


Abstract
The transcription factor BTB and CNC homology 1(BACH1) has been linked to coronary artery disease risk by human genome-wide association studies, but little is known about the role of BACH1 in vascular smooth muscle cell (VSMC) phenotype switching and neointima formation following vascular injury. Therefore, this study aims to explore the role of BACH1 in vascular remodeling and its underlying mechanisms. BACH1 was highly expressed in human atherosclerotic plaques and has high transcriptional factor activity in VSMCs of human atherosclerotic arteries. VSMC-specific loss of Bach1 in mice inhibited the transformation of VSMC from contractile to synthetic phenotype and VSMC proliferation and attenuated the neointimal hyperplasia induced by wire injury. Mechanistically, BACH1 suppressed chromatin accessibility at the promoters of VSMC marker genes via recruiting histone methyltransferase G9a and cofactor YAP and maintaining the H3K9me2 state, thereby repressing VSMC marker genes expression in human aortic smooth muscle cells (HASMCs). BACH1-induced repression of VSMC marker genes was abolished by the silencing of G9a or YAP. Thus, these findings demonstrate a crucial regulatory role of BACH1 in VSMC phenotypic transition and vascular homeostasis and shed light on potential future protective vascular disease intervention via manipulation of BACH1.
INTRODUCTION
Vascular smooth muscle cell (VSMC) phenotype switching plays a crucial role in vascular remodeling and in many cardiovascular diseases such as atherosclerosis progression, in-stent restenosis, or coronary allograft vasculopathy (CAV) (1). Neointimal lesions in vascular restenosis and CAV are largely composed of smooth muscle cells primarily originating from dedifferentiated VSMC (2), which are characterized by a significant reduction in contractile gene expression and increased cell proliferation and migration (3). Chromatin accessibility is important for cell fate determination in differentiation and multiple pathophysiological processes (4). Chromatin accessibility reflects a network of permissible physical genomes that is established through a dynamic interplay among histones, transcription factors (TFs), and chromatin remodelers (5). Although recent studies have identified transcription factors that are important for the regulation of VSMC identity located on open chromatin (6,7), few studies focused on the mechanisms of regulating chromatin accessibility in VSMC phenotype switch. How the transcription factor and histone modification regulate chromatin accessibility in the phenotypic modulation of VSMC has yet to be fully understood.
Histone modifications on chromatin are highly correlated with chromatin accessibility within regulatory genomic regions, which plays a critical role in activating or repressing gene transcription (8). H3K9me2 is reported to be enriched in the promoter of silenced genes and associated with decreased chromatin accessibility and transcriptional repression (9). H3K9me2 is catalyzed by histone methyltransferases euchromatic histone-lysine N-methyltransferase 2 (EHMT2) [also known as G9a] (10) and demethylated by Lysine-specific demethylase 1 (LSD1). G9a catalyzes H3K9me2 in mammals (9), and the histone methyl-transferase activity of G9a is more important than euchromatic histone methyltransferase 1(EHMT1) (11). G9a plays a crucial role in cell proliferation and migration. For instance, G9a increases cholangiocarcinoma proliferation via promoting H3K9me2 of target genes (12) and maintains T cell identity during differentiation (13). Previous studies have indicated that expression of G9a was increased in advanced human atherosclerotic carotid plaques (14), and an inhibitor for G9a (BIX-01294) suppressed arterial smooth muscle cell proliferation and migration (15). However, whether and how G9a participates in VSMC phenotype switching in vascular diseases remains unknown.
The transcription factor BTB and CNC homology 1 (BACH1) is a ubiquitously expressed member of the CNC and basic region leucine zipper (CNC-bZip) family and plays an essential role in the regulation of oxidative stress, heme oxidation, and cell cycle progression (16,17). Our previous studies revealed that BACH1 impaired both developmental angiogenesis in zebrafish embryos (18) and the angiogenic response to peripheral ischemic injury in adult mice (19). The BACH1 gene is shown to reside in the proximity of the genetic risk variant (rs2832227) of coronary artery disease (CAD) identified by genome-wide association studies (GWAS) (20). Our recent study suggests that deletion of endothelial BACH1 attenuated atherosclerosis by reducing endothelial inflammation (21). However, the underlying mechanism of how BACH1 contributes to vascular remodeling and the role of BACH1 on VSMC phenotype switching remains unclear.
Here, we aim to explore the role of BACH1 in mature VSMC phenotypic modulation and vascular injury-induced neointima formation. Our results suggest that VSMC-specific Bach1 deletion in mice inhibited injury-induced loss of VSMC identity and neointima formation in wire-injured femoral arteries. BACH1 facilitates the recruitment of G9a and YAP, maintains the state of H3K9me2, and decreases the chromatin accessibility at the promoter of VSMC marker genes, thereby repressing their expression and contributing to dedifferentiated VSMC phenotype. We have identified BACH1 as a novel regulator contributing to VSMC phenotypic switching to influence the risk of CAD by regulating chromatin accessibility.
MATERIALS AND METHODS
Human coronary artery samples
Six cases of normal heart tissues and seven cases of CAD heart tissues from patients who died suddenly were obtained from the Department of Forensic Medicine, School of Basic Medical Sciences, Fudan University. Control heart tissue samples were obtained from healthy individuals or patients without heart disease and CAD heart tissue samples were obtained from patients, who had coronary artery stenosis. The use of human samples was approved by the Ethics Committee Board at the School of Basic Medical Sciences, Fudan University (Ethic protocol approval number: 2021-C004). The information including age, sex, and clinically significant co-morbidities of the human coronary artery samples are in Supplementary Table S2.5
Animals
The Bach1 loxP mouse in C57/BL background was generated by flanking exon 3 and exon 4 of Bach1 with loxP sites from Gempharmatech. The Bach1 loxP mice were crossed with the Myh11 promoter-driven Cre recombinase line (22) to generate Bach1SMCKO mice for smooth muscle cell-specific deletion of Bach1 in the C57BL/6 background. All animal studies followed the guidelines of the Animal Care and Use Committee of the School of Basic Medical Sciences, Fudan University (approval number: 20160818). Smooth muscle-specific conditional Bach1 knockout mice Myh11-Cre-Bach1loxp/loxp (Bach1SMCKO) and Bach1loxp/loxp (Bach1WT) mice were used in the mouse femoral artery wire injury model.
Materials
Smooth Muscle Cell Medium (SMCM) (Sciencell, Faraday Ave, Carlsbad, #1101) was purchased from Sciencell. DAPI (Sigma Aldrich, St. Louis, MO, #D9542), Triton X-100 (Sigma Aldrich, St. Louis, MO, #T9284), and Formaldehyde solution (Sigma Aldrich, St. Louis, MO, # F8775) were from Sigma Aldrich Inc.; DMEM (Invitrogen, Carlsbad, CA, #12800082), Fetal Bovine Serum (Invitrogen, Carlsbad, CA, #10099-141), Trizol Regent (Invitrogen, Carlsbad, CA, #15596018), and Lipofectamine 3000 (Invitrogen, Carlsbad, CA, #L3000-015) were from Invitrogen; PMSF (Beyotime, Shanghai, China, #ST505) and Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Shanghai, China, # P0027) were from Beyotime; Protease inhibitor cocktail (MCE, Monmouth Junction, NJ, # HY-K0010) was from MCE, qPCR SYBR® Green Master Mix (Yeasen, Shanghai, China, #11201ES03) and Plus One Step Cloning Kit (Yeasen, Shanghai, China, #10911ES25) were from Yeasen.
Mouse femoral artery wire injury model
A murine femoral artery wire injury model for neointimal hyperplasia was performed as described previously (23). Before surgery, Bach1SMCKO and littermate control wild-type mice of 8–12 weeks of age were administrated with tamoxifen (100 mg/kg mice, every other day for a total of 4 times) to knock out Bach1 in VSMCs. Briefly, 8- to 12-week-old male Bach1SMCKO mutant and wild-type littermate mice were anesthetized with an intraperitoneal injection of 40 mg/kg pentobarbital sodium and the femoral artery on the left side was separated and looped proximally and distally with 6–0 silk suture for temporary blood flow cessation. A small branch between the rectus femoris and vastus medialis muscles was isolated, and a transverse arteriotomy was performed, then a flexible angioplasty wire (0.35-mm diameter; Cook Inc, Bloomington, IN) was inserted into the femoral artery for 5 min for vascular injury.
Cell culture
Primary human aortic smooth muscle cells (HASMC) (American Type Culture Collection, Manassas, VA, ATCC-PCS-100–012) were cultured in Smooth Muscle Cell Medium (SMCM), and passages 3 to 6 were used for further experiments. Human embryonic kidney (HEK) 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% Fetal Bovine Serum supplemented with 100 units/ml penicillin and streptomycin.
Lentivirus production and HASMCs infection
The YAP short hairpin RNAs (shRNAs) were kindly provided by Dr. Faxing Yu (Fudan University, Shanghai). For gene knockdown in HASMCs, YAP shRNAs and the control shRNAs were cloned into the pLKO.1 construct (Addgene, Watertown, MA, #8453). Lentiviral particles were generated by transfecting HEK293T cells with a pLKO.1 shRNA vector and packing vectors, psPAX2 and pMD2. G. The supernatant containing lentiviruses is concentrated at 1 105 G. To infect HASMCs, lentiviruses were mixed with SMCM, and the mixture was incubated with cells for 24 hours at the density of 2
105 G. To infect HASMCs, lentiviruses were mixed with SMCM, and the mixture was incubated with cells for 24 hours at the density of 2 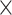 105 cells.
