center in Escherichia coli CueO and the Deletion Mutant Δα5—7 CueO. Biochemistry 2011, 50, 558–565. [DOI] [PubMed] [Google Scholar]
- (417).Kataoka K; Kogi H; Tsujimura S; Sakurai T Modifications of Laccase Activities of Copper Efflux Oxidase, CueO by Synergistic Mutations in the First and Second Coordination Spheres of the Type I Copper Center. Biochem. Biophys. Res. Commun 2013, 431, 393–397. [DOI] [PubMed] [Google Scholar]
- (418).Sekretaryova A; Jones SM; Solomon EI O2 Reduction to Water by High Potential Multicopper Oxidases: Contributions of the T1 Copper Site Potential and the Local Environment of the Trinuclear Copper Cluster. J. Am. Chem. Soc 2019, 141, 11304–11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (419).Sato K; Firbank SJ; Li C; Banfield MJ; Dennison C The Importance of the Long Type 1 Copper-Binding Loop of Nitrite Reductase for Structure and Function. Chem. Eur. J 2008, 14, 5820–5828. [DOI] [PubMed] [Google Scholar]
- (420).Hough MA; Ellis MJ; Antonyuk S; Strange RW; Sawers G; Eady RR; Samar Hasnain S High Resolution Structural Studies of Mutants Provide Insights into Catalysis and Electron Transfer Processes in Copper Nitrite Reductase. J. Mol. Biol 2005, 350, 300–309. [DOI] [PubMed] [Google Scholar]
- (421).Ellis MJ; Prudâncio M; Dodd FE; Strange RW; Sawers G; Eady RR; Hasnain SS Biochemical and Crystallographic Studies of the Met144Ala, Asp92Asn and His254Phe Mutants of the Nitrite Reductase from Alcaligenes xylosoxidans Provide Insight into the Enzyme Mechanism. J. Mol. Biol 2002, 316, 51–64. [DOI] [PubMed] [Google Scholar]
- (422).Zumft WG Cell Biology and Molecular Basis of Denitrification. Microbiol. Mol. Biol. Rev 1997, 61, 533–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (423).Horrell S; Kekilli D; Strange RW; Hough MA Recent Structural Insights into the Function of Copper Nitrite Reductases. Metallomics 2017, 9, 1470–1482. [DOI] [PubMed] [Google Scholar]
- (424).Pinho D; Besson S; Brondino CD; de Castro B; Moura I Copper-Containing Nitrite Reductase from Pseudomonas chlororaphis DSM 50135. Evidence for Modulation of the Rate of Intramolecular Electron Transfer through Nitrite Binding to the Type 2 Copper Center. Eur. J. Biochem 2004, 271, 2361–2369. [DOI] [PubMed] [Google Scholar]
- (425).Hunsicker-Wang LM; Pacoma RL; Chen Y; Fee JA; Stout CD A Novel Cryoprotection Scheme for Enhancing the Diffraction of Crystals of Recombinant Cytochrome Ba3 Oxidase from Thermus thermophilus. Acta Cryst. D 2005, 61, 340–343. [DOI] [PubMed] [Google Scholar]
- (426).Robinson H; Ang MC; Gao Y-G; Hay MT; Lu Y; Wang AH-J Structural Basis of Electron Transfer Modulation in the Purple CuA Center. Biochemistry 1999, 38, 5677–5683. [DOI] [PubMed] [Google Scholar]
- (427).Zitare UA; Szuster J; Santalla MC; Llases ME; Morgada MN; Vila AJ; Murgida DH Fine Tuning of Functional Features of the CuA Site by Loop-Directed Mutagenesis. Inorg. Chem 2019, 58, 2149–2157. [DOI] [PubMed] [Google Scholar]
- (428).Leguto AJ; Smith MA; Morgada MN; Zitare UA; Murgida DH; Lancaster KM; Vila AJ Dramatic Electronic Perturbations of CuA Centers via Subtle Geometric Changes. J. Am. Chem. Soc 2019, 141, 1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Zickermann V; Verkhovsky M; Morgan J; Wikstrom M; Anemuller S; Bill E; Steffens GCM; Ludwig B Perturbation ofthe CuA Site in Cytochrome c Oxidase of Paracoccus denitrificans by Replacement of Met227 with Isoleucine. Eur. J. Biochem 1995, 234, 686–693. [DOI] [PubMed] [Google Scholar]
- (430).Zhen Y; Schmidt B; Kang UG; Antholine W; Ferguson-Miller S Mutants of the Cu A Site in Cytochrome c Oxidase of Rhodobacter Sphaeroides : I. Spectral and Functional Properties. Biochemistry 2002, 41, 2288–2297. [DOI] [PubMed] [Google Scholar]
- (431).Wang K; Geren L; Zhen Y; Ma L; Ferguson-Miller S; Durham B; Millett F Mutants of the CuA Site in Cytochrome c Oxidase of Rhodobacter Sphaeroides : II. Rapid Kinetic Analysis of Electron Transfer. Biochemistry 2002, 41, 2298–2304. [DOI] [PubMed] [Google Scholar]
- (432).Slutter CE; Gromov I; Richards JH; Pecht I; Goldfarb D Mutations of the Weak Axial Ligand in the Thermus CuA Center Modulates Its Electronic Structure. J. Am. Chem. Soc 1999, 121, 5077–5078. [Google Scholar]
- (433).Slutter CE; Gromov I; Epel B; Pecht I; Richards JH; Goldfarb D Pulsed EPR/ENDOR Characterization of Perturbations of the CuA Center Ground State by Axial Methionine Ligand Mutations. J. Am. Chem. Soc 2001, 123, 5325–5336. [DOI] [PubMed] [Google Scholar]
- (434).Ledesma GN; Murgida DH; Ly HK; Wackerbarth H; Ulstrup J; Costa-Filho AJ; Vila AJ The Met Axial Ligand Determines the Redox Potential in CuA Sites. J. Am. Chem. Soc 2007, 129, 11884–11885. [DOI] [PubMed] [Google Scholar]
- (435).Blackburn NJ; Ralle M; Gomez E; Hill MG; Pastuszyn A; Sanders D; Fee JA Selenomethionine-Substituted Thermus thermophilus Cytochrome Ba3: Characterization of the CuA Site by Se and Cu K-EXAFS. Biochemistry 1999, 38, 7075–7084. [DOI] [PubMed] [Google Scholar]
- (436).Dennison C; Vijgenboom E; de Vries S; van der Oost J; Canters GW Introduction of a CuA Site into the Blue Copper Protein Amicyanin from Thiobacillus versutus. FEBS Lett. 1995, 365, 92–94. [DOI] [PubMed] [Google Scholar]
- (437).Andrew CR; Lappalainen P; Saraste M; Hay MT; Lu Y; Dennison C; Canters GW; Fee JA; Nakamura N; Sanders-Loehr J Engineered Cupredoxins and Bacterial Cytochrome c Oxidases Have Similar CuA Sites: Evidence from Resonance Raman Spectroscopy. J. Am. Chem. Soc 1995, 117, 10759–10760. [Google Scholar]
