


Abstract
Precise genome editing requires the resolution of nuclease-induced DNA double strand breaks (DSBs) via the homology-directed repair (HDR) pathway. In mammals, this is typically outcompeted by non-homologous end-joining (NHEJ) that can generate potentially genotoxic insertion/deletion mutations at DSB sites. Because of higher efficacy, clinical genome editing has been restricted to imperfect but efficient NHEJ-based approaches. Hence, strategies that promote DSB resolution via HDR are essential to facilitate clinical transition of HDR-based editing strategies and increase safety. Here we describe a novel platform that consists of a Cas9 fused to DNA repair factors to synergistically inhibit NHEJ and favor HDR for precise repairing of Cas-induced DSBs. Compared to canonical CRISPR/Cas9, the increase in error-free editing ranges from 1.5-fold to 7-fold in multiple cell lines and in primary human cells. This novel CRISPR/Cas9 platform accepts clinically relevant repair templates, such as oligodeoxynucleotides (ODNs) and adeno-associated virus (AAV)-based vectors, and has a lower propensity to induce chromosomal translocations as compared to benchmark CRISPR/Cas9. The observed reduced mutational burden, resulting from diminished indel formation at on- and off-target sites, provides a remarkable gain in safety and advocates this novel CRISPR system as an attractive tool for therapeutic applications depending on precision genome editing.
Graphical Abstract
Graphical Abstract.
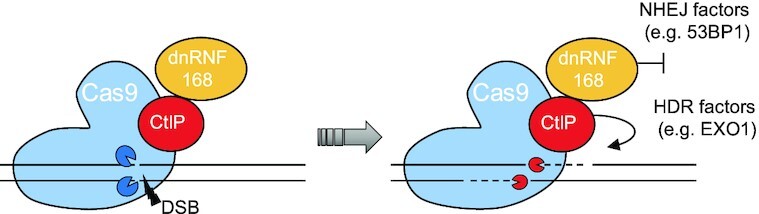
Upon the generation of a DNA double strand break (DSB) operated by the Cas9 protein, the dominant negative variant of RNF168 (dnRNF168) recruits but is unable to activate 53BP1 thus creating a barrier to the endorsement of non-homologous end-joining (NHEJ) DNA repair. The simultaneous presence of CtIP facilitates the recruitment of factors essential for engaging the homology-directed repair (HDR) pathway, such as EXO1, hence promoting DNA resection and facilitating the precise repair of the Cas9-induced DSB.
INTRODUCTION
Therapeutic genome editing using designer nucleases has made tremendous steps forward in the last decade. The introduction of a DNA double stranded break (DSB) within a desired genomic location can be exploited for achieving targeted editing of the cellular genome. On the one end, gene inactivation can be triggered by the deposition of insertion/deletion (indel) mutations at the DSB as a result of the activation of the error-prone non-homologous end-joining (NHEJ) DNA repair pathway. On the other end, precise genome editing, spanning from few nucleotide changes to the integration of large expression cassettes, requires the harnessing of homology directed repair (HDR) and the simultaneous availability of a properly designed DNA template that is used as a blueprint during DSB repair (1). With the inception of the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas) system, the precise manipulation of the cellular genetic material has been made possible with unprecedented ease (2). However, achieving high frequency of HDR-mediated DSB resolution is still challenging (3). In human cells this is mostly due to the reduced availability of key HDR components, with their expression being typically restricted to the S/G2-phases of the cell cycle, when this type of DSB repair pathways is active (1). As a consequence, most clinical genome editing applications exploit strategies that rely on imprecise NHEJ (4). Several concepts have been explored to increase the frequency of precise genome editing. These include the use of chemicals to inhibit NHEJ (5) or to enhance HDR (6), or the pharmacological synchronization of the cells to transiently keep them in cell cycle phases that support HDR (7). While promising, these approaches profoundly alter the normal behavior of the target cells (8) raising substantial safety concerns when explored in a therapeutic context. Thus, restricting the manipulation of DSB repair to the nuclease target site has been attempted to avoid global changes in DNA repair. To this end, several critical players involved in the resolution of a DSB via the HDR pathway have been tethered to the Cas9 endonuclease in order to increase the frequency of precision editing. Cas9 fusions to CtBP-interacting protein (CtIP) exonuclease, a minimal breast cancer 2 (BRCA2) motif (Brex27) or a dominant negative variant of the p53-binding protein 1 (53BP1) have shown promising increase in HDR-mediated DSB repair in different cell types (9–11). However, these reports largely disregarded functional validation in clinically relevant human cells, which is the ultimate target when exploring precision genome editing for therapeutic purposes. Furthermore, the unaltered NHEJ pathway contributes to a significant mutational burden at the on-target site as well as at off-target sites (12), eliciting substantial concerns with even greater impact when developing innovative therapeutics.
Here, we describe a novel CRISPR/Cas9 system that exploits the deposition of key DNA repair factors at the nuclease target site via their tethering to the Cas9 nuclease to alter the normal resolution of a DSB in favor of HDR and, thus, supporting precise genome editing. We demonstrate that this approach can be combined with different types of donor repair templates, including oligodeoxynucleotides (ODNs) and adeno-associated virus (AAV)-based vectors, emphasizing its high versatility and advocating its broad applicability. Importantly, we show that the novel CRISPR/Cas9 system is effective in cell lines as well as in clinically relevant human primary hematopoietic cells. In both T cells and hematopoietic stem and progenitor cells, we measured a reduced mutational burden at on-target and off-target sites, further endorsing the exploitation of this system for the development of novel precision therapeutics.
MATERIALS AND METHODS
Production of cas9 fusions
The Streptococcus pyogenes Cas9 (spCas9) coding sequence was amplified via PCR from lentiCRISPR_1516 (Addgene 49525) and cloned into a plasmid containing two nuclear localization signals (NLS) both at N- and C-terminus of the Cas9 and a flexible (G4S) linker at the C-terminus of the Cas9. The C-terminus fusions were generated by cleaving the resulting spCas9 expression plasmid with AvrII (NEB) and PmeI (NEB). The coding sequence of the DNA repair factors was amplified from cDNA using RNA isolated from HEK-293T cells and fused to the spCas9 via Gibson Assembly using the NEBuilder HiFi DNA Assembly kit (NEB) according to the manufacturer′s instructions. The dominant negative 53BP1 (dn53BP1) fragment (13) was PCR amplified from the cDNA mentioned above with primers #4723 (5′-GGAGAAGAAGAGTTTGATATGC) and #4724 (5′-CTGCTCTTCCAGGGCAGAG). For generating the dnRNF168 effector, lacking the RING domain, we amplified the N- and the C-terminal portions of RNF168 via PCR to bypass the RING coding sequence using the primers #4740 (5′-ATGGCTCTACCCAAAGAC) and #4745 (5′-GAGTCCACGACGAGCACTCGGACAGCGAGGGGATG) or #4746 (5′-CTGTCCGAGTGCTCGTCGTGGACTCGGTACCA-3′) and #4743 (5′-CTTTGTGCATCTCTGAAACATCTG) respectively. The two fragments, that included overlapping sequences, were fused to the spCas9 expression plasmid described above via Gibson Assembly. A schematic of all the Cas9-fusion proteins used in this study is reported in Supplementary Figure S1. The Cas9-CtIP-dnRNF168 expression plasmid is available through the non-profit plasmid repository Addgene. Other Cas9 fusions described in this study are available from the corresponding author on request.
Cell culture conditions
All cell lines used have been authenticated using a 16 DNA markers profile (Eurofins Genomics). HEK293T cells were kept in Dulbecco's Modified Eagle's Medium (Life Technologies) supplemented with 10% FCS (Thermo Fisher Scientific), 1% Penicillin/Streptomycin (Merck), 1mM sodium pyruvate (Merck) and grown in plates for adherent cells (Sarstedt) kept at 37°C in 5% CO2. K562 and Jurkat cells were kept in RPMI 1640 medium (Life Technologies) supplemented with 10% FCS (Thermo Fisher Scientific) and 1% Penicillin/Streptomycin (Merck) in plates for suspension cells (Sarstedt) kept at 37°C in 5% CO2. Human peripheral blood mononuclear cells (PBMCs) were isolated from leukocyte reduction system chambers by density gradient centrifugation (Ficoll-Paque). PBMCs were kept in RPMI 1640 medium (Life Technologies) supplemented with 10% FCS (Thermo Fisher Scientific) and 1% Penicillin/Streptomycin (Merck) for 4 h before activation. CD34+ hematopoietic stem and progenitor cells (HSPCs), were isolated from mobilized peripheral blood (IRB#329/10) by MACS using the CD34 MicroBead Kit UltraPure (Miltenyi Biotec) and cultured in CellGenix GMP SCGM (CellGenix) supplemented with recombinant human SCF (300 ng/ml; Immunotools), Flt3-L (300 ng/ml; Immunotools), TPO (100 ng/ml; Immunotools) and IL-3 (60 ng/ml; Preprotech) for 72 h before electroporation at a density of 0.5 × 106 cells/ml in a 24-well plate. After electroporation, cells were recovered in the aforementioned medium without IL-3 and cultured in a 96-well plate for 48h.
