

Abstract
While research into drug–target interaction (DTI) prediction is fairly mature, generalizability and interpretability are not always addressed in the existing works in this field. In this paper, we propose a deep learning (DL)-based framework, called BindingSite-AugmentedDTA, which improves drug–target affinity (DTA) predictions by reducing the search space of potential-binding sites of the protein, thus making the binding affinity prediction more efficient and accurate. Our BindingSite-AugmentedDTA is highly generalizable as it can be integrated with any DL-based regression model, while it significantly improves their prediction performance. Also, unlike many existing models, our model is highly interpretable due to its architecture and self-attention mechanism, which can provide a deeper understanding of its underlying prediction mechanism by mapping attention weights back to protein-binding sites. The computational results confirm that our framework can enhance the prediction performance of seven state-of-the-art DTA prediction algorithms in terms of four widely used evaluation metrics, including concordance index, mean squared error, modified squared correlation coefficient (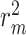 ) and the area under the precision curve. We also contribute to three benchmark drug–traget interaction datasets by including additional information on 3D structure of all proteins contained in those datasets, which include the two most commonly used datasets, namely Kiba and Davis, as well as the data from IDG-DREAM drug-kinase binding prediction challenge. Furthermore, we experimentally validate the practical potential of our proposed framework through in-lab experiments. The relatively high agreement between computationally predicted and experimentally observed binding interactions supports the potential of our framework as the next-generation pipeline for prediction models in drug repurposing.
) and the area under the precision curve. We also contribute to three benchmark drug–traget interaction datasets by including additional information on 3D structure of all proteins contained in those datasets, which include the two most commonly used datasets, namely Kiba and Davis, as well as the data from IDG-DREAM drug-kinase binding prediction challenge. Furthermore, we experimentally validate the practical potential of our proposed framework through in-lab experiments. The relatively high agreement between computationally predicted and experimentally observed binding interactions supports the potential of our framework as the next-generation pipeline for prediction models in drug repurposing.
Keywords: deep learning, drug–target affinity, binding sites, machine learning, drug–target interaction, SARS-CoV-2, DTA software, DTA database
INTRODUCTION
One critical step in drug designing and development is the identification of novel drug–target interactions (DTIs), which characterize the binding of innovative candidate drug compounds to particular protein targets. Such experiments are often highly automated with chemical libraries for test compounds screened for activity in high-throughput methodologies. However, considering the enormous chemical and proteomic spaces, identifying novel compounds with specific interactive characteristics toward a particular protein target(s), if feasible, is often expensive, time- and resource-consuming [1, 2]. To relieve this bottleneck, the use of computational methods is urgent to narrow down the search space for novel DTIs by predicting or estimating the interaction (strength) of novel drug–target (DT) pairs. A large body of studies proposed computational methods for DTI prediction tasks. Among these computational methods, docking simulations and machine learning methods are the two main approaches for in-silico prediction of DTI. Despite the accuracy and good interpretability of simulation and molecular docking, [3] in virtual screening tasks, the performance of these structure-based computational methods highly depends on the high-resolution 3D structure of protein data and their availability. Additionally, these methods typically require tremendous computational resources, which tends to be a bottleneck for computational speed and prevents any large-scale applications of the methods. To overcome some of these hurdles, artificial intelligence (AI), including machine learning (ML) and deep learning (DL) algorithms, have been introduced and applied across different stages of drug development and design pipelines. These models generally work by translating available knowledge about known drugs and their targets into some features, then used to train models that can predict interactions between new drugs or new targets. Earlier traditional ML methods use shallow models such as support vector machines, logistic regression, random forest, and shallow neural networks [4–7] to address the problem of DTI prediction/estimation. However, these shallow models are often inadequate in capturing crucial features needed in modeling of complex interactions. Recently and powered by the increase in data and high-capacity computing machines, DL models have gained increased attention to address diverse problems in bioinformatics/cheminformatics applications, including drug discovery, especially DTI prediction tasks. DL approaches advance traditional shallow ML models due to their ability to automatically learn and extract feature representation, therefore identifying, processing and extrapolating complex hidden interactions between drugs and targets [8–11]. Since the focus of this study is DL models, which have gained much momentum over the last decades, we dedicate the entire Section 3 to discussing the state-of-the-arts that use DL approaches to address the problem of DTI prediction. In this study, we propose a computational-experimental framework that enables the next-generation pipeline for developing generalizable, interpretable and more accurate prediction models in drug-repurposing applications. At the core of our framework is an augmented-DTA in-silico prediction module, which utilizes a graph convolutional neural network (GCNN)-based model called AttentionSiteDTI that acts as a detector to identify the most probable binding sites of the target protein. This critical information is then augmented into a DL-based DTA prediction model to predict the binding affinities between DT pairs to enhance their prediction performance by narrowing down the search space of binding sites to the most promising ones. This approach is significant due to depending on the protein-binding sites that are particularly sensitive and can be used to identify the ligands’ proper binding interactions [12]. As the computational results show, our framework leads to significantly improved performance of many state-of-the-art DTA prediction models in multiple evaluation metrics. We further design an in-vitro validation module, where we validate the practical application of our framework by comparing the computationally predicted DTA values with those experimentally observed (measured) in the laboratory for several candidate compounds interacting with several target proteins. Also, our in-lab experimental validation illustrates improved agreement between computationally predicted and experimentally observed binding affinities between candidate compounds and proteins. Encouraged by the computational and experimental results, we then utilize our framework to accelerate the process of hit compounds identification in drug repurposing with the case study of SARS-CoV-2. Visualization of the proposed computational–experimental framework is illustrated in section 1. It consists of three main modules: an end-to-end graph-based DL model, namely AttentionSiteDTI, to identify the most probable binding sites of the proteins. In the next step, this crucial piece of information is augmented with the DL model of our choice to perform in silico predictions of DTA values. Indeed, the role of AttentionSiteDTI is to narrow down the search space of regression models and to make the problem easier for them to learn, which leads to more accurate results. Finally in the third module, through an in-vitro validation, we validate the practical potential of the models in prediction of binding interactions in real-world applications.
Figure 1.

Our proposed framework includes three main modules: (1) in-silico prediction that consists of a DL-based DTA prediction model augmented with AttentionSiteDTI for finding the most probable binding sites of proteins; (2) in-vitro validation, where we compare our computationally-predicted results with experimentally measured DTA values in laboratory to test and validate the practical potential of our proposed framework and (3) drug-repurposing module that utilizes our prediction model to identify hit compounds in prioritizing the most potent interactions for further in-vitro or ex-vivo verification in the laboratory.
Over the last decade, most of the computational methods regarded DTI prediction as a binary classification problem, where the goal is to determine whether or not a drug–target (DT) pair interacts [13]. These methods ignore a vitally significant piece of information, namely the DT-binding affinity values, reflecting the strength of interaction in DT pairs [14]. Although there exist approaches that treat the problem of DTI prediction using a regression framework, we argue that the performance of these regression models can be improved using a more efficient approach, wherein we first narrow down the search space by identifying the most probable binding sites of the protein in a DT pair, and then use this auxiliary piece of information in prediction of binding affinity values. To achieve this, we introduce a novel DL-based framework, BindingSite-AugmentedDTA, which not only improves the generalization and interpretation capabilities of the DTA prediction task but also leads to enhanced performance of many state-of-the-art models.
Despite the significant efforts that have been devoted to the development of computational models in recent years, their prediction power is rarely evaluated through in-lab experiments, which makes their practical benefits unknown, and thus limits their potential application in drug discovery and repurposing. The work in [15] is the one of the few studies (if not the only one) that introduced a computational–experimental framework to evaluate the benefit of a kernel-based prediction model in prediction of DTI. Therefore, it is highly imperative to assess the prediction performance of in-silico models through experimental investigations in the laboratory in order to evaluate their practical power and to ensure their reliability in real-world predictions.
Contribution
The main contributions of our study are summarized as follows.
-
(i) We build an experimental–computational pipeline based on a graph convolutional neural network model, which is computationally and experimentally proven to lead to improved prediction performance when integrated with many state-of-the-art DL-based DTA prediction models. Also, our framework benefits from high generalizability and interpretability in prediction of DTA values:
(a) Our model is shown to significantly improve the prediction performance of many state-of-the-art models in terms of several performance metrics. The boost in the performance of these models is achieved by making them selectively focus on the most relevant parts of the input proteins when learning interaction relationships in DT pairs.