- (438).Hay M; Richards JH; Lu Y Construction and Characterization of an Azurin Analog for the Purple Copper Site in Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U.S.A 1996, 93, 461–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (439).Hwang HJ; Ang MC; Lu Y Determination of Reduction Potential of an Engineered CuA Azurin by Cyclic Voltammetry and Spectrochemical Titrations. JBIC, J. Biol. Inorg. Chem 2004, 9, 489. [DOI] [PubMed] [Google Scholar]
- (440).Hwang HJ; Berry SM; Nilges MJ; Lu Y Axial Methionine Has Much Less Influence on Reduction Potentials in a CuA Center than in a Blue Copper Center. J. Am. Chem. Soc 2005, 127, 7274–7275. [DOI] [PubMed] [Google Scholar]
- (441).New SY; Marshall NM; Hor TSA; Xue F; Lu Y Redox Tuning of Two Biological Copper Centers through Non-Covalent Interactions: Same Trend but Different Magnitude. Chem. Commun 2012, 48, 4217. [DOI] [PubMed] [Google Scholar]
- (442).Tsai M-L; Hadt RG; Marshall NM; Wilson TD; Lu Y; Solomon EI Axial Interactions in the Mixed-Valent CuA Active Site and Role of the Axial Methionine in Electron Transfer. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 14658–14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (443).Morgada MN; Abriata LA; Zitare U; Alvarez-Paggi D; Murgida DH; Vila AJ Control of the Electronic Ground State on an Electron-Transfer Copper Site by Second-Sphere Perturbations. Angew. Chem., Int. Ed 2014, 126, 6302–6306. [DOI] [PubMed] [Google Scholar]
- (444).Morgada MN; Llases M-E; Giannini E; Castro M-A; Alzari PM; Murgida DH; Lisa M-N; Vila AJ Unexpected Electron Spin Density on the Axial Methionine Ligand in CuA Suggests Its Involvement in Electron Pathways. Chem. Commun 2020, 56, 1223–1226. [DOI] [PubMed] [Google Scholar]
- (445).Shiga D; Funahashi Y; Masuda H; Kikuchi A; Noda M; Uchiyama S; Fukui K; Kanaori K; Tajima K; Takano Y; et al. Creation of a Binuclear Purple Copper Site within a De Novo Coiled-Coil Protein. Biochemistry 2012, 51, 7901–7907. [DOI] [PubMed] [Google Scholar]
- (446).Mirts EN; Dikanov SA; Jose A; Solomon EI; Lu Y A Binuclear CuA Center Designed in an All α-Helical Protein Scaffold. J. Am. Chem. Soc 2020, 142, 13779–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (447).Peacock AF Incorporating Metals into de Novo Proteins. Curr. Opin. Chem. Biol 2013, 17, 934–939. [DOI] [PubMed] [Google Scholar]
- (448).Rink WM; Thomas F De Novo Designed α-Helical Coiled-Coil Peptides as Scaffolds for Chemical Reactions. Chem. Eur. J 2019, 25, 1665–1677. [DOI] [PubMed] [Google Scholar]
- (449).Boyle AL; Rabe M; Crone NSA; Rhys GG; Soler N; Voskamp P; Pannu NS; Kros A Selective Coordination of Three Transition Metal Ions within a Coiled-Coil Peptide Scaffold. Chem. Sci 2019, 10, 7456–7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (450).Koebke KJ; Pecoraro VL Development of De Novo Copper Nitrite Reductases: Where We Are and Where We Need To Go. ACS Catal. 2018, 8, 8046–8057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Fujieda N; Hasegawa A; Ishihama K; Itoh S Artificial Dicopper Oxidase: Rational Reprogramming of Bacterial Metallo-β-Lactamase into a Catechol Oxidase. Chem. Asian J 2012, 7, 1203–1207. [DOI] [PubMed] [Google Scholar]
- (452).Mathieu E; Tolbert AE; Koebke KJ; Tard C; Iranzo O; Penner-Hahn JE; Policar C; Pecoraro V Rational De Novo Design of a Cu Metalloenzyme for Superoxide Dismutation. Chem. Eur. J 2020, 26, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (453).Tegoni M; Yu F; Bersellini M; Penner-Hahn JE; Pecoraro VL Designing a Functional Type 2 Copper Center That Has Nitrite Reductase Activity within α-Helical Coiled Coils. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 21234–21239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (454).Yu F; Penner-Hahn JE; Pecoraro VL De Novo-Designed Metallopeptides with Type 2 Copper Centers: Modulation of Reduction Potentials and Nitrite Reductase Activities. J. Am. Chem. Soc 2013, 135, 18096–18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (455).Koebke KJ; Yu F; Salerno E; Van Stappen C; Tebo AG; Penner-Hahn JE; Pecoraro VL Modifying the Steric Properties in the Second Coordination Sphere of Designed Peptides Leads to Enhancement of Nitrite Reductase Activity. Angew. Chem., Int. Ed 2018, 57, 3954–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (456).Mitra S; Prakash D; Rajabimoghadam K; Wawrzak Z; Prasad P; Wu T; Misra SK; Sharp JS; Garcia-Bosch I; Chakraborty S De Novo Design of a Self-Assembled Artificial Copper Peptide That Activates and Reduces Peroxide. ACS Catal. 2021, 11, 10267–10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (457).Berry SM; Strange JN; Bladholm EL; Khatiwada B; Hedstrom CG; Sauer AM Nitrite Reductase Activity in Engineered Azurin Variants. Inorg. Chem 2016, 55, 4233–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (458).Lancaster KM; George SD; Yokoyama K; Richards JH; Gray HB Type-Zero Copper Proteins. Nat. Chem 2009, 1, 711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Lancaster KM; Sproules S; Palmer JH; Richards JH; Gray HB Outer-Sphere Effects on Reduction Potentials of Copper Sites in Proteins: The Curious Case of High Potential Type 2 C112D/M121E Pseudomonas aeruginosa Azurin. J. Am. Chem. Soc 2010, 132, 14590–14595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (460).Mizoguchi TJ; Di Bilio AJ; Gray HB; Richards JH Blue to Type 2 Binding. Copper(II) and Cobalt(II) Derivatives of a Cys112Asp Mutant of Pseudomonas aeruginosa Azurin. J. Am. Chem. Soc 1992, 114, 10076–10078. [Google Scholar]
- (461).Faham S; Mizoguchi TJ; Adman ET; Gray HB; Richards JH; Rees DC Role of the Active-Site Cysteine of Pseudomonas aeruginosa Azurin. Crystal Structure Analysis of the CuII(Cys112Asp) Protein. JBIC, J. Biol. Inorg. Chem 1997, 2, 464–469. [Google Scholar]