Traffic light reporter (TLR) assay
HEK-TLR cells (14) were seeded in 24-well plates (Sarstedt) at a density of 150 000 cells/well. After 24 h, the cells were transfected with a DNA mixture containing 375 ng of the indicated nuclease expression plasmid, 375 ng of sgRNA expression plasmid, 375 ng of DNA repair template using polyethylenimine. To normalize for transfection efficiency, the DNA mixture contained 375 ng of a TagBFP expression plasmid. Three and six days post transfection, HEK-TLR cells were harvested and the quantification of DSB either resolved via NHEJ (TagRFP positive cells) or HDR (GFP positive cells) repair pathways were assessed in the TagBFP positive cells (to normalize for transfection efficiency) via flow cytometry using a BD-LSRFortessa (Becton Dickinson), by acquisition of a total of 10 000 events in the living cell population (SSC-A/FSC-A). Precision score was computed as the ratio of the HDR to NHEJ frequencies resulted by the flow cytometry analysis.
Gene conversion assay
HEK-BFP cells previously generated in our laboratory (15) were seeded in 24-well plates (Sarstedt) at a density of 150 000 cells/well. After 24 h, the cells were transfected using Lipofectamine-2000 (Thermo Fisher Scientific) with a DNA mixture containing 100 ng of the indicated nuclease expression plasmid, 100 ng of sgRNA expression plasmid and 1 pmol of the single stranded oligodeoxynucleotide (ssODN; Integrated DNA Technology) used as repair template (Supplementary Table S1). The extent of gene conversion, measured as the fraction of cells expressing GFP (GFP/FSC-A) was measured via flow cytometry using a BD-LSRFortessa (Becton Dickinson) 48 h later, by acquisition of a total of 10000 events in the living cell population (SSC-A/FSC-A).
Targeted integration of GFP transgene in human cell lines
For targeted integration at the AAVS1 locus using plasmid DNA as repair template, K562 and Jurkat cells were electroporated with 900 ng of the indicated nuclease expression plasmid, 450 ng of sgRNA expression plasmid targeting the AAVS1 gene (target site: ACCCCACAGTGGGGCCACTA; PAM:ggg) and 1350 ng of repair template plasmid. For both cell types, electroporation was performed using a 4D-Nucleofector system (Lonza) and cells were transferred in a single well of a 96-wells plate (Sarstedt). For K562 editing, 500 000 cells were electroporated using the SF Cell line 4D-Nucleofector Kit (Lonza) and the FF-100 program while for Jurkat cells editing, 500 000 cells were electroporated using the SE Cell line 4D-Nucleofector Kit (Lonza) and the CK-116 program. When using a recombinant AAV to deliver the repair template, the corresponding plasmid was omitted from the electroporation mix and K562 cells were transduced immediately after electroporation using 1 × 104 transducing unit (TU) per cell of an AAV2/6 containing the repair template produced as previously described (16). Twenty-four h after the delivery of genome editing components, cells were split into two wells of a 96-well plate, and the wells were filled with complete RPMI medium to let the cells expand for an additional day. Twenty-four hours later, the cells from the two wells were pooled together in a single well of a 48-wells plate containing complete RPMI medium. The cells were then cultured for nine days, changing the size of the wells accordingly to the cell number, harvested by centrifugation and resuspended in flow cytometry buffer for analysis. GFP + cells were measured by acquiring 10 000 total events in the living cell population (FSC-A/SSC-A) using a BD Accuri C6 Cytometer (Becton Dickinson). The HDR-mediated DSB repair assay at the LMNA gene was essentially performed as described (17). In brief, HEK293T cells were seeded in 24-well plates (Sarstedt) at a density of 120 000 cells/well. After 24 h, the cells were transfected using Lipofectamine-2000 (Thermo Fisher Scientific) with a DNA mixture containing 40 fmol of the indicated nuclease expression plasmid, 70 fmol of sgRNA expression plasmid targeting the LMNA gene (target site: GGTTGGCAGCGCTGCCCGCG; PAM:ggg) and 168.3 fmol of repair template plasmid (gift from Graham Dellaire; Addgene #122508). For indel analysis, genomic DNA was extracted 48-h later using the NucleoSpin® Tissue gDNA extraction kit (Machery Nagel) following the manufacturer's procedure. The target locus was amplified via PCR with the primers #8088 (5′-TCAAGGGTCTTGCGGGCATC) and #8087 (5′-CCAGAAGGTCTGAGGCAATGG) using the Q5® Hot Start High-Fidelity DNA Polymerase (New England Biolabs) according to manufacturer's procedure. The resulting PCR amplicon was purified using the QIAquick® PCR Purification Kit (Qiagen) and the product used for Sanger sequencing and TIDE analysis (https://tide.nki.nl/) to quantify the frequency of indel mutations. To measure the HDR-mediated repair of the nuclease induced DSB, the cells were cultured for 6 days and then harvested by centrifugation and resuspended in flow cytometry buffer for flow cytometry analysis. Clover+ cells were measured by acquiring 10 000 total events in the living cell population (FSC-A/SSC-A) using a BD Accuri C6 Cytometer (Becton Dickinson).
Gene editing outcome evaluation methods
T7 endonuclease 1 (T7E1) assay was used to identify the best-performing BFP-specific CRISPR/Cas9 (Supplementary Figure S2). The assay was performed and quantified as previously described (18). The correct integration of the GFP expression cassette at the AAVS1 locus (Supplementary Figure S4) was confirmed via in-out PCR. To this end, the 5′-junction was detected using the primers #1405 (5′-GACGTGAAGAATGTGCGAGA) and #1207 (5′-CCAGCTCCCATAGCTCAGTCTG) while the 3′-junction was detected using the primers #1208 (5′-GGGCTCAGTCTGAAGAGCAGAG) and #154 (5′-CTACGGCAAGCTGACCCTGAA), respectively. Amplicons were resolved on a 2% agarose gel. All the TIDE and TIDER (https://tide.nki.nl/) analysis reported were performed by amplifying the targeted locus using the Q5® Hot Start High-Fidelity DNA Polymerase (New England Biolabs) according to manufacturer's procedure. The AAVS1 target site was amplified using the primers #1567 (5′- CCTTCTTGTAGGCCTGCATCATCACC) and #1568 (5′-GGATCCTCTCTGGCTCCATCGTAAG) the CCR5#1 and CCR5#2 sites were amplified with the primers #985 (5′-AAGATGGATTATCAAGTGTCAAGTCC) and #3780 (5′-AGACCTTCTTTTTGAGATCTGG). The PCR reactions were purified using the QIAquick® PCR Purification Kit (Qiagen) and the sequencing outsourced (Genewiz). Next generation sequencing at the CCR5#1 on and off target sites has been performed as previously described (19). In brief, purified PCR amplicons, either obtained from control cells or from cells receiving the indicated nuclease, were combined in two separate pools and the NGS libraries constructed using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® in conjunction with NEBNext® Multiplex Oligos for Illumina® following the manufacturer's instructions. Libraries were quantified via droplet digital PCR using the ddPCR™ Library Quantification Kit for Illumina TruSeq (Bio-Rad) and subsequently sequenced on an Illumina MiSeq device using the Illumina MiSeq Reagent Kit v2 (300-cycles). NGS data was analyzed using the CRISPResso2 software package (20) and the positional INDEL distribution was determined using a custom script from CRISPResso2’s multiple alignment files.