(b) Our model is highly generalizable because of to two main reasons: first is that it can be easily integrated with any DL-based prediction model, and the second is due to our target protein input representation that uses protein pockets (i.e. molecular fragments of binding sites influencing binding character) encoded as graphs. This helps the model concentrate on learning generic topological features from protein pockets, which can be generalized to new proteins that are not similar to the ones in the training data.
(c) Our model is highly interpretable in terms of the language of protein-binding sites. Our model’s self-attention mechanism enables interpretability by understanding the prediction mechanism behind the model. In essence, this interpretability is enabled through learning which parts of the protein are more relevant in interacting with a given ligand. This is especially important in designing and developing new pharmaceutically active molecules, where it is crucial to know which parts of a molecule are essential for its biological properties.
(ii) We demonstrate the prediction power of our model in practical applications through conducting in-vitro experiments. To achieve this, we compare the computationally predicted binding affinities (between some candidate compounds and a target protein) against the experimentally measured binding affinities for the corresponding pairs. This is one of the main contributions of this work, as most of the in silico prediction models lack experimental validation, which is the ultimate determinant in assessing the validity of a computational model. Molecular dynamics (MD) simulations are additionally used as another computational yardstick to compare the in-lab experiments besides DL.
(iii) Finally, inspired by the good performance of our proposed framework, we utilize it for drug repurposing by predicting binding affinity values between FDA-approved drugs and key proteins of SARS-CoV-2 including spike, 3C-like protease, RNA-dependent RNA polymerase (RdRp), helicase as well as Spike/ACE2 complex. We then provide a reference list of the top-ranked FDA-approved antiviral drugs with good binding affinities targeting different proteins of SARS-CoV-2.
(iv) As a part of our work, we also contribute to four benchmarking datasets used for DTI prediction tasks. These datasets include KIBA and Davis, as the two most widely used datasets in the DTA prediction task, as well as the recent IDG-DREAM Drug-Kinase challenge dataset [16], as it contains activity data on understudied human kinomes, so-called dark kinases. (see supplementary material for more details on IDG-DREAM challenge data and results). These datasets lack information on the 3D structure of proteins, which might be required by many advanced prediction models, including ours. We extract this information from Protein Data Bank (PDB) files of proteins available at https://www.uniprot.org/, and we provide the community with more complete versions of these datasets.
BindingSite-AugmentedDTA FOR In-Silico PREDICTION
We utilize our recently developed model, called AttentionSiteDTI [17], to improve the prediction performance of state-of-the-art DTA regression models. Our model finds the probability with which each binding site (pocket) of the protein will interact with the drug. Finding the most probable binding sites of the protein, we can then integrate this critical auxiliary information with DTA prediction models to enhance their performance. We refer to this customized version as BindingSite-AugmentedDTA, which can utilize any DL-based regression model to perform DTA prediction task. Given the fact that intermolecular interactions between protein and ligand occur in pocket-like regions of the protein (binding sites), identification of the most probable binding sites can help narrow down the search space, which, in turn, leads to more efficient and accurate DTA predictions.
As the computational and experimental results confirm, our model not only improves the prediction performance of state-of-the-art models, but also our self-attention bidirectional long short-term memory (LSTM) mechanism is proven to be useful in capturing the desired interaction features, which provides interpretability to the predictions. The results illustrate that our regression-based framework offers a more realistic, interpretable, yet accurate formulation of the DTI prediction task in practical applications. A brief description of our model is provided in the supplementary material section 1.1, and our detailed approach is documented in [17]. Figure 2 provides visualizations of our attention-based interpretation module where it detects the most probable binding sites of the main proteins of SARS-CoV-2 in interaction with the drug named Remdesivir. The interpretability of AttentionSiteDTI is enabled through a self-attention mechanism, which makes the model learn which parts of the protein interact with the ligand in a given DT pair. The use of such mechanism is facilitated due to our novel formulation of DTI prediction problem as a sentence classification problem in NLP, where the drug–target complex is treated as a sentence with relational meaning between its biochemical entities a.k.a. protein pockets and drug molecule. That being said, the self-attention module works by comparing every binding site of the protein to the drug in the biochemical sentence of drug–target complex, and reweighing the embeddings of each binding pocket to include contextual relevance. The self-attention block is comprised of three main steps. Dot product to determine the alignment score by calculating similarities between each pair of entities (words) in the sentence; normalization of the alignment scores to obtain a set of scaled weights; reweighing of the original embeddings using the weights to return new contextualized word embeddings. We refer the interested readers to the original work in [17] for more detailed description on this approach. Moreover, we provide in the Appendix, a visualized description of how self-attention mechanism works to facilitate interpretability.
Figure 2.
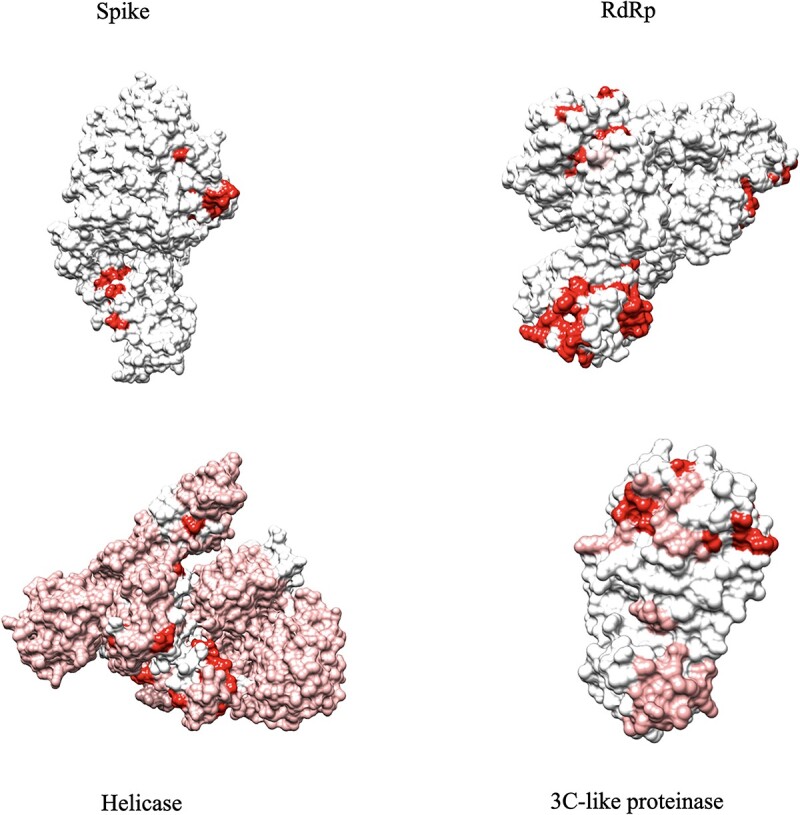
Pojected heatmap of self-attention weights on the 4 main proteins of SARS-CoV-2. This figure shows the interpretability of our model, which can give us the binding site that has the most probability of binding to the ligand.
Also, the overall architecture of our AttentionSiteDTI is shown in the Supplementary materials Figure 1.
RELATED WORK: DEEP LEARNING FOR DRUG–TARGET AFFINITY PREDICTION
Motivated by the success of the first studies that employed DL methods to model DTI, there has been an increasing number of later studies that adopted new DL architectures, such as convolutional neural networks (CNNs) and recurrent neural networks (RNNs) [18, 19] as well as stacked-autoencoders [20] to perform DTI binary interaction prediction using different input models for proteins and drugs. Apart from different types of neural networks, various representations of the input data for the drug and target have been applied to train DL models for DTI prediction. These models can be categorized into two main branches in terms of data representation. First is sequence representation-based approaches, which take as input the sequence information of drugs and targets (e.g. SMILES for drugs and amino acid sequence for proteins). The second category includes those approaches with graph representations for drug and/or protein (e.g. graph representations for drugs and amino acid sequence for proteins).