- (462).Rosenzweig AC Zeroing in on a New Copper Site. Nat. Chem 2009, 1, 684–685. [DOI] [PubMed] [Google Scholar]
- (463).Sieracki NA; Tian S; Hadt RG; Zhang J-L; Woertink JS; Nilges MJ; Sun F; Solomon EI; Lu Y Copper-Sulfenate Complex from Oxidation of a Cavity Mutant of Pseudomonas aeruginosa Azurin. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 924–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (464).Fujieda N; Schätti J; Stuttfeld E; Ohkubo K; Maier T; Fukuzumi S; Ward TR Enzyme Repurposing of a Hydrolase as an Emergent Peroxidase upon Metal Binding. Chem. Sci 2015, 6, 4060–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Mann SI; Heinisch T; Ward TR; Borovik AS Peroxide Activation Regulated by Hydrogen Bonds within Artificial Cu Proteins. J. Am. Chem. Soc 2017, 139, 17289–17292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Mann SI; Heinisch T; Weitz AC; Hendrich MP; Ward TR; Borovik AS Modular Artificial Cupredoxins. J. Am. Chem. Soc 2016, 138, 9073–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (467).Vaaje-Kolstad G; Westereng B; Horn SJ; Liu Z; Zhai H; Sørlie M; Eijsink VGH An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science 2010, 330, 219–222. [DOI] [PubMed] [Google Scholar]
- (468).Bissaro B; Røhr ÅK; Müller G; Chylenski P; Skaugen M; Forsberg Z; Horn SJ; Vaaje-Kolstad G; Eijsink VGH Oxidative Cleavage of Polysaccharides by Monocopper Enzymes Depends on H2O2. Nat. Chem. Biol 2017, 13, 1123–1128. [DOI] [PubMed] [Google Scholar]
- (469).Wang B; Johnston EM; Li P; Shaik S; Davies GJ; Walton PH; Rovira C QM/MM Studies into the H2O2-Dependent Activity of Lytic Polysaccharide Monooxygenases: Evidence for the Formation of a Caged Hydroxyl Radical Intermediate. ACS Catal. 2018, 8, 1346–1351. [Google Scholar]
- (470).Brantley RE; Smerdon SJ; Wilkinson AJ; Singleton EW; Olson JS The Mechanism of Autooxidation of Myoglobin. J. Biol. Chem 1993, 268, 6995–7010. [PubMed] [Google Scholar]
- (471).Rufo CM; Moroz YS; Moroz OV; Stöhr J; Smith TA; Hu X; DeGrado WF; Korendovych IV Short Peptides Self-Assemble to Produce Catalytic Amyloids. Nat. Chem 2014, 6, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (472).Makhlynets OV; Gosavi PM; Korendovych IV Short Self-Assembling Peptides Are Able to Bind to Copper and Activate Oxygen. Angew. Chem., Int. Ed 2016, 55, 9017–9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (473).Lengyel Z; Rufo CM; Moroz YS; Makhlynets OV; Korendovych IV Copper-Containing Catalytic Amyloids Promote Phosphoester Hydrolysis and Tandem Reactions. ACS Catal. 2018, 8, 59–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (474).Yamamura K; Kaiser ET Studies on the Oxidase Activity of Copper(II) Carboxypeptidase A. J. Chem. Soc., Chem. Commun 1976, 20, 830–831. [Google Scholar]
- (475).Wilson ME; Whitesides GM Conversion of a Protein to a Homogeneous Asymmetric Hydrogenation Catalyst by Site-Specific Modification with a Diphosphinerhodium(I) Moiety. J. Am. Chem. Soc 1978, 100, 306–307. [Google Scholar]
- (476).Yu F; Cangelosi VM; Zastrow ML; Tegoni M; Plegaria JS; Tebo AG; Mocny CS; Ruckthong L; Qayyum H; Pecoraro VL Protein Design: Toward Functional Metalloenzymes. Chem. Rev 2014, 114, 3495–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (477).Podtetenieff J; Taglieber A; Bill E; Reijerse EJ; Reetz MT An Artificial Metalloenzyme: Creation of a Designed Copper Binding Site in a Thermostable Protein. Angew. Chem., Int. Ed 2010, 49, 5151–5155. [DOI] [PubMed] [Google Scholar]
- (478).Ghattas W; Dubosclard V; Tachon S; Beaumet M; Guillot R; Réglier M; Simaan AJ; Mahy J-P CuII-Containing 1-Aminocyclopropane Carboxylic Acid Oxidase Is an Efficient Stereo-specific Diels-Alderase. Angew. Chem., Int. Ed 2019, 58, 14605–14609. [DOI] [PubMed] [Google Scholar]
- (479).Fischer J; Renn D; Quitterer F; Radhakrishnan A; Liu M; Makki A; Ghorpade S; Rueping M; Arold ST; Groll M; et al. Robust and Versatile Host Protein for the Design and Evaluation of Artificial Metal Centers. ACS Catal. 2019, 9, 11371–11380. [Google Scholar]
- (480).Rosati F; Roelfes G A Ligand Structure-Activity Study of DNA-Based Catalytic Asymmetric Hydration and Diels-Alder Reactions. ChemCatChem 2011, 3, 973–977. [Google Scholar]
- (481).Di Meo T; Ghattas W; Herrero C; Velours C; Minard P; Mahy J-P; Ricoux R; Urvoas A ARep A3: A Versatile Artificial Scaffold for Metalloenzyme Design. Chem. Eur. J 2017, 23, 10156–10166. [DOI] [PubMed] [Google Scholar]
- (482).Ghattas W; Cotchico-Alonso L; Maréchal J-D; Urvoas A; Rousseau M; Mahy J-P; Ricoux R Artificial Metalloenzymes with the Neocarzinostatin Scaffold: Toward a Biocatalyst for the Diels-Alder Reaction. ChemBioChem 2016, 17, 433–440. [DOI] [PubMed] [Google Scholar]
- (483).Osseili H; Sauer DF; Beckerle K; Arlt M; Himiyama T; Polen T; Onoda A; Schwaneberg U; Hayashi T; Okuda J Artificial Diels-Alderase Based on the Transmembrane Protein FhuA. Beilstein J. Org. Chem 2016, 12, 1314–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (484).Himiyama T; Sauer DF; Onoda A; Spaniol TP; Okuda J; Hayashi T Construction of a Hybrid Biocatalyst Containing a Covalently-Linked Terpyridine Metal Complex within a Cavity of Aponitrobindin. J. Inorg. Biochem 2016, 158, 55–61. [DOI] [PubMed] [Google Scholar]
- (485).Ghattas W; Dubosclard V; Wick A; Bendelac A; Guillot R; Ricoux R; Mahy J-P Receptor-Based Artificial Metalloenzymes on Living Human Cells. J. Am. Chem. Soc 2018, 140, 8756–8762. [DOI] [PubMed] [Google Scholar]
- (486).Reymond S; Cossy J Copper-Catalyzed Diels-Alder Reactions. Chem. Rev 2008, 108, 5359–5406. [DOI] [PubMed] [Google Scholar]
