

Abstract
Cigarette smoking remains the leading modifiable risk factor for cardiopulmonary diseases; however, the effects of nicotine alone on cardiopulmonary function remain largely unknown. Previously, we have shown that chronic nicotine vapor inhalation in mice leads to the development of pulmonary hypertension (PH) with right ventricular (RV) remodeling. The present study aims to further examine the cardiopulmonary effects of nicotine and the role of the α7 nicotinic acetylcholine receptor (α7-nAChR), which is widely expressed in the cardiovascular system. Wild-type (WT) and α7-nAChR knockout (α7-nAChR−/−) mice were exposed to room air (control) or nicotine vapor daily for 12 weeks. Consistent with our previous study, echocardiography and RV catheterization reveal that male WT mice developed increased RV systolic pressure with RV hypertrophy and dilatation following 12-week nicotine vapor exposure; in contrast, these changes were not observed in male α7-nAChR−/− mice. In addition, chronic nicotine inhalation failed to induce PH and RV remodeling in female mice regardless of genotype. The effects of nicotine on the vasculature were further examined in male mice. Our results show that chronic nicotine inhalation led to impaired acetylcholine-mediated vasodilatory response in both thoracic aortas and pulmonary arteries, and these effects were accompanied by altered endothelial nitric oxide synthase phosphorylation (enhanced inhibitory phosphorylation at threonine 495) and reduced plasma nitrite levels in WT but not α7-nAChR−/− mice. Finally, RNA sequencing revealed up-regulation of multiple inflammatory pathways in thoracic aortas from WT but not α7-nAChR−/− mice. We conclude that the α7-nAChR mediates chronic nicotine inhalation-induced PH, RV remodeling and vascular dysfunction.
Introduction
Cardiovascular and pulmonary diseases (CVPD) are the leading causes of death in the United States of America and worldwide, and smoking remains the most significant preventable risk factor [1]. While the steady decline of traditional cigarette smoking since the 1950s can be considered a health-care victory, this downward trend coincided with the advent and rise in popularity of electronic cigarettes (e-Cig) [2,3]. While e-Cig use is highest among current and former smokers [4], these products have proven increasingly attractive to never-smokers, especially among youth and young adults [5]. This appeal may be tied to the perception that these devices are “safer” than traditional cigarettes or even completely safe. However, e-Cig liquids and aerosols contain a mixture of potentially harmful chemicals [6], including nicotine, the addictive component of all tobacco products.
The extent of nicotine’s contribution to smoking-related CVPD remains generally unknown; however, there is increasing evidence from animal models that nicotine alone can exacerbate CVPD such as chronic hypertension [7], atherosclerosis [8], and lung cancer [9]. We recently demonstrated that chronic inhalation of vaporized nicotine alone is sufficient to induce pulmonary hypertension (PH) in male mice [10]. Mice exposed to nicotine vapor daily for 8 weeks exhibit increased pulmonary vascular resistance, elevated right ventricular systolic pressure (RVSP), and right ventricular (RV) hypertrophy. Additionally, we found that nicotine inhalation leads to acute increases in systemic blood pressure, consistent with previous reports [11–14]. These alterations in cardiopulmonary function are associated with angiotensin-converting enzyme (ACE) overexpression and mitogen-activated protein kinase (MAPK) activation in the RV. Subsequently, we demonstrated that angiotensin-II type 1 receptor (AT1R) antagonism attenuates inhaled nicotine-induced PH and RV remodeling [15].
While our previous investigations provide insight into nicotine’s impact on cardiopulmonary function, the precise pathogenic mechanisms involved in these changes have yet to be fully identified. Nicotine exerts many of its actions by interacting with nicotinic acetylcholine receptors (nAChR) expressed throughout the body, including the cardiovascular system. These receptors are pentameric ligand-gated ion channels which allow the flow of cations upon binding of the endogenous ligand acetylcholine (ACh) or exogenous nicotine [16]. Among the many subtypes of nAChR, the homomeric α7-nAChR has been linked with a variety of cardiovascular effects [17]. Importantly, Vang et al. recently demonstrated that the α7-nAChR mediates RV fibrosis and diastolic dysfunction in a rat Sugen/hypoxia model of PH [18]. However, the contribution of the α7-nAChR to cardiopulmonary dysfunction induced by inhaled nicotine remains unknown.
The current study aims to further examine the cardiopulmonary effects of nicotine and the role of the α7-nAChR using a well-established murine model of chronic nicotine inhalation [10,15].
Materials and methods
Animals
B6.129S7-Chrna7tm1Bay/J (α7-nAChR−/−) mice [19] were bred in-house in a colony established by breeding pairs purchased from the Jackson Laboratory (Bar Harbor, ME). At the time of experiments, age matched C57BL6/J wild-type (WT) mice were purchased from Jackson Laboratory. All mice were housed in a temperature- and humidity-controlled facility under a 12-h dark/light cycle. The mice were fed standard mouse chow (Teklab Extruded Rodent Diet 2019S; Envigo, Indianapolis, Indiana) and water ad libitum. All procedures conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Louisiana State University Health Sciences Center (LSUHSC) Institutional Animal Care and Use Committee (Protocol #3674). All animal works were performed at LSUHSC Animal Care Unit.
Chronic nicotine inhalation model
WT and α7-nAChR−/− mice were randomly assigned to air or nicotine vapor treatments. The nicotine inhalation model in this study was previously described by Oakes et al. [10]. Mice in the nicotine vapor group were housed in a nicotine inhalation chamber (La Jolla Alcohol Research, La Jolla, CA, U.S.A.). Nicotine vapor was produced by bubbling air at a flow rate of 3 L/min through a solution of pure nicotine (free base; Sigma-Aldrich, St. Louis, MO, U.S.A.). After the concentrated nicotine vapor was diluted by the addition of 60 L/min fresh air, the vapor was distributed to the nicotine chamber at a final flow rate of 7–8 L/min. The nicotine exposure was set on a 12 h on/12 h off schedule (9:00 pm to 9:00 am) that overlapped with the dark cycle (mouse active period; 6:00 pm to 6:00 am). The exhaust from the nicotine inhalation chamber was filtered through an activated carbon drum canister (Carbtrol with 90.7 kg of carbon) connected to the exhaust line of the research building. Mice in the air control group were housed in the same room, outside of the nicotine inhalation chamber.
Nicotine intake was monitored via biweekly measurements of serum cotinine, a stable metabolite of nicotine. Blood was obtained by submandibular vein puncture within 15 min following the end of the daily nicotine exposure period. The serum was separated, and cotinine levels were measured by enzyme-linked immunosorbent assay (ELISA; Calbiotech, El Cajon, CA, U.S.A.) per the manufacturer’s protocol.
Echocardiography
Cardiac structure and function were assessed at baseline and following 8 weeks of nicotine exposure via echocardiography using the Vevo 3100 Imaging System with a 30-MHz probe (VisualSonics, Toronto, Canada). Mice were anesthetized with 1.0–1.5% isoflurane and placed on a heating pad. B-mode and M-mode images of the long and short axis were recorded. Images of both the left ventricle (LV) and RV were analyzed using the leading-edge method in Vevo LAB 5.5.1 (VisualSonics). Group averages were calculated from measurements made during a minimum of three cardiac cycles per animal.
RV pressure measurement and heart weight
Mice underwent RV catheterization after 10–12 weeks of nicotine exposure. Mice were anesthetized with 2–3% isoflurane and placed on a heated pad. A high-fidelity pressure transducer (SPR-1000; Millar, Houston, TX, U.S.A.) was inserted into the right jugular vein and advanced through the right atrium into the RV. Systolic and diastolic pressure tracings from the RV were collected and analyzed with the PowerLab 8/35 acquisition system (ADInstruments, Colorado Springs, CO, U.S.A.). Deeply anesthetized mice were killed by exsanguination following RV catheterization. After removing the heart, the RV was dissected from LV and interventricular septal (S) tissues. Weights of the individual cardiac structures were obtained for calculation of Fulton index [RV/(LV+S)*100%]. Cardiac tissues were then snap frozen in liquid nitrogen and stored at −80°C for further analysis.
Aortic ring vascular reactivity
Thoracic aortas were dissected from fully anesthetized mice (2.5–3.0% isoflurane) and placed in cold modified Krebs-Henseleit buffer (KHB: 118 mM NaCl, 25 mM NaHCO3, 4.7 mM KCl, 1.2 mM KH2PO4, 1.25 mM CaCl2, 1.2 mM MgSO4, and 11 mM dextrose). Vessels were carefully cleaned of fat and cut transversely into rings. The vessels were suspended in an ex vivo tissue chamber with a Grass instruments pressure transducer system (Radnoti LLC, Covina, CA, U.S.A.). The rings were equilibrated in oxygenated KHB at 37°C for 1 h and set under tension with 0.5 g (4.90 mN) of force. Baseline tensions were recorded, then vessel responsiveness and maximal constriction were tested using 80 mM KCl. Changes in tension were recorded, and the vessels were rinsed with fresh KHB solution until tensions returned to baseline. Endothelium-dependent vasodilation was assessed by ACh-induced relaxation. The vessels were pre-constricted with 10−6 M L-phenylephrine (Phe) and allowed to stabilize before exposure to increasing concentrations of ACh (10−9 to 10−5 M). Concentration response curves were generated, and data were represented as percent (%) relaxation from Phe-induced precontraction. After rinsing with fresh KHB solution until tensions returned to baseline, the rings were then exposed to increasing concentrations of Phe (10−9 to 10−5 M), and data were analyzed as percentage of KCl maximal contraction normalized to baseline tension. After rinsing again with fresh KHB solution until tensions returned to baseline, endothelium-independent vasodilation was assessed by sodium nitroprusside (SNP)-induced relaxation of Phe precontracted rings. Experiments were performed as in ACh data collection, with increasing concentration of SNP (10−10 to 10−6 M).