A major limitation of the former is that these models use string representations for the drug compounds. However, this is not a natural and effective way to characterize molecules. Among all these approaches, we can refer to DeepDTA as the first approach that was introduced by [9] to predict binding affinity as opposed to a binary (interact, no-interact) value. This model uses SMILES to represent drug input data and amino acid sequences to represent protein input data. Also, Integer/label encoding was used to encode both drug SMILES and the protein sequences. A CNN model with three 1D convolutional layers followed by a max-pooling function (called the first CNN block) was then applied to each drug embedding. The latest features for each protein are also learned using an identical CNN block. The feature vectors for each DT pair are then concatenated and fed into three FC layers and finally regressed with the DTA scores. WideDTA is an attempt to improve upon DeepDTA, which was introduced by the same authors [11]. Along with SMILES and amino acid sequences, WideDTA uses two other text-based information sources, which are ligand maximum common substructure (LMCS) for drugs, and protein domains and motifs (PDM) based on PROSITE for proteins. Unlike DeepDTA, this model is a word-based model. That being said, both ligand SMILES and protein (amino acid) sequences are represented using a set of words instead of characters. More specifically, they represent a word in a drug SMILES using eight residues in the sequence and a protein with three residues in the sequence. A CNN model with four identical feature extracting blocks (extracting features from each of the text-based information sources) is then used to predict binding affinity scores. The concatenated features are then fed into 3 FC layers with two dropout layers to avoid overfitting. The output of the network is the scores for binding affinity for DT pairs. AttentionDTA [21] is another DL model that has been developed to predict compound–protein affinity using the semantic information of the drug’s SMILES string and protein’s amino acid sequence. This model uses two separate one-dimensional CNNsonvolution Neural Networks along with four different attention mechanisms to explore the relationship between drug and protein features.
As mentioned earlier, these sequence representations-based models fail at capturing the structural information of molecules, which in turn leads to the degraded predictive performance of the predictive model. On the other hand, a more natural way to describe the molecules seems to be graph representation, wherein the atom can be viewed as the nodes of the graph and chemical bonds as the edges. This form of data representation has inspired the use of DL models such as GCNNs that take graph structures as well as the characteristics of nodes or edges as their input data. Although GCNNs have already been used in drug discovery applications, including DTI, they are mainly focused on binary prediction task [22, 23] to address the problem of DTI. That being said, among the graph-based approaches that frame the prediction problem as a regression task, we can refer to GraphDTA [24] that predicts a continuous value for binding affinity. GraphDTA utilizes a GCCN-based module (with four different variants) on the molecular graph, describing drug molecules, along with a CCN-based module with three 1D convolutional layers, followed by a max pooling layer to extract feature representation of the input protein sequence. Finally, the concatenated vector of two representations is fed into several fully connected layers to predict the output DTA values. Inspired by GraphDTA, DGraphDTA [25] was also proposed for DTA prediction task. Similar to GraphDTA, DGraphDTA also uses graph representations for drug molecules. However, unlike GraphDTA, they use graph representation for protein sequences, as well. The focus of DGraphDTA (double graph DTA predictor) is on the protein graph representations. The protein graph construction is based on two sources of information. First is the protein residues as the nodes of the graphs; the Second is a representation called a contact map, which provides the spatial information of the interaction of residue pairs in the protein. More specifically, a contact map is a 2D representation of the 3D protein structure, usually in the form of a matrix consistent with the adjacency matrix in GNNs. The authors in [26] proposed a deep heterogeneous learning framework called DeepH-DTA, wherein three different modules have been used to extract features for drug molecules and protein sequences. Specifically, the drug molecules are encoded using two modules; one uses the SMILES representations along with a bidirectional ConvLSTM to model spatio-sequential information of the molecules, and the other one takes the topological drug information as input to generate drug representation using a heterogeneous graph attention (HGAT) network. The third module takes the amino acid sequence of a protein to learn an embedding using a squeezed-excited dense convolutional network. The output of these three modules is finally concatenated to estimate the final prediction score of DTA. DeepGS [27] in another graph-based approach, which consider both local chemical context and the molecular structure in DTA prediction task. With the help of some embedding techniques such as Smi2Vec and Prot2Vec, DeepGS encodes the amino acid sequences of proteins as well as the atoms of drug molecules to distributed representations. Similar to deep-DTA, DeepGS learns two representations for the drug using two different modules; one uses the molecular graph as the input to a graph attention network (GAT) that extracts the topological information of the drug, and the second one takes an atom matrix A of embedding vectors (based on a pre-trained dictionary) as the input to a 1-layer bi-directional gated recurrent unit (BiGRU) to captures the local chemical context of atoms in the drug. A similar process to the second module of drug encoding was used to encode proteins using a CNN that allows capturing local chemical information of the amino acids. Finally, the concatenated vector of the three latent representations is fed into a stack of fully connected layers to predict the binding affinity scores for DT pairs.
EXPERIMENTS AND RESULTS
Benchmark datasets
In this paper, two broadly used benchmark datasets for DTA, namely Davis [28], and KIBA [29] datasets, are used to compare the performance of our proposed model with the state-of-the-art models. The Davis dataset contains 442 proteins related to the kinase protein family along their inhibitors (68 compounds) and the dissociation constant (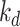 ) values corresponding to each assay. Similar to [9, 30] we convert the
) values corresponding to each assay. Similar to [9, 30] we convert the 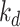 values to log space,
values to log space, 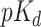 as explained in subsection 1
as explained in subsection 1
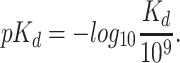 |
(1) |
KIBA dataset combines different kinase inhibitor bioactivities (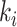 ,
, 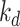 and IC50) to construct the KIBA score to optimize the consistency. [30] filtered this dataset to include the drugs and targets with at least 10 interactions, generating 229 unique proteins and 2111 unique drugs. subsection 1 provides the statistics of the two datasets.
and IC50) to construct the KIBA score to optimize the consistency. [30] filtered this dataset to include the drugs and targets with at least 10 interactions, generating 229 unique proteins and 2111 unique drugs. subsection 1 provides the statistics of the two datasets.
Table 1.
Benchmark datasets
| Dataset | Compounds | Proteins | Interactions |
|---|---|---|---|
| Davis | 68 | 442 | 30 056 |
| KIBA | 2111 | 229 | 118 254 |
Data collection and preprocessing
For the input of our model, we needed to extract PDB structures for the proteins. First, we extracted the identifier numbers for the proteins from https://www.uniprot.org/. Specifically, we restricted our search to human proteins, from which we chose the ones identified via the X-ray method. Although, there were a few proteins (total of 115 for Kiba and Davis, as shown in subsection 3) for which no experimental data (PDB structure) were available. For these proteins, we used the PDB structures predicted by Alphafold (https://alphafold.ebi.ac.uk/).
Figure 3.
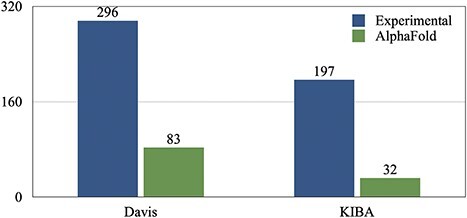
Number of experimentally validated vs AlphFold-predicted proteins’ 3D structures
Evaluation metrics
Four widely used metrics in regression tasks were used to evaluate the performance of the models. CI was first introduced by [31] and is a ranking metric that measures whether the predicted binding affinity value of two random DT pairs is in the same order as their corresponding true values or not. CI metric is defined as:
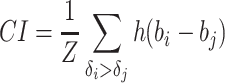 |
(2) |
where 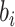 is the prediction value for the larger affinity
is the prediction value for the larger affinity 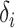 , and
, and 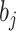 is the prediction value for the smaller affinity
is the prediction value for the smaller affinity 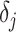 ,
, 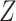 is a normalization constant,
is a normalization constant, 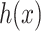 is the step function [32, 33] which is defined as:
is the step function [32, 33] which is defined as:
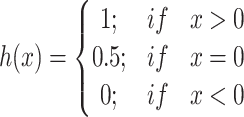 |
(3) |
MSE [34] is a commonly used loss function in regression-based models that measures the average squared difference between the predicted values and the actual values, and it is defined as:
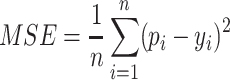 |
(4) |
where 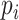 corresponds to the prediction value, and
corresponds to the prediction value, and 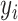 represents the actual output. Also,
represents the actual output. Also, 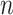 is the number of samples.
is the number of samples.
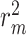 index [35] is another reported metric that was previously used to evaluate the external predictive performance of the models. The higher values (higher than 0.5) of this index determine whether a model is acceptable or not. As described in [36, 37], the metric is defined as below where
index [35] is another reported metric that was previously used to evaluate the external predictive performance of the models. The higher values (higher than 0.5) of this index determine whether a model is acceptable or not. As described in [36, 37], the metric is defined as below where 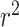 and
and 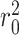 are the squared correlation coefficients with and without intercept:
are the squared correlation coefficients with and without intercept:
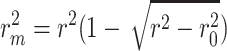 |
(5) |
The area under precision-recall (AUPR) is another reported metric previously used in many studies. In order to measure this metric, the quantitative values were converted into binary values. Similar to [9, 29, 30], the thresholds of 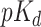 = 7 for Davis dataset and 12.1 for KIBA dataset were used to utilize binarization.