105 cells.
siRNA transfection
Small interfering RNA (siRNAs) against human BACH1, G9a and control siRNAs were designed and synthesized by Gene Pharma (Shanghai, China). HASMCs were transfected with the mixture of three siRNAs (20 nM) via Lipofectamine 3000 according to the manufacturer's protocol. The siRNA sequences are shown in Supplementary Table S2.4.
Luciferase assay
The luciferase assay was performed as described previously (19). Briefly, the cells were transfected with a β-galactosidase plasmid and the indicated DNA reporter plasmid or the pGL3-basic luciferase reporter plasmid. Transfection was performed with Lipofectamine 3000, and the transfected cells were cultured for 24 hours; then, the cells were harvested and luciferase activity was measured with a Luciferase Assay Kit (Promega, Madison, WI, #E1500). β-galactosidase (β-gal) activity was measured. Relative Luc activity was calculated as the ratio of Luc/β-gal activity.
RNA extraction, real-time PCR, and genome-wide gene expression
Total RNA was extracted using Trizol reagent and equal amounts (1 μg) were reverse-transcribed to complementary DNA (cDNA) by using the Rever Tra Ace qPCR RT Kit. Real-time quantitative PCR was performed with the qPCR SYBR® Green Master Mix, and the primer sequences are listed in Supplementary Table S2.2. The mixture was heated to 95°C for 5 min, cycled 40 times (95°C for 30 s, 58°C for 30 s, and 72°C for 30 s), and held at 72°C for 7 min. Melting curves were generated by increasing the temperature from 55°C to 95°C in 0.5°C increments at 10 s intervals and then visually to ensure that a single peak was present for each primer. Threshold amplification values (ct) were assigned by the CFX Manager analysis software (Bio-Rad). The mRNA levels were normalized to those of β-actin. For genome-wide expression analysis, non-ribosomal RNA was isolated from 1 μg total RNA by using a TrueLib Poly (A) mRNA Magnetic Isolation Module, and sequencing libraries were prepared by using the TrueLib mRNA Library Prep Kit for Illumina. Sequencing was performed on the NovaSeq 6000 (Illumina Inc, San Diego, CA) by Annoroad Company (Beijing, China)
Immunoblotting and immunoprecipitation
The cells were lysed with RIPA lysis buffer supplemented with 0.1 mmol/l PMSF and 1 mmol/l protease inhibitor cocktail. The cytoplasmic and nuclear protein extraction was performed according to the instructions described in the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Shanghai, China, P0028). For immunoprecipitation, the cells were lysed with NP-40 buffer supplemented with 0.1 mmol/l PMSF and 1 mmol/l protease inhibitor cocktail, 1 mg of total protein was incubated with the 3–5 μg antibody at 4°C overnight, and then with Protein A/G PLUS-Agarose (Santa Cruz, Santa Cruz, CA, #sc-2003) at 4°C for an additional 4 hours; then, the precipitates were washed once with RIPA and three times with PBST, resuspended in 40 μl 2 × SDS loading buffer, and immunoblotting was performed as previously described (24). Briefly, samples were heated at 100°C for 10 min and then resolved by SDS-PAGE on 8–12% polyacrylamide gels and transferred to a PVDF membrane by a wet transfer system. Chemiluminescence was detected on a Tanon-5500 Imaging System (Tanon Science & Technology Ltd, Shanghai, China). The intensity of the bands was measured via densitometric analysis with ImageJ software and normalized to the control. The following antibodies were used for Immunoblotting and/or Immunoprecipitation: BACH1 (Santa Cruz, Santa Cruz, CA, #sc-271211); β-actin (Santa Cruz, Santa Cruz, CA, #sc-47778); ACTA2 (Abcam, Cambridge, UK, #ab5694); CNN1 (Abcam, Cambridge, UK, #ab46794); TAGLN (Abcam, Cambridge, UK, #ab10135); YAP (Cell Signaling, Danvers, MA, #14074); G9a (Cell Signaling, Danvers, MA, #3306); H3K9me2 (Abcam, Cambridge, UK, #ab1220); Histone H3 (Cell Signaling, Danvers, MA, #4499); β-tubulin (Proteintech, Rosemont, IL, #10094–1-AP); Goat anti-Rabbit IgG (Invitrogen, Carlsbad, CA, #31460); Mouse Anti-Rabbit IgG, light chain specific (Jackson ImmunoResearch Laboratories, West Grove, PA, 211–032-171); Goat anti-Mouse IgG (Invitrogen, Carlsbad, CA, #31430); Rabbit anti-Goat IgG (Invitrogen, Carlsbad, CA, #31402).
Analysis of RNA-sequencing data
The FASTQ data were trimmed adaptor by trim galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), then mapped to human genome hg19 using HISAT2 (25), and assembled by Cufflinks (26) to get fragments per kilobase million (FPKM) for each gene. Differentially expressed genes were generated by DEseq2 (27) with fold change >2.0 and P < 0.05. Gene Ontology analysis (28) and KEGG Pathway analysis (29) were performed on David(https://david.ncifcrf.gov/) (30).
Chromatin immunoprecipitation (ChIP) and ChIP-seq
Chromatin immunoprecipitation (ChIP) and ChIP-seq were performed as described previously (31). Cells were cross-linked with 1% formaldehyde for 10 min, quenched with 0.125 M glycine for 10 min; then, the cell pellets were resuspended in low salt lysis buffer (50m MHEPES/KOH, pH 7.5, 150 mM NaCl, 1% Trixon-X 100, 0.05% SDS, 10 mM EDTA and proteinase inhibitors), and sonicated with a Q800R2 DNA shearing sonicator (Qsonica LLC, Newtown, CT) for 20 min in 30-s intervals to shear the chromatin into 200- to 400-bp lengths. Chromatin was immunoprecipitated with the indicated antibodies and Protein A/G PLUS-Agarose. For H3K9me2-ChIP, then cells were cross-linked, quenched, and washed as described above. Furthermore, the sonicated chromatin was incubated with anti-H3K9me2 overnight at 4°C. Then, beads were washed with low salt lysis buffer and low salt buffer (10 mM Tris–HCl pH 8.0, 250 mM LiCl, 1 mM EDTA, 0.5% NP40, 0.5% Na-Deoxycholate), and DNA was eluted in elution buffer (50 mM Tris–HCl pH 8.0, 10 mM EDTA, 1% SDS) with Proteinase K (Yeasen, Shanghai, China, # 10401ES60) and RNase A (Thermo Fisher, Waltham, MA, #EN0531) in 65°C for 4 h. The primers used in ChIP-qPCR assays are listed in Supplemental Supplementary Table S2.3. ChIP DNA libraries were prepared as directed by the protocol of VAHTS Universal DNA Library Prep Kit for Illumina (Vazyme Biotech, Nanjing, China, #ND617-01). Libraries were sequenced on NovaSeq 6000 by Annoroad Company.
Analysis of ChIP-sequencing and ATAC- sequencing data
Clean reads were mapped to the human genome hg19 with Bowtie2 (32). Peaks were called by the MACS2 call peak module with -g hs -q 0.05 –nomodel –keep-dup 1 (33). Differential peaks were called by MACS2 bdgdiff module with -l 200. One-sided binomial test was used to statistically confirm promoter enrichment as previously described (34). ChIP-seq and ATAC-seq motif and ChIP-seq visualization were performed by HOMER (35) and deepTools (36) and visualized on WashU Epigenome browser(http://epigenomegateway.wustl.edu/browser/). Heatmaps and signal plots were generated by deepTools (3.3.0).
Assay for transposase-accessible chromatin using sequencing (ATAC-seq)
Assay for transposase-accessible chromatin using sequencing (ATAC-seq) was performed as described previously (37). Cells were digested by trypsin (Invitrogen, Carlsbad, CA, #25200072) and then were pretreated with 200 U/ml DNase (Sigma-Aldrich, St. Louis, MO, #260913) for 30 min at 37°C to remove free-floating DNA and digest DNA from dead cells. Then 50000 cells were resuspended in 1 ml of cold RSB buffer (10 mM Tris–HCl pH 7.4, 10 mM NaCl, and 3 mM MgCl2). Cells were centrifuged at 500 rcf for 5 min and then resuspended in 50 μl of RSB buffer containing 0.5% NP40, 0.1% Tween-20, and 0.01% digitonin and incubated on ice for 10 min. After lysis, added 1 ml RSB buffer containing 0.1% Tween-20 and nuclei were centrifuged for 10 min at 500 rcf. Then the ATAC-seq libraries were prepared by using the TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme Biotech, Nanjing, China, #TD501-01). Libraries were sequenced on NovaSeq 6000 by Annoroad Company.