Myograph assessment of pulmonary artery and aorta reactivity
In a second cohort of mice, pulmonary arteries (PA) and thoracic aortas were analyzed using a wire myograph system. Vessels were dissected from fully anesthetized mice and placed in cold modified KHB. The vessels were carefully cleaned and cut transversely into rings. These rings were mounted on a wire myograph system (Multi Myograph System-610M, Danish Myo, Denmark), and bathed and equilibrated in 5 ml oxygenated KHB warmed to 37°C for 1h. The bathing solution was then changed to fresh KHB, and initial resting tensions were set to 3 mN for PA and 4 mN for aortic rings. Changes in tension were recorded by direct connection of the 620M Interface to a PC with Labchart Pro (Version 8). Vessel reactivity was assessed using ACh, SNP, and Phe, as described for the Radnoti system. For a subset of PA, the vessels were pre-treated with 1 μM of L-NAME (NG-Nitro-L-arginine Methyl Ester, Hydrochloride) for 30 min prior to Phe exposure.
Western blotting
Total proteins were extracted using RIPA buffer containing protease and phosphatase inhibitors (Cell Signaling, Danvers, MA, U.S.A.) and quantified using the bicinchoninic acid method (Pierce Biotechnology, Waltham, MA, U.S.A.). Equal amounts of proteins were analyzed by Western blotting as described [10]. Antibodies against total endothelial nitric oxide synthase (eNOS, #32027, Cell Signaling), phosphorylated eNOS at serine 1177 (#ab215717, Abcam, Waltham, MA, U.S.A.) and at threonine 495 (#9574, Cell Signaling), α7-nAChR (PA-77505, Invitrogen, Waltham, MA, U.S.A.) and GAPDH (#5174, Cell signaling) were used. The primary antibodies were used at 1:1000 dilutions and HRP-conjugated secondary antibodies (Cell Signaling) were used at 1:10,000 dilutions. Images were captured using the Amersham Imager 680 (GE Healthcare Bio-Sciences, Marlborough, MA, U.S.A.) and densitometry measurements were performed using ImageQuant™ TL software (GE Healthcare Bio-Sciences).
Plasma nitrite measurement
Plasma samples were isolated from venous blood obtained from the jugular vein and transferred into microtainer tubes containing lithium heparin (BD Biosciences, Franklin Lakes, NJ, U.S.A.). Plasma nitrite levels (NO2−) were measured using high performance liquid chromatography with a sensitivity to 0.1 pmol as previously described [20–22]. Plasma nitrite concentrations were quantified by ion chromatography using the Eicom ENO-20 NOx Analyzer (Amuza, San Diego, CA, U.S.A.).
RNA sequencing
RNA sequencing was performed at Tulane University School of Medicine Center for Translational Research in Infection and Inflammation NextGen Sequencing core. Briefly, total RNA was extracted from thoracic aortas isolated from WT and α7-nAChR−/− male mice following air or nicotine exposure (n = 3 per group), using RNeasy Plus mini kit (Qiagen, Ann Arbor, MI, U.S.A.) followed by library preparation using SMART-Seq v4 Ultra Low Input RNA Kit (Takara Bio, Mountain View, CA, U.S.A.). Illumina next generation sequencing was performed using the NextSeq 500 system with the high output flow cell (up to 800 M paired end reads). Ingenuity Pathway Analysis (IPA, Qiagen Bioinformatics) was used to identify differentially regulated genes and pathways.
Statistical analysis
Data were expressed as mean ± SEM and analyzed, where appropriate, by Student’s t test or two-way ANOVA followed by post hoc tests for multiple comparisons between means. Analyses were performed using GraphPad Prism 8 or later versions (GraphPad Software, San Diego, CA, U.S.A.), and values of P<0.05 were considered statistically significant.
Results
Chronic inhaled nicotine fails to induce PH in α7-nAChR knockout mice
After 10–12 weeks of nicotine exposure, RV systolic pressure (RVSP), an approximation of pulmonary artery pressure, was measured via RV catheterization. Consistent with our previous results [10,15], RVSP was significantly increased by chronic nicotine inhalation in WT male mice (31.4 ± 1.5 mmHg, n=10) versus the air controls (24.0 ± 1.3 mmHg, n=10, P<0.01) (Figure 1A). Nicotine-exposed α7-nAChR−/− males did not exhibit increased RVSP (25.7 ± 0.8 mmHg, n=10) compared with air-exposed α7-nAChR−/− littermates (21.6 ± 2.0 mmHg, n= 7) and had significantly lower RVSP when compared with nicotine-exposed WT males (P<0.05, Figure 1A). RVSP in air-exposed WT and α7-nAChR−/− males were comparable. The ratio of RV weight to LV and interventricular septum (LV+S) weight, referred to as the Fulton index, was measured to assess PH-induced RV hypertrophy [23]. Nicotine exposure in WT males induced a trend toward increased Fulton index (27.4 ± 0.7, n=10) versus air controls (25.4 ± 0.5, n=10, P=0.077) (Figure 1B). Fulton index was not significantly different between α7-nAChR−/− males exposed to air (25.5 ± 0.5.n=8) and nicotine (26.5 ± 0.5 n=10) (Figure 1B).
Figure 1. Chronic nicotine inhalation-induced pulmonary hypertension is mediated by α7-nAChR in male mice.
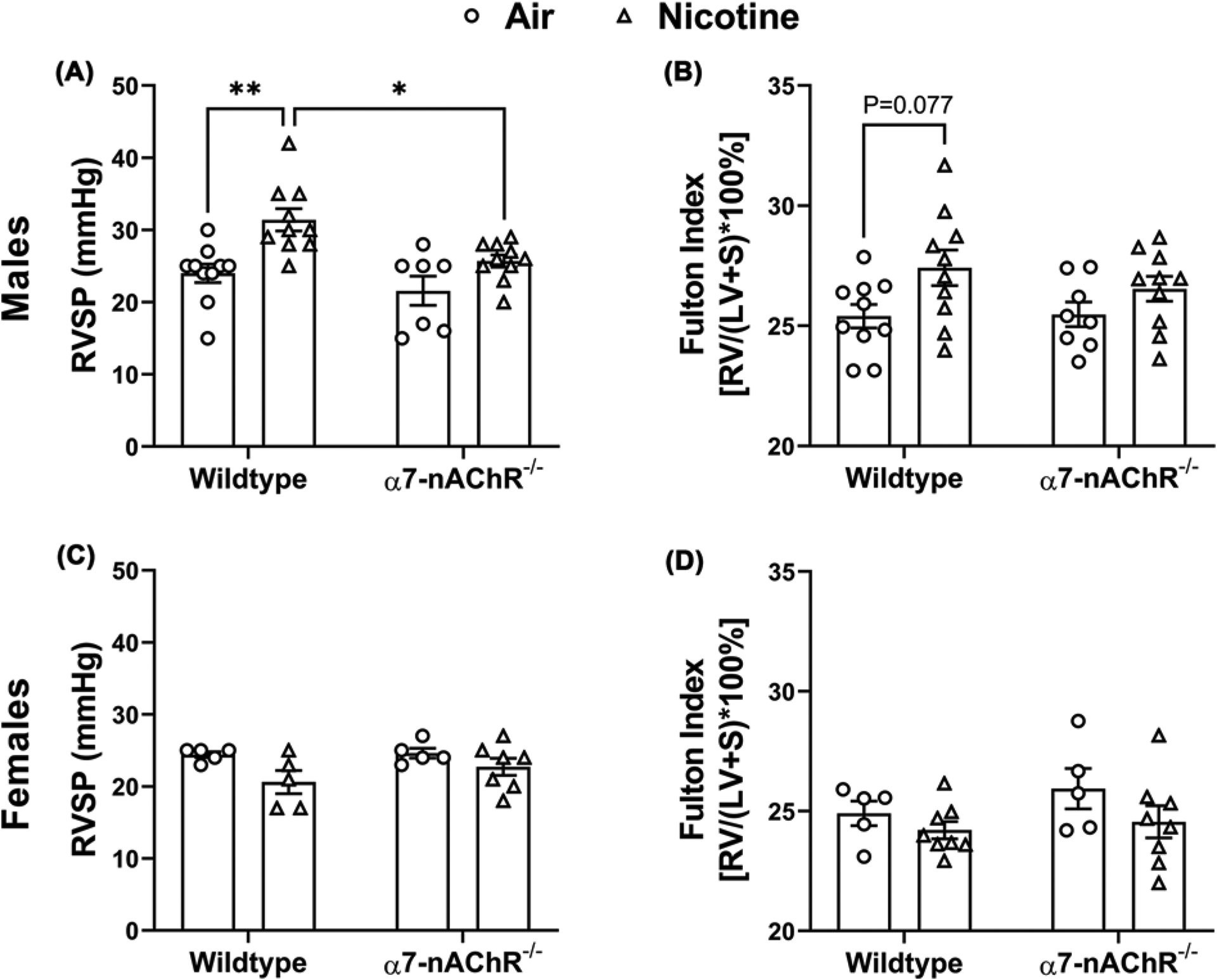
(A) Chronic nicotine inhalation leads to increased right ventricular systolic pressure (RVSP) in wild-type (n=10 air; n=10 nicotine) but not α7-nAChR−/− (n=7 air; n=10 nicotine) male mice. *P<0.05, **P<0.01. (B) Chronic nicotine inhalation results in a trend of increase in Fulton index (P=0.077) in wild-type (n=10 air; n=10 nicotine) but not α7-nAChR−/− (n=8 air; n=10 nicotine) male mice; LV, left ventricle; RV, right ventricle; S, interventricular septum. (C) Chronic nicotine inhalation does not alter RVSP in wild-type (n=5 air; n=5 nicotine) or α7-nAChR−/− (n=5 air; n=7 nicotine) female mice. (D) Chronic nicotine inhalation does not alter Fulton index in wild-type (n=5 air; n=8 nicotine) or α7-nAChR−/− (n=5 air; n=8 nicotine) female mice. The above data were analyzed by two-way ANOVA followed by Tukey–Kramer post hoc test.