= 7 for Davis dataset and 12.1 for KIBA dataset were used to utilize binarization.
Results and comparison
To assess the effectiveness of our proposed framework, we compare the predictive power of six cutting-edge binding affinity DL-based prediction approaches with and without their integration with our AttentionSiteDTI module. The benchmarking approaches include both sequence representation-based models such as DeepDTA, WideDTA and AttentionDTA, as well as graph representation-based approaches such as GraphDTA, DGraphDTA and DeepGS. We compare the performance of all these models with and without the help of our AttentionSiteDTI in finding the most probable binding sites of the proteins. All models’ performances are evaluated under the same experimental conditions on the two benchmark datasets, KIBA and Davis. We used CI, MSE, modified squared correlation coefficient (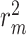 ) and the AUPC for model evaluation. Detailed definitions of these metrics are provided in the Supplementary material. As the results in subsection 4 and subsection 5 illustrate, all six models show improved performance when assisted with the AttentionSiteDTI model. The boost in models’ performance can be explained by the effectiveness of our model in finding the most probable binding sites of the proteins, which helps narrow down the search space of the prediction models by making them selectively concentrate on valuable parts of the proteins when learning the interactions between proteins and drugs; hence, enhancing the quality of the predictions. The missing values on the performance of the DeepH-DTA [26] is due to the missing information in the implementation of this model that was not provided in their code. Therefore, we were not able to produce the results of this model when combined with our AttentionSiteDTI. We gathered the results from the original paper on DeepH-DTA, as it was shown to outperform all other approaches in terms of all four metrics. However, its performance becomes very competitive with AttentionSite-augmented DGraphDTA and AttentionDTA on Kiba and Davis datasets, respectively.
) and the AUPC for model evaluation. Detailed definitions of these metrics are provided in the Supplementary material. As the results in subsection 4 and subsection 5 illustrate, all six models show improved performance when assisted with the AttentionSiteDTI model. The boost in models’ performance can be explained by the effectiveness of our model in finding the most probable binding sites of the proteins, which helps narrow down the search space of the prediction models by making them selectively concentrate on valuable parts of the proteins when learning the interactions between proteins and drugs; hence, enhancing the quality of the predictions. The missing values on the performance of the DeepH-DTA [26] is due to the missing information in the implementation of this model that was not provided in their code. Therefore, we were not able to produce the results of this model when combined with our AttentionSiteDTI. We gathered the results from the original paper on DeepH-DTA, as it was shown to outperform all other approaches in terms of all four metrics. However, its performance becomes very competitive with AttentionSite-augmented DGraphDTA and AttentionDTA on Kiba and Davis datasets, respectively.
Figure 4.
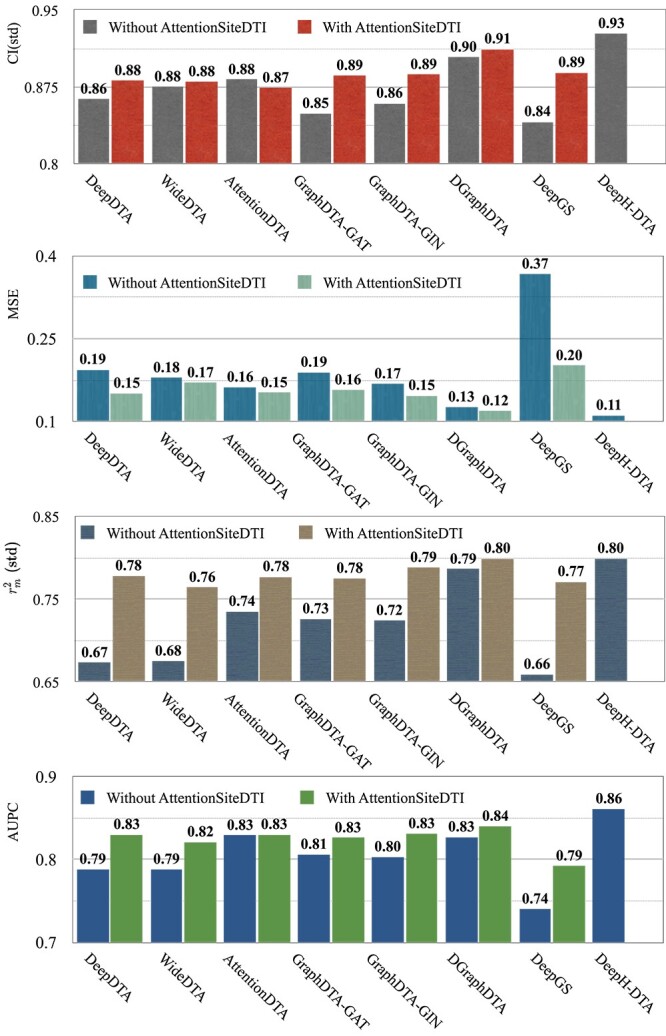
Performance of various DL-based DTA prediction models on KIBA dataset, with and without their integration with AttentionSiteDTI. Note that the scores of integrated DeepH-DTA do not show in the bar plots because of the missing information in the implementation of this model.
Figure 5.
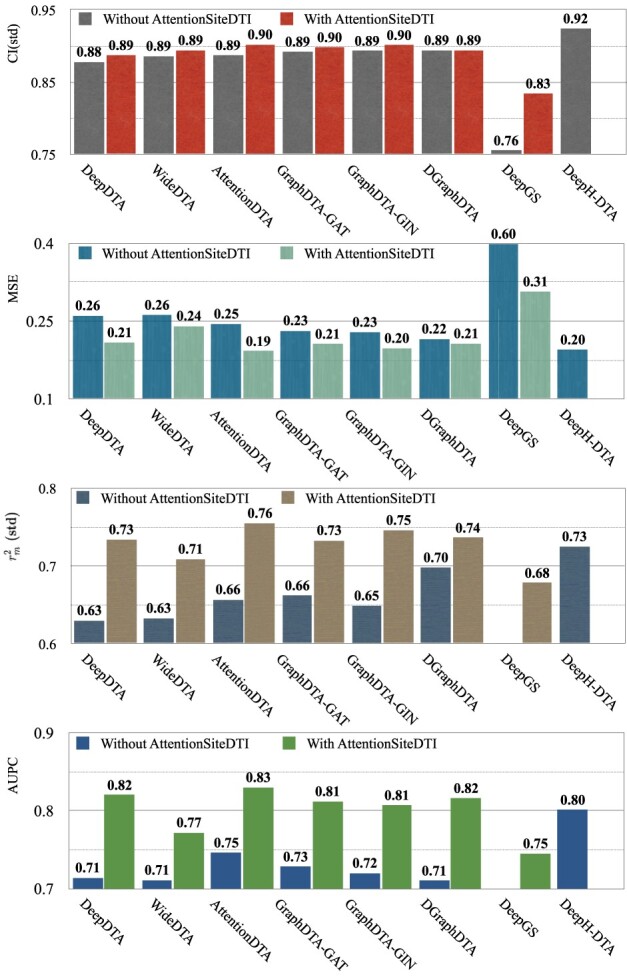
Performance of various DL-based DTA prediction models on Davis dataset, with and without their integration with AttentionSiteDTI. Note that the scores of integrated DeepH-DTA do not show in the bar plots because of the missing information in the implementation of this model. Also, the missing scores of DeepGS in the last two plots are because they are lower than the lower bound of the y-axis.
According to the results in subsection 2, in the KIBA dataset, all AttentionSite-augmented models exceed their own plain versions in all evaluation metrics, where DeepGS enjoys the most improvement in all metrics, achieving 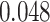 ,
, 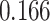 ,
, 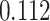 and
and 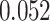 improvements for CI, MSE,
improvements for CI, MSE, 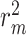 and AUPC, respectively. An exception is AttentionDTA, whose performance in CI metric in the augmented version is lower
and AUPC, respectively. An exception is AttentionDTA, whose performance in CI metric in the augmented version is lower 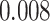 than vanilla AttentionDTA. Among all metrics,
than vanilla AttentionDTA. Among all metrics, 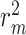 and
and 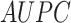 have the most noticeable changes, where their normalized average improvements are
have the most noticeable changes, where their normalized average improvements are 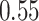 and
and 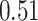 over all models. Note that we used min–max normalized of the difference scores to avoid the dependence on the choice of measurement units.
over all models. Note that we used min–max normalized of the difference scores to avoid the dependence on the choice of measurement units.
Table 2.