Cleavage under targets & tagmentation (CUT&tag)
Cleavage Under Targets & Tagmentation (CUT&Tag) was performed as described previously (38). Resuspended and withdrew enough of the concanavalin A (ConA) bead (10 μl/sample). 50 000 cells were harvested and immobilized to 10 μl ConA beads in at room temperature for 10 min. The bead-bound cells were incubated in 100 μl of primary antibody buffer with BACH1 (1:50) antibody or YAP (1:50) antibody at 4°C by rotating overnight. The next day, the primary antibody buffer was removed and cells were washed with 600 μl of dig-wash buffer three times. After washing, cells were resuspended in 100 ul of dig-wash buffer with 1:100 dilution of mouse anti-rabbit antibody or rabbit anti-mouse antibody and incubated at room temperature for 1 h with slow rotation. After a brief wash with dig-wash buffer as above, resuspended cells in 100 μl pG-Tn5 adapter complex (final 0.04 μM) and incubated at room temperature for 1 h with slow rotation. After incubation, wash the sample with 800 μl 300-wash buffer twice. followed by tagmentation in 300 μl tagmentation buffer and incubated at 37°C for 1 h. Then added 10 μl 0.5M EDTA, 3 μl 10% SDS and 2.5 μl 20 mg/ml Proteinase K to each sample and incubated at 50°C for 1 hour. Further extracted DNA by Phenol and chloroform (PCI). Sequencing libraries were prepared by using Hyperactive™ In-Situ ChIP Library Prep Kit for Illumina (Vazyme Biotech, Nanjing, China, #TD901-01). Libraries were sequenced on NovaSeq 6000 by Annoroad Company.
Analysis of scRNA-sequencing data
The Seurat (version 4.0) R package was used to quality filter and cluster cells (39). Then doublets were identified using DoubletFinder (40) and removed from downstream analysis. Pseudotime analysis was performed by Monocle 2 package, as described in the tutorials (http://cole-trapnell-lab.github.io/monocle-release/tutorials/) (41). SMCs were isolated and used as the input expression matrix for trajectory analysis. Transcription factor activities were calculated by DoRothEA (https://github.com/saezlab/dorothea) (42).
Analysis of snATAC-sequencing data
The R package Signac (version 1.2) was used for calculating and plotting differentially accessible regions for clusters. Then the motif activity was calculated for each cell by chromVAR method (43) with the human motifs from the JASPAR CORE motif collection. The co-accessibility scores between peaks were calculated by Cicero and detect cis-co-accessibility networks, and visualize co-accessibility connections between peaks (44).
Immunohistochemistry
Tissues were fixed in 4% neutralized paraformaldehyde (PFA). For antigen retrieval, the tissue sections were immersed in Tris-EDTA buffer (10 mM, pH 9.0) and heat retrieved for 20 min in 100°C water baths. The slices were permeabilized and blocked in TBS-T (0.03% Triton X-100) with 5% donkey serum (Jackson ImmunoResearch Laboratories, West Grove, PA, #017-000-121) for 1 h at RT. Immunostaining was performed using the following antibodies diluted in 1% donkey serum at 4 °C over-night: BACH1 (Proteintech, 14018-1-AP), ACTA2; TAGLN; KI-67 (Abcam, Cambridge, UK, #ab16667). After washing with TBST, sections were incubated with secondary antibodies: 488 nm-conjugated anti-rabbit secondary antibody(Jackson ImmunoResearch Laboratories, West Grove, PA, #711-005-152) or 594 nm-conjugated anti-goat secondary antibody(Jackson ImmunoResearch Laboratories, West Grove, PA, #705–005-147) or HRP-conjugated anti-rabbit secondary antibody or AP-conjugated anti-goat secondary antibody diluted in TBS-T (0.03% Triton X-100) for 2 hours, washed with TBS, then stained with DAPI for 10 min or incubated with DAB and Alkaline Phosphatase Kit (Sigma Aldrich, St. Louis, MO, #86R) for 30min sections were counterstained with hematoxylin. Images were captured with a fluorescence microscope (Olympus, Tokyo, Japan). The quantification was performed by NIH Image J software as previously described (45). The minimum threshold value was set at zero. The maximum threshold value was set so that the background signal is removed without removing the true signal. After setting the threshold, the image was converted to a black and white image. The mean grey value represents the quantified intensity. The relative protein expression value was normalized by the intensity value.
Immunofluorescence staining
Cells were fixed, permeabilized, incubated, stained, and photographed as described previously (19). Briefly, cells were fixed with 4% PFA and permeabilized with 0.3% Triton X-100. Then cells were blocked with 5% donkey serum and incubated with FITC-conjugated phalloidin (Sigma Aldrich, St. Louis, MO, #P5282) in blocking solution overnight. Nuclei were stained with DAPI, and then the cells were photographed under a fluorescence microscope (Olympus, Tokyo, Japan).
Ethynyl deoxyuridine (EdU) staining
HASMCs were treated with BACH1 siRNAs or Control siRNAs for 48 h, followed by treatment with 10 mM EdU (Beyotime, Shanghai, China, #C0071) in medium for an additional 2 h. Immunostaining and image analysis were performed as described in the instructions.
Statistical analysis
Data are expressed as mean ± SD. Differences between two groups were evaluated for significance via two-tailed Student's t-test, and differences among three or more groups were evaluated for significance via one-way analysis of variance (ANOVA) followed by Tukey post-hoc tests. Analyses were conducted with Prism software (Version 8.0). Biological experimental replicates in each group were shown in figure legends, and the exact p-values of the results were specified in figure, and P < 0.05 was considered significant.
RESULTS
BACH1 highly correlates with VSMC identity
To investigate the association of BACH1 gene with genetic variants linked with the risk of CAD, we reprocessed the published snATAC-seq dataset and the CAD/ myocardial infarction (MI) SNPs from human atherosclerotic lesions by Tiit Örd et al. (46) and applied Cicero to identify cis-co-accessibility networks (CCANs) to discover potential cis-regulatory elements of BACH1. The genetic variant rs73193808 was identified to be associated with the risk of CAD/MI (46). We found that the variant rs73193808, which is located in intron 7 of the MAP3K7CL gene, had VSMCs-specific co-accessibility with the BACH1 promoter in human atherosclerotic lesions (Figure 1A), indicating potential physical interactions between BACH1 promoter and rs73193808 in VSMCs. To further dissect the profile of the motif activity of BACH1 in human atherosclerotic lesions, we used chromVAR to identify transcription factor motifs from the JASPAR 2020 database (47) enriched within the accessible chromatin of VSMCs in human atherosclerotic lesions using snATAC-seq dataset from Tiit Örd et al. (46). Indeed, we observed high enrichment of BACH1, SRF, and KLF4 in VSMCs in these atherosclerotic lesions (Figure 1B). Analysis of the published scRNA-seq dataset from human atherosclerotic arteries (48) using DoRothEA further confirmed the high transcriptional factor activity of these transcriptional factors (TF) in VSMCs of human atherosclerotic arteries (Figure 1C). To examine the VSMC phenotype switching in human atherosclerotic arteries, we performed the pseudotime trajectory analysis with VSMCs expressing different levels of VSMC marker genes using scRNA-seq dataset from human atherosclerotic arteries (48). Synthetic SMCs were located at the end of the trajectory, indicating a presence of low ACTA2 and CNN1 expression and high KLF4 and BACH1 expression in human atherosclerotic arteries (Figure 1D). To determine whether the expression of BACH1 was altered in human coronary arteries with CAD, we analyzed the expression and localization of BACH1 in human atherosclerotic lesions by immunostaining. Of note, BACH1 expression in the neointima of the human coronary arteries with atherosclerotic plaques was higher than in normal human coronary arteries (Figure 1E and Online Figure IA). Taken together, these results suggest that BACH1 is associated with VSMCs’ identity in human atherosclerotic lesions.
Figure 1.

BACH1 highly correlates with VSMC identity. (A) Viewpoint plot of peak-promoter co-accessibility and the snATAC-Seq tracks centered at the CAD/MI SNP rs73193808 in VSMCs of human atherosclerotic lesion. (B) Motif enrichment z-scores were computed by chromVAR and enrichment z-scores for BACH1, KLF4, and SRF motifs in VSMCs of human atherosclerotic lesion projected onto UMAP coordinates. (C) Heatmap of selected TF activities inferred with DoRothEA from gene expression data generated via human atherosclerotic lesion 10X scRNA-Seq. (D) Heatmap of the expression of VSMC marker genes (ACTA2, CNN1), BACH1, and KLF4 along pseudotime in VSMCs of human atherosclerotic lesion. Contractile SMCs were located at origin of the trajectory (right) and Synthetic SMCs were located towards the termini of the trajectory (left). (E) The coronary artery sections from patients who had CAD or who died from non-cardiovascular diseases were immunostained for BACH1 (brown) and TAGLN (red). Representative images shown. Scale bar: 50μm. Quantitative and statistical analysis are shown in Online Figure I A (control n = 6, CAD n = 7, data are mean ± SD, unpaired towo-tail t-test).