- (487).Reetz MT; Jiao N Copper-Phthalocyanine Conjugates of Serum Albumins as Enantioselective Catalysts in Diels-Alder Reactions. Angew. Chem., Int. Ed 2006, 45, 2416–2419. [DOI] [PubMed] [Google Scholar]
- (488).Boersma AJ; Feringa BL; Roelfes G α,β-Unsaturated 2-Acyl Imidazoles as a Practical Class of Dienophiles for the DNA-Based Catalytic Asymmetric Diels-Alder Reaction in Water. Org. Lett 2007, 9, 3647–3650. [DOI] [PubMed] [Google Scholar]
- (489).Roelfes G LmrR: A Privileged Scaffold for Artificial Metalloenzymes. Acc. Chem. Res 2019, 52, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (490).Drienovská I; Rioz-Martínez A; Draksharapu A; Roelfes G Novel Artificial Metalloenzymes by In Vivo Incorporation of Metal-Binding Unnatural Amino Acids. Chem. Sci 2015, 6, 770–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (491).Basler S; Studer S; Zou Y; Mori T; Ota Y; Camus A; Bunzel HA; Helgeson RC; Houk KN; Jiménez-Osés G; et al. Efficient Lewis Acid Catalysis of an Abiological Reaction in a de Novo Protein Scaffold. Nat. Chem 2021, 13, 231–235. [DOI] [PubMed] [Google Scholar]
- (492).Villarino L; Chordia S; Alonso-Cotchico L; Reddem E; Zhou Z; Thunnissen AMWH; Maréchal J-D; Roelfes G Cofactor Binding Dynamics Influence the Catalytic Activity and Selectivity of an Artificial Metalloenzyme. ACS Catal. 2020, 10, 11783–11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (493).Gutiérrez de Souza C; Bersellini M; Roelfes G Artificial Metalloenzymes Based on TetR Proteins and Cu(II) for Enantiose-lective Friedel-Crafts Alkylation Reactions. ChemCatChem 2020, 12, 3190–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (494).Chordia S; Narasimhan S; Lucini Paioni A; Baldus M; Roelfes G In Vivo Assembly of Artificial Metalloenzymes and Application in Whole-Cell Biocatalysis. Angew. Chem., Int. Ed 2021, 60, 5913–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (495).Zhou Z; Roelfes G Synergistic Catalysis in an Artificial Enzyme by Simultaneous Action of Two Abiological Catalytic Sites. Nat. Catal 2020, 3, 289–294. [Google Scholar]
- (496).Fujieda N; Ichihashi H; Yuasa M; Nishikawa Y; Kurisu G; Itoh S Cupin Variants as a Macromolecular Ligand Library for Stereoselective Michael Addition of Nitroalkanes. Angew. Chem., Int. Ed 2020, 59, 7717–7720. [DOI] [PubMed] [Google Scholar]
- (497).Rimoldi I; Bucci R; Feni L; Santagostini L; Facchetti G; Pellegrino S Exploring the Copper Binding Ability of Mets7 HCtr-1 Protein Domain and His7 Derivative: An Insight in Michael Addition Catalysis. J. Pept. Sci 2021, 27, No. e3289. [DOI] [PubMed] [Google Scholar]
- (498).Bos J; García-Herraiz A; Roelfes G An Enantioselective Artificial Metallo-Hydratase. Chem. Sci 2013, 4, 3578. [Google Scholar]
- (499).Drienovská I; Alonso-Cotchico L; Vidossich P; Lledós A; Maréchal J-D; Roelfes G Design of an Enantioselective Artificial Metallo-Hydratase Enzyme Containing an Unnatural Metal-Binding Amino Acid. Chem. Sci 2017, 8, 7228–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (500).Renggli K; Sauter N; Rother M; Nussbaumer MG; Urbani R; Pfohl T; Bruns N Biocatalytic Atom Transfer Radical Polymerization in a Protein Cage Nanoreactor. Polym. Chem 2017, 8, 2133–2136. [Google Scholar]
- (501).Gold MH; Wariishi H; Valli K Extracellular Peroxidases Involved in Lignin Degradation by the White Rot Basidiomycete Phanerochaete Chrysosporium. In Biocatalysis in Agricultural Biotechnology; ACS Symposium Series, Vol. 389; American Chemical Society, 1989; pp 127–140. [Google Scholar]
- (502).Pereira MM; Santana M; Teixeira M A Novel Scenario for the Evolution of Haem-Copper Oxygen Reductases. Biochim. Biophys. Acta (BBA) - Bioenergetics 2001, 1505, 185–208. [DOI] [PubMed] [Google Scholar]
- (503).Wasser IM; de Vries S; Moënne-Loccoz P; Schröder I; Karlin KD Nitric Oxide in Biological Denitrification: Fe/Cu Metalloenzyme and Metal Complex NOx Redox Chemistry. Chem. Rev 2002, 102, 1201–1234. [DOI] [PubMed] [Google Scholar]
- (504).Rabus R; Venceslau SS; Wöhlbrand L; Voordouw G; Wall JD; Pereira IAC A Post-Genomic View of the Ecophysiology, Catabolism and Biotechnological Relevance of Sulphate-Reducing Prokaryotes. In Advances in Microbial Physiology, Vol. 66; Poole RK, Ed.; Academic Press, 2015; Chapter 2, pp 55–321. [DOI] [PubMed] [Google Scholar]
- (505).Reed CJ; Lam QN; Mirts EN; Lu Y Molecular Understanding of Heteronuclear Active Sites in Heme-Copper Oxidases, Nitric Oxide Reductases, and Sulfite Reductases through Biomimetic Modelling. Chem. Soc. Rev 2021, 50, 2486–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (506).Nastri F; Chino M; Maglio O; Bhagi-Damodaran A; Lu Y; Lombardi A Design and Engineering of Artificial Oxygen-Activating Metalloenzymes. Chem. Soc. Rev 2016, 45, 5020–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (507).Schiltz CJ; Lee A; Partlow EA; Hosford CJ; Chappie JS Structural Characterization of Class 2 OLD Family Nucleases Supports a Two-Metal Catalysis Mechanism for Cleavage. Nucleic Acids Res. 2019, 47, 9448–9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (508).Luo J; Du K-J; Yuan H; Wei C-W; Lang J-J; Wen G-B; Wang Y-H; Lin Y-W Rational Design of an Artificial Nuclease by Engineering a Hetero-Dinuclear Center of Mg-Heme in Myoglobin. ACS Catal. 2020, 10, 14359–14365. [Google Scholar]
- (509).Yeung BKS; Wang X; Sigman JA; Petillo PA; Lu Y Construction and Characterization of a Manganese-Binding Site in Cytochrome c Peroxidase: Towards a Novel Manganese Peroxidase. Chem. Biol 1997, 4, 215–221. [DOI] [PubMed] [Google Scholar]