In contrast our findings in male mice, chronic nicotine inhalation did not induce PH in either WT or α7-nAChR−/− females. Both RVSP and Fulton index were within normal physiologic parameters in air- or nicotine-exposed WT and α7-nAChR−/− females (Figure 1C,D). In our chronic nicotine inhalation model, serum cotinine levels were found to be consistently lower in females compared with males [24] (Supplementary Figure S1), consistent with published reports that females metabolize nicotine more rapidly than males in rodents [25], and similar to what has been observed in humans [26,27].
Chronic inhaled nicotine fails to induce RV remodeling in α7-nAChR knockout mice
RV remodeling, which is clinically associated with PH [28], was assessed by echocardiography following 8 weeks of nicotine exposure. Consistent with our previous studies [10,15], nicotine exposure in WT males resulted in an increase in RV free wall thickness during diastole (RV FWT; d, 0.43 ± 0.01 mm, n=19) versus air-exposed controls (0.30 ± 0.01 mm, n=15, P<0.0001) (Figure 2A). In contrast, nicotine exposure did not result in increased RV FWT; d in α7-nAChR−/− males (0.29 ± 0.01 mm, n=11) versus air-exposed littermates (0.31 ± 0.01 mm, n=7) (Figure 2A), and nicotine-exposed α7-nAChR−/− males had significantly lower RV FWT; d than their WT counterparts (P<0.0001). Nicotine exposure inWT males significantly increased RV internal diameter during diastole (RVID; d, 1.51 ± 0.02 mm, n=19) versus air-exposed WT males (1.26 ± 0.04 mm, n=15, P<0.0001) (Figure 2B). The above nicotine exposure associated RV dilatation was not observed in α7-nAChR−/− males (nicotine: 1.31 ± 0.05 mm, n=11; air: 1.42 ± 0.05 mm, n=7) (Figure 2B).
Figure 2. Chronic nicotine inhalation-induced RV remodeling is mediated by α7-nAChR in male mice.
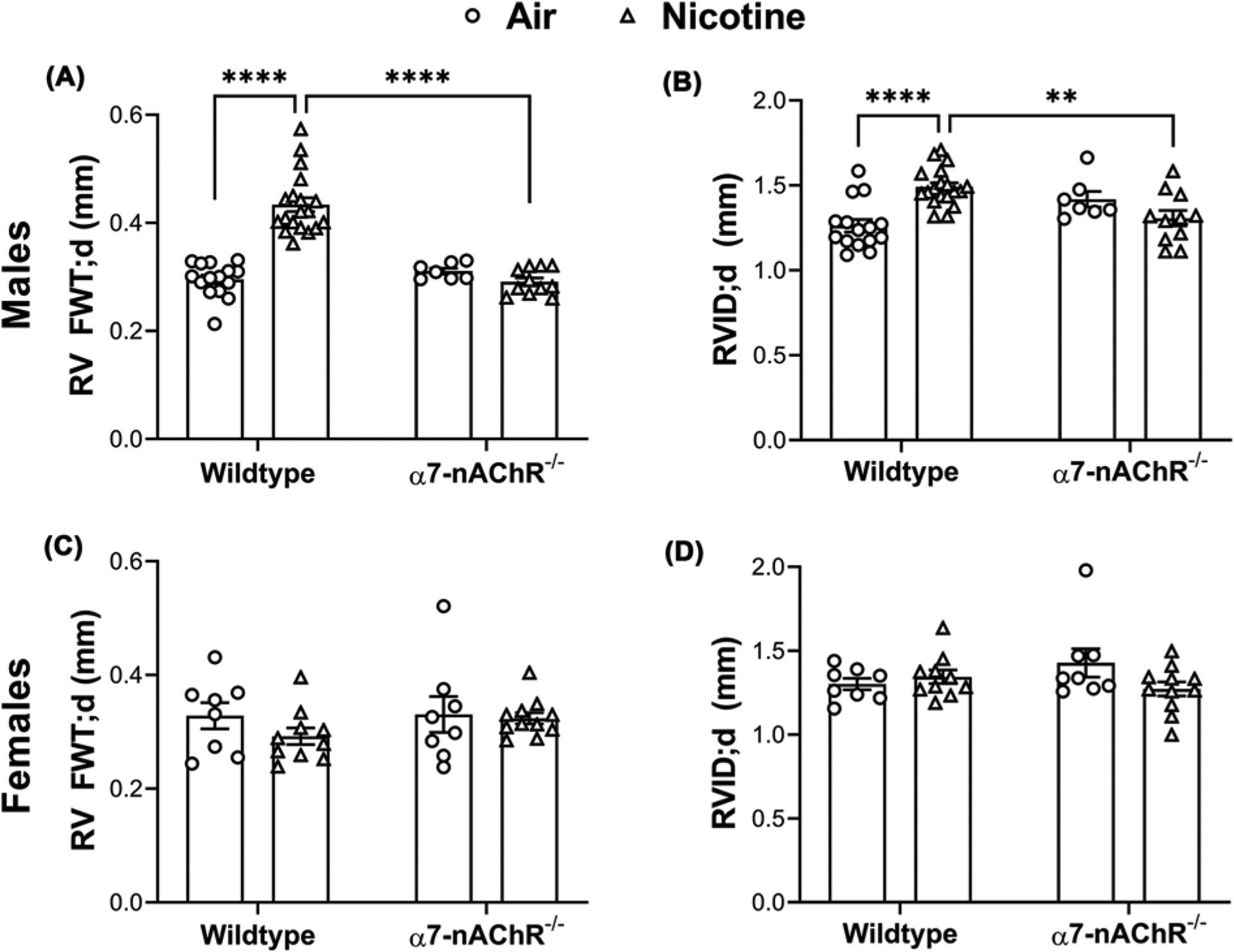
(A) Chronic nicotine inhalation leads to increased RV free wall thickness (FWT) at diastole (d) in wild-type (n=15 air; n=19 nicotine) but not α7-nAChR−/− (n=7 air; n=11 nicotine) male mice; ****P<0.0001. (B) Chronic nicotine inhalation leads to increased RV internal diameter (RVID) at diastole (d) in wild-type (n=15 air; n=19 nicotine) but not α7-nAChR−/− (n=7 air; n=11 nicotine) male mice; **P<0.01, ****P<0.0001. (C) Chronic nicotine inhalation does not alter RV free wall thickness (FWT) at diastole (d) in wild-type (n=8 air; n=10 nicotine) or α7-nAChR−/− (n=8 air; n=11 nicotine) female mice. (D) Chronic nicotine inhalation does not alter RV internal diameter (RVID) at diastole (d) in wild-type (n=8 air; n=10 nicotine) or α7-nAChR−/− (n=8 air; n=11 nicotine) female mice. The above data were analyzed by two-way ANOVA followed by Tukey–Kramer post hoc test.
In females, consistent with the absence of nicotine-induced PH, RV FWT; d and RVID; d were not altered by nicotine exposure in eitherWT or α7-nAChR−/− mice (Figure 2C,D).
LV structure and function as measured by echocardiography were unaffected by nicotine in either WT or α7-nAChR−/− mice (Table 1). There were no significant differences in LV internal diameter during systole and diastole, LV posterior wall thickness during systole and diastole, ejection fraction or fractional shortening in any of the study groups.
Table 1.
Left ventricular echocardiographic measurements in male and female mice
| Echocardiographic measurement | Air wild-type | Nicotine wild-type | Air α7-nAChR−/− | Nicotine α7-nAChR−/− |
|---|---|---|---|---|
| Males | n=15 | n=20 | n=7 | n=11 |
| LVID;s (mm) | 2.68 ± 0.10 | 2.63 ± 0.07 | 2.55 ± 0.15 | 2.52 ± 0.11 |
| LVID;d (mm) | 3.88 ± 0.10 | 3.87 ± 0.07 | 3.79 ± 0.20 | 3.81 ± 0.10 |
| LVPW;s (mm) | 1.29 ± 0.03 | 1.18 ± 0.06 | 1.18 ± 0.06 | 1.17 ± 0.04 |
| LVPW;d (mm) | 0.92 ± 0.03 | 0.83 ± 0.03 | 0.86 ± 0.06 | 0.82 ± 0.02 |
| EF (%) | 58.3 ± 2.91 | 60.9 ± 1.55 | 61.6 ± 2.41 | 63.4 ± 2.41 |
| FS (%) | 30.8 ± 1.96 | 32.3 ± 1.10 | 32.7 ± 1.72 | 34.0 ± 1.47 |
| Females | n=8 | n=10 | n=8 | n=11 |
| LVID;s (mm) | 2.76 ± 0.07 | 2.55 ± 0.05 | 2.56 ± 0.18 | 2.56 ± 0.07 |
| LVID;d (mm) | 3.80 ± 0.05 | 3.68 ± 0.04 | 3.75 ± 0.10 | 3.68 ± 0.05 |
| LVPW;s (mm) | 1.02 ± 0.02 | 1.06 ± 0.04 | 1.08 ± 0.04 | 1.02 ± 0.03 |
| LVPW;d (mm) | 0.79 ± 0.02 | 0.77 ± 0.04 | 0.73 ± 0.02 | 0.73 ± 0.01 |
| EF (%) | 53.7 ± 2.52 | 59.3 ± 1.43 | 60.4 ± 4.36 | 58.7 ± 1.97 |
| FS (%) | 27.4 ± 1.70 | 30.9 ± 0.96 | 32.3 ± 3.14 | 30.5 ± 1.28 |
Data are displayed as mean ± SEM and analyzed by two-way ANOVA; EF, ejection fraction; FS, fractional shortening; LV, left ventricle; LVID;s/d, LV internal diameter during systole/diastole; LVPW;s/d, LV posterior wall thickness during systole/diastole.