To identify the expression of BACH1 in neointima formation progression, we detected the mRNA expression of Bach1 in wire injured mouse femoral artery and found that Bach1 gene expression was upregulated at day 3, then declined from day 7 to day 28 after wire injury (Figure 2A). On the contrary, the expression of Bach2 was not significantly changed after wire injury (Online Figure IB). Analysis of the published microarray from mouse injured arteries (GEO accession no. GSE40637) (49) also confirmed the increase of Bach1 in the injured femoral artery (Figure 2B). Intriguingly, the gene expression of Bach1 was higher in the cultured aorta than in the fresh aorta (Figure 2C) and increased from passage 5 (P5) cultured VSMCs to P8 (Figure 2D). Similarly, BACH1 expression was induced by fetal bovine serum treatment in cultured VSMCs with the decreased expression of known regulated gene ACTA2 (Figure 2E), while the opposite effect was seen in VSMCs by transforming growth factor β (TGF-β) treatment (Figure 2F). Altogether, these results revealed that the expression of BACH1 was increased during wire injury of mouse femoral arteries and VSMC phenotype switching in vitro, indicating a strong correlation between BACH1 and VSMC identity.
Figure 2.

The expression of BACH1 is increased during VSMC phenotype switching progress. (A) Bach1 was significantly upregulated three days after injury in the murine femoral artery wire injury model. Total RNA was collected from the uninjured (day [D] 0) and injured femoral arteries (D3, D7, D14, and D28 postinjury) and RT-qPCR analyses were conducted to obtain relative expression levels. (n = 5 each group, Plots showing mean ± SD. One-way ANOVA followed by Tukey post-hoc tests). (B) Bach1 mRNA expression was upregulated comparing wire injured femoral artery with uninjured femoral artery in the public microarrays from mouse femoral artery (GEO accession no. GSE40637) (n = 4 each group, data are mean ± SD showing log2 mean ± SD. unpaired two-tail t-test). (C) RT-qPCR was used to examine the mRNA (Bach1, Yap, Acta2) levels in the freshly isolated aortas and the aortas cultured in DMEM containing 20% serum for 3 days (n = 4 independent experiments, data are mean ± SD. unpaired two-tail t-test). (D) HASMCs with the indicated passage number (P3, P5 or P8) were subjected to RT-qPCR analysis to obtain relative expression levels of BACH1, TAGLN, and ACTA2 (n = 4 independent experiments, data are mean ± SD. One-way ANOVA followed by Tukey post-hoc tests). (E, F) HASMCs subjected to serum starvation for 24 hours were used as the control and then harvested after the indicated stimulations and time points: Serum stimulation at 12 or 24 h (E), or TGF-β stimulation at 12 or 24 h (F). Total RNA was extracted and subjected to RT-qPCR analysis to obtain relative expression levels of BACH1 and ACTA2 (n = 4 independent experiments, data are mean ± SD. One-way ANOVA followed by Tukey post-hoc tests).
VSMC-specific loss of Bach1 in mice attenuates the neointima formation after wire injury
Based on the observed correlation between BACH1 and VSMC phenotype switching, we hypothesized that BACH1 might contribute to neointimal formation after vascular injury. To test this hypothesis, we studied the VSMC-specific role of Bach1 in vascular injury-induced neointimal hyperplasia in vivo. We generated the SMC-specific Bach1 inducible knockout mice (Bach1SMCKO) by crossing the Bach1flox/flox mice with transgenic mice expressing Cre-recombinase specifically in SMCs (Myh11-CreERT2 mice). Bach1flox/flox mice were generated by flanking exon 3 and exon 4 of Bach1 with loxP sites (Online Figure IC). We then developed a femoral artery wire injury model on Bach1SMCKO and Bach1WT littermate mice. The deletion of BACH1 in VSMCs was validated by immunofluorescence staining (Figure 3A), suggesting the specific deletion of Bach1 in VSMCs of Bach1SMCKO mice. We found that the intima area and the intima/media ratio in the Bach1SMCKO mice were decreased compared with wild-type littermate mice at day 28 after wire injury of the femoral artery (Figure 3B). Immunostaining showed an increase in the expression of ACTA2 (Figure 3C) but a significant reduction of KI67-positive VSMCs (Figure 3D) in the neointima of Bach1SMCKO mice compared with the wild-type littermates. These results suggest that VSMC-specific loss of Bach1 attenuates the neointimal hyperplasia and inhibits the transformation of VSMC from contractile to synthetic phenotype and the cell proliferation induced by femoral artery wire injury.
Figure 3.

VSMC-specific loss of Bach1 in mice attenuates the neointima formation after wire injury. (A) Bach1SMCKO mice exhibited specific loss of BACH1 expression in VSMCs of femoral artery following tamoxifen treatment by immunostaining (BACH1, green; TAGLN, red, and DAPI, blue). Scale bar: 100 μm. (B) Bach1SMCKO mice exhibited a significant decrease of injury-induced neointima formation compared with the wild-type (WT) mice, with representative HE staining images and quantification of neointima area, and intima-to-media ratio (n = 13 for each group, data are mean ± SD. unpaired two-tail t-test). Scale bar: 100 μm. (C) and (D) Bach1SMCKO mice exhibited a significant increase of ACTA2 expression (C) and a reduction of KI67-positive VSMCs (D) in the neointima at D28 after femoral artery wire injury compared with the wild-type mice, with representative images and quantification data (n = 6 for each group in (C); n = 10 for Bach1SMCKO mice and n = 9 for WT mice (D). Data are mean ± SD. unpaired two-tail t-test). Scale bar: 100 μm.
BACH1 deletion upregulates VSMC marker genes’ expression and reduces VSMC proliferation
To test whether BACH1 plays a role in VSMC phenotype switching under patho-physiological conditions, we transfected human aortic smooth muscle cells (HASMCs) with adenoviruses coding for BACH1 (AdBACH1), or GFP (AdGFP), or BACH1 siRNAs or a control siRNAs (ConsiRNA) and then subjected the transfected cells to various analyses. We utilized two independent siRNAs targeting different regions of the BACH1 gene. All two independent siRNAs efficiently downregulated BACH1 mRNA levels in HASMCs, concomitant with the increase in ACTA2 and CNN1 (Online Figure ID). Mixed BACH1 siRNA#1 and siRNA#2 were used in all subsequent BACH1 silencing experiments. BACH1 silencing significantly increased, while BACH1 overexpression decreased, the mRNA level of VSMC marker genes, including ACTA2 and CNN1 (Figure 4A), and concordant alteration in their protein expression (Online Figure IIA-IIB). We also isolated vascular smooth muscle cells from Bach1SMCKO mice and detected the mRNA level of VSMC marker genes. We found that loss of Bach1 in VSMCs significantly increased the mRNA level of VSMC marker genes (Figure 4B), consistent with our findings in HASMCs (Figure 4A). Furthermore, we found that, whereas serum treatment reduced the expression of ACTA2 and CNN1, BACH1 silencing increased the expression of these genes with serum treatment (Figure 4C). Conversely, overexpression of BACH1 significantly decreased TGF-β-induced expression of the same genes (Figure 4D). Similar results were also confirmed by qPCR analysis (Online Figure IID-IIE). It is important to note that BACH1 silencing decreased VSMC proliferation and growth under serum stimulation as demonstrated in ethynyl deoxyuridine (EdU) incorporation and CCK-8 analysis (Figure 4E-4F). Additionally, BACH1 silencing in serum-stimulation dedifferentiated VSMCs resulted in morphological changes from a polygonal synthetic phenotype to a spindle-like contractile phenotype (Online Figure IIF). Collectively, these results suggest that BACH1 silencing in HASMCs may promote the switch from a synthetic to a contractile phenotype and decrease the proliferation of HASMCs. These changes contribute to inhibiting neointima formation after vascular injury.
Figure 4.

BACH1 deletion upregulates VSMC marker genes’ expression and reduces VSMC proliferation. (A) The mRNA expression of ACTA2, CNN1 and BACH1 in HASMCs. RT-PCR showed that ACTA2 and CNN1 mRNA levels were increased in BACH1 silencing HASMCs compared with the control HASMCs, and decreased in Ad-BACH1-HASMCs compared with Ad-GFP-HASMCs (n = 5 independent experiments, data are mean ± SD. unpaired two-tail t-test). (B) The mRNA expression of Acta2, Cnn1 and Bach1 in primary mouse aortic smooth muscle cells. RT-PCR showed that Acta2 and Cnn1 mRNA levels were increased in Bach1SMCKO primary mouse aortic smooth muscle cells compared with Bach1WT primary mouse aortic smooth muscle cells (n = 4 independent experiments, data are mean ± SD. unpaired two-tail t-test). (C, D) Expression of ACTA2, CNN1 and BACH1 in HASMCs. Immunoblotting showed that ACTA2 and CNN1 protein levels were increased in BACH1 silencing HASMCs compared with the control HASMCs with serum stimulation for 24 h (C), and decreased in Ad-BACH1-HASMCs compared with Ad-GFP-HASMCs with TGF-β stimulation for 24 h (D) (n = 3 independent experiments, data are mean ± SD. One-way ANOVA followed by Tukey post-hoc tests). (E) Ethynyl deoxyuridine (EdU) analysis showed that EdU-positive VSMCs were decreased in BACH1 silencing HASMCs compared with the controls (n = 4 independent experiments, data are mean ± SD. unpaired two-tail t-test). Scale bar: 150 μm. (F) CCK-8 analysis showed that the cell proliferation was downregulated in BACH1 silencing HASMCs compared with the control HASMCs (n = 3 independent experiments, data are mean ± SD. unpaired 2-tail t-test).