Genotoxicity analysis
To quantify chromosomal aberrations derived from on- and off-target activities of the designer nucleases used, we applied CAST-Seq, a method previously described in our laboratory (19). NGS library preparation steps were essentially performed as previously described using the established CCR5#1Cen primer set (19). However, two minor changes to the original protocol were introduced: (i) PCR fragments obtained after the second, nested PCR were separated by agarose gel electrophoresis. DNA fragments with a size of 200–500 bp were subsequently isolated using the QIAquick® Gel Extraction Kit (Qiagen) and processed further; (ii) pooled NGS libraries were shipped to sequencing service providers (Azenta Life Sciences, formerly Genewiz, or Novogene). Sequencing was performed on a NovaSeq or Illumina HiSeq platform with 2 × 150 bp configuration. For bioinformatics analysis, our previously described pipeline for CRISPR/Cas9 nucleases was used (21). Translocations detected significantly in both technical replicates are shown. To measure the impact of the nucleases used in this study on chromosomal stability, we quantified the extent of terminal chromosomal loss via droplet digital PCR (ddPCR) as previously described (22,23). In brief, equimolar (80 fmol) amounts of an expression plasmid encoding either the unmodified Cas9 or the Cas9-CtIP-dnRNF168 were delivered in HEK293T cells together with 140 fmol of sgRNA expression plasmid targeting HBB (target site: CTTGCCCCACAGGGCAGTAA; PAM:cgg) using Lipofectamine-2000, according to manufacturer's recommendations. Genomic DNA was purified 48 h later using the NucleoSpin® Tissue gDNA extraction kit (Machery Nagel) following the manufacturer's procedure. For each ddPCR reaction mix, we used 50 ng of genomic DNA as input with the QX200TM EvaGreen ddPCR Supermix TM and complexed with 100 nM of corresponding primers (CARS1: Fw_5′-GGGCCAGGGAAGTGTATGATG and Rv_5′-ACAGACATCAGTGCCATTGCG; HBE: Fw_5′-CAGCTCACTCAGCTTAGCAAAGG and Rv_5′-GACAGCTTTGGAAACCTGTCGTC). Droplets were generated and data acquired with the QX200 Droplet Digital PCR System (Bio-Rad). Results were analyzed with QuantaSoftTM Analysis Pro (Bio-Rad). PCR conditions: lid preheat at 95°C for 5 min, 50 cycles of 95°C for 30 s, 65°C for 60 s, 72°C for 60 s, followed by 5 min at 4°C and 5 min at 90°C (ramping rate set to 2°C/s). The fold change compared to the mock sample of the ratio between the copy concentrations of CARS1 and HBE genes is indicative of terminal chromosomal loss and is calculated as follows: (CARS1copies/μl/HBEcopies/μl)sample/(CARS1copies/μl/ HBEcopies/μl)mock.
Immunofluorescent visualisation of DNA damage
HEK293T cells were seeded in 2-well chamber slide with removable wells (Thermo Fisher Scientific) at a density of 200 000 cells/well. After 24 h, cells were transfected with 80 fmol of an hemagglutinin- (HA)-tagged Cas9-CtIP-dnRNF168 expression plasmid using Lipofectamine-2000 as described above. After 24 h, cells were treated with γ-radiation (50 Gy) in order to induce DNA damage that was visualized 24 h later, by detecting the formation of γ-H2AX foci. In brief, cells were washed once with PBS buffer and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Subsequently, the fixed cells were washed three times with fresh PBS buffer and then permeabilized in 0.125% Triton X-100 (Sigma Aldrich) in PBS buffer for 15 min. Cells were then washed three times in fresh PBS buffer prior blocking in 5% bovine serum albumin (BSA; Sigma Aldrich) solution in PBS buffer for 1 h. The cells were then incubated overnight at 4°C with primary antibodies, either Alexa Fluor 488 mouse anti-H2AX (pS139, BD Pharmingen) antibody or rabbit anti-HA-Tag (NB600-363, Novus Biologicals) antibody, diluted 1:500 in 5% BSA solution. On the next day, the cells were washed three times with PBS buffer and HA-tagged Cas9 was visualized by incubation for 1 hour with Alexa Fluor 568 goat anti rabbit IgG (H + L; Thermo Fisher Scientific) secondary antibody, diluted 1:1000 in 5% BSA solution. Lastly, cells were washed five time with PBS buffer, and then coverslips were mounted with VECTASHIELD Antifade Mounting Medium containing DAPI, to counterstain cellular nuclei. Fluorescent images were obtained with an AxioObserver II inverted microscope (Zeiss) with 100× oil immersion objective.
Gene editing in human primary cells
PBMCs were activated 4 h after thawing using ImmunoCult™ Human CD3/CD28/CD2 T Cell Activator (5 μl/1 × 106; STEMCELL Technologies) and IL-2 (100U/μl; Immunotools), and kept at concentration of 2 × 106 cells/ml. Three days post-activation, 1 × 106 PBMCs were electroporated with a 4D-Nucleofector system (Lonza) using the P3 Primary Cell 4D-Nucleofector Kit (Lonza) and the EO-115 program. The DNA mixture contained 30 pmol of the nuclease expressing mRNA transcribed in vitro as previously described (24), 112.5 pmol of sgRNA (Synthego) and 25 pmol of ssODN (Integrated DNA Technologies). After electroporation, complete RPMI medium supplemented with IL-2 (1000 U/ml; Immunotools) was used to recover the cells before culturing them in a 96-well U-shaped-bottom plate (Falcon). Cells were harvested 5 days post electroporation and the genomic DNA was extracted using the NucleoSpin® Tissue gDNA extraction kit (Machery Nagel) following the manufacture's procedure and resuspended in 40 μl of Nuclease-free water. 200 000 HSPCs were electroporated with a 4D-Nucleofector system (Lonza) using the P3 Primary Cell 4D-Nucleofector Kit (Lonza) and the CA-137 program with a electroporation mix containing 30 pmol of nuclease expressing mRNA transcribed in vitro as previously described (25), 112.5 pmol of sgRNA (Synthego) and 50 pmol of the respective ODN (Integrated DNA Technologies). After electroporation, cells were recovered and cultured in a 96-well plate. Cells were harvested 2 days post electroporation and their genomic DNA was extracted using the NucleoSpin® Tissue gDNA extraction kit (Machery Nagel) following the manufacture's procedure and resuspended in 40 μl of Nuclease-free water.
Statistics
All data sets shown as bar graphs represent the average of at least three independent experiments. Biological replicates are represented as black dots in the figures and each dot results from the average of technical duplicates. Error bars indicate standard error of mean (SEM). Statistical significance was determined using a two-tailed, homoscedastic paired Student's t-test or multivariate ANOVA analysis as reported in the figure legends.