Difference scores on performance metrics between vanilla and augmented models for KIBA dataset. The values in the table reflect 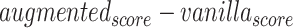 for different performance measures. To calculate average values, we used min–max normalized of the difference scores to avoid the dependence on the choice of measurement units
for different performance measures. To calculate average values, we used min–max normalized of the difference scores to avoid the dependence on the choice of measurement units
| Difference scores | ||||
|---|---|---|---|---|
| Methods | CI | MSE |
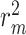
|
AUPC |
| String representation-based approaches | ||||
| DeepDTA | 0.018 | 0.043 | 0.105 | 0.042 |
| WideDTA | 0.005 | 0.009 | 0.089 | 0.033 |
| AttentionDTA | -0.008 | 0.01 | 0.042 | 0.001 |
| Graph representation-based approaches | ||||
| GraphDTA-GAT | 0.037 | 0.032 | 0.049 | 0.02 |
| GraphDTA-GIN | 0.028 | 0.021 | 0.064 | 0.028 |
| DGraphDTA | 0.008 | 0.007 | 0.013 | 0.013 |
| DeepGS | 0.048 | 0.166 | 0.112 | 0.052 |
| Min | –0.008 | 0.007 | 0.013 | 0.001 |
| Max | 0.048 | 0.166 | 0.112 | 0.052 |
| Normalized average | 0.022 | 0.215 | 0.553 | 0.510 |
For the Davis dataset, as the results show in subsection 3, the augmented models also achieve better results compared to their plain versions. similarly, DeepGS benefits the most from AttentionSiteDTI, where it attains 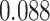
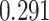 ,
, 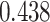 ,
, 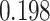 improvements for CI, MSE,
improvements for CI, MSE, 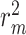 and AUPC, respectively. Again, the two performance metrics
and AUPC, respectively. Again, the two performance metrics 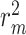 and
and 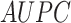 show the most average changes with
show the most average changes with 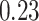 and
and 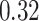 improvements across all models.
improvements across all models.
Table 3.
Difference scores on performance metrics between vanilla and augmented models for Davis dataset. The values in the table reflect 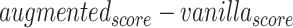 for different performance measures. To calculate average values, we used min–max normalized of the difference scores to avoid the dependence on the choice of measurement units
for different performance measures. To calculate average values, we used min–max normalized of the difference scores to avoid the dependence on the choice of measurement units
| Difference scores | ||||
|---|---|---|---|---|
| Methods | CI | MSE |
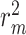
|
AUPC |
| String representation-based approaches | ||||
| DeepDTA | 0.009 | 0.052 | 0.104 | 0.106 |
| WideDTA | 0.008 | 0.021 | 0.076 | 0.061 |
| AttentionDTA | 0.015 | 0.053 | 0.098 | 0.084 |
| Graph representation-based approaches | ||||
| GraphDTA-GAT | 0.006 | 0.026 | 0.071 | 0.084 |
| GraphDTA-GIN | 0.008 | 0.031 | 0.097 | 0.087 |
| DGraphDTA | 0 | 0.009 | 0.038 | 0.116 |
| DeepGS | 0.088 | 0.291 | 0.438 | 0.198 |
| Min | 0 | 0.009 | 0.038 | 0.061 |
| Max | 0.088 | 0.291 | 0.438 | 0.198 |
| Normalized average | 0.218 | 0.213 | 0.234 | 0.322 |
It is worth mentioning that there exist other DL-based models such as [38–42], which were specifically proposed for binding site prediction task. However, none of them have been shown to be generalizable in integration with DL-based DTA prediction models. Indeed, to the best of our knowledge, we are the first to propose this unified framework, which also provides interpretability to the integrated models. From the figures in these two tables, we can also observe that sequence-based DL models have lower predictive power compared to graph-based models, which explains the effectiveness of graph representations in capturing structural information of drugs and/or proteins as deciding factors in the prediction of DTIs.
In-Vitro VALIDATION: SARS-CoV-2 CASE STUDY
We further experimentally tested and validated the performance of our proposed framework through experimentations in the laboratory by measuring the binding affinities between 13 candidate compounds and the Spike-ACE2 complex. These compounds include Darunavir, N-acetyl-neuraminic acid, N-Acetyllactosamine, 3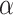 , 6
, 6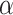 Mannopentaose, N-glycolylneuraminic acid, 2-Keto-3-deoxyoctonate ammonium salt, cytidine5-monophospho-N-acetylneuraminic acid sodium salt, Congo red, Direct Violet, Evans Blue, Calcomine scarlet 3B, Chlorazol Black and Methylene Blue as inhibitor molecules to bind to the Spike protein, or the ACE2 receptor protein (as the primary host factor recognized and targeted by SARS-CoV-2 Spike protein). Additional information on our choice of compounds is provided in the Supplementary material. We then assessed the prediction performance using three metrics: (1) Pearson correlation coefficient to measure the scoring power, indicating the binding affinity prediction capacity of the model, (2) CI to measure the ranking power, presenting the affinity-ranking capacity of the model and (3) MSE to measure the overall prediction error, showing the quality of the model in estimating the true values. To predict compound-protein binding affinities, we selected DeepDTA because of its wide adoption in previous studies as the first DL approach developed to predict drug–target binding affinity. DeepDTA is among state-of-the-art methods that have shown relatively good performance compared to many DL-based models with higher complexity in architecture and computation time. In order to show the effectiveness of our prediction framework, the performance measures were calculated under two settings; with and without the incorporation of AttentionSiteDTI with the DeepDTA regression model.
Mannopentaose, N-glycolylneuraminic acid, 2-Keto-3-deoxyoctonate ammonium salt, cytidine5-monophospho-N-acetylneuraminic acid sodium salt, Congo red, Direct Violet, Evans Blue, Calcomine scarlet 3B, Chlorazol Black and Methylene Blue as inhibitor molecules to bind to the Spike protein, or the ACE2 receptor protein (as the primary host factor recognized and targeted by SARS-CoV-2 Spike protein). Additional information on our choice of compounds is provided in the Supplementary material. We then assessed the prediction performance using three metrics: (1) Pearson correlation coefficient to measure the scoring power, indicating the binding affinity prediction capacity of the model, (2) CI to measure the ranking power, presenting the affinity-ranking capacity of the model and (3) MSE to measure the overall prediction error, showing the quality of the model in estimating the true values. To predict compound-protein binding affinities, we selected DeepDTA because of its wide adoption in previous studies as the first DL approach developed to predict drug–target binding affinity. DeepDTA is among state-of-the-art methods that have shown relatively good performance compared to many DL-based models with higher complexity in architecture and computation time. In order to show the effectiveness of our prediction framework, the performance measures were calculated under two settings; with and without the incorporation of AttentionSiteDTI with the DeepDTA regression model.
As the results in section 4 indicate, the AttentionSiteDTI helps enhance the performance of DeepDTA, especially in the two metrics, MSE and Pearson correlation coefficient, where the improvements are 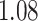 and
and 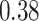 , respectively. As a matter of fact, when using DeepDTA, we observe a very low correlation of
, respectively. As a matter of fact, when using DeepDTA, we observe a very low correlation of 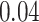 between the predicted and measured bioactivities, which is significantly boosted to
between the predicted and measured bioactivities, which is significantly boosted to 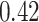 , considered a relatively good correlation as a result of higher quality binding affinity predictions by augmented DeepDTA. In fact, this illustrates the effectiveness of our proposed framework in improving the performance of DeepDTA when augmented with our AttentionSiteDTI. Note that section 6 reports the predicted and measured bioactivity profiles of Spike-ACE2 complex against 13 drug-like compounds tested in our experimental assay.
, considered a relatively good correlation as a result of higher quality binding affinity predictions by augmented DeepDTA. In fact, this illustrates the effectiveness of our proposed framework in improving the performance of DeepDTA when augmented with our AttentionSiteDTI. Note that section 6 reports the predicted and measured bioactivity profiles of Spike-ACE2 complex against 13 drug-like compounds tested in our experimental assay.
Table 4.
Performance comparison of the computational models on in-vitro experimental bioactivity results
| Methods | CI | MSE | Pearson Correlation |
|---|---|---|---|
| DeepDTA | 0.4358 | 3.7729 | 0.0463 |
| DeepDTA + AttentionSiteDTI | 0.4487 | 2.6907 | 0.4245 |
Figure 6.