Chronic nicotine inhalation impairs endothelium-dependent vasodilation in thoracic aortas
To further examine the effects of nicotine on the vasculature and the role of the α7-nAChR, we evaluated the vasoreactivity of both thoracic aortas and PA isolated from male WT and α7-nAChR−/− mice exposed to air or nicotine vapor for 12 weeks. Although our previous study revealed only acute increases in systemic blood pressure by inhaled nicotine (1–3 weeks of nicotine vapor exposure) [10], aortas isolated from WT mice following 12-week nicotine exposure exhibited reduced relaxation responses to the endothelium-dependent vasodilator ACh, as evidenced by the rightward and upward shift in the response curve compared with aortic rings isolated from the air control mice (Figure 3A). In particular, aortic ring relaxation was significantly reduced in response to ACh concentrations ranging from 10−7 to 10−5.5 M in aortas from the nicotine group compared with air controls. However, the EC50 of ACh was unaffected by nicotine (135.5 ± 50.9 nM of ACh in nicotine-exposed compared with 59.5 ± 12.1 nM of Ach in air controls, P=0.1776). In contrast with WT mice, aortas isolated from α7-nAChR−/− mice exposed to nicotine and air showed similar vasodilatory responses to ACh (Figure 3D), indicating that nicotine-induced impairment of endothelium-dependent vasodilation is indeed mediated by α7-nAChR.
Figure 3. Chronic nicotine inhalation impairs endothelium-dependent vasodilation of thoracic aorta in wild-type but not α7-nAChR−/− male mice.
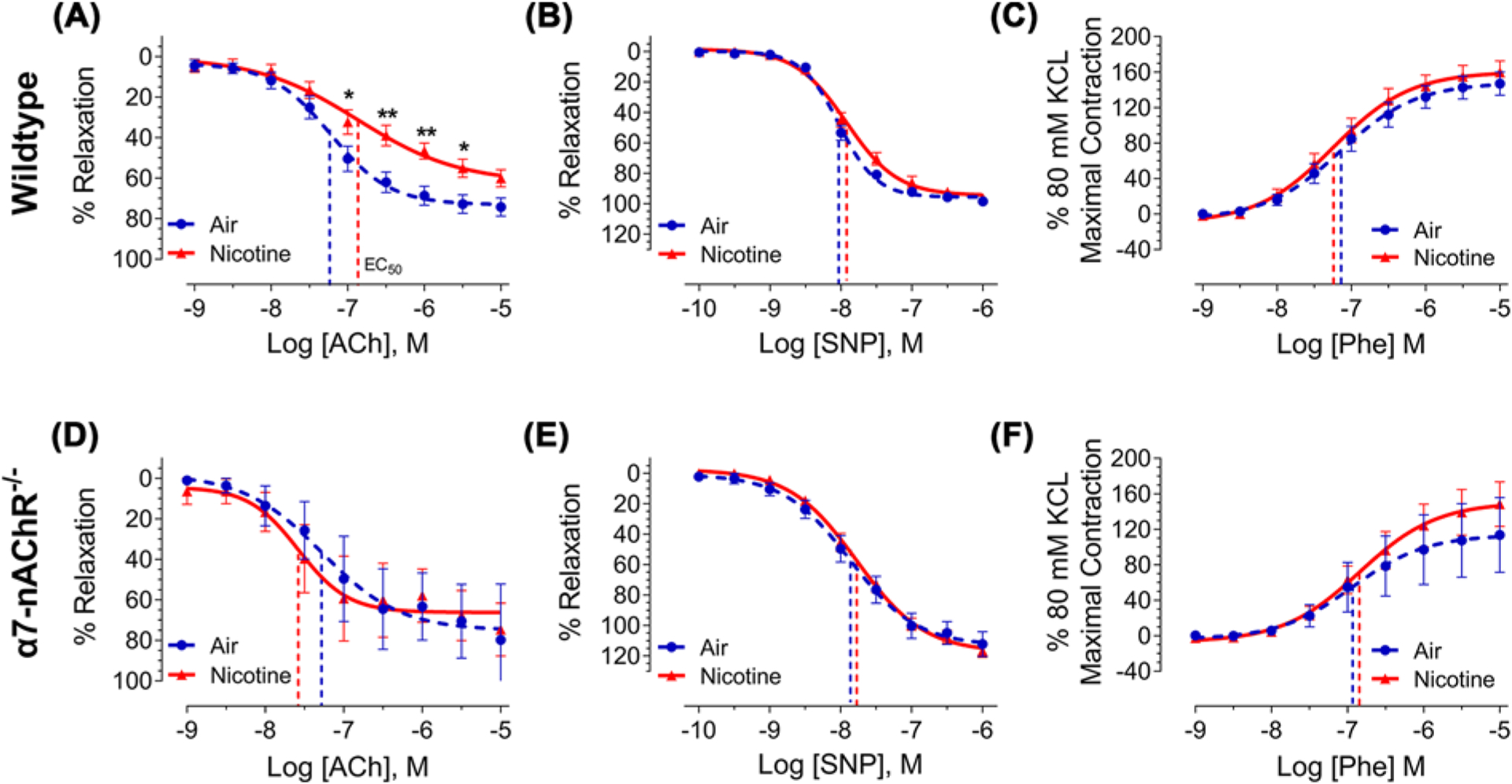
(A) Thoracic aortas isolated from wildtype male mice exposed to nicotine (n=12) exhibit reduced vasodilatory response to acetylcholine (ACh) compared with those from air-exposed males (n=11); *P<0.05, **P<0.01. The EC50 of ACh was not impacted by nicotine and is represented for each response curve as a dashed vertical line. (B and C) Chronic nicotine inhalation does not alter vasodilatory response to sodium nitroprusside (SNP) or vasoconstrictive response to phenylephrine (Phe) in thoracic aorta. Air, n=9–11; nicotine, n=10–12. (D–F) Thoracic aortas isolated from α7-nAChR−/− male mice exposed to nicotine (n=3–7) and air (n=3–6) exhibit similar vasoreactivity to ACh, SNP, and Phe. Response curves were analyzed by two-way ANOVA followed by Bonferroni’s multiple comparisons test and EC50 was analyzed by two-tailed Student’s t-test.
Aortic response to the endothelium-independent vasodilator SNP was not impacted by nicotine in either WT or α7-nAChR−/− mice (Figure 3B,E), indicating that nicotine does not alter vascular smooth muscle relaxation in response to the NO donor, SNP. Likewise, vascular smooth muscle constriction in response to Phe was not affected by nicotine in either WT or α7-nAChR−/− mice (Figure 3C,F).
Chronic nicotine inhalation alters vasoreactivity in pulmonary arteries
We further assessed the effects of nicotine on the pulmonary vasculature. Similar to our findings in thoracic aortas, the vasodilatory response of PA to ACh was impaired by nicotine in WT mice, with a rightward shift in the response curve as shown in Figure 4A. Relaxation was significantly reduced in response to 10−7.5 M and 10−7 M ACh in PA from nicotine-exposed animals compared to air controls. Furthermore, the EC50 of ACh was significantly increased (51.8 ± 5.8 nM of ACh in nicotine-exposed compared with 34.1 ± 3.1 nM of ACh in air controls, P=0.0144) (Figure 4A). Similar to thoracic aortas, PA response to SNP was not impacted by nicotine, with no change in the response curve or the EC50 (Figure 4B). Unlike thoracic aortas, however, nicotine inhalation exposure increased PA constriction in response to Phe, as evidenced by the upward and leftward shift in the response curve (Figure 4C). PA constriction was significantly increased in response to 10−5.5 M and 10−5 M of Phe in the nicotine group compared with air controls. Furthermore, the maximal contraction observed for 10−5 M Phe with respect to maximal KCl response was increased from 134% in PA from air controls to 197% in PA from nicotine-exposed mice.
Figure 4. Chronic nicotine inhalation impairs vasoreactivity of pulmonary artery in wild-type but not α7-nAChR−/− male mice.
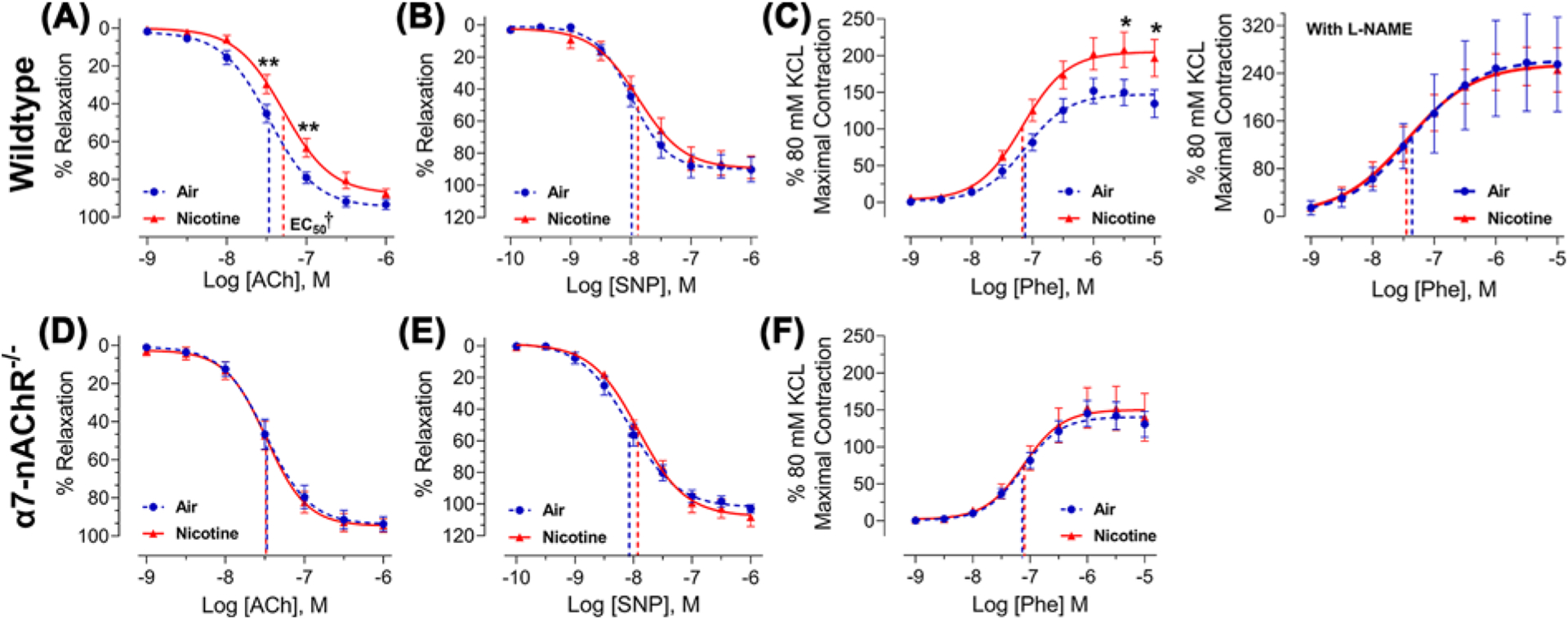
(A) Pulmonary arteries isolated from wild-type male mice exposed to nicotine (n=10) exhibit reduced vasodilatory response to acetylcholine (ACh) compared with those exposed to air (n=10); **P<0.01. The EC50 of ACh was significantly increased by nicotine and is represented for each response curve as a dashed vertical line; †P<0.05. (B) Pulmonary arteries isolated from wild-type male mice exposed to nicotine (n=6) exhibit similar vasodilatory response to sodium nitroprusside (SNP) compared with those exposed to air (n=6). (C) Pulmonary arteries isolated from wild-type male mice exposed to nicotine (n=10) exhibit enhanced vasoconstrictive response to phenylephrine (Phe) compared with those exposed to air (n=10), and pretreatment with L-NAME (10 μM) abolishes the heightened response (right, n=9/group); *P<0.05. (D–F) Pulmonary arteries isolated from α7-nAChR−/− male mice exposed to nicotine (n=7–8) and air (n=5–6) exhibit similar vasoreactivity to ACh, SNP, and Phe. Response curves were analyzed by two-way ANOVA followed by Bonferroni’s multiple comparisons test and EC50 was analyzed by two-tailed Student’s t-test.