RESULTS
Identification of the most efficient nuclease to promote precision gene editing
To alter the physiological repair of CRISPR/Cas9 induced DSBs, we sought to use the Cas9 nuclease as a scaffolding protein for the targeted deposition of key components of the DNA repair mechanism at the cleaved site. We reasoned that the presence of factors capable of either inhibiting NHEJ or promoting HDR during DSB repair would increase precision editing. We selected nine factors based on their crucial role in endorsing the early steps of HDR, such as the ability to stimulate DNA end resection or to promote the search of homologous sequences during DSB repair. In addition, since pharmacological inhibition of NHEJ is typically associated with increased HDR-mediated repair (5), we thought to explore this concept in a targeted manner and to inhibit NHEJ only at the DSB site (26). Specifically, we focused on 53BP1, a crucial component of DSB repair that is rapidly recruited to the nascent DSB where it promotes its resolution via NHEJ, and selected a previously characterized dominant negative (dn) 53BP1 mutant (10). Similarly, we explored the use of a dominant negative ring finger protein 168 (dnRNF168) lacking the RING domain which was implicated in recruitment of 53BP1 to the DSB both via direct protein–protein interaction and through the ubiquitylation of the chromatin surrounding the DSB (27). To achieve the synchronous presence of these factors during DSB formation, we fused them to the C-terminus of the Streptococcus pyogenes Cas9 protein via a flexible (G4S) linker. To assess the ability of these Cas9 fusions to promote HDR-mediated repair, we used the traffic light reporter (TLR) assay, a previously described molecular tool that allows the quantification of NHEJ- or HDR-mediated DSB repair events via flow cytometry (14). The TLR construct includes a fusion between mVenus and TagRFP genes, which is expressed from a transgene integrated into the AAVS1 locus in HEK293T cells (HEK-TLR cells). A mutation within the mVenus coding sequence combined with a shift in the reading frame of the TagRFP gene prevents the expression of both proteins (Figure 1A). Using a CRISPR/Cas9 nuclease targeted to the mutated mVenus gene results in a DSB that, in the absence of an appropriate repair template, is sealed via NHEJ. In principle, this leads to the formation of random indels that result in TagRFP expression in one third of the total repair events, potentially underestimating the extent of total NHEJ events. However, the propensity of Cas9 to create a common 1-bp indel at the DSB site (25), mitigates this bias. Simultaneous delivery of a repair matrix allows the precise correction of the mVenus gene via an HDR-mediated mechanism. Therefore, flow cytometry can easily monitor which DNA repair mechanism is engaged to resolve the nuclease-induced DSB (Figure 1A, left panel). We used this system to assess the impact of the different Cas9 fusions on DSB repair. The HEK-TLR cells were transfected with expression plasmids coding for either of the Cas9 fusion constructs indicated, together with a single guide RNA (sgRNA) targeting the mutated mVenus gene in proximity to the mutation. The repair template harbored the correct mVenus sequence flanked on either side by regions of homology to the genomic target site. To enable analysis of transfected cells, we co-transfected a plasmid expressing a blue fluorescent protein (BFP). Six days after transfection, cells were harvested and the extent of NHEJ- or HDR-mediated repair events were quantified by flow cytometry in the BFP positive cell population (Figure 1A, right panel). Using the unmodified CRISPR/Cas9 resulted in the vast majority of the total DSBs being resolved via NHEJ, with only about a sixth of the total events repaired via HDR, as indicated by HDR:NHEJ ratio (precision score) of 0.18 ± 0.01 (Figure 1B). In line with previous reports (9), use of a CRISPR/Cas system equipped with a Cas9-CtIP fusion resulted in reduction of NHEJ and a concomitant increase of HDR-mediated DSB repair events, giving rise to a 2.3-fold increase in the precision score while all the other Cas9 fusions showed only a minor effect (Figure 1B). Analysis of the NHEJ and HDR repair frequencies revealed that in most of the cases, the deposition of the indicated factors at the DSB site significantly reduced the extent of NHEJ-mediated repair with negligible increase of HDR-mediated DSB resolution (Figure 1C). To develop a single molecule capable of simultaneously inhibiting NHEJ and endorsing HDR, we generated five second generation Cas9 fusion proteins that combined the ability of CtIP to promote HDR with the NHEJ-inhibiting function of selected effectors tested above. These novel fusion proteins synergistically promoted precision editing with 3.7-fold gain in the precision score as compared to the Cas9-CtIP and 7.0-fold increase when compared to the benchmark Cas9 (Figure 1D). A more extensive analysis of the NHEJ- and HDR-mediated DSB repair frequencies, as measured with the TLR reporter, revealed that most of the second generation constructs were able to resolve DSB through either of the two DSB repair pathways with similar efficiency (Figure 1E). The most efficient fusion combining CtIP and dnRNF168 was capable of resolving a Cas9-induced DSB preferentially via HDR, i.e. with a precision score >1. This second generation Cas9 fusion, capable of hijacking the normal resolution of a nuclease-induced DSB, was selected for further characterization.
Figure 1.

CRISPR/Cas9-CtIP-dnRNF168 promotes HDR-mediated repair of a designer nuclease-induced DSB. (A) The Traffic Light Reporter (TLR) is integrated into the AAVS1 site of HEK293T cells (HEK-TLR) and includes a fusion of a mutated mVenus with an out-of-frame TagRFP genes. Depending on the engaged DSB repair pathway, the introduction of a CRISPR/Cas9 induced DSB results in the appearance of red or green cells that can be quantified via flow cytometry (left). Representative plots showing the gating strategy for the acquisition of the TagRFP+ (NHEJ) and mVenus+ (HDR) events within the transfected cell population (BFP+) is shown on the right. Δ: mutation abrogating mVenus expression. CAG: CMV enhancer, chicken beta-actin promoter. T2A: 2A self-cleaving peptide. (B, D) The bar graphs show the precision score, computed as the ratio between the HDR and NHEJ events measured as reported in A, for the various Cas9 fusion proteins. The fold change in relation to the unmodified Cas9 is indicated for the best variant. (C, E) The diagrams indicate the percentage of NHEJ or HDR events as determined by flow cytometry on day 6 post transfection. In panels B-E, each dot represents the average of experimental duplicates. Statistically significant differences, as compared to normal Cas9 nuclease, are indicated with asterisks and correspond to p-values calculated with ANOVA (*P< 0.05, **P< 0.01). Error bars indicate SEM.
Cas9 fused to CtIP and dnRNF168 promotes precision editing using short oligodeoxynucleotide templates
Since the TLR reporter system uses a plasmid-based donor template to repair the mVenus gene, we wondered whether the ability of Cas9-CtIP-dnRNF168 to promote precision editing was limited to this type of repair matrix. To this end, we designed a single stranded oligodeoxynucleotide (ssODN) capable of converting a BFP gene into a gene coding for green fluorescent protein (GFP) by means of two nucleotide changes (194 G > C and 196 C > T; Figure 2A). We employed HEK293T cells stably expressing BFP (HEK-BFP cells) and assayed the ability of selected first and second generation Cas9 fusions to promote BFP-to-GFP conversion using the ssODN repair template (Figure 2A and Supplementary Table S1). Cells were transfected with plasmids coding for the best performing BFP-targeting sgRNA (i.e. sgRNA#2; Supplementary Figure S2) and the different Cas9 fusions. The percentage of GFP positive cells, indicative of gene conversion, was evaluated two days later via flow cytometry. Differently from the TLR assay, the Cas9-CtIP fusion did not significantly increase the extent of precision editing. On the contrary, we measured a slight but significant increase in HDR when either RAD51 or RAD52 were deposited at the DSB, resulting in a 1.5-fold and 1.4-fold increase, respectively, in the number of cells expressing GFP when compared to standard Cas9 (Figure 2B). Again, targeted blockade of NHEJ resulted in the highest increase in gene conversion. Indeed, our novel CRISPR system equipped with the Cas9-CtIP-dnRNF168 fusion outperformed all other constructs and resulted in 13.5 ± 0.2% GFP-positive cells, corresponding to a 2.6-fold increase in gene conversion events (Figure 2B). Interestingly, Cas9-CtIP-dnRNF168 retained its ability to promote precise nucleotide exchange with high efficiency also when ODNs with different architectures were used. By varying the length of the ODN homology arms relative to the target site or by using double stranded ODNs, Cas9-CtIP-dnRNF168 outperformed the other constructs for their ability to promote BFP-to-GFP gene conversion (Supplementary Figure S3B and Supplementary Table S1).
Figure 2.

CRISPR/Cas9-CtIP-dnRNF168 promotes gene conversion using ssODN as repair template. (A) Co-delivery of the CRISPR/Cas9 together with single-stranded oligodeoxynucleotides (ssODN) harboring the nucleotide changes indicated in red (194 G > C and 196 C > T) results in BFP to GFP conversion (upper panel). Shown are exemplary plots that illustrate the extent of GFP+ cells resulting from gene conversion triggered by the indicated nucleases (lower panel). (B) The bar graph indicates the extent of BFP-to-GFP gene conversion measured via flow cytometry as percentage of GFP + cells. Experiments were performed in duplicate and pooled data are shown as black dots. Statistically significant differences, as compared to the unmodified Cas9 nuclease, are indicated with asterisks and correspond to p-values calculated with ANOVA (**P< 0.01). Error bars indicate SEM.
Cas9-CtIP-dnRNF168 promotes targeted integration of a large gene expression cassette
Next, we thought to validate our novel CRISPR system for targeted integration of a large DNA fragment containing a GFP-expression cassette in different target loci and multiple human cell lines. To this end, we generated a promoter-less GFP expression cassette for the targeted insertion of the GFP gene at the human AAVS1 locus. The donor template contained 700 base pairs homology regions flanking a promoter-trap cassette composed of a splice acceptor (SA) and the coding sequence for a T2A self-cleaving peptide fused to the GFP gene. Upon integration in the first intron of the target gene, the transcripts derived from the endogenous promoter undergo alternative splicing using the newly integrated SA thus securing GFP expression. We assessed the efficiency of our CRISPR system equipped with the Cas9-CtIP-dnRNF168 fusion in two human cell lines widely used as surrogate models for hematopoietic cells, namely the K562 erythroleukemic cell line and the Jurkat T lymphoblastic cell line. We delivered the genome editing components, including the plasmids encoding the Cas9 nuclease, the repair template and an AAVS1-targeting sgRNA, via electroporation. Nine days later, stable expression of GFP, indicative of targeted integration, was assessed via flow cytometry (Figure 3A, left side). In line with the previous experiments, the Cas9-CtIP-dnRNF168 fusion outperformed the unmodified Cas9 with an increase in GFP+ cells reaching up to 49.9 ± 3.0% and 13.8 ± 0.8% in K562 and Jurkat cells respectively (Figure 3B and Supplementary Figure S4A). The corresponding precision scores increased to 3.2- and to 24.8-fold in K562 or Jurkat cell lines, respectively, further underlying the ability of novel CRISPR system for promoting HDR-mediated DSB resolution in different cellular contexts (Figure 3C and Supplementary Figure S4B). Furthermore, we validated the performance of our novel CRISPR system in a third HDR-mediated DSB repair assay, designed to insert the coding sequence for the green fluorescent protein Clover, at the LMNA gene (17,28). Despite a similar ability to promote HDR-mediated repair at this target site, CRISPR/Cas9-CtIP-dnRNF168 was capable of dramatically reduce NHEJ-mediated repair thus resulting in a 5.3-fold increase in precision score as compared to the unmodified CRISPR system (Supplementary Figure S4C and D).