BACH1 binds at the VSMC marker genes promoter and inhibits their expression
To explore the mechanism of BACH1 in VSMC phenotype switching, we analyzed messenger RNA (mRNA) profiles in HASMCs transfected with BACH1 siRNAs (BACH1siRNA) or the control siRNAs (ConsiRNA). RNA-sequencing (RNA-seq) analysis indicated that 770 genes were upregulated, and 594 genes were downregulated in BACH1siRNA-HASMCs compared to ConsiRNA-HASMCs (fold change > 2, P < 0.05) (Online Supplementary Table S1). Consistent with our quantitative real-time PCR (qRT-PCR) results (Figure 4A), VSMC marker genes (e.g. CNN1, ACTA2) were identified in the list of upregulated genes, in parallel with the downregulation of cell proliferation-related genes such as CCND1 (cyclin D1) (Figure 5A). Furthermore, gene set enrichment analysis (GSEA) confirmed that genes related to smooth muscle cell maturation were upregulated in BACH1siRNA-HASMCs compared to ConsiRNA-HASMCs (Figure 5B). To further understand how BACH1 regulates VSMC marker genes expression, we performed the Cleavage Under Targets and Tagmentation (CUT&Tag) assay to assess the DNA-binding profiles of BACH1 in VSMCs. BACH1 was preferentially bound to gene promoters (55.8% versus 9%; P = 1.8E-16) (Online Figure IIIA-B), which was consistent with our recent study using ChIP-seq (50). Motif analysis showed that the top two motifs were BACH1 and its competitive factor NRF2 (nuclear factor erythroid 2–related factor 2) (Online Figure IIIC), which were in line with the previous report (16). The Gene Ontology (GO) analysis showed that BACH1-bound genes were associated with smooth muscle cell differentiation, proliferation, blood vessel development, and chromatin remodeling (Figure 5C). Furthermore, representative snapshots of the gene tracks show that BACH1 occupied genomic regions near VSMC marker genes (CNN1 and ACTA2) (Figure 5D), and ChIP-qPCR assays confirmed that BACH1 occupied these genes’ promoter regions (Figure 5E). The binding of BACH1 to the promoter regions of these genes was higher in fetal bovine serum-treated VSMCs than in control VSMCs, suggesting that increased occupancy of BACH1 on VSMC marker genes underlies the loss of VSMC identity in response to serum stimulation (Figure 5F). In contrast, the occupancy of BACH1 on VSMC marker genes declined in response to TGF-β1 stimulation (Figure 5F). We then conducted an experiment with luciferase reporter constructs containing the CNN1 promoter sequences with the BACH1 binding site. As expected, BACH1 overexpression significantly reduced luciferase activity of the CNN1 reporter in HASMCs (Figure 5G), consistent with our qRT-PCR results. We next overlapped CUT&Tag data of BACH1-binding genes with RNA-seq data from BACH1siRNA-versus ConsiRNA-HASMCs and identified 302 BACH1-directly regulated genes (Figure 5H). GO analysis indicated that BACH1-directly regulated genes are enriched in angiogenesis, smooth muscle cell differentiation and artery morphogenesis (Online Figure IIID). Overall, these results suggest that BACH1 directly regulates VSMC identity gene expression.
Figure 5.

BACH1 binds at the VSMC marker genes’ promoter and inhibits their expression. (A) Heatmap showed that ACTA2 and CNN1 were upregulated and CCND1 was down-regulated in BACH1 silencing HASMCs compared with the control HASMCs by RNA-seq analysis. (B) Gene Set Enrichment Analysis (GSEA) of differentially expressed genes between BACH1 silencing HASMCs and the control HASMCs among genes associated with mature smooth muscle cell (NES = −1.12, Q =0.14). (C) The Gene Ontology analysis of BACH1 enriched genes. (D) Representative snapshots of CUT&Tag tracks for BACH1 on the promoter of ACTA2 and CNN1. (E) ChIP-qPCR analysis validated the enrichment of BACH1 at the promoter of ACTA2 and CNN1, HO-1 was used as a positive control (n = 4 independent experiments, data are mean ± SD. unpaired two-tail t-test). (F) ChIP-qPCR analysis validated the increased enrichment of BACH1 under serum stimulation and the decreased enrichment of BACH1 under TGF-β1 stimulation on BACH1-targeting genes’ promoter (n = 4 independent experiments, data are mean ± SD. unpaired two-tail t-test). (G) The CNN1 luciferase reporter or a pGL3-basic luciferase reporter was co-transfected with the control vector or BACH1-Flag into HASMCs and luciferase activity was evaluated 24 h later (n = 3 independent experiments, data are mean ± SD. unpaired two-tail t-test). (H) Venn diagram showed the corresponding numbers of BACH1-binding genes analyzed by CUT&Tag, and genes whose expression was affected by BACH1 knockdown analyzed by RNA-seq.
BACH1 facilitates G9a and YAP occupancy and decreases chromatin accessibility at VSMC marker genes’ promoter
Next, we explored whether BACH1 recruits some cofactors to inhibit VSMC marker genes’ expression. Since a previous study has shown that YAP plays a crucial role in the phenotypic modulation of VSMC by inhibiting VSMC marker gene expression (51), we hypothesized that BACH1 might inhibit VSMC marker genes’ expression by facilitating the recruitment of YAP. Analysis of our BACH1 binding heatmaps and our YAP Cut&Tag data showed that YAP was highly co-occupied with BACH1 binding genes loci (Figure 6A). BACH1-bound genes were associated with chromatin remodeling. To determine whether BACH1 affects chromatin accessibility in HASMCs, we performed an assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) to assess the chromatin opening in HASMCs. We found significant colocalization of BACH1 and YAP in HASMC regions of open chromatin (Figure 6A). We have identified 6201peaks for BACH1, 15 720 peaks for YAP, and 170 635 peaks for ATAC-Seq. Furthermore, we found that 4760 enriched genes for YAP and BACH1 were overlapping, and BACH1-binding sites were strongly enriched at genes with open chromatin (Figure 6B). The TFs of bZIP family have a core DNA binding sites TGAG(C)TC. Motif analysis of ATAC-seq peaks revealed that most of the top motifs in the accessible chromatins in HASMCs are bZIP family TFs consisting of BACH1 (Online Figure IVA), suggesting BACH1 may regulate chromatin accessibility and genes expression in HASMC. H3K9me2 is reported to be enriched in the promoter of silenced genes and associated with decreased chromatin accessibility and transcriptional repression (9). To assess whether BACH1 affects the chromatin opening and the enrichment of H3K9me2 in HASMCs, we performed ATAC-seq and H3K9me2 ChIP-seq in Control (Con) or BACH1-overexpressing HASMCs (BACH1-OE). Heatmaps revealed that overexpression of BACH1 resulted in decreased chromatin accessibility, but increased binding of H3K9me2 at the promoters of target genes (Figure 6C). Similar results were also observed in the signal profiles of ATAC-seq and H3K9me2 (Figure 6D-E), while the global levels of H3K9me2 did not significantly change between Con and BACH1-OE HASMCs (Online Figure IVB). KEGG pathway analysis indicated that the genes of closed chromatin in BACH1-OE HASMCs were enriched in the pathways of the Hippo signaling pathway (Online Figure IVC). We applied qPCR to validate whether some randomly selected genes (CENPF, FOXK1, and LMNA), which were linked to decreased chromatin accessibility upon BACH1 overexpression, are regulated by BACH1. qPCR assays confirmed that BACH1 silencing significantly increased the mRNA expression of these genes (Figure 6F). Histone methyltransferase G9a has been shown to regulate H3K9me2 levels (11). To determine whether BACH1 recruits YAP and G9a to the VSMC marker gene promoters and regulates chromatin accessibility, we performed Co-immunoprecipitation (co-IP) in HASMCs and indicated that BACH1 interacted with YAP and G9a (Figure 6G). Representative snapshots of ChIP-seq tracks showed the increased enrichment of H3K9me2 and the decreased chromatin accessibility at the BACH1 binding sites of CNN1, ACTA2 and MYH11 promoters in BACH1-OE HASMCs compared to Controls (Figure 7A and Online Figure VA). Next, ChIP-qPCR further confirmed that the chromatin occupancies of YAP, G9a, and H3K9me2 near the promoters of ACTA2 and CNN1 were lower in BACH1siRNA-HASMCs than in ConsiRNA-HASMCs, and higher in AdBACH1-HASMCs than in AdGFP-HASMCs (Figure 7B–D), suggesting a critical role of BACH1 in facilitating G9a and YAP recruitment in VSMC marker genes. Moreover, serum treatment promoted the nuclear localization of BACH1 and YAP in HASMCs (Online Figure VB) and increased the enrichment of G9a and H3K9me2 near the promoters of ACTA2 and CNN1 in HASMCs (Online Figure VC-VD). We applied a small-molecule TEAD⋅YAP inhibitor (TEAD-347) to inhibit YAP-TEAD protein-protein interaction. TEAD-347 has been shown to form a covalent complex with TEAD4, to inhibit its binding to YAP, to block its transcriptional activity, and to suppress the expression of connective tissue growth factor (CTGF) (52). In our study, ChIP-qPCR assay indicated that TEAD-347 blocked the recruitment of YAP to the YAP-TEAD target gene (CTGF), but failed to block the recruitment of YAP to the promoters of ACTA2 and CNN1 in BACH1-overexpressed HASMCs (Figure 7E). These results suggest that TEAD binding is not involved in the recruitment of YAP to the promoters of these genes (i.e. ACTA2 and CNN1) in HASMCs. To determine the role of Hippo signaling in the recruitment of YAP to the same target genes, we applied a selective MST1/2 inhibitor (XMU-MP-1) to inhibit Hippo signaling in HASMCs that had been transfected with BACH1. XMU-MP-1 is known to block MST1/2 kinase activities, thereby activating the downstream effector YAP and enhancing the nuclear accumulation of YAP and TAZ (53). By ChIP-qPCR assay, we showed that XMU-MP-1 increased the recruitment of YAP to both canonical target gene (CTGF) and VSMC marker genes (i.e. ACTA2 and CNN1) under baselines conditions, and BACH1 overexpression facilitated the recruitment of YAP to these target genes when the cells were cultured with XMU-MP-1(Figure 7E). This experiment indicates that the recruitment of YAP by BACH1 is increased when Hippo signaling is inactive. Together, these results suggest that BACH1 may facilitate the recruitment of G9a and YAP at VSMC marker genes and decrease chromatin accessibility.