The endothelium-dependent vasodilatory response to ACh relies on eNOS-mediated nitric oxide (NO) production in endothelial cells, and disruption in eNOS/NO signaling has been shown to also enhance vasoconstrictive response to adrenergic agonists, such as Phe [29,30]. To test this hypothesis, we analyzed Phe-induced PA vasoconstriction in the presence of L-NAME, an arginine analog that competitively inhibits eNOS-mediated NO production [31,32]. As expected (Figure 4C), pre-treatment with L-NAME increased PA response to Phe in the air control group (maximal contraction in response to 10−5 M Phe increased from 134% in the absence of L-NAME to 255% in the presence of L-NAME) due to reduced basal eNOS-mediated NO production. Importantly, pre-treatment with L-NAME abolished the difference in Phe-induced vasoconstriction between the air and nicotine groups (Figure 4C). These data indicate that the enhanced vasoconstrictive response to Phe in PA isolated from nicotine-exposed mice is likely secondary to changes in eNOS activity/NO bioavailability. Furthermore, compared with the thoracic aorta, which is not considered a reactive vessel, it is likely that the PA is more sensitive to changes in vasoreactive substances.
Similar to thoracic aortas, PA isolated from α7-nAChR−/− mice exposed to air or nicotine exhibited similar responses to ACh, SNP, and Phe (Figure 4D–F), confirming that the effects of nicotine on PA vasoreactivity are mediated by α7-nAChR.
Chronic nicotine inhalation impairs eNOS/NO signaling pathway
To directly assess the effects of nicotine on eNOS/NO signaling pathway, we performed Western blotting to examine total eNOS protein and the phosphorylation status of eNOS subunits. Activity of eNOS can be modulated through a multitude of post-translational regulatory mechanisms, including both inhibitory and stimulatory phosphorylation [33,34]. In RV samples from WT male mice chronically exposed to nicotine vapor, there was an increase in the ratio of phosphorylated eNOS at Thr495 (inhibitory phosphorylation) to total eNOS (Figure 5A), and increased Thr495 phosphorylation is associated with decreased eNOS activity [34]. In contrast, the ratio of phosphorylated Ser1177 (stimulatory phosphorylation site) to total eNOS was not significantly altered by nicotine (Figure 5A). The above changes in eNOS phosphorylation also led to an increase in the ratio of inhibitory phosphorylation (at Thr495) to stimulatory phosphorylation (at Ser1177) in WT male mice by nicotine (Figure 5A). No changes in eNOS phosphorylation at either Thr495 or Ser1177 were observed in RV from α7-nAChR−/− male mice exposed to nicotine compared with the air controls (Figure 5B), indicating that nicotine-induced changes in eNOS phosphorylation are also mediated by the α7-nAChR. Consistent with these findings, plasma nitrite levels, a biomarker of NO bioavailability and vascular function [35], were decreased in WT male mice exposed to nicotine compared with air controls (Figure 5C). In contrast, plasma nitrite levels were similar in α7-nAChR−/− male mice exposed to air or nicotine (Figure 5D).
Figure 5. Chronic nicotine inhalation leads to altered eNOS phosphorylation and reduced plasma nitrite levels in wild-type but not α7-nAChR−/− male mice.
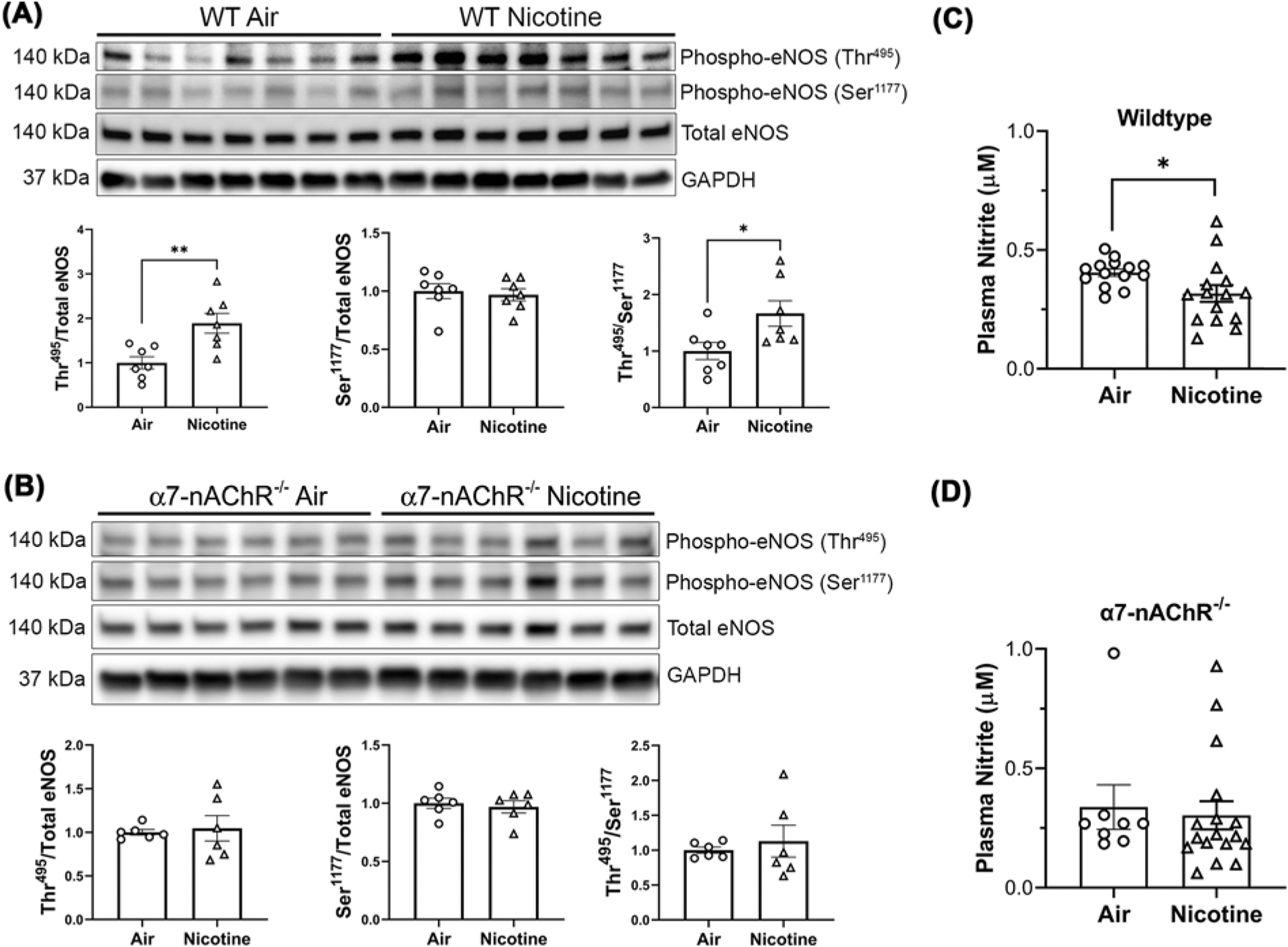
(A) Chronic nicotine inhalation leads to increased inhibitory phosphorylation of eNOS at Thr495 without significant alteration of the stimulatory phosphorylation at Ser1177 in wildtype male mice; **P<0.01. (B) Chronic nicotine inhalation does not alter the levels of total or phosphorylated eNOS in α7-nAChR−/− male mice. (C and D) Chronic nicotine inhalation leads reduced plasma nitrite levels in wild-type (n=14 air; n=15 nicotine) but not α7-nAChR−/− (n=8 air; n=17 nicotine) male mice; *P<0.05. Data were analyzed by two-tailed Student’s t-test.
To gain insight into why female mice are protected from nicotine-induced cardiopulmonary dysfunction, we examined the levels of total and phosphorylated eNOS, as well as the expression levels of α7-nAChR, in RV samples from WT females in comparison with WT males. The expression levels of α7-nAChR or total eNOS were not significantly different between WT males and females, in either nicotine exposed animals or air controls (Supplementary Figure S2). Consistent with the absence of nicotine-induced PH and RV remodeling, nicotine did not alter eNOS phosphorylation at either Thr495 or Ser1177 in WT females (Supplementary Figure S2). The above data indicate that the protection observed in females is not due to the differential expression of α7-nAChR or total eNOS between the sexes; instead, nicotine fails to alter eNOS activity (phosphorylation status) in females.