Figure 3.

CRISPR/Cas9-CtIP-dnRNF168 promotes the targeted integration of large expression cassettes. (A) Experimental design. K562 cells were electroporated with the corresponding CRISPR/Cas9 expression plasmids together with a plasmid as repair template (left) or transduced immediately upon electroporation with an adeno associated virus (AAV)-based vector containing the HDR template (right). Nine days later, NHEJ and HDR frequencies were measured via TIDE or flow cytometry, respectively. (B, D) The graphs show the frequency of gene edited K562 cells that underwent either NHEJ- or HDR-mediated DSB resolution, measured by TIDE (NHEJ) or flow cytometry (HDR), respectively. (C, E) Graphs show the precision score, calculated as the ratio of HDR to NHEJ events of the results shown in panels B and D, respectively. Fold change, as compared to the unmodified CRISPR/Cas9 nuclease, is indicated within the graph. Each dot represents biological replicates. Statistically significant differences, as compared to the unmodified CRISPR/Cas9 nuclease, are indicated with asterisks within the graphs and correspond to P-values calculated with a two-tailed, paired Student's t-test (**P< 0.01). Error bars indicate SEM.
For therapeutic genome editing, AAV vectors are commonly used to deliver the DNA repair template into the target cells (29,30). We therefore tested the ability of CRISPR/Cas9-CtIP-dnRNF168 to promote targeted genomic alterations in the presence of an AAV-based repair matrix. Following our established pipeline (31), we generated an AAV serotype 6 (AAV6) vector containing a GFP expression cassette driven by the phosphoglycerate kinase (PGK) promoter. K562 cells were electroporated with the two plasmids encoding the Cas9 nucleases or the sgRNA, respectively, and immediately transduced with the AAV donor template (Figure 3A, right side). Cells receiving only the AAV donor showed a transient GFP expression that was undetectable by day 9 (Supplementary Figure S5A). In contrast, the two nucleases tested promoted targeted integration of the GFP expression cassette, resulting on average in 68.2 ± 0.7% of the cells expressing GFP, 9 days after electroporation (Figure 3D and Supplementary Figure S5A). In-out PCR using a primer binding the genomic region outside of the homology arm of the repair template paired with a primer binding inside the donor cassette confirmed the targeted integration of the GFP expression cassette (Supplementary Figure S5B). Since NHEJ-mediated DSB repair results in genotoxic indel mutations at the nuclease target site, we sought to compare side-by-side the genotoxic potential of the nucleases used. To this end, we profiled the mutational landscape at the AAVS1 target site using TIDE (Tracking of Indels by Decomposition) (32). Interestingly, despite a similar HDR frequency, using Cas9-CtIP-dnRNF168 resulted in a significantly lower mutational burden, with almost 2-fold reduction in the number of indels at the target site as compared to canonical Cas9 (16% versus 37.7%; Figure 3D and Supplementary Figure S5C). As a consequence, the computed precision score was 2.2-fold higher as compared to unmodified Cas9 (Figure 3E). Importantly, the use of CRISPR/Cas9-CtIP-dnRNF168 resulted in a robust reduction of +1 insertions, which is the predominant outcome of CRISPR/Cas9 based genome editing events and is potentially dangerous when targeting protein coding regions as it results in frameshift mutations (25,33) (Supplementary Figure S5C).
CRISPR/cas9-CtIP-dnRNF168 stimulates precise gene editing in primary human T lymphocytes and hematopoietic stem and progenitor cells
Having established CRISPR/Cas9-CtIP-dnRNF168 as an efficient tool for promoting precise gene editing in cell lines, we sought to assess its ability to install a point mutation in clinically relevant primary human hematopoietic cells, such as T lymphocytes and hematopoietic stem and progenitor cells (HSPCs). Since these cells do not tolerate plasmid DNA for the expression of genome editing tools, we delivered the nucleases in form of in vitro transcribed mRNA alongside with a chemically modified sgRNA targeted to site #2 in the exon 3 of the CCR5 gene (i.e. CCR5#2) previously characterized in our laboratory (19). We designed both ssODN and dsODN (Supplementary Table S1) to introduce a silent nucleotide change to abolish the protospacer-adjacent motif (PAM) and avoid Cas9 cleavage of the target site upon editing (Figure 4A). Gene editing components were electroporated in activated HSPCs or T cells (24,34) and the frequencies of DSBs resolved either via NHEJ or HDR measured via TIDER (35) two and five days later, respectively. In both cell types tested, CRISPR/Cas9-CtIP-dnRNF168 promoted an increase in precision score ranging from 1.8-fold to 2.1-fold in primary HSPC or T cells, respectively, as compared to canonical CRISPR/Cas9 (Figure 4B and Supplementary Figure S7). TIDER analysis indicated that both CRISPR/Cas9 and CRISPR/Cas9-CtIP-dnRNF168 supported similar levels of HDR-mediated DSB repair, installing the desired nucleotide change with similar efficiencies in the two cell types analyzed (Figure 4C and D, left panels). However, as compared to the canonical Cas9, the use of our novel Cas9 variant ensued a robust reduction of indel mutations at the target site, including the predominant +1 insertion which results in an out-of-frame mutation at this site (Figure 4C and D, right panels).
Figure 4.

CRISPR/Cas9-CtIP-dnRNF168 stimulates precise genome editing in primary human hematopoietic cells using an ssODN repair template. (A) Experimental design. Genome editing components are electroporated in primary human hematopoietic stem and progenitor cells (HSPC) or T lymphocytes and the extent of precision editing is measured via TIDER or next generation sequencing two and five days later, respectively. (B) The graph shows the precision score of the indicated nuclease targeted to CCR5 site #2, computed as the ratio between the HDR and NHEJ events measured via TIDER, either in primary human T cells (left) or stem and progenitor cells (right). Fold change, as compared to the unmodified Cas9 nuclease, are reported within the graphs. (C, D) The bar graphs indicate the frequency of NHEJ- or HDR-mediated DSB resolution (left panels) and the nature of the different indel mutation identified (right panels) via TIDER. The extent of unmodified sequence is indicated in blue. (E) The diagram shows the precision score measured via next generation sequencing and calculated as in panel B for the indicated nucleases targeted to CCR5 site #1. Fold change, as compared to the unmodified Cas9 nuclease, are reported within the graph. (F) Extent of indel formation at the CCR5 site #1 target site (On-target) and at a previously identified off target site (Off-target) in the CENPJ gene, using the indicated nucleases. (G) Frequency of the +1 indel measured at the on- and off-target sites indicated in panel F. (H) The graph shows the chromosomal aberrations identified within a window of 2.4 kb surrounding the CCR5 site #1 target site (on-target) via CAST-seq. The normalized read count is indicated. (I) Bar graph displays normalized CAST-seq reads that indicate the number of translocations between the on-target site and an off-target site on the indicated chromosomes. In panels B–G, each dot represents a biological replicate. Statistically significant differences, as compared to the unmodified Cas9 nuclease, are indicated with asterisks and correspond to p-values calculated with a two-tailed, paired Student's t-test (*P< 0.05, **P< 0.01). Error bars indicate SEM.