- (510).Gengenbach A; Syn S; Wang X; Lu Y Redesign of Cytochrome c Peroxidase into a Manganese Peroxidase: Role of Tryptophans in Peroxidase Activity. Biochemistry 1999, 38, 11425–11432. [DOI] [PubMed] [Google Scholar]
- (511).Gengenbach A; Wang X; Lu Y Designing a Manganese Peroxidase. In Oxidative Delignification Chemistry: Fundamentals and Catalysis; ACS Symposium Series, Vol. 785; American Chemical Society, 2001; Chapter 31, pp 487–500. [Google Scholar]
- (512).Feng M; Tachikawa H; Wang X; Pfister TD; Gengenbach AJ; Lu Y Resonance Raman Spectroscopy of Cytochrome c Peroxidase Variants That Mimic Manganese Peroxidase. JBIC, J. Biol. Inorg. Chem 2003, 8, 699–706. [DOI] [PubMed] [Google Scholar]
- (513).Sundaramoorthy M; Kishi K; Gold MH; Poulos TL Preliminary Crystallographic Analysis of Manganese Peroxidase from Phanerochaete Chrysosporium. J. Mol. Biol 1994, 238, 845–848. [DOI] [PubMed] [Google Scholar]
- (514).Pfister TD; Mirarefi AY; Gengenbach AJ; Zhao X; Danstrom C; Conatser N; Gao Y-G; Robinson H; Zukoski CF; Wang AH-J; Lu Y Kinetic and Crystallographic Studies of a Redesigned Manganese-Binding Site in Cytochrome c Peroxidase. JBIC, J. Biol. Inorg. Chem 2006, 12, 126. [DOI] [PubMed] [Google Scholar]
- (515).Hosseinzadeh P; Mirts EN; Pfister TD; Gao Y-G; Mayne C; Robinson H; Tajkhorshid E; Lu Y Enhancing Mn(II)-Binding and Manganese Peroxidase Activity in a Designed Cytochrome c Peroxidase through Fine-Tuning Secondary-Sphere Interactions. Biochemistry 2016, 55, 1494–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (516).Tiefenbrunn T; Liu W; Chen Y; Katritch V; Stout CD; Fee JA; Cherezov V High Resolution Structure of the Ba3 Cytochrome c Oxidase from Thermus thermophilus in a Lipidic Environment. PLOS ONE 2011, 6, No. e22348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (517).Miner KD; Mukherjee A; Gao Y-G; Null EL; Petrik ID; Zhao X; Yeung N; Robinson H; Lu Y A Designed Functional Metalloenzyme That Reduces O2 to H2O with Over One Thousand Turnovers. Angew. Chem., Int. Ed 2012, 51, 5589–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (518).Sigman JA; Kwok BC; Lu Y From Myoglobin to Heme-Copper Oxidase: Design and Engineering of a CuB Center into Sperm Whale Myoglobin. J. Am. Chem. Soc 2000, 122, 8192–8196. [Google Scholar]
- (519).Sigman JA; Kim HK; Zhao X; Carey JR; Lu Y The Role of Copper and Protons in Heme-Copper Oxidases: Kinetic Study of an Engineered Heme-Copper Center in Myoglobin. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 3629–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (520).Kaila VRI; Verkhovsky MI; Wikström M Proton-Coupled Electron Transfer in Cytochrome Oxidase. Chem. Rev 2010, 110, 7062–7081. [DOI] [PubMed] [Google Scholar]
- (521).Yu Y; Mukherjee A; Nilges MJ; Hosseinzadeh P; Miner KD; Lu Y Direct EPR Observation of a Tyrosyl Radical in a Functional Oxidase Model in Myoglobin During Both H2O2 and O2 Reactions. J. Am. Chem. Soc 2014, 136, 1174–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (522).Petrik ID; Davydov R; Ross M; Zhao X; Hoffman B; Lu Y Spectroscopic and Crystallographic Evidence for the Role of a Water-Containing H-Bond Network in Oxidase Activity of an Engineered Myoglobin. J. Am. Chem. Soc 2016, 138, 1134–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (523).Vojtěchovský J; Chu K; Berendzen J; Sweet RM; Schlichting I Crystal Structures of Myoglobin-Ligand Complexes at Near-Atomic Resolution. Biophys. J 1999, 77, 2153–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (524).Mukherjee S; Mukherjee A; Bhagi-Damodaran A; Mukherjee M; Lu Y; Dey A A Biosynthetic Model of Cytochrome c Oxidase as an Electrocatalyst for Oxygen Reduction. Nat. Commun 2015, 6, 8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (525).Yu Y; Cui C; Liu X; Petrik ID; Wang J; Lu Y A Designed Metalloenzyme Achieving the Catalytic Rate of a Native Enzyme. J. Am. Chem. Soc 2015, 137, 11570–11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (526).Durley RCE; Mathews FS Refinement and Structural Analysis of Bovine Cytochrome B5 at 1.5 Å Resolution. Acta Cryst. D 1996, 52, 65–76. [DOI] [PubMed] [Google Scholar]
- (527).Bhagi-Damodaran A; Petrik ID; Marshall NM; Robinson H; Lu Y Systematic Tuning of Heme Redox Potentials and Its Effects on O2 Reduction Rates in a Designed Oxidase in Myoglobin. J. Am. Chem. Soc 2014, 136, 11882–11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (528).Mukherjee S; Mukherjee M; Mukherjee A; Bhagi-Damodaran A; Lu Y; Dey A O2 Reduction by Biosynthetic Models of Cytochrome c Oxidase: Insights into Role of Proton Transfer Residues from Perturbed Active Sites Models of CcO. ACS Catal. 2018, 8, 8915–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (529).Lin Y-W; Yeung N; Gao Y-G; Miner KD; Tian S; Robinson H; Lu Y Roles of Glutamates and Metal Ions in a Rationally Designed Nitric Oxide Reductase Based on Myoglobin. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (530).Petrik ID; Davydov R; Kahle M; Sandoval B; Dwaraknath S; Ädelroth P; Hoffman B; Lu Y An Engineered Glutamate in Biosynthetic Models of Heme-Copper Oxidases Drives Complete Product Selectivity by Tuning the Hydrogen-Bonding Network. Biochemistry 2021, 60, 346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (531).Bu Y; Cukier RI Structural Character and Energetics of Tyrosyl Radical Formation by Electron/Proton Transfers of a Covalently Linked Histidine-Tyrosine: A Model for Cytochrome c Oxidase. J. Phys. Chem. B 2005, 109, 22013–22026. [DOI] [PubMed] [Google Scholar]