Chronic nicotine inhalation robustly alters vascular gene expression
Given the impact of nicotine inhalation exposure on vascular reactivity, we next analyzed gene expression in thoracic aortas from WT and α7-nAChR−/− male mice (n=3 per group) exposed to air or nicotine. Gene expression was robustly altered in aortas from WT male mice exposed to nicotine vapor compared with air controls, with 179 differentially expressed genes (adjusted P-value < 0.1). Among these genes, 149 were found to be up-regulated, whereas 30 were down-regulated (Figure 6A and Supplementary Table S1). In contrast, only 7 non-overlapping genes were found to be differentially expressed in α7-nAChR−/− male mice exposed to nicotine. (Figure 6A and Supplementary Table S2). The above data indicate that nicotine-induced changes in vascular gene expression is largely mediated by the α7-nAChR.
Figure 6. Chronic nicotine inhalation alters aortic gene expression in male mice.
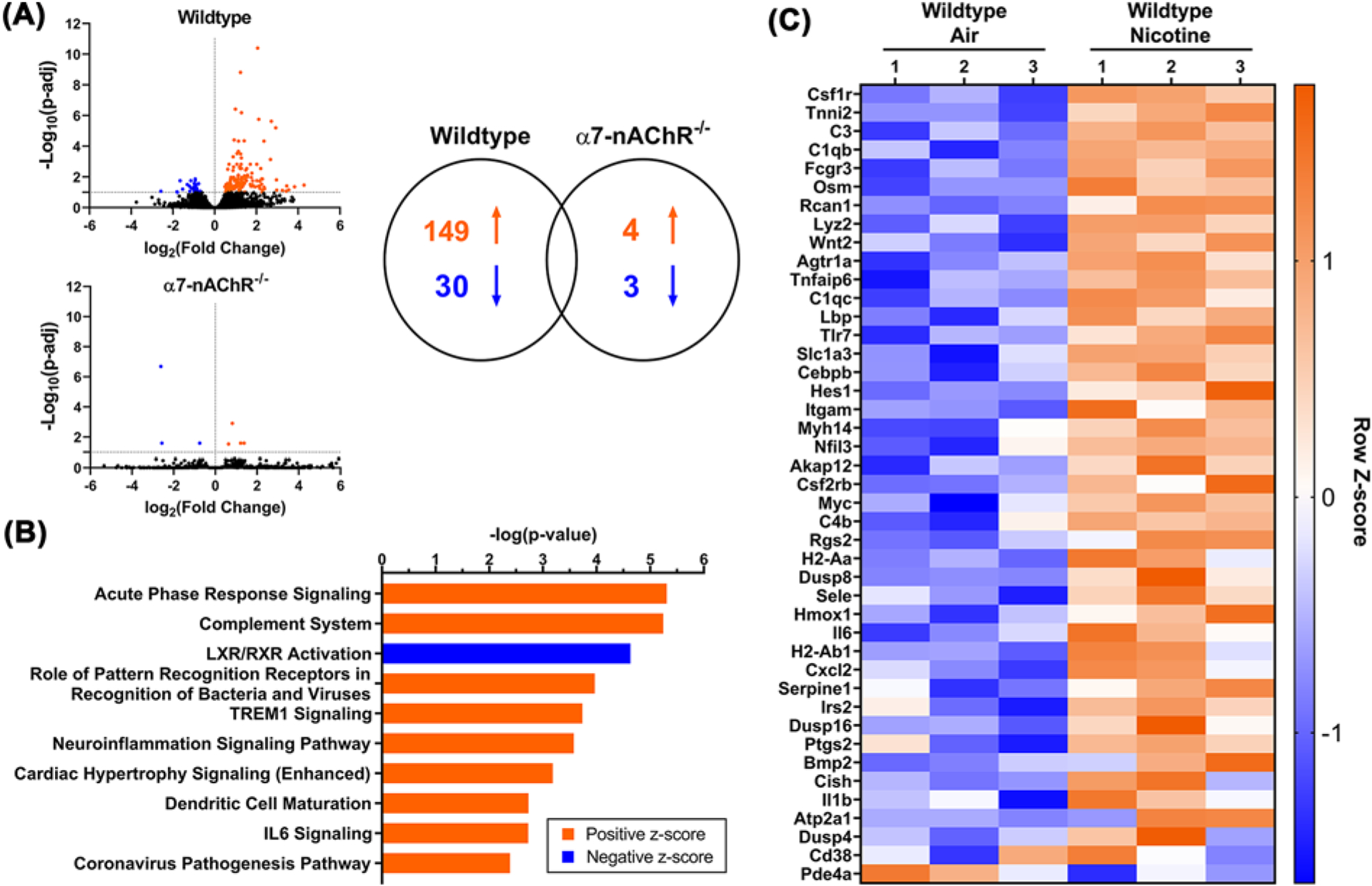
(A) RNA sequencing (n=3/group) reveals that chronic nicotine inhalation in wild-type male mice results in 179 differentially regulated genes (149 up-regulated and 30 down-regulated), while only 7 non-overlapping genes are found to be differentially regulated by nicotine in α7-nAChR−/− male mice. (B) Top-10 signaling pathways differentially regulated by nicotine in wild-type male mice. (C) Heatmap of genes identified in the pathways differentially regulated by nicotine in wild-type male mice.
Ingenuity pathway analysis revealed 21 pathways differentially regulated by nicotine in WT males, including 18 up-regulated and 3 down-regulated pathways (Supplementary Table S3). Figure 6B displays the top 10 differentially regulated pathways. Nicotine exposure activates multiple inflammatory pathways, including acute phase response (P=4.91E-06), complement system (P=5.71E-06), pathogen recognition (P=1.08E-04), IL-6 signaling (P=1.88E-03), and cardiac hypertrophy signaling (P=6.54E-04). Of the 149 genes up-regulated by nicotine inhalation exposure, the following genes are involved in three or more of the up-regulated inflammatory pathways: C3 (1.45-fold), Il6 (interleukin 6, 11.68-fold), Il1b (interleukin 1B, 5.21-fold), Osm (oncostatin M, 14.14-fold), Serpine1 (2.40-fold), Tlr7 (Toll-like receptor 7, 2.87-fold), and Ptgs2 (prostaglandin-endoperoxide synthase 2, 2.49-fold). Furthermore, Agtr1a, which encodes AT1R, was up-regulated 4.48-fold by nicotine. Figure 6C shows the heatmap of genes involved in the differentially regulated pathways in WT male mice by nicotine. None of the above gene expression changes were found in the α7-nAChR−/− male mice exposed to nicotine compared with air controls.
Discussion
Nicotine exerts its actions by binding and signaling through various nAChRs, which are expressed throughout the body in addition to the central nervous system. The α7-nAChR is widely expressed in the cardiovascular system [17]; however, the role of this receptor in nicotine-induced cardiovascular dysfunction is largely unknown. The current study fills this gap in knowledge, showing that chronic nicotine inhalation-induced PH, RV remodeling and vascular dysfunction are mediated by the α7-nAChR. Our findings are consistent with recent reports in support of a role for this receptor in PH pathogenesis and RV remodeling. Indeed, Vang et al. showed that the α7-nAChR mediates RV fibrosis and diastolic dysfunction in the Sugen/hypoxia model of PH in rats [18]. In RV samples from human patients with pulmonary arterial hypertension, α7-nAChR expression was increased, accompanied by reduced acetylcholinesterase activity compared with RV samples from control subjects [36]. The above findings indicate that α7-nAChR could serve as a novel therapeutic target in the treatment of PH and RV remodeling.
Consistent with our previous studies [10,15], chronic inhaled nicotine exposure led to the development of PH, characterized by elevated RVSP, in WT male mice. Elevated RVSP was associated with thickening of the RV wall and dilatation of the RV chamber, which predispose patients to RV failure and mortality in clinical PH [28]. These findings were absent in α7-nAChR−/− males, demonstrating an essential role of this receptor in the pathogenesis of nicotine-induced PH. The α7-nAChR is expressed on multiple cell lineages within the heart, including cardiomyocytes and cardiac fibroblasts [37], and α7-nAChR-mediated communication between these cell types is an important mechanism underlying RV fibrosis and dysfunction in Sugen/hypoxia-induced PH in rats [18]. Further studies are required to elucidate the relationship between nicotinic receptor signaling and cardiac remodeling pathways in the pathogenesis of nicotine-dependent and nicotine-independent PH. In contrast with our current and previous findings in male mice [10,15], females appear protected from nicotine-induced PH and RV remodeling following chronic inhalation exposure to nicotine. Nicotine metabolism in both rodents and humans is driven by sex-dependent differences in cytochrome P450 activity [26], and in our study, we observed consistently lower cotinine levels, suggesting more rapid nicotine metabolism, in females compared with males. A recent study in mice exposed to e-Cig containing 20.2 mg/ml of nicotine demonstrates LV dysfunction in adolescent male mice but not in adolescent or adult female mice [38]. Consistent with our study, this group also found lower serum cotinine levels in females, which were associated with a 3-fold increase in cytochrome P450 2A5 mRNA expression, further supporting more rapid nicotine metabolism in females [38]. In addition to the possible dose-dependent effect of nicotine, female sex hormones have been shown to have direct protective effects on the pulmonary vasculature [39]. The mechanisms underlying the female protection from nicotine-induced PH warrant future investigation.
Endothelial dysfunction with reduced NO bioavailability is considered an early event and a key underlying feature in most forms of PH and cardiovascular diseases. Reduced NO production by eNOS leads to increased vascular smooth muscle tone, increased vascular resistance, and vascular inflammation. It is well-documented that chronic cigarette smoke exposure in rodent models or in cultured endothelial cells results in reduced eNOS/NO signaling [40–43]. However, the mechanisms responsible for the impairment of the eNOS/NO pathway appear to be different between cigarette smoke and nicotine only exposure. Chronic cigarette smoke exposure both in vivo and in vitro leads to decreases in both total eNOS and stimulatory eNOS phosphorylation at Ser1177 [40–43]. In contrast, our study reveals that chronic exposure to inhaled nicotine alone results in elevated inhibitory eNOS phosphorylation at Thr495, without significant changes in the levels of total eNOS or stimulatory eNOS phosphorylation at Ser1177. Consistent with the increase in inhibitory eNOS phosphorylation, plasma NO bioavailability (i.e., plasma nitrite levels) was significantly reduced following nicotine exposure compared to the air controls. The above alteration of eNOS phosphorylation and NO availability are mediated by the α7-nAChR as these effects were not observed in α7-nAChR−/− mice. Vascular endothelial cells express α7-nAChR [44], and additional studies are needed to uncover the mechanisms underlying α7-nAChR-mediated alterations in eNOS phosphorylation and NO bioavailability.