Figure 6.
BACH1 facilitates G9a and YAP occupancy and decreases chromatin accessibility at VSMC marker genes’ promoters. (A) Heatmaps showed the genome-wide CUT&Tag binding profiles of BACH1 and YAP, ATAC-seq of opening chromatin and at TSS (TSS ± 5 kb). (B) Venn diagram of genes enriched by BACH1, genes enriched by YAP, and genes with opening of the chromatin. (C) Heatmaps of H3K9me2 and opening chromatin at BACH1 enriched regions in Con- and BACH1-overexpressing (BACH1-OE) HASMCs (peak center ± 4kb for H3K9me2 and peak center ± 3kb for ATAC-seq). (D) Signal profiles of ATAC-seq signal at BACH1 enriched regions in Con and BACH1-OE HASMCs. (E) Signal profiles of H3K9me2 with ChIP-seq signal at BACH1 enriched regions in Con and BACH1-OE HASMCs. (F) The mRNA expression of CENPF, FOXK1, LMNA, and BACH1 in HASMCs. RT-PCR showed that CENPF, FOXK1, and LMNA mRNA levels were increased in BACH1 silencing HASMCs compared with the control HASMCs (n = 4 independent experiments, data are mean ± SD. unpaired two-tail t-test). (G) Co-immunoprecipitation of BACH1, YAP and G9a in HASMCs. BACH1 was immunoprecipitated from HASMCs; the amount of YAP and G9a present in the precipitate was evaluated via Western blot.
Figure 7.

BACH1 facilitates G9a and YAP occupancy at VSMC marker genes’ promoter. (A) Representative snapshots of H3K9me2 and ATAC-seq in Con and BACH1-OE HASMCs at the promoter of ACTA2 and CNN1. (B) ChIP-qPCR analysis validated the decrease of H3K9me2 and G9a in BACH1-silencing HASMCs at the promoter of ACTA2 and CNN1 (n = 5 independent experiments, data are mean ± SD. unpaired two-tail t-test). (C) ChIP-qPCR analysis validated the increase of H3K9me2 and G9a in BACH1-OE HASMCs at the promoter of ACTA2 and CNN1 (n = 5 independent experiments, data are mean ± SD. unpaired two-tail t-test). (D) ChIP-qPCR analysis validated the decrease of YAP in BACH1-silencing HASMCs and the increase of YAP in BACH1-OE HASMCs at the promoter of ACTA2 and CNN1 (n = 4 independent experiments, data are mean ± SD. unpaired 2-tail t-test). (E) ChIP-qPCR analysis validated the increase of YAP in BACH1-OE HASMCs at the promoter of ACTA2 and CNN1, when were treated with the TEAD⋅YAP inhibitor (TEAD-347) or MST1/2 inhibitor (XMU-MP-1), CTGF was used as a positive control (n = 4 independent experiments, data are mean ± SD. One-way ANOVA followed by Tukey post-hoc tests).
G9a and YAP are crucial for BACH1-induced repression of VSMC marker genes
G9a and Yap gene expression was also upregulated at day 3, then declined from day 7 to day 28 after wire injury in the mouse femoral artery (Online Figure VIA). We then investigated whether BACH1 repressed VSMC marker genes expression by G9a and YAP in HASMCs. In BACH1-overexpressing HASMCs we observed that the mRNA and/or protein levels of ACTA2 and CNN1 declined. Moreover, this phenomenon was abolished by the use of G9a inhibitor BIX-01294 and G9a, or YAP silencing (Figure 8A–C, Online Figure VIB–VID), indicating that G9a and YAP are crucial for BACH1-induced repression of VSMC marker genes.
Figure 8.

G9a and YAP are crucial for BACH1-induced repression of VSMC marker genes. (A) Immunoblotting showed that BACH1-induced decreases of ACTA2 and CNN1 protein were abolished by the G9a inhibitor BIX-01294 (n = 4 independent experiments, data are mean ± SD. One-way ANOVA followed by Tukey post-hoc tests). (B) Immunoblotting showed that BACH1-induced decreases of ACTA2 and CNN1 protein were abolished by silencing of G9a. Quantitative and statistical analysis are shown in Online Figure VIB. (n = 4 independent experiments, data are mean ± SD. One-way ANOVA followed by Tukey post-hoc tests). (C) Immunoblotting showed that BACH1-induced decreases of ACTA2 and CNN1 protein were abolished by silencing of YAP. Quantitative and statistical analysis are shown in Online Figure VIB (n = 3 independent experiments, data are mean ± SD. One-way ANOVA followed by Tukey post-hoc tests). (D) Schematic review showed that BACH1 suppresses the VSMC contractile phenotype and regulates the chromatin accessibility by the recruitment of G9a and YAP and maintains the H3K9me2 state on VSMC marker genes’ promoters.
DISCUSSION
A previous study demonstrated that global knockout of Bach1 in mice attenuated SMC proliferation and neointimal formation, independently of HO-1 (54). The underlying mechanism of how BACH1 in VSMCs contributes in VSMCs to neointima formation following vascular injury remains unclear. In this study, we have identified an essential role of BACH1 in suppressing the VSMC contractile phenotype and maintaining the enrichment of H3K9me2 by the recruitment of G9a and YAP, and thus reduced chromatin accessibility on promoters of VSMC marker genes such as ACTA2 and CNN1 (Figure 8D). VSMC-specific loss of Bach1 in mice inhibited the transformation of VSMC from contractile to synthetic phenotype and VSMC proliferation and attenuated the neointimal hyperplasia in wire-injured femoral arteries. Our findings provide crucial insights into the regulatory role of BACH1 in the VSMC phenotypic transition and vascular homeostasis.
Recently, the CAD-associated genetic variant rs73193808 has been identified to have allele-specific enhancer activity in HASMCs (46). Rs73193808 is the nearest CAD SNP to the BACH1 gene observed, so we explored the association between rs73193808 and BACH1. We identified that the site containing rs73193808 was co-accessible with the BACH1 promoter in VSMCs, suggesting a potential correlation of this SNP and the expression of BACH1 in atherosclerotic lesions. Moreover, we observed the high transcriptional factor activity of BACH1 in VSMCs of atherosclerotic arteries, which is in accordance with the increased expression of BACH1 of VSMCs in human coronary atherosclerotic lesions. Mechanistically, we observed an increased BACH1 expression in the injured femoral arteries of mice, and BACH1 was induced and translocated into the nucleus in HASMCs upon serum stimulation. The reasons for the increase of BACH1 in response to serum have yet to be determined. One possibility is that serum may induce the expression of SP1 (55), which is an activator for BACH1 (56). Our previous study described the association of the CAD-linked risk variant rs2832227 with BACH1 gene expression in carotid plaques from patients. In line with this notion, endothelial BACH1 was an essential inducer of vascular inflammation and atherogenesis (21). Both our studies focusing previously on endothelial cells and now on VSMCs support a link between BACH1 and atherosclerosis-related diseases.
Chromatin accessibility is critical for controlling cell identity and regulating the activity of super-enhancers and promoters (57). YAP was shown to partially reprogram chromatin accessibility and induce a more proliferative state in adult cardiomyocytes (58). G9a plays a vital role in maintaining cell identity during differentiation (13). H3K9me2 has been documented to be involved in regulating the proinflammatory VSMC phenotype (59). However, how the transcription factors and histone modification regulate chromatin accessibility to maintain VSMC identity is largely unknown. A recent study of our group shows that BACH1 recruits the essential pluripotency regulator NANOG and the MLL/SET1 complexes to maintain the activity of promoters and (super-) enhancers in embryonic stem cells (50). Here we identified that BACH1 binds G9a which is another member of SET domain-containing family. Once G9a is recruited to the promoters of VSMC marker genes, the state of enriched H3K9me2 at the promoters of these genes is maintained. This state suppresses chromatin accessibility and the transcription of these genes. Thus, BACH1 serves as a critical regulator in modulating the chromatin accessibility and dedifferentiated phenotype of VSMCs during VSMC phenotype switching.