- (532).Liu X; Yu Y; Hu C; Zhang W; Lu Y; Wang J Significant Increase of Oxidase Activity through the Genetic Incorporation of a Tyrosine-Histidine Cross-Link in a Myoglobin Model of Heme-Copper Oxidase. Angew. Chem., Int. Ed 2012, 51, 4312–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (533).Yu Y; Lv X; Li J; Zhou Q; Cui C; Hosseinzadeh P; Mukherjee A; Nilges MJ; Wang J; Lu Y Defining the Role of Tyrosine and Rational Tuning of Oxidase Activity by Genetic Incorporation of Unnatural Tyrosine Analogs. J. Am. Chem. Soc 2015, 137, 4594–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (534).Yu Y; Zhou Q; Wang L; Liu X; Zhang W; Hu M; Dong J; Li J; Lv X; Ouyang H; et al. Significant Improvement of Oxidase Activity through the Genetic Incorporation of a Redox-Active Unnatural Amino Acid. Chem. Sci 2015, 6, 3881–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (535).Yeung N; Lin Y-W; Gao Y-G; Zhao X; Russell BS; Lei L; Miner KD; Robinson H; Lu Y Rational Design of a Structural and Functional Nitric Oxide Reductase. Nature 2009, 462, 1079–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (536).Matsumura H; Chakraborty S; Reed J; Lu Y; Moënne-Loccoz P Effect of Outer-Sphere Side Chain Substitutions on the Fate of the Trans Iron-Nitrosyl Dimer in Heme/Nonheme Engineered Myoglobins (FeBMbs): Insights into the Mechanism of Denitrifying NO Reductases. Biochemistry 2016, 55, 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (537).Bhagi-Damodaran A; Michael MA; Zhu Q; Reed J; Sandoval BA; Mirts EN; Chakraborty S; Moënne-Loccoz P; Zhang Y; Lu Y Why Copper Is Preferred over Iron for Oxygen Activation and Reduction in Haem-Copper Oxidases. Nat. Chem 2017, 9, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (538).Bhagi-Damodaran A; Reed JH; Zhu Q; Shi Y; Hosseinzadeh P; Sandoval BA; Harnden KA; Wang S; Sponholtz MR; Mirts EN; et al. Heme Redox Potentials Hold the Key to Reactivity Differences between Nitric Oxide Reductase and Heme-Copper Oxidase. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (539).Mirts EN; Petrik ID; Hosseinzadeh P; Nilges MJ; Lu Y A Designed Heme-[4Fe-4S] Metalloenzyme Catalyzes Sulfite Reduction like the Native Enzyme. Science 2018, 361, 1098–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (540).Ueno T; Koshiyama T; Ohashi M; Kondo K; Kono M; Suzuki A; Yamane T; Watanabe Y Coordinated Design of Cofactor and Active Site Structures in Development of New Protein Catalysts. J. Am. Chem. Soc 2005, 127, 6556–6562. [DOI] [PubMed] [Google Scholar]
- (541).Carey JR; Ma SK; Pfister TD; Garner DK; Kim HK; Abramite JA; Wang Z; Guo Z; Lu Y A Site-Selective Dual Anchoring Strategy for Artificial Metalloprotein Design. J. Am. Chem. Soc 2004, 126, 10812–10813. [DOI] [PubMed] [Google Scholar]
- (542).Zhang J-L; Garner DK; Liang L; Chen Q; Lu Y Protein Scaffold of a Designed Metalloenzyme Enhances the Chemoselectivity in Sulfoxidation of Thioanisole. Chem. Commun 2008, 14, 1665–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (543).Zhang J-L; Garner DK; Liang L; Barrios DA; Lu Y Noncovalent Modulation of pH-Dependent Reactivity of a Mn-Salen Cofactor in Myoglobin with Hydrogen Peroxide. Chem. Eur. J 2009, 15, 7481–7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Buvelot H; Posfay-Barbe KM; Linder P; Schrenzel J; Krause K-H Staphylococcus aureus, Phagocyte NADPH Oxidase and Chronic Granulomatous Disease. FEMS Microbiol. Rev 2017, 41, 139–157. [DOI] [PubMed] [Google Scholar]
- (545).Miller A-F Redox Tuning over Almost 1 V in a Structurally Conserved Active Site: Lessons from Fe-Containing Superoxide Dismutase. Acc. Chem. Res 2008, 41, 501–510. [DOI] [PubMed] [Google Scholar]
- (546).Barwinska-Sendra A; Garcia YM; Sendra KM; Baslé A; Mackenzie ES; Tarrant E; Card P; Tabares LC; Bicep C; Un S; et al. An Evolutionary Path to Altered Cofactor Specificity in a Metalloenzyme. Nat. Commun 2020, 11, 2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (547).Barwinska-Sendra A; Baslé A; Waldron KJ; Un S A Charge Polarization Model for the Metal-Specific Activity of Superoxide Dismutases. Phys. Chem. Chem. Phys 2018, 20, 2363–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (548).Matthews RG Cobalamin-Dependent Methyltransferases. Acc. Chem. Res 2001, 34, 681–689. [DOI] [PubMed] [Google Scholar]
- (549).Morita Y; Oohora K; Sawada A; Doitomi K; Ohbayashi J; Kamachi T; Yoshizawa K; Hisaeda Y; Hayashi T Intraprotein Transmethylation via a CH3-Co(III) Species in Myoglobin Reconstituted with a Cobalt Corrinoid Complex. Dalton Trans. 2016, 45, 3277–3284. [DOI] [PubMed] [Google Scholar]
- (550).Hayashi T; Morita Y; Mizohata E; Oohora K; Ohbayashi J; Inoue T; Hisaeda Y Co(II)/Co(I) Reduction-Induced Axial Histidine-Flipping in Myoglobin Reconstituted with a Cobalt Tetradehydrocorrin as a Methionine Synthase Model. Chem. Commun 2014, 50, 12560–12563. [DOI] [PubMed] [Google Scholar]
- (551).Sommer DJ; Vaughn MD; Ghirlanda G Protein Secondary-Shell Interactions Enhance the Photoinduced Hydrogen Production of Cobalt Protoporphyrin IX. Chem. Commun 2014, 50, 15852–15855. [DOI] [PubMed] [Google Scholar]
- (552).Brucker EA; Olson JS; Phillips GN; Dou Y; Ikeda-Saito M High Resolution Crystal Structures of the Deoxy, Oxy, and Aquomet Forms of Cobalt Myoglobin. J. Biol. Chem 1996, 271, 25419–25422. [DOI] [PubMed] [Google Scholar]
- (553).Key HM; Dydio P; Clark DS; Hartwig JF Abiological Catalysis by Artificial Haem Proteins Containing Noble Metals in Place of Iron. Nature 2016, 534, 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (554).Ward TR Directed Evolution of Iridium-Substituted Myoglobin Affords Versatile Artificial Metalloenzymes for Enantiose-lective C-C Bond-Forming Reactions. Angew. Chem., Int. Ed 2016, 55, 14909–14911. [DOI] [PubMed] [Google Scholar]