Our finding that nicotine-induced changes in aortic gene expression is mediated by α7-nAChR further supports a role for this receptor in nicotine’s impact on the vasculature. While seemingly contradictory to the well-established cholinergic anti-inflammatory effects of acute nicotine, our study shows that chronic exposure to inhaled nicotine in mice elicits multiple proinflammatory pathways in the vasculature. Our results are consistent with the well-documented association between cigarette smoking and the development of atherosclerosis, where nicotine has been shown to enhance atherosclerotic progression by increasing inflammation, reactive oxygen species production, and immune cell activation [45]. Down-regulation of LXR/RXR (liver X receptor/retinoid X receptor) also supports a proinflammatory role for nicotine in the vasculature, as LXR has been found to inhibit inflammation and the development of atherosclerosis [46]. In addition, Agtr1a, which encodes AT1R, was up-regulated 4.48-fold by nicotine, indicating that altered local activity of the renin–angiotensin system in the vasculature may also play a role in nicotine-induced vascular dysfunction. This finding is in line with our previous study demonstrating that AT1R mediates the development of PH and RV remodeling following chronic nicotine inhalation [15].
In conclusion, our study reveals a pivotal role for the α7-nAChR in chronic nicotine inhalation-induced PH, RV remodeling, and vascular dysfunction including alterations in eNOS phosphorylation/NO bioavailability and impaired endothelium-dependent vasodilation. While these insights are crucial to delineating the long-term effects of nicotine exposure, there are some limitations to this study. In our nicotine exposure model, the mice are subjected to 12 h of continuous nicotine vapor inhalation each day, with serum cotinine levels comparable to mice exposed to e-Cig containing 50–60 mg/ml of nicotine [47]. While products containing similar nicotine concentrations are commercially available, including JUUL Pods with 5% nicotine (59 mg/ml), our exposure time frame may not recapitulate typical usage patterns in humans. Furthermore, while inhaled nicotine impairs cardiopulmonary function as shown in our study, it remains unknown whether nicotine replacement therapies (e.g., nicotine patches or gums) are safe as promoted or would have similar cardiovascular manifestations as inhaled nicotine. Another limitation of our study is the use of the global α7-nAChR−/− mice, which does not allow us to dissect cell type-specific contribution of the α7-nAChR in nicotine-induced cardiopulmonary dysfunction. Future studies using targeted α7-nAChR−/− mice could provide further insights. Despite the above limitations, our study strongly suggests that targeting α7-nAChR could have therapeutic potential in treating cardiopulmonary dysfunction resulting from nicotine inhalation associated with tobacco product use.
Supplementary Material
Clinical perspectives.
The popularity of e-Cig and the recent increase in conventional cigarette sales (the first time in the past 20 years primarily due to the COVID-19 pandemic) are alarming public health concerns. The general lack of knowledge of the harmful effects of nicotine on the cardiopulmonary system renders both cigarette smokers and e-Cig users vulnerable to nicotine-induced cardiopulmonary dysfunction. The present study was undertaken to further elucidate the cardiopulmonary effects of nicotine and the role of the α7-nAChR.
Our study reveals that the α7-nAChR mediates chronic nicotine inhalation-induced PH, RV remodeling, and vascular dysfunction, including alterations in gene expression, changes in eNOS phosphorylation/NO bioavailability, and impaired endothelium-dependent vasodilation.
The present study clearly demonstrates the adverse consequences of chronic nicotine inhalation on the cardiopulmonary system and the essential role of α7-nAChR in mediating the deleterious effects of nicotine. The present study furthers our understanding of the significant health risks of inhaled nicotine and strongly suggests that targeting α7-nAChR could have therapeutic potential in treating cardiopulmonary dysfunction associated with tobacco product use.
Acknowledgements
The authors thank Ms. Tamara M. Morris for her excellent technical assistance and mouse husbandry.
Funding
This study was supported by research grants from the National Institute of Health [grant numbers HL135635 (to J.D.G., E.L. and X.Y.); HL146098, HL146514 and HL151398 (to D.J.L.); and F30HL160071 (to A.K.W.)]; and from the American Heart Association [Award 829761 (to A.K.W.) and Award 20POST35200075 (to Z.L.)].
Abbreviations
- α7-nAChR
α7 nicotinic acetylcholine receptor
- ACE
angiotensin-converting enzyme
- ACh
acetylcholine
- AT1R
angiotensin-II type 1 receptor
- CVPD
cardiovascular and pulmonary diseases
- e-Cig
electronic cigarette
- eNOS
endothelial nitric oxide synthase
- LV
left ventricle
- MAPK
mitogen-activated protein kinase
- NO
nitric oxide
- PA
pulmonary arteries
- PH
pulmonary hypertension
- Phe
phenylephrine
- RVSP
right ventricular systolic pressure
- RV
right ventricular/right ventricle
- SNP
sodium nitroprusside
- WT
wild-type
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
CRediT Author Contribution
Anna K. Whitehead: Formal analysis, Funding acquisition, Investigation, Visualization, Writing—original draft, Writing—review & editing. Nicholas D. Fried: Formal analysis, Investigation, Visualization, Writing—original draft, Writing—review & editing. Zhen Li: Formal analysis, Funding acquisition, Investigation, Writing—review & editing. Kandasamy Neelamegam: Investigation, Writing—review & editing. Charlotte S. Pearson: Investigation, Writing—review & editing. Kyle B. LaPenna: Formal analysis, Investigation, Writing—review & editing. Thomas E. Sharp: Formal analysis, Investigation, Writing—review & editing. David J. Lefer: Conceptualization, Resources, Funding acquisition, Writing—review & editing. Eric Lazartigues: Conceptualization, Resources, Formal analysis, Funding acquisition, Methodology, Project administration, Writing—review & editing. Jason D. Gardner: Conceptualization, Resources, Formal analysis, Funding acquisition, Methodology, Project administration, Writing—review & editing. Xinping Yue: Conceptualization, Resources, Formal analysis, Supervision, Funding acquisition, Investigation, Visualization, Methodology, Writing—original draft, Project administration, Writing—review & editing.
Data Availability
The data underlying this article are available in the article and in its supplementary material online, and additional information is available from the corresponding authors upon request. RNA sequencing data have been deposited to the NCBI Gene Expression Omnibus (GEO) with the accession number GSE196184 [48].
References
- 1.Cornelius ME, Wang TW, Jamal A, Loretan CG and Neff LJ (2020) Tobacco product use among adults - United States, 2019. MMWR Morb. Mortal. Wkly. Rep 69, 1736–1742, 10.15585/mmwr.mm6946a4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Breland A, Soule E, Lopez A, Ramoa C, El-Hellani A and Eissenberg T (2017) Electronic cigarettes: what are they and what do they do? Ann. N. Y. Acad. Sci 1394, 5–30, 10.1111/nyas.12977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dai H and Leventhal AM (2019) Prevalence of e-cigarette use among adults in the United States, 2014–2018. JAMA 322, 1824–1827, 10.1001/jama.2019.15331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hedman L, Backman H, Stridsman C, Bosson JA, Lundback M, Lindberg A et al. (2018) Association of electronic cigarette use with smoking habits, demographic factors, and respiratory symptoms. JAMA Netw. Open 1, e180789, 10.1001/jamanetworkopen.2018.0789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammond D, Reid JL, Rynard VL, Fong GT, Cummings KM, McNeill A et al. (2019) Prevalence of vaping and smoking among adolescents in Canada, England, and the United States: repeat national cross sectional surveys. BMJ 365, l2219, 10.1136/bmj.l2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eshraghian EA and Al-Delaimy WK (2021) A review of constituents identified in e-cigarette liquids and aerosols. Tob. Prev. Cessat 7, 10, 10.18332/tpc/131111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bui LM, Keen CL and Dubick MA (1995) Comparative effects of 6-week nicotine treatment on blood pressure and components of the antioxidant system in male spontaneously hypertensive (SHR) and normotensive Wistar Kyoto (WKY) rats. Toxicology 98, 57–63, 10.1016/0300-483X(95)91102-U [DOI] [PubMed] [Google Scholar]
- 8.Lau PP, Li L, Merched AJ, Zhang AL, Ko KW and Chan L (2006) Nicotine induces proinflammatory responses in macrophages and the aorta leading to acceleration of atherosclerosis in low-density lipoprotein receptor(−/−) mice. Arterioscler. Thromb. Vasc. Biol 26, 143–149, 10.1161/01.ATV.0000193510.19000.10 [DOI] [PubMed] [Google Scholar]
- 9.Davis R, Rizwani W, Banerjee S, Kovacs M, Haura E, Coppola D et al. (2009) Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS ONE 4, e7524, 10.1371/journal.pone.0007524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oakes JM, Xu J, Morris TM, Fried ND, Pearson CS, Lobell TD et al. (2020) Effects of chronic nicotine inhalation on systemic and pulmonary blood pressure and right ventricular remodeling in mice. Hypertension 75, 1305–1314, 10.1161/HYPERTENSIONAHA.119.14608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cellina GU, Honour AJ and Littler WA (1975) Direct arterial pressure, heart rate, and electrocardiogram during cigarette smoking in unrestricted patients. Am. Heart J 89, 18–25, 10.1016/0002-8703(75)90004-6 [DOI] [PubMed] [Google Scholar]