CRISPR/cas9-CtIP-dnRNF168 is less genotoxic than CRISPR/cas9 in cell lines and in primary human cells
Despite being less detrimental at the DSB site, with a more favorable indel distribution, we sought to investigate whether the overexpression of Cas9-CtIP-dnRNF168 compromises genomic stability through unwanted interaction with key endogenous DNA repair factors. We used a previously described CRISPR/Cas9 nuclease targeting the HBB gene, located on the p-arm of chromosome 11 at a distance of 5.2 Mb from the telomere (36), and measured the tendency of this nuclease to induce terminal chromosomal loss via droplet digital PCR (22,23). To this end, we determined the copy numbers of two genes, CARS1 and HBE, located 2.2 Mb (on the telomeric side) and 43 kb (on the centromeric side) away from the HBB gene, respectively (Supplementary Figure S6A). When targeting HBB using the standard CRISPR/Cas9, we observed a trend towards reduction in terminal chromosome 11 signal (based on CARS1:HBE ratio), suggesting the presence of chromosomal truncation. Using the Cas9-CtIP-dnRNF168 fusion did not change the frequency of truncation events (Supplementary Figure S6B). Having established the accuracy in targeted genome editing, we sought to determine the effect of CRISPR/Cas9-CtIP-dnRNF168 at off-target sites in primary HSPCs. Since the CCR5#2-specific CRISPR/Cas system described above did not show any sign of off-target cleavage in our previous report (19), we profiled the specificity of CRISPR/Cas9-CtIP-dnRNF168 using an sgRNA targeting the CCR5 site #1 (i.e. CCR5#1) for which we previously identified the occurrence of indel mutations at one prominent off-target site in the CENPJ gene (19). Genomic DNA from HSPCs that were edited with either the unmodified CRISPR/Cas9 or the CRISPR/Cas9-CtIP-dnRNF168 at the CCR5#1 site was extracted 2 days after electroporation. Targeted amplicon next generation sequencing (NGS) encompassing the CCR5#1 on-target site and the CENPJ off-target site, respectively, revealed a 1.3-fold increase in the precision score (Figure 4E), supporting the reproducibility of CRISPR/Cas9-CtIP-dnRNF168 performance at different targets. In particular, CRISPR/Cas9-CtIP-dnRNF168 resulted in overall reduced NHEJ-mediated mutagenesis at the analyzed off-target site (Figure 4F) with a significant reduction of the +1 insertion (Figure 4G). We recently showed that concomitant on- and off-target activities result in genomic rearrangements, including translocations, and that a subset of these aberrations is dependent on the HDR machinery. We therefore sought to investigate whether the use of novel Cas9 variant results in a different profile of large genomic aberration as compared to canonical CRISPR/Cas9. CAST-seq analysis was performed 2 days after electroporation of T cells with either unmodified CRISPR/Cas9 or CRISPR/Cas9-CtIP-dnRNF168 targeting the CCR5#1 site. In line with our previous report, we confirmed previously identified translocations between the CCR5#1 target site and off-target cleavage sites on chromosomes 1, 13, 16, 19 and 22 (Supplementary Table S2). Analysis of the large deletions at the CCR5#1 site revealed no differences between the two nucleases (Figure 4H). Similarly, the frequencies of translocations between the CCR5#1 on-target site and the chromosomes harboring off-target sites were comparable, except for a site on chromosome 22 that was retrieved with higher frequency for CRISPR/Cas9-CtIP-dnRNF168 edited cells (Figure 4I). Taken together, considering the identical off-targeting profile retrieved and the similar impact on chromosomal truncation events obtained using the two CRISPR/Cas9 system tested, we conclude that the reduced mutational burden at both the on- and the off-target sites analyzed (Supplementary Table S2), highlights the increased safety profile of genome editing using CRISPR/Cas9-CtIP-dnRNF168.
DISCUSSION
Precision genome editing combines high efficiency in targeted gene modification with reduced mutagenic potential of the editing tools. Both of these attributes are crucial from a clinical perspective. However, clinical exploitation of genome editing is mostly limited to disruptive strategies based on NHEJ to inactivate regulatory elements or genes. Use of chemicals to promote HDR by either inhibiting NHEJ or stalling the cells in specific phases of the cell cycle has been promising, but the broad and non-specific effects of those compounds on cellular physiology preclude clinical translation. To alter the standard resolution of a designer nuclease induced DSB, we tethered rationally selected key players of DNA repair to the Cas9 protein. We used an established traffic light reporter system to monitor the extent of DSB repair either via NHEJ or HDR. In line with previous reports (9–11), we confirmed that local deposition of CtIP or dn53BP1 increased HDR (Figure 1C), and observed that the number of total repair events (i.e. NHEJ + HDR) was somewhat reduced when using CRISPR/Cas9-CtIP-dnRNF168 as compared to the canonical CRISPR/Cas9 (Figure 1E; CRISPR/Cas9 = 48.5%; CRISPR/Cas9-CtIP-dnRNF168 = 28.6%). Since the TagRFP coding sequence is placed on a 1bp frameshift (14), the predominant +1 indel typically induced by the canonical CRISPR/Cas9 and its reduction when using CRISPR/Cas9-CtIP-dnRNF168 might explain this result by restoring TagRFP frame more frequently when using the unmodified Cas9. Alternatively, increased sealing of the nuclease induced DSB without indel formation, similarly to the results obtained in primary cells (Figure 4F), might also explain this phenomenon. Interestingly, a recent report showed that fusion of the C-terminal portion of EXO1 to the Cas9 promoted HDR-mediated DSB repair in different cell lines and reduced p53 mediated cytotoxicity (37). On the contrary, our data show that fusing the full-length EXO1 to Cas9 had no effect on DSB resolution, suggesting that tethering of the entire EXO1 protein to the DSB site impairs precise editing. To our surprise, deposition of RAD51 did not result in increased HDR-mediated DSB resolution even though previous reports have shown that its overexpression positively impacts on CRISPR/Cas mediated targeted gene addition (38). This suggests that supraphysiological levels of RAD51 are necessary to promote RAD51-mediated repair of a nuclease induced DSB via HDR. However, high levels of RAD51 are often found in cancer cells and provide increased resistance to DNA damage, thus jeopardizing the efficacy of chemotherapy (39). This supports the concept that, as opposed to overexpression, a strategy that promotes the local deposition of critical HDR effectors at the DSB site, such as the one adopted herein, is preferable. Our data show that the use of Cas9 fused to single selected factors resulted mostly in reduced frequency of NHEJ-mediated indel formation, which is either due to steric hindrance that restrains the recruitment of factors involved in NHEJ, or because these factors promote seamless DSB resolution. Conversely, the local deposition of multiple synergistic effectors fused to the Cas9 protein, supports precision gene editing by simultaneously decreasing NHEJ and increasing HDR frequency in cell lines. Our best Cas9 fusion included CtIP together with a dominant negative RNF168 (dnRNF168) and resulted in the preferential resolution of the Cas9-induced DSB via HDR. We hypothesize that the presence of dnRNF168 at the DSB creates a barrier to the engagement of NHEJ. Indeed, dnRNF168 lacks the RING domain which is responsible for ubiquitinating 53BP1, an essential step for DSB-induced signalling and 53BP1’s role in NHEJ (27). On the other hand, dnRNF168 retains the multiple 53BP1 interaction regions, implicating that this NHEJ-promoting factor might be recruited to the DSB where, however, it is unable to trigger NHEJ as it is not ubiquitinated. In the absence of NHEJ induction, the local presence of CtIP promotes DNA resection (21,40), so accelerating HDR engagement. Conversely, the interaction of the dnRNF168 with endogenous 53BP1 could be in principle deleterious if it would lead to the accumulation of the Cas9-CtIP-dnRNF168 fusion protein at naturally occurring DSBs where it would compromise proper DNA repair. To rule out this hypothesis, we overexpressed our Cas9 variant in the absence of a sgRNA in cells with irradiation-induced DNA damage. The absence of co-localization of the Cas9-fusion to sites of DNA damage foci, visualized by phosphorylated γ-H2AX, suggests that proper resolution of natural DSBs should not be altered (Supplementary Figure S8). While further analyses are necessary to define the interactions of the Cas9-CtIP-dnRNF168 fusion protein with other DNA repair factors within the cells, our results suggest that combining NHEJ-inhibition with HDR-endorsement is sgRNA-dependent and therefore restricted to the nuclease target site. We believe that this feature is crucial to promote precision gene editing and it is particularly relevant for the safe editing of clinically relevant primary hematopoietic cells.