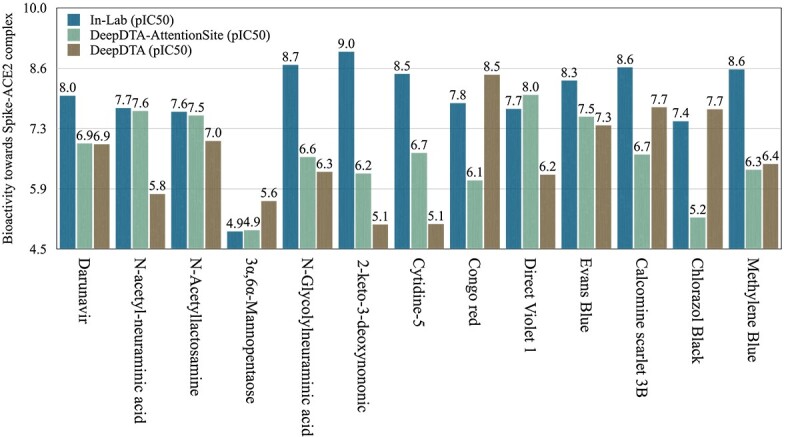
Predicted and measured binding affinity values of Spike-ACE2 complex with 13 drug-like compounds in our experimental assay. The predicted values are reported for DeepDTA model with and without its integration with AttentionSiteDTI. We chose DeepDTA for computational convenience, as we needed to pre-train the model on the BindingDB dataset due to the small size of our experimental data.
DRUG REPURPOSING: SARS-Cov-2 CASE STUDY
Encouraged by the excellent performance of our framework, we now apply it to predict binding affinity values between 3445 commercially available FDA-approved drugs (including 85 antiviral drugs) and key proteins of SARS-CoV-2, including 3C-like protease, RNA-dependent RNA polymerase(RdRp), helicase as well as Spike/ACE2 complex. We use AttentionSite-augmented DeepDTA, which is pre-trained on the BindingDB dataset to predict binding affinities between genome sequences of SARS-CoV-2—extracted from the National Center for Biotechnology Information (NCBI) database—with 85 FDA-approved antiviral drugs from DrugBank. The exhaustive list of drugs is provided in the Supplementary material.
As the results show in section 5, the Spike protein–ACE2 interface was predicted to have the highest binding affinity with Oseltamivir phosphate, Etravirine and Fosamprenavir calcium, all with PIC50 values > 8 nM, which are followed by other antiviral drugs with predicted binding affinities of PIC50 > 7 nM potency (we only included top 10 antiviral drugs in section 5 with highest binding affinity). Among the prediction results, Fosamprenavir calcium, Simeprevir, Elvitegravir, Simeprevir sodium, elvitegravir and ritonavir have shown good inhibition towards SARS-CoV-2 helicase, with predicted binding affinity values > 7 nM. Furthermore, Fosamprenavir calcium, elvitegravir, Tipranavir, Asunaprevir and Elvitegravir were predicted to have a potential affinity (PIC50 > 7 nM) to SARS-CoV-2 3C-like Proteinase. Finally, elvitegravir and Fosamprenavir calcium were the only inhibitors that were predicted to have binding affinities 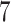 nM in binding with RNA-dependent RNA polymerase. These verifying studies are available in section 5. Despite the existence of any strong real-world evidence on the effectiveness of these drugs (except for the FDA-approved drug Remdesivir) against COVID-19, we found that some of these candidate drugs have already been suggested or introduced by other studies, including in-silico, preclinical and clinical trials.
nM in binding with RNA-dependent RNA polymerase. These verifying studies are available in section 5. Despite the existence of any strong real-world evidence on the effectiveness of these drugs (except for the FDA-approved drug Remdesivir) against COVID-19, we found that some of these candidate drugs have already been suggested or introduced by other studies, including in-silico, preclinical and clinical trials.
Table 5.
Top 10 FDA-approved antiviral drugs predicted by AttentionSite-Augmented DeepDTA model to have highest affinity scores with 4 genome sequences related to SARS-CoV-2
| Drug Name | pIC50 | Rank in 3416 FDA-Approved Drugs | Rank in 85 Antiviral Drugs |
|---|---|---|---|
| Spike-ACE2 | |||
| Oseltamivir phosphate | 8.29 | 42 | 1 |
| Etravirine | 8.22 | 52 | 2 |
| Fosamprenavir calcium | 8.20 | 57 | 3 |
| Tipranavir | 7.94 | 108 | 4 |
| elvitegravir | 7.88 | 141 | 5 |
| ganciclovir | 7.84 | 162 | 6 |
| Simeprevir | 7.71 | 221 | 7 |
| Rimantadine | 7.67 | 248 | 8 |
| saquinavir | 7.65 | 259 | 9 |
| danoprevir | 7.57 | 314 | 10 |
| helicase | |||
| Fosamprenavir calcium (FPV) | 7.51 | 21 | 1 |
| Simeprevir | 7.47 | 24 | 2 |
| Elvitegravir | 7.28 | 52 | 3 |
| Simeprevir sodium | 7.11 | 87 | 4 |
| elvitegravir | 7.09 | 97 | 5 |
| ritonavir | 7.01 | 120 | 6 |
| Indinavir sulfate | 6.98 | 132 | 7 |
| Amprenavir | 6.93 | 155 | 8 |
| nelfinavir | 6.88 | 182 | 9 |
| Tenofovir alafenamide fumarate | 6.86 | 190 | 10 |
| 3C-like | |||
| Fosamprenavir calcium | 7.72 | 5 | 1 |
| elvitegravir | 7.25 | 29 | 2 |
| Tipranavir | 7.17 | 37 | 3 |
| Asunaprevir | 7.08 | 46 | 4 |
| Elvitegravir | 7.06 | 47 | 5 |
| Atazanavir sulfate | 6.92 | 68 | 6 |
| Daclatasvir | 6.88 | 77 | 7 |
| Saquinavir mesylate | 6.77 | 97 | 8 |
| Simeprevir | 6.71 | 121 | 9 |
| Simeprevir sodium | 6.68 | 129 | 10 |
| RdRp | |||
| elvitegravir | 7.60 | 8 | 1 |
| Fosamprenavir calcium | 7.24 | 24 | 2 |
| ritonavir | 6.96 | 53 | 3 |
| Asunaprevir | 6.90 | 66 | 4 |
| Simeprevir sodium | 6.87 | 71 | 5 |
| Daclatasvir | 6.85 | 74 | 6 |
| Simeprevir | 6.82 | 79 | 7 |
| nelfinavir | 6.81 | 82 | 8 |
| Amprenavir | 6.79 | 93 | 9 |
| Saquinavir mesylate | 6.78 | 96 | 10 |
A more interesting observation in our prediction results can be seen in section 6. There are eight antiviral drugs that are currently in clinical trials to be assessed for their efficacy against COVID-19. These drugs include Atazanavir, Daclatasvir, Danoprevir, Darunavir, Elvitegravir, Lopinavir, Oseltamivir and Ritonavir, which are also listed in the present prediction results as suitable potential inhibitors to all four subunits of SARS-CoV-2 (Spike-ACE2 interface, helicase, 3C-like protease and RdRp), mostly with binding affinity values > 6 nM in PIC50. Also, Remdesivir, which is the only FDA-approved drug for the treatment of COVID-19, shows great predicted potency to all subunits of SARS-CoV-2 as follows: against Spike-ACE interface (PIC50 7.47 nM), RNA-dependent RNA polymerase (PIC50 6 nM), helicase (PIC50 6 nM) and 3C-like protease(PIC50 5.68 nM).
Table 6.
Binding affinity values of selected antiviral drugs in clinical trials to treat COVID-19, binding with different SARS-CoV-2 related receptors. The ranks are listed among 85 antiviral drugs*
| Drug Name | # Clinical | # Supporting | Spike-ACE2 | helicase | 3C-like | RdRp | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Trial | References | pIC50 | rank | pIC50 | rank | pIC50 | rank | pIC50 | rank | |
| Atazanavir | 3 | 6 | 6.89 | 36 | 6.36 | 32 | 6.18 | 23 | 6.10 | 21 |
| Daclatasvir | 8 | 5 | 7.23 | 24 | 6.57 | 21 | 6.88 | 7 | 6.85 | 6 |
| Danoprevir | 2 | 3 | 7.57 | 10 | 5.68 | 54 | 6.46 | 17 | 5.71 | 33 |
| Darunavir | 2 | 6 | 6.40 | 50 | 6.72 | 15 | 5.85 | 39 | 6.05 | 24 |
| Elvitegravir | 7 | 2 | 7.43 | 14 | 7.28 | 3 | 7.06 | 5 | 6.61 | 12 |
| Lopinavir | 39 | 26 | 7.44 | 12 | 5.55 | 60 | 5.26 | 60 | 5.18 | 53 |
| Oseltamivir | 9 | 5 | 6.75 | 41 | 5.24 | 70 | 5.27 | 59 | 5.12 | 57 |
| Remdesivir | 67 | 70 | 7.47 | 11 | 6.00 | 44 | 5.68 | 43 | 6.00 | 26 |
| ritonavir | 57 | 27 | 7.16 | 26 | 7.01 | 6 | 6.62 | 11 | 6.96 | 3 |
*The information on the number of clinical trials and supporting references are gathered from https://covid19-help.org/ as of July 2022.