- (555).Dydio P; Key HM; Nazarenko A; Rha JY-E; Seyedkazemi V; Clark DS; Hartwig JF An Artificial Metalloenzyme with the Kinetics of Native Enzymes. Science 2016, 354, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (556).Key HM; Dydio P; Liu Z; Rha JY-E; Nazarenko A; Seyedkazemi V; Clark DS; Hartwig JF Beyond Iron: Iridium-Containing P450 Enzymes for Selective Cyclopropanations of Structurally Diverse Alkenes. ACS Cent. Sci 2017, 3, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (557).Gu Y; Natoli SN; Liu Z; Clark DS; Hartwig JF Site-Selective Functionalization of (sp3)C-H Bonds Catalyzed by Artificial Metalloenzymes Containing an Iridium-Porphyrin Cofactor. Angew. Chem., Int. Ed 2019, 58, 13954–13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (558).Watson HC The Stereochemistry of the Protein Myoglobin. Protein Stereochem. 1969, 4, 299. [Google Scholar]
- (559).Park S-Y; Yamane K; Adachi S; Shiro Y; Weiss KE; Maves SA; Sligar SG Thermophilic Cytochrome P450 (CYP119) from Sulfolobus solfataricus: High Resolution Structure and Functional Properties. J. Inorg. Biochem 2002, 91, 491–501. [DOI] [PubMed] [Google Scholar]
- (560).Selvan D; Prasad P; Farquhar ER; Shi Y; Crane S; Zhang Y; Chakraborty S Redesign of a Copper Storage Protein into an Artificial Hydrogenase. ACS Catal. 2019, 9, 5847–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (561).Slater JW; Shafaat HS Nickel-Substituted Rubredoxin as a Minimal Enzyme Model for Hydrogenase. J. Phys. Chem. Lett 2015, 6, 3731–3736. [DOI] [PubMed] [Google Scholar]
- (562).Vita N; Platsaki S; Baslé A; Allen SJ; Paterson NG; Crombie AT; Murrell JC; Waldron KJ; Dennison C A Four-Helix Bundle Stores Copper for Methane Oxidation. Nature 2015, 525, 140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (563).Slater JW; Marguet SC; Gray ME; Monaco HA; Sotomayor M; Shafaat HS Power of the Secondary Sphere: Modulating Hydrogenase Activity in Nickel-Substituted Rubredoxin. ACS Catal. 2019, 9, 8928–8942. [Google Scholar]
- (564).Lewis LC; Shafaat HS Reversible Electron Transfer and Substrate Binding Support [NiFe3S4] Ferredoxin as a Protein-Based Model for [NiFe] Carbon Monoxide Dehydrogenase. Inorg. Chem 2021, 60, 13869–13875. [DOI] [PubMed] [Google Scholar]
- (565).Khare SD; Kipnis Y; Greisen PJ; Takeuchi R; Ashani Y; Goldsmith M; Song Y; Gallaher JL; Silman I; Leader H; et al. Computational Redesign of a Mononuclear Zinc Metalloenzyme for Organophosphate Hydrolysis. Nat. Chem. Biol 2012, 8, 294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (566).Wilchek M; Bayer EA Introduction to Avidin-Biotin Technology. In Methods in Enzymology; Wilchek M, Bayer EA, Eds.; Academic Press, 1990; Vol. 184, pp 5–13. [DOI] [PubMed] [Google Scholar]
- (567).Lin C-C; Lin C-W; Chan ASC Catalytic Hydrogenation of Itaconic Acid in a Biotinylated Pyrphos-Rhodium(I) System in a Protein Cavity. Tetrahedron: Asymmetry 1999, 10, 1887–1893. [Google Scholar]
- (568).Collot J; Gradinaru J; Humbert N; Skander M; Zocchi A; Ward TR Artificial Metalloenzymes for Enantioselective Catalysis Based on Biotin-Avidin. J. Am. Chem. Soc 2003, 125, 9030–9031. [DOI] [PubMed] [Google Scholar]
- (569).Skander M; Humbert N; Collot J; Gradinaru J; Klein G; Loosli A; Sauser J; Zocchi A; Gilardoni F; Ward TR Artificial Metalloenzymes: (Strept)Avidin as Host for Enantioselective Hydro-genation by Achiral Biotinylated Rhodium-Diphosphine Complexes. J. Am. Chem. Soc 2004, 126, 14411–14418. [DOI] [PubMed] [Google Scholar]
- (570).Klein G; Humbert N; Gradinaru J; Ivanova A; Gilardoni F; Rusbandi UE; Ward TR Tailoring the Active Site of Chemzymes by Using a Chemogenetic-Optimization Procedure: Towards Substrate-Specific Artificial Hydrogenases Based on the Biotin-Avidin Technology. Angew. Chem., Int. Ed 2005, 44, 7764–7767. [DOI] [PubMed] [Google Scholar]
- (571).Reetz MT; Peyralans JJ-P; Maichele A; Fu Y; Maywald M Directed Evolution of Hybrid Enzymes: Evolving Enantioselectivity of an Achiral Rh-Complex Anchored to a Protein. Chem. Commun 2006, 41, 4318–4320. [DOI] [PubMed] [Google Scholar]
- (572).Weber PC; Ohlendorf DH; Wendoloski JJ; Salemme FR Structural Origins of High-Affinity Biotin Binding to Streptavidin. Science 1989, 243, 85–88. [DOI] [PubMed] [Google Scholar]
- (573).Letondor C; Humbert N; Ward TR Artificial Metal-loenzymes Based on Biotin-Avidin Technology for the Enantioselective Reduction of Ketones by Transfer Hydrogenation. Proc. Natl. Acad. Sci. U.S.A 2005, 102, 4683–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (574).Letondor C; Pordea A; Humbert N; Ivanova A; Mazurek S; Novic M; Ward TR Artificial Transfer Hydrogenases Based on the Biotin-(Strept)Avidin Technology: Fine Tuning the Selectivity by Saturation Mutagenesis of the Host Protein. J. Am. Chem. Soc 2006, 128, 8320–8328. [DOI] [PubMed] [Google Scholar]
- (575).Dürrenberger M; Heinisch T; Wilson YM; Rossel T; Nogueira E; Knörr L; Mutschler A; Kersten K; Zimbron MJ; Pierron J; et al. Artificial Transfer Hydrogenases for the Enantioselective Reduction of Cyclic Imines. Angew. Chem., Int. Ed 2011, 50, 3026–3029. [DOI] [PubMed] [Google Scholar]
- (576).Creus M; Pordea A; Rossel T; Sardo A; Letondor C; Ivanova A; LeTrong I; Stenkamp RE; Ward TR X-Ray Structure and Designed Evolution of an Artificial Transfer Hydrogenase. Angew. Chem., Int. Ed 2008, 47, 1400–1404. [DOI] [PubMed] [Google Scholar]
- (577).Pordea A; Creus M; Letondor C; Ivanova A; Ward TR Improving the Enantioselectivity of Artificial Transfer Hydrogenases Based on the Biotin-Streptavidin Technology by Combinations of Point Mutations. Inorg. Chim. Acta 2010, 363, 601–604. [Google Scholar]