YAP is a key effector molecule downstream of the Hippo signaling pathway and the nuclear accumulation of YAP is regulated by Hippo signaling pathway. YAP was shown to attenuate the differentiation of VSMC from cardiovascular progenitor cells (60) and to promote VSMC proliferation (61), but whether YAP directly regulates VSMC marker gene expression in VSMCs is still unknown. In the current study, we demonstrated that YAP was recruited by BACH1 to the promoters of VSMC marker genes ACTA2 and CNN1, leading to their decrease expression, and the recruitment of YAP was regulated by Hippo signaling in HASMCs. TEADs are reported to interact with YAP to regulate gene transcription (62). However, a previous study has shown that STAT3 recruits YAP independent of TEADs in endothelial cells (63). Similarly to what was described for STAT3, we also found in HASMCs that BACH1 recruited YAP to the promoters of ACTA2 and CNN1 independently of TEADs in HASMCs. Thus, these results suggest that BACH1 is essential for the recruitment of YAP to the promoters of target genes in HASMCs. Our recently published investigations suggest that BACH1 activates the expression of YAP and its recruitment to the promoter of inflammatory genes, leading to the increase expression of these genes (21). The different regulatory roles of BACH1 in ECs and VSMCs may be depending on recruiting distinct cofactors (coactivators or corepressors) that can activate or repress gene expression. Thus, BACH1 may exert context-dependent roles as a repressor or activator of gene expression.
The effect of BACH1 on cell proliferation seems to differ profoundly among cell types. Unlike the effect on VSMCs shown here, we previously reported that BACH1 deficiency increased EC proliferation and migration (19). Endothelial regeneration reduces the risk of restenosis after endothelial denudation injury associated with coronary artery interventions. Thus, loss of BACH1 might reduce neointimal formation through at least two mechanisms. One is mediated by maintaining the differentiated VSMC phenotype and the antiproliferative effect in VSMCs, another probably involves promoting endothelial proliferation and reendothelialization. The reasons for the differences in the proliferation between VSMCs and ECs are not clear, but they may be due to differences in cofactors and chromatin accessibility in different cell types.
Taken together, the results presented here show that BACH1 serves as a regulator of VSMC phenotype switching and vascular neointima lesion formation. BACH1 is essential to regulate chromatin accessibility by recruiting G9a and YAP to the promoter of VSMC marker genes and by maintaining the di-methylated state of H3K9, which contribute to a dedifferentiated phenotype of VSMCs. The critical role of BACH1 in neointimal formation that we undercover here, set the stage for the development of protective strategies targeting BACH1 against atherosclerotic cardiovascular disease onset and progression and in-stent restenosis.
DATA AVAILABILITY
The RNAseq, CUT&Tag, ATAC-seq, and ChIP-seq of H3K9me2 raw and processed data were deposited in the Gene Expression Omnibus (GEO) repository (GSE184391).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Yuzhen Zhang (Shanghai East Hospital, Tongji University School of Medicine) for providing Myh11-CreERT2 mice and Dr Faxing Yu (Fudan University, Shanghai) for providing the YAP shRNA plasmid. We appreciate Yue Qin and Xianlong Ng for providing editorial assistance.
Author contributions: M.D., W.X.H. and J.Q. conceived and designed the project. G.J.Y., Q.J.J., J.M.P., L.Q.H., W.X.X., L.L.L., P.Q., J.J.Y., G.F., M.S.Y., H.Y. Q., L.J.Y., L.Y.B., M.J.H. and J.N. performed experiments and analyzed the data. M.D. and G.J.Y wrote the manuscript. Z.X.L., J.L.D., Z.J.Y. and E.O. provided valuable comments and interpreted the results. M.D. supervised the whole study.
Contributor Information
Jieyu Guo, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Jingjing Qiu, CAS Key Laboratory of Tissue Microenvironment and Tumor, Shanghai Institutes of Nutrition and Health, Innovation Center for Intervention of Chronic Disease and Promotion of Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai 200031, China.
Mengping Jia, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Qinhan Li, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Xiangxiang Wei, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Liliang Li, Department of forensic medicine, School of basic medical sciences, Fudan University, Shanghai 200032, China.
Qi Pan, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Jiayu Jin, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Fei Ge, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Siyu Ma, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Yunquan He, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Jiayi Lin, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Yongbo Li, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Jinghua Ma, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Nan Jiang, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Xiuling Zhi, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Lindi Jiang, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Jianyi Zhang, Department of Biomedical Engineering, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Elena Osto, University and University Hospital Zurich, Institute of Clinical Chemistry and Swiss Federal Institute of Technology, Laboratory of Translational Nutrition Biology, Zurich, CH 8952, Switzerland.
Qing Jing, CAS Key Laboratory of Tissue Microenvironment and Tumor, Shanghai Institutes of Nutrition and Health, Innovation Center for Intervention of Chronic Disease and Promotion of Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai 200031, China.
Xinhong Wang, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
Dan Meng, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Department of Rheumatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Great Program [92068202, 82220108020, 92168206] of the National Natural Science Foundation of China, Program of Shanghai Academic/Technology Research Leader [20XD1400600]; Shanghai Science and Technology Commission of China [22QA1408900]; National Key Research and Development Program of China [2018YFC2000202]; Young Elite Scientist Sponsorship Program by CAST [2021QNRC001]. Funding for open access charge: Great Program [92068202] of the National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Basatemur G.L., Jorgensen H.F., Clarke M.C.H., Bennett M.R., Mallat Z.. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019; 16:727–744. [DOI] [PubMed] [Google Scholar]
- 2. Owens G.K., Kumar M.S., Wamhoff B.R.. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004; 84:767–801. [DOI] [PubMed] [Google Scholar]
- 3. Bennett M.R., Sinha S., Owens G.K.. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016; 118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Denny S.K., Yang D., Chuang C.H., Brady J.J., Lim J.S., Gruner B.M., Chiou S.H., Schep A.N., Baral J., Hamard C.et al.. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell. 2016; 166:328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Klemm S.L., Shipony Z., Greenleaf W.J.. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019; 20:207–220. [DOI] [PubMed] [Google Scholar]
- 6. Nagao M., Lyu Q., Zhao Q.Y., Wirka R.C., Bagga J., Nguyen T., Cheng P., Kim J.B., Pjanic M., Miano J.M.et al.. Coronary disease-associated gene TCF21 inhibits smooth muscle cell differentiation by blocking the myocardin-serum response factor pathway. Circ. Res. 2020; 126:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim J.B., Zhao Q.Y., Nguyen T., Pjanic M., Cheng P., Wirka R., Travisano S., Nagao M., Kundu R., Quertermous T.. Environment-snsing aryl hydrocarbon receptor inhibits the chondrogenic fate of modulated smooth muscle cells in atherosclerotic lesions. Circulation. 2020; 142:575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Etchegaray J.P., Mostoslavsky R.. Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol. Cell. 2016; 62:695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tachibana M., Sugimoto K., Nozaki M., Ueda J., Ohta T., Ohki M., Fukuda M., Takeda N., Niida H., Kato H.et al.. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Gene Dev. 2002; 16:1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hou Q.Q., Xiao Q., Sun X.Y., Ju X.C., Luo Z.G.. TBC1D3 promotes neural progenitor proliferation by suppressing the histone methyltransferase G9a. Sci. Adv. 2021; 7:eaba8053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shinkai Y., Tachibana M.. H3K9 methyltransferase G9a and the related molecule GLP. Gene Dev. 2011; 25:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma W.B., Han C., Zhang J.Q., Song K., Chen W.N., Kwon H., Wu T.. The histone methyltransferase G9a promotes cholangiocarcinogenesis through regulation of the hippo pathway kinase LATS2 and YAP signaling pathway. Hepatology. 2020; 72:1283–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Antignano F., Burrows K., Hughes M.R., Han J.M., Kron K.J., Penrod N.M., Oudhoff M.J., Wang S.K.H., Min P.H., Gold M.J.et al.. Methyltransferase G9A regulates T cell differentiation during murine intestinal inflammation. J. Clin. Invest. 2014; 124:1945–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Greissel A., Culmes M., Napieralski R., Wagner E., Gebhard H., Schmitt M., Zimmermann A., Eckstein H.H., Zernecke A., Pelisek J.. Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thromb. Haemostasis. 2015; 114:390–402. [DOI] [PubMed] [Google Scholar]