CONCLUSION
In this paper, we proposed a framework that can enhance DTA predictions by first finding the most probable binding sites of the protein, thus, making the binding affinity prediction more efficient and accurate. Our AttentionalSiteDTA is not only highly generalizable as it can be combined with any DL-based regression model but also provides interpretability to integrated models due to its attention mechanism, which enables learning which binding sites of a protein interact with a given ligand. The computational results confirm that our framework leads to improved prediction performance of seven state-of- the-art DTA prediction algorithms in terms of four widely used evaluation metrics, including CI, MSE, the modified, squared coefficient of correlation ( ) and the AUPC. Furthermore, we experimentally validated the prediction potential of our framework by conduction in-vitro experiments. To achieve this, we measured the binding affinities between 13 drug-like compounds and the Spike-ACE2 interface and compared these experimental results against binding affinity values that we predicted by our computational models. The results of our in-lab validation illustrated the potential of our framework to significantly enhance the performance of the widely adopted DeepDTA model in the prediction of binding affinity values in real-world applications. Also, following the analysis of our drug screening, we proposed several FDA-approved antivirals that could display antiviral activities against SARS-CoV-2. Some of these compounds are currently undergoing clinical trials, and some others are also suggested by other studies for further evaluation in experimental assays and clinical trials to investigate their actual activity against COVID-19. Another contribution of this work is to provide the community with extended versions of the two most commonly used datasets, Kiba and Davis, for which we manually extracted information on the 3D structure of all proteins in these two datasets from the PDB files of proteins available in https://www.uniprot.org/.
) and the AUPC. Furthermore, we experimentally validated the prediction potential of our framework by conduction in-vitro experiments. To achieve this, we measured the binding affinities between 13 drug-like compounds and the Spike-ACE2 interface and compared these experimental results against binding affinity values that we predicted by our computational models. The results of our in-lab validation illustrated the potential of our framework to significantly enhance the performance of the widely adopted DeepDTA model in the prediction of binding affinity values in real-world applications. Also, following the analysis of our drug screening, we proposed several FDA-approved antivirals that could display antiviral activities against SARS-CoV-2. Some of these compounds are currently undergoing clinical trials, and some others are also suggested by other studies for further evaluation in experimental assays and clinical trials to investigate their actual activity against COVID-19. Another contribution of this work is to provide the community with extended versions of the two most commonly used datasets, Kiba and Davis, for which we manually extracted information on the 3D structure of all proteins in these two datasets from the PDB files of proteins available in https://www.uniprot.org/.
Key Points
We developed an experimental–computational pipeline based on a graph neural networks, which improves prediction performance, when combined with many state-of-the-art DTA prediction models. Our proposed framework outperforms many state-of-the-art models, in terms of several performance metrics. These models are improved by focusing on the most relevant parts of input proteins when learning interaction relationships between drugs and targets.
Our approach van be easily integrated with any DL-based predictin model.
Our model is highly generalizable due to the use of protein pockets (i.e. molecular fragments of binding sites influencing binding character) encoded as graphs, representing input proteins to the model. Also, our framework enables interpretability by learning which parts of the protein interact most efficiently with a given ligand.
We validated our result through conducting in-vitro experiments. We also utilized our method for drug repurposing by predicting binding affinity values between FDA-approved drugs and key proteins of SARS-CoV-2.
We contributed to the two most widely used benchmarking datasets in the DTA prediction task, namely Kiba and Davis, by manually extracting 3D structures of the proteins in these two datasets.
DATA AND CODE AVAILABILITY
All datasets and codes are publicly available at https://github.com/yazdanimehdi/BindingSite-AugmentedDTA.
Supplementary Material
Niloofar Yousefi, PhD (University of Central Florida), is a postdoctoral research associate at UCF’s Complex Adaptive Systems (CAS) laboratory in the collage of Engineering and Computer Science. Her research areas include machine learning, artificial intelligence and statistical learning theory to develop data analytics solutions with more transparency and explainability.
Mehdi Yazdan-Jahromi is a third-year PhD student in computer science at the University of Central Florida. His current research interests include computer vision, drug–target interaction and algorithmic fairness.
Aida Tayebi is a third year PhD student at University of Central Florida. Her current research interests include algorithmic fairness and bias mitigation techniques in DTI.
Elayaraja Kolanthai, PhD (Anna University), is a postdoctoral research associate at UCF’s Materials Science and Engineering. His current research interests include Development of nanoparticles, layer-by-layer antimicrobial/antiviral nanoparticle coating, polymer composites for tissue engineering and gene/drug delivery application.
Craig J. Neal, PhD (University of Central Florida), is a postdoctoral research associate at UCF’s Materials Science and Engineering. His current research interests include wet chemical synthesis and surface engineering of nanoparticles for biomedical applications and electrochemical devices. Electroanalysis of nanomaterials and bio-nano interactions.
Tanumoy Banerjee is a second year PhD student at Lehigh University. His current research interest is developing energy storage technologies and computational modeling of hydrogen-based energy storage systems.
Agnivo Gosai did his PhD from Iowa State University in Mechanical Engineering. His research explored the development of point of care biosensors for Ebola and other diseases. He is currently employed at Corning Inc. and is working on Telecom device, biomass reactor R&D. Dr. Gosai has multiple publications and patent applications.
Ganesh Balasubramanian is an associate professor of Mechanical Engineering & Mechanics at Lehigh University. He received his BME degree in Mechanical Engineering from Jadavpur University (India) in 2007, his PhD in Engineering Mechanics from Virginia Tech in 2011, and was a postdoctoral research associate in Theoretical Physical Chemistry at TU Darmstadt (Germany) till fall of 2012. His research and teaching interests are in advanced energy and structural materials, nanoscale transport and mechanics, and predictive engineering. Some of his recognitions include the NSF CAREER award, ASEE Outstanding New ME Educator award, AFRL Summer Faculty Fellowship, Graduate Man of the Year and Liviu Librescu Scholarship at Virginia Tech, and Young Engineering Fellowship from the Indian Institute of Science.
Sudipta Seal is currently the chair of the Department of Materials Science and Engineering at University of Central Florida, as well as a Pegasus Professor and a University distinguished professor. He joined the Advanced Materials Processing and Analysis Center and UCF in 1997. He has been consistently productive in research, instruction and service to UCF since 1998. He has served as the Nano Initiative coordinator for the vice president of research and commercialization. He served as the director of AMPAC and the NanoScience Technology Center from 2009 to 2017.
Ozlem Ozmen Garibay is an assistant professor of Industrial Engineering and Management System at the University of Central Florida where she directs the Human-Centered Artificial Intelligence Research Lab (Human-CAIR Lab). Prior to that, she served as the Director of Research Technology. Her areas of research are big data, social media analysis, social cybersecurity, artificial social intelligence, human-machine teams, social and economic networks, network science, STEM education analytics, higher education economic impact and engagement, artificial intelligence, evolutionary computation, and complex systems.
Contributor Information
Niloofar Yousefi, Industrial Engineering and Management Systems, University of Central Florida, 32816, 4000 Central Florida Blvd., Orlando, FL, USA.
Mehdi Yazdani-Jahromi, Computer Science, University of Central Florida, 32816, 4000 Central Florida Blvd., Orlando, FL, USA.
Aida Tayebi, Industrial Engineering and Management Systems, University of Central Florida, 32816, 4000 Central Florida Blvd., Orlando, FL, USA.
Elayaraja Kolanthai, College of Medicine, Bionix Cluster, University of Central Florida, 4000 Central Florida Blvd., Orlando 32816, FL, USA.
Craig J Neal, College of Medicine, Bionix Cluster, University of Central Florida, 4000 Central Florida Blvd., Orlando 32816, FL, USA.
Tanumoy Banerjee, Department of Mechanical Engineering and Mechanics, Lehigh University, Bethlehem 18015, PA, USA.
Agnivo Gosai, Independent Researcher, Corning, NY, USA.