- (578).Schwizer F; Köhler V; Dürrenberger M; Knörr L; Ward TR Genetic Optimization of the Catalytic Efficiency of Artificial Imine Reductases Based on Biotin-Streptavidin Technology. ACS Catal. 2013, 3, 1752–1755. [Google Scholar]
- (579).Zhao J; Rebelein JG; Mallin H; Trindler C; Pellizzoni MM; Ward TR Genetic Engineering of an Artificial Metalloenzyme for Transfer Hydrogenation of a Self-Immolative Substrate in Escherichia Coli’s Periplasm. J. Am. Chem. Soc 2018, 140, 13171–13175. [DOI] [PubMed] [Google Scholar]
- (580).Wu S; Zhou Y; Rebelein JG; Kuhn M; Mallin H; Zhao J; Igareta NV; Ward TR Breaking Symmetry: Engineering Single-Chain Dimeric Streptavidin as Host for Artificial Metalloenzymes. J. Am. Chem. Soc 2019, 141, 15869–15878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (581).Satoh T; Miura M Oxidative Coupling of Aromatic Substrates with Alkynes and Alkenes under Rhodium Catalysis. Chem. Eur. J 2010, 16, 11212–11222. [DOI] [PubMed] [Google Scholar]
- (582).Hyster TK; Knörr L; Ward TR; Rovis T Biotinylated Rh(III) Complexes in Engineered Streptavidin for Accelerated Asymmetric C-H Activation. Science 2012, 338, 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (583).Jeschek M; Reuter R; Heinisch T; Trindler C; Klehr J; Panke S; Ward TR Directed Evolution of Artificial Metalloenzymes for in Vivo Metathesis. Nature 2016, 537, 661–665. [DOI] [PubMed] [Google Scholar]
- (584).Heinisch T; Schwizer F; Garabedian B; Csibra E; Jeschek M; Vallapurackal J; Pinheiro VB; Marlière P; Panke S; Ward TR E. Coli Surface Display of Streptavidin for Directed Evolution of an Allylic Deallylase. Chem. Sci 2018, 9, 5383–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (585).Pierron J; Malan C; Creus M; Gradinaru J; Hafner I; Ivanova A; Sardo A; Ward TR Artificial Metalloenzymes for Asymmetric Allylic Alkylation on the Basis of the Biotin-Avidin Technology. Angew. Chem., Int. Ed 2008, 47, 701–705. [DOI] [PubMed] [Google Scholar]
- (586).Chatterjee A; Mallin H; Klehr J; Vallapurackal J; Finke AD; Vera L; Marsh M; Ward TR An Enantioselective Artificial Suzukiase Based on the Biotin-Streptavidin Technology. Chem. Sci 2016, 7, 673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (587).Vornholt T; Christoffel F; Pellizzoni MM; Panke S; Ward TR; Jeschek M Systematic Engineering of Artificial Metalloenzymes for New-to-Nature Reactions. Sci. Adv 2021, 7, No. eabe4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (588).Köhler V; Mao J; Heinisch T; Pordea A; Sardo A; Wilson YM; Knörr L; Creus M; Prost J-C; Schirmer T; et al. OsO4· Streptavidin: A Tunable Hybrid Catalyst for the Enantioselective Cis-Dihydroxylation of Olefins. Angew. Chem., Int. Ed 2011, 50, 10863–10866. [DOI] [PubMed] [Google Scholar]
- (589).Pordea A; Creus M; Panek J; Duboc C; Mathis D; Novic M; Ward TR Artificial Metalloenzyme for Enantioselective Sulfoxidation Based on Vanadyl-Loaded Streptavidin. J. Am. Chem. Soc 2008, 130, 8085–8088. [DOI] [PubMed] [Google Scholar]
- (590).Srivastava P; Yang H; Ellis-Guardiola K; Lewis JC Engineering a Dirhodium Artificial Metalloenzyme for Selective Olefin Cyclopropanation. Nat. Commun 2015, 6, 7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (591).Yang H; Swartz AM; Park HJ; Srivastava P; Ellis-Guardiola K; Upp DM; Lee G; Belsare K; Gu Y; Zhang C; et al. Evolving Artificial Metalloenzymes via Random Mutagenesis. Nat. Chem 2018, 10, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (592).Upp DM; Huang R; Li Y; Bultman MJ; Roux B; Lewis JC Engineering Dirhodium Artificial Metalloenzymes for Diazo Coupling Cascade Reactions. Angew. Chem., Int. Ed 2021, 60, 23672–23677. [DOI] [PubMed] [Google Scholar]
- (593).Kato S; Onoda A; Grimm AR; Tachikawa K; Schwaneberg U; Hayashi T Incorporation of a Cp*Rh(III)-Dithiophosphate Cofactor with Latent Activity into a Protein Scaffold Generates a Biohybrid Catalyst Promoting C(sp2)-H Bond Functionalization. Inorg. Chem 2020, 59, 14457–14463. [DOI] [PubMed] [Google Scholar]
- (594).Kato S; Onoda A; Taniguchi N; Schwaneberg U; Hayashi T Directed Evolution of a Cp*RhIII-Linked Biohybrid Catalyst Based on a Screening Platform with Affinity Purification. ChemBioChem 2021, 22, 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (595).Monnard FW; Nogueira ES; Heinisch T; Schirmer T; Ward TR Human Carbonic Anhydrase II as Host Protein for the Creation of Artificial Metalloenzymes: The Asymmetric Transfer Hydrogenation of Imines. Chem. Sci 2013, 4, 3269. [Google Scholar]
- (596).Fleishman SJ; Leaver-Fay A; Corn JE; Strauch E-M; Khare SD; Koga N; Ashworth J; Murphy P; Richter F; Lemmon G; et al. RosettaScripts: A Scripting Language Interface to the Rosetta Macromolecular Modeling Suite. PLOS ONE 2011, 6, No. e20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (597).Heinisch T; Pellizzoni M; Dürrenberger M; Tinberg CE; Köhler V; Klehr J; Häussinger D; Baker D; Ward TR Improving the Catalytic Performance of an Artificial Metalloenzyme by Computational Design. J. Am. Chem. Soc 2015, 137, 10414–10419. [DOI] [PubMed] [Google Scholar]
- (598).Stein A; Chen D; Igareta NV; Cotelle Y; Rebelein JG; Ward TR A Dual Anchoring Strategy for the Directed Evolution of Improved Artificial Transfer Hydrogenases Based on Carbonic Anhydrase. ACS Cent. Sci 2021, 7, 1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (599).Fischer S; Ward TR; Liang AD Engineering a Metathesis-Catalyzing Artificial Metalloenzyme Based on HaloTag. ACS Catal. 2021, 11, 6343–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (600).Lutz S Beyond Directed Evolution—Semi-Rational Protein Engineering and Design. Curr. Opin. Biotechnol 2010, 21, 734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]