- 12.Groppelli A, Giorgi DM, Omboni S, Parati G and Mancia G (1992) Persistent blood pressure increase induced by heavy smoking. J. Hypertens 10, 495–499, 10.1097/00004872-199205000-00014 [DOI] [PubMed] [Google Scholar]
- 13.Haass M and Kubler W (1997) Nicotine and sympathetic neurotransmission. Cardiovasc. Drugs Ther 10, 657–665, 10.1007/BF00053022 [DOI] [PubMed] [Google Scholar]
- 14.Najem B, Houssiere A, Pathak A, Janssen C, Lemogoum D, Xhaet O et al. (2006) Acute cardiovascular and sympathetic effects of nicotine replacement therapy. Hypertension 47, 1162–1167, 10.1161/01.HYP.0000219284.47970.34 [DOI] [PubMed] [Google Scholar]
- 15.Fried ND, Morris TM, Whitehead A, Lazartigues E, Yue X and Gardner JD (2021) Angiotensin II type 1 receptor mediates pulmonary hypertension and right ventricular remodeling induced by inhaled nicotine. Am. J. Physiol. Heart Circ. Physiol 320, H1526–H1534, 10.1152/ajpheart.00883.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albuquerque EX, Pereira EF, Alkondon M and Rogers SW (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev 89, 73–120, 10.1152/physrev.00015.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitehead AK, Erwin AP and Yue X (2021) Nicotine and vascular dysfunction. Acta. Physiol. (Oxf.) 231, e13631, 10.1111/apha.13631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vang A, da Silva Goncalves Bos D, Fernandez-Nicolas A, Zhang P, Morrison AR, Mancini TJ et al. (2021) alpha7 Nicotinic acetylcholine receptor mediates right ventricular fibrosis and diastolic dysfunction in pulmonary hypertension. JCI Insight 6, e142945, 10.1172/jci.insight.142945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orr-Urtreger A, Goldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M et al. (1997) Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J. Neurosci 17, 9165–9171, 10.1523/JNEUROSCI.17-23-09165.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bryan NS, Calvert JW, Elrod JW, Gundewar S, Ji SY and Lefer DJ (2007) Dietary nitrite supplementation protects against myocardial ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U. S. A 104, 19144–19149, 10.1073/pnas.0706579104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elrod JW, Calvert JW, Gundewar S, Bryan NS and Lefer DJ (2008) Nitric oxide promotes distant organ protection: evidence for an endocrine role of nitric oxide. Proc. Natl. Acad. Sci. U. S. A 105, 11430–11435, 10.1073/pnas.0800700105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calvert JW, Condit ME, Aragon JP, Nicholson CK, Moody BF, Hood RL et al. (2011) Exercise protects against myocardial ischemia-reperfusion injury via stimulation of beta(3)-adrenergic receptors and increased nitric oxide signaling: role of nitrite and nitrosothiols. Circ. Res 108, 1448–1458, 10.1161/CIRCRESAHA.111.241117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Schreier DA, Hacker TA and Chesler NC (2013) Progressive right ventricular functional and structural changes in a mouse model of pulmonary arterial hypertension. Physiol. Rep 1, e00184, 10.1002/phy2.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitehead AK, Meyers MC, Taylor CM, Luo M, Dowd SE, Yue X et al. (2022) Sex-dependent effects of inhaled nicotine on the gut microbiome. Nicotine Tob. Res, 10.1093/ntr/ntac064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen K, Kanamori K, Shin CS, Hamid A and Lutfy K (2020) The impact of sex on changes in plasma corticosterone and cotinine levels induced by nicotine in C57BL/6J Mice. Brain Sci. 10, 705, 10.3390/brainsci10100705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benowitz NL, Lessov-Schlaggar CN, Swan GE and Jacob P III (2006) Female sex and oral contraceptive use accelerate nicotine metabolism. Clin. Pharmacol. Ther 79, 480–488, 10.1016/j.clpt.2006.01.008 [DOI] [PubMed] [Google Scholar]
- 27.Kandel DB, Hu MC, Schaffran C, Udry JR and Benowitz NL (2007) Urine nicotine metabolites and smoking behavior in a multiracial/multiethnic national sample of young adults. Am. J. Epidemiol 165, 901–910, 10.1093/aje/kwm010 [DOI] [PubMed] [Google Scholar]
- 28.Lan NSH, Massam BD, Kulkarni SS and Lang CC (2018) Pulmonary arterial hypertension: pathophysiology and treatment. Diseases 6, 38, 10.3390/diseases6020038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carrier GO and White RE (1985) Enhancement of alpha-1 and alpha-2 adrenergic agonist-induced vasoconstriction by removal of endothelium in rat aorta. J. Pharmacol. Exp. Ther 232, 682–687 [PubMed] [Google Scholar]
- 30.Young MA and Vatner SF (1986) Enhanced adrenergic constriction of iliac artery with removal of endothelium in conscious dogs. Am. J. Physiol 250, H892–H897, 10.1152/ajpheart.1986.250.5.H892 [DOI] [PubMed] [Google Scholar]
- 31.Pfeiffer S, Leopold E, Schmidt K, Brunner F and Mayer B (1996) Inhibition of nitric oxide synthesis by NG-nitro-L-arginine methyl ester (L-NAME): requirement for bioactivation to the free acid, NG-nitro-L-arginine. Br. J. Pharmacol 118, 1433–1440, 10.1111/j.1476-5381.1996.tb15557.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Langen J, Fransen P, Van Hove CE, Schrijvers DM, Martinet W, De Meyer GR et al. (2012) Selective loss of basal but not receptor-stimulated relaxation by endothelial nitric oxide synthase after isolation of the mouse aorta. Eur. J. Pharmacol 696, 111–119, 10.1016/j.ejphar.2012.09.016 [DOI] [PubMed] [Google Scholar]
- 33.Dudzinski DM and Michel T (2007) Life history of eNOS: partners and pathways. Cardiovasc. Res 75, 247–260, 10.1016/j.cardiores.2007.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michell BJ, Chen Z, Tiganis T, Stapleton D, Katsis F, Power DA et al. (2001) Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J. Biol. Chem 276, 17625–17628, 10.1074/jbc.C100122200 [DOI] [PubMed] [Google Scholar]
- 35.Lauer T, Preik M, Rassaf T, Strauer BE, Deussen A, Feelisch M et al. (2001) Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc. Natl. Acad. Sci. U. S. A 98, 12814–12819, 10.1073/pnas.221381098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.da Silva Goncalves Bos D, Van Der Bruggen CEE, Kurakula K, Sun XQ, Casali KR, Casali AG et al. (2018) Contribution of impaired parasympathetic activity to right ventricular dysfunction and pulmonary vascular remodeling in pulmonary arterial hypertension. Circulation 137, 910–924, 10.1161/CIRCULATIONAHA.117.027451 [DOI] [PubMed] [Google Scholar]
- 37.Dvorakova M, Lips KS, Bruggmann D, Slavikova J, Kuncova J and Kummer W (2005) Developmental changes in the expression of nicotinic acetylcholine receptor alpha-subunits in the rat heart. Cell Tissue Res. 319, 201–209, 10.1007/s00441-004-1008-1 [DOI] [PubMed] [Google Scholar]
- 38.Neczypor EW, Saldana TA, Mears MJ, Aslaner DM, Escobar YH, Gorr MW et al. (2022) e-Cigarette aerosol reduces left ventricular function in adolescent mice. Circulation 145, 868–870, 10.1161/CIRCULATIONAHA.121.057613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriguez-Arias JJ and García-Álvarez A (2021) Sex differences in pulmonary hypertension. Front. Aging 2, 727558, 10.3389/fragi.2021.727558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horinouchi T, Mazaki Y, Terada K and Miwa S (2020) Cigarette smoke extract and its cytotoxic factor acrolein inhibit nitric oxide production in human vascular endothelial cells. Biol. Pharm. Bull 43, 1804–1809, 10.1248/bpb.b20-00522 [DOI] [PubMed] [Google Scholar]
- 41.Kim SY, Kim HJ, Park MK, Huh JW, Park HY, Ha SY et al. (2016) Mitochondrial E3 ubiquitin protein ligase 1 mediates cigarette smoke-induced endothelial cell death and dysfunction. Am. J. Respir. Cell Mol. Biol 54, 284–296, 10.1165/rcmb.2014-0377OC [DOI] [PubMed] [Google Scholar]
- 42.Marwick JA, Edirisinghe I, Arunachalam G, Stevenson CS, Macnee W, Kirkham PA et al. (2010) Cigarette smoke regulates VEGFR2-mediated survival signaling in rat lungs. J. Inflamm. (Lond.) 7, 11, 10.1186/1476-9255-7-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edirisinghe I, Yang SR, Yao H, Rajendrasozhan S, Caito S, Adenuga D et al. (2008) VEGFR-2 inhibition augments cigarette smoke-induced oxidative stress and inflammatory responses leading to endothelial dysfunction. FASEB J. 22, 2297–2310, 10.1096/fj.07-099481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heeschen C, Weis M, Aicher A, Dimmeler S and Cooke JP (2002) A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J. Clin. Invest 110, 527–536, 10.1172/JCI0214676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu X, Zong T, Yang P, Li L, Wang S, Wang Z et al. (2021) Nicotine: regulatory roles and mechanisms in atherosclerosis progression. Food Chem. Toxicol 151, 112154, 10.1016/j.fct.2021.112154 [DOI] [PubMed] [Google Scholar]
- 46.Zelcer N and Tontonoz P (2006) Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Invest 116, 607–614, 10.1172/JCI27883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El-Mahdy MA, Mahgoup EM, Ewees MG, Eid MS, Abdelghany TM and Zweier JL (2021) Long-term electronic cigarette exposure induces cardiovascular dysfunction similar to tobacco cigarettes: role of nicotine and exposure duration. Am. J. Physiol. Heart Circ. Physiol 320, H2112–H2129, 10.1152/ajpheart.00997.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yue X and Whitehead A (2022) Alpha7 nicotinic acetylcholine receptor mediates chronic nicotine inhalation-induced cardiopulmonary dysfunction. NCBI Gene Expression Omnibus, GSE196184 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its supplementary material online, and additional information is available from the corresponding authors upon request. RNA sequencing data have been deposited to the NCBI Gene Expression Omnibus (GEO) with the accession number GSE196184 [48].