Ganesh Balasubramanian, Department of Mechanical Engineering and Mechanics, Lehigh University, Bethlehem 18015, PA, USA.
Sudipta Seal, College of Medicine, Bionix Cluster, University of Central Florida, 4000 Central Florida Blvd., Orlando 32816, FL, USA; Advanced Materials Processing and Analysis Center, Department of Materials Science and Engineering, University of Central Florida, 4000 Central Florida Blvd., Orlando 32816, FL, USA.
Ozlem Ozmen Garibay, Industrial Engineering and Management Systems, University of Central Florida, 32816, 4000 Central Florida Blvd., Orlando, FL, USA.
References
- 1. Cohen P. Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov 2002; 1(4): 309–15. [DOI] [PubMed] [Google Scholar]
- 2. Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science 2004; 303(5665): 1800–5. [DOI] [PubMed] [Google Scholar]
- 3. Salsbury FR, Jr. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr Opin Pharmacol 2010; 10(6): 738–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yu H, Chen J, Xu X, et al.. A systematic prediction of multiple drug–target interactions from chemical, genomic, and pharmacological data. PloS One 2012; 7(5): e37608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tabei Y, Yamanishi Y. Scalable prediction of compound-protein interactions using minwise hashing. BMC Syst Biol 2013; 7(6): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shi Y, Zhang X, Liao X, et al.. Protein-chemical interaction prediction via kernelized sparse learning svm. World Scientific, 2013, 41–52. [PubMed] [Google Scholar]
- 7. Cheng Z, Zhou S, Wang Y, et al.. Effectively identifying compound-protein interactions by learning from positive and unlabeled examples. IEEE/ACM Trans Comput Biol Bioinform 2016; 15(6): 1832–43. [DOI] [PubMed] [Google Scholar]
- 8. Lee I, Keum J, Nam H. Deepconv-dti: prediction of drug–target interactions via deep learning with convolution on protein sequences. PLoS Comput Biol 2019; 15(6): e1007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Öztürk H, Özgür A, Ozkirimli E. Deepdta: deep drug–target binding affinity prediction. Bioinformatics 2018; 34(17): i821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karimi M, Wu D, Wang Z, Shen Y. Deepaffinity: interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 2019; 35(18): 3329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Öztürk H, Ozkirimli E, Özgür A. Widedta: prediction of drug–target binding affinity arXiv preprint arXiv:1902.04166. 2019. [DOI] [PMC free article] [PubMed]
- 12. HajiEbrahimi A, Ghafouri H, Ranjbar M, Sakhteman A. Protein ligand interaction fingerprints. In: Pharmaceutical Sciences: Breakthroughs in Research and Practice. Hershey, PA, IGI Global, 2017, 1072–91. [Google Scholar]
- 13. Ezzat A, Wu M, Li XL, Kwoh CK. Computational prediction of drug–target interactions using chemogenomic approaches: an empirical survey. Brief Bioinform 2019; 20(4): 1337–57. [DOI] [PubMed] [Google Scholar]
- 14. Thafar M, Raies AB, Albaradei S, et al.. Comparison study of computational prediction tools for drug–target binding affinities. Front Chem 2019; 7:782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cichonska A, Ravikumar B, Parri E, et al.. Computational-experimental approach to drug–target interaction mapping: a case study on kinase inhibitors. PLoS Comput Biol 2017; 13(8): e1005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cichońska A, Ravikumar B, Allaway RJ, et al.. Crowdsourced mapping of unexplored target space of kinase inhibitors. Nat Commun 2021; 12(1): 3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yazdani-Jahromi M, Yousefi N, Tayebi A, et al.. Attentionsitedti: an interpretable graph-based model for drug–target interaction prediction using nlp sentence-level relation classification. Brief Bioinform 2022; 23(4): bbac272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gómez-Bombarelli R, Wei JN, Duvenaud D, et al.. Automatic chemical design using a data-driven continuous representation of molecules. ACS Central Science 2018; 4(2): 268–76PMID: 29532027. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jastrzebski S, Leśniak D, Marian Czarnecki W. Learning to smile(s). 2016. arXiv preprint arXiv:1602.06289v2. 10.48550/ARXIV.1602.06289. [DOI]
- 20. Wang L, You ZH, Chen X, et al.. A computational-based method for predicting drug–target interactions by using stacked autoencoder deep neural network. J Comput Biol 2018; 25(3): 361–73. [DOI] [PubMed] [Google Scholar]
- 21. Zhao Q, Xiao F, Yang M, et al.. Attentiondta: prediction of drug–target binding affinity using attention model. 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 2019, pp. 64–9. 10.1109/BIBM47256.2019.8983125. [DOI]
- 22. Gao KY, Fokoue A, Luo H, et al.. Interpretable drug target prediction using deep neural representation. IJCAI. 2018, 3371–7. [Google Scholar]
- 23. Mayr A, Klambauer G, Unterthiner T, et al.. Large-scale comparison of machine learning methods for drug target prediction on chembl. Chem Sci 2018; 9(24): 5441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nguyen T, Le H, Quinn TP, et al.. Graphdta: predicting drug–target binding affinity with graph neural networks. Bioinformatics 2021; 37(8): 1140–7. [DOI] [PubMed] [Google Scholar]
- 25. Jiang M, Li Z, Zhang S, et al.. Drug–target affinity prediction using graph neural network and contact maps. RSC Adv 2020; 10(35): 20701–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abdel-Basset M, Hawash H, Elhoseny M, et al.. Deeph-dta: deep learning for predicting drug–target interactions: a case study of covid-19 drug repurposing. IEEE Access 2020; 8:170433–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin X. Deepgs: deep representation learning of graphs and sequences for drug-target binding affinity prediction arXiv preprint arXiv:2003.13902. 2020.
- 28. Davis MI, Hunt JP, Herrgard S, et al.. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 2011; 29(11): 1046–51. [DOI] [PubMed] [Google Scholar]
- 29. Tang J, Szwajda A, Shakyawar S, et al.. Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. J Chem Inf Model 2014; 54(3): 735–43. [DOI] [PubMed] [Google Scholar]
- 30. He T, Heidemeyer M, Ban F, et al.. Simboost: a read-across approach for predicting drug–target binding affinities using gradient boosting machines. J Chem 2017; 9(1): 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gönen M, Heller G. Concordance probability and discriminatory power in proportional hazards regression. Biometrika 2005; 92(4): 965–70. [Google Scholar]
- 32. Davies B. Integral Transforms and Their Applications, Vol. 41. Springer Science & Business Media, New York, NY, 2002. [Google Scholar]
- 33. Pahikkala T, Airola A, Pietilä S, et al.. Toward more realistic drug–target interaction predictions. Brief Bioinform 2015; 16(2): 325–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wackerly D, Mendenhall W, Scheaffer RL. Mathematical statistics with applications. Cengage Learning, 2014. [Google Scholar]
- 35. Roy PP, Roy K. On some aspects of variable selection for partial least squares regression models. QSAR & Combinatorial Science 2008; 27(3): 302–13. [Google Scholar]
- 36. Pratim Roy P, Paul S, Mitra I, Roy K. On two novel parameters for validation of predictive qsar models. Molecules 2009; 14(5): 1660–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roy K, Chakraborty P, Mitra I, et al.. Some case studies on application of “rm2” metrics for judging quality of quantitative structure–activity relationship predictions: emphasis on scaling of response data. J Comput Chem 2013; 34(12): 1071–82. [DOI] [PubMed] [Google Scholar]
- 38. Cui Y, Dong Q, Hong D, Wang X. Predicting protein-ligand binding residues with deep convolutional neural networks. BMC Bioinformatics 2019; 20(1): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xia CQ, Pan X, Shen HB. Protein–ligand binding residue prediction enhancement through hybrid deep heterogeneous learning of sequence and structure data. Bioinformatics 2020; 36(10): 3018–27. [DOI] [PubMed] [Google Scholar]
- 40. Stepniewska-Dziubinska MM, Zielenkiewicz P, Siedlecki P. Improving detection of protein-ligand binding sites with 3d segmentation. Sci Rep 2020; 10(1): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mylonas SK, Axenopoulos A, Daras P. Deepsurf: a surface-based deep learning approach for the prediction of ligand binding sites on proteins. Bioinformatics 2021; 37(12): 1681–90. [DOI] [PubMed] [Google Scholar]
- 42. Kandel J, Tayara H, Chong KT. Puresnet: prediction of protein-ligand binding sites using deep residual neural network. J Chem 2021; 13(1): 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets and codes are publicly available at https://github.com/yazdanimehdi/BindingSite-AugmentedDTA.