Therapeutic genome editing includes various strategies, such as single nucleotide exchanges or the integration of large transgene expression cassettes. We explored the ability of our novel CRISPR system to promote targeted genome editing using different repair templates, such as small ODNs or AAV-based vectors. Using a BFP to GFP gene conversion assay, we demonstrated that Cas9-CtIP-dnRNF168 outperformed the other Cas9 fusion constructs for successful gene conversion by 2.6-fold. In line with previous reports showing that ODN-mediated gene editing is HDR-independent but relies on single-strand template repair (SSTR) (41), Cas9 fusions that included key HDR players, i.e. CtIP, RAD51 or RAD52 alone, did not show major effect on the fraction of GFP positive cells as compared to canonical Cas9. However, inhibiting the first steps of NHEJ-mediated DSB repair through the deposition of dnRNF168 to the break site increased gene conversion significantly, which is further promoted by the simultaneous presence of CtIP. This suggests a novel implication for end resection in SSTR-mediated DSB resolution or that local inhibition of NHEJ forces DSB-resolution via HDR also in the presence of ODNs. Further experiments will be necessary to elucidate this fascinating mechanism. As compared to standard CRISPR/Cas9 approaches, CRISPR/Cas9-CtIP-dnRNF168 also promoted the targeted addition of large inserts using an AAV repair template in different human hematopoietic cell lines, highlighting the high versatility of this novel editing system. A platform capable of endorsing targeted genome editing by increasing HDR and/or reducing the mutational burden derived from unwanted mutagenesis at both on- and off-target sites, would be of high value for therapeutic applications. We hence evaluated the performance of CRISPR/Cas9-CtIP-dnRNF168 also in clinically relevant human primary T lymphocytes and HSPCs. In line with a recent report (42), our data emphasize that localization of key DNA repair factors at a DSB is not sufficient to promote HDR in primary cells but that it is essential to reduce NHEJ, so limiting the mutational burden associated with genome editing and resulting in a substantial increase in the precision score. Importantly, CAST-seq and ddPCR analysis revealed that altering the resolution of DSBs with CRISPR/Cas9-CtIP-dnRNF168 does not change the distribution of large genomic rearrangements or chromosomal truncations that occur in similar numbers as when using the unmodified Cas9. Indeed, large deletions at the CCR5#1 target site or truncation of chromosome 11 were similar in the samples treated with either nuclease. Similarly, in-depth analysis of the retrieved translocations revealed a generally reduced number of both homology-mediated and off-target mediated translocations when using CRISPR/Cas9-CtIP-dnRNF168 (Supplementary Table S2). Previous reports showed that the mutagenic potential of genome editing strategies both at on-target and off-target sites is significant (12,19). Consequently, CRISPR/Cas9-CtIP-dnRNF168, that mitigates these detrimental effects by reducing the mutational burden and in particular the occurrence of out-of-frame indels, such as the predominant +1 insertion, are invaluable and might contribute to alleviate the clinical risks associated with therapeutic genome editing. Robust NHEJ inhibitors, such as M3814, can also significantly improve HDR and reduce the occurrence of out-of-frame +1 insertions through the transient inhibition of DNA-PKcs activity (43). However, it is important to keep in mind that small molecule NHEJ inhibitors act globally, i.e. they also inhibit NHEJ at natural DSBs. This emphasizes the advantage of using CRISPR/Cas9-CtIP-dnRNF168 that results in similar effects but in a localized manner.
Despite its efficacy, our tethering strategy resulted in large Cas9 proteins that reached 330 kDa for our best performing Cas9-CtIP-dnRNF168. The large coding sequence prevents the use of well-established viral delivery vehicles, such as AAV vectors. Furthermore, the production of large recombinant Cas9 fusion proteins for delivery as ribonucleoprotein (RNP) might be cumbersome. We therefore established a protocol to efficiently transfer Cas9-CtIP-dnRNF168 to primary human hematopoietic cells in the form of in vitro transcribed mRNA. To solve this issue, we anticipate that further refinement of the Cas9 fusion partners is essential. For instance, reducing the size of Cas9-CtIP-dnRNF168 to a minimal functional unit, will further streamline the application of this novel technology. Reducing the protein size would also facilitate the purification of the Cas9-CtIP-dnRNF168 protein and enable its delivery as RNPs, which is typically favored for clinical translation due to the reduced persistence of the nuclease.
In conclusion, we established a novel platform that is capable of promoting precision editing in two clinically relevant human cell types. Considering the reduced mutagenic potential, CRISPR/Cas9-CtIP-dnRNF168 offers a substantial safety benefit and opens novel avenues for further exploring the use of CRISPR/Cas9 system as a novel tool for the next generation precision medicine.
DATA AVAILABILITY
CAST-seq and next generation sequencing datasets have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE225452.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Maria Silvia Roman Azcona and Jamal Alzubi for productive discussions, Yongxing Fang for generating the HEK-BFP cell line, and Melina el Gaz for the technical support. We are grateful to Ralf Kühn (MDC, Berlin) for providing the HEK-TLR cells, the Lighthouse Core Facility (Medical Center – University of Freiburg) for help with flow cytometry, and the Blood Donation Center, Medical Center – University of Freiburg, for providing LRS chambers. The article processing charge was funded by the Baden-Wuerttemberg Ministry of Science, Research and Art and the University of Freiburg in the funding program Open Access Publishing.
Notes
Present address: Antonio Carusillo, Vector BioPharma AG, Basel, Switzerland.
Present address: Laura Mosti, AGC Biologics, Milan, Italy.
Contributor Information
Antonio Carusillo, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany; Ph.D. Program, Faculty of Biology, University of Freiburg, Freiburg 79106, Germany.
Sibtain Haider, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany; Ph.D. Program, Faculty of Biology, University of Freiburg, Freiburg 79106, Germany.
Raul Schäfer, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany.
Manuel Rhiel, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany.
Daniel Türk, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany.
Kay O Chmielewski, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany; Ph.D. Program, Faculty of Biology, University of Freiburg, Freiburg 79106, Germany.
Julia Klermund, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany.
Laura Mosti, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany.
Geoffroy Andrieux, Institute of Medical Bioinformatics and Systems Medicine, Medical Center – University of Freiburg, Freiburg 79106, Germany; Faculty of Medicine, University of Freiburg, 79106, Germany.
Richard Schäfer, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany; Institute for Transfusion Medicine and Immunohaematology, German Red Cross Blood Donor Service Baden-Württemberg-Hessen gGmbH, Goethe University Hospital, Frankfurt am Main, 60596, Germany.
Tatjana I Cornu, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany; Faculty of Medicine, University of Freiburg, 79106, Germany.
Toni Cathomen, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany; Faculty of Medicine, University of Freiburg, 79106, Germany.
Claudio Mussolino, Institute for Transfusion Medicine and Gene Therapy, Medical Center - University of Freiburg, Freiburg 79106, Germany; Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Freiburg 79106, Germany; Faculty of Medicine, University of Freiburg, 79106, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement IMGENE [765269]; German Research Foundation (DFG) [3861/3-1 to C.M.]; G.A is supported by the Medical Informatics Funding Scheme (EkoEstMed–FKZ 01ZZ2015). Funding for open access charge: University of Freiburg.
Conflict of interest statement. T.C., T.I.C. and C.M. have funded research collaborations with Cellectis, Cimeio Therapeutics and Lepton Pharmaceuticals.