- 15. Yang Q., Lu Z., Singh D., Raj J.U.. BIX-01294 treatment blocks cell proliferation, migration and contractility in ovine foetal pulmonary arterial smooth muscle cells. Cell Proliferat. 2012; 45:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Warnatz H.J., Schmidt D., Manke T., Piccini I., Sultan M., Borodina T., Balzereit D., Wruck W., Soldatov A., Vingron M.et al.. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J. Biol. Chem. 2011; 286:23521–23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang X.Y., Guo J.Y., Wei X.X., Niu C., Jia M.P., Li Q.H., Meng D.. Bach1: function, regulation, and involvement in disease. Oxid Med Cell Longev. 2018; 2018:1347969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang L., Yin M., Xu J., Jia M., Sun S., Wang X., Zhang J., Meng D.. The transcription factor Bach1 suppresses the developmental angiogenesis of zebrafish. Oxid Med. Cell Longev. 2017; 2017:2143875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang L., Yin M., Wei X.X., Liu J.X., Wang X.H., Niu C., Kang X.L., Xu J., Zhou Z.W., Sun S.Y.et al.. Bach1 represses Wnt/beta-catenin signaling and Angiogenesis. Circ. Res. 2015; 117:364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van der Harst P., Verweij N.. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ. Res. 2018; 122:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jia M.P., Li Q.H., Guo J.Y., Shi W.H., Zhu L., Huang Y.J., Li Y.B., Wang L., Ma S.Y., Zhuang T.et al.. Deletion of BACH1 attenuates atherosclerosis by reducing endothelial inflammation. Circ. Res. 2022; 130:1038–1055. [DOI] [PubMed] [Google Scholar]
- 22. Wirth A., Benyo Z., Lukasova M., Leutgeb B., Wettschureck N., Gorbey S., Orsy P., Horvath B., Maser-Gluth C., Greiner E.et al.. G(12)-G(13)-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 2008; 14:64–68. [DOI] [PubMed] [Google Scholar]
- 23. Wang M.A., Ihida-Stansbury K., Kothapalli D., Tamby M.C., Yu Z., Chen L.H., Grant G., Cheng Y., Lawson J.A., Assoian R.K.et al.. Microsomal prostaglandin E-2 synthase-1 modulates the response to vascular injury. Circulation. 2011; 123:631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Litovchick L. Resolving proteins for immunoblotting by gel electrophoresis. Cold Spring Harb. Protoc. 2018; 2018: 10.1101/pdb.prot098434. [DOI] [PubMed] [Google Scholar]
- 25. Kim D., Landmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L.. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012; 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Binns D., Dimmer E., Huntley R., Barrell D., O’Donovan C., Apweiler R.. QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics. 2009; 25:3045–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M.. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016; 44:D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sherman B.T., Hao M., Qiu J., Jiao X.L., Baseler M.W., Lane H.C., Imamichi T., Chang W.Z.. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022; 50:W216–W221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shen H.J., Xu W.Q., Guo R., Rong B.W., Gu L., Wang Z.T., He C.X., Zheng L.J., Hu X., Hu Z.et al.. Suppression of enhancer overactivation by a RACK7-histone demethylase complex. Cell. 2016; 165:331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Feng J.X., Liu T., Qin B., Zhang Y., Liu X.S.. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012; 7:1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laurent B., Ruitu L., Murn J., Hempel K., Ferrao R., Xiang Y., Liu S.C., Garcia B.A., Wu H., Wu F.Z.et al.. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol. Cell. 2015; 57:957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B.et al.. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017; 14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kaya-Okur H.S., Janssens D.H., Henikoff J.G., Ahmad K., Henikoff S.. Efficient low-cost chromatin profiling with CUT&Tag. Nat. Protoc. 2020; 15:3264–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y.H., Stoeckius M., Smibert P., Satija R.. Comprehensive integration of single-cell data. Cell. 2019; 177:1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McGinnis C.S., Murrow L.M., Gartner Z.J.. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019; 8:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S.Q., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L.. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014; 32:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holland C.H., Tanevski J., Perales-Paton J., Gleixner J., Kumar M.P., Mereu E., Joughin B.A., Stegle O., Lauffenburger D.A., Heyn H.et al.. Robustness and applicability of transcription factor and pathway analysis tools on single-cell RNA-seq data. Genome Biol. 2020; 21:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schep A.N., Wu B.J., Buenrostro J.D., Greenleaf W.J.. chromVAR : inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017; 14:975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pliner H.A., Packer J.S., McFaline-Figueroa J.L., Cusanovich D.A., Daza R.M., Aghamirzaie D., Srivatsan S., Qiu X.J., Jackson D., Minkina A.et al.. Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility Data. Mol. Cell. 2018; 71:858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Crowe A.R., Yue W.. Semi-quantitative determination of protein expression using immunohistochemistry staining and analysis: an integrated protocol. Bio-Protocol. 2019; 9:e3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ord T., Ounap K., Stolze L.K., Aherrahrou R., Nurminen V., Toropainen A., Selvarajan I., Lonnberg T., Aavik E., Yla-Herttuala S.et al.. Single-cell epigenomics and functional fine-mapping of atherosclerosis GWAS loci. Circ. Res. 2021; 129:240–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranasic D.et al.. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020; 48:D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wirka R.C., Wagh D., Paik D.T., Pjanic M., Nguyen T., Miller C.L., Kundu R., Nagao M., Coller J., Koyano T.K.et al.. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019; 25:1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bae Y.H., Mui K.L., Hsu B.Y., Liu S.L., Cretu A., Razinia Z., Xu T.N., Pure E., Assoian R.K.. A FAK-Cas-Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci. Signal. 2014; 7:ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Niu C., Wang S.Q., Guo J.Y., Wei X.X., Jia M.P., Chen Z.X., Gong W.X., Qin Y., Wang X.H., Zhi X.L.et al.. BACH1 recruits NANOG and histone H3 lysine 4 methyltransferase MLL/SET1 complexes to regulate enhancer-promoter activity and maintains pluripotency. Nucleic Acids Res. 2021; 49:1972–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang X.B., Hu G.Q., Gao X.W., Wang Y., Zhang W., Harmon E.Y., Zhi X., Xu Z.P., Lennartz M.R., Barroso M.et al.. The induction of Yes-associated protein expression after arterial injury is crucial for smooth muscle phenotypic modulation and neointima formation. Arterioscl. Throm. Vas. 2012; 32:2662–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bum-Erdene K., Zhou D.H., Gonzalez-Gutierrez G., Ghozayel M.K., Si Y.B., Xu D., Shannon H.E., Bailey B.J., Corson T.W., Pollok K.E.et al.. Small-molecule covalent modification of conserved cysteine leads to allosteric inhibition of the TEAD center dot Yap protein-protein interaction. Cell Chem Biol. 2019; 26:378–389. [DOI] [PubMed] [Google Scholar]
- 53. Fan F.Q., He Z.X., Kong L.L., Chen Q.H., Yuan Q., Zhang S.H., Ye J.J., Liu H., Sun X.F., Geng J.et al.. Pharmacological targeting of kinases MST1 and MST2 augments tissue repair and regeneration. Sci. Transl. Med. 2016; 8:352ra108. [DOI] [PubMed] [Google Scholar]
- 54. Omura S., Suzuki H., Toyofuku M., Ozono R., Kohno N., Igarashi K.. Effects of genetic ablation of bach1 upon smooth muscle cell proliferation and atherosclerosis after cuff injury. Genes Cells. 2005; 10:277–285. [DOI] [PubMed] [Google Scholar]
- 55. Wamhoff B.R., Hoofnagle M.H., Burns A., Sinha S., McDonald O.G., Owens G.K.. A G/C element mediates repression of the SM22 alpha promoter within phenotypically modulated smooth muscle cells in experimental atherosclerosis. Circ. Res. 2004; 95:981–988. [DOI] [PubMed] [Google Scholar]
- 56. Sun J.Y., Muto A., Hoshino H., Kobayashi A., Nishimura S., Yamamoto M., Hayashi N., Ito E., Igarashi K.. The promoter of mouse transcription repressor bach1 is regulated by Sp1 and trans-activated by Bach1. J. Biochem. 2001; 130:385–392. [DOI] [PubMed] [Google Scholar]
- 57. Carter B., Zhao K.J.. The epigenetic basis of cellular heterogeneity. Nat. Rev. Genet. 2021; 22:235–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Monroe T.O., Hill M.C., Morikawa Y., Leach J.P., Heallen T., Cao S.Y., Krijger P.H.L., de Laat W., Wehrens X.H.T., Rodney G.G.et al.. YAP partially reprograms chromatin accessibility to directly induce adult cardiogenesis in vivo. Dev. Cell. 2019; 48:765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Harman J.L., Dobnikar L., Chappell J., Stokell B.G., Dalby A., Foote K., Finigan A., Freire-Pritchett P., Taylor A.L., Worssam M.D.et al.. Epigenetic regulation of vascular smooth muscle cells by histone H3 lysine 9 dimethylation attenuates target gene-induction by inflammatory signaling. Arterioscl. Throm. Vas. 2019; 39:2289–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang L., Qiu P., Jiao J., Hirai H., Xiong W., Zhang J., Zhu T., Ma P.X., Chen Y.E., Yang B.. Yes-associated protein inhibits transcription of myocardin and attenuates differentiation of vascular smooth muscle cell from cardiovascular progenitor cell lineage. Stem Cells. 2017; 35:351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang Y., Hu G., Liu F., Wang X., Wu M., Schwarz J.J., Zhou J.. Deletion of yes-associated protein (YAP) specifically in cardiac and vascular smooth muscle cells reveals a crucial role for YAP in mouse cardiovascular development. Circ. Res. 2014; 114:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lin K.C., Park H.W., Guan K.L.. Regulation of the hippo pathway transcription factor TEAD. Trends Biochem. Sci. 2017; 42:862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. He J.L., Bao Q.K., Zhang Y., Liu M.M., Lv H.Z., Liu Y.J., Yao L., Li B.C., Zhang C.H., He S.et al.. Yes-associated protein promotes angiogenesis via signal transducer and activator of transcription 3 in endothelial cells. Circ. Res. 2018; 122:591–605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNAseq, CUT&Tag, ATAC-seq, and ChIP-seq of H3K9me2 raw and processed data were deposited in the Gene Expression Omnibus (GEO) repository (GSE184391).