REFERENCES
- 1. Carusillo A., Mussolino C.. DNA damage: from threat to treatment. Cells. 2020; 9:1665–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rodriguez-Rodriguez D.R., Ramirez-Solis R., Garza-Elizondo M.A., Garza-Rodriguez M.L., Barrera-Saldana H.A.. Genome editing: a perspective on the application of CRISPR/Cas9 to study human diseases (review). Int. J. Mol. Med. 2019; 43:1559–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shin J.J., Schroder M.S., Caiado F., Wyman S.K., Bray N.L., Bordi M., Dewitt M.A., Vu J.T., Kim W.T., Hockemeyer D.et al.. Controlled cycling and quiescence enables efficient HDR in engraftment-enriched adult hematopoietic stem and progenitor cells. Cell Reports. 2020; 32:108093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hirakawa M.P., Krishnakumar R., Timlin J.A., Carney J.P., Butler K.S.. Gene editing and CRISPR in the clinic: current and future perspectives. Biosci. Rep. 2020; 40:4–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fu Y.W., Dai X.Y., Wang W.T., Yang Z.X., Zhao J.J., Zhang J.P., Wen W., Zhang F., Oberg K.C., Zhang L.et al.. Dynamics and competition of CRISPR-Cas9 ribonucleoproteins and AAV donor-mediated NHEJ, MMEJ and HDR editing. Nucleic Acids Res. 2021; 49:969–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Song J., Yang D., Xu J., Zhu T., Chen Y.E., Zhang J.. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016; 7:10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin S., Staahl B.T., Alla R.K., Doudna J.A.. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014; 3:e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vartak S.V., Swarup H.A., Gopalakrishnan V., Gopinatha V.K., Ropars V., Nambiar M., John F., Kothanahally S.K.S., Kumari R., Kumari N.et al.. Autocyclized and oxidized forms of SCR7 induce cancer cell death by inhibiting nonhomologous DNA end joining in a Ligase IV dependent manner. FEBS J. 2018; 285:3959–3976. [DOI] [PubMed] [Google Scholar]
- 9. Charpentier M., Khedher A.H.Y., Menoret S., Brion A., Lamribet K., Dardillac E., Boix C., Perrouault L., Tesson L., Geny S.et al.. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018; 9:1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jayavaradhan R., Pillis D.M., Goodman M., Zhang F., Zhang Y., Andreassen P.R., Malik P.. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019; 10:2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ma L., Ruan J., Song J., Wen L., Yang D., Zhao J., Xia X., Chen Y.E., Zhang J., Xu J.. MiCas9 increases large size gene knock-in rates and reduces undesirable on-target and off-target indel edits. Nat. Commun. 2020; 11:6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thomas M., Burgio G., Adams D.J., Iyer V.. Collateral damage and CRISPR genome editing. PLoS Genetics. 2019; 15:e1007994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xie A., Hartlerode A., Stucki M., Odate S., Puget N., Kwok A., Nagaraju G., Yan C., Alt F.W., Chen J.et al.. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol. Cell. 2007; 28:1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chu V.T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K., Kuhn R.. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015; 33:543–548. [DOI] [PubMed] [Google Scholar]
- 15. Fang Y., Stroukov W., Cathomen T., Mussolino C.. Chimerization enables gene synthesis and lentiviral delivery of customizable TALE-based effectors. International J. Mol. Sci. 2020; 21:795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van den Berg F.T., Makoah N.A., Ali S.A., Scott T.A., Mapengo R.E., Mutsvunguma L.Z., Mkhize N.N., Lambson B.E., Kgagudi P.D., Crowther C.et al.. AAV-mediated expression of broadly neutralizing and vaccine-like antibodies targeting the HIV-1 envelope V2 region. Mol. Ther. Methods Clin. Dev. 2019; 14:100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pinder J., Salsman J., Dellaire G.. Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015; 43:9379–9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mussolino C., Morbitzer R., Lutge F., Dannemann N., Lahaye T., Cathomen T.. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011; 39:9283–9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Turchiano G., Andrieux G., Klermund J., Blattner G., Pennucci V., El Gaz M., Monaco G., Poddar S., Mussolino C., Cornu T.I.et al.. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell. 2021; 28:1136–1147. [DOI] [PubMed] [Google Scholar]
- 20. Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E.et al.. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019; 37:224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ira G., Pellicioli A., Balijja A., Wang X., Fiorani S., Carotenuto W., Liberi G., Bressan D., Wan L., Hollingsworth N.M.et al.. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004; 431:1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boutin J., Rosier J., Cappellen D., Prat F., Toutain J., Pennamen P., Bouron J., Rooryck C., Merlio J.P., Lamrissi-Garcia I.et al.. CRISPR-Cas9 globin editing can induce megabase-scale copy-neutral losses of heterozygosity in hematopoietic cells. Nat. Commun. 2021; 12:4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nahmad A.D., Reuveni E., Goldschmidt E., Tenne T., Liberman M., Horovitz-Fried M., Khosravi R., Kobo H., Reinstein E., Madi A.et al.. Frequent aneuploidy in primary human T cells after CRISPR-Cas9 cleavage. Nat. Biotechnol. 2022; 40:1807–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Romito M., Juillerat A., Kok Y.L., Hildenbeutel M., Rhiel M., Andrieux G., Geiger J., Rudolph C., Mussolino C., Duclert A.et al.. Preclinical evaluation of a novel TALEN targeting CCR5 confirms efficacy and safety in conferring resistance to HIV-1 infection. Biotechnol. J. 2021; 16:e2000023. [DOI] [PubMed] [Google Scholar]
- 25. Lemos B.R., Kaplan A.C., Bae J.E., Ferrazzoli A.E., Kuo J., Anand R.P., Waterman D.P., Haber J.E.. CRISPR/Cas9 cleavages in budding yeast reveal templated insertions and strand-specific insertion/deletion profiles. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E2040–E2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Panier S., Boulton S.J.. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014; 15:7–18. [DOI] [PubMed] [Google Scholar]
- 27. Bohgaki M., Bohgaki T., El Ghamrasni S., Srikumar T., Maire G., Panier S., Fradet-Turcotte A., Stewart G.S., Raught B., Hakem A.et al.. RNF168 ubiquitylates 53BP1 and controls its response to DNA double-strand breaks. Proc. Natl. Acad. Sci. U. S. A. 2013; 110:20982–20987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nambiar T.S., Billon P., Diedenhofen G., Hayward S.B., Taglialatela A., Cai K., Huang J.W., Leuzzi G., Cuella-Martin R., Palacios A.et al.. Stimulation of CRISPR-mediated homology-directed repair by an engineered RAD18 variant. Nat. Commun. 2019; 10:3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bonafont J., Mencia A., Chacon-Solano E., Srifa W., Vaidyanathan S., Romano R., Garcia M., Hervas-Salcedo R., Ugalde L., Duarte B.et al.. Correction of recessive dystrophic epidermolysis bullosa by homology-directed repair-mediated genome editing. Mol. Ther. 2021; 29:2008–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cromer M.K., Camarena J., Martin R.M., Lesch B.J., Vakulskas C.A., Bode N.M., Kurgan G., Collingwood M.A., Rettig G.R., Behlke M.A.et al.. Gene replacement of alpha-globin with beta-globin restores hemoglobin balance in beta-thalassemia-derived hematopoietic stem and progenitor cells. Nat. Med. 2021; 27:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mosti L., Langner L.M., Chmielewski K.O., Arbuthnot P., Alzubi J., Cathomen T.. Targeted multi-epitope switching enables straightforward positive/negative selection of CAR T cells. Gene Ther. 2021; 28:602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brinkman E.K., Chen T., Amendola M., van Steensel B.. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014; 42:e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shi X., Shou J., Mehryar M.M., Li J., Wang L., Zhang M., Huang H., Sun X., Wu Q.. Cas9 has no exonuclease activity resulting in staggered cleavage with overhangs and predictable di- and tri-nucleotide CRISPR insertions without template donor. Cell Discov. 2019; 5:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patsali P., Turchiano G., Papasavva P., Romito M., Loucari C.C., Stephanou C., Christou S., Sitarou M., Mussolino C., Cornu T.I.et al.. Correction of IVS I-110(G>A) beta-thalassemia by CRISPR/Cas-and TALEN-mediated disruption of aberrant regulatory elements in human hematopoietic stem and progenitor cells. Haematologica. 2019; 104:e497–e501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brinkman E.K., Kousholt A.N., Harmsen T., Leemans C., Chen T., Jonkers J., van Steensel B.. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 2018; 46:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S.et al.. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature. 2016; 539:384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hackley C.R. A novel set of Cas9 fusion proteins to stimulate homologous recombination: cas9-hrs. CRISPR J. 2021; 4:253–263. [DOI] [PubMed] [Google Scholar]
- 38. Kurihara T., Kouyama-Suzuki E., Satoga M., Li X., Badawi M., Thiha, Baig D.N., Yanagawa T., Uemura T., Mori T.et al.. DNA repair protein RAD51 enhances the CRISPR/Cas9-mediated knock-in efficiency in brain neurons. Biochem. Biophys. Res. Commun. 2020; 524:621–628. [DOI] [PubMed] [Google Scholar]
- 39. Xu Y., Chen K., Cai Y., Cheng C., Zhang Z., Xu G.. Overexpression of Rad51 predicts poor prognosis and silencing of Rad51 increases chemo-sensitivity to doxorubicin in neuroblastoma. Am. J. Transl. Res. 2019; 11:5788–5799. [PMC free article] [PubMed] [Google Scholar]
- 40. Sartori A.A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S.P.. Human CtIP promotes DNA end resection. Nature. 2007; 450:509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Richardson C.D., Kazane K.R., Feng S.J., Zelin E., Bray N.L., Schafer A.J., Floor S.N., Corn J.E.. CRISPR-Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat. Genet. 2018; 50:1132–1139. [DOI] [PubMed] [Google Scholar]
- 42. Kuo C.Y., Long J.D., Campo-Fernandez B., de Oliveira S., Cooper A.R., Romero Z., Hoban M.D., Joglekar A.V., Lill G.R., Kaufman M.L.et al.. Site-specific gene editing of Human hematopoietic stem cells for X-linked hyper-IgM syndrome. Cell Reports. 2018; 23:2606–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Riesenberg S., Chintalapati M., Macak D., Kanis P., Maricic T., Paabo S.. Simultaneous precise editing of multiple genes in human cells. Nucleic Acids Res. 2019; 47:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CAST-seq and next generation sequencing datasets have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE225452.